
Photocatalytic decarboxylative alkenylation of α-amino and α-hydroxy acid-derived redox active esters by NaI/PPh3 catalysis†
Ya-Ting
Wang
a,
Ming-Chen
Fu
a,
Bin
Zhao
a,
Rui
Shang
 *ab and
Yao
Fu
*a
*ab and
Yao
Fu
*a
aHefei National Laboratory for Physical Sciences at the Microscale, CAS Key Laboratory of Urban Pollutant Conversion, Anhui Province Key Laboratory of Biomass Clean Energy, iChEM, University of Science and Technology of China, Hefei 230026, China. E-mail: fuyao@ustc.edu.cn
bDepartment of Chemistry, School of Science, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: rui@chem.s.u-tokyo.ac.jp
First published on 17th January 2020
Abstract
Herein, we report the photocatalytic decarboxylative alkenylation reactions of N-(acyloxy)phthalimide derived from α-amino and α-hydroxy acids with 1,1-diarylethene, and with cinnamic acid derivatives through double decarboxylation, using sodium iodide and triphenylphosphine as redox catalysts. The reaction proceeds under mild irradiation conditions with visible blue light (440 nm or 456 nm) in an acetone solvent without recourse to transition-metal or organic dye based photoredox catalysts. The reaction proceeds via photoactivation of a transiently self-assembled chromophore from N-(acyloxy)phthalimide and NaI/PPh3. Solvation plays a crucial role in the reactivity.
While most of the currently reported photoredox catalysis1 reactions entail the use of transition-metal-2 or organic dye-3 based photoredox catalysts to harvest photon energy of visible light, our recent work revealed that sodium iodide and triphenyl phosphine in combination with suitable substrates [e.g. N-(acyloxy)phthalimide or redox active esters (RAEs)]4 form a transiently assembled chromophore in a suitable solvent system, which absorbs visible light to deliver high energy radical species useful for organic synthesis.5 We recently reported the photocatalytic decarboxylative alkylation of silyl enol ether and N-heteroarene using the NaI/PPh3 photoredox catalytic system with broad substrate scope. In this work, we report the application of NaI/PPh3 photoredox catalysis for direct decarboxylative alkenylation6 of α-amino and α-hydroxy acid-derived redox active esters with 1,1-diarylethenes,7 and with cinnamic acid derivatives via double decarboxylation.8 These transformations generate structure motifs of allylic amine9 and allylic ether without using any transition metal under redox neutral conditions. Compared with the previously reported catalytic condition using ruthenium-, iridium-, or organic dye-based photoredox catalysts for similar transformations,10 the NaI/PPh3 system has the advantages of easy accessibility and low cost, as well as the green solvent system using low boiling-point acetone.11
Our working hypothesis using the NaI/PPh3 photoredox system for decarboxylative alkenylation of α-amino acid-derived RAEs is depicted in Fig. 1. It is reported that NaI, PPh3, and RAEs assemble to form a chromophore that absorbs visible light of blue range in an acetone solvent.5 We envisaged that the photoactivation of this transiently formed chromophore will induce electron transfer from iodide to an N-phthalimide moiety to further generate α-amino alkyl radicals through extrusion of carbon dioxide for alkenylation reaction. PPh3 will stabilize iodine radicals to generate ˙I-PPh3 species,12 which resemble a photoredox catalyst in their oxidized form. The formed α-amino alkyl radical attacks 1,1-diarylethene and cinnamic acid to generate relatively stable benzylic radicals which were subsequently oxidized by ˙I-PPh3 to produce the alkenylation products after proton elimination and to regenerate NaI/PPh3. Thus, the process delivers the product of decarboxylative alkenylation, in which the carboxylate group in a carboxylic acid derivative is replaced by an alkene moiety.6a In this work, we successfully developed this transformation with substantial substrate scope and good functional group compatibility. The reaction reported herein further extended the synthetic scope of the NaI/PPh3 catalytic system for low-cost photoredox catalysis.
