
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsights into the role of noncovalent interactions in distal functionalization of the aryl C(sp2)–H bond†
Anju
Unnikrishnan
 and
Raghavan B.
Sunoj
*
and
Raghavan B.
Sunoj
*
Department of Chemistry, Indian Institute of Technology Bombay, Powai, Mumbai 400076, India. E-mail: sunoj@chem.iitb.ac.in
First published on 18th February 2019
Abstract
Burgeoning interest in distal functionalization of aryl C–H bonds led to the development of iridium-catalyzed borylation reactions. The significance and inadequate mechanistic understanding of C(sp2)–H borylations motivated us to investigate the key catalytic steps and the origin of a directing-group-free regiocontrol in the reaction between aryl amides and B2pin2 (bis(pinacolato)diboron). An Ir(III)(ubpy)tris(boryl) complex, generated from the pre-catalyst [Ir(OMe)(cod)]2 by the action of a bipyridine-urea ligand (ubpy) and B2pin2, is considered as the most likely active catalyst. The meta C–H activation of N,N-dihexylbenzamide is energetically more favorable over the para isomer. The origin of this preference is traced to the presence of a concerted action of noncovalent interactions (NCIs), primarily between the catalyst and the substrate, in the regiocontrolling transition states (TSs). Molecular insights into such TSs revealed that the N–H⋯O interaction between the tethered urea moiety of the Ir-bound ubpy ligand of the catalyst and the amide carbonyl of the substrate is a critical interaction that helps orient the meta C–H bond nearer to iridium. Other NCIs such as C–H⋯π between the substrate and the catalyst, C–H⋯O involving the substrate C–H and the oxygen of the B2pin2 ligand and C–H⋯N between the substrate and the N atom of the Ir-bound ubpy confirm the significance of such interactions in providing the desirable differential energies between the competing TSs that form the basis of the extent of regioselectivity.
Introduction
Selective activation of thermodynamically strong and kinetically inert C–H bonds has garnered the attention of chemists for decades. Among the several activation strategies available, functionalization via C–H bond activation using a borylation reaction is a promising one due to the wider utility of the borylated products.1 The C–H borylation reactions witnessed a number of interesting developments encompassing a range of transition metals such as Co, Ni, Ru, Rh, Pd and Ir.2 Some of the most important examples in this genre employ iridium catalysts in conjunction with the prototypical B2pin2 (bis(pinacolato)diboron) as the borylating agent. In this regard, use of pre-catalysts such as [Ir(X)(cod)]2 (where X = Cl and OMe) and bipyridine ligands in the activation of the C(sp2)–H bond is noteworthy.3A lot of effort has been expended toward developing selective activation of aryl C–H bonds, wherein one typically strives to achieve control over ortho, meta and para functionalization. The functional group (FG) directed borylation is an effective protocol for imparting ortho selectivity.4 Along the similar lines, efforts for achieving meta selectivity5 continued to receive attention due to the synthetic value of the meta functionalized products.6 Development of a functionalization strategy without having to use an additional directing group (DG) on the substrate, is certainly a great advantage.7 It will, therefore, be of inherent value if the catalyst could perform the directing role such that the method can be utilized for a broader range of substrates.8 Such an approach would help reduce the proportion of a DG from stoichiometric to catalytic levels. A number of such endeavors where the catalyst is tailored to perform the role of a directing group rely on the careful control/use of weak noncovalent interactions (NCIs).9 Harnessing NCIs as a handle to gain regiocontrol in transition metal catalyzed C–H activation reactions remains much less explored at this stage of development. NCIs when operating in a concerted manner are known to impact the stereochemical outcome of reactions,10 which is an idea that could be exploited in the catalyst design for regioselective transformations as well. Within the NCI directed C–H functionalization strategies, two distinct methods employing [Ir(OMe)(cod)]2 and bipyridine-derived ligands as the catalytic system have been reported very recently. While the approach developed concurrently by Kanai11 and Chattopadhyay12 proposes a secondary interaction as responsible for directed C–H functionalization, the other one by Phipps demonstrated an ion-pair directed regiocontrol.13
In keeping with our continued efforts in probing the mechanism and selectivity controlling factors in transition metal catalyzed C–H functionalization reactions,14 we became interested in examining the important meta C(sp2)–H borylation of aryl amides using [Ir(OMe)(cod)]2 (Scheme 1). The observed regioselective borylation was proposed to arise from a hydrogen bonding interaction between the catalyst and the substrate.15 Although the experimental studies on iridium catalyzed ortho borylation reactions are widely available, the current mechanistic understanding of meta C–H borylation is inadequate. Furthermore, rationalization of regioselectivity in these reactions typically invoked qualitative geometric features of certain putative intermediates or that of a transition state (TS) in a proposed mechanistic pathway. Hence, a number of vital geometric and energetic aspects responsible for the observed product distribution remain vague at this stage. Using DFT computations (SMD(p-xylene)/B3LYP-D3/6-31G**,SDD(Ir)), we aim to gain insights into (a) the energetic details of the catalytic cycle, (b) molecular geometry as well as electronic features of the critical intermediates and TSs, (c) the origin of regioselectivity, and (d) how changes in the substituents of the catalyst and/or substrate could impact the regiochemical outcome. The knowledge on the origin of regioselectivity would help make more rational modifications to the substrate and/or ligand as well as to expand the scope of such catalytic reactions.
