
Metal phosphonates incorporating metalloligands: assembly, structures and properties
Song-Song
Bao
,
Ming-Feng
Qin
and
Li-Min
Zheng
 *
*
State Key Laboratory of Coordination Chemistry, School of Chemistry and Chemical Engineering, Collaborative Innovation Centre of Advanced Microstructures, Nanjing University, Nanjing 210023, China. E-mail: lmzheng@nju.edu.cn
First published on 28th August 2020
Abstract
Metal phosphonates are an important class of metal–organic hybrid materials that exhibit versatile structures, intriguing functions and high water and thermal stability. Despite a large number of metal phosphonates achieved over the past few decades, those incorporating metalloligands are rather limited. The metalloligand approach can provide a unique opportunity in constructing homo- or mixed-metal coordination polymers with rationally immobilized functional moieties for various applications in gas storage and separation, heterogeneous catalysis, sensing and multifunctional materials. In this feature article, we shall introduce the current status of a special subclass of metal phosphonates, namely, metal–metalloligand phosphonates (MMPs), including synthetic strategies, crystal structures, and properties of those based on paddlewheel diruthenium, metallo-polyazamacrocycle, metalloporphyrin and metallo-tris-bipyridine ligands. Future challenges in this field are discussed.
 Song-Song Bao | Song-Song Bao completed his PhD degree at Nanjing University in 2008 under the supervision of Prof. Li-Min Zheng. He then studied at Goethe University Frankfurt as a Post-Doctoral Research Associate in the research group of Prof. Matthias Wagner. He is currently working as associate professor at the School of Chemistry and Chemical Engineering, Nanjing University. His current interests involve the design and synthesis of multifunctional metal phosphonates by using the metalloligand approach. |
 Ming-Feng Qin | Ming-Feng Qin received his bachelor's degree and master's degree from Jiangsu University in 2015 and 2018, respectively. He is currently working on his PhD under the supervision of Prof. Li-Min Zheng at the School of Chemistry and Chemical Engineering at Nanjing University. His current research interests focus on photocatalytic properties of metal phosphonates containing metalloligands. |
 Li-Min Zheng | Li-Min Zheng received her PhD degree from Nanjing University in 1992 under the guidance of Prof. Xinquan Xin. Then she became a lecturer (1992–1997), associate professor (1997–2002) and professor (2002–) at the same University. She spent two years with Prof. Silvio Decurtins as a postdoctoral fellow at the University of Zurich (1994–1996) and one year with Prof. Allan J. Jacobson at the University of Houston (1999–2000). Her research group seeks to develop new crystalline and low-dimensional materials based on metal phosphonates with interesting physical and chemical properties. |
1. Introduction
Metal–organic hybrid materials, in particular metal–organic frameworks (MOFs), have been developed explosively over the past few decades.1–3 These materials are commonly constructed by a combination of metal-containing nodes and organic linkers, wherein a large number of organic linkers with different shapes have been designed and synthesized. Aside from enormous efforts devoted to homometallic MOFs with beautiful structures and fascinating properties, heterometallic systems containing metalloligands have attracted more and more attention. Metalloligands are defined as metal complexes with appended functional groups that are able to coordinate with second metal ions. Within the context of the metalloligand approach, it is possible to place the auxiliary functional groups in a pre-organized conformation.4 Hence this approach offers opportunities to rationally immobilize functionalized or catalytically active metal sites within the frameworks. As a result, enhanced performance in gas storage and separation, heterogeneous catalysis, sensing, or multifunction can be achieved in metal–metalloligand systems.5Metal phosphonates are an important class of metal–organic hybrid materials, showing promising applications in ion-exchange and sorption, catalysis, and materials science.6 Compared with many other metal–organic systems, metal phosphonates often show high water and thermal stability,7,8 attributed to the presence of inorganic components in the structure such as clusters, chains or layers. The organic moieties of the phosphonate ligands RPO32− can be modified by other functional groups resulting in a number of metal phosphonates with versatile structures and functions. For more information, the interested reader is directed to a book which covers most aspects of this field,9 and several review articles concerned with the synthesis and structures,10 optical,11 magnetic,12 and proton conductive properties,13 as well as their application in catalysis, adsorption, energy storage, drug delivery, etc.14 In contrast to the rapid progress of metal phosphonate chemistry, utilization of metalloligands in synthesis has not been well explored. The incorporation of metalloligands in metal phosphonates can not only confer additional function to the material but also provide possibilities to tune the properties through different metal components. However, the number of known metal–metalloligand phosphonates (MMPs) are rather limited. The obstacle could arise from the synthetic challenges in isolating phosphonate-based metalloligands, and the fast precipitation of the MMP products leading to the difficulties in analysing their structures using the conventional single crystal determination method. To solve the structures of MMPs with extremely small crystal sizes, Rietveld refinement using powder X-ray diffraction data as well as single-crystal electron diffraction method has been employed.15
In this feature article, we shall introduce the current status of metal phosphonates incorporating metalloligands including the general synthetic strategies, crystal structures and properties. It is worth mentioning that the metalloligands discussed herein are restricted to those with pendant phosphonate groups such that each phosphonate group can provide at least one oxygen donor for further coordination (Scheme 1). As far as we are aware, there are only a few types of such metalloligands reported so far, including paddlewheel diruthenium diphosphonate, metallo-polyazamacrocycle-phosphonate, metalloporphyrin–phosphonate and metallo-tris-bipyridine-phosphonate ligands. These metalloligands share a common feature in that they can be isolated as discrete species and used as linkers to connect second metal ions. But not all MMPs containing these metalloligands were synthesized using metalloligands as the precursor. In some cases, they were obtained through direct reaction of metal salt and phosphonic acid. In order to give readers a whole picture of these systems, all MMPs containing these metalloligands will be summarized, and their structures and properties will be described. Finally, this article will end up with a discussion of future challenges in this field.
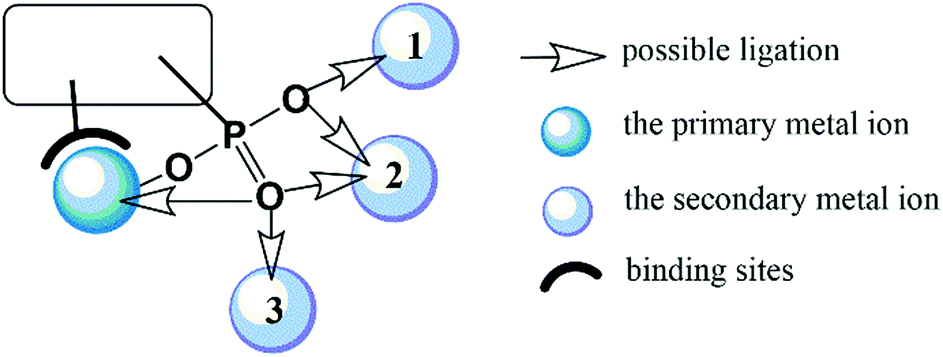 | ||
| Scheme 1 The coordination modes of a phosphonate-based metalloligand. | ||
2. Synthetic strategies
There are two general synthetic strategies to produce MMPs based on the different precursors as linkers. First, a one-step procedure using phosphonic acid as the precursor. The MMPs can be obtained by direct reaction of appropriate metal salts with phosphonic acids in solution under ambient or hydro/solvothermal conditions. This approach is simple but challenging in controlling the final products. In particular, when there are more than one kind of metal ions present, it can be difficult to obtain heterometallic MMPs in pure form. Further, the metalloligand could not be formed as anticipated under these one-pot reaction conditions. Second, a two-step procedure using metalloligands as the precursor. This approach involves the preparation of the desired metalloligands, followed by the reaction of the metalloligands with suitable metal salts. The advantage of this approach is that the pre-formed metalloligand can not only immobilize the central metal ion but also provide coordination donors with different orientations, thus allowing the formation of MMPs in a more controllable manner. However, considering the strong coordination capabilities of the phosphonate groups, it could be a challenging task to isolate these phosphonate-based metalloligands as linkers for further assembling with other metal ions. It should be emphasized that in both strategies, other reaction parameters such as the molar ratio and concentration of the metal ion and linkers, the pH of the reaction mixture, the reaction temperature and time as well as the solvents are also critical for achieving crystalline materials of MMPs suitable for single-crystal X-ray diffraction studies.3. Crystal structures
3.1. Diruthenium paddlewheel units as metalloligands
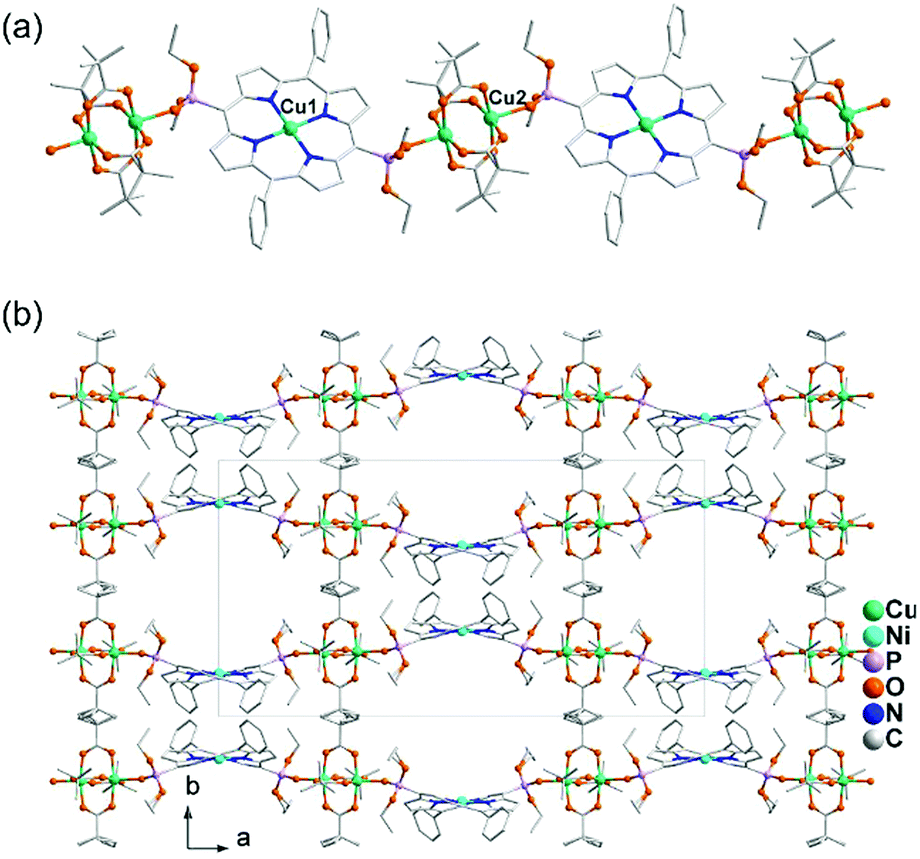
Diruthenium Ru2 (4+ to 6+) cores with paddlewheel structures are attractive metalloligands for functional MOFs owing to their variable electronic configurations and magnetic and catalytic properties.16 Successful examples include those with the paddlewheel dimers constructed from chelating and bridging O,O′-donor ligands, such as carboxylate,17 carbonate18 and phosphate.19 The Ru2–diphosphonate system has superiority over carbonate because the organic groups of the phosphonate ligand can be modified with additional functions. In 2003, we reported the first example of metal phosphonates with extended structures which contain a diruthenium diphosphonate paddlewheel core, (NH4)3Ru2(hedp)2·2H2O (1),20 where hedpH4 is 1-hydroxyethylidenediphosphonic acid [CH3C(OH)(PO3H2)2]. This compound was synthesized by a one-pot reaction of RuCl3 and hedpH4 under hydrothermal conditions. It has a 2D layer structure in which the negatively charged RuII,III2(hedp)23− paddlewheels are inter-connected by filling the axial sites of ruthenium with the phosphonate oxygen atoms from the equivalent Ru2(hedp)23− units. The interlayer space is filled with NH4+ cations and lattice water molecules. Here, the diruthenium motif is not only an acceptor but also a donor (Scheme 2a). A similar layer topology is also found in Na3Ru2(hedp)2·4H2O (2).21 In this case, the layers are further connected by {NaO6} octahedra, forming a 3D framework structure.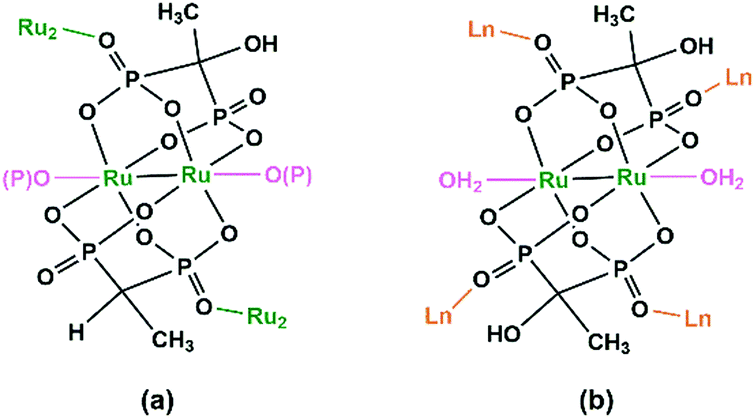 | ||
| Scheme 2 Coordination modes of Ru2(hedp)2n− in 1 (a) and 3–15 (b). | ||
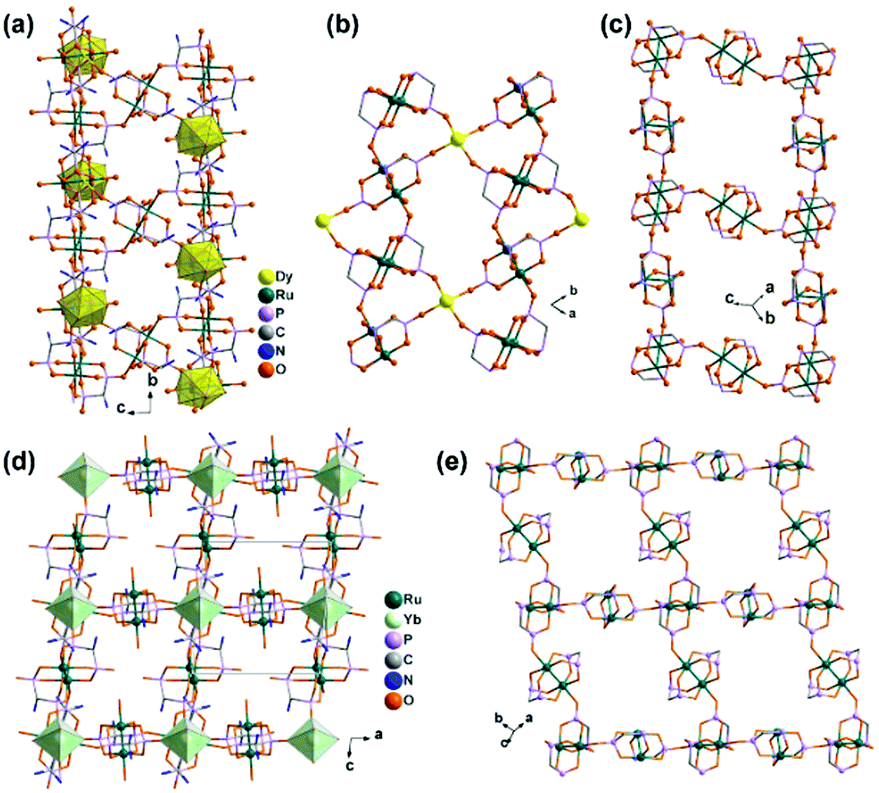
Interestingly, compound 1 is soluble in an aqueous solution of salt with the dissociation of the framework structure and the formation of stable dinuclear species of Ru2(hedp)23−, confirmed by the isolation of K3[Ru2(hedp)2(H2O)2]·6H2O.22 The pre-formed Ru2(hedp)23− paddlewheel is stable in aqueous solution and can provide four pendant phosphonate oxygen atoms and two hydroxyl groups as potential donors. Thus it can serve as a metalloligand to assemble with second metal ions to construct MMPs. Indeed, the direct reaction of Ru2(hedp)23− solution with Ln(NO3)3 (Ln = La, Ce, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er) at room temperature led to two types of Ln–Ru2 compounds with the formula Ln(H2O)4[Ru2(hedp)2(H2O)2]·5.5H2O [Ln = La (3), Ce (4)] and Ln(H2O)4[Ru2(hedp)2(H2O)2]·8H2O [Ln = La (5), Ce (6), Pr (7), Nd (8), Sm (9), Eu (10), Gd (11), Tb (12), Dy (13), Ho (14), Er (15)].23 In all cases, Ru2(hedp)2(H2O)23− acts as a tetra-dentate metalloligand, coordinating to four LnIII ions using its four pendant phosphonate oxygen atoms (Scheme 2b). Compounds 3 and 4 are isostructural. They have a square-grid layer structure in which each Ru2(hedp)2(H2O)23− binds to four LnIII ions and vice versa (Fig. 1a). Compounds 5–15 are also isostructural, but possess a 3D PtS-type open-framework structure (Fig. 1b). The lanthanide ions are eight-coordinated by four phosphonate oxygen (OP) atoms and four water molecules (Ow) and the hydroxyl groups remain pendant. Despite similar connectivity of Ru2(hedp)2(H2O)23− and LnIII ions, the significant structural difference between 3–4 and 5–15 originates from the slight difference in the geometry of {LnOp4Ow4} polyhedra. In particular, the geometry of {LnOp4} is essential in determining the topology of the final products, which is a distorted square plane in 3–4, but closer to a distorted tetrahedron in 5–15. Fig. 1 shows the single layer and the framework structures of 3 and 5, respectively. 5 contains three kinds of channels, A (4.3 × 3.5 Å along the [330]), B (3.6 × 4.9 Å along the [330]) and C (3.5 × 3.5 Å along the [001]) (van der Waals radii accounted), which host the lattice water molecules. As far as we are aware, these are the first examples of Ln–Ru2 heterometallic compounds containing paddlewheel Ru2n+ cores.
