
Spin crossover modulation in a coordination polymer with the redox-active bis-pyridyltetrathiafulvalene (py2TTF) ligand†
Lisa
Zappe
a,
Sophie
Schönfeld
a,
Gerald
Hörner
 a,
Katrina A.
Zenere
b,
Chanel F.
Leong
b,
Cameron J.
Kepert
b,
Deanna M.
D'Alessandro
b,
Birgit
Weber
*a and
Suzanne M.
Neville
*c
a,
Katrina A.
Zenere
b,
Chanel F.
Leong
b,
Cameron J.
Kepert
b,
Deanna M.
D'Alessandro
b,
Birgit
Weber
*a and
Suzanne M.
Neville
*c
aDepartment of Chemistry, University of Bayreuth, Universitätsstraße 30, 95448 Bayreuth, Germany. E-mail: weber@uni-bayreuth.de
bSchool of Chemistry, The University of Sydney, Sydney, New South Wales 2006, Australia
cSchool of Chemistry, University of New South Wales, Sydney, NSW 2052, Australia. E-mail: s.neville@unsw.edu.au
First published on 7th August 2020
Abstract
A one-dimensional FeII coordination polymer (CP) has been formed which includes the redox-active ligand bis-pyridyltetrathiafulvalene (py2TTF) and a Schiff base-like N2O2 ligand. This CP is both spin crossover (SCO) and redox-active in the solid-state, and chemical oxidation results in SCO modification.
The structural modularity of coordination polymers (CPs) offers many opportunities for ligand-, metal- or multi-functionality.1 There are many CPs which have been rationally designed towards achieving redox-active and redox-switchable CPs.2 Of these, the electron-rich tetrathiafulvalene (TTF) core is a commonly utilised bridging organic ligand component as it shows two reversible and easily accessible oxidation processes to its radical cation (TTF˙+) and dication (TTF2+) states. Since the TTF moiety is flexible and the difference in ligand geometry between the neutral planar and boat-like oxidised states is not large,3 flexible CPs may be expected to accommodate the redox-induced structural change without framework collapse. In such CPs, the redox activity of the TTF core can be manipulated to modulate secondary properties such as charge transfer,4 optical and magnetic behaviour,5,6 as well as guest adsorption.2,7
Apart from the commonly used perturbation approaches of temperature, pressure and light irradiation,8 there is considerable interest in finding new ways to control spin crossover (SCO) properties. One proposal is to employ redox-active ligands to influence the ligand field strength and therefore alter the SCO properties. These frameworks could conceivably be used for smart gas sorption applications, switchable by temperature, pressure, light or redox-state of the ligand.9 Recently, this concept was reported for the first time whereby the spin-state and conductivity of a framework material were tuned through guest-induced redox-state switching of TTF moieties.6 In this material, chemical oxidation by iodine doping resulted in a deactivation of the temperature and light-activated SCO properties. It was concluded that oxidation of the TTF core perturbed the FeII ligand field strength and hence the SCO properties. SCO can also be triggered by ligand-based redox processes.10,11
Encouraged by these approaches to obtain multifunctional materials with parallel redox- and spin-state switchable properties, we investigate here the structural, magnetic and redox properties of a one-dimensional CP which incorporates the bis-pyridyl tetrathiafulvalene bridging ligand, py2TTF. To entice the formation of a 1-D CP, a N2O2 coordinating Schiff base-like ligand has been employed;12 in contrast to classic Schiff base ligands (e.g., salen), the equilibrium between the two possible tautomeric structures, enol-imine and keto-enamine, is shifted to the keto-enamine structure. Upon coordination of a metal centre, the two amino nitrogen atoms of the ligand are deprotonated, and the negative charge is delocalised over the six membered chelate ring. In an octahedral ligand field with two additional N-donor ligands in axial positions, these systems offer a relatively rare N4O2 coordination sphere, which is favourable for FeII SCO.13,14 Importantly, the ligand field strength of such materials can be tuned by variation of substituents on the Schiff base-like ligand, and axial ligand choice offers flexibility to the type of complex formed, be it mononuclear, dinuclear, or 1-D chain-type structures.13 Due to the neutral charge of the complexes, strong supramolecular interaction networks are prevalent in this family and afford a wide variety of SCO behaviours, including wide hysteresis loops15 and multi-stepped transitions.16,17 Here, through the use of the bridging and redox-active ligand py2TTF, we probe the structure and SCO properties of this novel 1-D chain structural motif, including the spin-state and structural effects of chemical oxidation via iodine infusion.
