
An ESIPT-based fluorescent switch with AIEE, solvatochromism, mechanochromism and photochromism†
Xuedan
Liu
a,
Aisen
Li
b,
Weiqing
Xu
 b,
Zhiyong
Ma
*a and
Xinru
Jia
c
b,
Zhiyong
Ma
*a and
Xinru
Jia
c
aBeijing State Key Laboratory of Organic–Inorganic Composites, College of Chemical Engineering, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: mazhy@mail.buct.edu.cn
bState Key Laboratory for Supramolecular Structure and Materials, Institute of Theoretical Chemistry, Jilin University, Changchun 130012, China
cBeijing National Laboratory for Molecular Sciences, Key Laboratory of Polymer Chemistry and Physics of the Ministry of Education, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China
First published on 30th January 2019
Abstract
In this study, we report a newly synthesized molecule 1via linking a benzothiazole to rhodamine, which could exhibit AIEE, unique acidichromism and solvatochromism in solution, fascinating photochromism in a polymer matrix, intriguing mechanochromism in solid state and remarkable piezochromism in the crystal. 1 is a rarely reported organic molecule, which is sensitive to multiple stimuli such as acid/base, force, pressure, light and solvent polarity due to its distinct intramolecular hydrogen bonding and spiro-lactam structure. 1 demonstrated AIEE property in a THF/H2O system because of the water-induced aggregation and aggregation-suppressed nonradiative relaxation of the excited states. In the solution, 1 afforded yellowish-green emission at 520 nm (keto emission) in a non-polar solvent and blue emission at 447 nm (enol emission) in a polar solvent. Surprisingly, a blue shift in the emission peak was observed in a DCM solution upon treatment with acid or base, which is quite different from that of traditional rhodamine. In the solid state, the emission color of 1 changed from yellowish-green to red upon grinding or applying hydrostatic pressure, owing to the force-induced ring-opening reaction. In the polymer matrix, 1 manifested reversible photochromic behaviour and could serve as a good photoprinting material. This study is expected to contribute to the development of multistimuli-responsive fluorescent switch with multiple functions in different states.
Introduction
Fluorescent molecular switches with tunable luminescent behavior using external stimuli,1–10 especially in their solid state,11–14 have attracted considerable attention due to their potential applications in optical data recording, displays, chemical sensors and fluorescent probes.15–29 Rhodamines and their derivatives are well known as molecular switches and have been widely studied in intelligent materials, owing to the remarkable absorption and emission change that occurs during the reversible isomerization between the closed-ring and open-ring forms.30,31 However, aggregation-induced quenching of rhodamines limits its applications in the solid state. Highly emissive organic fluorescent materials with multistimuli-responsive behavior in different states are in high demand. Therefore, it is of great significance to fabricate sensor materials with multiple functions to further widen the scope of application in different fields.32–37 However, reports on such materials are very rare.The photo-induced proton transfer through an intramolecular hydrogen bond is defined as excited-state intramolecular proton transfer (ESIPT).38 Organic ESIPT-based fluorescent molecules have been objects of interest in the past decades because they exhibit remarkably large Stokes shift, unique dual emission and special sensitivity to the external microenvironment due to their four-level photophysical cycles.39 Because of the transient changes involved in the ESIPT process, ESIPT emission is quite easily affected by external stimuli, resulting in the change of fluorescence properties.40–44 Therefore, making good use of the ESIPT process is an effective way to fabricate smart luminescent materials that are sensitive to external stimuli such as solvent polarity, pH, heat, mechanical force and pressure.45–50 The most studied ESIPT system consists of the derivatives of 2-(2′-hydroxyphenyl)benzimidazole (HBI), 2-(2′-hydroxyphenyl)benzoxazole (HBO) and 2-(2′-hydroxyphenyl)benzothiazole (HBT), where the hydroxyl linked to its conjugated group acts as the proton donor and the imidazole/oxazole/thiazole ring acts as the proton acceptor.51 Hence, the combination of rhodamine with ESIPT is predicted to yield a new type of multi-responsive luminescent material both in solution and solid state. Prof. Xiao-Feng Yang has reported a benzothiazole-substituted rhodamine, which exhibited ESIPT and ratiometric response towards copper ion.52 However, the study neglected the responses to other stimuli and the fluorescent properties in the solid state.
In this study, we report the synthesis of a new molecule 1 (Scheme 1) by linking a benzothiazole to rhodamine, which is shown to exhibit AIEE, unique acidichromism and solvatochromism in solution, fascinating photochromism in the polymer matrix, intriguing mechanochromism in solid state and remarkable piezochromism in the crystal due to its distinct intramolecular hydrogen bonding and spiro-lactam structure. The single crystal was developed successfully, which showed an antiparallel slip-stacked dimer structure. 1 showed AIEE property in the THF/H2O system because of the water-induced aggregation and aggregation-suppressed nonradiative relaxation ways of the excited states. In solution, 1 exhibited a yellowish-green emission at 520 nm (keto emission) in the non-polar solvent and blue emission at 447 nm (enol emission) in the polar solvent in general. Surprisingly, a blue shift and enhancement of emission were observed in the DCM solution upon treatment with acid or base, which was quite different from that of traditional rhodamine. In the solid state, on grinding or applying hydrostatic pressure, the emission color of 1 changed from yellowish-green to red owing to the force-induced ring-opening reaction. In the polymer matrix, 1 manifested a reversible photochromic behaviour and could serve as a good photo-printing material. To the best of our knowledge, 1 is a rarely reported organic molecule that is sensitive to multiple stimuli such as acid/base, force, pressure, light and solvent polarity. This study would contribute to the development of a multistimuli-responsive fluorescent switch with multiple functions in different states.