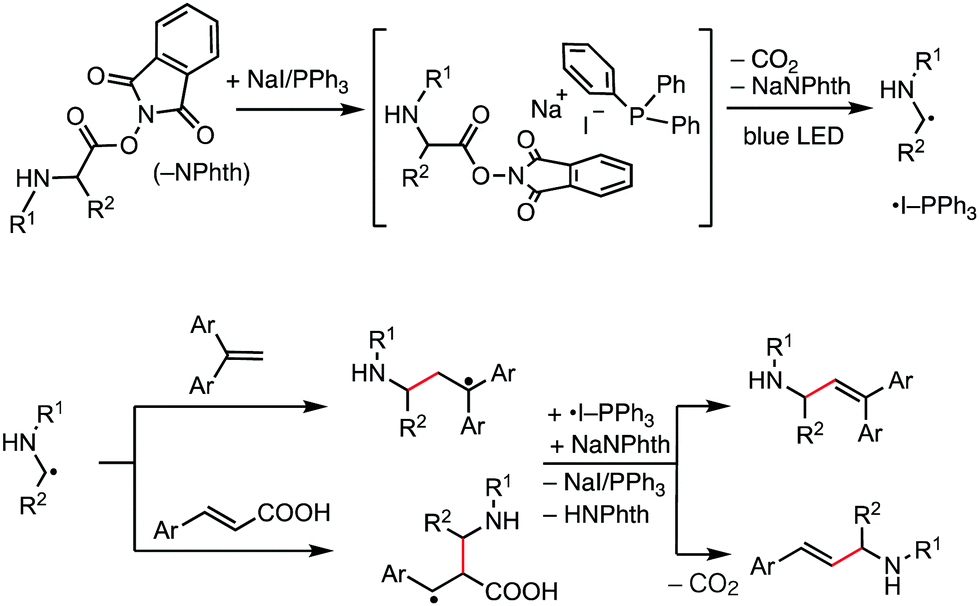 | ||
| Fig. 1 Working hypothesis of photocatalytic decarboxylative alkenylation using NaI/PPh3 catalysis. | ||
Decarboxylative alkenylation was first performed using 1,1-diphenylethylene (1) and RAEs of Cbz-protected alanine (2) as coupling partners. After careful optimization of all reaction parameters, the desired product 3 was obtained in 84% isolated yield using a combination of NaI (10 mol%) and PPh3 (10 mol%) as photocatalysts in acetone under 456 nm blue LED irradiation at room temperature. Measuring the UV-vis absorption spectrum revealed that a chromophore with absorption onset overlapping with the wavelength of blue LEDs was formed in the reaction mixture (see the ESI,† Fig. S2 for details). Control experiments revealed that iodide, phosphine, and irradiation were all essential parameters. As noted in a previous report,5,13 solvation has a heavy influence on the assembly of the charge-transfer complex (CTC) through non-covalent interactions. As shown in Scheme 1 (second row), the reaction also proceeded in THF, NMP, DMA or DMF, albeit in lower yields. 1,4-Dioxane and PhCF3, which were suitable solvents in NaI/PPh3-catalyzed decarboxylative alkylation of N-heterocycles,5 were totally ineffective. Because phosphine stabilizes iodine radicals in the form of R3P-I˙ species and facilitates intermolecular charge transfer, a series of phosphines with different electronic natures were examined (Scheme 1, third row). Tris(4-methoxyphenyl)phosphine and tris(4-fluorophenyl)phosphine gave no significant change in the yield of 3 compared with PPh3. When tricyclohexylphosphine was used instead of PPh3, the isolated yield of 3 was reduced to 70%. The reaction did not proceed when BINAP or tri(2-furanyl)phosphine was used as the catalyst. Investigation of other alkali iodides (see the ESI† for further details) showed that LiI, KI, RbI, and CsI gave slightly lower yields compared to NaI. ZnI2 failed to serve as a catalyst for this transformation as ZnI2 did not form a chromophore with 2 to absorb blue light under the reaction conditions (see the ESI,† Fig. S2 for further details). It is interesting to note that tetrabutylammonium iodide, which is entirely ineffective in the decarboxylative alkylation of silyl enol ethers,5 also produced the desired product in 70% yield. Although we could not fully rationalize the observed cation effect on reaction efficiency, we speculate that the cation affects the assembly of the transient chromophore, electron transfer, and further diffusion of the radical pair in a solvent cage. The reaction can be easily scaled up to gram scale without reducing its efficiency.