The [FeLa(py2TTF)]n coordination polymer was synthesised by ligand substitution (Scheme 1). By this method, the precursor, [FeLa(MeOH)2], was added to 1.2 equivalents of the py2TTF ligand and heated under reflux. The coordination polymer formed as a red, fine-crystalline product. The purity of the coordination polymer was confirmed by elemental analysis and IR spectroscopy (ESI†).
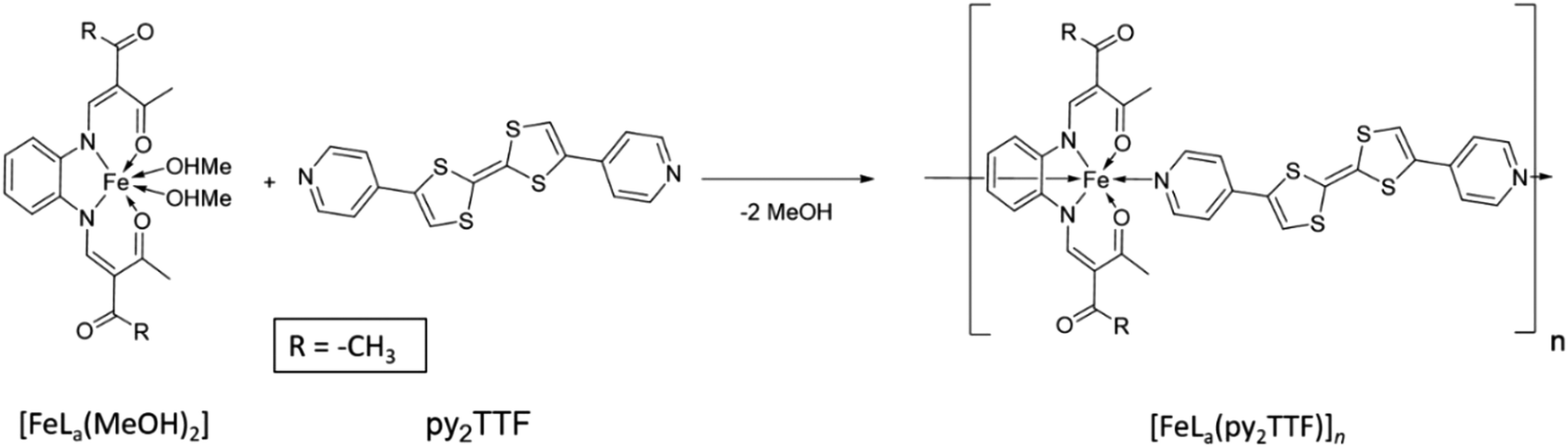 | ||
| Scheme 1 Synthetic scheme of the iron(II) coordination polymer [FeLX(py2TTF)]n: R = CH3 (La) | ||
A single crystal X-ray diffraction structure was obtained on a small crystal of [FeLa(py2TTF)]n at 100 K at the Australian Synchrotron (Table S1, ESI†). The complex crystallises with triclinic symmetry (space group: P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ). The asymmetric unit contains two distinct FeII centres (Fe1 and Fe2), two py2TTF ligands (L1 and L2) and two La equatorial ligands (La1 and La2; Fig. 1). As can be seen in Fig. 1b, the La2 ligand is strongly disordered over two positions. Fig. 1 shows the asymmetric unit fragments composed from Fe1-L1-La1 (Fig. 1a) and Fe2-L2-La2 (Fig. 1b). Fig. 2 shows the 1-D chains formed by Fe1-L1-La1 (Fig. 2a) and Fe2-L2-La2 (Fig. 2b). These distinct asymmetric unit fragments form two unique 1-D CP chains of the formula [FeLa(py2TTF)]n (Fig. 2).
). The asymmetric unit contains two distinct FeII centres (Fe1 and Fe2), two py2TTF ligands (L1 and L2) and two La equatorial ligands (La1 and La2; Fig. 1). As can be seen in Fig. 1b, the La2 ligand is strongly disordered over two positions. Fig. 1 shows the asymmetric unit fragments composed from Fe1-L1-La1 (Fig. 1a) and Fe2-L2-La2 (Fig. 1b). Fig. 2 shows the 1-D chains formed by Fe1-L1-La1 (Fig. 2a) and Fe2-L2-La2 (Fig. 2b). These distinct asymmetric unit fragments form two unique 1-D CP chains of the formula [FeLa(py2TTF)]n (Fig. 2).
