
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiomolecule–polymer hybrid compartments: combining the best of both worlds
Claire E.
Meyer†
,
Sarah-Luise
Abram†
 ,
Ioana
Craciun
and
Cornelia G.
Palivan
*
,
Ioana
Craciun
and
Cornelia G.
Palivan
*
Department of Chemistry, University of Basel, Mattenstrasse 24a, 4058, Basel, Switzerland
First published on 14th April 2020
Abstract
Compartmentalization is a fundamental principle in biology that is needed for the temporal and spatial separation of chemically incompatible reactions and biomolecules. Nano- or micro-sized compartments made of synthetic polymers are used to mimick this principle. The self-assembly of these polymers into vesicular objects is highly compatible with the integration of biomolecules, either into the lumen, the membrane or onto the surface of the vesicles. Thus, a great variety of biohybrid nano- and microscaled compartments has been developed exploiting the specific function and properties of targeting peptides, antibodies, enzymes, nucleic acids or lipids. Such biohybrid compartments have moved from simple systems encapsulating e.g. a model protein into complex multicompartmentalized structures that are able to combine the activity of different biomolecular cargos getting closer to the realization of artifical organelles or cells. Encapsulation of medically relevant cargos combined with careful design of the polymeric scaffold and specific surface functionalization have led to a significant progress in therapeutical applications such as targeted drug delivery or enzyme replacement therapy.
Introduction
The most prominent example of a biological compartment is a cell, as it encloses an aqueous volume within its thin phospholipidic membrane to form a micrometer-sized vesicle. The compartmentalization of chemical reactions is a fundamental characteristic of cellular life, which has been the centre of numerous studies aiming to mimic some of the biological functions. Liposomes, spherical vesicles made of phospholipids, have been the spearhead for the development of micrometre-sized artificial vesicles used as protocells. Beyond that, their nanometre-sized analogues have gained particular interest in drug delivery as therapeutic payloads can be encapsulated inside the vesicles’ lumen. At the same time, stealth or targeting agents can be anchored in their membrane or attached to the surface of the vesicles. Later, the formation of nano- or micro-sized vesicles was also demonstrated with synthetic polymers.1–3 These polymeric vesicles position themselves as an attractive alternative as they demonstrate an enhanced robustness and a greater chemical versatility while, when appropriately selected, they retain the liposomes’ typical softness. The progress in polymer chemistry gave access to a variety of copolymers with tailored properties and excellent biocompatibility that can compete with the intrinsic biocompatibility and biodegradability of natural polymers. The precise chemical tuning (e.g. length, charge, or responsiveness) and the possibility of easy functionalization with reactive moieties for bioconjugation makes synthetic copolymers great building blocks for biohybrid compartments. Such biohybrid compartments integrate biomolecules for their specific functionality into their polymeric scaffold and thus inherit the advantages of both biological and synthetic systems. The different biological components can be (I) attached to the compartment's surface, (II) encapsulated inside the cavity or (III) integrated into its membrane (Fig. 1). Thereby, different classes of biomolecules can be exploited for their specific functions in different positions of the compartment. A peptide for targeting on the surface can for instance be combined with encapsulated siRNA as cargo or a channel protein embedded inside the membrane allows the access of substrates to an enzyme encapsulated in the cavity.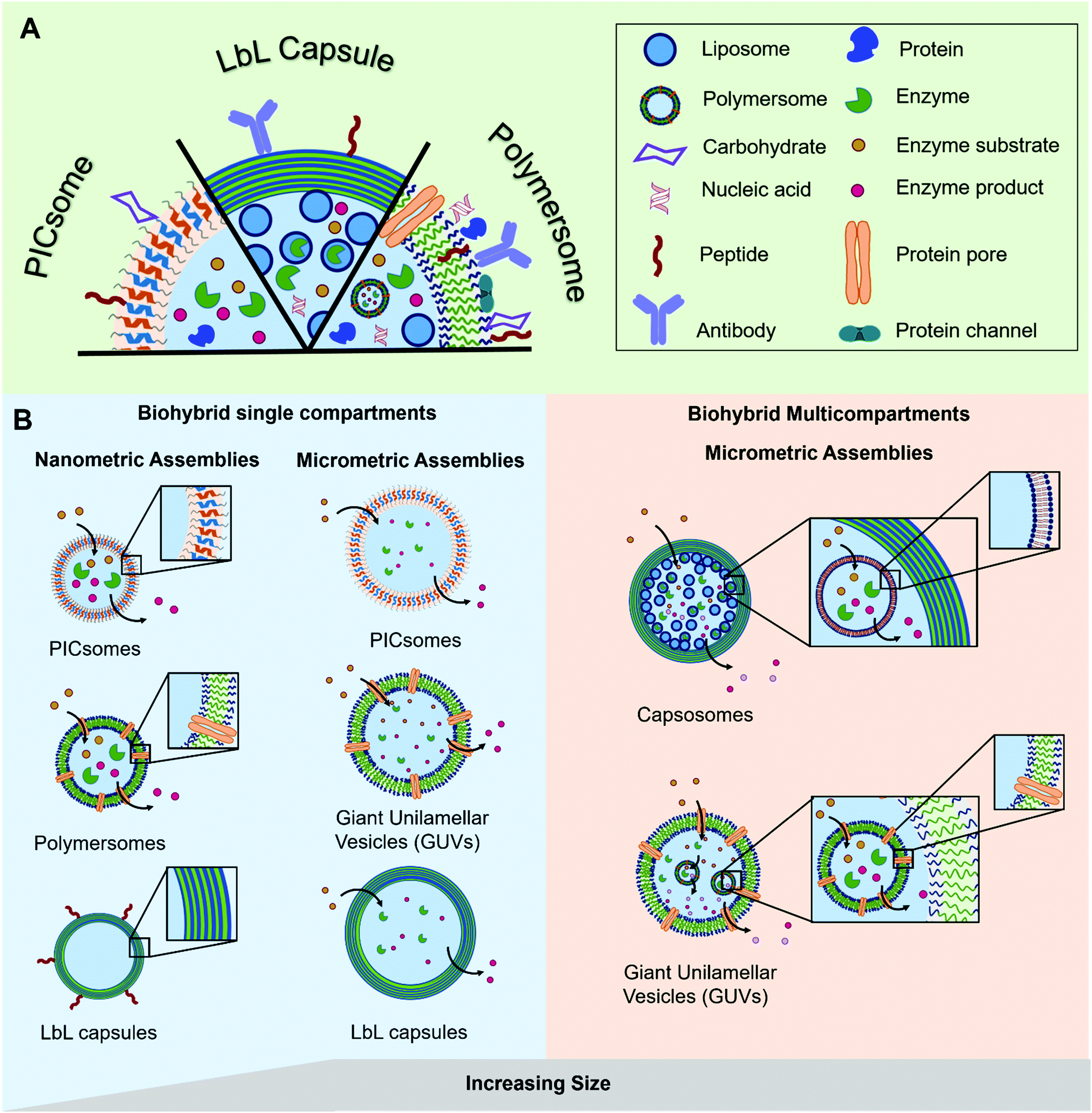 | ||
| Fig. 1 Schematic representation of (A) the different types of biomolecules and their possible locations within the polymeric assemblies and (B) the different types of hybrid polymer/biomolecule compartments (polymersomes, PICsomes, LbL capsules), showing their diversity in terms of size and arrangement, modified with permission from ref. 65. | ||
In this review we explore the various approaches in which biomolecules can be combined with synthetic polymeric systems to obtain superior biohybrid materials. Specifically, we highlight the recent advances in the development of nano- and micro-sized polymeric compartments combined with peptides, antibodies, nucleic acids, sugars, proteins/enzymes or lipids as biological components. We omit assemblies of natural polymers like chitosan or alginate or polymeric biomolecules like peptides or DNA. We will first cover nano-sized assemblies, then compartments in the micrometre size range and finally the combination of both in multicompartment systems. An overview regarding the used polymers for capsules and vesicles, their properties and the methods for self-assembly and capsule preparation will be given beforehand.
Synthetic nano/micro-sized polymer compartments
Self-assembly and compartment formation
In general, there are two ways to prepare nano-/micro-sized compartments from synthetic polymers: either their intrinsic properties allow the self-assembly in aqueous solution into hollow structures or the cavity is obtained by their deposition on a sacrificial template particle that is dissolved afterwards to result in a hollow polymeric capsule. Self-assembly can be achieved on one hand by hydrophobic/hydrophilic interactions using amphiphilic molecules such as synthetic block-copolymers or peptides. At certain ratios of the hydrophilic to hydrophobic blocks they form ordered vesicular structures in aqueous solution that are called (in analogy to their lipid counterparts) polymersomes at the nanoscale and giant unilamellar vesicles (GUVs) at the microscale. On the other hand, ionic/electrostatic interactions of oppositely charged polymers (polyelectrolytes) can also result in the formation of nano- and micro-sized vesicles referred to as PICsomes (Poly Ion Complex).Amphiphilic block-copolymers
Amphiphilic block-copolymers can consist of two (AB), three (ABA/ABC) or even more (ABABAB…) blocks with one being hydrophilic (A and C) and the other one being hydrophobic (B). Either the two polymeric blocks are prepared separately and connected afterwards with a covalent bond (e.g. by a click reaction) or the first block is functionalized after its polymerisation to become a “macroinitiator”.4 The first block can thus be extended just by addition of the second type of monomer. Commonly used polymerizations include controlled radical polymerisation (e.g. ATRP, RAFT) and ionic (ring-opening) polymerisations and their combinations. More details can be found in the specific literature on block-copolymer synthesis.5 Popular hydrophilic blocks include PMOXA, PEG, PGMA, PAA and PIAT; examples for hydrophobic blocks comprise PDMS, PS, PMA, PB, PCL, PHPMA, or PNIPAM. The Tables 1 and 2 provide all polymers used for the preparation of the biohybrid compartments referenced in this review. In case one of the used blocks responds to a certain chemical or physical stimulus, the vesicles present a high potential for applications in triggered drug release or the field of sensors. Thus, the properties of the vesicles can be fine-tuned by a careful selection of characteristics of the hydrophobic and hydrophilic polymer blocks. The membrane thickness can for instance be varied in the range of 5–30 nm depending on the molecular weight of the block-copolymer.6 Permeability and mechanical stability are as well influenced by the choice of the two building blocks and their molecular weight (MW). As an example, PDMS–PMOXA polymersomes are typically considered to be impermeable even for small molecules whereas assemblies from PS–PIAT provide semipermeable features.7| Polymer | Location of biomolecules | Associated biomolecules | Application | Ref. | |
|---|---|---|---|---|---|