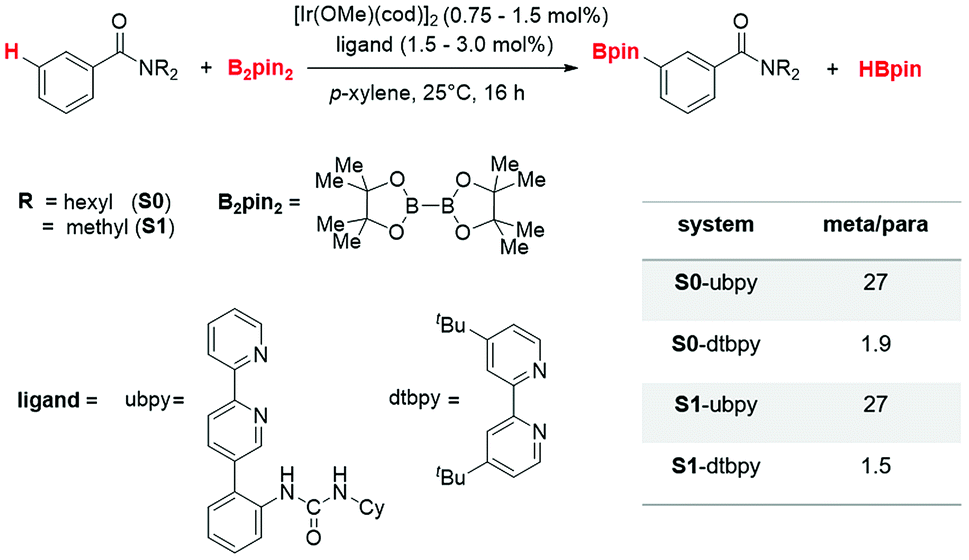 | ||
| Scheme 1 Iridium(III)-catalyzed meta C(sp2)–H borylation of aryl amides using B2pin2. | ||
Computational methods
All computations were performed using the Gaussian09 (Revision D.01) suite of quantum chemical programs.16 We employed the hybrid density functional B3LYP17 with Grimme's dispersion correction (D3)18 in combination with the Stuttgart-Dresden double-ζ zeta basis set (SDD)19 with an effective core potential for 60 inner electrons out of 77 total electrons for iridium and the 6-31G** basis set20 for all the other elements. Similar computational methods were successfully employed in the study of transition metal catalyzed reactions.21 All stationary points identified as TSs were characterized by one and only one imaginary frequency that corresponds to the expected reaction coordinate. The intrinsic reaction coordinate (IRC) calculations22 were also done at the same level of theory to examine whether the TS is connected to the reactant/product as desired. The effect of the solvent was taken into account using the SMD solvation model in para-xylene as the continuum dielectric (ε = 2.27).23 The free energies of all the TSs and intermediates reported in the manuscript were obtained by adding the thermal and entropic corrections with the quasi rigid-rotor harmonic oscillator approximation24 to the electronic energies in the condensed phase. Thus, the results and discussion are presented using the Gibbs free energies obtained at the SMD(p-xylene)/B3LYP-D3/6-31G**,SDD(Ir) level of theory at 298.15 K and 1 atm pressure, unless stated otherwise. Topological analyses of the electron densities were performed using Bader's Atoms-in-Molecules (AIM) using the AIM2000 software so as to analyze the weak inter-atomic interactions in various TSs.25 Further, the regions of attractive and repulsive interactions are identified through the generation of NCI plots.26 The energy span of the catalytic cycle has been calculated using the energetic span model developed by Shaik and Kozuch.27Results and discussion
The catalytic regioselective borylation of C(sp2)–H bonds of aromatic amides using B2pin2 (bis(pinacolato)diboron), employing [Ir(OMe)(cod)]2 as the pre-catalyst (cod = 1,5-cyclooctadiene) in the presence of a bipyridine-derived ligand, is examined (Scheme 1). The ligand employed here is a urea-bpy (ubpy) system tethered via an ortho-phenylene linker. The Ir(III)(ubpy)tris(boryl) complex formed by the action of the ubpy ligand and the borylating agent B2pin2 on the pre-catalytic Ir(I) species is considered as the active catalyst.3b,28 While different possible configurations of the active catalyst as well as that of the catalyst–substrate complex are first examined using a model system, all species involved in the catalytic cycle presented in the manuscript employ only the real system.291. Important details of the catalytic cycle
The key mechanistic steps in the overall catalytic cycle, starting with the formation of a catalyst–substrate complex are shown in Scheme 2. To generate adequate space to accommodate the substrate near the iridium center, a higher energy configuration (A2) of the active catalyst is considered.30 Depending on the site of interaction of the substrate with the catalyst, three distinct coordination modes, such as an Ir⋯π (when the aromatic amide is coordinated through the aryl π ring), Ir⋯N (nitrogen of the amide) and Ir⋯O (carbonyl oxygen of the amide) binding, are examined.31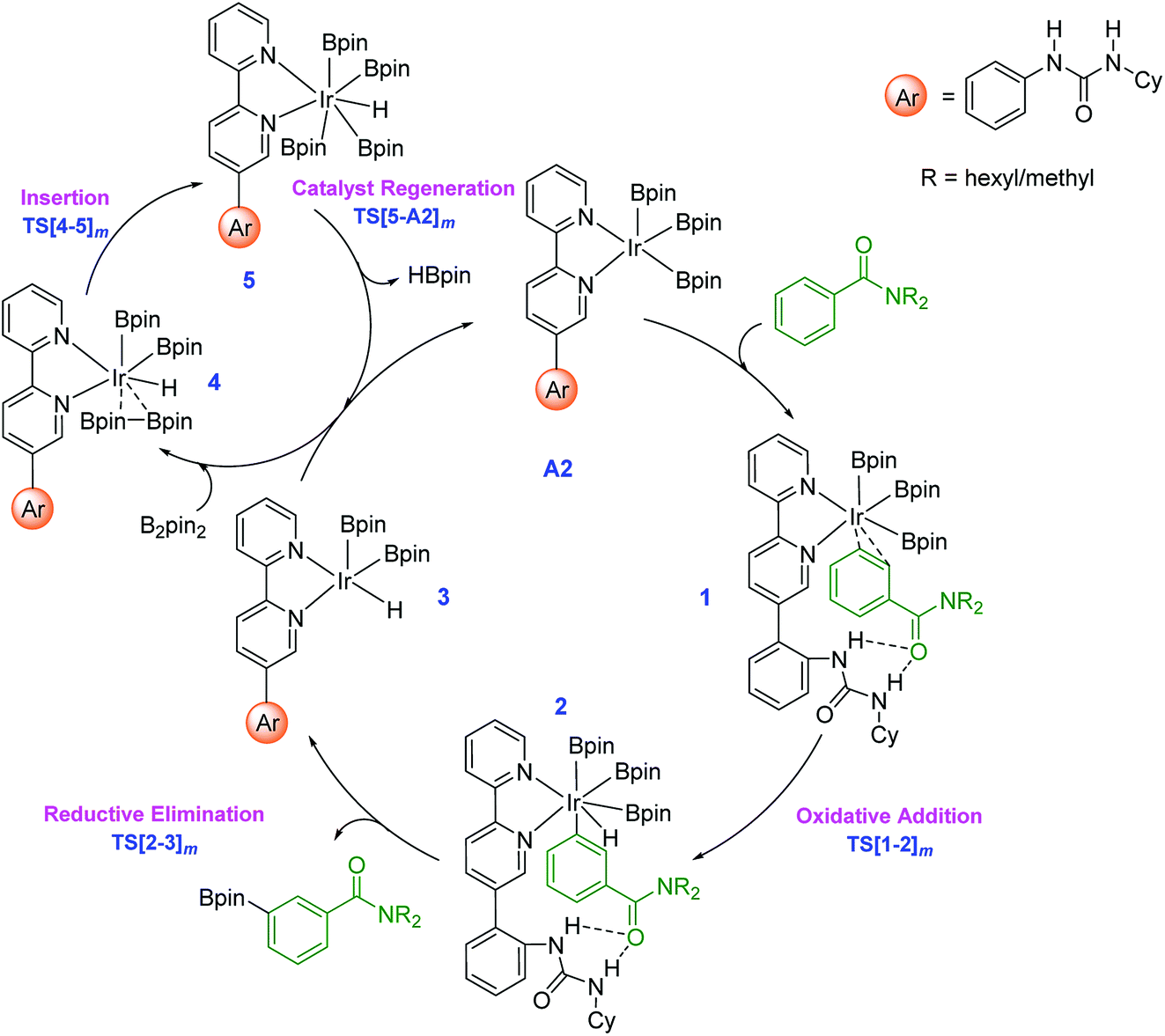 | ||
| Scheme 2 Important mechanistic steps in the catalytic cycle for the Ir(III)-catalyzed borylation of aryl amides through C–H activation. The subscript m in the TS notation indicates the meta isomer. | ||
The computed energies suggest that the substrate coordination to the iridium center via oxygen or nitrogen atom of the amide group is equally feasible as they differ only by a kcal mol−1.32 The optimized geometries of the catalyst–substrate complexes in both these binding modes convey that only the ortho aryl C–H bond is close enough to the iridium center for any effective interaction. In other words, when N,N-dihexylbenzamide is bound to the catalyst through its amide moiety, the meta and para positions remain far from the iridium center to afford C–H bond activation.33 In the π-binding mode, on the other hand, all the three aryl C–H bonds, including the meta enjoy enhanced proximity to the iridium center that can lead to effective functionalization.34 Thus, the π-binding mode in the catalyst–substrate complex is considered as the reactive conformer in our study as shown in Scheme 2.