 | ||
| Scheme 1 Key reaction parameters. Reaction conditions: 1 (0.2 mmol, 1.0 eq.), 2 (0.3 mmol, 1.5 eq.), NaI (10 mol%), PPh3 (10 mol%), acetone (2.0 mL), irradiation with blue LEDs (456 nm) at room temperature for 15 h under an argon atmosphere. Yield of the isolated product. | ||
With the optimized reaction conditions in hand, the scope of the reaction was investigated. As shown in Scheme 2, a wide range of natural and unnatural α-amino acid-derived RAEs and 1,1-diarylethylenes were found to be suitable substrates with moderate to good yields obtained. A variety of functional groups, such as phthalimide (4), benzyloxycarbonyl (5, 13), tert-butyloxycarbonyl (6), ester (7), aryl halides (8, 20, 21, 22), and sulfide (9) were all well-tolerated. Terminal alkyne could also be tolerated affording the desired product (14) in 61% yield. RAEs derived from proline (10, 11) and 2-azetidinecarboxylic acid (12) reacted smoothly. 1,1-Diarylethylenes of different electronic properties were examined. Both electron-rich (19) and electron-deficient (20, 21) alkenes could be used. It is worth noting that dipeptide-derived RAEs generated the desired alkenylation products in good yields (16, 17, 23), suggesting the potential to use this method for the modification of bioactive peptides.14 For certain substrates, the catalyst loading was increased to 20 mol% (4) and the solvent was modified (6) to improve the yields. The reaction did not proceed well with styrene and 1,1-dialkylethylene, probably because of the much slower rate of radial addition to the double bond of these alkenes compared with 1,1-diarylethylene.
