
S-Mg2(dobpdc): a metal–organic framework for determining chirality in amino acids†
Hui Min
Tay
 a,
Aditya
Rawal
b and
Carol
Hua
*a
a,
Aditya
Rawal
b and
Carol
Hua
*a
aSchool of Chemistry, The University of Melbourne, Parkville, Victoria 3010, Australia. E-mail: carol.hua@unimelb.edu.au
bMark Wainwright Analytical Centre, University of New South Wales, Kensington, New South Wales 2052, Australia
First published on 2nd November 2020
Abstract
Chirality is a key aspect of amino acids and is essential for life. Here, a chiral metal–organic framework, S-Mg2dobpdc, is used to determine the chirality of three BOC protected amino acids (alanine, valine and proline) by 13C solid-state NMR with chemical shift differences of up to 1.3 ppm observed between enantiomers. The chiral sensitivity persists upon in situ deprotection of the amino acids by thermolysis of the BOC group.
Amino acids are the building blocks of proteins and an important component in chemical and biological systems. The different enantiomers of amino acids can lead to vastly different physiological and metabolic outcomes. The growing use of amino acid derivatives in pharmacology, chemical science and biochemistry highlights the need for efficient methods to determine chiral purity. The elucidation of chirality in amino acids can be challenging due to their lack of strong chromophoric groups, leading to a weak chiro-optical response in circular dichroism spectroscopy,1 or the need for multiple protection, derivation and deprotection steps. The majority of chiral elucidation methods involve the use of chromatographic techniques, including HPLC and GC,2 or optical methods, including fluorescence and luminescence.3 Solid-state NMR offers a direct means of identifying and quantifying the enantiopurity of a sample. The use of solid-state NMR for the quantification of chirality is underdeveloped, with the main focus in this area concerning the use of liquid crystals as a chiral matrix.8,9
Coordination Polymers (CPs) and Metal Organic Frameworks (MOFs) are crystalline solid materials comprising of metal ions bridged by organic ligands in 1D, 2D or 3D to create a scaffold containing pores.4 Chiral MOFs and CPs are typically used as stationary chiral phases for HPLC5 or as heterogenous asymmetric catalysts.6 Considerably less attention has been focussed on the use of MOFs and CPs for the detection of chiral purity. Developments in this area with MOFs and CPs primarily utilise optical spectroscopic techniques, including fluorescence and UV-Vis.7 CPs and MOFs can potentially act as solid-state chiral solvating agents due to their tunable pores, which can form strong intermolecular interactions with chiral guests, resulting in a diastereomeric complex observable by NMR. While solid-state NMR spectra can have large peak widths, making the small chemical shifts between diastereomers difficult to resolve, MOFs and CPs are crystalline materials that typically have narrow peak widths and sharp NMR resonances.
While three previous reports have used MOFs for chiral elucidation, none of these were able to differentiate between enantiomers from a racemic mixture.10–12 Only small chemical shift differences of up to 0.4 ppm were observed in the 13C NMR spectra of a chiral analogue of the UMCM-1 framework11 upon loading with enantiopure 1-phenyl-2,2,2-trifluoroethanol (TFPE), or when alanine and leucine were loaded into [Zn2(L-Phe)(bpe)2]n (L-Phe = L-phenylalanine, bpe = bis(4-pyridyl)ethylene).12
Long et al. recently reported the synthesis of a chiral form of Mg2dobpdc (H4dobpdc = 4,4′-dioxidobiphenyl-3,3′-dicarboxylic acid).13 The axial chirality of Mg2dobpdc results from the rotation of the two phenyl rings in dobpdc4− relative to each other, yielding enantiomerically pure domains of solely right-handed or left-handed helices. The grafting of chiral 1,2-diaminocyclohexane enabled the solid-state NMR characterisation of the ammonium carbamate chains formed upon the adsorption of CO2 to yield chemical shift differences of up to 2.0 ppm. Mg2(dobpdc) (and its ZnII analogue) have been extensively studied by solid-state NMR,14–19 including in experiments that probe molecular dynamics using 15N NMR20 and investigations of local disorder by 25Mg NMR.21,22 The breadth of information concerning the behaviour of Mg2(dobpdc) makes it a valuable target for the study of chiral elucidation by solid-state NMR.