Since the N-hexyl chains are conformationally flexible, as many as 8 conformers of all the important TSs are identified.35 The results presented herein are on the basis of the most favorable geometry for both the meta and para C–H activation TSs. The key mechanistic events in the catalytic cycle, as shown in Scheme 2, start with an oxidative addition in the catalyst–substrate complex 1 wherein Ir(III) inserts into the meta C–H bond via transition state TS[1-2]m. The ensuing Ir(V)-aryl intermediate 2 then undergoes a reductive elimination (RE) through TS[2-3]m to generate the borylated product and an Ir(III)-hydride intermediate (3). Uptake of one molecule of B2pin2 by 3 can then lead to a weakly interacting complex between 3 and B2pin2, which is denoted as 4. In the following step, insertion of B2pin2 into 4 generates a hepta-coordinate Ir(V) species (5) viaTS[4-5]m. The catalyst regeneration can be completed by the expulsion of a molecule of HBpin through the RE transition state TS[5-A2]m. The active catalyst A2, thus generated, can then sustain the catalytic cycle by the uptake of another molecule of the substrate.36
The Gibbs free energy profile for the formation of the meta borylated product is provided in Fig. 1. The formation of the borylated product exhibits notable barriers of 21.6 and 21.5 kcal mol−1, respectively, for the C–H activation and the RE. According to the energetic span model,27 intermediate 1 and TS[1-2]m are, respectively, the turnover determining intermediate (TDI) and turnover determining transition state (TDTS). The activation span (δE), calculated as the energy difference between the TDTS and TDI, is found to be 21.6 kcal mol−1.37 The subsequent steps in the mechanism, such as the B2pin2 insertion and the catalyst regeneration, are much less energy demanding, as indicated by their relatively lower barriers.
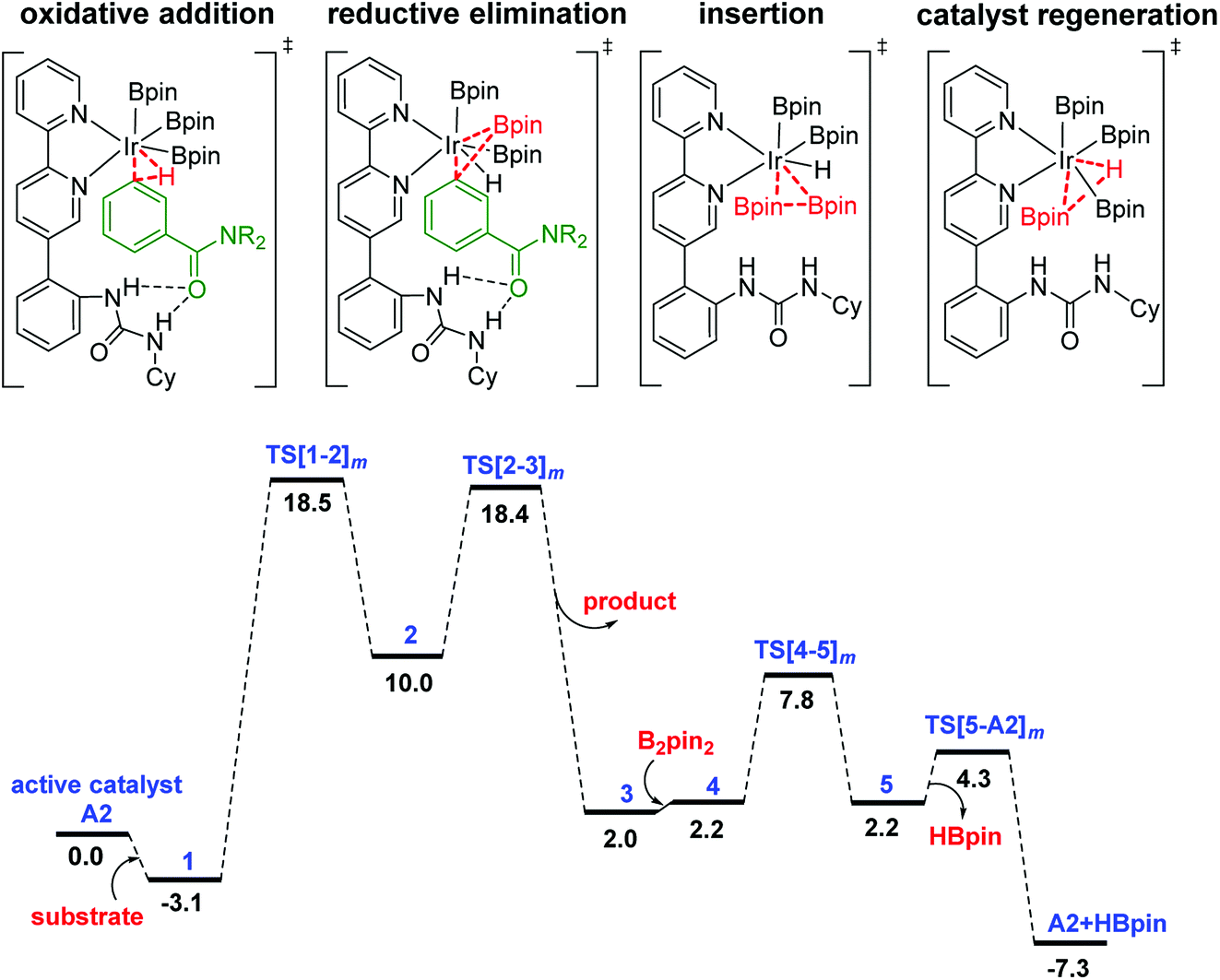 | ||
| Fig. 1 Free energy profile of the Ir(III)-catalyzed meta C–H activation reaction. The subscript m in the TS notation indicates the meta isomer. | ||
2. Factors controlling regioselectivity
The knowledge of energetics, geometry, and electronic features of the important TSs involved in a catalytic cycle would be valuable toward developing a better understanding of the catalytic transformation. For the present example, such features of the regio-controlling TSs enabling meta C–H activation are not established yet. Kanai's insightful working hypothesis,15 on the other hand, placed a significant emphasis on the H-bonding between N,N-dialkylbenzamide and the urea moiety of the Ir-bound ubpy ligand as the key factor in their proposed TSs. The observed meta to para ratio of 27 corresponds to a selectivity of 93% in favor of meta borylation. To probe this important observation in greater depth, TSs for the meta and para C–H activation of substrates such as N,N-dihexylbenzamide (S0) and N,N-dimethylbenzamide (S1) are identified. Different variations in the catalyst (ubpy), and the borylating agent (B0) are also considered in this study. The original catalyst–substrate combination is referred to as ubpy-S0 while that with a modified substrate is denoted as ubpy-S1. Optimized geometries of the regiocontrolling TSs for S0 and S1 are provided in Fig. S2 in the ESI.† Interestingly, two crucial N–H⋯O H-bonding interactions between the N–H of the urea and the oxygen atom of the amide (shown as v and w in Fig. 2) are noticed in the meta C–H activation transition states TS[1-2]m(S0) and TS[1-2]m(S1). However, only one such interaction (v) is found in the case of para C–H activation TSs (TS[1-2]p(S0) and TS[1-2]p(S1)) with both the substrates. More important clues on regioselectivity could be gathered from these TSs through a comparison of other noncovalent interactions such as C–H⋯π, C–H⋯O and C–H⋯N besides the N–H⋯O H-bonding. An approximate estimate of the strength of these weak NCIs is calculated by using the values of the electron densities at the respective bond critical points (ρbcp) as obtained through Bader's AIM analysis.25 | ||
| Fig. 2 Optimized geometries of the TSs for the meta and para C–H activation of ubpy-S0 and ubpy-S1 catalyst–substrate combinations. The distances are in Å and electron densities (ρ × 10−2 au) at the bond critical points are given in parentheses. The hydrogen atoms that are not involved in any noticeable interaction are removed for improved clarity. | ||
A summary of topological analysis of the electron density for TS[1-2]m and TS[1-2]p in the case of both ubpy-S0 and ubpy-S1 catalyst–substrate combinations, together with important interatomic distances, is given in Fig. 2. It can be noticed that in TS[1-2]m(S0) both the N-hexyl arms of the substrate are involved in multiple C–H⋯π interactions (shown as a, b, c, d, and e) with the catalyst through the aryl π-face of the Ir-bound ligands (i.e., bpy ligand as well as with the phenyl-urea linker). Additionally, the cyclohexyl ring (Cy) attached to the urea moiety also participates in C–H⋯π interaction (f) with the substrate in TS[1-2]m(S0). On the other hand, in the para C–H activation transition state TS[1-2]p(S0), the relative orientation of the substrate is such that the C–H⋯π interaction with the catalyst is absent. Another significant NCI in TS[1-2]m(S0) is the C–H⋯O interaction between the aryl C–H of the substrate and B2pin2 (shown as h). Other C–H⋯O contacts denoted as g, i, j, k and l in TS[1-2]m(S0) represent the interactions between the ligands (Ir-bound bpy, phenyl-urea linker and Bpin). An equally good number of C–H⋯O interactions (g, i, m, n, o and p) are also noticed in the TS[1-2]p(S0). However, the overall number of NCIs is found to be higher and they are relatively more efficient in TS[1-2]m(S0) than those in TS[1-2]p(S0) for the ubpy-S0 combination.