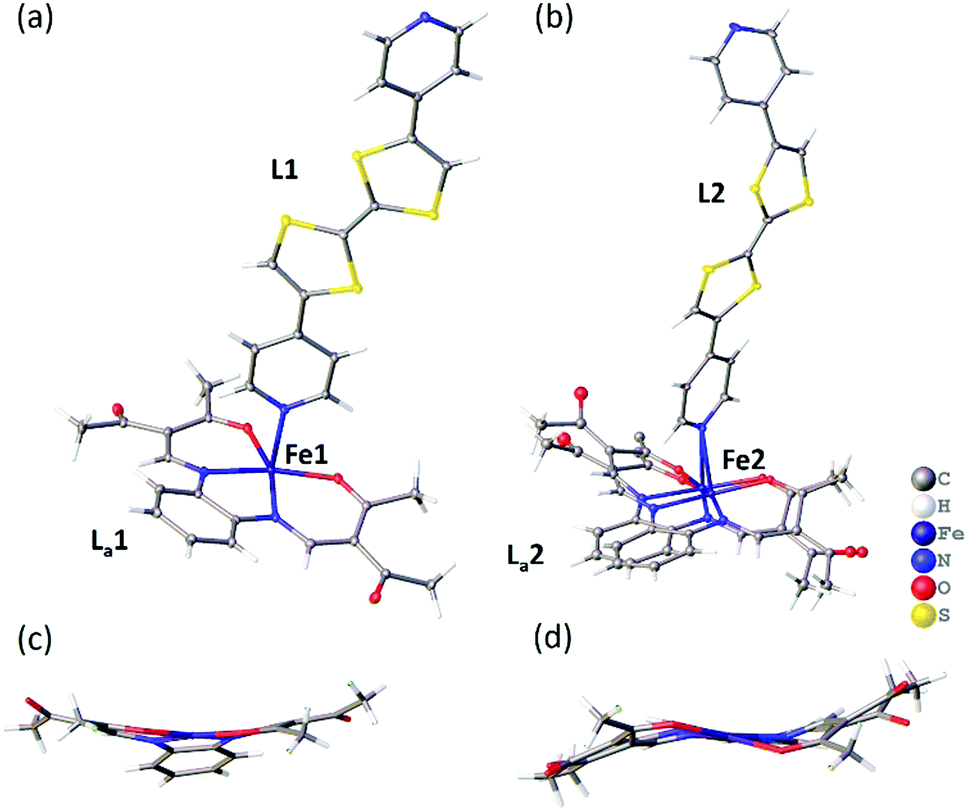 | ||
| Fig. 1 Structural image of the asymmetric unit (100 K) of [FeLa(py2TTF)]n showing the distinct fragments composed of (a) Fe1-L1-La1 and (b) Fe2-L2-La2. (c & d) show the umbrella and meso conformations of the La1 and La2 equatorial ligands. Thermal ellipsoid plot (50% probability); solvent molecules omitted for clarity. | ||
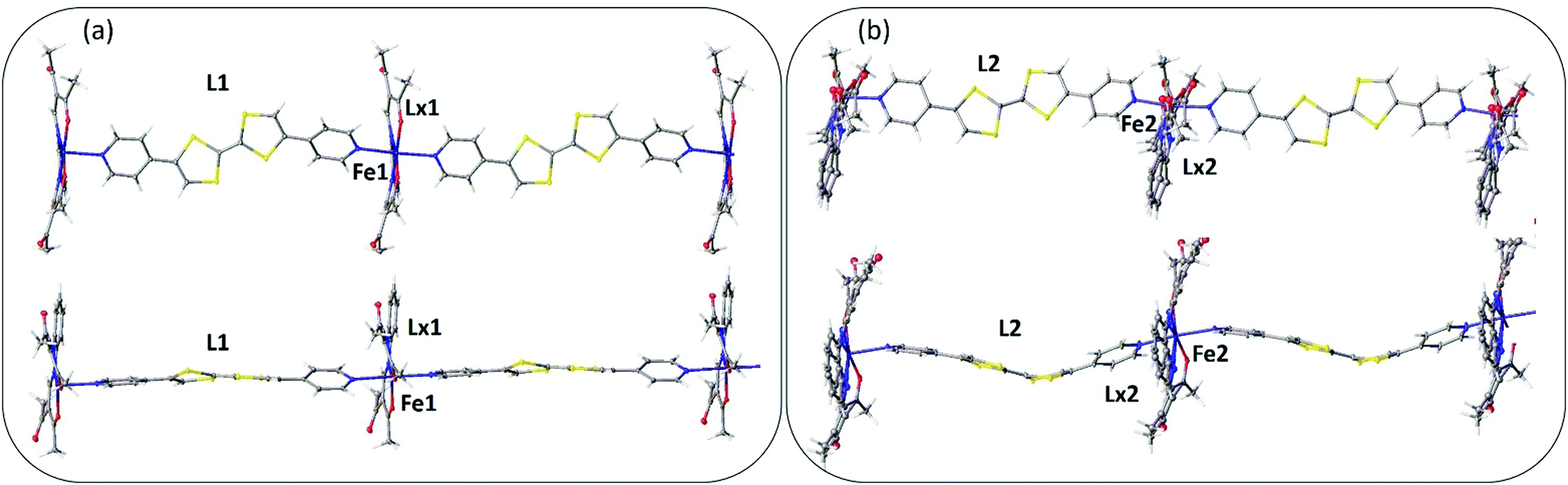 | ||
| Fig. 2 Structural representation (100 K) of the coordination polymer chains of [FeLa(py2TTF)]n formed by (a) Fe1-L1-La1 and (b) Fe2-L2-La2, highlighting the linear versus undulating chain motifs directed by the difference in L1 and L2 geometries. Each chain is shown in two different orientations. | ||
There are several points of substantial difference between these two distinct 1-D chains. Firstly, the py2TTF ligand (L1) is relatively planar (Z-like conformation) in Fe1-L1-La1 and hence an approximately linear 1-D chain structure is formed (Fig. 2a). In contrast, the py2TTF ligand (L2) in Fe2-L2-La2 is distinctly bent (U-like conformation), resulting in an undulating 1D chain structure (Fig. 2b). Both the ligand geometry and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond length of the TTF core are indicators of the ligand redox state.2,3 Here, while the geometries of the py2TTF ligands differ, the CC bond lengths are consistent with both L1 (1.348(10) Å) and L2 (1.341(11) Å) being in the neutral state. The neutral ligand state was also confirmed by EPR spectroscopy whereby no signal was observed in the 3300–3550 G region in the absence of an applied potential (Fig. S4, ESI†).