 | ||
| Scheme 1 Molecular structures of the target molecule 1. The ring-closed form undergoes ring opening on application of force. | ||
Results and discussion
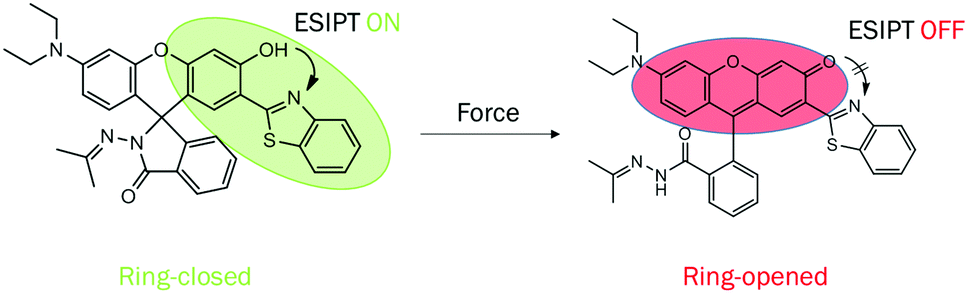
Compound 2 was acquired from 2,4-dihydroxybenzaldehyde and 2-aminobenzenethiol by a typical two-step condensation reaction, according to the literature.52 The target molecule 1 was obtained via Schiff base reaction between compound 2 and acetone (white powder, 53% yield). 1H NMR, 13C NMR, HR-MS and crystallographic analysis were performed to verify the molecular structures and product purity. The detailed synthetic procedures and characterization processes are supplied in the ESI† (Scheme S1 and Fig. S1–S3).Firstly, we studied the photophysical properties of 1 in different organic solvents. Interestingly, 1 showed unique sensitivity to different polarities of the solvents. For example, in nonpolar solvents such as cyclohexane and toluene, the emission peak of 1 was centered around 520 nm due to ESIPT facilitated by the strong intramolecular hydrogen bonding (Fig. 1a and c). With increasing polarity, another emission peak was detected at 447 nm in acetone and methanol, which was ascribed to the excited states of the enol form. It is worth mentioning that a dual-fluorescence was observed in ethanol, which suggested that both enol and keto forms coexisted in the excited states. Unexpectedly, a new emission band appeared at 457 nm only in DMSO, DMF and acetonitrile solvents. Correspondingly a new broad absorption band was located in the range of 375–450 nm in the absorption spectra (Fig. 1b), which was attributed to the phenolic anion generated via deprotonation induced by the intermolecular hydrogen bonding between the phenolic hydroxyl groups and the solvent molecules as reported previously.53 Thus, 1 remarkably exhibited triple emissions with varying solvent polarity.
 | ||
| Fig. 1 (a) Fluorescence spectra, (b) absorption spectra and (c) optical images of 1 in different organic solvents (20 μM, λex = 365 nm, from left to right: cyclohexane, toluene, DCM, THF, ethyl acetate, chloroform, acetone, acetonitrile, DMF, ethanol, methanol, DMSO). | ||
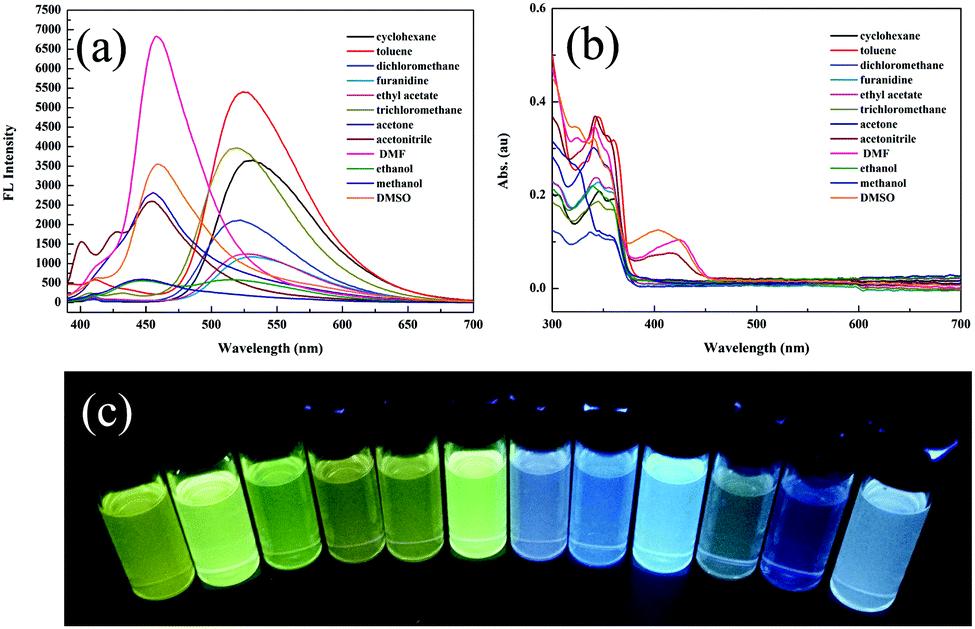
The aggregation-induced emission enhancement (AIEE) property of 1 was studied in the THF/H2O system. Interestingly, 1 exhibited AIEE behavior though it did not contain typical AIEE structures such as the TPE moiety. In the diluted THF solution of 1, weak emission bands were detected at 405 nm and 430 nm (Fig. 2a and c), which were attributed to the xanthene moiety. The typical emission band ascribed to ESIPT was of low intensity with a low quantum yield of 2.78% due to the free rotation of thiazole ring. The fluorescent behavior of 1 changed with the water fraction in the system. The target molecule 1 began to aggregate and a yellow emission centered at 520 nm emerged when the water proportion (fw) reached 70%. The emissive intensity increased continuously with the water proportion. When fw was above 90%, 1 was highly emissive at 520 nm with a high quantum yield of 20.79% because the aggregation of 1 molecules prohibited the nonradiative relaxation channels and facilitated the ESIPT process. With increasing water proportion, the absorption bands red-shifted on account of Mie scattering effect (Fig. 2b), which further verified the aggregation process of 1. These results reveal that 1 is AIEE active.