Although the above-mentioned analysis, based on the difference in the number of NCIs, offers a qualitative insight into the origin of meta selectivity, it does not provide room for quantitative assessment. For instance, a lesser number of more efficient interactions can outweigh the influence of a greater number of weaker interactions. Hence, we endeavored to quantify the important NCIs using the topological parameters such as the electron density at the bond critical point (ρbcp), the corresponding Laplacian of the electron density (∇2ρ), and kinetic energy (G) using Espinosa's formulation.38 Although Espinosa's formulation was proposed for the quantification of isolated pairwise intermolecular H⋯F interactions, we have extended the same to various intramolecular weak NCIs operating in important TSs in the present study. Even though the formulation has not been applied to complex intramolecular interactions such as that prevail in regiocontrolling TSs, we believe that it could provide a reasonable measure of the relative strengths of the NCIs such as C–H⋯π, C–H⋯O, C–H⋯N and N–H⋯O. While a detailed mapping of the NCIs in TS[1-2]m and TS[1-2]p for both the ubpy-S0 and ubpy-S1 pairs is given in the ESI,†39 herein a succinct representation of the major NCIs such as C–H⋯π, C–H⋯O, C–H⋯N and N–H⋯O is provided. It can be gleaned from the bar diagram, as given in Fig. 3, that each type of NCI has a different contribution in different catalyst–substrate systems.
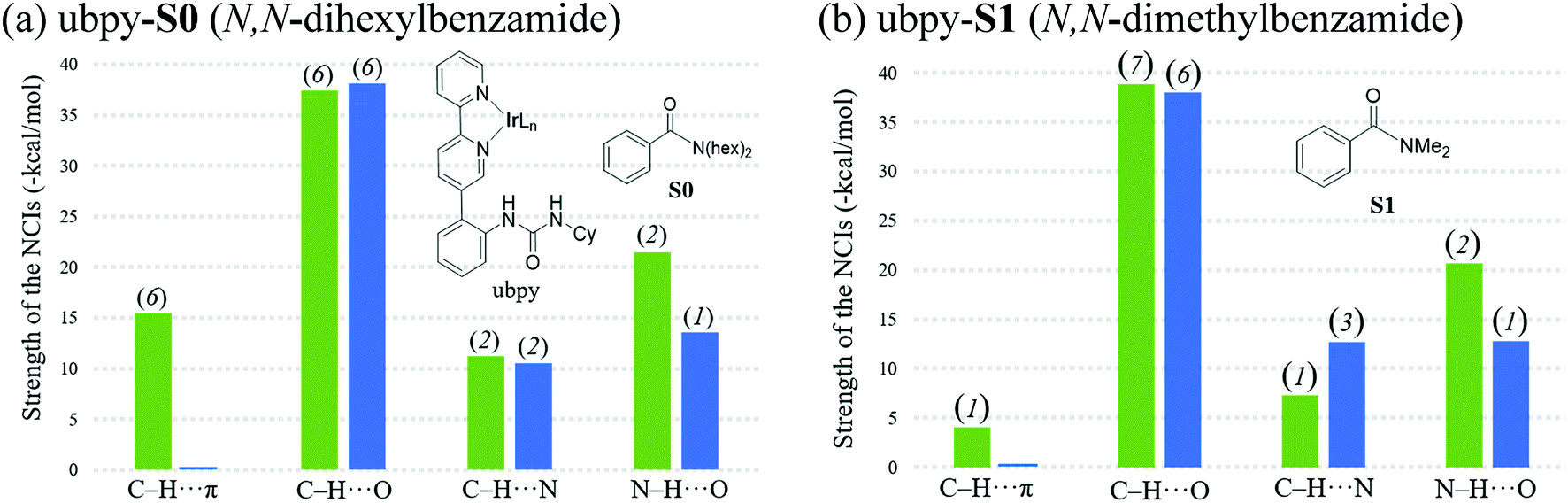 | ||
| Fig. 3 Comparison of the sum of interaction energies of the NCIs in the meta (green) and para (blue) C–H activation TSs for (a) substrates S0 and (b) S1. Values in parentheses above the bar diagrams are the number of noncovalent interactions. | ||
Since the dihexyl chain of the substrate engages in a good number of C–H⋯π interactions with the catalyst, we wanted to examine whether changes in such interactions might affect the extent of regioselectivity. To this end, a modified substrate S1 is considered wherein the N,N-dihexyl group on the amido nitrogen is replaced with a N,N-dimethyl substituent. The optimized transition state geometries are devoid of C–H⋯π interactions, except one such prominent contact between the cyclohexane of the urea moiety and the aryl group of the substrate (shown as f in Fig. 2). This interaction is present only in the case of TS[1-2]m(S1) but not in TS[1-2]p(S1). Interestingly, a number of other NCIs such as C–H⋯O and C–H⋯N interactions are found to be present in both the meta and para TSs for N,N-dimethyl amide as the substrate. It is also of interest to note that the NCIs in the case of the S1 system are not as pronounced as those in S0 (Fig. 3). Further, the total strength of all the important NCIs is estimated to be −70.9 kcal mol−1 for TS[1-2]m(S1) and only −63.5 kcal mol−1 for TS[1-2]p(S1). The regioselectivity, calculated using the energy difference of 7.4 kcal mol−1 between the competing meta and para C–H activation TSs for the N,N-dimethylamide is strongly in favor of the meta isomer.
The summary of noncovalent interactions shown in Fig. 3 conveys that except for the C–H⋯O contacts, all the other NCIs are more dominant in TS[1-2]m(S0) (shown in green) than in TS[1-2]p(S0) (blue) for the ubpy-S0 catalyst–substrate pair. It should be noted that TS[1-2]m(S0) is 6.6 kcal mol−1 lower in energy than TS[1-2]p(S0), suggesting that the NCIs do have a direct influence on the extent of regioselectivity. In the case of the S1 system, though the number of C–H⋯π contacts is much smaller, other NCIs such as C–H⋯O and N–H⋯O interactions are able to effectively make TS[1-2]m(S1) lower in energy by 7.4 kcal mol−1 as compared to TS[1-2]p(S1). Thus, it appears that the C–H⋯π interactions may not solely be responsible for the observed high regioselectivity if the cumulative effect of other weak interactions is able to compensate.