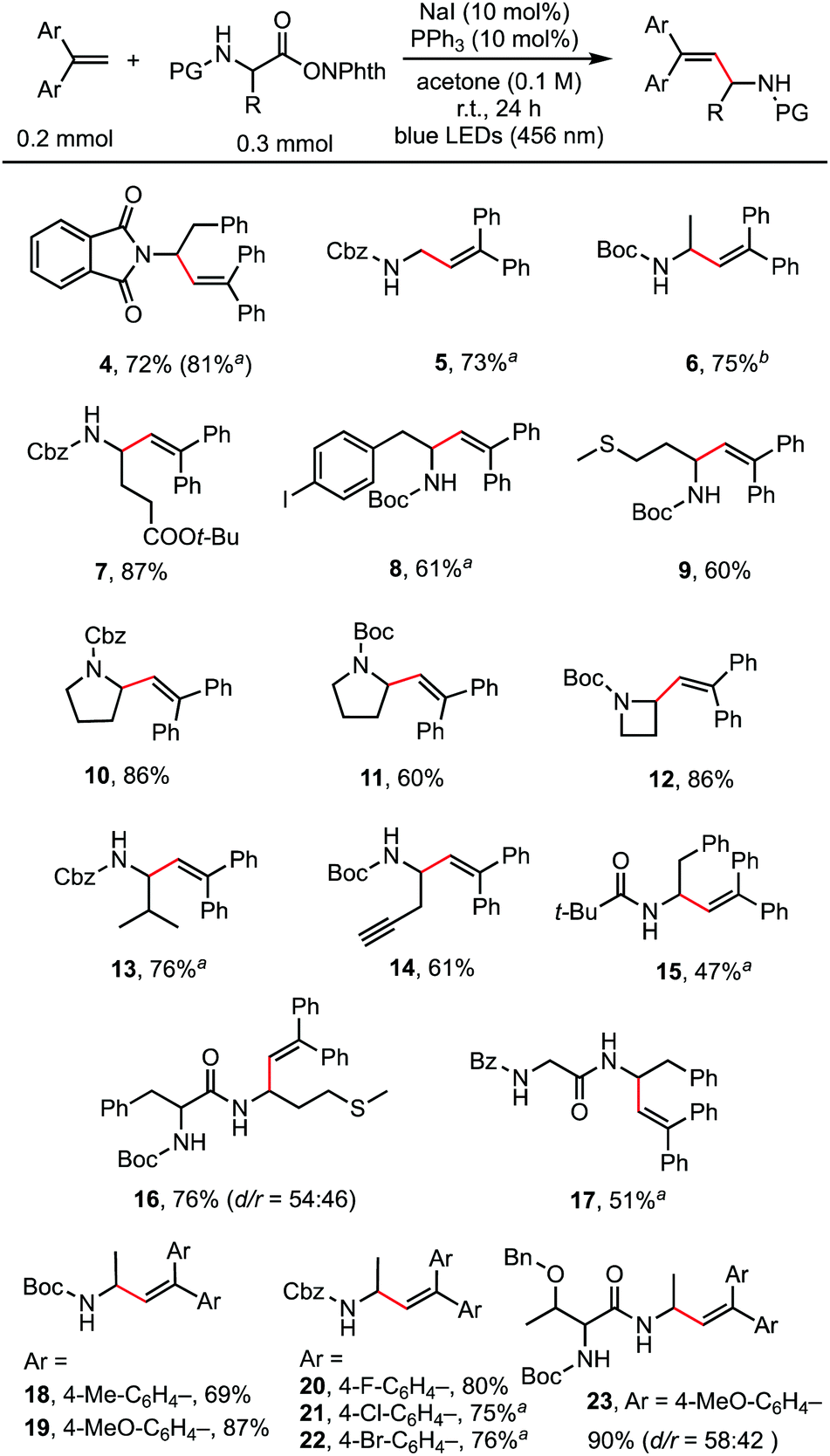 | ||
Scheme 2 Decarboxylative alkenylation of α-amino acid-derived RAEs. Reaction conditions: 1,1-diarylethylenes (0.2 mmol, 1.0 eq.), RAEs (0.3 mmol, 1.5 eq.), NaI (10 mol%), PPh3 (10 mol%), acetone (2.0 mL), irradiation with blue LEDs (456 nm) at room temperature for 24 h under an argon atmosphere. Yield of isolated product. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) NaI (20 mol%), PPh3 (20 mol%). bNaI (20 mol%), PPh3 (20 mol%), THF (2.0 mL). NaI (20 mol%), PPh3 (20 mol%). bNaI (20 mol%), PPh3 (20 mol%), THF (2.0 mL). | ||
Besides α-amino acids, α-hydroxy acids were also amenable coupling partners (Scheme 3). RAEs derived from various O-protected α-hydroxy acids, including O-aryl, O-alkyl and O-benzyl substituents, reacted readily with 1,1-diphenylethylene to give the alkenylation product in moderate to good yields (66–95%). Functionalities such as aryl chloride, aryl iodide, and even aryl pinacol boronate, which were useful for subsequent cross-coupling reactions,15 were well tolerated as shown in entries 25, 26, and 27. Cyclic α-alkoxy acids (30, 31) also reacted well. This reaction is easily scaled up to gram scale without decreasing the yield. Thioglycolic acid-derived redox-active ester was also suitable for this transformation to construct an allylic sulphide structure (32).
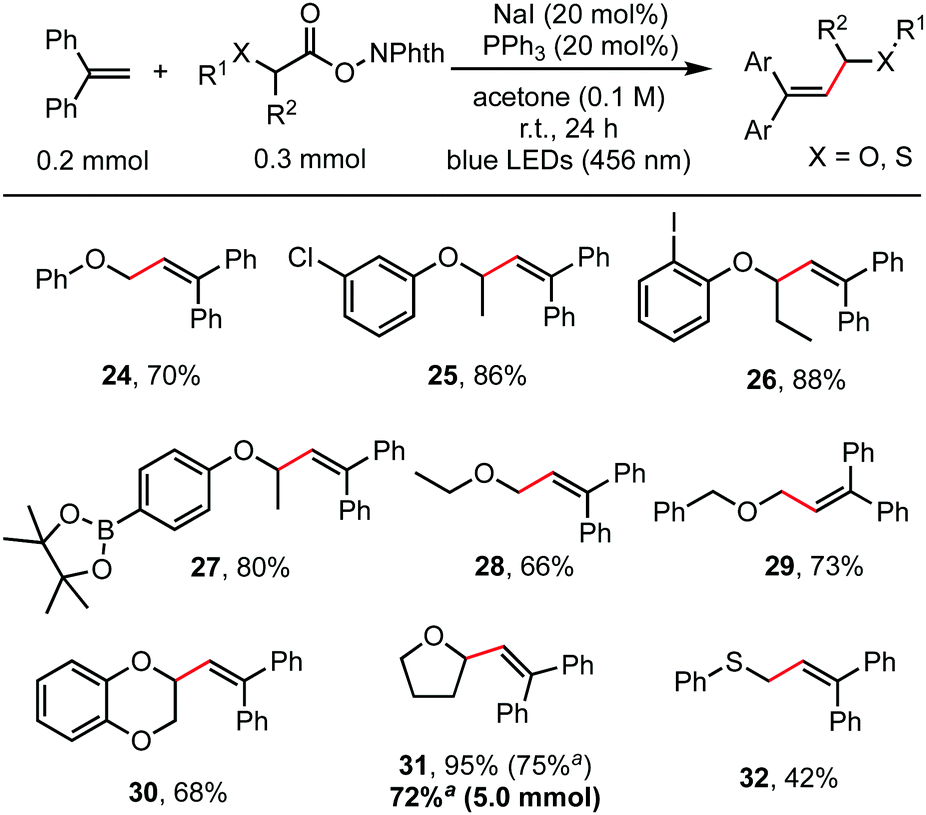 | ||