 | ||
| Fig. 2 (a) Fluorescence spectra, (b) absorption spectra and (c) optical images of 1 in different THF/water (v/v) mixtures (20 μM, λex = 365 nm). | ||
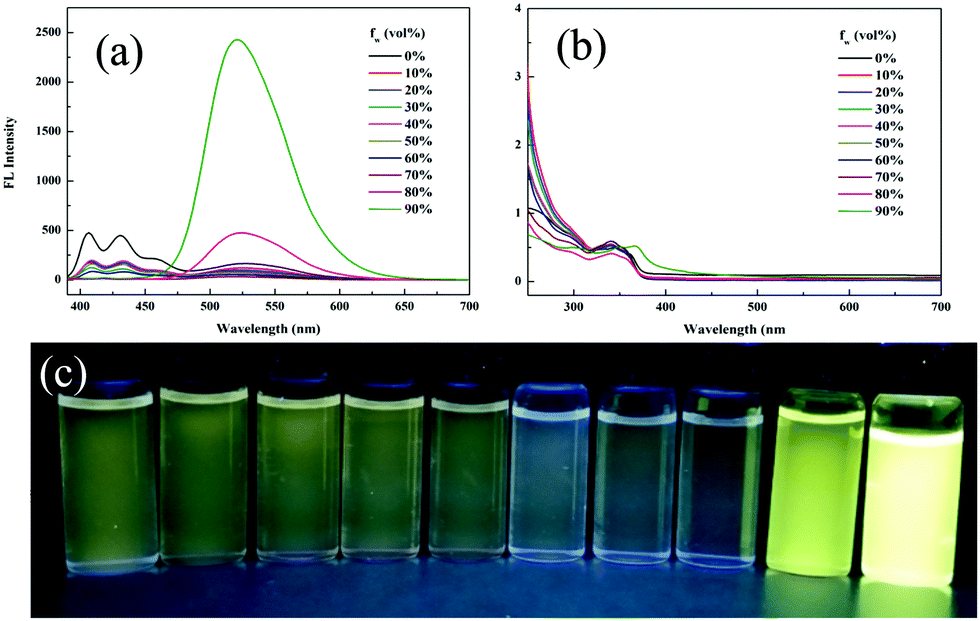
The reversible ring-opening/closing isomerization of 1 triggered by acid and base was investigated in dichloromethane (DCM) solution. In contrast to rhodamine lactam, the fluorescence spectra of the fluorescent switch 1 showed a blue shift upon stimulation by acid or base. In the original DCM solution, the emission peak was located at 520 nm due to ESIPT. On addition of 0.01 M NaOH ethanol solution, the DCM solution turned red under visible light and changed to bright blue under 365 nm UV light, unexpectedly. Two new absorption bands at 385 nm and 565 nm were detected (Fig. 3a and Fig. S4, ESI†), which confirmed the deprotonation process and the ring-opening reaction. The new emission peak intensity at 457 nm gradually strengthened with the addition of base, which was quite similar to that in DMSO and provided solid evidence that it corresponded to the phenolic anion. As for the acid stimulus, the DCM solution turned red under visible light and the emission red-shifted from 520 nm to 477 nm and further to 415 nm with an enhancement in the intensity on increasing the volume of TFA (Fig. 3b and Fig. S5, ESI†). Three new absorption bands were detected at 370 nm, 508 nm and 546 nm (Fig. S5, ESI†), thus indicating the occurrence of the ring-opening reaction. In acidic condition, the 477 nm and 415 nm emissions could probably be ascribed to the enol forms. It is worth mentioning that this is the first rhodamine derivative to show both emission blue shift and emission enhancement when induced with an acid or base.
 | ||
| Fig. 3 Fluorescence spectra of 1 after adding (a) 0.01 M NaOH ethanol solution and (b) TFA. | ||
Tuning the fluorescent behavior of solid-state materials through external stimuli has been in the limelight in recent years. 1 displayed reversible mechanochromic behavior in the solid state. The pristine powder was obtained after removing the solvent completely. Initially, it showed an emission peak at 520 nm with yellowish-green color (QY: 46.12%, τ = 4.27 ns) (Fig. 4a) due to ESIPT. Even after slight shearing, the emission peak at 520 nm was retained and no shift was observed. We conducted XRD and DSC to gain insight into the microstructural changes after applying force. For the pristine powder, numerous sharp scattering peaks were detected, indicating that the pristine powder was highly crystalline. After slight shearing, the scattering peaks disappeared (Fig. 4c), implying a force-induced phase transition from ordered stacking to an amorphous state. In the DSC curves, there were two sharp endothermic peaks at 172 °C (assigned to the solid-to-solid transition) and 258 °C (melting point) for the pristine powder (Fig. S6, ESI†); however, only a glass transition process was observed for the ground powder, verifying that the crystal structure was damaged by external force. The above results disclosed that the ESIPT-based emission was independent of the physical molecular stacking and relied totally on the strong intramolecular hydrogen bonding in this system. Further shearing endowed the powder with a reddish emission at 600 nm, which was assigned to the ring-open form of 1. We tested the solid-state absorption spectra of the ground red powder, which showed a broad absorption band at 590 nm (Fig. S7, ESI†), thus verifying the ring-open form of compound 1. After heavy grinding, the QY decreased drastically to 10.12% and the lifetime was 2.08 ns, which implied that the ESIPT process was prohibited by the shearing force. After annealing, the reddish color powder regained the original yellowish-green color. In addition, no scattering peaks was observed in the XRD spectra (Fig. S8, ESI†), suggesting that the ring-closed molecule structure of 1 could be recovered by heating but the ground sample could not regain the original crystal state by heating.