Herein, we present the use of S-Mg2dobpdc for the chiral elucidation of amino acids using 13C solid-state NMR. S-Mg2dobpdc was grafted post-synthetically with enantiopure BOC alanine, valine and proline to the MgII metal site. Unprotected amino acids were obtained by thermal deprotection of the BOC groups post-grafting. The 13C NMR chemical shift differences between the L- and D-enantiomers of the BOC protected and free amino acids as well as the S-Mg2dobpdc framework were determined and the mode of binding discussed.
S-Mg2(dobpdc) was made according to the procedure reported by Long et al.13 Enantiomerically pure di-tert-butyl dicarbonate (BOC) protected alanine, valine and proline were then grafted onto the framework to yield S-Mg2(dobpdc)@BOC-L-Xxx (Xxx = Ala, Val, Pro) and S-Mg2(dobpdc)@BOC-D-Xxx (Xxx = Ala, Val, Pro), as white powders (Fig. 1). The BOC group ensured that coordination to the MgII metal centre only occurred through the carboxylic acid instead of the primary amine of the amino acid. Alanine, valine and proline were chosen for incorporation to minimise the steric bulk present, ensuring that a high percentage loading and strong intermolecular interactions to the framework could be achieved. Amino acids with aliphatic side chains enabled easy identification from the aromatic dobpdc4− ligand in NMR studies. The PXRD of S-Mg2(dobpdc)@BOC-Xxx (Xxx = Ala, Val, Pro) verified that the crystallinity and structure of the S-Mg2(dobpdc) framework had been retained (ESI,† Fig. S1–S4). The incorporation of the BOC amino acid guest into S-Mg2dobpdc was verified by the BOC carbonyl peak in the FT-IR spectrum at 1670 cm−1 (ESI,† Fig. S5–S7) and via solution-state 1H NMR spectroscopy of the digested samples of S-Mg2(dobpdc)@BOC-Xxx (Xxx = Ala, Val, Pro) (ESI,† Fig. S8–S13).
 | ||
| Fig. 1 Schematic of the appendage of BOC protected amino acids to the open MgII metal sites on S-Mg2dobpdc to yield S-Mg2dobpdc@BOC-Xxx (Xxx = Ala, Val, Pro) where R = CH3 (alanine), iPr (valine), CHCH2CH2CH2 (proline) followed by the thermal deprotection of the BOC group to yield S-Mg2dobpdc@Xxx (Xxx = Ala, Val, Pro). The appendage of amino acids to 50% of MgII metal sites is shown here for clarity. The atoms are represented as follows: magnesium (purple), carbon (black), oxygen (red). | ||
The thermal gravimetric analyses (TGA) of S-Mg2(dobpdc)@BOC-Xxx (Xxx = Ala, Val, Pro) displayed three stepwise mass losses (ESI,† Fig. S14–S22). The first mass loss below 100 °C was attributed to the loss of water adsorbed from the atmosphere. The second mass loss between 100 and 200 °C was due to the thermal deprotection of the BOC group from the appended BOC amino acids. The post-synthetic deprotection of the BOC group from proline within a MOF has previously been reported.23–25 The third mass loss between 220 and 280 °C is due to thermal decomposition of the amino acids into gaseous products and can be correlated to the size of the side chain, with S-Mg2(dobpdc)@BOC-Val having the largest mass loss.26,27
The thermal BOC deprotection of the amino acids was confirmed by FT-IR measurements after heating the S-Mg2(dobpdc)@BOC-Xxx (Xxx = Ala, Val, Pro) samples at 200 °C for 30 mins to yield S-Mg2(dobpdc)@Xxx, where the BOC carbonyl peak at 1670 cm−1 disappeared (ESI,† Fig. S23–S25). The conversion of BOC-Ala to Ala resulted in red shifts of the FT-IR absorption bands to wavelengths similar to those for the Mg2dobpdc framework. Remarkably, PXRD measurements (ESI,† Fig. S26–S28) confirmed that crystallinity was retained after BOC deprotection. In the TGA of S-Mg2(dobpdc)@Xxx (Xxx = Ala, Val, Pro), the mass loss between 100 and 200 °C was absent, confirming the successful deprotection of the BOC group (ESI,† Fig. S29–S31).