C bond length of the TTF core are indicators of the ligand redox state.2,3 Here, while the geometries of the py2TTF ligands differ, the CC bond lengths are consistent with both L1 (1.348(10) Å) and L2 (1.341(11) Å) being in the neutral state. The neutral ligand state was also confirmed by EPR spectroscopy whereby no signal was observed in the 3300–3550 G region in the absence of an applied potential (Fig. S4, ESI†).
Aside from the axial ligand differences, the equatorial ligand conformations are different in both chains. An umbrella-like arrangement is observed in La1 (Fig. 1c), which is typical of HS species in these types of complexes,13 whereas the La2 ligand shows a meso-conformation (Fig. 1d). Equatorial ligands of this type tend to be more planar in the LS state – hence, the meso-conformation may be an indicator of a subtle LS character, but may also arise here due to crystal packing effects. The O–Fe–O angles (Fe1: 112°, Fe2: 112–115°) and axial ligand bond lengths (d〈Fe-Npy〉: Fe1: 2.279(2) Å, Fe2: 2.271(2) and 2.256(2) Å) are sensitive to spin-state and all are in the range typical of HS FeII sites in these systems.12,13 However, we note that these parameters are less reliable in Fe2 due to the substantial equatorial ligand disorder at this site. Additionally, we note the possibility of difference in solvent inclusion, and therefore SCO behaviour, between the diffraction and magnetometry samples.17,18 Supporting the HS assignment at Fe1 and a degree of LS character at Fe2 are DFT calculations (Fig. S9 and S10, ESI†) which show an idealised HS structure with an umbrella shaped equatorial ligand and a subtle twist in the py2TTF ligand, as seen in the Fe1-L1-La1 chain, and a LS structure with a planar equatorial ligand and a more bent py2TTF ligand. The single crystal structure of the Fe2-L2-La2 chain is intermediate between these two optimised calculated structures.