 | ||
| Fig. 1 (a) One square-grid layer of structure 3. (b) 3D PtS-type open-framework structure of 5. All H atoms are omitted for clarity. Adapted from ref. 23. | ||
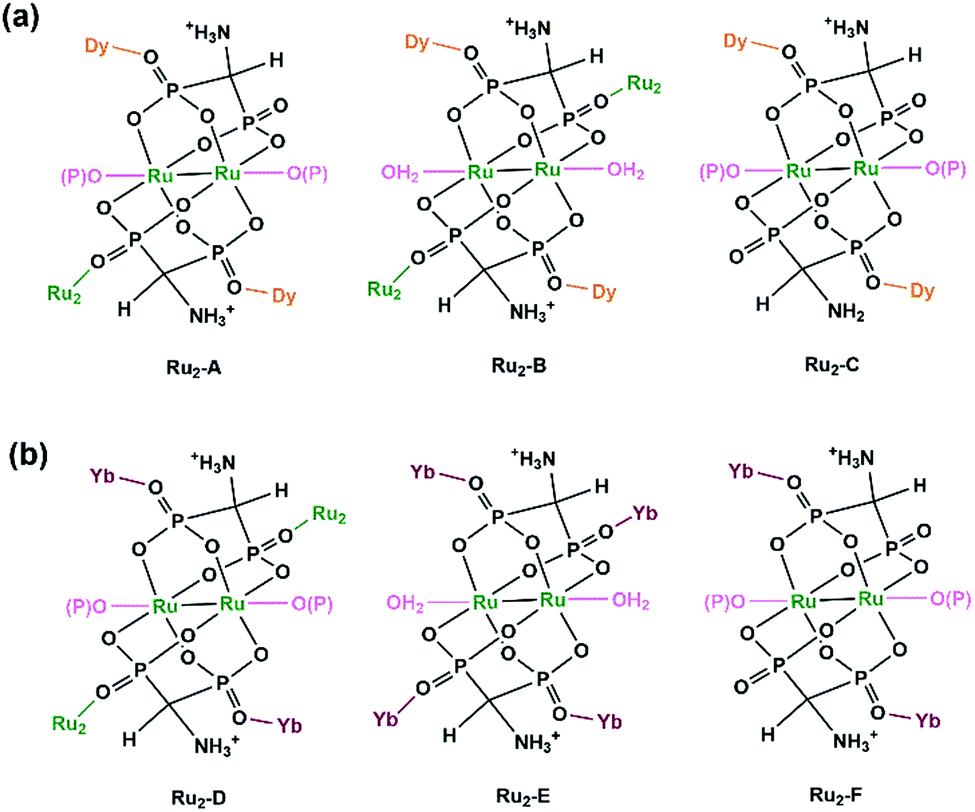
By functionalizing the methylenediphosphonate ligand with an amino group, we obtained a 1D Ru2(II,II) compound (H3O)2[Ru2(Hamdp)2] (16), where amdp4− represents 1-ammoniummethylene-diphosphonate [NH2CH(PO3)24−]. 16 is soluble in aqueous solution with the formation of discrete species of Ru2(Hamdp)2. The assembly of Ru2(Hamdp)2 with Ln(NO3)3 (Ln = Dy, Yb) in aqueous solution at room temperature resulted in mixed-valent compound Dy(H2O)3[Ru2(Hamdp)2][Ru2(Hamdp)2(H2O)2][Ru2(Hamdp)(amdp)]0.5·12H2O (17) with a 2D double layer structure and Yb[Ru2(Hamdp)2]2[Ru2(Hamdp)2(H2O)2]·15H2O (18) with a 3D porous framework structure (Fig. 2).24 The mixed-valent nature of the Ru2 units is suggested by their Ru–Ru distances and confirmed by magnetic studies.
 | ||
| Fig. 2 (a) A double layer structure of 17. (b) The kagomé layer made up of Ru2(Hamdp)2− and DyIII ions in structure 17. (c) The ladder-like chain of pure Ru2 in structure 17. (d) 3D open-framework structure of 18. (e) The Ru2 layer in structure 18. All H atoms, –NH3+ groups, uncoordinated phosphonate O atoms and coordination water molecules are omitted in (b, c and e) for clarity. Adapted from ref. 24. | ||
The structures of 17 and 18 are unique compared to other known diruthenium coordination polymers, and hence deserve further description. In 17, three types of Ru2(Hxamdp)2 units are present serving as bi-, tri- and tetra-dentate metalloligands, respectively (Scheme 3a). The coordination of Ru2(Hamdp)2− (tri- and tetra-dentate) with DyIII ions leads to a kagomé layer (Fig. 2b), and two such layers are further pillared by the third type of Ru2(Hamdp)(amdp)2− (bi-dentate) forming a double layer structure (Fig. 2a and c). In 18, there exist three types of Ru2(Hxamdp)2 units acting as bi- and tetra-dentate metalloligands (Scheme 3b). Although the coordination mode of the bi-dentate Ru2 (Ru2-F, Scheme 3b) is similar to that in 17 (Ru2-C, Scheme 3a), that of the tetra-dentate ones is remarkably different. There are two kinds of tetra-dentate Ru2(Hamdp)2− metalloligands in 18. One of them binds to two other Ru2 units and two YbIII ions and accepts two phosphonate oxygen atoms in the axial positions (Ru2-D), while the other binds to four YbIII ions with the axial positions filled with water molecules (Ru2-E). These three types of Ru2 units are cross-linked to form a layer containing rhombic eight-member rings of Ru2 (Fig. 2e), unlike four-member rings in 1. The Ru2 layers are stacked by six-coordinated Yb into a 3D framework (Fig. 2d). The framework contains two kinds of narrow channels, A (approximately 4.1 × 6.7 Å along the c-axis) and B (approximately 6.6 × 10.6 Å along the a-axis) (van der Waals radii not accounted), which host the lattice water molecules.
 | ||
| Scheme 3 Coordination modes of Ru2(Hxamdp)2 dimers in 17 (a) and 18 (b). Reprinted with permission from ref. 24. Copyright 2019, American Chemical Society. | ||
Apparently, paddlewheel diruthenium diphosphonates can be used as metalloligands to successfully assemble Ln–Ru2 MMPs. Compared with the Ru2(hedp)2/Ln complexes,23 however, compounds 17 and 18 exhibit entirely new structural topologies, attributed to the different charges of the Ru2(Hxamdp)2 unit due to the presence of protonated amino group in the diphosphonate ligand. This opens a new route to design and synthesize Ru2-based materials with new architectures by modifying the organic groups of the diphosphonate ligand. Table 1 summarizes MMPs containing Ru2(hedp)2 and Ru2(Hxamdp)2 metalloligands.
| Compound | Ru2 core | Space group | Ru–Ru (Å) | N | Structural feature | Magnetic property D (cm−1)/θ (K)/zJ (cm−1) | Ref. |
|---|---|---|---|---|---|---|---|
| a N: dimension of the structure, n.a.: not available. | |||||||
| Na3Ru2(hedp)2·4H2O (2) | Ru25+ | C2/c | 2.354 | 3D | Open framework | 96.0/0.64/n.a. | 21 |
| {La(H2O)4[Ru2(hedp)2(H2O)2]}·5.5H2O (3) | Ru25+ | P21/n | 2.357 | 2D | Square-grid layer | 101.6/n.a./−0.044 | 23 |
| {Ce(H2O)4[Ru2(hedp)2(H2O)2]}·5.5H2O (4) | Ru25+ | P21/n | 2.357 | 2D | Square-grid layer | 101.6/n.a./−0.044 | 23 |
| {La(H2O)4[Ru2(hedp)2(H2O)2]}·8H2O (5) | Ru25+ | C2/c | 2.356 | 3D | PtS type open-framework | 103.2/n.a./−0.023 | 23 |
| {Ce(H2O)4[Ru2(hedp)2(H2O)2]}·8H2O (6) | Ru25+ | C2/c | 2.356 | 3D | PtS type open-framework | n.a. | 23 |
| {Pr(H2O)4[Ru2(hedp)2(H2O)2]}·8H2O (7) | Ru25+ | C2/c | 2.418 | 3D | PtS type open-framework | n.a. | 23 |
| {Nd(H2O)4[Ru2(hedp)2(H2O)2]}·8H2O (8) | Ru25+ | C2/c | 2.351 | 3D | PtS type open-framework | n.a. | 23 |
| {Gd(H2O)4[Ru2(hedp)2(H2O)2]}·8H2O (11) | Ru25+ | C2/c | 2.354 | 3D | PtS type open-framework | n.a. | 23 |
| {Tb(H2O)4[Ru2(hedp)2(H2O)2]}·8H2O (12) | Ru25+ | C2/c | 2.346 | 3D | PtS type open-framework | n.a. | 23 |
| {Dy(H2O)4[Ru2(hedp)2(H2O)2]}·8H2O (13) | Ru25+ | C2/c | 2.360 | 3D | PtS type open-framework | n.a. | 23 |
| {Ho(H2O)4[Ru2(hedp)2(H2O)2]}·8H2O (14) | Ru25+ | C2/c | 2.353 | 3D | PtS type open-framework | n.a. | 23 |
| {Er(H2O)4[Ru2(hedp)2(H2O)2]}·8H2O (15) | Ru25+ | C2/c | 2.342 | 3D | PtS type open-framework | n.a. | 23 |
| {Dy(H2O)3[Ru2(Hamdp)2][Ru2(Hamdp)2(H2O)2][Ru2(Hamdp)(amdp)]0.5}·12H2O (17) | Ru25+ |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
2.346–2.358 | 2D | Pillared-bilayer | n.a./−16.2/n.a. | 24 |
| {Yb[Ru2(Hamdp)2]2[Ru2(Hamdp)2(H2O)2]}·15H2O (18) | Ru25+ |
P |
2.354–2.362 | 3D | Open framework | n.a./−30.0/n.a. | 24 |
3.2. Metallo-polyazamacrocycle units as metalloligands
Polyazamacrocycles functionalized by phosphonate groups offer multiple metal binding sites of two distinct types of chemical nature, one at the phosphonate pendant arms and the other within the polyazamacrocycle ring. Table 2 gives the MMPs containing metallo-polyazamacrocycle ligands. 1,4,7-Triazacyclononane-1,4,7-triyl-tris(methylenephosphonic acid) (notpH6) contains the smallest polyazamacrocycle with three nitrogen and nine oxygen atoms as potential coordination donors. It is ideally suited to form six-coordinate complexes with two groups of facial donors: the macrocyclic nitrogen atoms on one side and the pendant phosphonate oxygen atoms on the other side, thus leading to mononuclear species such as MIII(notpH3) (M = Fe, Co)25 and CuII(notpH4)26 (Scheme 4). These mononuclear species are polar and can serve as metalloligands, by using the remaining six uncoordinated phosphonate oxygen atoms, to connect other metal ions into extended networks. Moreover, the octahedrally coordinated FeIII or CoIII centers are chiral showing Δ or Λ configurations. Unfortunately, enantioenriched MMPs containing M(notpHx) metalloligands have not been reported so far. Only in rare cases, racemic conglomerates were obtained which contain an equal amount of opposite enantiomers.| Compound | Space group | N | Structure | Proton conductivity (S cm−1) and other propertiesa | Ref. |
|---|---|---|---|---|---|
| a N: dimension of the structure, n.a.: not available, AF: antiferromagnetic. | |||||
| [Mn3(notp)(H2O)6]·1.5H2O (19) |
R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) |
2D | Layer containing 6-rings | Weak AF coupling | 27 |
| [Zn2Fe(notp)Cl(H2O)] (20) | P63 | 2D | Layer containing 6-rings | 4.3 × 10−7, 95% RH, 25 °C | 29 |
| [ZnCo(notpH)(H2O)]·2H2O (21) | Pca21 | 2D | Layer containing 10-rings | 1.0 × 10−6, 95% RH, 25 °C, dielectric | 29 |
| [LaCo(notp)(H2O)4]·nH2O (22) | P21/n | 2D | Layer containing 6-, 8-rings | 3.0 × 10−6, 95% RH, 25 °C, magnetic | 30 |
| [LaCo(notpH)(H2O)6]ClO4·5H2O (23) | P21/n | 2D | Layer containing 12-rings | 3.5 × 10−6, 95% RH, 25 °C | 31 |
| [NdFe(notpH)(H2O)4]ClO4·5H2O (24) | P21/n | 2D | Layer containing 4-, 8-rings | Paramagnetic | 32 |