| Polymersome | PDMS–PMOXA-based block copolymers (PMDS–PMOXA–PDMS, PEO–PDMS–PMOXA) | Cavity | Enzyme (SOD) | Antioxidant | 66 |
| Cavity, membrane | Protein (Hb, OmpF) | ROS detoxification | 67 | ||
| Protein (OmpF), enzyme (HRP) | Enzyme kinetic in crowded space | 77 | |||
| Model of pH-responsive nanocompartment | 82 | ||||
| 83 | |||||
| Protein (OmpF), enzyme (penicilin acylase) | Model for self-defending surface | 71 | |||
| Protein (OmpF), enzyme (LPO, SOD) | Artificial peroxisome | 70 | |||
| Protein (OmpF), enzyme (HRP or uricase) | Detoxification of uric acid | 68 | |||
| Protein (α-HL, PGM) | Model for glycolysis completion | 85 | |||
| Membrane | Peptide (gA) | Stimuli-responsive nanocompartment | 36 | ||
| Protein (proteorhodopsin), lipid (DOPC) | Model for protein reconstitution study | 88 | |||
| Protein (Aqp Z) | Model for water treatment/drug delivery | 87 | |||
| Protein (Aqp) | Model for sensitive bio-device | 84 | |||
| Membrane, Surface | Peptide (fusion eGFP-peptides) | Model for surface functionalisation | 60 | ||
| Surface | Peptide (NLS) | Targeting/delivery | 55 | ||
| DNA (single strands) | Interconnected organelles mimicking | 37 | |||
| PEG/PEO-based block copolymers (PEG–PCL, PEG–PLA, PEG–PTMC, PEG–PTMC–DTC, PEG–PLGA, PEG–PGA, PEG–PDTC–TMC, PEG–PS, PEG–PLC. PEO–PDPA–PAA, PEO–PCMA–DEA, PEO–PDMS–PEO, PEO–PB) | Cavity, surface | Antibody (anti-EpCAM), siRNA | Targeting/delivery | 45 | |
| Peptide (cRGD, GALA), protein (cytC) | 47 | ||||
| Peptide (cNGQGEQc), siRNA | 48 | ||||
| Peptide (ApoE), protein (saporin) | 54 | ||||
| Peptide (S14G-humanin), protein (lactoferrin) | 58 | ||||
| Surface | Peptide (aptamer AS1411) | Targeting/drug delivery | 61 | ||
| Peptide (GE11) | 51 | ||||
| Peptide (cell penetrating) | 56 | ||||
| Peptide (Tet-1), Protein (transferrin) | 53 | ||||
| Peptide (iRG) | 49 | ||||
| Protein (transferrin) | 57 | ||||
| Peptide (iRGD) | 46 | ||||
| Peptide (LinTT1) | 50 | ||||
| Peptide (NLS) | 52 | ||||
| Membrane | Peptide (MT1) | Enzyme detection | 78 | ||
| Protein (Mb/HRP) | Model for catalyst development | 81 | |||
| Lipid (POPC) | Model for drug delivery | 100 | |||
| Lipid (POPC) | 99 | ||||
| Lipid (HSPC) | 103 | ||||
| Protein (BR), enzyme (ATP synthase) | Model for protein reconstitution | 86 | |||
| Protein (Cyt bo3), lipid (POPC) | 102 | ||||
| Lipid (DOPC) | Model for artificial organelles | 101 | |||
| Membrane/surface | Protein (lectin), carbohydrate (glucopyranosyl) | Anti-diabetes therapy | 59 | ||
| Other block copolymers (PTMC–PGA, PEP, PMPC–PDPA, PS–PAA, PS–PIAT, PCL, PHPMA, PB–PS, DEGMA, PGG, PNIPAM, PNIPAM–CMA–DEA, DMAEMA, PEOGA–PLA) | Cavity | DNA (plasmid) | Gene delivery | 63 | |
| DNA (plasmid)/siRNA | 64 | ||||
| Enzyme (trypsin) | Enzyme kinetic in confined space | 75 | |||
| Cavity, membrane | Protein (HSA, GFP) | Intracellular delivery | 80 | ||
| Carbohydrate (matooligosaccharide), enzyme (β-gal) | Enzyme prodrug therapy | 74 | |||
| Cavity, membrane, surface | Enzyme (GOx, CalB, HRP) | Enzyme positioning in polymersomes | 73 | ||
| Membrane | Carbohydrate (glucose) | Cell glycocalyx mimicking | 90 | ||
| Carbohydrate (pyranoside/glucoside) | Model for carbohydrate–protein interaction study | 92 | |||
| Carbohydrate (hyluronic acid) | Bacteria detection | 91 | |||
| Carbohydrate (thioglucose) | Drug carrier | 89 | |||
| Lipid (cholesterol) | Drug delivery | 93 | |||
| Lipid (cholesterol) | Drug delivery | 95 | |||
| Protein (cytC), enzyme (cytC oxidase) | Model for artificial chloroplast | 69 | |||
| Lipid (HSPC/DPPC) | Model for drug delivery | 104 | |||
| Membrane, surface | Peptide (aptamer AS1411) | Targeting/drug delivery | 60 | ||
| Protein (BSA), antibody (cetuximab) | 79 | ||||
| Surface | Antibody (Trastuzumab) | Targeting/imaging | 44 | ||
| PICsomes | PEG–P(Asp), P(Asp-AP) | Cavity | Enzyme (ASNase) | Therapeutic catalytic nanocarrier | 106 |
| Surface | Peptide (cRGD) | Drug delivery, imaging | 105 | ||
| LbL capsules | PDPA–PEG | Surface | Peptide (GLGYGWS) | Drug delivery | 107 |
| Polymer | Location of biomolecules | Associated biomolecules | Application | Ref. | |
|---|---|---|---|---|---|
| GUVs | PMOXA–PDMS–PMOXA block copolymers | Cavity, membrane | Peptide (melittin), enzyme (GOx) | Model for biosensors, enzyme delivery | 126 |
| Protein (OmpF), enzyme (HRP) | Model for cell mimicking | 128 | |||
| Membrane | Peptide (gA) | Stimuli-responsive nanocompartment | 36 | ||
| Peptide (lasalocid, Alamethicin, ETH5234) | Model for biomineralization study | 124 | |||
| PEO-based block copolymers (PEO–PB, PEO–PDMS, PEO–PI) | Cavity | Enzyme (Cat) | Model for payload release | 130 | |
| Membrane | Lipid (DPPC, POPC) | Model for delivery platform | 98 | ||
| Lipid (DPPC, POPC, cholesterol) | 96 | ||||
| Lipid (DPPC, POPC) | Model for lipid/polymer hybrids study | 113 | |||
| Lipid (DPPC) | 116 | ||||
| Lipid (POPC) | Model for cell mimicking | 17 | |||
| Protein (OmpF), Lipid (DPPC) | Model for membrane protein reconstitution | 127 | |||
| Other block copolymers (PNIPAM, BA and PDMS–biomolecule conjugates) | Cavity, membrane | Protein (BSA, ferritin, Mb), enzyme (RNApol, ALP, LPL) | Model for cell mimicking | 133 | |
| Protein (BSA), DNA, enzyme (ALP) | 134 | ||||
| Membrane | Carbohydrate (glucose/galactose) | 119 | |||
| Carbohydrate (glucose) | 120 | ||||
| PICsomes | PEG–P(Asp), P(Asp-AP) | Cavity | Protein (Mb) | Drug delivery | 129 |
| PEG–PLG, PLL–carbohydrate conjugate | Carbohydrate (mannose) | Membrane/surface | Model for intercellular recognition | 122 | |
| LbL capsules | PNIPAM, PDEAEMA–PBMA | Cavity | Protein (Mb), enzyme (GOx) | Synthetic biology | 137 |
| PDEAEMA–PDSM | Protein (Mb) | 138 | |||
| Multicompartments | PMOXA–PDMS–PMOXA block copolymers | Cavity | Protein (GFP), antibody (IgG) | Model for cell mimicking | 156 |
| Cavity, membrane | Protein (OmpF), enzyme (HRP, GOx) | 76 | |||
| Cavity, membrane, surface | Peptide (gA), enzyme (LPL), carbohydrate (heparin) | 118 | |||
| PLL, PMA, PVP based polymers | Cavity | DNA (plasmid) | Model for drug delivery | 145 | |
| Peptide (Cys-KP9) | 24 | ||||
| Cavity, membrane (inner compartment) | Enzyme (β-lac), lipid (DMPC, DPPC) | 146 | |||
| Enzyme (β-lac), lipid (DOPC, DOPS, DMPC, DPPC, DPPS) | 25 | ||||
| Enzyme (β-lac), lipid (DOPC, DOPS, DPPC) | Model for therapeutic artificial cell | 144 | |||
| Enzyme (trypsin), lipid (PC, PE) | Biomedical applications | 148 | |||
| Enzyme (GR), peptide (KP9), lipid (DPPC, cholesterol) | Model for drug release and enzyme therapy | 150 | |||
| Enzyme (PAL), lipid (DMPC, DPPC, PC) | Therapeutic microreactor | 151 | |||
| Enzyme (subtilisin), lipid (DOPC, DOPS, DMPC, DPPC, DPPS) | Model for cell mimicking | 147 | |||
| Enzyme (Gox, HRP), lipid (DMPC, DPPC) | 154 | ||||
| Enzyme (TRP, HRP), lipid (DMPC, DPPC) | 155 | ||||
| Membrane | Lipid (DOPC, DOPS) | 149 | |||
| Other polymers (PS–PIAT, PEO–PB, PAH, PSS) | Cavity | Enzyme (CalB, PAMO, ADH, Alc) | Model for cell mimicking/microreactor | 7 | |
| Protein (HSA), enzyme (GOx, POD) | Possible biomedical applications | 143 | |||
| Cavity, membrane (inner compartment) | Enzyme (TYR), lipid (DMPC, DPPC, PC) | Therapeutic microreactor | 152 | ||
| Membrane (inner compartment) | Lipid (POPC, DMPC, DPPC, PC) | Model for microreactor | 18 | ||
The different types of block-copolymers can organise in different types of membranes (see Fig. 2A), e.g. an AB–BA bilayer with the hydrophobic blocks within the membrane and the hydrophilic ones directing to the aqueous solution outside and inside the vesicle. Triblock copolymers can form monolayered (ABA) structures as well as bilayers with a U type configuration of the polymer.8 In case of immiscible hydrophilic blocks (A and C), they can form domains within the vesicle's membrane.9 Moreover, a selective orientation of the longer block towards the outside and the shorter one towards the vesicle's interior can be achieved by careful design of the block length in ABC type copolymers.10 Such asymmetric membranes can favour for instance the insertion of a membrane protein with preferential orientation.