With respect to each other, the Fe1-L1-La1 and Fe2-L2-La2 chains are arranged in a grid array with the Fe1 and Fe2 chains approximately 90° to each other (Fig. S1, ESI†). In this grid arrangement the TTF cores are packed in 1-D columns (Fig. 3). The S⋯S distances between adjacent TTF units are typical for Intervalence Charge Transfer (IVCT).4,19 This TTF core stacking array also suggests that the bending in L2 is driven by optimising these interactions.
 | ||
| Fig. 3 Structural image showing the 1-D stacked TTF cores in [FeLa(py2TTF)]n. | ||
A Mössbauer spectrum was collected at room temperature to rule out any μ-oxido impurities which are a thermodynamic sink for these complexes. In Fig. 4(inset), the spectrum reveals one doublet with a quadrupole splitting and an isomer shift in the expected range for FeII HS complexes of this ligand type (δ = 0.903 mm s−1; ΔEQ = 2.15(3) mm s−1). Once the absence of μ-oxido impurities was confirmed by both Mössbauer spectroscopy and elemental analysis, magnetic susceptibility measurements were conducted to assess any spin-state changes with temperature variation. Data were collected over the range 300–50–300 K (Fig. 4). The χMT values around room temperature are ca. 3.43 cm3 K mol−1, typical for HS FeII sites. Between 200 and 100 K a gradual decrease in χMT values occurs with a minimum of ca. 2.65 cm3 K mol−1 achieved by 50 K. This is indicative of ca. 25% conversion of HS FeII sites to the LS FeII state below 100 K. This incomplete SCO behaviour is in line with the observations of single crystal analysis at 100 K, whereby the Fe1-L1-La1 chains maintain a HS character and the Fe2-L2-La2 chains a partial SCO.
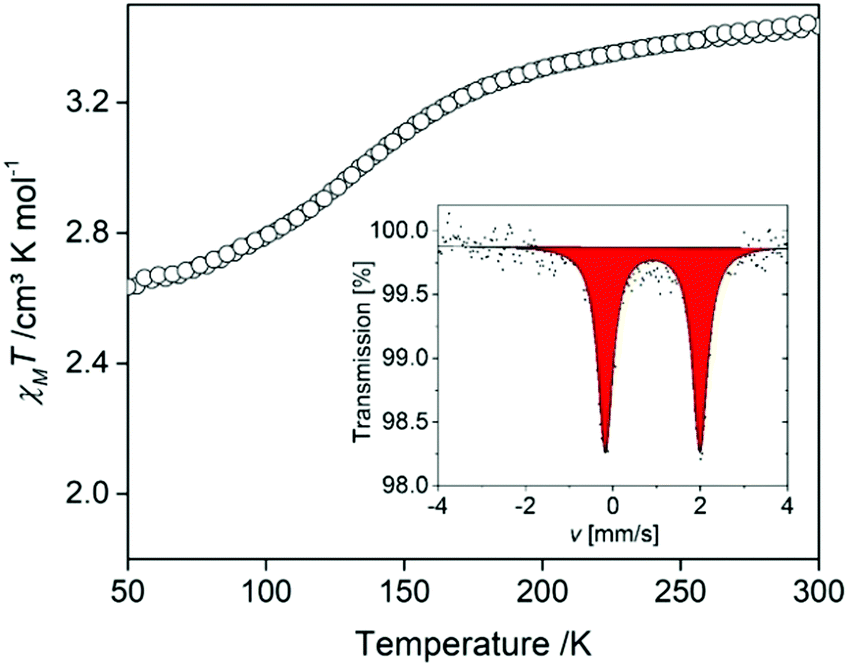 | ||
| Fig. 4 χ M T vs. T plot of [FeLa(py2TTF)]n over the range 300–50–300 K. Inset: Mössbauer spectra at room temperature. | ||
Cyclic voltammetry (CV) on a solid sample of [FeLa(py2TTF)]n was performed to investigate the potential for redox modulation of the SCO CP (Fig. S2 and S3, ESI†). The CV exhibited two quasi-reversible processes at 0.17 and 0.54 V in the anodic region which are in good agreement with potentials previously reported for the first and second oxidation of the TTF core (TTF/TTF˙+ and TTF˙+/TTF2+, respectively).2 Additional irreversible processes were observed at 0.87 and 0.99 V; these are tentatively assigned to oxidation processes for the equatorial ligand. The presence of a FeII/III process in this region was discounted, as previous literature reports for Schiff-base FeII polymers show that these processes occur at very negative potentials.20
In an attempt to assign the electrochemical processes, UV/VIS/NIR spectroelectrochemistry (SEC) was performed on [FeLa(py2TTF)]n over an applied potential range from 0–0.9 V (Fig. S7, ESI†). Application of the anodic potential resulted in the appearance of two bands in the visible and NIR regions (∼6000 cm−1 or ∼1667 nm). The latter is tentatively attributed to IVCT as it occurs in a similar region to previous reports.4,21 The oxidation process was also followed by EPR SEC in a field between 3300 and 3550 G (equivalent to g values in the range 2.12 to 1.96), whereby a signal emerges corresponding to the radical cation TTF˙+ as the applied potential is increased up to 0.75 V (Fig. S4, ESI†).