| [SmFe(notpH)(H2O)4]ClO4·5H2O (25) | P21/n | 2D | Layer containing 4-, 8-rings | Paramagnetic | 32 |
| [EuFe(notpH)(H2O)4]ClO4·5H2O (26) | P21/n | 2D | Layer containing 4-, 8-rings | Paramagnetic | 32 |
| [GdFe(notpH)(H2O)4]ClO4·5H2O (27) | P21/n | 2D | Layer containing 4-, 8-rings | Weak AF coupling | 32 |
| [CaCo(notpH2)(H2O)2]ClO4·4H2O (28) | P21/n | 2D | Layer containing 4-, 8-rings | 1.55 × 10−5, 95% RH, 25 °C | 33 |
| [CaCo(notpH2)(H2O)2]Cl·5H2O (30) | P21/c | 2D | Layer containing 4-, 8-rings | 1.40 × 10−4, 95% RH, 25 °C | 34 |
| [CaCo(notpH2)(H2O)2]NO3·4H2O (31) | P21/n | 2D | Layer containing 4-, 8-rings | 4.37 × 10−5, 95% RH, 25 °C | 34 |
| [CaCo(notpH2)(H2O)2]PF6·2H2O (32) | P21/n | 2D | Layer containing 4-, 8-rings | 1.39 × 10−5, 95% RH, 25 °C | 34 |
| [Ca2Co2(notpH2)2(H2O)8](IrCl6)·2H2O (33) |
P |
1D | {CaO7} dimer linked by {PO3C} | 1.27 × 10−6, 95% RH, 25 °C | 35 |
| [CaCo2(notpH3)2(H2O)2](IrCl6)·4H2O (34) | C2/c | 1D | {CaO7} linked by {PO3C} | 6.1 × 10−6, 95% RH, 25 °C; magnetic | 35 |
| [Co2(notpH4)2](IrCl6)·6H2O (35) |
P |
0D | Discrete monomer | 1.43 × 10−5, 95% RH, 25 °C | 35 |
| [LaCu(notpH2)(H2O)2]ClO4·3H2O (36) |
P |
1D | {LaO9} linked by μ-O and {PO3C} | Weak AF coupling | 32 |
| [GdCu(dotpH4)Cl]·4.5H2O (37) |
P |
3D | Open-framework with channels | Relaxometry | 37 |
| [{Na13(OH)3(H2O)29}{Gd(dotp)}2]·7H2O·2CH3CH2OH (38) | P4/n | 2D | Double layer structure | n.a. | 38 |
| [Mn2Gd(dota-4ampH)(H2O)7]·21H2O (39) | P21/c | 2D | Square-grid layer | Breathing effect | 40 |
| [Cu2Cl2(Me2do2pH2)(H2O)]·0.5C3H6O·2.5H2O (41) | P21/n | 2D | Rhombic layer | AF coupling | 41 |
| Li[Nd(do3apH)(H2O)]·11.5H2O (42) |
P |
1D | Polar linear chain | n.a. | 42 |
| Li[Tb(do3apH)(H2O)]·0.5HCl·5H2O (43) | Iba2 | 1D | Zigzag chain | n.a. | 43 |
| [Cu(H2O)2{Cu(L1)}](ClO4)·2H2O·0.5Me2O (44) | P21/c | 1D | Zigzag chain | n.a. | 45 |
| [Cd2.75(L2)(H2O)7]·1.5NO3·7H2O·MeOH (45) | P21/c | 3D | Open-framework with channels | n.a. | 46 |
 | ||
| Scheme 4 Molecular structures of metalloligands MIII(notpH3) and MII(notpH4). | ||
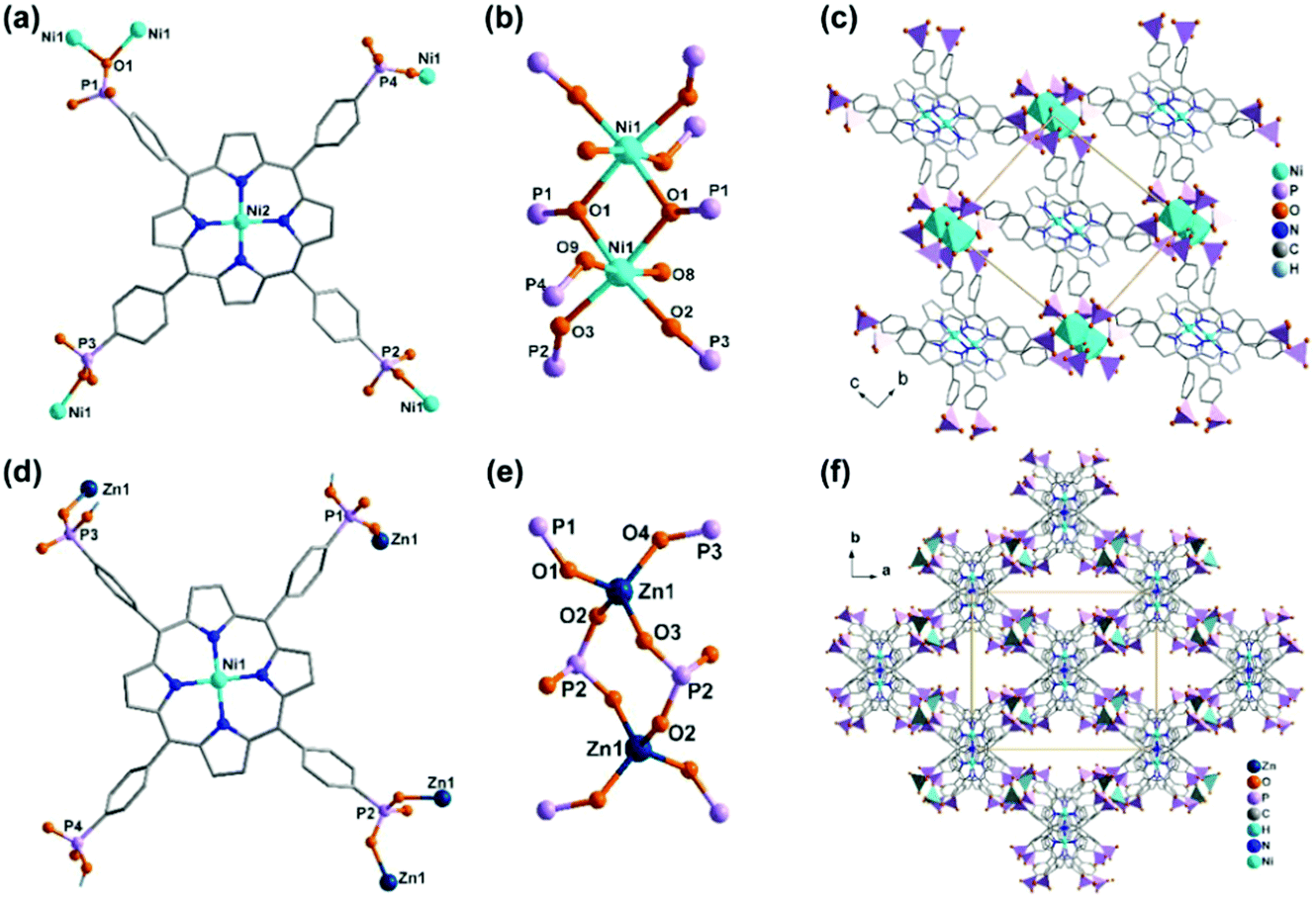
In 2006 we reported the first example of coordination polymers containing the M(notp) unit with the formula Mn3(notp)(H2O)6·1.5H2O (19),27 which was obtained by direct reaction of manganese salt and notpH6 under hydrothermal conditions. This compound crystallizes in a highly symmetrical trigonal system with space group R. Each MnII(notp)4− binds to six MnII ions via corner-sharing of {PO3C} tetrahedra and {MnO6} octahedra (Scheme 5a), while each {MnO6} octahedron is corner-shared with three {PO3C} tetrahedra from three equivalent MnII(notp)4− units, thus leading to an inorganic layer containing six-member rings of Mn3P3 (Fig. 3a). A unique feature of the layer structure lies in the alignment of the hydrophobic triazacyclononane moieties on one side and the hydrophilic coordinated water molecules on the other side, which results in a polar layer. Two such polar layers are connected by hydrogen bond interactions between the coordination water molecules, leading to a supramolecular double layer structure which is centrosymmetric (Fig. 3b). Notably, direct reactions of uranyl or lanthanide ions with notpH6 at low pH (ca. 1) resulted in UO2(noptH3)·H2O27 and a series of Ln-notp compounds28 in which the macrocycle rings are partially protonated without the presence of M(notp) unit.
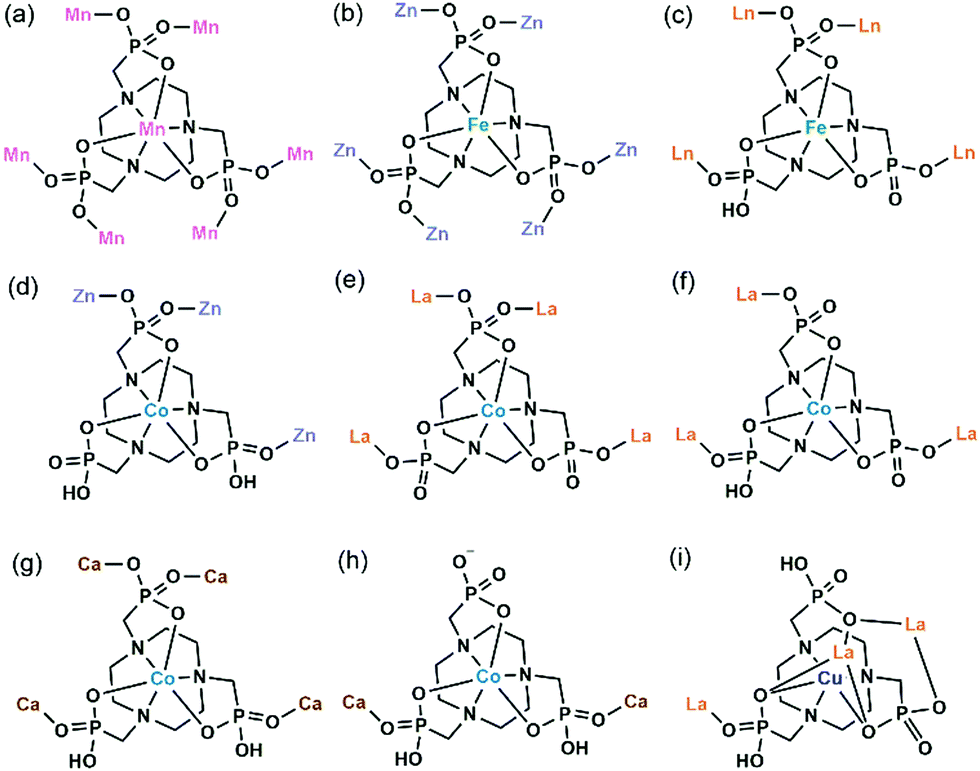 | ||
| Scheme 5 Coordination modes of M(notpHx) metalloligands. | ||
 | ||
| Fig. 3 (a) Single layer and (b) supramolecular double layer of structure 19. (c) Packing diagram of structure 20. (d) Packing diagram of structure 21. All H atoms except those attached to oxygen in (d) are omitted for clarity. Adapted from ref. 27 and reproduced from ref. 29 with permission from the Royal Society of Chemistry. | ||
Although the overall structure of compound 19 is non-polar, the single layer polarity is retained and extended to the whole structure in compounds Zn2Fe(notp)Cl(H2O) (20) and [ZnCo(notpH)(H2O)]·2H2O (21),29 where FeIII(notp)3− and CoIII(notpH)2− are metalloligands. The two compounds were synthesized by a bottom-up approach using pre-formed FeIII(notpH3) or CoIII(notpH3) monomers as precursors. 20 crystallizes in the hexagonal P63 chiral space group which is also polar. Although the single crystal of 21 is chiral, the bulk sample is a conglomerate and is circular dichroism silent. Within the structure, the six phosphonate oxygen atoms in Fe(notp)3− are each coordinated to a Zn atom (Scheme 5b), whereas each Zn atom is tetrahedrally coordinated by three phosphonate oxygen atoms from three equivalent Fe(notp)3− ligands and one Cl− anion. Such arrangement results in a polar layer containing six-member rings of Zn2FeP3 with the organic components all residing on the same side of the layer. These polar layers are stacked unidirectionally in an ABAB manner, leading to a polar 3D network structure (Fig. 3c).
The polar structure is also observed in 21. This compound crystallizes in an orthorhombic system, polar space group Pca21. The Co(notpH)2− metalloligand is tri-dentate (Scheme 5d). Only three of its six phosphonate oxygen atoms are involved in coordination with three equivalent Zn atoms, while each Zn is tetrahedrally coordinated by three phosphonate oxygen atoms from three Co(notpH)2− ligands and one water molecule. The resulted layer is undulated along the a-axis containing ten-member rings of Zn3Co2P5. The layer is racemic due to the presence of both Δ and Λ configurations of the cobalt centre. It is polar, however, with the organic components aligning in the same direction along the c-axis. The layers are packed unidirectionally to form a polar 3D network (Fig. 3d). The polar nature is confirmed by the dielectric measurements on single crystals of 21.
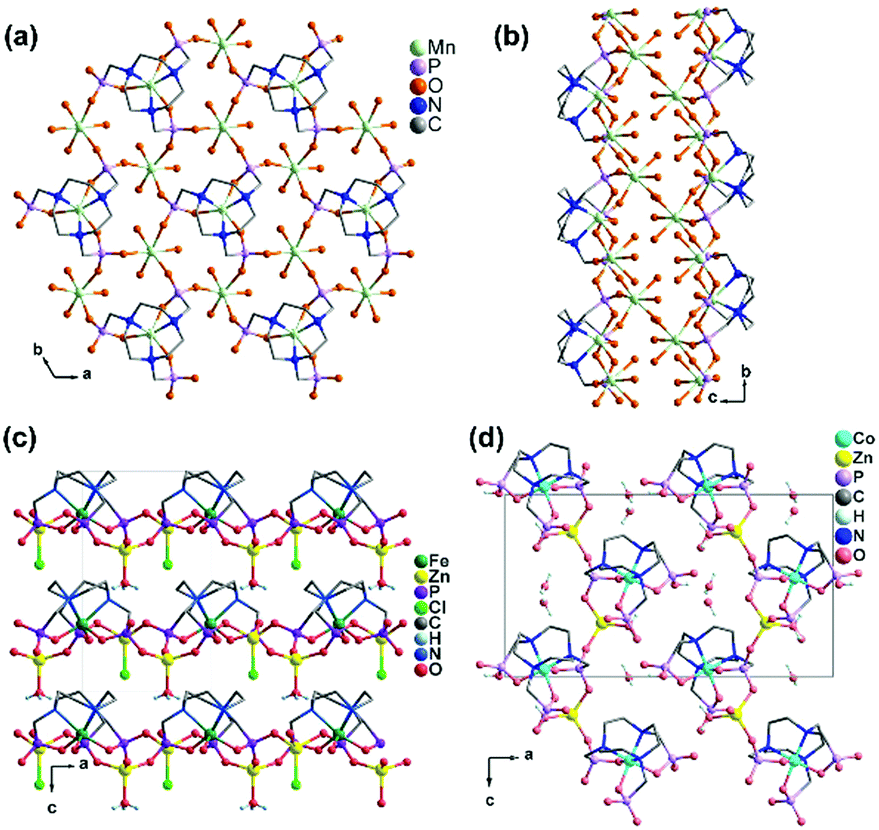
In contrast, the bottom-up assembly of MIII(notpH3) (M = Co, Fe) metalloligands with lanthanide or calcium ions resulted in products crystallizing in centrosymmetric space groups, possibly due to the fact that both LnIII and CaII ions have no significant stereochemical influence on coordination. [LaCo(notp)(H2O)4]·nH2O (22)30 and [LaCo(notpH)(H2O)6]ClO4·5H2O (23)31 can be isolated by reacting CoIII(notpH3) and La(ClO4)3 at different pH, more acidic in the latter case. They crystallize in monoclinic space groups P21/c and P21/n, respectively. In 22, LaIII is eight-coordinated by four phosphonate oxygen atoms and four water molecules. Each Co(notp)3− acts as a tetradentate metalloligand and links to four LaIII ions through four phosphonate oxygen atoms (Scheme 5e), while each La is connected to four Co(notp)3− and an equivalent La atom through phosphonate linkages. This leads to a 2D waved layer containing 6-member rings of CoLa2P3 and 8-member rings of Co2La2P4 (Fig. 4a). The interlayer space is filled with lattice water molecules.
 | ||
| Fig. 4 Layer structures of compounds (a) [LaCo(notp)(H2O)4]·nH2O (22), (b) [LaCo(notpH)(H2O)6]ClO4·5H2O (23), (c) [NdFe(notpH)(H2O)4]ClO4·5H2O (24), and (d) [CaCo(notpH2)(H2O)2](ClO4)·4H2O (28). All C, N and H atoms as well as coordination water molecules are omitted for clarity. Adapted from ref. 30–33. | ||
In 23, LaIII is nine-coordinated by three phosphonate oxygen atoms from three Co(notpH)2−, unlike that in 22. Further, one phosphonate group in Co(notpH)2− is protonated. Each Co(notpH)2− serves as a tridentate metalloligand connecting to three LaIII ions (Scheme 5f). As a result, a layer structure containing 12-member rings of Co3La3P6 is constructed, which is completely different from that of 22 (Fig. 4b). The layer is positively charged with the ClO4− counter-anions and lattice water molecules residing in the interlayer space.