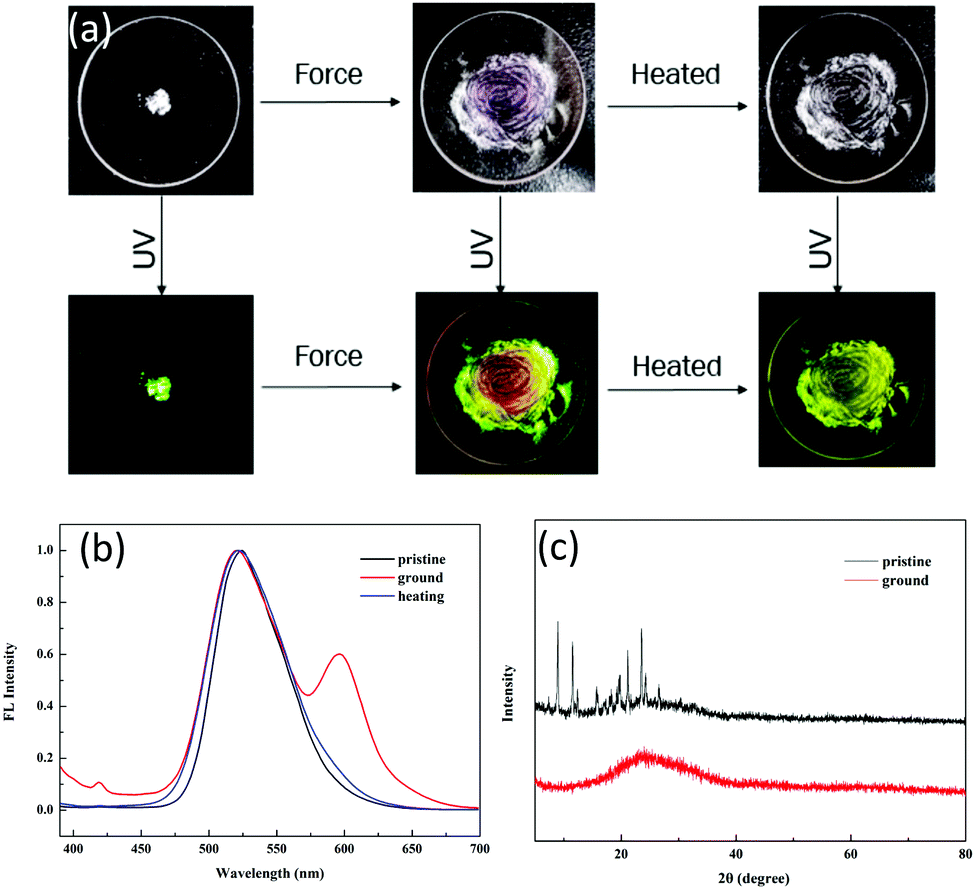 | ||
| Fig. 4 (a) Optical images of the color-changing process of 1 due to shearing and heating under room light and 365 UV light. (b) Fluorescence spectra of the pristine powder, powder after heavy grinding and the heated powder. (c) XRD patterns of the pristine powder and powder after slight grinding. | ||
In order to dynamically monitor the color-changing process in situ and quantify the relationship between the emission transformation and the external force, single crystals of 1 were subjected to hydrostatic pressure in a DAC equipment, where silicone oil was used as the pressure transmitting medium. Notably, it exhibited a clear color change when a pressure was applied. The original single crystal had bright green emission at 520 nm under 365 nm UV light due to the ESIPT process. When the pressure was increased to 10.08 GPa, the single crystal turned dark-red with an emission peak at 590 nm (Fig. 5a and b), which was due to the pressure-induced ring-opening reaction of the spiro-lactam. At the same time, the emission wavelength red-shifted gradually (Fig. S9, ESI†) and the fluorescence intensity weakened correspondingly. Once the pressure was released, the single crystal could be restored to its original green color, indicating that the ring-open species of 1 recovered to the ring-closed form (Fig. 5c). Hence, the pressure-responsive emission color change was completely reversible.