 | ||
| Fig. 2 (A) Schematic representation of different membrane architectures resulting from different copolymer structures. (B) Influence of the copolymer's f-ratio on the membrane curvature and the morphology of the self-assembly. | ||
However, the self-organisation of amphiphilic block copolymers does not always result in the desired vesicular structures, but spherical, cylindrical, gyroidal or lamellar ones instead.4 The ratio of the hydrophilic to hydrophobic blocks determines to a great extend the curvature of the membrane structures formed during self-assembly (see Fig. 2B).11–13 The f-ratio i.e. the ratio of the hydrophilic block's MW to the total MW of the block-copolymer, is used to express that influence. Vesicular structures are generally favoured at f-ratios of 35 ± 10%11 but depend as well on the specific copolymer.14,15 Increased f-ratios lead to a higher membrane curvature and thus towards the formation of spherical micelles. Another way to describe the influence of the copolymer design on the morphology of self-assembled structures is the packing parameter p. It is defined as p = v/aOlc with v = volume of the hydrophobic part, aO = contact area of the head group and lc length of the hydrophobic part.12,13
Besides the chemical composition, the MW and the dispersity of the block-copolymer, the self-assembly is influenced as well by several external factors like the used solvents, the concentration, the presence of acids, bases or salts, and the method used for the preparation of the vesicles.12
Several techniques are used to support the self-assembly process. Film rehydration methods expose an anhydrous copolymer film to a rehydration solvent (usually water or an aqueous buffer) that induces film swelling and self-assembly into vesicular structures. The mutual diffusion between the rehydration solvent and the polymer film dictates the formation of the polymersomes: high concentration gradients result in the formation of nano-sized vesicles while mild diffusion conditions lead to micro-sized vesicles.12,16 If micro-sized vesicles are desired, the rehydration can also be supported by an oscillating electric field (electroformation of GUVs). Film rehydration does not result in homogeneous size distributions. Nano-sized polymersomes can be extruded through a membrane with defined nanopores in order to narrow the size distribution to smaller sizes. In case of micro-sized vesicles, microfluidic tools are needed to achieve homogenous size distributions. The copolymers are dissolved in the oil phase of w/o/w double emulsions produced by the microfluidic device. Removal of the oil phase results in the formation of highly monodisperse GUVs. W/o/w double emulsions are also used in emulsion centrifugation which is the phase transfer of emulsion droplets over an interface. This method is for instance used for the preparation of lipid–copolymer hybrid GUVs17 and for the encapsulation of liposomes into GUVs.18 Furthermore, the slow exposure of a copolymer solution in an organic solvent to an aqueous environment can induce self-assembly into polymersomes or GUVs (solvent exchange and solvent injections methods). The copolymer solution can be either added to water or water can be added slowly to the organic copolymer solution. Subsequently, the organic solvent is either removed by evaporation or by dialysis against water. The presence of the organic solvent during self-assembly limits the application of these methods for biohybrid vesicles as it might harm the functionality of the biomolecular cargo. More detailed information on the preparation methods and their comparison are provided by other reviews.4,14,19
Polyelectrolytes
The second possibility for the self-assembly of polymers into membranes is based on the ionic interaction of oppositely charged polymers forming polyion complex (PIC) vesicles called PICsomes. The two interacting polymers are not covalently linked together. Usually, one anionic and one cationic PEG–poly(amino acid)-block copolymer are simply mixed in aqueous solution and assemble into semipermeable unilamellar membranes with PEG units on both sides.20 The vesicular structure and size can be controlled by variations of the PEG content and the concentrations used for the self-assembly. For nano-sized PICsomes PEG–poly(amino acid)-block copolymers should be mixed with pure poly(amino acids) in order to lower the PEG content further.21 Mechanical stress like vortexing and changes in pH or ionic strength can cause the reversible disassembly of the vesicular structure or increase its permeability. PICsomes offer a range of properties that are beneficial for applications in medicine. The simple preparation in purely aqueous environment together with the intrinsic biocompatibility of the poly(amino acids) are good prerequisites for the encapsulation of active biomolecular cargos. Stability in physiological conditions was achieved by crosslinking, which also allows a fine tuning of the permeability.22Polymeric capsules from LbL deposition
In contrast to the previously described methods, the layer-by-layer (LbL) technique requires the use of a colloidal particle, mostly silica or CaCO3, as a sacrificial template for the creation of the inner cavity.23 Two polymers interacting by electrostatic forces or hydrogen bonding are deposited alternately on the template before it is dissolved to obtain the hollow sphere. Thus, the size of the template particle, which can be tuned from nano- to micrometre size, reflects the size of the capsule's inner cavity. Typically, PVP and PMA which interact via hydrogen bonding at pH values below the pKa of PMA are used for this technique. Using thiol functionalized PMASH, the stability of these capsules can be extended to physiologically relevant pH by crosslinking.24 The resulting pure PMA hydrogel capsules are biodegradable, nontoxic, semipermeable and thus well suited for biomedical applications. Depending on the used crosslinking chemistry, also non-degradable or selectively degradable polymer shells can be prepared.24 More recent studies replace the labour intense LbL assembly of PMASH/PVP capsules by polydopamine shells that are deposited on the template in a single step.25Physico-chemical characterization of biohybrid polymer compartments
In order to fully exploit the potential of biohybrid polymeric compartments their physico-chemical properties need to be fully understood. Thus, reliable and reproducible methods should be used to characterize their dimensions, shape, and morphology as well as the properties of the synthetic membranes such as polarity, surface charge, elasticity, thickness, permeability and lamellarity. Additionally, the properties and functionality of the incorporated biomolecules (e.g. enzyme activity, selectivity of pores, specificity of antibodies) must be shown.Methods for size and morphology determination of biohybrid compartments can be roughly divided into techniques for direct visualization, mainly microscopy, and techniques based on the scattering of radiation. Nano-sized vesicles are often investigated by transmission electron microscopy (TEM). Due to the organic nature of the samples, negative staining with electron-rich heavy metal atoms is needed to improve contrast. The vacuum applied during TEM measurements results in deformations that can only be avoided by cryo-techniques where the samples are rapidly plunge-frozen in liquid alkanes in order to preserve their original morphology by embedding them in a thin layer of vitrified ice. Thus, membrane properties like lamellarity and bilayer thickness get accessible (Fig. 3). Besides electron microscopy, atomic force microscopy (AFM) belongs to the frequently applied techniques for size and topography of immobilized polymeric vesicles with the advantage that the measurements can be performed in liquid and thus provide information about the sample characteristics in a biologically relevant environment while still having a nanometre resolution. Mechanical properties like Young's modulus or bending modulus can be derived at the same time by force spectroscopy.26,27
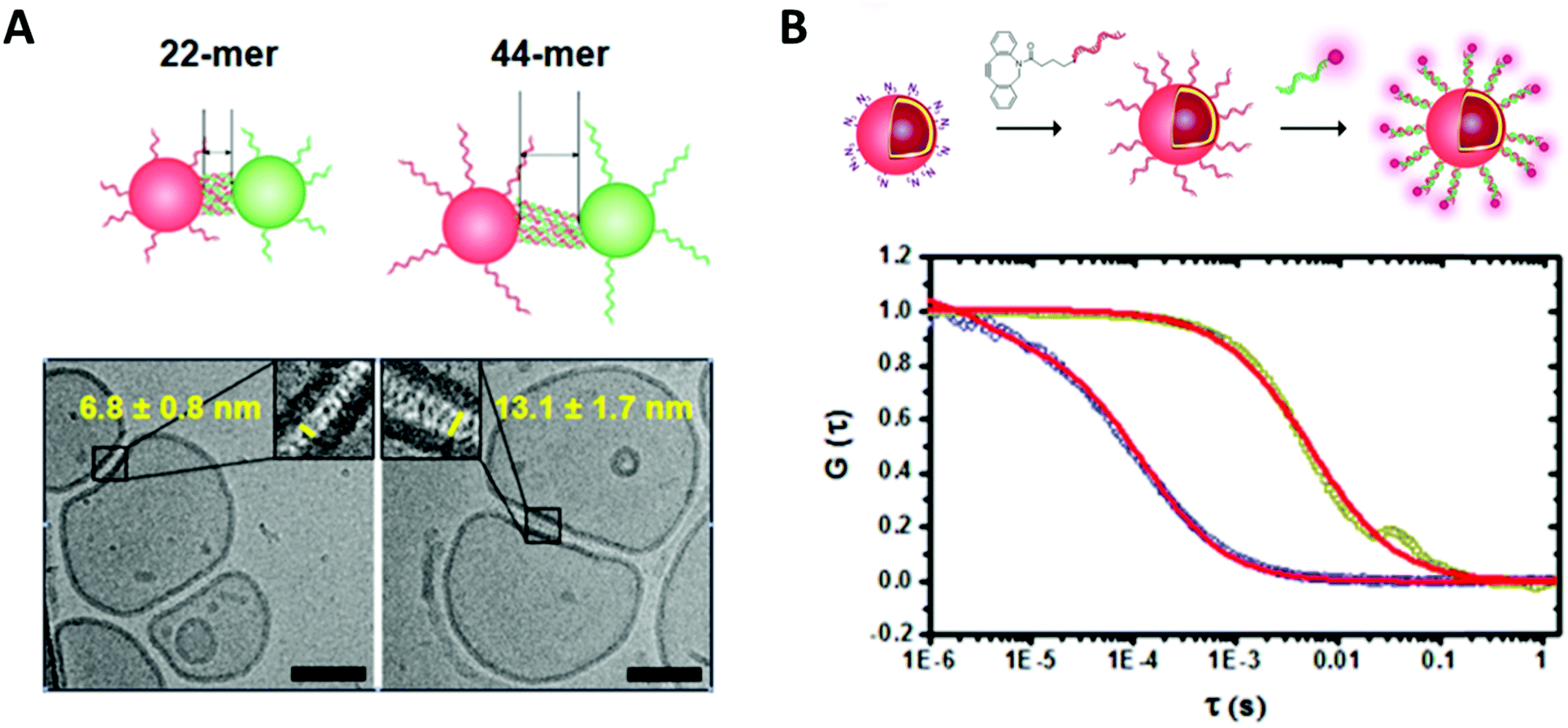 | ||
| Fig. 3 (A) Cryo transmission electron micrograph of DNA functionalized polymersome clusters showing clearly the morphology, the membrane thickness and the gap created by the attached DNA strands. (B) Normalized fluorescence correlation spectroscopy autocorrelation curve of polymersomes functionalized with ssDNA and the hybridized fluorescently labelled complementary strand (dark yellow, red fit) and the free fluorescently labelled ssDNA (blue, red fit). Reprinted with permission from ref. 37. Copyright (2016) American Chemical Society. | ||
Larger vesicles in the micron range can be visualized easily in their liquid environment by light/fluorescence/confocal laser scanning microscopy. Several super-resolution techniques are used to overcome the diffraction limit of photons to expand fluorescence microscopy to nanoscale compartments,28,29e.g. SIM (structured illumination microscopy),30 SMLM (single-molecule localization microscopy) combined with sPAINT (spectroscopic point accumulation for imaging in nanoscale topography)31 or STORM (stochastic optical reconstruction microscopy) combined with single molecule tracking.32
Fluorescence correlation spectroscopy (FCS) is another fluorescence-based technique that is of high value for the characterization of biohybrid compartments. This method analyses the fluctuations in fluorescence intensity of a small number of fluorescent particles/molecules within a very tiny volume due to diffusion (e.g. Brownian motion) by using temporal autocorrelation. Diffusion coefficients, hydrodynamic radii and concentrations can be derived from these data, which is especially useful to quantify and to investigate the behavior of fluorescently labelled biomolecules within the environment of the polymeric membranes or inside compartments (Fig. 3).33–37
Apart from direct visualization by TEM, laser light scattering is one of the routine measurements for simultaneous size and morphology determination of nanosized compartments. For dynamic light scattering (DLS), the intensity fluctuations of the scattered laser light due to the Brownian motion of the vesicles are used to determine the hydrodynamic radius by Stokes–Einstein relation. However, the data interpretation for polydisperse or anisotropic samples gets difficult. Static light scattering (SLS), usually measured at multiple angles, gives information about the morphology by the radius of gyration.
Similarly, the elastic scattering of neutrons (SANS) or X-rays (SAXS) at small angles can be used to get more detailed information about the morphology, shape and structure of polymeric membranes and compartments, but is limited by the need of appropriate radiation facilities.38,39
Nanoparticle tracking analysis (NTA) combines visualization with scattering. The light scattered by single vesicles is tracked by a CCD camera and used to calculate the vesicle concentration and the hydrodynamic diameter by Stokes–Einstein relation.40,41 Moreover, various other techniques are applied for sorting and purification of biohybrid compartments (e.g. flow cytometry, size exclusion chromatography (SEC), asymmetrical flow field-flow fractionation (AF4)). Structural and mechanical properties of polymeric vesicles are also subject of computational studies using coarse-grained simulations such as dissipative particle dynamics.42,43 Shape transformation, rupture, fusion, and membrane characteristics could be simulated and used to explain experimental results. However, the interaction or integration of biomolecules within polymer membranes is not covered extensively by this field yet.
Nano-sized single compartment biohybrid vesicles
Biohybrid polymersomes with surface functionalisation
Nano-sized polymer vesicles (polymersomes) position themselves as an attractive platform for biological applications as they allow the encapsulation of hydrophilic compounds within their internal cavity, the insertion of hydrophobic small molecules in their membrane and the attachment of a wide variety of compounds on their external surface.In this way, polymersomes combined with the biomolecule of interest have been widely exploited in medical applications, especially for targeting applications and drug delivery. Polymersomes surface-functionalized with cell specific targeting ligands like antibodies enable to combine the robustness and loading capacity of the polymeric compartments with the high binding affinity of antibodies.44,45 For example, Trastuzumab, the first monoclonal humanized antibody approved by FDA, was chosen as a ligand to direct maghemite nanoparticle-filled polymersomes to specific breast cancer cells in order to develop a novel MRI contrast agent.44 However, antibodies are expensive to produce, large in size and often immunogenic which motivates the use of alternatives.