| Scheme 3 Decarboxylative alkenylation of α-hydroxy acid and thioglycolic acid derivatives. Reaction conditions: 1,1-diphenylethylenes (0.2 mmol, 1.0 eq.), RAEs (0.3 mmol, 1.5 eq.), NaI (20 mol%), PPh3 (20 mol%), acetone (2.0 mL), irradiation by blue LEDs (456 nm) at room temperature for 24 h under an argon atmosphere. Yield of the isolated product. aNaI (10 mol%), PPh3 (10 mol%). | ||
As shown in the working hypothesis, alkenylation between α-amino acid-derived RAEs and cinnamic acids via dual decarboxylation also proceeded successfully without further optimization of the reaction conditions. As shown in Scheme 4, cinnamic acid and its analogues reacted readily with RAEs derived from N-Boc-protected phenylalanine furnishing the desired products in moderate to good yields. A methoxy substituent on the para- (33), meta- (34), and ortho-position (35) of cinnamic acid has no obvious effect on the reaction outcome. Dimethylamino (36) and diphenylamino (37) groups, which are sensitive to oxidant, were also compatible. This protocol is also applicable to acrylic acids possessing β-heteroarene-substituents, such as thiophene (38), benzothiophene (39), benzofuran (40), pyridine (41), and furan (42).
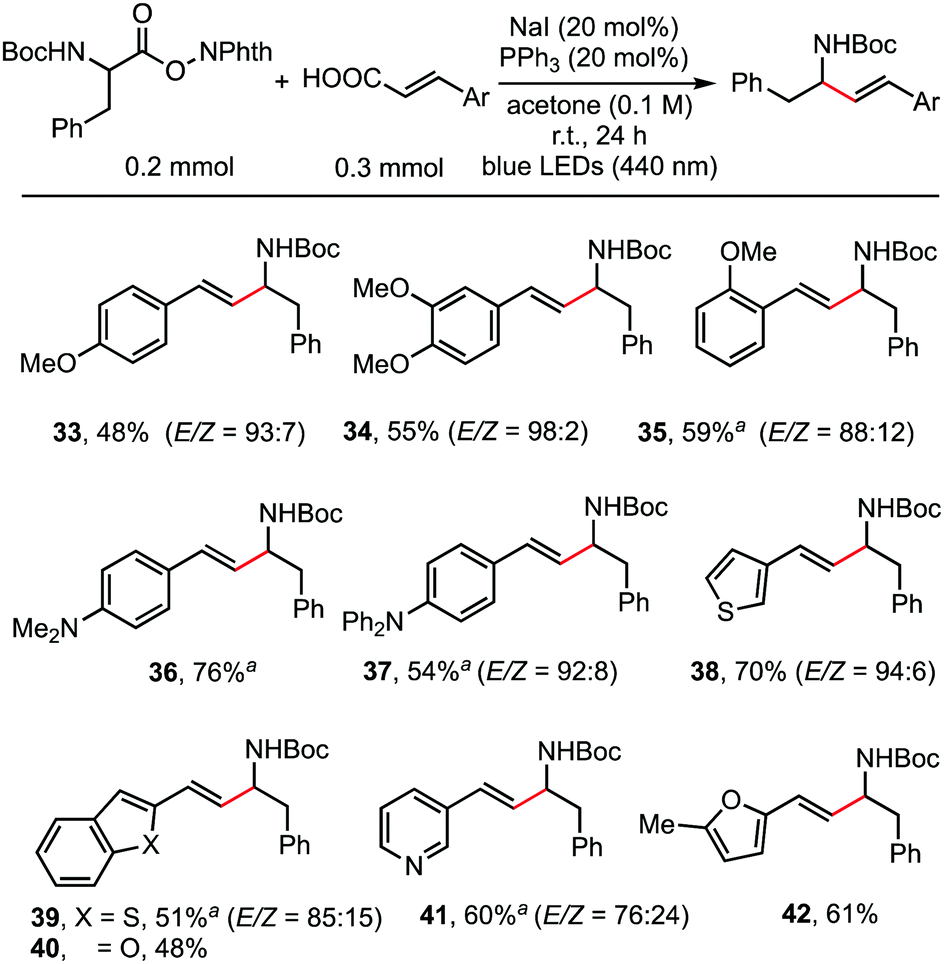 | ||
| Scheme 4 NaI/PPh3-catalyzed alkenylation of α-amino-acid derived RAEs via double decarboxylation. Reaction conditions: RAEs (0.2 mmol, 1.0 eq.), cinnamic acid derivatives (0.3 mmol, 1.5 eq.), NaI (20 mol%), PPh3 (20 mol%), acetone (2.0 mL), irradiation with blue LEDs (440 nm) at room temperature for 24 h under an argon atmosphere. Yield of the isolated product. aDMA (2 mL) instead of acetone (2 mL). | ||