Interestingly, similar reactions using FeIII(notpH3) instead of CoIII(notpH3) with lanthanide perchloride resulted in layered compounds [LnFe(notpH)(H2O)4]ClO4·5H2O [Ln = Nd (24), Sm (25), Eu (26), Gd (27)] with different structures.32 Compounds 24–27 are isostructural, crystallizing in the monoclinic P21/n space group. The coordination environment of the LnIII ion is similar to that in 22, which is eight-coordinated by four phosphonate oxygen atoms and four water molecules. While the Fe(notpH)2− metalloligand is analogous to Co(notpH)2− in 23 with one phosphonate group singly protonated, their coordination modes are different. Fe(notpH)2− is tetra-dentate (Scheme 5c), whereas Co(notpH)2− is tri-dentate. Consequently, a different type of layer structure containing 4- and 8-member rings is built up, as seen in Fig. 4c.
The replacement of LaIII by CaII leads to a new layered compound [CaCo(notpH2)(H2O)2](ClO4)·4H2O (28),33 where the two phosphonate groups are each singly protonated in order to maintain the overall charge balance. The CaII ion prefers an octahedral coordination geometry with the six sites provided by four phosphonate oxygen atoms and two water molecules. Each Ca is connected to four [Co(notpH2)]− and vice versa, leading to a 2D layer with similar topology to that in 24–27 (Fig. 4d and Scheme 5g). Humidity dependent single crystal to single crystal structural transformation is observed for 28, forming a new phase [CaCo(notpH2)(H2O)2](ClO4)·2H2O (29) at low atmosphere humidity, accompanied by a glide of the layer relative to the adjacent layers and a re-arrangement of the hydrogen bond network. The ClO4− in 28 can be replaced by other counter-anions forming [CaCo(notpH2)(H2O)2]X·nH2O [X = Cl− (30, n = 5), NO3− (31, n = 4), PF6− (32, n = 2)] with the same layer topology but different number of lattice water molecules.34
The counter-anion can be paramagnetic. By incorporating IrCl62−, trimetallic CoIII–CaII–IrIV compounds [Ca2Co2(notpH2)2(H2O)8](IrCl6)·2H2O (33) and [CaCo2(notpH3)2(H2O)2](IrCl6)·4H2O (34) as well as [Co2(notpH4)2](IrCl6)·6H2O (35) were isolated at pH 3.5, 2.5 and 1.7, respectively.35 In 33, edge-sharing dimers of {CaO7} are connected by the bidentate [Co(notpH2)]− metalloligands (Scheme 5h) into infinite chains. 34 also shows a chain structure, made up of {CaO6} octahedra and Co(notpH3) linkages. In the case of 35, discrete Co(notpH4)+ species is found together with the IrCl62− anion.
Despite the dominant octahedral geometries of the metal centres in M(notpHx) (M = CoIII, FeIII, MnII), the CuII ion favours a square or square pyramidal environment due to the Jahn–Teller distortion. In [LaCu(notpH2)(H2O)2]ClO4·3H2O (36),32 the CuII ion in Cu(notpH2)2− has a distorted square pyramidal geometry with the basal sites occupied by two O and two N donors from the same notpH24−, and axial site by the third nitrogen atom. Cu(notpH2)2− serves as a hepta-dentate metalloligand binding to three equivalent La atoms (Scheme 5i). Each La atom is nine-coordinated by seven phosphonate oxygen and two water molecules. Neighbouring La atoms are bridged by Cu(notpH2)2− metalloligands through μ3-O and O–P–O units alternatively, forming an infinite chain (Fig. 5).
 | ||
| Fig. 5 Chain structure of 36. All H atoms are omitted for clarity. Adapted from ref. 32. | ||
Other polyazamacrocycle phosphonate ligands that have been commonly studied are 1,4,7,10-tetraazacyclododecane (cyclen) and 1,4,8,11-tetrazacyclotetradecane (cyclam) with one-, two-, three- or four-pendant phosphonate arms. However, most studies were limited to their coordination chemistry in solution due to their utilization in medicine and molecular biology as contrast agents in magnetic resonance imaging (MRI) or metal radioisotope carriers for diagnostic and/or therapeutic purposes.36 Coordination polymers incorporating cyclen- or cyclam-phosphonate metalloligands are rare.
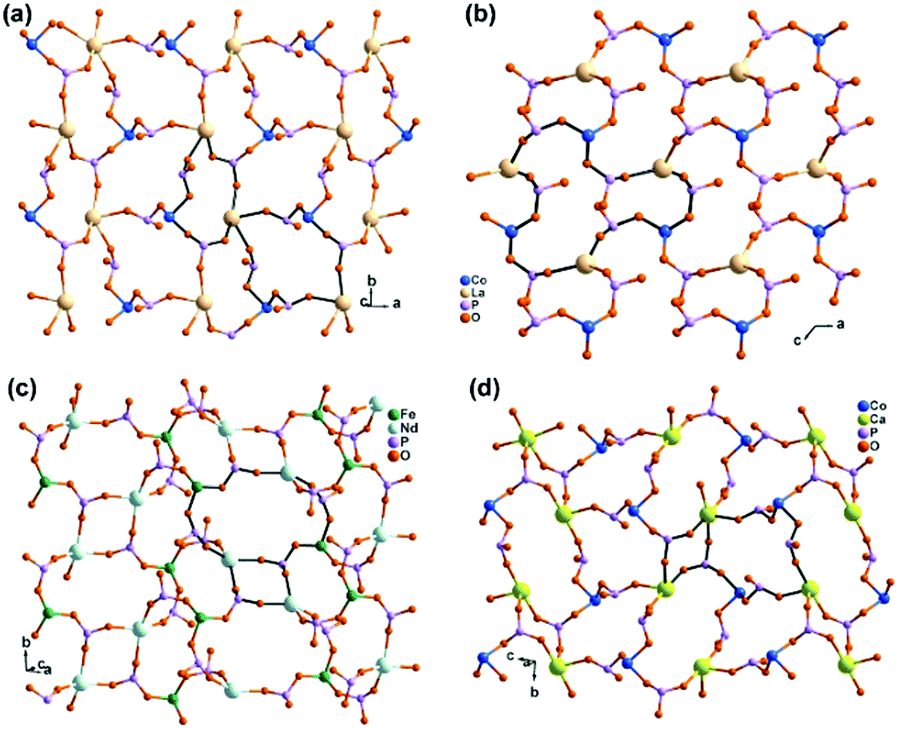
An interesting example is [GdCu(dotpH4)Cl]·4.5H2O (37)37 reported by Maspoch and co-workers, where dotpH8 represents 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetramethylenephosphonic acid. The compound was synthesized by direct reaction of CuCl2, Gd(NO3)3 and dotpH8 in aqueous solution at 85 °C. Within the structure, there are two crystallographically distinguished Cu atoms (Cu1, Cu2), each having a square pyramidal geometry. The basal sites are provided by four N atoms from the dotpH44− ligand and the axial site by the Cl− anion. Each Cu(dotpH4)2− serves as a hexa-dentate metalloligand binding to six Gd atoms (Scheme 6a). There are also two kinds of Gd atoms, Gd1 and Gd2, each having a distorted octahedral coordination environment. The equivalents of each Gd are doubly bridged by O–P–O units forming Gd12 and Gd22 dimers. These dimers are further connected by O–P–O units alternatively forming infinite chains. The Gd chains are cross-linked by Cu(dotpH4)2− to construct a 3D open framework structure with one dimensional channels (ca. 5 × 5 Å, van der Waals radii not accounted) (Fig. 6a and b). Notably, the same structure cannot be obtained by replacing Cu(II) by Zn(II), Ni(II), Co(II), Mn(II), Fe(III) and Mg(II), likely because they have less preference for nitrogen donors compared to Cu(II).
 | ||
| Scheme 6 Coordination modes of metalloligands based on cyclen-, cyclam- and 1,10-diaza-18-crown-6-phosphonate. | ||
 | ||
| Fig. 6 (a) The 3D open framework structure and (b) the Gd chain connection of 37. (c) Single layer in structure 38. (d) Layer structure made up of Mn2(PO3)4(H2O)6 dimers and Gd(DOTA-4ampH) linkages in 39. Adapted from ref. 37, 38 and 40. | ||
Based on the same dotp ligand, Avecilla and co-workers reported a Na–Gd compound, [{Na13(OH)3(H2O)29} {Gd(dotp)}2]·7H2O·2CH3CH2OH (38).38 As shown in Fig. 6c, the Gd(dotp) metalloligands in 38 are dispersed in the {NaOx} polyhedral network by coordinating to four Na atoms through their eight pendant phosphonate oxygen atoms forming a layer (Scheme 6b). Two such layers are connected by {NaOx} polyhedra to form a double layer structure. If notpH8 reacted with lanthanide or transition metal salts at low pH, discrete or polymeric compounds can be isolated in which the macrocycle nitrogen atoms are partially protonated and not involved in coordination with metal ions.39
1,4,7,10-Tetraazacyclododecane-1,4,7,10-tetraacetamido methylenephosphonic acid (DOTA-4ampH8) has four phosphonic acid arms with an additional acetamido moiety. It is ideally suited to wrap up the lanthanide ion using four N and four acetoamido O atoms to form a metalloligand, leaving the four phosphonate groups for further coordination. Maspoch et al. successfully isolated a two-dimensional Mn–Gd compound [Mn2Gd(DOTA-4ampH)(H2O)7]·21H2O (39),40 using the pre-formed Gd(DOTA-4ampHx) metalloligand as the precursor. It crystallizes in monoclinic space group P21/c. The Gd(III) ion is nine-coordinated by four N, four acetoamido O atoms and one water molecule. The metalloligand Gd(DOTA-4ampH) is hexa-dentate, using four phosphonate oxygen atoms, each from one phosphonate group (Scheme 6c). Two of the four phosphonate oxygen atoms act as μ3-O bridging two Mn atoms, thus forming Mn2(PO3)4(H2O)6 dimers. The dimers are connected by the metalloligands in four directions, leading to a layer structure in the ac plane (Fig. 6d). The layers are stacked along the c-axis generating 1D channels with dimensions of approximately 14.8 × 9.5 Å (van der Waals radii not accounted) where the lattice water molecules reside. Compound 39 shows interesting humidity dependent single-crystal to single-crystal structural transformation, forming [Mn2Gd(DOTA-4ampH)(H2O)7]·5H2O (40) at RH < 40%. The transformation causes the layers changing from corrugated to flatter, but the coordination network remains almost the same. Water sorption isotherms show a gate-opening effect at a RH of ca. 85% upon water adsorption, and a gate-closure effect at RH 55–77% upon water desorption.
Vizza and co-workers synthesized a cyclen derivative with two phosphonic acid arms, e.g. 4,10-dimethyl-1,4,7,10-tetraazacyclododecane-1,7-bis(methanephosphonic acid) (Me2do2pH4). The reaction of Me2do2pH4 with Cu(OAc)2 at room temperature results in the formation of compound {[Cu2Cl2(Me2do2pH2)(H2O)]·0.5C3H6O·2.5H2O} (41).41 Analogous to that in 37, the Cu(II) ion also has a distorted square-pyramidal coordination geometry with the four basal positions occupied by macrocycle N atoms and the axial position by Cl− anion. However, the Cu(Me2do2pH2) metalloligand is tri-dentated (Scheme 6d). Its two phosphonate arms each provide one phosphonate oxygen to bind three Cu(II) ions. One phosphonate oxygen serves as μ3-O bridging two equivalent Cu atoms to form the {Cu2O2} dimer. The dimers are connected by Cu(Me2do2pH2) in four directions forming a 2D layer containing rhombic rings made up of four metalloligands, four Cu atoms from {Cu2O2} dimers, and eight {PO3C} groups (Fig. 7a). The layers are stacked forming a 3D supramolecular network, generating 1D channels along the a-axis. The channel size is ca. 5.2 × 5.8 Å (van der Waals radii not accounted).
 | ||
| Fig. 7 (a) Single layer of structure 41. (b) Chain structure of 42. (c) Chain structure of 55. | ||
By employing a mono(methylphosphonate)-cyclen derivative, 1,4,7,10-tetraazacyclododecane-4,7,10-tris(carboxymethyl)-1-methylphosphonic acid (H5do3aP), Lukeš and co-workers isolated two Li–Ln compounds with chain structures, e.g. Li[Nd(Hdo3ap)(H2O)]·11.5H2O (42)42 and Li[Tb(Hdo3ap)(H2O)]·0.5HCl·5H2O (43).43 In both cases, the Ln(III) ions are nine-coordinated by four macrocycle N atoms, three carboxylate O atoms and one phosphonate O atom as well as one coordination water molecule. However, the metalloligand Nd(Hdo3ap)− in 42 is bi-dentate linking to two Li atoms via one carboxylate O atom and one phosphonate O atom (Scheme 6e). But the Tb(Hdo3ap)− in 43 is tri-dentate, chelating and bridging to two equivalent Li atoms (Scheme 6f). As a consequence, the tetrahedrally coordinated Li(I) ions in 42 are connected by Nd(Hdo3ap)− into a linear chain which is polar (Fig. 7b). On the other hand, the Li(I) ions in 43 are connected by Tb(Hdo3ap)− into a zigzag chain via two phosphonate oxygen atoms which is non-polar (Fig. 7c). It should be mentioned that the overall structure of 42 is non-polar due to the centrosymmetric packing of the polar chains.