As the removal of the BOC group can be directly observed from the mass loss in the TGA traces, this was used to determine the percentage incorporation of the amino acid, which was verified by elemental analysis. The highest percentage loading was achieved with BOC-Ala (57%), followed by BOC-Pro (55%) and BOC-Val (39%). It is likely that both the degree of steric bulk and the conformation of the amino acid had an effect on the amount of amino acid incorporated into S-Mg2(dobpdc). DMF solvent molecules remained coordinated to the MgII metal sites that have no amino acid appended, even after heating the samples at 200 °C, as observed in the 13C CPMAS spectra (vide infra).
The S-Mg2dobpdc framework and the amino acid appended samples were analysed by 13C solid-state NMR (Fig. 2). The 13C CPMAS spectrum of S-Mg2dobpdc contained well-defined spectral lines with widths at half height of 0.7 ppm, enabling all the 13C resonances arising from the dobpdc4− ligand to be assigned (ESI,† Fig. S33). A 13C non-quaternary suppression (NQS) experiment was used to assign the quaternary carbon resonances (ESI,† Fig. S34–S36).
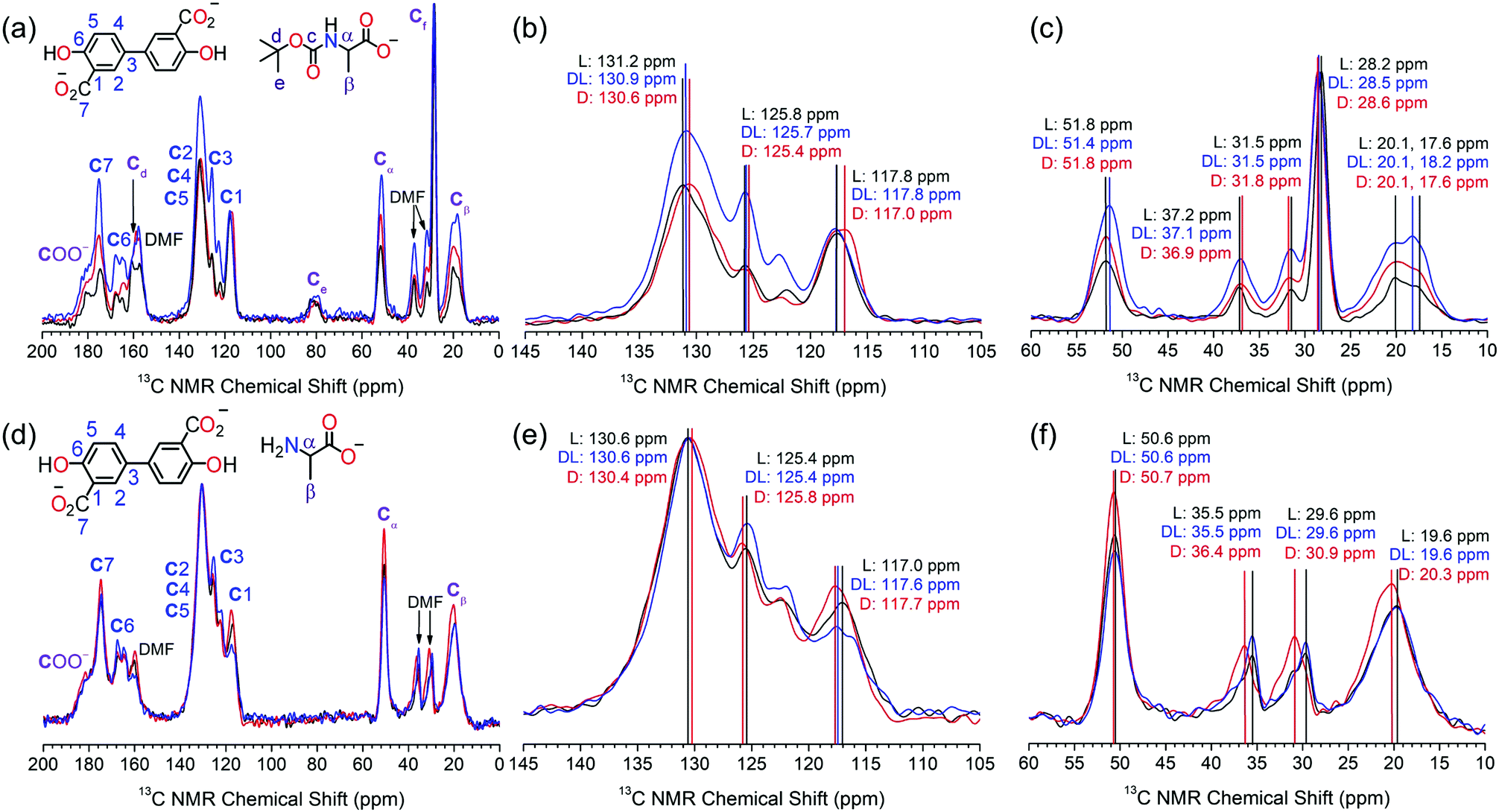 | ||
| Fig. 2 13C solid state CPMAS spectra of Mg2dobpdc@BOC-L-Ala (black), Mg2dobpdc@BOC-D-Ala (red) and Mg2dobpdc@BOC-DL-Ala (blue) acquired at 175 MHz showing (a) the full spectrum between 200–0 ppm, (b) zoomed in section between 145–105 ppm, and (c) zoomed in section between 60–10 ppm. The 13C solid state CPMAS spectra of Mg2dobpdc@L-Ala (black), Mg2dobpdc@D-Ala (red) and Mg2dobpdc@DL-Ala (blue) acquired at 175 MHz showing (d) the full spectrum between 200–0 ppm, (e) zoomed in section between 145–105 ppm, and (f) zoomed in section between 45–10 ppm. All spectra are scaled to the same maximum intensity. | ||
The 13C CPMAS spectra of S-Mg2(dobpdc)@BOC-Xxx (Xxx = Ala, Val, Pro) showed distinct chemical shift differences of up to 0.6 ppm in specific 13C NMR resonances between the L- and D-amino acids (Fig. 2a–c and Fig. S37–S42, ESI†). The largest chemical shift occurred at C2, C3, C4, C5 and Ce, suggesting that the methyl groups from the BOC group are involved in CH–π intermolecular interactions with the phenyl rings of the dobpdc4− ligand. Remarkably, the C2–C5 signals of dobpdc4− can be used to discriminate between the L and D enantiomers of BOC-Ala by the 13C NMR chemical shift. Mg2dobpdc@BOC-DL-Ala (comprised of 60% BOC-L-Ala, 40% BOC-D-Ala) has a chemical shift of 130.9 ppm, which is intermediate between that of Mg2dobpdc@BOC-L-Ala (131.2 ppm) and Mg2dobpdc@BOC-D-Ala (130.6 ppm) (Fig. 2b). 