The reaction in Scheme 1 was totally suppressed when 2,2,6,6-tetramethylpiperding-1-oxyl (TEMPO) was added in 2 equivalent amounts as a radical scavenger (see the ESI†). A radical-clock experiment was performed as shown in Scheme 5. A ring-closed product (43) was obtained in 57% yield. These results indicated that the decarboxylative alkenylation proceeds through a free radical pathway.16 Finally, besides reaction with tertiary alkyl as demonstrated in our previous work,5 the reaction also proceeded well with RAEs that deliver secondary and primary normal alkyls (eqn (2) and (3)), though the reaction with primary normal alkyl proceeded in a relatively lower yield. The reaction did not proceed well with RAE derived from phenyl acetic acid.
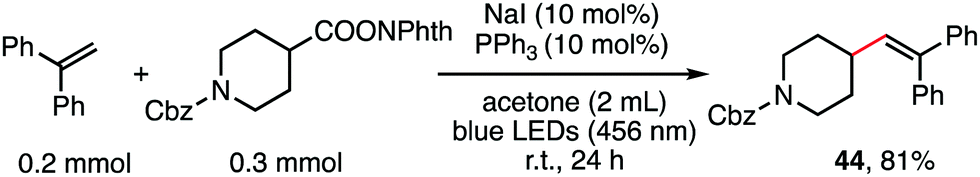 | (2) |
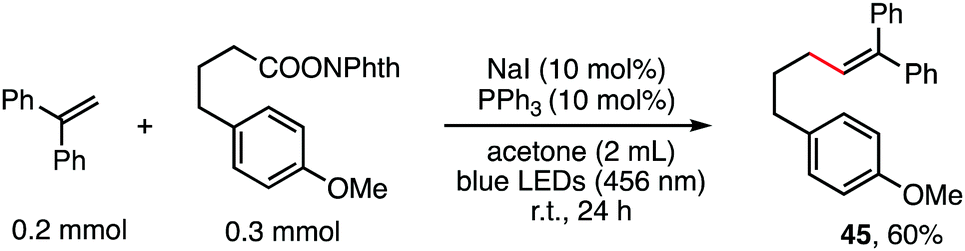 | (3) |
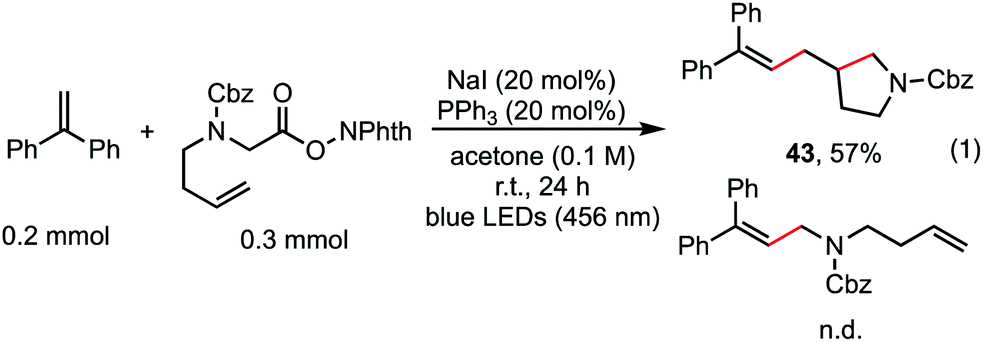 | ||
| Scheme 5 Radical-clock experiment. | ||
In conclusion, NaI/PPh3 works as an easily accessible and low-cost redox catalyst for decarboxylative alkenylation of various RAEs derived from α-amino acids, α-hydroxy acids, and also thioglycolic acid with 1,1-diarylethenes. Cinnamic acid and its analogues were also used as alkenylation reagents through double decarboxylation. The reactions reported herein offer a low-cost and operationally simple method to synthesize compounds containing allylic amine and allylic ether structures from biomass-derived carboxylic acids.