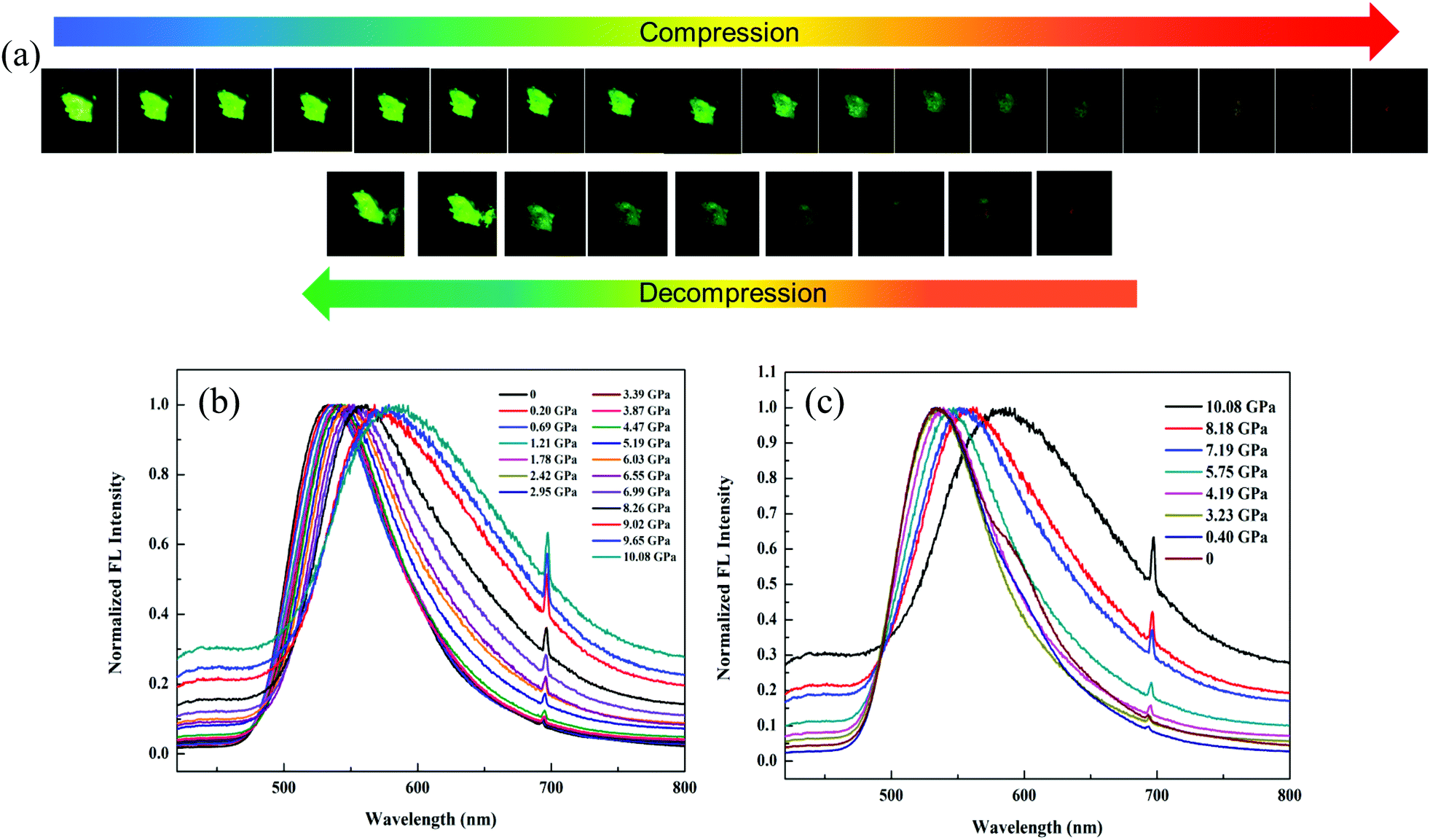 | ||
| Fig. 5 (a) In situ fluorescence images of a single crystal of 1 during the compression and decompression process (λex = 365 nm); in situ fluorescence spectra (λex = 365 nm) during the (b) compression and (c) decompression process (λex = 365 nm). | ||
To further explore the interdependence of the molecular structure and spectroscopic behavior, and gain insights into the mechanochromic mechanism, the single crystal structure of 1 was analyzed (Table S1, ESI†). Interestingly, the single crystals of 1 were obtained accidentally during a trial to cultivate single crystals of 2 in acetone. The single crystal belongs to the monoclinic crystal system with the space group of P 21/c (No. 14). Each unit cell contained four molecules (Fig. S10, ESI†). The rhodamine moiety was highly twisted due to the spiro-carbon atom. The HBT unit and the phenol part were located almost in the same plane with a small dihedral angle of 7.77° (Fig. 6a), owing to the strong intramolecular hydrogen bonding between the –OH group and the N atom, which provides direct evidence for the ESIPT process. Interestingly, the single crystal of 1 adopted an antiparallel slip-stacked dimer structure (3.35 Å) with a considerable offset by intermolecular π–π interactions (Fig. 6b). In the single crystal, one molecule interacted with at least seven adjacent molecules via the π–π interactions, C–H⋯π and C–H⋯H–C interactions, which suggests that the molecule is highly constrained in the crystal state. When subjected to high pressure, the pressure would be conducted to the spiro-lactam structure and result in the ring-opening reaction.