13C chemical shift differences also occur for C3 and Cβ. It is surprising that no changes in the chemical shift were observed for Cα, given this is where the stereocentre is located, however, chiral elucidation in S-Mg2dobpdc likely involves the different orientations of the amino acid relative to the aromatic rings of dobpdc4−. A similar interaction was observed for UMCM where the 13C resonances of the framework, instead of the stereocentre, changed upon the incorporation of chiral guest molecules.11
The largest chemical shift changes were observed for alanine, followed by proline (ESI,† Fig. S37–S39), then valine (ESI,† Fig. S40–S42). This trend is attributed to the size of the amino acid side chain, with the small methyl side chain in alanine enabling the amino acid to effectively interact with S-Mg2dobpdc. The valine isopropyl side chain has greater steric bulk, precluding the formation of strong intermolecular interactions required for effective chiral discrimination. The orientation of the side chain relative to the carboxylate and BOC groups of the amino acid is also an important consideration. The restricted movement of the five membered pyrrolidene ring in proline likely helps to yield a geometry that allows enhanced interaction with the dobpdc4− ligand.
The steric effect of the BOC group on chiral discrimination by 13C solid state NMR was additionally investigated by analysis of the deprotected amino acid samples of S-Mg2dobpdc@Ala. The 13C CPMAS spectra of S-Mg2(dobpdc)@L-Ala and S-Mg2(dobpdc)@D-Ala displayed enhanced 13C chemical shift differences of up to 1.3 ppm when compared with the BOC-protected samples, with the largest chemical shift differences observed for the CH3 groups of DMF, followed by that of C1 and Cβ (Fig. 2d–f). The removal of the bulky BOC group enables a stronger interaction between the amino acid and the framework components, thereby enhancing chiral elucidation. The broad Cβ peak of alanine becomes narrower when the BOC group is removed, indicating that the BOC group may cause some conformational disorder. When the framework was loaded with DL-Ala, the 13C chemical shifts more closely resembled that of Mg2(dobpdc)@L-Ala than Mg2(dobpdc)@D-Ala. This is consistent with the composition of DL-Ala, which consists of a 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 ratio of L- to D-Ala.