As far as we are aware, there are only two coordination polymers containing cyclam-phosphonate metalloligands. One is Ni(cyclam1p),44 where cyclam-p represents [(1,4,8,11-tetraazacyclotetradecan-1-yl)methylene]phosphonic acid. Within the structure, Ni(cyclam1p) serves as both donor and acceptor using one phosphonate oxygen to bind the Ni atom from the neighbouring unit (Scheme 6g), thus forming an infinite chain. The other is {Cu(H2O)2[Cu(L1)]}(ClO4)·2H2O·0.5acetone (44),45 isolated by Kotek and co-workers, where the cyclam derivative H3L1 contains a single diphosphonate arm. In this case, Cu(L1) serves as a tri-dentate metalloligand (Scheme 6h), chelating and bridging the Cu(II) ions into infinite zigzag chains. The chain consists of corner sharing {CuO5} square pyramids and {PO3C} tetrahedra. The cyclam moieties are pendant on the two sides of the inorganic chain (Fig. 8a).
 | ||
| Fig. 8 (a) Chain structure of 44. (b) The Cd5{Cd4(L2)4} cluster and (c) packing diagram of structure 45. The C, N and H atoms in (c) are omitted for clarity. Colour codes: {CdO8} and {CdO6} green, {PO3C} purple. | ||
Clearfield and co-workers employed N,N′-bis(phosphonomethyl)-1,10-diaza-18-crown-6 (H4L2), and isolated compound Cd2.75(L2)(H2O)7·1.5NO3·7H2O·MeOH (45) with an open framework structure.46 Within the metalloligand Cd(L2)2−, the Cd atom is eight-coordinated by four O and two N atoms from the crown ring and two O atoms from the phosphonate arms. The metalloligand is hepta-dentate, chelating and bridging five Cd atoms (Scheme 6i). A pentanuclear Cd5 cluster is found, where the central {CdO8} is edge-shared with four {CdO6} octahedra. Each Cd5 cluster is connected to four Cd(L2)2−, forming a Cd5{Cd4(L2)4} cluster (Fig. 8b). The Cd5{Cd4(L2)4} clusters are cross-linked via {Cd2O2} dimers, forming a 3D framework structure (Fig. 8c). The channel sizes are ca. 4.2 × 4.2 Å along the c axis or 9.3 × 14.1 Å towards (0 5 −4) (van der Waals radii not accounted), respectively.
To summarize, metallo-polyazamacrocycle-phosphonates can be used as efficient metalloligands for the construction of MMPs with structures ranging from 1D chain, 2D layer to 3D framework. For metallo-polyazamacrocyles with four, three, or two phosphonate arms, they usually form 2D layer structures with second metal ions. Only in a few cases, 1D chain or 3D open framework structures are observed. In contrast, metallo-polyazamacrocycles with one phosphonate arm tend to form a 1D chain structure. This statement is not rigorous, however, as the given examples are still extremely limited. The formation of a particular structure is highly dependent on the reaction conditions, the coordination geometries of the second metal ions and the involvement of metal clusters as nodes. In addition, these metalloligands possess a unique feature of polarity that distinguishes them from many other phosphonate ligands. This feature together with their structural diversity and tuneable composition endows them with advantages in the design and synthesis of materials with multifunction.
3.3. Metalloporphyrin units as metalloligands
Porphyrins and their complexes have attracted intensive research interest across many fields due to their rich coordination chemistry and photophysical, redox and catalytic properties.47 In recent years a number of metal–organic frameworks (MOFs) have been obtained using metalloporphyrins as building units.48 Especially Zr-MOFs containing H2TCPP4− are highly porous and stable.49 However, it is still a great challenge to construct MMPs incorporating metalloporphyrins, especially those based on metalloporphyrin-phosphonic acids, due to the difficulties in synthesizing the metalloligands and in forming crystalline materials with sufficient size for structural determination. Table 3 gives the known MMPs based on the metalloporphyrins shown in Scheme 7.| Compound | Space group | N | Structure | Proton conductivity (S cm−1) and other propertiesa | Ref. |
|---|---|---|---|---|---|
| a N: dimension of the structure, n.a.: not available. UV-vis absorption spectra and cyclic voltammetry (CV) measurements were carried out in solution. | |||||
| Cu2(tBuCOO)4{Cu(DEPPP)} (46) |
P |
1D | Linear chain | n.a. | 54 |
| Cu2(tBuCOO)4{Pd(DEPPP)} (47) |
P |
1D | Linear chain | n.a. | 54 |
| Cu2(tBuCOO)4{Ni(DEPPP)} (48) | C2/c | 1D | Zigzag chain | n.a. | 54 |
| MnNi(TPPPH6)(H2O) (49) |
P |
3D | Framework isostructural to 51 | N2 and H2O sorption | 60 |
| CoNi(TPPPH6)(H2O) (50) |
P |
3D | Framework isostructural to 51 | N2 and H2O sorption | 60 |
| NiNi(TPPPH6)(H2O) (51) |
P |
3D | Open-framework with channels | 5.6 × 10−6, 90% RH, 80 °C | 60 |
| CdNi(TPPPH6)(H2O) (52) |
P |
3D | Framework isostructural to 51 | N2 and H2O sorption | 60 |
| Zn3[Ni(TPPPH3)3]·9(CH3)2NH2·3DMF·17H2O (53) | C2/c | 3D | Open-framework with channels | 1.55 × 10−3, 80% RH, 75 °C | 61 |
| [Co2Ni(TPPPH4)]·2DABCO·6H2O (54) |
P![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) c2 c2 |
3D | Open-framework with channels | N2 sorption, BET: 700 m2 g−1 | 62 |
| [Zr2Ni(TPPPH2)(OH/F)2] (55) | I41/acd | 3D | Open-framework with channels | BET: 1070 m2 g−1, CV, UV-vis | 63 |
| [Hf2Ni(TPPPH2)(OH/F)2] (56) | I41/acd | 3D | Open-framework with channels | BET: 1030 m2 g−1, UV-vis | 63 |
| [Na2Cu(TPPPH4)]·(NH2(CH3)2)2 (57) |
P |
3D | Open-framework with channels | BET: 697 m2 g−1 | 64 |
| [EuIr6(ppy)12(bpp)2(bppH)4](CF3SO3)·xH2O (58) |
P |
0D | Supramolecular framework | Phosphorescence | 65 |
| [TbIr6(ppy)12(bpp)2(bppH)4](CF3SO3)·xH2O (59) |
P |
0D | Supramolecular framework | Phosphorescence | 65 |
| [DyIr6(ppy)12(bpp)2(bppH)4](CF3SO3)·xH2O (60) |
P |
0D | Supramolecular framework | Phosphorescence, magnetic | 65 |
| [ErIr6(ppy)12(bpp)2(bppH)4](CF3SO3)·xH2O (61) |
P |
0D | Isostructural to 58–60 | Phosphorescence, magnetic | 65 |
| [YbIr6(ppy)12(bpp)2(bppH)4](CF3SO3)·xH2O (62) |
P |
0D | Isostructural to 58–60 | Phosphorescence, magnetic | 65 |
| [La3(H2O)9{Ru(4,4′-dpbpy)3H5.5}2]·nH2O (63) | P21/n | 3D | Isostructural to 64 | 2.8 × 10−6, 95% RH, 25 °C | 66 |
| Pr3[Ru(DPBPY)3H5.5]2 (64) | P21/n | 3D | Open-framework with channels | 3.7 × 10−6, 95% RH, 25 °C | 66 |
| [GdCoIII(5,5′-dpbpyH2)3(H2O)2]·6.5H2O (65) | C2/c | 2D | Isostructural to 67 | Magnetic | 67 |
| [DyCoIII(5,5′-dpbpyH2)3(H2O)2]·6.5H2O (66) | C2/c | 2D | Isostructural to 67 | Magnetic | 67 |
| [TbCoIII(5,5′-dpbpyH2)3(H2O)2]·6.5H2O (67) | C2/c | 2D | Layer with (4,4) connectivity | Magnetic | 67 |
| [Gd{NiII(5,5′-dpbpy)3H7}(H2O)3]NaCl·6H2O (68) | C2 | 3D | Open-framework with channels | Magnetic | 67 |
 | ||
| Scheme 7 Molecular structures of metalloporphyrin–phosphonate ligands. | ||
Gorbunova and co-workers synthesized a meso-phosphorylporphyrin, e.g. 5,15-bis(diethoxyphosphoryl)-10,20-diphenylporphyrin (DEPPP, Scheme 7a), through Pd-catalyzed coupling reactions. The solution reactions of H2-DEPPP with metal salts resulted in compounds M(DEPPP) (M = Zn,50 Cu,51 Cd52) with similar layer structures, where DEPPP serves as both donor and acceptor. For M(DEPPP) (M = Pt, Pd, Ni), layered coordination polymers cannot be obtained because the central metal ions favour a four-coordinated square-planar geometry.52 Another related 2D polymeric MMP is Zn(CN-DEPPP) based on 5,15-bis(diethoxyphosphoryl)-10,20-bis(4′-cyanophenyl) porphyrin (CN-DEPPP, Scheme 7a), described by Kadish and co-workers.53 The electrochemical properties were studied for all these compounds.
Direct reactions of Cu2(tBuCOO)4(CH3CN)2 and M(DEPPP) (M = Cu, Pd, Ni) in chloroform resulted in the formation of compounds Cu2(tBuCOO)4{M(DEPPP)} [M = Cu (46), Pd (47), Ni (48)].54 These compounds display similar zigzag chain structures, where the paddlewheel dimers of Cu2(tBuCOO)4 are connected by the M(DEPPP) metalloligands (Fig. 9a). In 46, there exist two kinds of Cu atoms. One locates at the centre of the porphyrin cavities adopting a square-planar CuN4 environment. The other incorporates in the paddlewheel unit showing a square-pyramidal CuO5 geometry with the apical site occupied by phosphoryl O atom. It is noted that the geometry of the porphyrin macrocycle can be rather different depending on the central metal ion. A strong nonplanarity of the porphyrin core is observed only for Ni(DEPPP). This leads to the formation of a zigzag chain in 48, instead of a linear chain as in 46 and 47. The packing of the zigzag chains results in regular pores in the crystal with dimensions of ca. 10 × 12 Å (van der Waals radii not accounted) (Fig. 9b).
 | ||
| Fig. 9 (a) Linear chain structure of 46. (b) Zigzag chain structure of 48. Adapted from ref. 50 and 54. | ||
Recently, Ermakova et al. developed synthetic routes to accessing mono(diethoxyphosphorylporphyrins) with different substitutes. Compounds Zn(Por-L1)55 (Scheme 7b) and Zn(Por-L2)56 (Scheme 7c) show chain structures, where the porphyrin macrocycle units are connected to each other through coordination of zinc with phosphoryl O atoms. Apart from these polymers, discrete dimeric or tetrameric compounds were also reported based on zinc complexes of 10-(dialkoxyphosphoryl)-5,15-diarylporphyrinates or β-dialkoxyphosphoryl-5,10,15,20-tetraphenylporphyrins (Por-L3, Scheme 7d),57 and gallium(III) and indium(III) complexes of meso-mono(diethoxyphosphoryl)porphyrins (Por-L4, Scheme 7e).58
Noting that the diethoxyphosphoryl group can provide only one oxygen donor for coordination and the resulted M–O(P) bond is weak, it is highly desired to prepare metal phosphonates using porphyrin-phosphonic acids as ligands. A typical ligand is tetra(4-phosphonophenyl)porphyrin (H10TPPP). Although it was known early in 1992,59 coordination polymers containing HxTPPP-based metalloligands were reported only recently. Stock and co-workers employed a high-throughput method and synthesized a series of metal–organic frameworks using Ni(HxTPPP) metalloporphyrins (Scheme 7f) as the precursor. Compounds M(Ni-H6TPPP)(H2O) [M = Mn (49), Co (50), Ni (51), Cd (52)]60 are isostructural, and the structure of Ni(Ni-H6TPPP)(H2O)·11H2O (51) is shown in Fig. 10. Ni-H6TPPP2− acts as a penta-dentate metalloligand, binding five equivalent Ni1 atoms via four phosphonate oxygen atoms, each from a different phosphonate group (Fig. 10a). The phosphonate oxygen O1 serves as μ3-O bridging two Ni1 atoms forming an edge-sharing {Ni12O2} dimer (Fig. 10b). Each dimer is connected to eight Ni-H6TPPP2− metalloligands and each Ni-H6TPPP2− links to four {Ni12O2} dimers, therefore constructing a 3D porous structure containing two kinds of rhombic channels with dimensions 2.8 × 5.0 and 3.8 × 3.8 Å (van der Waals radii accounted) or 5.5 × 8.0 and 7.0 × 7.0 Å (van der Waals radii not accounted) that host water molecules (Fig. 10c). Sorption measurements revealed that 51 shows the highest water uptake of 181 mg g−1. Moreover, it is stable in the pH range between 1 and 11.
 | ||
| Fig. 10 (a–c) The structure of compound 51: (a) coordination mode of Ni-H6TPPP2−, (b) dimeric building unit, (c) packing diagram. (d–f) The structure of compound 53: (d) coordination mode of Ni-H6TPPP2−, (e) dimeric building unit, (f) packing diagram. Adapted from ref. 60 and 61. | ||
A dinuclear building unit is also found in compound Zn3(Ni-H3TPPP)3·9(CH3)2NH2·3DMF·17H2O (53).61 Here the metalloligand Ni-H3TPPP5− acts as a tetra-dentate ligand, binding to four Zn atoms (Fig. 10d). The Zn atoms have tetrahedral geometries, unlike the octahedral geometry of the Ni(II) ion in 51. The Zn atoms are doubly bridged by O–P–O units forming {Zn2(OPO)2} dimers (Fig. 10e). Each dimer is connected to six Ni-H3TPPP5−, while each Ni-H3TPPP5− is connected to three {Zn2(OPO)2} dimers. Although the connecting topology in 53 is quite different from that in 51, a 3D open framework containing rhombic channels is again constructed in 51 (Fig. 10f). The channel size is ca. 4.5 × 10 Å (van der Waals radii not accounted). The anionic framework is charge-balanced by (CH3)2NH2+ which, together with the solvent molecules, occupies the channel space.
Another example containing the Ni-HxTPPP metalloligand is [Co2(Ni-H4TPPP)]·2DABCO·6H2O (54 or Co-CAU-36) (DABCO = 1,4-diazabicyclo[2.2.2]octane),62 the structure of which was determined by single-crystal electron diffraction using continuous rotation electron diffraction (cRED) data collected at low temperature (96 K). In this compound, Ni-H4TPPP4− is octa-dentate, coordinating to eight Co atoms using eight of its twelve phosphonate oxygen atoms (Fig. 11a). The Co atom has a distorted tetrahedral geometry. The equivalent Co atoms are doubly bridged by phosphonate groups through corner-sharing of {CoO4} and {PO3C} forming an inorganic chain (Fig. 11b). The chains are cross-linked by the Ni-H4TPPP4− moieties to form a 3D open framework with 1D rectangular channels generated along the c-axis (Fig. 11c). The channel has a pore diameter of ca. 9 Å (van der Waals radii accounted) when taking the DABCO guest molecules into account, and a diameter of ca. 12.5 Å (van der Waals radii accounted) when the pores are empty.
 | ||
| Fig. 11 (a–c) The structure of compound 54: (a) coordination mode of Ni-H6TPPP2−, (b) dimeric building unit, (c) packing diagram. (d–f) The structure of compound 55: (d) coordination mode of Ni-H6TPPP2−, (e) dimeric building unit, (f) packing diagram. Adapted from ref. 62 and 63. | ||
Interestingly, a similar framework structure with rectangular channels is also observed for tetravalent metal compounds [M2(Ni-H2TPPP)(OH/F)2]·H2O [M = Zr (55), Hf (56); (M-CAU-30)].6355 and 56 are isostructural. Like that in 54, the metalloligand Ni-H2TPPP6− acts as an octa-dentate ligand, binding to eight ZrIV (or HfIV) ions (Fig. 11d). A significant difference between 55 and 54 is the coordination geometry of second metal ions, which is tetrahedral for Co(II) in 54 but octahedral for Zr(IV) in 55. The adjacent Zr atoms are bridged not only by two phosphonate groups but also by OH−/F− anions, hence leading to an infinite chain containing corner-sharing {ZrO6} octahedra (Fig. 11e). The chains are cross-linked by the Ni-H2TPPP6− ligands to form a 3D framework (Fig. 11f). The channel diameters are 1.3 × 2.0 nm (van der Waals radii accounted). M-CAU-30 show the highest specific surface area among metal phosphonates of 1070 and 1030 m2 g−1 for 55 and 56, respectively, derived from the experimental data for the corresponding compounds contaminated with ZrO2 or HfO2 impurities. They are chemically stable in the pH range 0–12 and thermally stable up to 420 °C in air. The remarkable stability of M-CAU-30 makes them promising candidates for many applications such as gas separation and storage, catalysis and proton conduction.