Targeting or cell penetrating peptides, less expensive and smaller than antibodies, have been widely exploited to target polymersomes to locations of interest within cells or living organisms. For example, cyclic (RGD and NGQ),46–49 pH sensitive fusogenic (GALA)47 and other targeting (GE11, NLS, LinTT1, ApoE, Tet-1)50–54 peptides have been mainly used to deliver smart polymer vehicles to specific cells or organelles and/or to improve their tumour penetration for therapeutic or diagnostic applications. Recently, targeting of the nuclear interior has been achieved using polymersomes functionalized with nuclear localization signal (peptide NLS), which position themselves as promising nanocarriers for drug delivery to cell nuclei.55 The most frequent approach to prepare such vesicular peptide/polymer biohybrids consists in the preparation of polymer compartments followed by the decoration of their surface via the chemical ligation of the chosen targeting peptide.44,47,49,52,53,56
Beyond that, the peptides can also be coupled to the block copolymers prior to vesicle formation via mixing of the non-functionalized polymer with a peptide-functionalized counterpart that self-assemble together to form peptide-functionalized polymersomes.46,48,50,51,54 Nevertheless, even if this approach enables the simultaneous usage of different types of polymer, e.g. by mixing a triblock co-polymer with a peptide-functionalized diblock copolymer, several features like the use of polymers with matching lengths of hydrophilic and hydrophobic parts is crucial in order to achieve their co-self-assembly into polymersomes.48 It is also common to use a peptide-functionalized co-polymer with a slightly longer hydrophilic block to enhance the exposure of the peptide and potentiate the targeting/cell penetrating effect.54
Just like their shorter counterparts, proteins like transferrin can be covalently attached to the surface of preformed polymersomes for targeting purposes.53,57,58 For other applications like sensors and regulating systems, the surface of polymersomes can be enriched with proteins via electrostatic interactions prior to stable immobilization using photo-crosslinking.59 This way, surface immobilized proteins that exhibit specific binding properties are able to bind and release their compound of affinity from the external environment, e.g. as anti-diabetes system: Concanavalin A immobilized on the polymersome's surface able to bind glucose from the exterior. Surface immobilized fluorescently labelled proteins are also exploited as fluorescent tags to monitor processes occurring within the membrane. For example the membrane insertion of a non-fluorescent anchor of interest can be detected by linking fluorescent eGFP to it.60
The surface of polymersomes has also been decorated with nucleic acid based compounds for targeting applications. Aptamers, which are DNA or RNA single strands with high binding affinity to specific targets, are particularly interesting for this goal. The 26-mer DNA aptamer AS1411 binds for example selectively to nucleolin, a compound overexpressed in certain types of cancer cells. Thus, the surface functionalization of polymer vesicles with this aptamer can significantly improve the endocytosis in these cancer cells lines.61,62 For further applications, the specificity of the hybridization of single-stranded DNA (ssDNA) towards their complementary strands were exploited to selectively connect distinct polymersomes that are surface-functionalized with ssDNA. This approach enables different spatial organizations of polymersomes into clusters in order to mimic the interconnections between natural cellular organelles.37
Among other possible applications like sensors, spatial organisation or attachment, the main objective of surface functionalisation of polymersomes is targeting, which explains the recurrence of the selection of biomolecules (antibody, peptides, DNA) able to direct the nanocompartments to the location of choice. However, targeted polymersomes possess a real interest only if they deliver a specific function, which is usually obtained by encapsulation of compounds of interest or potent biomolecules inside their cavity.
Biohybrid polymersomes with encapsulated payload
Polymer vesicles are appealing carriers for hydrophilic compounds and biomolecules, especially for catalytic and sensitive molecules like enzymes, as they offer a confined environment inside their aqueous cavity and are thus able to protect the payload from the external milieu.Polymersomes encapsulating nucleic acid cargos offer protection from nuclease degradation, which motivated recent work on their potency as non-viral, so potentially safer, gene delivery systems.45,63,64 Plasmid DNA or siRNA can be entrapped within the aqueous cavity of the vesicles via physical encapsulation during polymersome formation.45,63 On one hand, slightly positively charged polymers can support the challenging encapsulation of large nucleic acids like plasmids or genes by binding the negatively charged DNA via electrostatic forces.63,64 On the other hand, such interactions can also destabilize the polymersome suspensions or prevent their self-assembly. Thus, the length of the polymer block containing protonatable groups as well as the pH have to be carefully optimized to match the chosen nucleic acids with the self-assembly conditions.63,64
The encapsulation of enzymes inside polymersomes’ cavity to develop catalytic nanocompartments, also called nanoreactors, able to produce or degrade compounds of interest have been studied.65 Depending on the loaded enzymes, such biohybrid systems aim to suit different applications. For example, polymersomes loaded with superoxide dismutase or haemoglobin act as antioxidant nanocompartments as they enable the in situ detoxification from reactive oxygen species (ROS).66,67 The presence of two kinds of catalytic polymersomes working in tandem (one encapsulating uricase and the other one horseradish peroxidase (HRP)) enables to cumulate the effects of both catalytic nanocompartments to achieve simultaneous detoxification and uric acid degradation for therapeutic applications (Fig. 4A).68 Additionally, such system, composed of two kinds of catalytic polymersomes working in tandem, mimics the communication within bioassemblies like cells and organelles to investigate the effects of distance on the overall cascade reaction efficiency (Fig. 4B).68 Interestingly, the ratio between the mean intercompartment distance and the diameter of the compartments is about 10, which reflects the order of magnitude of the ratio between cell–cell communication distance (for cells with 1 μm distance typical for autocrine signaling). Besides therapeutic applications, catalytic polymersomes are also used as model for artificial organelles (chloroplast,69 peroxisome70), building blocks of antimicrobial surfaces71 or biosensors.72 The main role of the polymeric nanocompartment is the segregation of the enzymes to protect them from proteolytic degradation. However, the interaction between the encapsulated enzymes and the external environment is essential for the catalytic activity of the nanocompartment. The majority of the polymersomes, assembled from PS–PIAT73 or PGG74 for example, are inherently permeable to small molecules and thus permit a supply in substrate for the encapsulated enzyme and also the exit of a resulting product to the external environment. On the opposite, polymersomes made of PDMS–PMOXA block copolymers are impermeable to small molecules. Selective permeability has been achieved via incorporation of pores or channels in the polymeric membrane to establish a molecular flow through the polymersomes (see next section).75 To study the permeabilization of the vesicles as well as the diffusion of compounds through their polymeric membrane, catalytic compartments encapsulating very stable enzymes like HRP or glucose oxidase (GOx) were used as basic research, before to aim at therapeutic applications. HRP and GOx benefit from the well-established, accessible and convenient monitoring of their reaction kinetics, which makes these enzymes suitable for the development of artificial cells,76 models of molecular crowding77 or enzyme positioning within the polymersomes.73 With the help of these stable model enzymes, it has been demonstrated that the combined effect of enzyme confinement coupled to a crowded milieu inside polymersomes affects the enzyme kinetics by lowering the Michaelis–Menten constant (Km) compared to non-encapsulated enzymes.77 For example, the Km value for non-encapsulated HRP (55 × 10−6 M) decreases to 25 × 10−6 M when encapsulated in PMOXA–PDMS–PMOXA polymersomes (Fig. 5). The co-encapsulation of a substance simulating molecular crowding such as PEG together with HRP results in even lower Km (5 × 10−6 M) (Fig. 5).77 This decrease of Michaelis–Menten constant for encapsulated enzymes when co-encapsulated with their substrates, highlights a greater enzyme–substrate affinity because of the molecular confinement inside the compartment that increases the collision frequencies between the reagents.77 Additionally, an increased viscosity inside the confined environment via encapsulation of crowding agents to mimic the viscosity of cells or organelles, also decreases the Km and thus improves the enzyme activity.77
 | ||
| Fig. 4 (A) Scheme of the two catatylic compartments working in tandem. (B) Conversion of substrate Amplex Red by cascade reaction inside the catalytic compartments in tandem representing the cascade reaction efficiency, at different mean intervesicles distances. Reprinted with permission from ref. 68. Copyright (2018) American Chemical Society. | ||
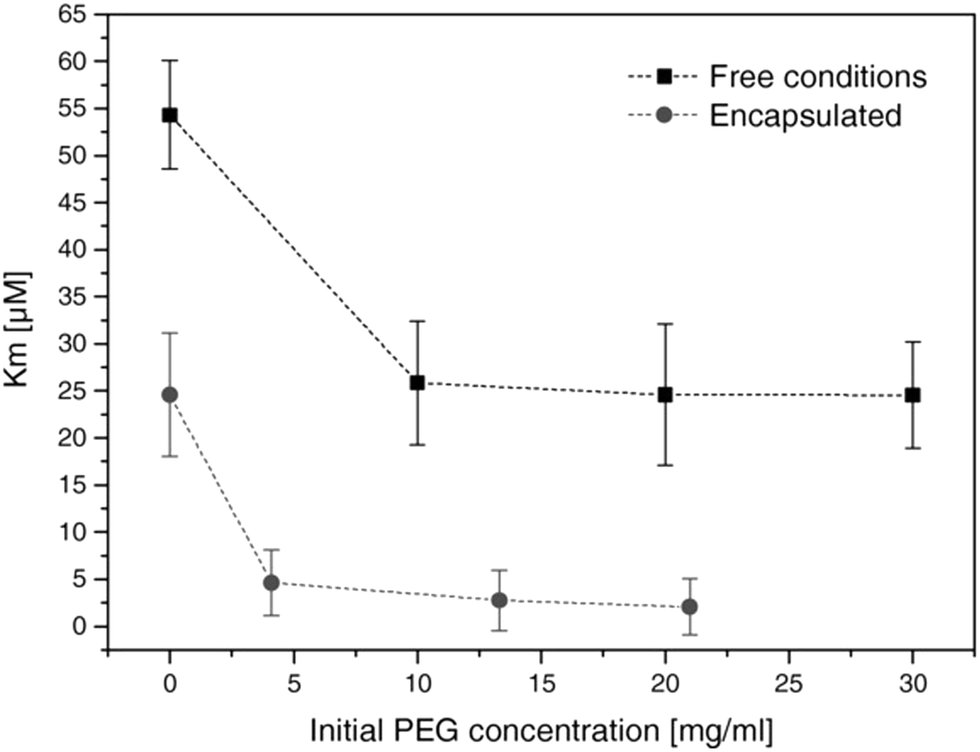 | ||
| Fig. 5 Michaelis–Menten constant of HRP (free in solution and encapsulated in polymersomes) in presence of different concentrations of PEG. Km decreases with increasing concentrations of PEG. Reprinted with permission from ref. 77. Copyright (2017) Wiley-VCH. | ||
However, in a different system where enzymes are encapsulated inside impermeable PMOXA–PDMS–PMOXA polymersomes, but the substrate is not co-encapsulated but has to diffuse through the pore to reach the enzymes, this diffusion effect is the limiting factor of enzyme efficiency. In fact, if the substrate can easily encounter the enzyme once in the compartment (low Km), the slower efflux of substrate hinders the enzyme activity, according to the decrease of catalytic efficiency (kcat/Km).68 These parameters (Km, kcat/Km, viscosity) are key factors to control to reach optimal enzyme activity as desirable to develop efficient catalytic nanocompartments.77 However, when such catalytic nanocompartments are working in tandem and support cascade reactions it is not anymore possible to use Michaelis–Menten model to get information about the kinetic parameters.