The introduction of iodine is an effective synthetic strategy for chemical oxidation. The iodine doped species was achieved by slow diffusion with iodine vapour. The colour of the compound changed from red to black after 2 days. The structure shows solvent accessible channels that the iodine could readily diffuse through (pore volume 15%; Fig. S1, ESI†). To determine the oxidation state of the TTF cores in the oxidised species, EPR measurements were performed (Fig. 5: inset). The EPR spectrum in a field between 1900 and 4500 G (g values 3.67–1.55) showed a strong signal for an organic radical at 3437 G (g value of 2.03), which provides evidence that the TTF cores are oxidised to their radical cation by iodine doping. Powder X-ray diffraction data (Fig. S8, ESI†) indicate a retention of crystallinity and general structure in the oxidised species compared to [FeLa(py2TTF)]n; a shift in the most intense reflections to higher 2θ angle indicates a contraction in the cell dimensions, but severe peak overlap prevented quantitative determination of the structural change.
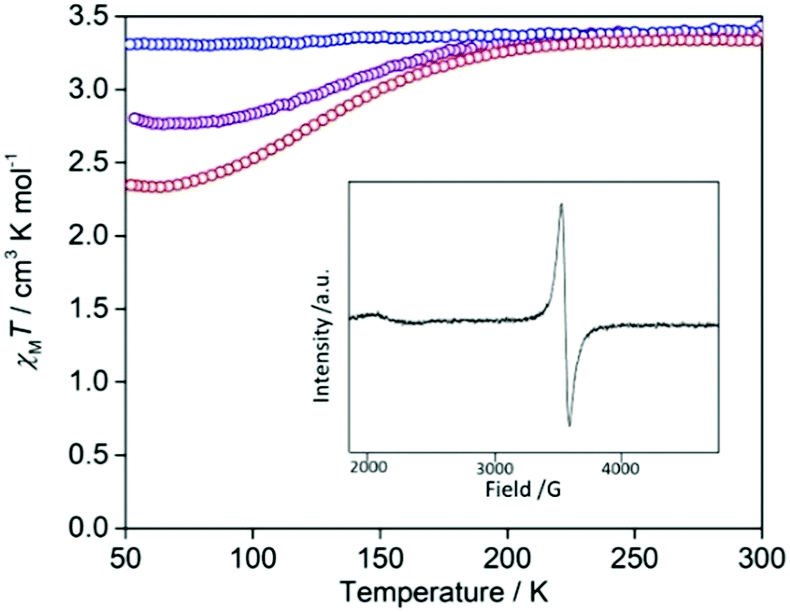 | ||
| Fig. 5 χ M T vs. T plot of [FeLa(py2TTF)]n (red) and doped with iodine for 2 days (purple) and 2 weeks (blue). Inset: EPR spectrum of [FeLa(py2TTF)]n infused with iodine for 2 weeks showing an oxidised TTF signal. | ||
The SCO properties of the iodine infused crystals show a gradual decrease in HS to LS conversion with time exposed to iodine, with a complete HS character achieved after exposure for 2 weeks (Fig. 5). This same effect of HS stabilisation upon TTF oxidation was similarly observed in the redox-active SCO complex reported by Wang et al.6 and was attributed to oxidative ligand field strength variation.
In summary, a 1-D chain CP with both SCO and redox activity has been successfully prepared by the combination of a bis-pyridyl functionalised TTF core and a Schiff base-like N2O2 ligand. Multifunctionality is achieved in this CP via SCO at the FeII centres and redox activity of the TTF-based ligand. Chemical oxidation of the TTF cores, achieved by iodine infusion, results in modification of the SCO properties. Thereby this material provides a very rare example of redox-modulated SCO properties and dual SCO and redox functionality.