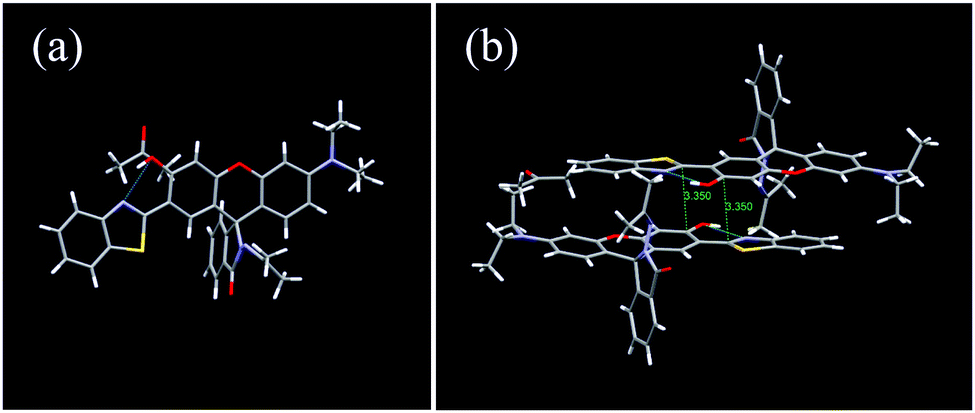 | ||
| Fig. 6 (a) Monomer and (b) antiparallel slip-stacked dimer unit with close contacts in the single crystal of 1. | ||
Generally, the rhodamine amide can undergo a UV light-promoted heterolytic bond fissure to form a normal colored zwitterionic architecture. However, its very short lifetime in the ring-open state limits its application as a photochromic molecule. In this system, 1 displayed very fine photochromic property in the matrix. Firstly, we distributed the powder of 1 on a piece of filter paper by immersing in the THF solution (Fig. S11, ESI†). The treated filter paper exhibited fascinating photo-activity with a distinct color change from yellow to red. The fluorescence spectra showed that a new emission peak emerged at 595 nm after UV irradiation (Fig. S12, ESI†). Secondly, we prepared a polyurethane film containing 1 in the weight ratio of 1%. The PU film demonstrated reversible light-patterning behaviour and was able to serve as a material for photo-printing. Initially, we covered the PU film with a butterfly pattern and then positioned the film under 365 UV light. As a result, a red butterfly was visualized in the film (Fig. 7). When the film was placed on a hot-plate at 150 °C, the red-colored pattern disappeared without any trace in five minutes. The UV light-induced emission “green-red” switch could be repeated for multiple cycles, implying fast photo-reversibility and good photo-stability of 1. After annealing, the film was covered with a new pattern and positioned again under UV light. Consequently, the new pattern appeared clearly, suggesting the easy “writing–erasing” nature of the photochromic PU film. This operation could be cycled several times. Fig. 7 shows the pictures of the same film patterned repeatedly through three cycles of UV light irradiation and heating treatment.
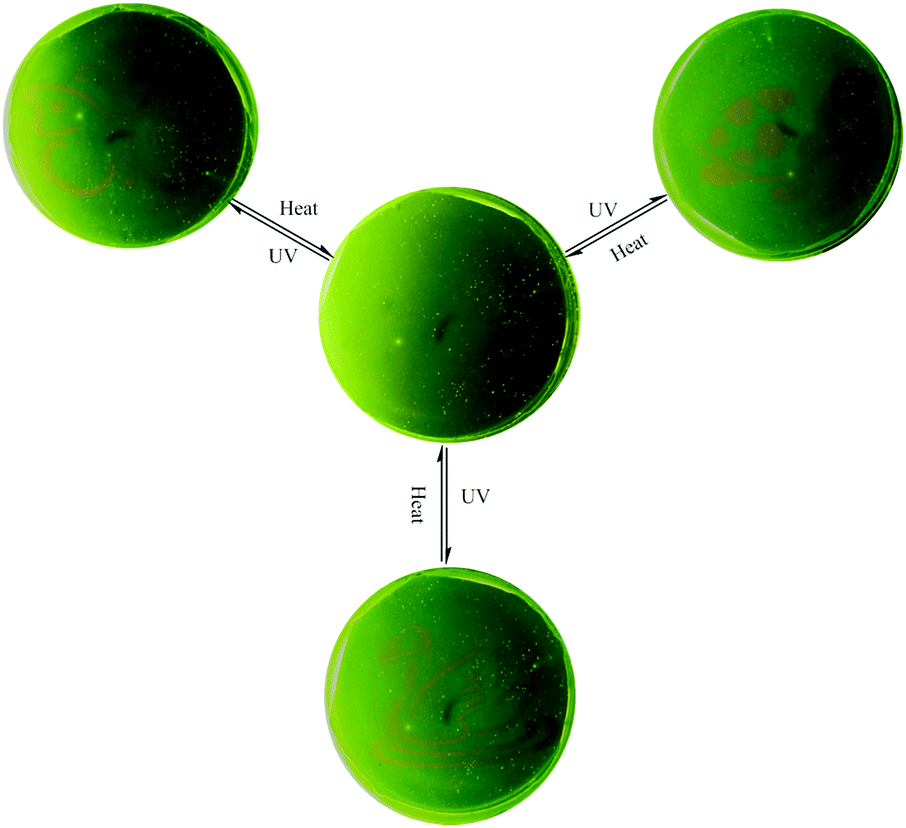 | ||
| Fig. 7 Three photo-printing cycles of UV light irradiation and heating treatment are demonstrated with three different patterns (butterfly, tortoise and swan) printed on the same PU film (containing 1 wt% of 1). | ||
Conclusions
In conclusion, we have reported a new molecule 1 synthesized by fusing rhodamine with HBT, which successfully combines the properties of both molecules and exhibits new properties. 1 is endowed with multiple features such as AIEE, acidichromism, solvatochromism, mechanochromism, piezochromism and photochromism, owing to its distinct intramolecular hydrogen bonding and spiro-lactam structure. 1 is an unusual organic molecule that responds differently to multiple external stimuli and can lay the foundation for the preparation of smart materials. This study also provides a new strategy to exploit a broad perspective for the development of multistimuli-responsive fluorescent switch.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was financially supported by the National Natural Science Foundation of China (21704002), the Natural Science Foundation of Beijing Municipality (2182054) and the Fundamental Research Funds for the Central Universities (buctrc201723&ZY1701) to Z. Y. Ma.Notes and references
- Z. Chi, X. Zhang, B. Xu, X. Zhou, C. Ma, Y. Zhang, S. Liu and J. Xu, Chem. Soc. Rev., 2012, 41, 3878–3896 RSC.