From therapeutic compounds to catalytic biomolecules (enzymes), encapsulated cargos often determine the application of the biohybrid nanocompartment. However, the encapsulated payloads can only fulfil their active/therapeutic role when in contact with the external milieu, which is usually achieved via tuning of the permeability/responsiveness of the polymeric membrane.
Biohybrid polymersomes with membrane functionalisation
Biomolecules containing a hydrophobic domain can be inserted within the polymeric membrane to add new features to the bio-hybrid nanocompartments such as stimuli-responsiveness.In the case of the impermeable PDMS–PMOXA membrane, the insertion of channel-forming antibiotic peptides like gramicidin leads to an ion-selective membrane permeability.36 The use of amphiphilic peptides can also be interesting to achieve responsiveness. Such peptides self-assembling with block-copolymers lead to polymer/peptide hybrid compartments, so called peptiSomes, where the peptide part of the membrane can be cleaved in the presence of proteinases. This allows the proteolytic disruption of the compartments’ membranes and thus the release of the payload.78 Some peptides also spontaneously insert into the polymeric membrane of preformed polymersomes. These peptides are generally derived from the transmembrane domains of membrane proteins from viral, yeast or bacteriophage sources.60 They can also originate from other natural proteins like rabbit Cytochrome b5, or be artificial and engineered to adopt an α-helical conformation.60 This approach permits to avoid chemical ligation as their insertion appears to be a concentration-dependent process governed by hydrophobic driving forces. Furthermore, their anchoring properties permit the functionalization of polymersomes with other molecules without destabilizing or rupturing the polymersome membranes.60
More complex amino acid based biomolecules like proteins have been used to constitute a major structural component of biohybrid polymer vesicles: conjugates made of hydrophilic human serum albumin and a hydrophobic polymer block self-assembled into biodegradable protein–polymer vesicles that show great potential as drug delivery vehicles.79 Another amphiphilic BSA–polymer conjugate self-assembled into structures with tuneable and ‘’evolving’’ morphologies that changed from sphere to worm and then to vesicles due to the increasing molecular weight during polymerization. This process of morphology change also enables the encapsulation of GFP, proving potential for drug delivery.80 Asymmetric conjugates made of ABC triblock copolymers containing either HRP or Myoglobin, have been shown to form vesicles but also other nanoassemblies like micelles or rods.81 However, only a few examples of such protein–polymer conjugates have been reported to form desirable nanoassemblies as the inherent complexity of these systems impedes the control over their architecture. Moreover, membrane proteins have been reconstituted in the polymeric membrane. In case of impermeable polymersomes, the permeabilization that is necessary for the confection of catalytic nanocompartments for example can be achieved via the membrane insertion of protein pores like Outer membrane protein F (OmpF). This porin has a cut-off of 600 Da that permits the passage of small molecules into and from the inner space of polymersomes while retaining bigger encapsulated macromolecules like enzymes inside.77 OmpF can also be chemically modified prior to the insertion to obtain pores that open upon pH change82 or biovalves able to control the opening and closing of the pore.83 Interestingly, the successful incorporation of membrane proteins can be achieved even if the polymeric membrane is thicker than biological lipid membranes and thicker than the hydrophobic part of the protein, which suggests a conformational adaptation between the polymer and the protein.34 However, this phenomenon of adaption requires specific properties of the polymeric membrane: high flexibility of block-copolymers is essential to achieve membrane fluidity that is similar to natural phospholipidic bilayers.34 The hydrophobic mismatch between the membrane thickness and the size of the protein also influences the insertion process as it should range from 3.5 to 5 times to achieve a successful protein reconstitution. Finally, block-copolymers with high polydispersity index (PDI) also favour protein insertion as they enhance the ability of the polymer chains to adapt to the dimensions of protein.34 To go one step further, the asymmetry of biological membranes has been mimicked by the design of asymmetric ABC triblock copolymers, which gives access to a preferential orientation of the inserted protein.84 The orientation of membrane proteins is of great interest to achieve a directional functionality,84e.g. to control the influx/outflux through polymersomes in case of active diffusion of compounds via channel porins, or to control the exposition of the protein towards the cavity or the surface of vesicles.
Recent advances in polymer chemistry moved these synthetic membranes closer to their biological inspiration, thus allowing the successful insertion of several proteins like α-hemolysin,85 Bacteriorhodopsin86 or AquaporinZ87 into polymeric membranes. Nevertheless, the reconstitution of membrane proteins in polymer membranes remains extremely complex as it requires a high stability of the protein, adapted surfactant and pH conditions88 and strategies to deal with the hydrophobic size mismatch between the protein and the synthetic membrane. The development of a mathematical framework based on design of experiments (DoE) is a particularly interesting approach to identify the requirements for a successful reconstitution process of membrane proteins into the membrane of polymersomes.88 This DoE method enables to devise an experimental plan that samples a given parameter space optimally, allowing a low number of experimental runs to detect interactions and estimate factors’ effects. For example, the effects of several factors (pH-values, detergent concentration and polymer/protein ratio) on the biohybrid protein–polymersomes characteristics (size, homogeneity, functionality) were identified via the reconstitution of proton pump proteorhodopsin in polymersomes, using such an approach (Fig. 6).88
 | ||
| Fig. 6 (A) Interaction plot of the biohybrid proteorhodopsin/polymersome formation, showing the behavior (size, polydispersity index (PdI)) of formed biohybrids toward changing conditions (pH, percentage of n-octyl-β-D-glucopyranoside (OG) surfactant, polymer/protein ratio (PPR). (B) Functionality of the proteorhodopsin/polymersome hybrids: the proton gradient is dependent of the number of pumps reconstituted in the polymer membrane (as well as their orientation and integrity). Highest proton-pumping activity should be found within the derived region yielding homogenous and large enough vesicles: the modeled response surfaces of the pH gradient depending on factors influencing the formation of the hybrids like the two most influential factors (pH, PPR), and third factor that is fixed at its central value (OG = 1.12%), shows that the formed pH gradient becomes larger at basic pH values and higher PPR values. Reprinted from ref. 88. Creative Commons CC BY. | ||
Besides proteins also carbohydrates can be inserted into the membranes of polymersomes. Considering their variety of roles in biological processes, notably in cell–cell and cell–matrix communication, such biohybrids have been exploited for applications like cell-surface recognition and cell signalling. Only very few studies have been devoted to block-copolymers containing sugar moieties with the ability to self-assemble into vesicles.74,89,90 Notably, the development of enzyme responsive vesicles involving the incorporation of enzyme-degradable sugars within the block-copolymer: hyaluronic acid containing block-copolymers have been used to set up hyaluronidase responsive vesicles that were used for the detection of pathogenic bacteria like Staphylococcus aureus that are secreting this enzyme.91 In this sense, polymersomes with a glycosylated surface can be considered as mimics of cells with their associated pericellular matrix (glycocalix).90 Towards a similar goal, so called glycosomes have been prepared via the co-self-assembly of a phenylboronic acid-functionalized synthetic block-copolymer with hydrophilic glycopolymers made of either galactoside or glucoside.92 The assembly process is driven by the dynamic covalent bond between the phenylboronic acid and sugars. Glycopolymersomes assembled from glycosylated block copolymers acted as “sugar sponges” to regulate the blood glucose level. The binding of carbohydrate-binding proteins like lectin enabled the dynamic sugar replacement between the free glucose of the external milieu and the sugars exposed at the surface of vesicles.59 Additionally, carbohydrate-conjugated polymers containing maltooligosaccharide formed vesicles with interesting molecular-weight-dependent permeability catalytic nanocompartments proposed in enzyme prodrug therapy.74 The permeability coefficient is correlated to the partition coefficient, and as small hydrophobic molecules have high partition coefficient, they are more likely to pass the membrane than larger and more hydrophilic molecules.
The combination of block copolymers and phospholipids has gained interest for the formation of hybrid vesicles with membrane properties closer to biological ones. Phospholipid-block copolymer conjugates were used for the preparation of hybrid vesicles that were particularly studied for their cellular interactions.93,94 They demonstrated the relevance of shear stress in cell uptake and how shorter hydrophobic extensions are enhancing their internalization by macrophages.93 Compared to liposomes they also showed an enhanced lysosomal escape.95 Instead of using lipid–polymer conjugates, both types of macromolecules eventually also form hybrid vesicles if they are just blended. The successful formation of such hybrid assemblies that bring together the biocompatibility and softness of liposomes and robustness and chemical versatility of polymersomes depends on the correct choice of the amphiphilic building blocks. Molar fraction, molecular weight and block size have to be optimized in order to achieve a successful assembly. Block-copolymers that closely match the dimensions of phospholipids have been shown to favourably assemble into biohybrid vesicles.96–99 These hybrids possess a great potential for drug delivery as the efficacy of payload loading and release can be modulated by the polymer/lipid ratio and the length of the diblock copolymer.99,100 In addition, such hybrid vesicles exhibit higher permeability compared to polymersomes and liposomes, which has been further enhanced by the insertion of ionophores or ion channels.101 However, the biggest potential of hybrid lipid/polymer vesicles has been shown for the reconstitution of membrane proteins: they form a durable environment that lowers the initial enzyme activity but enables a significant extension of the functional lifetime of the protein compared to its reconstitution in liposomes.102 Furthermore, the incorporation of lipids like phosphatidylcholine into polymeric membranes can enhance the targeting properties of functionalized vesicles. The insertion of a lipid as additional compound into the membrane is diluting the density of polymer brushes on the vesicle's surface that are obstructing targeting ligands due to steric effects.103 Such chimeric vesicles exhibit particularly low toxicity in vitro and in vivo, especially at high ratio of polymer and even at high concentrations of vesicles.104 The tunability and particular properties offered by such hybrid systems is of great interest for the development of drug delivery nanocarriers, imaging tools and catalytic nanocompartments.
The incorporation of biomolecules within the polymersome's membrane mainly enhances the tunability of the membrane, especially permeability and responsiveness. However, further investigations of the complex behaviour of the polymers with biomolecules within these hybrid membranes are required, which are often performed by the mean of GUVs considering the more convenient characterization of their structure compared to their nano-sized counterparts.99
Biohybrid PICsomes and layer-by-layer capsules
Other types of polymer-based nanocompartments have been gaining interest as well: PICsomes and LbL capsules. Although they have not been studied as extensively as polymersomes, a small number of biohybrid PICsomes and LbL capsules has been reported.Surface-functionalization of PICsomes has been achieved using peptides (cRGD) for neovascular targeting. Before their functionalization, PICsomes were assembled and crosslinked using EDC reaction to couple PEG-based block aniomers with homo catiomers.105 This strategy of surface-functionalization, rather than pre-functionalization of block-copolymers with peptides before self-assembly, has been selected to avoid peptide aggregation. The resulting peptide-functionalized nanocompartments showed enhanced cell uptake and tumour accumulation and aim for drug delivery and targeting applications. The inner cavity of PICsomes has also been exploited to enclose biomolecules: therapeutic-relevant L-asparaginase loaded into PICsomes has been shown to retain the catalytic activity even in vivo, which positions this system as an interesting application of nanocompartments for cancer therapy.106
Finally, a nano-sized LbL capsule functionalized with a peptide was used to target atherosclerotic plaques in vivo.107 However, these nanocompartments degraded in human plasma, and their stabilization via deposition of poly-histidine on the core particle before deposition of the polymers did not totally prevent this phenomenon.107
One of the main differences between these nanocompartments and polymersomes resides in the organization of their shell/capsule, which resemble less to biological membranes. Thus, the possibilities to mimic membrane-oriented biological processes like membrane protein reconstitution are limited. According to our knowledge, no examples of nano-sized PICsomes or LbL capsules with biomolecules directly embedded within the shell/capsules have been reported. In this way, polymersomes remain more polyvalent due to the tunability of their inner cavity, surface, but also of their membrane using biomolecules. However, innovations in polymer chemistry and development of polymer–biomolecule conjugates as part of the structure of these nanocompartments could replace membrane proteins to achieve responsiveness and selective permeability. Additionally, the lack of inherent robustness of PICsomes and LbL capsules requires additional steps like crosslinking or deposition of stabilization compounds, highlighting the need for further research.