:40 ratio of L- to D-Ala.
The linewidths in the NMR spectra of the amino acid loaded frameworks were broader than for S-Mg2(dobpdc), with linewidths at half height of 1.8 ppm. This can be attributed to the local disorder of the MgII environment upon guest loading. Previous studies of 25Mg NMR on rac-Mg2dobpdc and Mg2dobdc (dobdc4− = 2,5-dihydroxyterephthalate) have shown that although long-range ordering is maintained upon dehydration and guest loading, slight changes in the orientation of the ligands result in small changes in the Mg–O bond length and O–Mg–O bond angle to yield local disorder.21,22
The porous and robust S-Mg2dobpdc framework was shown to be highly effective for chiral elucidation by 13C solid state NMR with BOC-alanine, BOC-valine, BOC-proline and alanine with up to a 1.3 ppm difference. The chemical shift changes between the L- and D-amino acids were attributed to intermolecular interactions between the amino acid and the aromatic protons of S-Mg2dobpdc. The largest changes in the 13C chemical shift between the L- and D-enantiomers occur in alanine-loaded frameworks due to the low steric bulk of the side chain which enables strong intermolecular interactions to be formed with S-Mg2dobpdc. It is noted that S-Mg2dobpdc is locally sensitive to the chirality of the amino acid which is detected by 13C NMR, but is unable to be detected by PXRD, which measures long range ordering. Future studies include the analysis of other amino acids, including those containing aromatic side chains and additional Hydrogen bonding sites, as well as computational calculations to determine the origin of the 13C chemical shift difference between enantiomers. This study has demonstrated that solid-state NMR spectroscopy together with chiral MOFs is a versatile and elegant approach to overcoming the challenges of chiral elucidation for amino acids.
We thank Dr Doug Lawes for assistance with 1H solution state NMR, Dr Marc-Antione Sani for preliminary solid-state NMR experiments, Ms Lisa Hua for experimental assistance and A/Prof. Brendan Abrahams for support during this work. H. M. T. thanks the Australian Commonwealth Government and the University of Melbourne for a Research Training Programme Scholarship, Rowden White Scholarship and an Elizabeth and Vernon Puzey Scholarship. C. H. gratefully acknowledges the University of Melbourne for a McKenzie Fellowship.
Conflicts of interest
There are no conflicts to declare.Notes and references
- C. Wolf and K. W. Bentley, Chem. Soc. Rev., 2013, 42, 5408–5424 RSC
.
- M. Dołowy and A. Pyka, Biomed. Chromatogr., 2014, 28, 84–101 CrossRef
- J. Wang, H.-B. Liu, Z. Tong and C. S. Ha, Coord. Chem. Rev., 2015, 303, 139–184 CrossRef CAS
- H. Furukawa, K. E. Cordova, M. O’Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef
- T. Duerinck and J. F. M. Denayer, Chem. Eng. Sci., 2015, 124, 179–187 CrossRef CAS
- C. Wang, M. Zheng and W. Lin, J. Phys. Chem. Lett., 2011, 2, 1701–1709 CrossRef CAS
- X. Zhang, J. Yin and J. Yoon, Chem. Rev., 2014, 114, 4918–4959 CrossRef CAS
-
T. J. Wenzel, Discrimination of Chiral Compounds Using NMR Spectroscopy, John Wiley & Sons, Hoboken, New Jersey, 2007 Search PubMed