Yücesan, Zorlu and co-workers reported an alkali phosphonate MOF containing the Cu-H4TPPP4− metalloligand, formulated as [Na2(Cu-H4TPPA)]·(NH2(CH3)2)2 (57).6457 crystallizes in a triclinic space group P. It also shows a 3D open framework structure with rectangular shaped channels (7.6 × 7.6 Å, van der Waals radii not accounted), composed of Na–O–P–O inorganic chains and Cu-H4TPPP4− linkages. Each Cu-H4TPPP4− binds to eight NaI ions. The NaI ions are either four-coordinated (for Na1) or five-coordinated (for Na2), each of which is edge-shared with its equivalent ones forming {Na12O2} or {Na22O2} dimers. The dimers are connected via phosphonate oxygen and O–P–O units in an alternative manner, forming an infinite chain. These chains are cross-linked by Cu-H4TPPP4− metalloligands into a 3D framework (Fig. 12). After activation, the compound shows a surface area of 698 m2 g−1 which is dramatically larger than that of the other Na-MOFs, but close to that of [Co2(Ni-H4TPPP)]·2DABCO·6H2O (700 m2 g−1).
 | ||
| Fig. 12 Packing diagram of structure 57. Adapted from ref. 64. | ||
To summarize, metalloporphyrins with one or two esterified phosphonate groups form 1D chains with second metal ions, while those with four phosphonic acid groups form 3D open framework structures with different channel sizes. These MMPs show very high thermal and chemical stability and a large surface area, especially those containing tetravalent ZrIV and HfIV ions. By retaining chemically transformable groups like esters in some of these porphyrin-based MMPs, there exist further opportunities to diversify their structures and functions via crystal to crystal transformations. All these features are remarkable for the development of functional MOFs for future applications.
3.4. Cyclometalate or metal–polypyridine complexes as metalloligands
Metalloligands such as cyclometalated Ir(III) complexes and Ru(II) polypyridine complexes are very attractive for constructing functional coordination polymers because they can act not only as a chromophore or luminophore but also as a reversible redox reaction centre. By using Ir(ppy)2(bppH) (ppy = 2-phenylpyridine, bppH2 = 2-pyridylphosphonic acid) as the metalloligand, we isolated isostructural heptanuclear LnIr6 clusters [LnIr6(ppy)12(bpp)2(bppH)4](CF3SO3)·xH2O [Ln = Eu (58), Tb (59), Dy (60), Er (61), Yb (62); ppy = 2-phenylpyridine; bpp = 2-pyridylphosphonate].65 The clusters are packed in the lattice, forming a supramolecular network containing channels in the a- and c-directions, where the lattice water molecules and the CF3SO3− anions reside (Fig. 13). The two edge distances of the quadrilateral window in the ab plane are 15.877 and 16.958 Å, respectively. However, coordination polymers incorporating Ir(III) cyclometalate-phosphonate metalloligands have not been documented.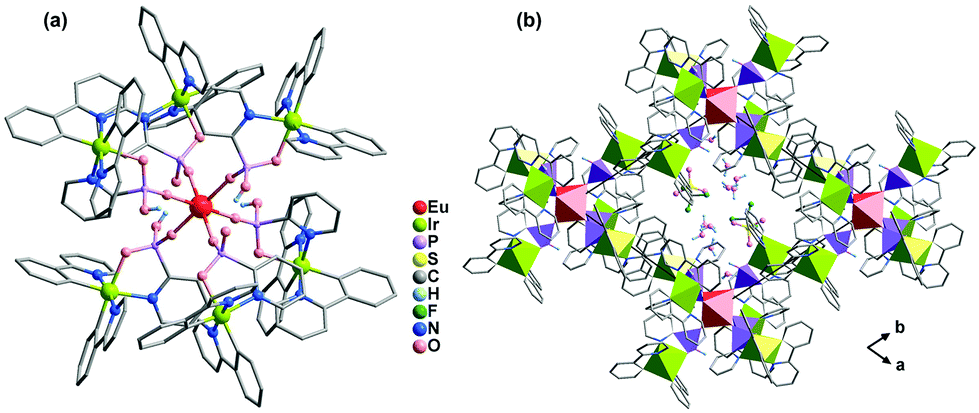 | ||
| Fig. 13 (a) The heptanuclear cluster of 58. (b) Packing diagram of 58 viewed along the c-axis. Color codes: EuO6, red; IrC2N3O, yellow green; PO3C, purple. Adapted from ref. 65. | ||
MMPs containing Ru(II)-polypyridine metalloligands are also rare. The only examples are Ln3(H2O)9[Ru(4,4′-dpbpy)3H5.5]2·nH2O [Ln = LaIII (63), PrIII (64)],66 where dpbpyH4 is 2,2′-bipyridinyl-4,4′-diphosphonic acid (Scheme 8). The two compounds are isostructural. Single crystal structural analysis of 64 reveals that it crystallizes in monoclinic space group P21/n. There are two kinds of Pr atoms in the structure. The Pr1 atom is coordinated by five phosphonate oxygen atoms and three water molecules to form a square antiprismatic geometry. The disordered Pr2 atom (half/half) is surrounded by five phosphonate oxygen atoms and three water molecules. RuII(4,4′-dpbpy)3H5.51.5− serves as an octa-dentate metalloligand, binding to eight Pr atoms via eight of its eighteen phosphonate oxygen atoms (Scheme 9a). Two of the six phosphonate groups are involved in bridging the Pr1 atoms into {Pr12(OPO)2} dimers. The remaining four phosphonate groups are involved in coordination with one Pr1 and three Pr2 atoms. As a result, the Pr atom and {Pr12(OPO)2} dimers are cross-linked by the [Ru(4,4′-dpbpy)3H5.5]4.5− metalloligands forming a three-dimensional open framework (Fig. 14). The framework contains two kinds of narrow porous channels along the b-axis with dimensions of ca. 4.4 × 6.0 Å and 3.2 × 4.6 Å (van der Waals radii not accounted).
 | ||
| Scheme 8 Molecular structures of metalloligands RuII(4,4′-dpbpyH4)32+ and [M(5,5′-dpbpyH4)3]n+. | ||
 | ||
| Scheme 9 Coordination modes of metalloligands RuII(4,4′-dpbpyH4)32+ and [M(5,5′-dpbpyH4)3]n+. | ||
 | ||
| Fig. 14 Packing diagram of structure 64. Adapted from ref. 66. | ||
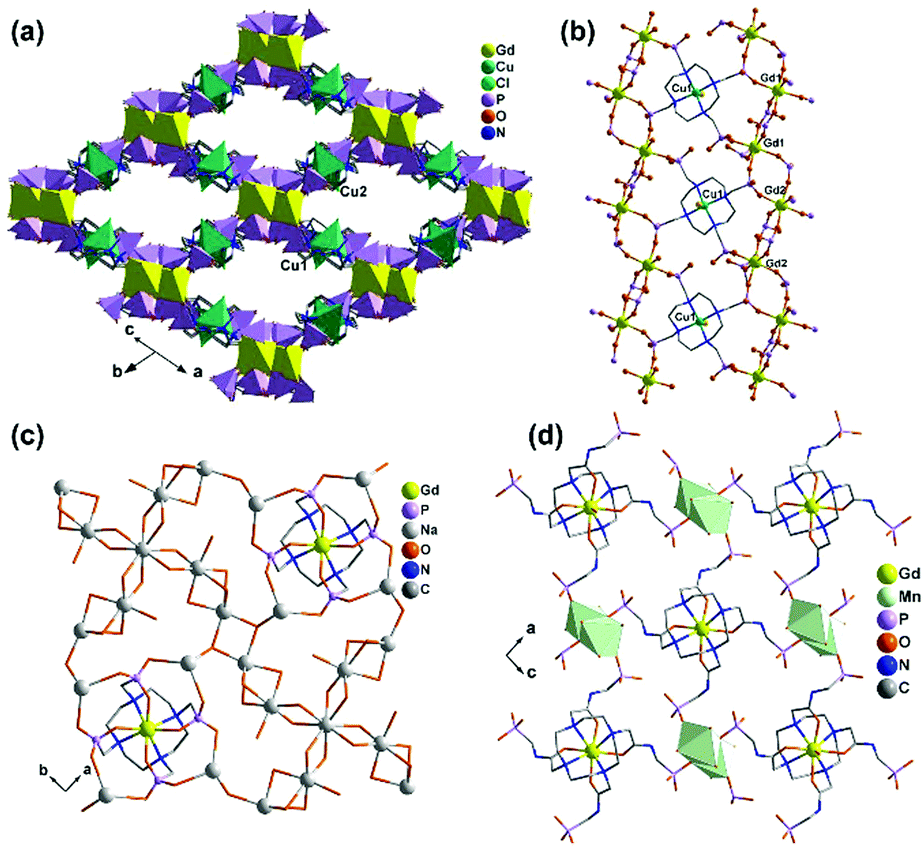
The central metal ions in RuII(bpy)3-based metalloligands can be substituted by other metal ions such as Co(II) or Ni(II). Rentschler and co-workers employed 2,2′-bipyridinyl-5,5′-diphosphonic acid (5,5′-dpbpyH4) and obtained [LnCoIII(5,5′-dpbpyH2)3(H2O)2]·6.5H2O [Ln = GdIII (65), DyIII (66) and TbIII (67)] and [GdNiII(5,5′-dpbpyH2)3(H2O)3]NaCl·6H2O (68),67 where M(5,5′-dpbpyH4)3 (M = CoIII, NiII) act as metalloligands (Scheme 8). Compounds 65–67 are isostructural, crystallizing in the space group C2/c. The lanthanide atom is seven-coordinated with a capped trigonal prismatic geometry, which is rare for lanthanide ions. The seven sites are occupied by five oxygen atoms from separate phosphonate groups of four Co(5,5′-dpbpyH2)33− and two water molecules. All phosphonates are mono protonated in the Co(5,5′-dpbpyH2)33− unit, which serves as a penta-dentate metalloligand and links four equivalent lanthanide atoms through its five phosphonate oxygen atoms (Scheme 9b). Therefore, each Ln(III) is connected by four Co(5,5′-dpbpyH2)33− units and vice versa, leading to a two-dimensional layer (Fig. 15a). The uncoordinated phosphonate groups are pendant on the two sides of the layer, and are involved in the interlayer hydrogen bonds.
 | ||
| Fig. 15 (a) One layer and (b) layer topology of structure 67. (c) Packing diagram and framework topology of structure 68. The topologies in (b) and (d) are presented by the connections between the metalloligand centers and the lanthanide atoms. Adapted from ref. 67. | ||
Interestingly, different connectivity and dimensionality are observed in 68 when the metal center of CoIII(5,5′-dpbpyH2)33− is replaced by the divalent Ni ion. In this case, each Gd atom in 68 again has a capped trigonal prismatic coordination. But the seven coordination sites are filled with only four phosphonate oxygen atoms and three water molecules. Another significant difference for the metalloligand Ni(5,5′-dpbpyH2)34− is that it is tetra-dentate, binding to four Gd atoms via its four phosphonate groups. The two phosphonate groups of one 5,5′-dpbpyH2 remain uncoordinated (Scheme 9c). As a result, each Gd is connected to four Ni(5,5′-dpbpyH2)34− and vice versa. Although this kind of connectivity is similar to that in 67, the geometry of GdNi4 in 68 differs from that of TbCo4 in 67. The four Ni atoms in GdNi4 are arranged closer to a trigonal pyramid, unlike the four Co atoms in TbCo4 which are closer to a square plane. Thus a three-dimensional open framework is constructed in 68 which contains rhombic channels along the c-axis with dimensions of ca. 6.4 × 10.8 Å (van der Waals radii not accounted) (Fig. 15c). The sodium and chloride ions are occupying the space within the channels.
To summarize, the use of cyclometalate or metal–polypyridine complexes as metalloligands to construct MMPs brings not only internal functions such as luminescence and redox but also more diverse structures. The cyclometalated Ir(III) complex containing 2-pyridylphosphonate was used to assemble a series of LnIr6 heptanuclear species. On the other hand, M(bpy)3-based ligands were employed to build MMPs with 2D layer and 3D open framework structures. Moreover, the phosphonate groups in bipyridine can be placed in different substitution positions. The varied number of phosphonate groups in these metalloligands together with their different pre-organized conformation offers more coordination modes, leading to MMPs with different architectures. Although only a handful of examples have been given in this aspect, the result is encouraging in exploring new MMPs based on cyclometalate or metal–polypyridine complexes with versatile structures and interesting properties.
4. Properties and applications
Metal–metalloligand phosphonates offer several advantages over other metal phosphonates not only in structural complexity but also in functions. The presence of different metal ions could affect their physical properties and, in some cases, manipulate the property synergistically. The immobilization of the pre-organized functional metalloligand within metal phosphonate frameworks also provides opportunities in exploring new materials with high thermal and water stability, interesting magnetic, optical, and proton conductive properties and efficient heterogeneous catalytic performance.4.1. Magnetic and optical properties
For metal–metalloligand phosphonates, the magnetic moment can originate from either the metalloligand, or the second metal ion, or both. Typical paramagnetic metalloligands are metal–metal bonded Ru2(hedp)2n− species with paddlewheel structures. Early studies on diruthenium tetracarboxylates have demonstrated that the homovalent RuII,II2(O2CR)4 and mixed-valent [RuII,III2(O2CR)4]+ contain two (S = 1) and three unpaired electrons (S = 3/2) in electron configurations of σ2π4δ2(π*)2(δ*)2 and σ2π4δ2π*2δ*1.68 Accordingly, the ground states for RuII,II2(hedp)24− and RuII,III2(hedp)23− are S = 1 and 3/2, respectively. As already described, a series of MMPs containing Ru25+ units have been obtained. They show large and positive zero-field splitting (ZFS) with the D parameters being 78.3–103.2 cm−1 (Table 1). The interactions between the Ru2 dimers are usually weak, which can be ferromagnetic as observed in layer compounds 1–2, or antiferromagnetic as observed in Ln–Ru2 frameworks. The introduction of the lanthanide ion into the Ru2 system can bring an additional magnetic source and multiple exchange pathways. For Dy–Ru2 and Yb–Ru2 compounds 17 and 18, field-induced slow magnetic relaxation characteristic of single molecule magnet (SMM) behaviour was observed. But no peaks of in-phase and out-of-phase ac signals appear down to 2 K. Lanthanide complexes are well known to be promising systems for SMMs owing to their large single-ion magnetic anisotropy arising from the unquenched orbital angular momentum and strong spin–orbit coupling.69 The observation of magnetization relaxation in 17 and 18 can be mainly attributed to the presence of Kramers ions DyIII or YbIII. Though the Ru25+ dimer also shows strong spin–orbit coupling, its contribution to magnetic relaxation remains unclear.The magnetic properties of one metal ion could be affected by another metal ion in MMPs. An interesting example is [LaCo(notp)(H2O)4]·nH2O (22)30 which is diamagnetic with the octahedral CoIII ion in a low spin state. It has a layer structure where the metalloligand Co(notp)3− is quadra-dentated binding to four LaIII ions. Each La atom is eight-coordinated by four phosphonate oxygen atoms from four Co(notp)3− and four water molecules. Thermal treatment up to 220 °C causes the release of both lattice and coordination water molecules. However, the coordination number of La remains the same as confirmed by EXAFS measurements. The vacant sites of La are filled with the phosphonate oxygen atoms from neighbouring Co(notp)3−, leading to the elongation of the Co–O bond length. The elongation of the Co–O length weakens the ligand field strength and stabilizes the high-spin CoII. A thermally induced electron transfer occurs with the switching of the cobalt oxidation state, from diamagnetic low-spin CoIII to paramagnetic high-spin CoII (Fig. 16). Moreover, the dehydrated H[hs-CoIILaIII(notp)] can turn back to the original [ls-CoIIILaIII(notp)(H2O)4]·nH2O upon rehydration in air. Compound 22 serves as a good example of metal–organic systems showing dehydration induced synergistic change of the coordination spheres of two kinds of metal ions linked by a polyazamacrocycle ligand and its impact on the magnetic properties.