Micro-sized single compartment biohybrid vesicles
To gain a comprehensive understanding of biohybrid membrane properties and composition, GUVs within the micrometre range are used. Using such large, cell sized vesicles, allows for the visualization of the membrane structure or reactions that take place within the inner cavity of the GUVs in real time, elucidating whether domains are formed and providing information regarding membrane fluidity, elasticity and integrity. These systems are primarily used as artificial cells or cell models to study enzymatic reactions within a controlled simplified surrounding that still retains selected characteristics of the cells. To create an artificial cell the membrane composition can be adapted to contain lipid/glycopolymer/polymer mixtures or biomolecule/polymer hybrids and the inner cavity can be loaded with enzymes to study membrane and crowding effects on biorelevant reactions.Biohybrid GUVs: membrane properties, composition and functionalization
Historically the first GUVs designed as artificial cells consisted of a lipidic membrane, as these are considerably easier to form since synthetic lipids will typically assemble into unilamellar vesicles.19 However, larger lipid based GUVs are quite unstable and cannot be stored for long periods of time, thus limiting their applicability for biotechnological applications or as cell models for reactions that have extended time periods.19 A solution consists in designing lipid–polymer hybrid membranes that possess the strength and elasticity of the polymeric membrane combined with the increased fluidity and thinness of the lipid membrane (Fig. 7A).108,109 Obtaining hybrid membranes that do indeed blend the best mechanical properties of lipids and polymers is not trivial. It has been shown that mixing PEO–PDMS–PEO triblock GUVs with increasing content of lipids results in the formation of membranes with increased stretching moduli however at the expense of toughness.110 The mismatch between the length of the triblock copolymer and the lipid as well as the stretched chain conformation of the polymer within the membrane lead to destabilization of the membrane at the lipid/polymer boundaries.110 Using triblock copolymers with a higher molecular weight and longer hydrophobic block can promote a U-shape conformation of the polymer within the hybrid membrane.111 The entanglement induced by the U-shape conformation results in hybrid GUVs with increased stretching modulus and improved structural stability.111 Using a PDMS–PEO diblock copolymers that self-assemble into a bilayer does indeed overcome the limitations of the triblock copolymer and when mixed with lipids, a hybrid membrane with increased lysis strain and cohesive energy density, as compared to the pure lipid GUVs, was obtained.112 | ||
| Fig. 7 (A) Diffusion of fluorescently labelled proteins through lipid membranes (squares) compared to triblock polymeric membranes (circles) as a function of membrane thickness. Reprinted with permission from ref. 34. Copyright (2015) American Chemical Society. (B) Overview of polymer/lipid hybrid GUVs with different molar ratios highlighting domain formation and phase separation. Reprinted with permission from ref. 114. Copyright (2013) Elsevier. | ||
Recently, it has been shown that mixing block copolymers with lipids results in formation of lipid nano-domains, which can grow into micrometre sized domains culminating in complete phase separation.113 This effect depends on the mol% of lipid within the final assembly, the main chain transition temperature, the hydrophobic mismatch between the block copolymer and the lipid and temperature (Fig. 7B).113,114 By controlling these parameters, domains with precise sizes can be obtained. For example, low temperatures and mol% of lipid within the assembly lead to small nano-meter sized domains. Also, from a thermodynamic aspect, at low lipid mol% entropy tends to dominate leading to a uniform distribution of the lipid within the membrane.115 Increasing the mol% of incorporated lipids resulted in a growth of the domains followed by complete phase separation at 50% molar lipid compositions, budding and fission.113,114 At higher lipid fractions, enthalpy dominates due to the hydrophobic mismatch between the polymer and lipid, thus leading to phase separation and domain formation.115 The membrane properties are also governed by the molar composition and is more fluid-like at high polymer concentrations or more elastic-like at high lipid composition.116 However, it is important to note that formation of domains can lead to membranes with decreased toughness with destabilization occurring at the lipid/polymer boundaries.110,111
These types of hybrid compartments not only attempt to amalgamate the top characteristics of polymers and lipids into one cohesive unit, but can also be used to design asymmetric membranes.17 Such membranes are obtained using a 2-step procedure, where first the polymer or lipids are assembled into a giant monolayered structure followed by addition of a second monolayer of lipid or polymer, respectively. These membranes will be used as tools for cell biomimicry to study the effects of membrane asymmetry and heterogeneity found in natural cell membranes.
While lipid/polymer hybrid membranes can be used as tools to better understand the fluidity, elasticity, domain formation, asymmetry and heterogeneity of natural membranes, a different approach focuses on adding cell like recognition patterns to artificial membranes. The cell membrane is covered by a glycosaminoglycan layer that can interact with multiple proteins leading to activation of receptors, signalling pathways, as well as pathogen infections.117 One approach to mimic the cell membrane composition is by incorporating glycopolymers within a polymeric membrane. This has been accomplished elegantly by coupling the hydrophobic PDMS blocks with a hydrophilic heparin block118 or a glycopolymer block.119,120 In the case where heparin was used as a mimic for heparan sulphate, small nanometre sized polymersomes121 or GUVs118 with exposed heparin on the surface were obtained. They were shown to bind protamine, a heparin binding protein, thus these GUVs can be used as a platform to study interactions of heparan sulphate binding proteins with the cell surface. The interactions between lectins and the cell surface glycan layer play key roles in multiple biological processes including pathogen invasion. To study these interactions biohybrid glycosylated polymers have been used.119,120 These polymers were shown to selectively interact with lectin functionalized beads or fimH positive E. coli, thus mimicking natural cellular interactions. Along similar lines, PICsomes were also tethered to carbohydrates to create a synthetic glycosaminoglycan layer.122 In this case the aggregation kinetics of PICsomes bound to lectins could be studied demonstrating the availability and selectivity of multiple binding sites.
Biohybrid GUVs: membrane protein insertion
Membrane proteins have become important functional components for biotechnical devices such as sensors, nanoreactors and artificial cells as they enable a selective communication between the inner cavity and the surroundings.123 One of the first examples describing the insertion of membrane proteins within a polymeric GUV was published in the early 2000s, where 3 different ionophores were used for selective or unselective transport of calcium.124 Still to this day, formation of biohybrid polymeric GUVs equipped with inserted proteins is considerably more complex compared to polymersomes, as the decreased membrane curvature of these larger GUVs, in some cases impedes the functional insertion of membrane pores or peptide forming pores.125 The strain of insertion can be overcome by using PMOXA–PDMS–PMOXA block copolymers with tailored block lengths, where the soft and flexible character of the PDMS hydrophobic unit moulds to accommodate small hydrophobic pores resulting in a stable assembly.36 By employing GUVs, the fluidity of the membrane and diffusion of membrane pores through the polymeric membrane has been measured by fluorescence correlation spectroscopy (FCS), revealing key molecular parameters that play a role in polymeric biohybrid membrane formation. One such factor that governs the functional insertion of pores within a membrane is the block length of the block copolymer. Increasing the block length, results in a thicker membrane where the inserted membrane pores exhibit a decrease in lateral diffusion as they travel through the membrane.6 However, the flexible PDMS hydrophobic block enables the contraction and compression of the membrane around the inserted protein, to accommodate and surpass the hydrophobic mismatch.34 The increased polydispersity of the block copolymer also contributes to the polymer flexibility, where the shorter polymeric chains within the membrane can readily adjust to accommodate the inserted membrane protein. In a very recent study it was also shown that insertion of melittin, a small pore forming peptide, within a polymeric membrane can be facilitated by adding the peptide during the early phases of the self-assembly process.126 The interdependency of membrane curvature and successful protein insertion was also explored and revealed that increased membrane curvature promotes protein insertion, most likely due to changes in packing as well as conformation of the polymer when assembled within as vesicular structure as opposed to a planar membrane.126 Using polymer–lipid hybrid membranes that possess thin lipid domains could support improved membrane protein insertion. In one example OmpF was indeed successfully reconstituted in a polymer–lipid hybrid membrane.127 However, this field is still in its infancy with most of the research focused on the membrane properties as opposed to membrane protein insertion.Biohybrid GUVs: encapsulating enzymes
To obtain giant catalytic compartments, biohybrid polymeric membranes with inserted membrane pores can be used as an approach to study catalytic reactions in real time. One such system consists of incorporating OmpF within the membrane of a PMOXA–PDMS–PMOXA triblock copolymer.128 Using this type of catalytic compartment it was possible to study the conversion of the substrate Amplex red to the fluorescent substrate resorufin using confocal laser scanning microscopy. Also due to the fact that GUVs are similarly sized to cells, it is possible to analyse them via flow cytometry, thus obtaining crucial information related to the entire sample population and not only each GUV in particular. Giant PICsomes are also exceptional candidate for the design of microreactors and can be loaded with biologically relevant enzymes such as metmyoglobin.129 The semipermeable nature of PICsome together with the addition of a PEG biocompatible shell, promotes the use of PICsomes as enzyme or drug carriers for biomedical applications. Furthermore, cellular functions such as cell secretion, can also be mimicked using GUVs by incorporating a cascade reaction that leads to the controlled rupture of the polymeric membrane.130 To increase the encapsulation efficiency and introduce a mixture of enzymes and substrates within giants, microfluidics has been used to generate artificial cells capable of expressing proteins in situ.131As an alternative to encapsulating functional enzymes or proteins, proteinosomes composed of a protein–polymer membrane have been introduced.132 In this type of assembly the membrane is composed of a protein inner leaflet, typically bovine serum albumin functionalized with a pNIPAAm polymer.133 The compartments assemble in water droplets emulsified in a continuous oil phase forming micrometre sized giants with an aqueous inner cavity. The inner cavity can be loaded with enzymes, DNA or machinery needed for protein expression to obtain biocompatible and functional micro-reactors.133,134
Biohybrid LbL microreactors
The second most prevalent category of micrometre sized vesicles are the giants fabricated through an LbL deposition of polyelectrolytes onto a core substrate.135,136 As these types of polymeric LbL membranes are permeable towards small molecules, enzymatic reactions can take place within their inner cavity without the need for membrane inserted pores. Permeability of these microreactors has been also tuned through the use of temperature and/or pH responsive polymers such as poly(N-isopropylacrylamide) and polydiethylaminoethylmethacrylate, respectively.135,137,138 Combining the stimuli responsiveness with enzyme encapsulation, mostly revolving around GOx and catalase as well as others,139 supports applications such as enzyme replacement therapy, biocatalysis and biosensors.Multicompartment biohybrid vesicles
The field of biological cell mimicry is further advanced by the development of compartment-in-compartment biohybrid vesicles. Such multicompartments are seen as a step forward towards functional models for eukaryotic cells and their cellular organelles, which are able to perform multiple chemically incompatible enzymatic reactions simultaneously by separating them in subcompartments. Besides the fundamental interest in the creation of artificial cell mimics, compartmentalized vesicles gained also increased attention for enzyme replacement therapy. They could serve as biocompatible delivery vehicles preventing administered enzymes from fast degradation in biological environments while offering at the same time space for additional cargos and responsiveness for triggered catalytic activity or release, thus mimicking the spatio-temporal control of cells over their metabolic reactions.Different multicompartment architectures
Multicompartment vesicles were pioneered with the development of so called vesosomes where liposomes were encapsulated inside bigger liposomes.140 This approach was rapidly transferred to synthetic polymeric materials such as polymeric vesicles or LbL capsules resulting in all possible combinations.141 They are mainly classified by the bigger, outer compartment that is either a LbL capsule, a polymeric vesicle or, as very recently realized, a giant plasma membrane vesicle (GPMV).142The encapsulation of the subcompartments, such as small polymersomes, micelles or liposomes, inside polymeric vesicles is usually achieved during their self-assembly by film rehydration, in microfluidics or by double emulsions and emulsion centrifugation methods, often resulting in mixtures of single and multicompartment structures that cannot be separated.