-
A. Kumar and N. Suryaprakash, Two-Dimensional NMR of Molecules Oriented in Liquid Crystals-Recent Developments, John Wiley & Sons, Ltd, 2007 Search PubMed
- C. Kutzscher, H. C. Hoffmann, S. Krause, U. Stoeck, I. Senkovska, E. Brunner and S. Kaskel, Inorg. Chem., 2015, 54, 1003–1009 CrossRef CAS
- H. C. Hoffmann, S. Paasch, P. Müller, I. Senkovska, M. Padmanaban, F. Glorius, S. Kaskel and E. Brunner, Chem. Commun., 2012, 48, 10484–10486 RSC
- X. Ma, Y. Zhang, Y. Gao, X. Li, C. Wang, H. Yuan, A. Yu, S. Zhang and Y. Cui, Chem. Commun., 2020, 56, 1034–1037 RSC
- J. D. Martell, L. B. Porter-Zasada, A. C. Forse, R. L. Siegelman, M. I. Gonzalez, J. Oktawiec, T. Runčevski, J. Xu, M. Srebro-Hooper, P. J. Milner, K. A. Colwell, J. Autschbach, J. A. Reimer and J. R. Long, J. Am. Chem. Soc., 2017, 139, 16000–16012 CrossRef CAS
- A. C. Forse, S. A. Altobelli, S. Benders, M. S. Conradi and J. A. Reimer, J. Phys. Chem. C, 2018, 122, 15344–15351 CrossRef CAS
- A. C. Forse, M. I. Gonzalez, R. L. Siegelman, V. J. Witherspoon, S. Jawahery, R. Mercado, P. J. Milner, J. D. Martell, B. Smit, B. Blümich, J. R. Long and J. A. Reimer, J. Am. Chem. Soc., 2018, 140, 1663–1673 CrossRef CAS
- A. C. Forse, K. A. Colwell, M. I. Gonzalez, S. Benders, R. M. Torres-Gavosto, B. Blümich, J. A. Reimer and J. R. Long, Chem. Mater., 2020, 32, 3570–3576 CrossRef CAS
- P. J. Milner, R. L. Siegelman, A. C. Forse, M. I. Gonzalez, T. Runčevski, J. D. Martell, J. A. Reimer and J. R. Long, J. Am. Chem. Soc., 2017, 139, 13541–13553 CrossRef CAS
- A. C. Forse, P. J. Milner, J. H. Lee, H. N. Redfearn, J. Oktawiec, R. L. Siegelman, J. D. Martell, B. Dinakar, L. B. Porter-Zasada, M. I. Gonzalez, J. B. Neaton, J. R. Long and J. A. Reimer, J. Am. Chem. Soc., 2018, 140, 18016–18031 CrossRef CAS
- R. L. Siegelman, P. J. Milner, A. C. Forse, J. H. Lee, K. A. Colwell, J. B. Neaton, J. A. Reimer, S. C. Weston and J. R. Long, J. Am. Chem. Soc., 2019, 141, 13171–13186 CrossRef CAS
- J. Xu, Y. M. Liu, A. S. Lipton, J. Ye, G. L. Hoatson, P. J. Milner, T. M. McDonald, R. L. Siegelman, A. C. Forse, B. Smit, J. R. Long and J. A. Reimer, J. Phys. Chem. Lett., 2019, 10, 7044–7049 CrossRef CAS
- J. Xu, V. V. Terskikh and Y. Huang, J. Phys. Chem. Lett., 2013, 4, 7–11 CrossRef CAS
- J. Xu, E. S. M. Blaakmeer, A. S. Lipton, T. M. McDonald, Y. M. Liu, B. Smit, J. R. Long, A. P. M. Kentgens and J. A. Reimer, J. Phys. Chem. C, 2017, 121, 19938–19945 CrossRef CAS
- X. Zhou, F. Xu, Z. Wu, H. Li and S. Yang, ACS Omega, 2019, 4, 8588–8597 CrossRef CAS
- C. Kutzscher, G. Nickerl, I. Senkovska, V. Bon and S. Kaskel, Chem. Mater., 2016, 28, 2573–2580 CrossRef CAS
- D. J. Lun, G. I. N. Waterhouse and S. G. Telfer, J. Am. Chem. Soc., 2011, 133, 5806–5809 CrossRef CAS
- Y. C. Lien and W. W. Nawar, J. Food Sci., 1974, 39, 911–913 CrossRef CAS
- Y. C. Lien and W. W. Nawar, J. Food Sci., 1974, 39, 914–916 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Thermal gravimetric analysis (TGA) traces, powder X-ray diffraction (PXRD) patterns, ATR infrared spectra (IR), solution state NMR spectra and additional 13C CPMAS spectra. See DOI: 10.1039/d0cc05539e |
| This journal is © The Royal Society of Chemistry 2020 |