- Z. Ma, Z. Wang, M. Teng, Z. Xu and X. Jia, ChemPhysChem, 2015, 16, 1811–1828 CrossRef CAS PubMed.
- A. Lavrenova, D. W. R. Balkenende, Y. Sagara, S. Schrettl, Y. C. Simon and C. Weder, J. Am. Chem. Soc., 2017, 139, 4302–4305 CrossRef CAS PubMed.
- K. Nagura, S. Saito, H. Yusa, H. Yamawaki, H. Fujihisa, H. Sato, Y. Shimoikeda and S. Yamaguchi, J. Am. Chem. Soc., 2013, 135, 10322–10325 CrossRef CAS PubMed.
- Q. Qi, C. Li, X. Liu, S. Jiang, Z. Xu, R. Lee, M. Zhu, B. Xu and W. Tian, J. Am. Chem. Soc., 2017, 139, 16036–16039 CrossRef CAS PubMed.
- T. Seki, N. Tokodai, S. Omagari, T. Nakanishi, Y. Hasegawa, T. Iwasa, T. Taketsugu and H. Ito, J. Am. Chem. Soc., 2017, 139, 6514–6517 CrossRef CAS.
- M. Tanioka, S. Kamino, A. Muranaka, Y. Ooyama, H. Ota, Y. Shirasaki, J. Horigome, M. Ueda, M. Uchiyama, D. Sawada and S. Enomoto, J. Am. Chem. Soc., 2015, 137, 6436–6439 CrossRef CAS PubMed.
- Y. Sagara and T. Kato, Nat. Chem., 2009, 1, 605–610 CrossRef CAS PubMed.
- L. Huang, C. Wu, L. Zhang, Z. Ma and X. Jia, ACS Appl. Mater. Interfaces, 2018, 10, 34475–34484 CrossRef CAS PubMed.
- C. Wang and Z. Li, Mater. Chem. Front., 2017, 1, 2174–2194 RSC.
- Z. Ma, X. Meng, Y. Ji, A. Li, G. Qi, W. Xu, B. Zou, Y. Ma, G.-C. Kuang and X. Jia, Dyes Pigm., 2019, 162, 136–144 CrossRef CAS.
- A. Li, Z. Ma, J. Wu, P. Li, H. Wang, Y. Geng, S. Xu, B. Yang, H. Zhang, H. Cui and W. Xu, Adv. Opt. Mater., 2018, 6, 1700647 CrossRef.
- Y. Liu, Q. Zeng, B. Zou, Y. Liu, B. Xu and W. Tian, Angew. Chem., Int. Ed., 2018, 57, 15670–15674 CrossRef CAS PubMed.
- Y.-B. Gong, P. Zhang, Y.-r. Gu, J.-Q. Wang, M.-M. Han, C. Chen, X.-J. Zhan, Z.-L. Xie, B. Zou, Q. Peng, Z.-G. Chi and Z. Li, Adv. Opt. Mater., 2018, 6, 1800198 CrossRef.
- J. Xiong, K. Wang, Z. Yao, B. Zou, J. Xu and X.-H. Bu, ACS Appl. Mater. Interfaces, 2018, 10, 5819–5827 CrossRef CAS PubMed.
- K. Dojin, K. J. Eon and P. S. Young, Adv. Funct. Mater., 2018, 28, 1706213 CrossRef.
- S. K. Park, I. Cho, J. Gierschner, J. H. Kim, J. H. Kim, J. E. Kwon, O. K. Kwon, D. R. Whang, J.-H. Park, B.-K. An and S. Y. Park, Angew. Chem., Int. Ed., 2016, 55, 203–207 CrossRef CAS PubMed.
- H.-J. Kim, D. R. Whang, J. Gierschner, C. H. Lee and S. Y. Park, Angew. Chem., Int. Ed., 2015, 54, 4330–4333 CrossRef CAS PubMed.
- N. Mizoshita, T. Tani and S. Inagaki, Adv. Mater., 2012, 24, 3350–3355 CrossRef CAS PubMed.
- M. Martínez-Abadía, R. Giménez and M. B. Ros, Adv. Mater., 2018, 30, 1704161 CrossRef PubMed.
- Y. Sagara, S. Yamane, M. Mitani, C. Weder and T. Kato, Adv. Mater., 2016, 28, 1073–1095 CrossRef CAS.
- A. Julià-López, J. Hernando, D. Ruiz-Molina, P. González-Monje, J. Sedó and C. Roscini, Angew. Chem., Int. Ed., 2016, 55, 15044–15048 CrossRef PubMed.
- Z. Ma, M. Teng, Z. Wang, S. Yang and X. Jia, Angew. Chem., Int. Ed., 2013, 52, 12268–12272 CrossRef CAS PubMed.
- Z. Ma, Z. Wang, X. Meng, Z. Ma, Z. Xu, Y. Ma and X. Jia, Angew. Chem., Int. Ed., 2016, 55, 519–522 CrossRef CAS PubMed.
- Z. Mao, Z. Yang, Y. Mu, Y. Zhang, Y.-F. Wang, Z. Chi, C.-C. Lo, S. Liu, A. Lien and J. Xu, Angew. Chem., Int. Ed., 2015, 54, 6270–6273 CrossRef CAS PubMed.