It is also interesting to note that the difference in the total strength of NCIs between meta and para C–H activation TSs exhibits a good correlation with the predicted regioselectivity. The predicted preference toward the meta C–H activation for ubpy-S0 and ubpy-S1 is, respectively, 6.6 and 7.4 kcal. This is in line with the experimentally observed meta to para ratio of 27 for both the catalyst–substrate pairs. Appreciable difference in the collective strength of NCIs between the meta and para C–H activation TSs is noted in the case of ubpy-S0 (23.4 kcal mol−1) and ubpy-S1 (7.4 kcal mol−1), thus favoring meta C–H activation over the alternative para pathway. Hence, a combination of C–H⋯π, C–H⋯N and C–H⋯O interactions together with the N–H⋯O H-bonding makes the meta C–H activation TS lower in energy than the corresponding para position.40
While it is prudent to acknowledge that various noncovalent interactions described above impact the predicted relative energy order between the regiocontrolling TSs, analysis of the effect of distortion in such TSs is equally important. The Distortion–Interaction/Activation-Strain (DI-AS) analysis41 was therefore carried out on these TSs to gain additional insights into the factors responsible for the observed regioselectivity. It can be noticed from the data provided in Table 1 that in both ubpy-S0 and ubpy-S1 catalyst–substrate combinations, the para C–H activation TSs experience a higher total distortion relative to the corresponding meta analogue. The extent of distortion in the substrate, as well as the catalyst in the para TS, is found to be higher than that in the meta case for ubpy-S1.42 Thus, due to the combined effect of the higher number of NCIs operating in concert as well as the relatively lower distortion experienced by both the meta TSs, we could rationalize the preference toward the high meta regioselectivity in both the above-mentioned examples.
| System | ΔEdis (ζ) | ||
|---|---|---|---|
| Substrate | Catalyst | Total in the TS | |
| ubpy-S0 (meta) | 58.9 | 23.4 | 107.8 |
| ubpy-S0 (para) | 58.8 | 25.4 | 109.2 |
| ubpy-S1 (meta) | 56.0 | 22.8 | 100.0 |
| ubpy-S1 (para) | 62.5 | 25.5 | 107.3 |
3. Rational modifications to change the pattern of NCIs
After having understood the critical role of various NCIs as well as the N–H⋯O H-bonding in the C–H activation step, we considered two new modifications of the parent system. As the N–H⋯O interactions were also found to play a vital role in imparting selectivity, the parent ligand ubpy is modified by removing the phenylene-urea linker to a simple bpy. Similarly, instead of B2pin2 (B0), we have considered B2(OMe)4 (B1) as the borylating agent.43 The change in the regioselectivity due to these modifications is predicted for the substrate S0. The bpy ligand led to no energy difference between the meta and para C–H activation TSs, implying no regioselectivity. In other words, turning off the vital N–H⋯O interactions between the substrate and the catalyst, by way of removing the urea moiety diminishes the energetic advantage toward the meta C–H activation as compared to the competing para analogue.44 Similar to the case with the bpy ligand, the B1 modification also leads to a relatively smaller energy difference between the meta and para TSs (2.6 kcal mol−1) compared to the unmodified system (6.6 kcal mol−1).45 A similar trend in selectivity is also noticed when computed using relative enthalpy differences between the meta and para TSs for the aforesaid modifications.46 A detailed analysis of the meta and para C–H activation TSs of these modified systems based on Espinosa's formulation47 and the DI-AS analysis48 is thus performed to assess how NCIs impact the regiochemical outcome of this reaction.We note that the prominent NCIs other than the N–H⋯O H-bonding in these modified systems are the C–H⋯O and C–H⋯N interactions. A quantified NCI, as given in Table 2, conveys that for the bpy modification, the total strength of the C–H⋯O interactions in TS[1-2]p(bpy) (−36.7 kcal mol−1) is higher than that in the meta TS (−31.3 kcal mol−1), while TS[1-2]m(bpy) enjoys improved C–H⋯N interaction (−8.9 kcal mol−1) than in the para counterpart (−5.5 kcal mol−1) (Table 2).47 While the predicted strength of the C–H⋯O interactions appears to be overestimated, it serves the present purpose wherein we intend to compare the relative strengths of such interactions in chemically identical meta and para C–H activation TSs. Another interaction, namely, the N–H⋯O interaction is absent in both the TS geometries, and four more C–H⋯π interactions (−9.8 kcal mol−1) are found in the meta TS than that in the para TS. Thus, the absence of some of the important NCIs in TS[1-2]m(bpy) and TS[1-2]p(bpy) appears to result in comparable energies for both these TSs, which in turn, leads to very low regioselectivity.47 In the case of B1 modification, equal numbers of C–H⋯O interactions are identified in both meta and para TSs, albeit the cumulative strength of such interactions is higher in para (−48.1 kcal mol−1) than that in meta (−37.8 kcal mol−1) TS. However, a greater number of stronger N–H⋯O interactions are found in the meta TS than in para (Table 2). The C–H⋯N interactions, on the other hand, are found to be of comparable strengths in the meta (−11.9 kcal mol−1) and para C–H activation TSs (−9.3 kcal mol−1). The lack of notable differences in the noncovalent interactions can be regarded as the origin of the small energy difference (2.6 kcal mol−1) between the two competing TSs.47
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
Since clarity on whether ortho C–H activation is more likely than that at the para position under the experimental conditions could not be sought on the basis of the computed energetics of the C–H activation step, we have carefully examined the ensuing steps of the catalytic cycle in greater detail. Interestingly, the Ir(V)-aryl intermediate (2) formed as a result of the C–H activation at the ortho as well as para positions is found to be more reversible than the one derived at the meta position.50 Furthermore, a comparative study of the reductive elimination (RE) leading to the formation of the borylated product is undertaken for the ubpy-S0 catalyst–substrate combination. An elementary step barrier of 21.5 kcal mol−1 is found for the RE to a meta borylated product, which is in concert with the experimental preference toward the meta product. However, the RE elimination barrier at the para position is 27.4 kcal mol−1 while that at the ortho position is found to be 32.8 kcal mol−1, suggesting that the para product would be the next most likely product other than the meta, in accord with the experimental observations. Though the pathways appear to compete in the initial oxidative addition step, the application of the energetic span model also helped us understand the regioselectivity more convincingly.51 While the TDI is the respective catalyst–substrate complex (1) in all these pathways, the TDTS for the meta pathway is found to be the oxidative addition and that for the para and ortho pathways it is the reductive elimination. The corresponding energetic span δE for para and ortho is, respectively, 27.3 and 32.8 kcal mol−1. In other words, the catalytic turnover toward ortho is the least favored, followed by the para product. It is also interesting to note that the relative enthalpies of the relevant TSs also convey a similar trend in the predicted selectivities.52 Thus, the overall energetic features of various borylation pathways suggest the formation of the meta product as the major and para as the minor regioisomer in this catalytic transformation.53
Conclusion