We are grateful for the support from the National Key R&D Program of China (2018YFB1501600), the National Natural Science Foundation of China (21572212, 21732006, 51821006, 51961135104), the Strategic Priority Research Program of CAS (XDB20000000), HCPST (2017FXZY001), KY (2060000119), the China Postdoctoral Science Foundation (2018M642519), the Fundamental Research Funds for the Central Universities (WK2060120005) and the USTC Research Funds of the Double First-Class Initiative (YD2060002003).
Conflicts of interest
The authors declare no competing interests.Notes and references
- (a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed; (b) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898 CrossRef CAS; (c) M. N. Hopkinson, A. Tlahuext-Aca and F. Glorius, Acc. Chem. Res., 2016, 49, 2261 CrossRef CAS; (d) J. J. Douglas, M. J. Sevrin and C. R. J. Stephenson, Org. Process Res. Dev., 2016, 20, 1134 CrossRef CAS; (e) J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS; (f) M. D. Kärkäs, ACS Catal., 2017, 7, 4999 CrossRef.
- (a) J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828 CrossRef CAS PubMed; (b) J. C. Tellis, D. N. Primer and G. A. Molander, Science, 2014, 345, 433 CrossRef CAS PubMed; (c) J. Jin and D. W. C. MacMillan, Nature, 2015, 525, 87 CrossRef CAS; (d) E. B. Corcoran, M. T. Pirnot, S. Lin, S. D. Dreher, D. A. DiRocco, I. W. Davies, S. L. Buchwald and D. W. C. MacMillan, Science, 2016, 353, 279 CrossRef CAS; (e) C. P. Johnston, R. T. Smith, S. Allmendinger and D. W. C. MacMillan, Nature, 2016, 536, 322 CrossRef CAS PubMed; (f) C. Le, Y. Liang, R. W. Evans, X. Li and D. W. C. MacMillan, Nature, 2017, 547, 79 CrossRef CAS; (g) E. R. Welin, C. Le, D. M. Arias-Rotondo, J. K. McCusker and D. W. C. MacMillan, Science, 2017, 355, 380 CrossRef CAS PubMed.
- (a) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075 CrossRef CAS PubMed; (b) J. Luo and J. Zhang, ACS Catal., 2016, 6, 873 CrossRef CAS; (c) M. K. Bogdos, E. Pinard and J. A. Murphy, Beilstein J. Org. Chem., 2018, 14, 2035 CrossRef CAS PubMed.
- (a) T. Qin, J. Cornella, C. Li, L. R. Malins, J. T. Edwards, S. Kawamura, B. D. Maxwell, M. D. Eastgate and P. S. Baran, Science, 2016, 352, 801 CrossRef CAS PubMed; (b) W.-M. Cheng, R. Shang and Y. Fu, ACS Catal., 2017, 7, 907 CrossRef CAS; (c) W.-M. Cheng, R. Shang, M.-C. Fu and Y. Fu, Chem. – Eur. J., 2016, 22, 11161 CrossRef PubMed; (d) S. Murarka, Adv. Synth. Catal., 2018, 360, 1735 CrossRef CAS; (e) J. Schwarz and B. König, Green Chem., 2018, 20, 323 RSC; (f) J.-Q. Liu, A. Shatskiya, B. S. Matsuura and M. D. Kärkäs, Synthesis, 2019, 51, 2759 CrossRef CAS.