LbL multicompartments exist with either one smaller LbL capsule as single subcompartment (shell-in-shell structure)143 or thousands of subcompartments that are deposited in a controlled way onto the template during the preparation of the micron sized outer capsule (Fig. 8).144 In this case, the possible subcompartments comprise small LbL capsules,24 polymersomes,145 or liposomes,146 with the last ones being used for the majority of all LbL biohybrid multicompartments. The LbL approach allows the control over the spatial positioning of the subcompartments. Depending on the polymers used for the precursor or separation layer (Fig. 8), they either stay attached to the inner walls of the LbL capsule or start to move freely within the cavity after template removal.147 If only one hemisphere of the template is accessible for the subunit deposition, even Janus type multicompartments can be prepared by the LbL approach.148
 | ||
| Fig. 8 (A) Schematic illustration of capsosome assembly. Reproduced with permission from ref. 144. Copyright (2010) American Chemical Society. (B) Capsosomes as intracellular microreactors loaded with GOx and HRP for the enzymatic cascade conversion of β-D-glucose into resorufin. (C) Differential interference contrast (DIC) and confocal laser scanning (CLSM) microscopy images of macrophages with internalized capsosomes showing the cascade's red fluorescent end product resorufin after 24 h. Control images in the absence of capsosomes are shown at the bottom. (C) and (D) are reprinted with permission from ref. 154. Copyright (2017) American Chemical Society. | ||
As for single compartments, the biological component can be encapsulated as cargo inside the subunits or the lumen of the main compartment, but it can be also found within or outside of the membranes. Most of the biohybrid multicompartments are multifunctional systems that comprise different classes of biomolecules in different places, e.g. enzymes inside the subcompartment and channel proteins within the membranes or lipids forming the subunits that encapsulate enzymes inside.
Biohybrid multicompartment LbL capsules
LbL assembled polymeric capsules are semipermeable by nature, and thus able to keep larger (biomacro-)molecules or subunits inside while allowing the diffusion of smaller ones, i.e. substrates or analytes. For that reason, LbL capsules show great potential for encapsulated enzymatic catalysis which was demonstrated first by the enzymatic cascade between GOx and peroxidase that have been separately encapsulated in an inner and outer LbL compartment of a shell-in-shell structure.143 Their potential for applications in drug delivery is also related to the selective degradation of LbL capsules upon different chemical stimuli dependent on the crosslinking chemistry. A polymer peptide conjugate as model vaccine was for instance released from LbL subunits responsive to intracellular relevant glutathione (GSH) concentrations.24As already mentioned, most LbL derived multicompartments are so-called capsosomes with liposomal subunits. Liposomes offer protection for fragile encapsulated biomolecules or other hydrophobic or hydrophilic substrates whereas the polymeric LbL carrier capsule serves as structural scaffold. Cholesterol modified polymer layers are necessary to immobilize the preformed liposomes on the templates as pure electrostatic interactions are often not enough to keep them in place when covered with the subsequent polymer layer.146 Deposition in several layers allows the delivery of high doses of encapsulated complementary cargos in a controlled ratio within one carrier capsule.149
If capsosomes are used as microreactors, accessibility of the enzyme inside the liposomal subunits is crucial and usually achieved by permeabilizing them either with the help of detergents146 or by an increase in temperature above the lipids’ melting temperature Tm. Hence, for medical applications, the composition of the liposomal subunits is often tuned for a Tm close to 37 °C. This concept allowed for instance the combination of enzyme therapy (the continuous production of GSH as antioxidant) with triggered drug release (here a small oligopeptide cleaved from its linker by GSH) in the same system.150 Cascade or parallel reactions using up to three different enzymes were advanced using PDA based capsosomes (e.g. uricase, HRP and ascorbate oxidase)25 which were later further optimized for the encapsulation of medically relevant enzymes (e.g. for the treatment of phenylketonuria151 or melanoma152). As a proof of concept, the enzymatic activity of such extracellular microreactors was shown in microfluidic setups mimicking the dynamic conditions of tumor or intestine tissues.151,152
Another step forward to the application of capsosomes in enzyme replacement therapy was achieved with the demonstration of intracellular activity after supporting cell uptake with PEG surface grafting and RGD functionalization.153 Intracellular activity in macrophages could also be shown for two enzymes encapsulated in distinct liposomal subunits working as cascade154 or in parallel.155 Fluorescent Au nanoclusters were additionally incorporated into the systems to monitor cellular uptake.
Despite this encouraging progress some challenges remain, such as a prolonged stability of the liposomal subcompartments to ensure a sustained activity of the encapsulated enzyme as well as an improved encapsulation efficiency. Replacement of the liposomal subcompartments with polymersomes offers a possibility to address these challenges. However, examples for polymersome subunits in LbL capsules remain scarce. In one of the rare examples, plasmid DNA has been released from polymersome subunits by a double switch in pH.145
Biohybrid multicompartment polymeric vesicles
Multicompartment vesicles for the encapsulation of biomacromolecules have also been prepared solely on the basis of polymersomes. As proof of principle, fluorescent proteins were encapsulated inside the distinct compartments of a polymersome-in-polymersome structure with 45% of the polymersomes being multicompartments.156 With the introduction of the channel protein OmpF into the membrane of the inner polymersome, a cascade reaction was enabled between HRP in the subcompartment and GOx in the lumen of the semipermeable outer polymersome, thus advancing this system to a cell mimic.76 Later, a cofactor dependent three enzyme cascade was realized inside a GUV encapsulating one enzyme, the substrates and a cofactor in its lumen and two other enzymes inside its semipermeable polymersome subcompartments using an emulsion-centrifugation process.7Recently, a multicompartmentalized GUV containing polymersomes, micelles and reduction sensitive nanoparticles as three different subcompartments was reported for mimicking a cellular signal transduction strategy (Fig. 9).118 First, Heparin was used as receptor like surface modification and allowed the targeted interaction with protamine. Second, the reaction of a hydrophobic enzyme, immobilized within the membrane of the GUVs, was started on demand by releasing its substrate from the reduction sensitive nanoparticles. In a third scenario, the GUVs were selectively permeabilized for sodium ions by directing the peptide ion channel gramicidin A into the GUV membrane after its stimuli responsive release from the encapsulated nanoparticles.
 | ||
| Fig. 9 (A) Schematic representation of the modular multicompartment system able to mimic a cellular signal transduction strategy. (B) DTT triggered gramicidin gA ion channel recruitment from subcompartments to the polymer membrane visualized by activation of a sodium ion sensitive dye (green fluorescence in CLSM images) either by gA mediated Na+ efflux (upper part) or gA mediated Na+ influx (lower part). Reprinted with permission from ref. 118. Copyright (2019) Wiley-VCH. | ||
Beyond polymersomes also liposomes serve as subcompartments inside synthetic GUVs. The tunability of their melting temperature by changing the lipid composition was exploited for a temperature triggered sequential release of dyes from liposomal subcompartments inside a GUV with the future aim of starting cascade reactions on demand inside the polymersome as microreactor.18
An early example for a multicompartment vesicle showed as well the inverse configuration by encapsulating polymersomes inside a bigger liposome without providing any specific functionality.157 In contrast, polymersomes were introduced very recently as subcompartments into giant plasma membrane vesicles (GPMVs) to form molecular factories.142 GPMVs are giant vesicles directly isolated from cells. As such they provide the lipid and protein complexity of intact cell plasma membranes together with the cytoplasm inside. The reported molecular factories are the first example of functional artificial cells with the outer membrane complexity of a cell, internal cytoplasm and synthetic artificial organelles (enzyme filled polymersomes equipped with channel porins) that are functional even under in vivo conditions in a zebrafish vertebrate animal model without showing toxicity.
Conclusion and perspectives
The examples given in this review demonstrate the great progress in the field of micro- or nano-sized biohybrid compartments. During the past decade, these systems advanced from simple polymeric vesicles encapsulating simply dyes as proof of principle or just one type of biomolecule to complex multifunctional and even multicompartmentalized biohybrid materials ready for medical applications. Most systems exploit the chemical robustness of synthetic polymers for protection of their delicate biomolecular cargos and use the polymers’ chemical tunability to adapt their properties for the desired application and functionalization. A great variety of techniques are nowadays available for vesicle or polymeric capsule production at the micro- and nanoscale and the controlled incorporation of biomolecular cargos.It was shown for all classes of biomolecules that their intrinsic functionality can be preserved and specifically exploited in such biohybrid compartments: transmembrane proteins introduce selective permeability to polymeric vesicles, lipids tune their biophysical properties for improved biocompatibility, peptide functionalization allows selective targeting while encapsulation of enzymes supports spatially confined catalytic activity.
Research in such biohybrid compartments is motivated on one hand by direct applications in drug delivery of e.g. encapsulated proteins, nucleic acids or hydrophobic molecules entrapped within the membrane. On the other hand, it is driven by the fundamental interest in advancing biohybrids to artificial cell mimics. Several principles like the compartmentalized structure, confined enzymatic catalysis or simple cellular signal transduction and biological permeabilization strategies have been successfully implemented in biohybrid compartments.
However, selective permeabilization and responsiveness to specific stimuli are still under intense investigation as for instance the reconstitution of membrane proteins is not fully understood and cannot be generally achieved yet. Further studies of the required membrane properties, with the help of lipid/polymer biohybrid membranes, will promote that field. Furthermore, improvements in the encapsulation efficiency of biomacromolecular cargos inside the polymeric compartments would have a great impact on drug delivery applications as well as on nano/microreactors exploiting the catalytic activity of enzymes. Similar strategies are also required for the controlled assembly of multicompartmentalized biohybrids if different types of subunits in precise amounts and ratios should be used. Finally, such complex biohybrid systems still lack extensive investigations of their fate in contact with a real biological environment, such as living tissues or organisms. In this respect, also a tuneable biodegradability would be favourable. All in all, there is still plenty of research needed before fully functional cell models or complex artificial organelles with therapeutic relevance can be realized.