Density functional theory investigation of important Ir(III)-catalyzed meta selective aryl C(sp2)–H borylation revealed that the key mechanistic steps in the lower energy pathway are (i) oxidative addition (C–H activation), (ii) reductive elimination (borylation), and (iii) catalyst regeneration. A comparison of energies and stereoelectronic factors operating in the C–H activation transition states for meta and para functionalization of N,N-dihexylbenzamide helped us gain significant new insights into the role of various noncovalent interactions, particularly between the catalyst and the substrate. The catalyst Ir(III)(ubpy)tris(boryl), decorated with a phenylene-urea tether on the bpy ligand is found to play an important role in positioning the aryl amide through N–H⋯O H-bonding interactions such that the C–H activation at the meta position is rendered energetically more favorable over that at para. However, we noted that the presence or absence of this H-bonding interaction could not solely account for the regioselectivity. A good number of noncovalent interactions between the catalyst (Ir-bound ligands) and the substrate are found to be vital toward bringing about the energy difference between the meta and para C–H activation TSs. These interactions, operating between the C–H bonds of the substrate (hexyl and aryl moieties) and (i) the bipyridyl nitrogen atoms as well as the π-face of the Ir-bound ubpy ligand and (ii) the oxygen atoms of B2pin2, are found to be more prominent in the meta C–H activation transition state, thereby making it 6.6 kcal mol−1 lower in energy than the para analogue. This energy difference is fully consistent with the experimental observation of the high meta to para ratio of 27.Additional series of computations on modified systems obtained by changing the substrate (replacing dihexyl with dimethyl), catalysts without the urea moiety on the Ir-bound ubpy ligand, and the use of B2(OMe)4 instead of B2pin2 as the borylating agent further helped us conclude that the balance between C–H⋯π, C–H⋯O and C–H⋯N NCIs that operate between the catalyst and the substrate is more important than the primary N–H⋯O H-bonding contact that binds the substrate to the catalyst. For instance, the meta C–H activation TS for the N,N-dimethylbenzamide is noted to enjoy a larger number of relatively better NCIs thereby maintaining high regioselectivity even though the C–H⋯π interactions are not as much as those in N,N-dihexylbenzamide. For the catalysts devoid of N–H⋯O interactions (bpy and dtbpy), the low selectivity could be attributed to the absence of differentiating NCIs between the meta and the para TSs. With the modified borylating agent, the predicted lower selectivity relative to the parent system is found to be due to the presence of similar efficiencies in the C–H⋯O and C–H⋯N interactions in the para TS to those in the meta TS. The regioselectivity of the borylation reaction thus hinges upon a set of NCIs that operate in concert and hence could be fine-tuned by making a rational choice of the ligand on the catalyst and suitable reactants. These conclusions are expected to have broader applicability in developing catalytic regioselective protocols using noncovalent interactions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Computing time from the SpaceTime supercomputing at IIT Bombay is gratefully acknowledged. A. U. acknowledges UGC (New Delhi) for a senior research fellowship. We thank Bhaskararao B., Reddi Y. and Kisan H. (IIT Bombay) for valuable discussions during the course of this work.Notes and references
- (a) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS; (b) J. Simon, S. Salzbrunn, G. K. S. Prakash, N. A. Petasis and G. A. Olah, J. Org. Chem., 2001, 66, 633–634 Search PubMed; (c) J. F. Hartwig, Acc. Chem. Res., 2012, 45, 864–873 CrossRef CAS PubMed.
- (a) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754–8786 CrossRef CAS PubMed; (b) R.-Y. Zhu, M. E. Farmer, Y.-Q. Chen and J.-Q. Yu, Angew. Chem., Int. Ed. Engl., 2016, 55, 10578–10599 CrossRef CAS PubMed; (c) Z. Dong, Z. Ren, S. J. Thompson, Y. Xu and G. Dong, Chem. Rev., 2017, 117, 9333–9403 CrossRef CAS PubMed; (d) Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247–9301 CrossRef CAS PubMed; (e) J. R. Hummel, J. A. Boerth and J. A. Ellman, Chem. Rev., 2017, 117, 9163–9227 CrossRef CAS PubMed; (f) C. Patel, V. Abraham and R. B. Sunoj, Organometallics, 2017, 36, 151–158 CrossRef CAS.
- (a) T. Ishiyama, J. Takagi, K. Ishida, N. Miyaura, N. R. Anastasi and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 390–391 CrossRef CAS PubMed; (b) H. Tamura, H. Yamazaki, H. Sato and S. Sakaki, J. Am. Chem. Soc., 2003, 125, 16114–16126 CrossRef CAS PubMed; (c) G. A. Chotana, B. A. Vanchura II, M. K. Tse, R. J. Staples, R. E. Maleczka Jr and M. R. Smith III, Chem. Commun., 2009, 0, 5731–5733 RSC.
- (a) T. A. Boebel and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 7534–7535 CrossRef CAS PubMed; (b) T. Ishiyama, H. Isou, T. Kikuchi and N. Miyaura, Chem. Commun., 2010, 46, 159–161 RSC; (c) P. C. Roosen, V. A. Kallepalli, B. Chattopadhyay, D. A. Singleton, R. E. Maleczka and M. R. Smith, J. Am. Chem. Soc., 2012, 134, 11350–11353 CrossRef CAS PubMed; (d) A. Ros, R. Fernández and J. M. Lassaletta, Chem. Soc. Rev., 2014, 43, 3229–3243 RSC.
- (a) J.-Y. Cho, M. K. Tse, D. Holmes, R. E. Maleczka and M. R. Smith, Science, 2002, 295, 305–308 CrossRef CAS PubMed; (b) T. Ishiyama, J. Takagi, J. F. Hartwig and N. Miyaura, Angew. Chem., Int. Ed. Engl., 2002, 41, 3056–3058 CrossRef CAS; (c) R. J. Phipps and M. J. Gaunt, Science, 2009, 323, 1593–1597 CrossRef CAS PubMed; (d) B. Chen, X.-L. Hou, Y.-X. Li and Y.-D. Wu, J. Am. Chem. Soc., 2011, 133, 7668–7671 CrossRef CAS PubMed; (e) N. Hofmann and L. Ackermann, J. Am. Chem. Soc., 2013, 135, 5877–5884 CrossRef CAS PubMed; (f) C. Cheng and J. F. Hartwig, Science, 2014, 343, 853–857 CrossRef CAS PubMed; (g) Y.-F. Yang, G.-J. Cheng, P. Liu, D. Leow, T.-Y. Sun, P. Chen, X. Zhang, J.-Q. Yu, Y.-D. Wu and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 344–355 CrossRef CAS PubMed; (h) Q. Yu, L. Hu, Y. Wang, S. Zheng and J. Huang, Angew. Chem., Int. Ed. Engl., 2015, 54, 15284–15288 CrossRef CAS PubMed; (i) Z. Dong, J. Wang and G. Dong, J. Am. Chem. Soc., 2015, 137, 5887–5890 CrossRef CAS PubMed; (j) S. Li, L. Cai, H. Ji, L. Yang and G. Li, Nat. Commun., 2016, 7, 10443 CrossRef PubMed.
- J. Li, S. Warratz, D. Zell, S. De Sarkar, E. E. Ishikawa and L. Ackermann, J. Am. Chem. Soc., 2015, 137, 13894–13901 CrossRef CAS PubMed.
- (a) K. M. Engle and J.-Q. Yu, J. Org. Chem., 2013, 78, 8927–8955 CrossRef CAS PubMed; (b) P. Wang, P. Verma, G. Xia, J. Shi, J. X. Qiao, S. Tao, P. T. W. Cheng, M. A. Poss, M. E. Farmer, K.-S. Yeung and J.-Q. Yu, Nature, 2017, 551, 489–493 CrossRef CAS PubMed.
- Applicable to C(sp3)–H activation: (a) G. Chen, Z. Zhuang, G.-C. Li, T. G. Saint-Denis, Y. Hsiao, C. L. Joe and J.-Q. Yu, Angew. Chem., Int. Ed. Engl., 2017, 56, 1506–1509 CrossRef CAS PubMed; (b) R.-Y. Zhu, T. G. Saint-Denis, Y. Shao, J. He, J. D. Sieber, C. H. Senanayake and J.-Q. Yu, J. Am. Chem. Soc., 2017, 139, 5724–5727 CrossRef CAS PubMed.
- (a) K. Müller-Dethlefs and P. Hobza, Chem. Rev., 2000, 100, 143–168 CrossRef; (b) R. R. Knowles and E. N. Jacobsen, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20678–20685 CrossRef CAS PubMed; (c) O. Takahashi, Y. Kohno and M. Nishio, Chem. Rev., 2010, 110, 6049–6076 CrossRef CAS PubMed; (d) M. J. Plevin, D. L. Bryce and J. Boisbouvier, Nat. Chem., 2010, 2, 466–471 CrossRef CAS PubMed; (e) L. M. Salonen, M. Ellermann and F. Diederich, Angew. Chem., Int. Ed. Engl., 2011, 50, 4808–4842 CrossRef CAS PubMed.