- M.-C. Fu, R. Shang, B. Zhao, B. Wang and Y. Fu, Science, 2019, 363, 1429 CrossRef CAS PubMed.
- (a) J. T. Edwards, R. R. Merchant, K. S. McClymont, K. W. Knouse, T. Qin, L. R. Malins, B. Vokits, S. A. Shaw, D.-H. Bao, F.-L. Wei, T. Zhou, M. D. Eastgate and P. S. Baran, Nature, 2017, 545, 213 CrossRef CAS PubMed; (b) G.-Z. Wang, R. Shang and Y. Fu, Org. Lett., 2018, 20, 888 CrossRef CAS PubMed; (c) M. Koy, F. Sandfort, A. Tlahuext-Aca, L. Quach, C. G. Daniliuc and F. Glorius, Chem. – Eur. J., 2018, 24, 4552 CrossRef CAS PubMed.
- (a) A. J. Borah and G. Yan, Org. Biomol. Chem., 2015, 13, 8094 RSC; (b) C. Wang, Y. Lei, M. Guo, Q. Shang, H. Liu, Z. Xu and R. Wang, Org. Lett., 2017, 19, 6412 CrossRef CAS PubMed.
- (a) K. Xu, Z. Tan, H. Zhang, J. Liu, S. Zhang and Z. Wang, Chem. Commun., 2017, 53, 10719 RSC; (b) J.-J. Zhang, J.-C. Yang, L.-N. Guo and X.-H. Duan, Chem. – Eur. J., 2017, 23, 10259 CrossRef CAS PubMed.
- M.-Y. Li, M. González-Esguevillas, S. Berritt, X.-D. Yang, A. Bellomo and P. J. Walsh, Angew. Chem., Int. Ed., 2016, 55, 2825 CrossRef CAS PubMed.
- (a) C. Hu and Y. Chen, Org. Chem. Front., 2015, 2, 1352 RSC; (b) H. Cao, H. Jiang, H. Feng, J. M. C. Kwan, X. Liu and J. Wu, J. Am. Chem. Soc., 2018, 140, 16360 CrossRef CAS PubMed; (c) X. Jiang, M.-M. Zhang, W. Xiong, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 2402 CrossRef CAS PubMed.
- M. C. R. Symons and R. L. Petersen, J. Chem. Soc., Faraday Trans. 2, 1979, 75, 210 RSC.
- (a) A. Noble and V. K. Aggarwa, Sci. China: Chem., 2019, 62, 1083 CrossRef CAS; (b) B. List and Y. Li, Synfacts, 2019, 15, 0791 CrossRef.
- O. Kysel, G. Juhász, P. Mach and G. Košík, Chem. Pap., 2007, 61, 66 CAS.
- (a) S. Bloom, C. Liu, D. K. Kölmel, J. X. Qiao, Y. Zhang, M. A. Poss, W. R. Ewing and D. W. C. MacMillan, Nat. Chem., 2017, 10, 205 CrossRef PubMed; (b) J. Wang, H. Lundberg, S. Asai, P. Martín-Acosta, J. S. Chen, S. Brown, W. Farrell, R. G. Dushin, C. J. O’Donnell, A. S. Ratnayake, P. Richardsonc, Z. Liu, T. Qin, D. G. Blackmond and P. S. Baran, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E6404 CrossRef CAS PubMed.
- (a) H. C. Brown, G. W. Kramer, A. B. Levy and M. M. Midland, Organic Syntheses via Boranes, Wiley-Interscience, New York, 1975 Search PubMed; (b) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS.
- (a) Ł. Woźniak, J. J. Murphy and P. Melchiorre, J. Am. Chem. Soc., 2015, 137, 5678 CrossRef PubMed; (b) D. Wang, N. Zhu, P. Chen, Z. Lin and G. Liu, J. Am. Chem. Soc., 2017, 139, 15632 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc09654j |
| This journal is © The Royal Society of Chemistry 2020 |