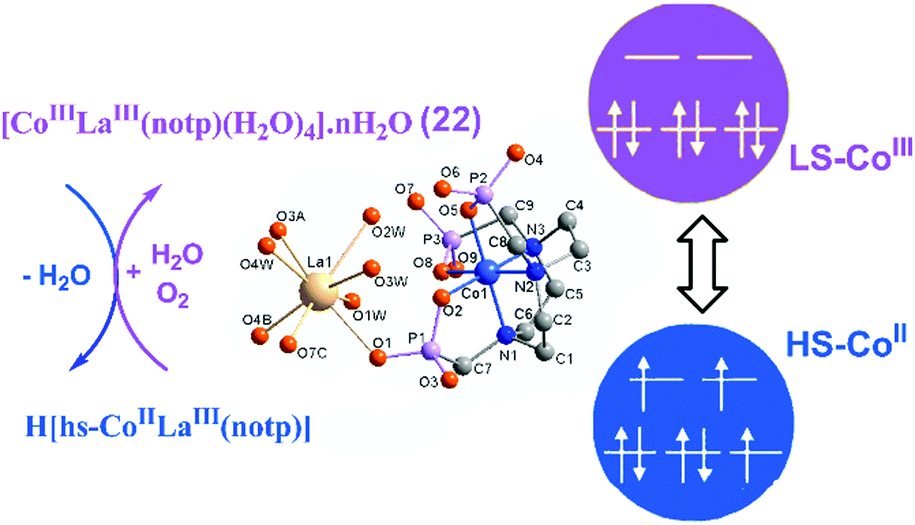 | ||
| Fig. 16 The scheme of the valence change between 22 and H[hs-CoIILaIII(notp)]. Adapted from ref. 30. | ||
As a comparison, the LnIII–FeIII and CaII–CoIII compounds 24–28 based on metalloligands M(notp)3− (M = Fe, Co) cannot undergo such valence change. This can be explained by the fact that the oxidation state of FeIII is not so sensitive to the ligand field, the coordination number of CaII is less than that of LaIII, and the Ca–O bonds are weaker than La–O bonds. By incorporating paramagnetic IrCl62− as a counter-anion, [CaCo2(notpH3)2(H2O)2](IrCl6)·4H2O (34) exhibits slow magnetic relaxation originating from isolated IrIVCl62− species.35
Paramagnetic materials can be potential contrast agents (CAs) for magnetic resonance imaging (MRI). However, most of the research work has been focused on mononuclear GdIII complexes using polyazamacrocycle derivatives as ligands due to their high stability in aqueous solution. Maspoch and co-workers immobilized GdIII ions in the framework structure of [GdCu(dotpH4)Cl]·4.5H2O (37),37 which is stable in physiological saline solution and cell culture media. This material can be made into colloidal nanoparticles and shows interesting relaxometric properties with r1 at a high field (500 MHz) of 5 mM−1 s−1 and a maximum r1 = 15 mM−1 s−1 at 40 MHz. Furthermore, it can be used in a broad pH range, and the performance increases with temperature.
Reports on optically active MMPs are quite limited. For those containing metalloporphyrins, their UV-visible absorption spectra usually show a strong Soret band located at 410–440 nm (S0 → S2 transition) and less intensive Q bands centered at 520–650 nm (S0 → S1 transition).50–52,56,57,63 Heptanuclear clusters [LnIr6(ppy)12(bpp)2(bppH)4](CF3SO3)·xH2O [Ln = Dy (60), Er (61), Yb (62)]65 are multifunctional materials showing both phosphorescence originating from the cyclometalated Ir(III) component and field-induced single molecule magnet behaviour arising from the anisotropic lanthanide ions. Compounds Ln3(H2O)9[Ru(4,4′-dpbpy)3H5.5]2·nH2O [Ln = LaIII (56), PrIII (57)] provide rare examples of MMPs incorporating the luminescent Ru(II) polypyridine metalloligand.66 Interestingly, they exhibit a two-step vapochromism triggered by humidity. The triplet metal-to-ligand charge-transfer (3MLCT) emission is blue-shifted on increasing the relative humidity (RH) in the low RH region, and red-shifted in the high RH region, resulting in the highest-energy 3MLCT emission at medium RH. The origin is ascribed to the water-adsorption triggered reconstruction of the porous structure and the proton release from the metalloligand to the water filled channels.
4.2. Proton conductive properties
Proton conductive materials play critical roles in many renewable energy and electronics technologies.70 Metal phosphonates are promising candidates owing to their high thermal and water stability, their ability to provide phosphonic acid groups as the proton source within extensive H-bond networks and their insulating nature, which are essential for high performance proton conductors.13 Since the number of MMPs is quite limited, only a few of them have been subjected to proton conductivity measurements. As shown in Tables 2 and 3, most compounds show conductivity in the range of 10−7–10−4 S cm−1 at 95% RH and 25 °C. The conductivity can increase with increasing temperature, reaching 10−3 S cm−1 as observed for compound 53 at 90% RH and 80 °C.It is interesting to observe the enhancement of proton conductivity of [CoLa(notpH)(H2O)6]ClO4·5H2O (23)31via in situ solid state phase transition. This compound is closely related to [CoLa(notp)(H2O)4]·nH2O (22) except for one phosphonate group protonated and hence an extra perchloride anion for charge compensation. Proton conductivity measurements on pellets of the powder samples of 22 and 23 show identical conductivity at 95% RH and 25 °C, being 3.0 × 10−6 and 3.5 × 10−6 S cm−1, respectively. Remarkably, 23 undergoes a solid state phase transition at 45 °C and 93% RH, forming a new phase (H3O)[CoLa(notp)(H2O)4]ClO4·xH2O which has the same layer structure as that of 22 but with additional hydronium ions and ClO4− anions between the neutral layers. Consequently, this new phase shows an enhanced proton conductivity of 4.2 × 10−5 S cm−1 at 25 °C and 95% RH, one order of magnitude higher than that of 22 (3.00 × 10−6 S cm−1) and 23 (3.50 × 10−6 S cm−1).
The single crystal proton conductivity measurement can offer a chance to understand the proton transport mechanism. We used a single crystal of [CoCa(notpH2)(H2O)2]ClO4·4H2O (28)33 for AC impedance measurements. The result demonstrates that the [010] direction of H-bond extension is the preferred proton conduction pathway showing the highest conductivity of 1.00 × 10−3 S cm−1 at 25 °C and 95% RH. On the other hand, the [20−1] direction, which involves the phosphonate oxygen atoms in the H-bond network, shows the lowest conductivity of 4.35 × 10−8 S cm−1 at 25 °C and 95% RH. Moreover, compound 28 experiences a reversible humidity dependent SC–SC structural transformation forming [CoCa(notpH2)(H2O)2]ClO4·2H2O (29) below 70% RH at room temperature with the continuous H-bond network interrupted (Fig. 17a and b). The structural transformation leads to a drastic decrease in proton conductivity by ∼5 orders of magnitude (Fig. 17c). The process is reversible and the response is quick (Fig. 17d), and thus the material can be used as an impedance humidity sensor for future applications.
 | ||
| Fig. 17 (a and b) Packing diagrams of structures [CaCo(notpH2)(H2O)2](ClO4)·4H2O (28) (a) and [CaCo(notpH2)(H2O)2](ClO4)·2H2O (29) (b). (c) Proton conductivity and adsorption isotherms of 28. (d) Impedance response and recovery time to RH variation between 55% and 95% for a single crystal of 28 along the [010] direction. Reprinted with permission from ref. 33. Copyright 2015, American Chemical Society. | ||
To increase the proton conductivity of MMPs, one way is to increase the concentration of protons within the extensive H-bond network by providing more protonated phosphonate groups or acidic counterions. Another way is to produce composites by introducing salts into the framework or combine the MMP with suitable polymers. All these remain to be further explored.
4.3. Catalytic properties
MMPs can rationally immobilize catalytically active metal sites within the robust frameworks. Metalloporphyrins are prominent candidates for catalysis. Although the catalytic properties of MOFs containing metalloporphyrins have been well-explored,71 those of MMPs still remain rarely described due to the limited examples of MMPs. Manganese porphyrins Mn(porphyrin)Cl are active homogeneous catalysts for many oxidation reactions with H2O2 as the oxidizing agent in the presence of nitrogen base cocatalysts. By functionalizing manganese porphyrin with four phosphonate groups, Bujoli and co-workers reported the first examples of insoluble zinc phosphonate hybrid materials incorporating manganese porphyrins.72 They act as efficient heterogeneous oxidation catalysts in the epoxidation of cyclooctene with PhIO showing similar yields to their mononuclear precursor. Moreover, these catalysts show a different behaviour from their homogeneous counterparts in the competitive hydroxylation of alkane mixtures using PhIO as the oxidant, attributed to shape selectivity imposed by the phosphonate support.73The introduction of chirality into catalytically active MMPs can extend their application in enantioselective catalysis. Lin and co-workers designed and synthesized a series of chiral porous zirconium phosphonates embedding enantiopure and catalytically active Ru species (Scheme 10). Compounds [Zr{Ru(L3)(DMF)2Cl2}]·2MeOH and [Zr{Ru(L4)(DMF)2Cl2}]·2MeOH are found to be highly active catalysts for asymmetric hydrogenation of β-ketoesters.74 Zr[Ru(L3)(DPEN)Cl2]·4H2O and Zr[Ru(L4)(DPEN)Cl2]·4H2O are excellent catalysts for enantioselective hydrogenation of unfunctionalized aromatic ketones.75
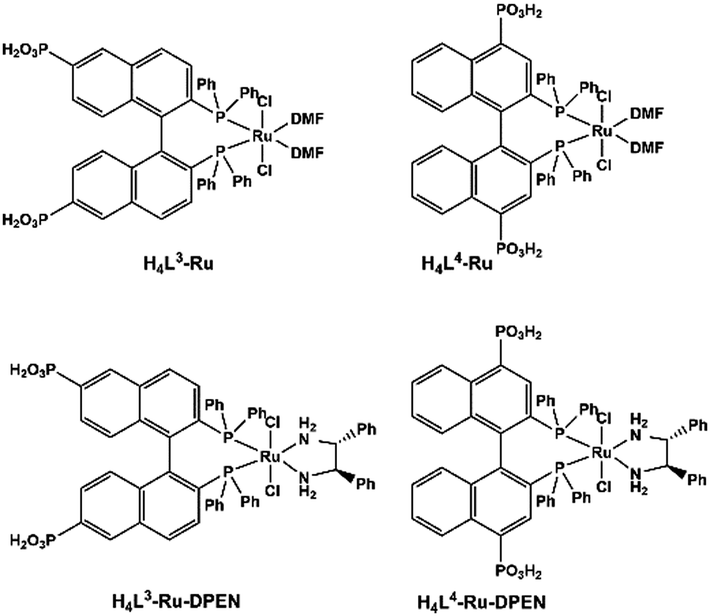 | ||
| Scheme 10 Molecular structures of Ru complexes of binaphthalene-bisphosphonic acids. Reprinted with permission from ref. 10f. Copyright 2020, Springer Nature. | ||
5. Summary and outlook
In this feature article, we have presented the current status of metal–metalloligand phosphonates (MMPs) from syntheses, structures to properties. It is clear that using the pre-formed metalloligand as the reaction precursor is a more efficient and controllable approach to construct MMPs. Further, the metalloligand can bring multiple phosphonate groups as coordination donors with different orientations, resulting in MMPs with new structure types and special functions such as polarity. Moreover, the immobilized metalloligand with magnetic, optical, and catalytic properties can offer additional functions of MMPs for potential applications.Given the limited types of known metallo-phosphonate ligands, the limited number of MMPs and the limited studies on their properties, the development of this field is still in its early stage.