- (a) A. J. Neel, M. J. Hilton, M. S. Sigman and F. D. Toste, Nature, 2017, 543, 637–646 CrossRef CAS PubMed; (b) J. P. Wagner and P. R. Schreiner, Angew. Chem., Int. Ed. Engl., 2015, 54, 12274–12296 CrossRef CAS PubMed; (c) S. E. Wheeler, T. J. Seguin, Y. Guan and A. C. Doney, Acc. Chem. Res., 2016, 49, 1061–1069 CrossRef CAS PubMed; (d) R. B. Sunoj, Acc. Chem. Res., 2016, 49, 1019–1028 CrossRef CAS PubMed.
- (a) Y. Kuninobu, H. Ida, M. Nishi and M. Kanai, Nat. Chem., 2015, 7, 712–717 CrossRef CAS PubMed; (b) H. L. Li, Y. Kuninobu and M. Kanai, Angew. Chem., Int. Ed. Engl., 2017, 56, 1495–1499 CrossRef CAS PubMed.
- (a) R. Bisht and B. Chattopadhyay, J. Am. Chem. Soc., 2016, 138, 84–87 CrossRef CAS PubMed; (b) M. E. Hoque, R. Bisht, C. Haldar and B. Chattopadhyay, J. Am. Chem. Soc., 2017, 139, 7745–7748 CrossRef CAS PubMed; (c) M. R. Smith, R. Bisht, C. Haldar, G. Pandey, J. E. Dannatt, B. Ghaffari, R. E. Maleczka and B. Chattopadhyay, ACS Catal., 2018, 8, 6216–6223 CrossRef CAS PubMed.
- (a) H. J. Davis, M. T. Mihai and R. J. Phipps, J. Am. Chem. Soc., 2016, 138, 12759–12762 CrossRef CAS PubMed; (b) H. J. Davis and R. J. Phipps, Chem. Sci., 2017, 8, 864–877 RSC; (c) M. T. Mihai, H. J. Davis, G. R. Genov and R. J. Phipps, ACS Catal., 2018, 8, 3764–3769 CrossRef CAS.
- (a) M. Anand, R. B. Sunoj and H. F. Schaefer, ACS Catal., 2016, 6, 696–708 CrossRef CAS; (b) U. Dutta, A. Modak, B. Bhaskararao, M. Bera, S. Bag, A. Mondal, D. W. Lupton, R. B. Sunoj and D. Maiti, ACS Catal., 2017, 7, 3162–3168 CrossRef CAS; (c) S. Singh, S. K and R. B. Sunoj, J. Org. Chem., 2017, 82, 9619–9626 CrossRef CAS PubMed; (d) A. Deb, S. Singh, K. Seth, S. Pimparkar, B. Bhaskararao, S. Guin, R. B. Sunoj and D. Maiti, ACS Catal., 2017, 7, 8171–8175 CrossRef CAS; (e) C. Athira and R. B. Sunoj, Org. Biomol. Chem., 2016, 15, 246–255 RSC; (f) C. Athira, A. Changotra and R. B. Sunoj, J. Org. Chem., 2018, 83, 2627–2639 CrossRef CAS PubMed.
- Y. Kuninobu, H. Ida, M. Nishi and M. Kanai, Nat. Chem., 2015, 7, 712–717 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- (a) A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- (a) P. Fuentealba, H. Stoll, L. von Szentpaly, P. Schwerdtfeger and H. Preuss, J. Phys. B: At. Mol. Phys., 1983, 16, L323–L328 CrossRef CAS; (b) D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- (a) P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS; (b) M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS; (c) V. A. Rassolov, J. A. Pople, M. A. Ratner and T. L. Windus, J. Chem. Phys., 1998, 109, 1223–1229 CrossRef CAS; (d) V. A. Rassolov, M. A. Ratner, J. A. Pople, P. C. Redfern and L. A. Curtiss, J. Comput. Chem., 2001, 22, 976–984 CrossRef CAS.
- (a) B. Chattopadhyay, J. E. Dannatt, I. L. Andujar-De Sanctis, K. A. Gore, R. E. Maleczka, D. A. Singleton and M. R. Smith, J. Am. Chem. Soc., 2017, 139, 7864–7871 CrossRef CAS PubMed; (b) A. Guđmundsson, K. P. J. Gustafson, B. K. Mai, B. Yang, F. Himo and J.-E. Bäckvall, ACS Catal., 2018, 8, 12–16 CrossRef; (c) B. Tutkowski, S. Kerdphon, E. Limé, P. Helquist, P. G. Andersson, O. Wiest and P.-O. Norrby, ACS Catal., 2018, 8, 615–623 CrossRef CAS; (d) S. Chen, X. Huang, E. Meggers and K. N. Houk, J. Am. Chem. Soc., 2017, 139, 17902–17907 CrossRef CAS PubMed.
- (a) K. Fukui, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS; (b) C. Gonzalez and H. B. Schlegel, J. Chem. Phys., 1998, 90, 2154 CrossRef.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- S. Grimme, Chem.–Eur. J., 2012, 18, 9955–9964 CrossRef CAS PubMed.
- (a) R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, Oxford, New York, 1994 Search PubMed; (b) AIM2000 Version 2.0, Buro fur Innovative Software, SBK Software: Bielefeld, Germany, 2002 Search PubMed; (c) F. Biegler-König and J. Schönbohm, J. Comput. Chem., 2001, 22, 545–559 CrossRef; (d) F. Biegler-König and J. Schönbohm, J. Comput. Chem., 2002, 23, 1489–1494 CrossRef PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- S. Kozuch and S. Shaik, Acc. Chem. Res., 2011, 44, 101–110 CrossRef CAS PubMed.
- For more details of Ir(III)(bpy)tris(boryl) as an active catalyst, see: (a) T. M. Boller, J. M. Murphy, M. Hapke, T. Ishiyama, N. Miyaura and J. F. Hartwig, J. Am. Chem. Soc., 2005, 127, 14263–14278 CrossRef CAS PubMedB. A. Vanchura II, S. M. Preshlock, P. C. Roosen, V. A. Kallepalli, R. J. Staples, R. E. Maleczka Jr, D. A. Singleton and M. R. Smith III, Chem. Commun., 2010, 46, 7724–7726 RSC; (b) P. C. Roosen, V. A. Kallepalli, B. Chattopadhyay, D. A. Singleton, R. E. Maleczka and M. R. Smith, J. Am. Chem. Soc., 2012, 134, 11350–11353 CrossRef CAS PubMed; (c) Q. Li, C. W. Liskey and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 8755–8765 CrossRef CAS PubMed.
- (a) In the model system, the cyclohexane (Cy) of the ligand and the N-hexyl chain of the substrate are replaced by a methyl group and the borylating reagent B2pin2 is replaced by bis(ethyleneglycolato)diboron (B2eg2); (b) More details are provided in Fig. S1 in the ESI.†; (c) In the most favored geometry of the active catalyst, N–H···O interactions between the oxygen atom of the Beg (from B2eg2) ligand and the N–Hs of the urea bring the urea moiety closer to the Beg; (d) Two key configurations of the active catalyst, with and without the N–H···O interactions differ by about 6 kcal mol−1. See Fig. S2 in the ESI† for further details.
- In addition, the meta C–H activation TS obtained from the energetically favored geometry of the active catalyst (A1) differed by around 12 kcal mol−1 relative to the one obtained from A2. See Fig. S8 in the ESI.†.