- J. Yang, Z. Ren, Z. Xie, Y. Liu, C. Wang, Y. Xie, Q. Peng, B. Xu, W. Tian, F. Zhang, Z. Chi, Q. Li and Z. Li, Angew. Chem., Int. Ed., 2017, 56, 880–884 CrossRef CAS PubMed.
- J.-A. Li, J. Zhou, Z. Mao, Z. Xie, Z. Yang, B. Xu, C. Liu, X. Chen, D. Ren, H. Pan, G. Shi, Y. Zhang and Z. Chi, Angew. Chem., Int. Ed., 2018, 57, 6449–6453 CrossRef CAS PubMed.
- X. Wu, J. Guo, Y. Cao, J. Zhao, W. Jia, Y. Chen and D. Jia, Chem. Sci., 2018, 9, 5270–5277 RSC.
- Q. Li and Z. Li, Adv. Sci., 2017, 4, 1600484 CrossRef PubMed.
- X. Chen, T. Pradhan, F. Wang, J. S. Kim and J. Yoon, Chem. Rev., 2011, 112, 1910–1956 CrossRef PubMed.
- H. N. Kim, M. H. Lee, H. J. Kim, J. S. Kim and J. Yoon, Chem. Soc. Rev., 2008, 37, 1465–1472 RSC.
- Q. Qi, J. Qian, S. Ma, B. Xu, S. X.-A. Zhang and W. Tian, Chem. – Eur. J., 2015, 21, 1149–1155 CrossRef CAS PubMed.
- X. Su, Y. Ji, W. Pan, S. Chen, Y.-M. Zhang, T. Lin, L. Liu, M. Li, Y. Liu and S. X.-A. Zhang, J. Mater. Chem. C, 2018, 6, 6940–6948 RSC.
- Q. Yu, X. Su, T. Zhang, Y.-M. Zhang, M. Li, Y. Liu and S. X.-A. Zhang, J. Mater. Chem. C, 2018, 6, 2113–2122 RSC.
- L. Huang, Y. Qiu, C. Wu, Z. Ma, Z. Shen and X. Jia, J. Mater. Chem. C, 2018, 6, 10250–10255 RSC.
- Z. Ma, Y. Ji, Y. Lan, G.-C. Kuang and X. Jia, J. Mater. Chem. C, 2018, 6, 2270–2274 RSC.
- Y. Li, Z. Ma, A. Li, W. Xu, Y. Wang, H. Jiang, K. Wang, Y. Zhao and X. Jia, ACS Appl. Mater. Interfaces, 2017, 9, 8910–8918 CrossRef CAS PubMed.
- A. P. Demchenko, K.-C. Tang and P.-T. Chou, Chem. Soc. Rev., 2013, 42, 1379–1408 RSC.
- J. E. Kwon and S. Y. Park, Adv. Mater., 2011, 23, 3615–3642 CrossRef CAS PubMed.
- F. Wu, L. Wang, H. Tang, D. Cao and X. Jiang, Chem. – Asian J., 2018, 13, 950–954 CrossRef CAS PubMed.
- S. Tsuchiya, K.-i. Sakai, K. Kawano, Y. Nakane, T. Kikuchi and T. Akutagawa, Chem. – Eur. J., 2018, 24, 5868–5875 CrossRef CAS PubMed.
- Y. Niu, R. Wang, P. Shao, Y. Wang and Y. Zhang, Chem. – Eur. J., 2018, 24, 16670–16676 CrossRef CAS PubMed.
- T. He, N. Niu, Z. Chen, S. Li, S. Liu and J. Li, Adv. Funct. Mater., 2018, 28, 1706196 CrossRef.
- F. Borbone, A. Tuzi, B. Panunzi, S. Piotto, S. Concilio, R. Shikler, S. Nabha and R. Centore, Cryst. Growth Des., 2017, 17, 5517–5523 CrossRef CAS.
- V. S. Padalkar, D. Sakamaki, K. Kuwada, A. Horio, H. Okamoto, N. Tohnai, T. Akutagawa, K.-i. Sakai and S. Seki, Asian J. Org. Chem., 2016, 5, 938–945 CrossRef CAS.
- V. S. Padalkar, D. Sakamaki, N. Tohnai, T. Akutagawa, K.-i. Sakai and S. Seki, RSC Adv., 2015, 5, 80283–80296 RSC.
- R. Wei, P. Song and A. Tong, J. Phys. Chem. C, 2013, 117, 3467–3474 CrossRef CAS.
- D. Yao, S. Zhao, J. Guo, Z. Zhang, H. Zhang, Y. Liu and Y. Wang, J. Mater. Chem., 2011, 21, 3568–3570 RSC.
- Y. Qian, S. Li, G. Zhang, Q. Wang, S. Wang, H. Xu, C. Li, Y. Li and G. Yang, J. Phys. Chem. B, 2007, 111, 5861–5868 CrossRef CAS PubMed.
- H. Lin, X. Chang, D. Yan, W.-H. Fang and G. Cui, Chem. Sci., 2017, 8, 2086–2090 RSC.
- V. S. Padalkar and S. Seki, Chem. Soc. Rev., 2016, 45, 169–202 RSC.
- H. Wen, Q. Huang, X.-F. Yang and H. Li, Chem. Commun., 2013, 49, 4956–4958 RSC.
- J. Cheng, D. Liu, W. Li, L. Bao and K. Han, J. Phys. Chem. C, 2015, 119, 4242–4251 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of experimental sections, including synthesis, characterization, spectra, SAXSS patterns and optical images. CCDC 1883880. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8qm00633d |
| This journal is © the Partner Organisations 2019 |