Future challenges remain in several aspects. First, the exploration of new MMPs. This exploration can be based on the known phosphonate-based metalloligands. For example, the assembly of paddlewheel diruthenium diphosphonate with transition metal ions or clusters has not been investigated. For metalloligands based on polyazamacrocycles, the numbers of MMPs and the structural types are still very limited. Even less has been studied for metalloporphyrin and metallo-tris-bipyridine-phosphonate ligands. So there is plenty of room for these already known metalloligands to construct new MMPs by optimizing reaction conditions and introducing different metal ions or clusters as nodes. The exploration of new MMPs can also be based on new phosphonate-based metalloligands by pre-design and synthesis. In this regard, there are a great many choices considering the vast reservoir of organic chelating and bridging ligands. Second, the characterization of MMPs. Metal phosphonates are well recognized by their low solubility and easy precipitation. It is often difficult to grow single crystals with sufficiently large size for conventional single crystal determination. The situation is particularly true for MMPs because the metalloligands usually possess more than one phosphonate groups which makes the solubility of MMPs even lower. Apart from the synthetic efforts to grow crystals with bigger size, a promising solution to solve the structures is by Rietveld refinement using powder X-ray diffraction data and, in some cases, combining with the single-crystal electron diffraction method. Third, the application of MMPs. The properties of MMPs originate from both the metalloligands and the second metal ions or clusters. By designing and synthesizing specific metalloligands, it is possible to construct MMPs with desired functions. For instance, the combination of paddlewheel diruthenium diphosphonate with paramagnetic transition metal ions may result in magnetic materials with long-range ordering, like that found for diruthenium phosphates.76 The MMPs containing polar metallo-polyazamacrocycles may be designed and synthesized to possess polarity or even chirality for further applications. The incorporation of luminescent cyclometalate or metal–polypyridine complexes in MMPs brings optical functions for application in sensors and multifunctional materials. By taking the advantages of metalloporphyrin, the related MMPs are expected to show interesting electrical and optical properties that can be utilized in photocatalysis and photodynamic therapy. Transformation of the channels may lead to switching in electrical and optical properties of these porphyrin-based materials. Some compounds exhibit conductance from combined mechanism such as proton conductivity together with inherent semiconducting properties, thus showing potential in multifunctional materials. Finally, the functional MMPs may be fabricated into nanoparticles, nanowires or nanosheets which may lead to applications in catalysis, molecular devices and nanotechnologies.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (21731003 and 21671098) and the National Key R&D Program of China (2018YFA0306004) is acknowledged.Notes and references
-
(a) H. Furukawa, K. E. Cordova, M. O’Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 Search PubMed
; (b) S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695–704 Search PubMed
-
(a) B. B. Lin, S. C. Xiang, H. B. Xing, W. Zhou and B.-L. Chen, Coord. Chem. Rev., 2019, 378, 87–103 Search PubMed
-
(a) Y. Cui, B. Chen and G. Qian, Coord. Chem. Rev., 2014, 273, 76–86 Search PubMed
-
(a) G. Kumar and R. Gupta, Chem. Soc. Rev., 2013, 42, 9403–9453 Search PubMed
-
(a) M. C. Das, S. Xiang, Z. Zhang and B. Chen, Angew. Chem., Int. Ed., 2011, 50, 10510–10520 Search PubMed
-
A. Clearfield, Progress in Inorganic Chemistry, Wiley, New York, 1998, vol. 47, pp. 371–510 Search PubMed
-
(a) A. D. G. Firmino, F. Figueira, J. P. C. Tomé, F. A. Almeida Paz and J. Rocha, Coord. Chem. Rev., 2018, 355, 133–149 Search PubMed
-
(a) T. Zheng, Z. Yang, D. Gui, Z. Liu, X. Wang, X. Dai, S. Liu, L. Zhang, Y. Gao, L. Chen, D. Sheng, Y. Wang, J. Diwu, J. Wang, R. Zhou, Z. Chai, T. E. Albrecht-Schmitt and S. Wang, Nat. Commun., 2017, 8, 15369 Search PubMed
-
Metal Phosphonate Chemistry: From Synthesis to Applications, ed. A. Clearfield and K. Demadis, The Royal Society of Chemistry, 2012 Search PubMed
-
(a) A. Clearfield, Curr. Opin. Solid State Mater. Sci., 2002, 6, 495–506 Search PubMed
- J.-G. Mao, Coord. Chem. Rev., 2007, 251, 1493–1520 Search PubMed
- S.-S. Bao and L.-M. Zheng, Coord. Chem. Rev., 2016, 319, 63–85 Search PubMed
- S.-S. Bao, G. K. H. Shimizu and L.-M. Zheng, Coord. Chem. Rev., 2019, 378, 577–594 Search PubMed
-
(a) S. J. Shearan, N. Stock, F. Emmerling, J. Demel, P. A. Wright, K. D. Demadis, M. Vassaki, F. Costantino, R. Vivani, S. Sallard, I. R. Salcedo, A. Cabeza and M. Taddei, Crystals, 2019, 9, 270 Search PubMed
-
(a) N. Hermer, H. Reinsch, P. Mayer and N. Stock, CrystEngComm, 2016, 18, 8147–8150 Search PubMed
-
(a)
F. A. Cotton, C. A. Murillo and R. A. Walton, Multiple Bonds Between Metal Atoms, Springer Science and Business Media, New York, 3rd edn, 2005 Search PubMed
-
(a) H. Miyasaka, Acc. Chem. Res., 2013, 46, 248–257 Search PubMed
- J.-H. Yang, R.-M. Cheng, Y.-Y. Jia, J. Jin, B.-B. Yang, Z. Cao and B. Liu, Dalton Trans., 2016, 45, 2945–2954 Search PubMed
-
(a) F. A. Cotton, T. Datta, L. Labella and M. Shang, Inorg. Chim. Acta, 1993, 203, 55–60 Search PubMed
- X.-Y. Yi, L.-M. Zheng, W. Xu and S.-H. Feng, Inorg. Chem., 2003, 42, 2827–2829 Search PubMed
- B. Liu, Y.-Z. Li and L.-M. Zheng, Solid State Sci., 2006, 8, 1041–1045 Search PubMed
-
(a) X.-Y. Yi, B. Liu, R. Jiménez-Aparicio, F. A. Urbanos, S. Gao, W. Xu, J.-S. Chen, Y. Song and L.-M. Zheng, Inorg. Chem., 2005, 44, 4309–4314 Search PubMed
- B. Liu, B.-L. Li, Y.-Z. Li, Y. Chen, S.-S. Bao and L.-M. Zheng, Inorg. Chem., 2007, 46, 8524–8532 Search PubMed
- S. M. Elahi, Q.-H. Lai, M. Ren, S.-S. Bao, M. Kurmoo and L.-M. Zheng, Inorg. Chem., 2019, 58, 14034–14045 Search PubMed
- M. Yu Antipin, A. P. Baranov, M. I. Kabachnik, T. Ya Medved, Yu. M. Polikarpov, Yu. T. Struchkov and B. K. Shcherbakov, Dokl. Akad. Nauk SSSR, 1986, 287, 130 Search PubMed
- M. I. Kabachnik, M. Yu Antipin, B. K. Shcherbakov, A. P. Baranov, Yu. T. Struchkov, T. YaMedved and Yu. M. Polikarpov, Koord. Khim., 1988, 14, 536 Search PubMed
- S.-S. Bao, G.-S. Chen, Y. Wang, Y.-Z. Li, L.-M. Zheng and Q.-H. Luo, Inorg. Chem., 2006, 45, 1124–1129 Search PubMed
-
(a) S.-S. Bao, L.-F. Ma, Y. Wang, L. Fang, C.-J. Zhu, Y.-Z. Li and L.-M. Zheng, Chem. – Eur. J., 2007, 13, 2333–2343 Search PubMed
- S.-B. Liu, S.-S. Bao and L.-M. Zheng, Dalton Trans., 2020, 49, 3758–3765 Search PubMed
- S.-S. Bao, Y. Liao, Y.-H. Su, X. Liang, F.-C. Hu, Z. Sun, L.-M. Zheng, S. Wei, R. Alberto, Y.-Z. Li and J. Ma, Angew.
Chem., Int. Ed., 2011, 50, 5504–5508 Search PubMed
- S.-S. Bao, K. Otsubo, J. M. Taylor, Z. Jiang, L.-M. Zheng and H. Kitagawa, J. Am. Chem. Soc., 2014, 136, 9292–9295 Search PubMed
- Y.-H. Su, S.-S. Bao and L.-M. Zheng, Inorg. Chem., 2014, 53, 6042–6047 Search PubMed
- S.-S. Bao, N.-Z. Li, J. M. Taylor, Y. Shen, H. Kitagawa and L.-M. Zheng, Chem. Mater., 2015, 27, 8116–8125 Search PubMed
- S.-S. Bao, Y.-X. Wu, N.-Z. Li and L.-M. Zheng, Eur. J. Inorg. Chem., 2016, 4476–4482 Search PubMed
- S.-B. Liu, X.-D. Huang, S.-S. Bao, M. Kurmoo and L.-M. Zheng, Cryst. Growth Des., 2019, 19, 4836–4843 Search PubMed
-
(a) M. P. C. Campello, S. Lacerda, I. C. Santos, G. A. Pereira, C. F. G. C. Geraldes, J. Kotek, P. Hermann, J. Vaněk, P. Lubal, V. Kubíček, É. Tóth and I. Santos, Chem. – Eur. J., 2010, 16, 8446–8465 Search PubMed
- A. Carné-Sánchez, C. S. Bonnet, I. Imaz, J. Lorenzo, É. Tóth and D. Maspoch, J. Am. Chem. Soc., 2013, 135, 17711–17714 Search PubMed
- F. Avecilla, J. A. Peters and C. F. G. C. Geraldes, Eur. J. Inorg. Chem., 2003, 4179–4186 Search PubMed
-
(a) T.-H. Yang, K. Zhou, S.-S. Bao, C.-J. Zhu and L.-M. Zheng, Inorg. Chem. Commun., 2008, 11, 1075–1078 Search PubMed
- J. Aríñez-Soriano, J. Albalad, C. Vila-Parrondo, J. Pérez-Carvajal, S. Rodríguez-Hermida, A. Cabeza, J. Juanhuix, I. Imaz and D. Maspoch, Chem. Commun., 2016, 52, 7229–7232 Search PubMed
- G. Giambastiani, W. Oberhauser, C. Bianchini, F. Laschi, L. Sorace, P. Brueggeller, R. Gutmann, A. Orlandini and F. Vizza, Eur. J. Inorg. Chem., 2005, 2027–2031 Search PubMed
- J. Rudovský, P. Cígler, J. Kotek, P. Hermann, P. Vojtíšek, I. Lukeš, J. A. Peters, L. V. Elst and R. N. Muller, Chem. – Eur. J., 2005, 11, 2373–2384 Search PubMed
- P. Vojtíšek, P. Cígler, J. Kotek, J. Rudovský, P. Hermann and I. Lukeš, Inorg. Chem., 2005, 44, 5591–5599 Search PubMed
- G. Neri, M. Forster, J. J. Walsh, C. M. Robertson, T. J. Whittles, P. Farràs and A. J. Cowan, Chem. Commun., 2016, 52, 14200–14203 Search PubMed
- M. Paúrová, T. David, I. Císařová, P. Lubal, P. Hermann and J. Kotek, New J. Chem., 2018, 42, 11908–11929 Search PubMed
- J.-G. Mao, Z. Wang and A. Clearfield, Inorg. Chem., 2002, 41, 3713–3720 Search PubMed
-
Handbook of Porphyrin Science, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific Press, Singapore, 2010–2014 Search PubMed
-
(a) W.-Y. Gao, M. Chrzanowski and S. Ma, Chem. Soc. Rev., 2014, 43, 5841–5866 Search PubMed
-
(a) D. Feng, W.-C. Chung, Z. Wei, Z.-Y. Gu, H.-L. Jiang, Y.-P. Chen, D. J. Darensbourg and H.-C. Zhou, J. Am. Chem. Soc., 2013, 135, 17105–17110 Search PubMed
- Y. Y. Enakieva, A. G. Bessmertnykh, Y. G. Gorbunova, C. Stern, Y. Rousselin, A. Y. Tsivadze and R. Guilard, Org. Lett., 2009, 11, 3842–3845 Search PubMed
- A. A. Sinelshchikova, S. E. Nefedov, Y. Yu. Enakieva, Y. G. Gorbunova, A. Yu. Tsivadze, K. M. Kadish, P. Chen, A. Bessmertnykh-Lemeune, C. Stern and R. Guilard, Inorg. Chem., 2013, 52, 999–1008 Search PubMed
- R. I. Zubatyuk, A. A. Sinelshchikova, Y. Y. Enakieva, Y. G. Gorbunova, A. Y. Tsivadze, S. E. Nefedov, A. Bessmertnykh-Lemeune, R. Guilard and O. V. Shishkin, CrystEngComm, 2014, 16, 10428–10438 Search PubMed
- Y. Fang, X. Jiang, K. M. Kadish, S. E. Nefedov, G. A. Kirakosyan, Y. Y. Enakieva, Y. G. Gorbunova, A. Y. Tsivadze, C. Stern, A. Bessmertnykh-Lemeune and R. Guilard, Inorg. Chem., 2019, 58, 4665–4678 Search PubMed
- M. A. Uvarova, A. A. Sinelshchikova, M. A. Golubnichaya, S. E. Nefedov, Y. Yu. Enakieva, Y. G. Gorbunova, A. Yu. Tsivadze, C. Stern, A. Bessmertnykh-Lemeune and R. Guilard, Cryst. Growth Des., 2014, 14, 5976–5984 Search PubMed
- E. V. Ermakova, Y. Yu. Enakieva, S. E. Nefedov, V. V. Arslanov, Y. G. Gorbunova, A. Yu. Tsivadze, C. Stern and A. Bessmertnykh-Lemeune, Eur. J. Org. Chem., 2019, 3146–3162 Search PubMed
- A. Yu. Mitrofanov, Y. Rousselin, V. N. Khrustalev, A. V. Cheprakov, A. Bessmertnykh-Lemeune and I. P. Beletskaya, Eur. J. Inorg. Chem., 2019, 1313–1328 Search PubMed
-
(a) S. E. Nefedov, K. P. Birin, A. Bessmertnykh-Lemeune, Y. Y. Enakieva, A. A. Sinelshchikova, Y. G. Gorbunova, A. Y. Tsivadze, C. Stern, Y. Fang and K. M. Kadish, Dalton Trans., 2019, 48, 5372–5383 Search PubMed
- Y. Y. Enakieva, M. V. Volostnykh, S. E. Nefedov, G. A. Kirakosyan, Y. G. Gorbunova, A. Y. Tsivadze, A. G. Bessmertnykh-Lemeune, C. Stern and R. Guilard, Inorg. Chem., 2017, 56, 3055–3070 Search PubMed
- S. B. Ungashe, W. L. Wilson, H. E. Katz, G. R. Scheller and T. M. Putvinski, J. Am. Chem. Soc., 1992, 114, 8717–8719 Search PubMed
- T. Rhauderwiek, K. Wolkersdörfer, S. Øien-Ødegaard, K.-P. Lillerud, M. Wark and N. Stock, Chem. Commun., 2018, 54, 389–392 Search PubMed
- Y. Y. Enakieva, A. A. Sinelshchikova, M. S. Grigoriev, V. V. Chernyshev, K. A. Kovalenko, I. A. Stenina, A. B. Yaroslavtsev, Y. G. Gorbunova and A. Y. Tsivadze, Chem. – Eur. J., 2019, 25, 10552–10556 Search PubMed
- B. Wang, T. Rhauderwiek, A. K. Inge, H. Xu, T. Yang, Z. Huang, N. Stock and X. Zou, Chem. – Eur. J., 2018, 24, 17429–17433 Search PubMed
- T. Rhauderwiek, H. Zhao, P. Hirschle, M. Döblinger, B. Bueken, H. Reinsch, D. De Vos, S. Wuttke, U. Kolb and N. Stock, Chem. Sci., 2018, 9, 5467–5478 Search PubMed
- M. Maares, M. M. Ayhan, K. B. Yu, A. O. Yazaydin, K. Harmandar, H. Haase, J. Beckmann, Y. Zorlu and G. Yücesan, Chem. – Eur. J., 2019, 25, 11214–11217 Search PubMed
-
(a) D. Zeng, M. Ren, S.-S. Bao, L. Li and L.-M. Zheng, Chem. Commun., 2014, 50, 8356–8359 Search PubMed
- A. Kobayashi, K. Shimizu, A. Watanabe, Y. Nagao, N. Yoshimura, M. Yoshida and M. Kato, Inorg. Chem., 2019, 58, 2413–2421 Search PubMed
- C. Köhler and E. Rentschler, Dalton Trans., 2016, 45, 12854–12861 Search PubMed
-
(a) F. A. Cotton, V. M. Miskowski and B. Zhong, J. Am. Chem. Soc., 1989, 111, 6177 Search PubMed
-
(a) D. N. Woodruff, R. E. P. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110–5148 Search PubMed
- K. D. Kreuer, S. J. Paddison, E. Spohr and M. Schuster, Chem. Rev., 2004, 104, 4637–4678 Search PubMed
-
(a) M. M. Pereira, L. D. Dias and M. J. F. Calvete, ACS Catal., 2018, 8, 10784–10808 Search PubMed
- D. Demand, B. Schollorn, D. Mansuy, J. Rouxel, P. Battioni and B. Bujoli, Chem. Mater., 1995, 7, 995–1000 Search PubMed
- D. Deniaud, G. A. Spyroulias, J.-F. Bartoli, P. Battioni, D. Mansuy, C. Pinel, F. Odobel and B. Bujoli, New J. Chem., 1998, 22, 901–905 Search PubMed
- A. Hu, H. L. Ngo and W. Lin, Angew. Chem., Int. Ed., 2003, 115, 6182–6185 Search PubMed
- A. Hu, H. L. Ngo and W. Lin, J. Am. Chem. Soc., 2003, 125, 11490–11491 Search PubMed
- B.-B. Yang, L.-N. Feng, X.-M. Fan, K.-X. Zhang, J.-H. Yang and B. Liu, Inorg. Chem. Front., 2017, 4, 1061–1065 Search PubMed
| This journal is © The Royal Society of Chemistry 2020 |