- See Fig. S3 in the ESI.†.
- The most favorable arrangement of the catalyst-substrate complex along with their computed energies is shown in Fig. S4 in the ESI.†.
- (a) The optimized geometries of the catalyst-substrate complex in different binding modes are provided in Fig. S5 in the ESI.†; (b) Interesting strategies, such as template assisted distal functionalization, have been developed to access distal meta and para positions of aryl rings, see: ; (c) D. Leow, G. Li, T.-S. Mei and J.-Q. Yu, Nature, 2012, 486, 518–522 CrossRef CAS PubMed; (d) A. Maji, B. Bhaskararao, S. Singha, R. B. Sunoj and D. Maiti, Chem. Sci., 2016, 7, 3147–3153 RSC; (e) Y. Wu, Y.-Q. Chen, T. Liu, M. D. Eastgate and J.-Q. Yu, J. Am. Chem. Soc., 2016, 138, 14554–14557 CrossRef CAS PubMed; (f) X.-H. Liu, H. Park, J.-H. Hu, Y. Hu, Q.-L. Zhang, B.-L. Wang, B. Sun, K.-S. Yeung, F.-L. Zhang and J.-Q. Yu, J. Am. Chem. Soc., 2017, 139, 888–896 CrossRef CAS PubMed; (g) H.-J. Xu, Y. Lu, M. E. Farmer, H.-W. Wang, D. Zhao, Y.-S. Kang, W.-Y. Sun and J.-Q. Yu, J. Am. Chem. Soc., 2017, 139, 2200–2203 CrossRef CAS PubMed; (h) Z. Zhang, K. Tanaka and J.-Q. Yu, Nature, 2017, 543, 538–542 CrossRef CAS PubMed; (i) A. Maji, S. Guin, S. Feng, A. Dahiya, V. K. Singh, P. Liu and D. Maiti, Angew. Chem., Int. Ed. Engl., 2017, 56, 14903–14907 CrossRef CAS PubMed.
- (a) The energies of the C–H activation TSs in the solvent phase for the model system with Ir···π and Ir···N modes of binding are, respectively, 1.1 and 0.7 kcal mol−1 less favored over the Ir···O binding. A similar trend is also found for the corresponding activation barriers (∼26 to 27 kcal mol−1), suggesting that all the three binding modes could facilitate C–H activation at the ortho position; (b) The optimized TS geometries for the ortho C–H activation in all three modes of the model system are given in Fig. S6 in the ESI.†.
- The details of conformational sampling of the transition state geometries, particularly arising due to the long hexyl chains, are presented in Tables S1–S5 in the ESI.†.
- The optimized TS geometries for all the steps are provided in the ESI (Fig. S7†).
- (a) To obtain the maximum δE, different likely stationary points involved in the catalytic cycle are considered as the TDTS and TDI. More details are provided in Table S6 in the ESI.†; (b) The turn-over frequency estimated using the calculated δE is found to be 4.8 × 10−4 s−1; (c) S. Kozuch and J. M. L. Martin, ACS Catal., 2012, 2, 2787–2794 CrossRef CAS.
- E. Espinosa, I. Alkorta, J. Elguero and E. Molins, J. Chem. Phys., 2002, 117, 5529 CrossRef CAS.
- The details of the important NCIs as identified through the AIM analysis along with their energetic contribution calculated using Espinosa's formulation for the systems, ubpy-S0 and ubpy-S1, are presented in Table S7 in the ESI.†.
- Additional depiction of all these noncovalent interactions is provided in the form of NCI plots in Fig. S9 in the ESI.†.
- F. M. Bickelhaupt and K. N. Houk, Angew. Chem., Int. Ed. Engl., 2017, 56, 10070–10086 CrossRef CAS PubMed.
- The details of the DI-AS analysis are provided in Fig. S10 and Table S8 in the ESI.†.
- (a) P. T. Brain, A. J. Downs, P. Maccallum, D. W. H. Rankin, H. E. Robertson and G. A. Forsyth, J. Chem. Soc., Dalton Trans., 1991, 0, 1195–1200 RSC; (b) H. Braunschweig and A. Damme, Chem. Commun., 2013, 49, 5216–5218 RSC; (c) H. Kinuta, H. Takahashi, M. Tobisu, S. Mori and N. Chatani, Bull. Chem. Soc. Jpn., 2014, 87, 655–669 CrossRef CAS; (d) F. Haeffner, Comput. Theor. Chem., 2018, 1131, 90–98 CrossRef CAS.
- (a) The predicted low regioselectivity is comparable to the closest experimental observation wherein Ir-dtbpy, devoid of the phenyl-urea linker yielded a meta to para ratio of 1.9. Our calculations of the dtbpy-S0 system also indicated an energy difference of only 0.4 kcal mol−1 in favor of meta C–H activation; (b) The meta and para C–H activation TS geometries for the dtbpy-S0 system are given in Fig. S11 in the ESI.†.
- The meta and para C–H activation TS geometries with a modified Ir-bound ligand (bpy) as well as with a modified borylating agent (B1) are given in Fig. S12 in the ESI.†.
- Relative enthalpy and free energy differences between the meta and para C–H activation TSs for the modified and unmodified systems are presented in Table S9 in the ESI.†.
- (a) The relative strengths of all the important NCIs in terms of their individual energetic contribution for the ligand bpy and the borylating agent B1 are provided in Table S10 in the ESI.†; (b) The bar diagrams for bpy and B1 systems are given in Fig. S13 in the ESI.†.
- The details of the DI-AS analysis for dtbpy, bpy and B1 modifications are provided in Tables S11–S13 in the ESI.†.
- (a) The calculations were also performed with a much larger basis set, 6-311++G**, for all atoms except for iridium, for which LANL2TZ(f) was used; (b) The details are provided in Table S14 in the ESI.† Various levels of theories were consistent in conveying the meta C–H activation as the most favored site of activation followed by ortho and para positions.
- (a) See Fig. S14 and S15 in the ESI† for Gibbs free energy profiles of para and ortho borylations; (b) The C–H activation step in the ortho and para pathways is found to be more reversible toward the reactants. The barriers for the reversal of the Ir(V)-aryl intermediate (2) are 6.7 and 4.6 kcal mol−1, respectively, for ortho and para intermediates. The corresponding value of the meta intermediate is 8.5 kcal mol−1. The elementary step barriers for the forward reaction (i.e., the reductive elimination from 2o and 2p) in the ortho and para pathways are, respectively, 15.7 and 6.6 kcal mol−1, suggesting that ortho borylation is least likely. Thus, para borylation is the only alternative possibility other than the meta product.
- (a) To obtain a maximum δE, different likely stationary points involved in the catalytic cycle for both the para and ortho pathways are considered as the TDTS and TDI. See Table S15 and S16 in the ESI† for additional details; (b) The turn-over frequency estimated using the calculated δE is found to be 5.6 × 10−8 and 5.3 × 10−12 s−1, respectively, for the para and ortho isomers.
- The enthalpies of formation of meta, ortho and para borylated products are summarized in Table S17 in the ESI.†.
- (a) Energy profile diagram of the oxidative addition and RE steps for three isomers are given in Fig. S16 in the ESI.†; (b) The energy of the catalyst-substrate complex in each case differs by about 4 kcal mol−1. See Fig. S15 in the ESI† for additional details; (c) Calculations at other higher levels of theories for RE in the case of meta, ortho and para isomers are presented in Table S18 in ESI.†.
Footnote |
| † Electronic supplementary information (ESI) available: Optimized geometries and additional schemes, figures, and tables as described in the text along with the Cartesian coordinates are provided. See DOI: 10.1039/c8sc05335a |
| This journal is © The Royal Society of Chemistry 2019 |