This work was supported by Fellowships and Discovery Project funding from the Australian Research Council. Access and use of the facilities of the Australian Synchrotron was supported by ANSTO, using the MX1 beamline.
Conflicts of interest
There are no conflicts to declare.References
-
(a) H.-C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef PubMed
; (b) D. J. Tranchemontagne, J. L. Mendoza-Cortés, M. O’Keeffe and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1257–1283 RSC
- H.-Y. Wang, L. Cui, J.-Z. Xie, C. F. Leong, D. M. D’Alessandro and J.-L. Zuo, Coord. Chem. Rev., 2017, 345, 342–361 CrossRef CAS
- F. Gao, F.-F. Zhu, X.-Y. Wang, Y. Xu, X.-P. Wang and J.-L. Zuo, Inorg. Chem., 2014, 53, 5321–5327 CrossRef CAS PubMed
- D. A. Sherman, R. Murase, S. G. Duyker, Q. Gu, W. Lewis, T. Lu, Y. Liu and D. M. D’Alessandro, Nat. Commun., 2020, 11, 2808 CrossRef CAS
-
(a) H.-Y. Wang, J. Su, J.-P. Ma, F. Yu, C. F. Leong, D. M. D’Alessandro, M. Kurmoo and J.-L. Zuo, Inorg. Chem., 2019, 58, 8657–8664 CrossRef CAS
- H.-Y. Wang, J.-Y. Ge, C. Hua, C.-Q. Jiao, Y. Wu, C. F. Leong, D. M. D’Alessandro, T. Liu and J.-L. Zuo, Angew. Chem., Int. Ed., 2017, 56, 5465–5470 CrossRef CAS
- J. Su, S. Yuan, H.-Y. Wang, L. Huang, J.-Y. Ge, E. Joseph, J. Qin, T. Cagin, J.-L. Zuo and H.-C. Zhou, Nat. Commun., 2017, 8, 2008 CrossRef
-
(a) M. A. Halcrow, Chem. Soc. Rev., 2011, 40, 4119–4142 RSC
-
(a) Z.-P. Ni, J.-L. Liu, M. N. Hoque, W. Liu, J.-Y. Li, Y.-C. Chen and M.-L. Tong, Coord. Chem. Rev., 2017, 335, 28–43 CrossRef CAS
- M. Gruber, V. Davesne, M. Bowen, S. Bouari, E. Beaurepaire, W. Wulfhekel and T. Miyamachi, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 195415 CrossRef
- S. Karan, C. Garcia, M. Karolak, D. Jacob, N. Lorente and R. Berndt, Nano Lett., 2018, 18, 88–93 CrossRef CAS PubMed
- B. Weber and E.-G. Jäger, Eur. J. Inorg. Chem., 2009, 465–477 CrossRef CAS
- B. Weber, Coord. Chem. Rev., 2009, 253, 2432–2449 CrossRef CAS
-
(a) K. Senthil Kumar, Y. Bayeh, T. Gebretsadik, F. Elemo, M. Gebrezgiabher, M. Thomas and M. Ruben, Dalton Trans., 2019, 48, 15321–15337 RSC
-
(a) W. Bauer, C. Lochenie and B. Weber, Dalton Trans., 2014, 43, 1990–1999 RSC
- W. Bauer, T. Pfaffeneder, K. Achterhold and B. Weber, Eur. J. Inorg. Chem., 2011, 3183–3192 CrossRef CAS
- W. Bauer, W. Scherer, S. Altmannshofer and B. Weber, Eur. J. Inorg. Chem., 2011, 2803–2818 CrossRef
- K. Dankhoff, C. Lochenie, F. Puchtler and B. Weber, Eur. J. Inorg. Chem., 2016, 2136–2143 CrossRef
- C. F. Leong, C.-H. Wang, C. D. Ling and D. M. D’Alessandro, Polyhedron, 2018, 154, 334–342 CrossRef
-
(a) B. Weber, H. Görls, M. Rudolph and E.-G. Jäger, Inorg. Chim. Acta, 2002, 247–265 CrossRef
- P. M. Usov, C. Fabian and D. M. D’Alessandro, Chem. Commun., 2012, 48, 3945–3947 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, synthesis, single crystal diffraction analysis, magnetic measurements, electrochemical data. CCDC 2004198. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc03788e |
| This journal is © The Royal Society of Chemistry 2020 |