Abbreviations
Polymers
| BA | Butyl acrylate |
| CMA | 4-Methyl-[7-methacryloyl) oxyethyloxy] coumarin |
| DEA | 2-(Diethylamino)ethyl methacrylate |
| DEGMA | (Diethylene glycol) methyl ether methacrylate |
| DMAEMA | Dimethylaminoethyl acrylate |
| PDTC/DTC | Poly(2,2-dimethyltrimethylene carbonate) |
| P(ASP) | Poly(α,β-aspartic acid) |
| P(Asp-AP) | Poly[(5-aminopentyl)-α,β-aspartamide] |
| PAA | Poly(acrylic acid) |
| PAH | Polyallylamine |
| PB | Poly(butadiene) |
| PBMA | Poly(butyl methacrylate) |
| PCL | Poly(caprolactone) |
| PCMA | Poly(2-cinnamoylethyl methacrylate) |
| PDPA | Poly(2-(diisopropylamino)ethyl methacrylate) |
| PDEAEMA | Poly(2-(diethylamino)ethyl methacrylate) |
| PDMS | Poly(dimethylsiloxane) |
| PCL | Poly(caprolactone) |
| PEP | Isopropyl ethylene phosphate |
| PEG/PEO | Poly(ethylene glycol)/poly(ethylene oxide) |
| PEOGA | Poly(oligoethylene glycol acrylate) |
| PGA | Poly(glutamic acid) |
| PGG | Poly(L-γ-glutamyl-glutamine) |
| PHPMA | Poly(2-hydroxypropyl methacrylate) |
| PI | Poly(isoprene) |
| PIAT | Polyisocyanoalanine(2-thiophene-3-yl-ethyl)amide |
| PLA | Polylactic acid |
| PLG | Poly(L-glutamate) |
| PLGA | Poly(lactic-glycolic acid) |
| PLL | Poly(L-lysine) |
| PMA | Poly(methacrylic acid) |
| PMOXA | Poly(2-methyloxazoline) |
| PMPC | Poly(2-(methacryloyloxy)ethyl phosphorylcholine) |
| PNIPAM | Poly(N-isopropylacrylamide) |
| POEGMA | Poly[2-(2-methoxyethoxy)ethyl methacrylate-co-oligo (ethylene glycol)methyl ether methacrylate] |
| PS | Polystyrene |
| PSS | Poly(styrene-4-sulfonic acid) |
| PTMC/TMC | Poly(trimethylene carbonate) |
| PVP | Poly(vinylpyrrolidone) |
| PVPON | poly(N-vinyl pyrrolidone) |
Lipids
| DOPC | 1,2-Dioleoyl-sn-glycero-3-phosphocholine |
| DOPS | 1,2-Dioleoyl-sn-glycero-3-phospho-L-serine |
| DMPC | 1,2-Dimyristoyl-sn-glycero-3-phosphocholine |
| DPPC | 1,2-Dipalmitoyl-sn-glycero-3-phosphocholine |
| DPPS | 1,2-Dipalmitoyl-sn-glycero-3-phosphoserine |
| HSPC | Hydrogenated soybean phosphatidylcholine |
| PC | Phosphatidylcholines |
| PE | Phosphatidylethanolamine |
| POPC | 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine |
Proteins and Peptides
| ADH | Alcohol dehydrogenase |
| Alc | Alcalase |
| ALP | Alkaline phosphatase |
| ApoE | Apolipoprotein E |
| Aqp Z | Aquaporin Z |
| ASNase | Asparaginase |
| BR | Bacteriorhodopsin |
| BSA | Bovine serum albumin |
| CalB | Lipase B from Candida antarctica |
| Cat | Catalase |
| cRGD | CyclicRGD |
| cytC | Cytochrome c |
| gA | Gramicidin A |
| GALA | Glutamic acid-alanine-leucine-alanine |
| GFP | Green fluorescent protein |
| GOx | Glucose oxidase |
| GR | Glutathione reductase |
| HAS | Human serum albumin |
| Hb | Hemoglobin |
| HRP | Horseradish peroxidase |
| iRGD | InternalizingRGD |
| LPO | Lactoperoxidase |
| LPL | Lipoprotein lipase |
| Mb | Myoglobin |
| NLS | Nuclear localization sequence |
| OmpF | Outer membrane protein F |
| PAL | Phenylalanine ammonia lyase |
| PAMO | Phenylacetone Monooxygenase |
| PGM | Phosphoglucomutase |
| POD | Peroxidase |
| RNApol | RNA polymerase |
| SOD | Superoxide dismutase |
| TRP | Trypsin |
| TYR | Tyrosinase |
| α-HL | α-Haemolysin |
| β-gal | β-Galactosidase |
| β-lac | β-Lactamase |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support provided by the Swiss National Science Foundation, the Swiss Nanoscience Institute, the University of Basel and the National Centre of Competence in Research-Molecular Systems Engineering (NCCR-MSE).Notes and references
- L. Zhang and A. Eisenberg, Science, 1995, 268, 1728 CrossRef CAS PubMed.
- J. Ding and G. Liu, J. Phys. Chem. B, 1998, 102, 6107–6113 CrossRef CAS.
- B. M. Discher, Y.-Y. Won, D. S. Ege, J. C. M. Lee, F. S. Bates, D. E. Discher and D. A. Hammer, Science, 1999, 284, 1143 CrossRef CAS PubMed.
- M. Garni, R. Wehr, S. Y. Avsar, C. John, C. Palivan and W. Meier, Eur. Polym. J., 2019, 112, 346–364 CrossRef CAS.
- H. Feng, X. Lu, W. Wang, N. G. Kang and J. W. Mays, Polymers, 2017, 9, 494 CrossRef PubMed.
- F. Itel, M. Chami, A. Najer, S. Lörcher, D. Wu, I. A. Dinu and W. Meier, Macromolecules, 2014, 47, 7588–7596 CrossRef CAS.
- R. J. Peters, M. Marguet, S. Marais, M. W. Fraaije, J. C. van Hest and S. Lecommandoux, Angew. Chem., Int. Ed., 2014, 53, 146–150 CrossRef CAS PubMed.
- C. LoPresti, H. Lomas, M. Massignani, T. Smart and G. Battaglia, J. Mater. Chem., 2009, 19, 3576–3590 RSC.
- C. LoPresti, M. Massignani, C. Fernyhough, A. Blanazs, A. J. Ryan, J. Madsen, N. J. Warren, S. P. Armes, A. L. Lewis, S. Chirasatitsin, A. J. Engler and G. Battaglia, ACS Nano, 2011, 5, 1775–1784 CrossRef CAS PubMed.
- E. V. Konishcheva, U. E. Zhumaev and W. P. Meier, Macromolecules, 2017, 50, 1512–1520 CrossRef CAS.
- D. E. Discher and F. Ahmed, Annu. Rev. Biomed. Eng., 2006, 8, 323–341 CrossRef CAS PubMed.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef CAS PubMed.
- M. Dionzou, A. Morère, C. Roux, B. Lonetti, J. D. Marty, C. Mingotaud, P. Joseph, D. Goudounèche, B. Payré, M. Léonetti and A. F. Mingotaud, Soft Matter, 2016, 12, 2166–2176 RSC.
- D. Wu, M. Spulber, F. Itel, M. Chami, T. Pfohl, C. G. Palivan and W. Meier, Macromolecules, 2014, 47, 5060–5069 CrossRef CAS.
- G. Battaglia and A. J. Ryan, J. Phys. Chem. B, 2006, 110, 10272–10279 CrossRef CAS PubMed.
- A. Peyret, E. Ibarboure, J. F. Le Meins and S. Lecommandoux, Adv. Sci., 2018, 5, 1700453 CrossRef PubMed.
- A. Peyret, E. Ibarboure, N. Pippa and S. Lecommandoux, Langmuir, 2017, 33, 7079–7085 CrossRef CAS PubMed.
- E. Rideau, R. Dimova, P. Schwille, F. R. Wurm and K. Landfester, Chem. Soc. Rev., 2018, 47, 8572–8610 RSC.
- A. Kishimura, Polym. J., 2013, 45, 892–897 CrossRef CAS.
- Y. Anraku, A. Kishimura, Y. Yamasaki and K. Kataoka, J. Am. Chem. Soc., 2013, 135, 1423–1429 CrossRef CAS PubMed.
- Y. Anraku, A. Kishimura, M. Oba, Y. Yamasaki and K. Kataoka, J. Am. Chem. Soc., 2010, 132, 1631–1636 CrossRef CAS PubMed.
- L. L. del Mercato, P. Rivera-Gil, A. Z. Abbasi, M. Ochs, C. Ganas, I. Zins, C. Sönnichsen and W. J. Parak, Nanoscale, 2010, 2, 458–467 RSC.
- O. Kulygin, A. D. Price, S.-F. Chong, B. Städler, A. N. Zelikin and F. Caruso, Small, 2010, 6, 1558–1564 CrossRef CAS PubMed.
- L. Hosta-Rigau, M. J. York-Duran, Y. Zhang, K. N. Goldie and B. Stadler, ACS Appl. Mater. Interfaces, 2014, 6, 12771–12779 CrossRef CAS PubMed.
- G. Battaglia, C. LoPresti, M. Massignani, N. J. Warren, J. Madsen, S. Forster, C. Vasilev, J. K. Hobbs, S. P. Armes, S. Chirasatitsin and A. J. Engler, Small, 2011, 7, 2010–2015 CrossRef CAS PubMed.
- K. Jaskiewicz, M. Makowski, M. Kappl, K. Landfester and A. Kroeger, Langmuir, 2012, 28, 12629–12636 CrossRef CAS PubMed.
- A. Aloi and I. K. Voets, Curr. Opin. Colloid Interface Sci., 2018, 34, 59–73 CrossRef CAS.
- S. Pujals, N. Feiner-Gracia, P. Delcanale, I. Voets and L. Albertazzi, Nat. Rev. Chem., 2019, 3, 68–84 CrossRef.
- S. J. Roobol, T. A. Hartjes, J. A. Slotman, R. M. de Kruijff, G. Torrelo, T. E. Abraham, F. Bruchertseifer, A. Morgenstern, R. Kanaar, D. C. van Gent, A. B. Houtsmuller, A. G. Denkova, M. E. van Royen and J. Essers, Nanotheranostics, 2020, 4, 14–25 CrossRef PubMed.
- J. L. Davis, Y. Zhang, S. Yi, F. Du, K.-H. Song, E. A. Scott, C. Sun and H. F. Zhang, Langmuir, 2020, 36, 2291–2299 CrossRef CAS PubMed.
- A. Boreham, P. Volz, D. Peters, C. M. Keck and U. Alexiev, Eur. J. Pharm. Biopharm., 2017, 110, 31–38 CrossRef CAS PubMed.
- L.-H. Dieu, D. Wu, C. G. Palivan, V. Balasubramanian and J. Huwyler, Eur. J. Pharm. Biopharm., 2014, 88, 316–324 CrossRef CAS PubMed.
- F. Itel, A. Najer, C. G. Palivan and W. Meier, Nano Lett., 2015, 15, 3871–3878 CrossRef CAS PubMed.
- K. Kiene, S. H. Schenk, F. Porta, A. Ernst, D. Witzigmann, P. Grossen and J. Huwyler, Eur. J. Pharm. Biopharm., 2017, 119, 322–332 CrossRef CAS PubMed.
- M. Lomora, M. Garni, F. Itel, P. Tanner, M. Spulber and C. G. Palivan, Biomaterials, 2015, 53, 406–414 CrossRef CAS PubMed.
- J. Liu, V. Postupalenko, S. Lörcher, D. Wu, M. Chami, W. Meier and C. G. Palivan, Nano Lett., 2016, 16, 7128–7136 CrossRef CAS PubMed.
- E. Di Cola, I. Grillo and S. Ristori, Pharmaceutics, 2016, 8, 10 CrossRef PubMed.
- L. Ruiz-Pérez, L. Messager, J. Gaitzsch, A. Joseph, L. Sutto, F. L. Gervasio and G. Battaglia, Sci. Adv., 2016, 2, e1500948 CrossRef PubMed.
- J. Habel, A. Ogbonna, N. Larsen, S. Cherré, S. Kynde, S. R. Midtgaard, K. Kinoshita, S. Krabbe, G. V. Jensen, J. S. Hansen, K. Almdal and C. Hèlix-Nielsen, RSC Adv., 2015, 5, 79924–79946 RSC.
- E. Scarpa, J. L. Bailey, A. A. Janeczek, P. S. Stumpf, A. H. Johnston, R. O. C. Oreffo, Y. L. Woo, Y. C. Cheong, N. D. Evans and T. A. Newman, Sci. Rep., 2016, 6, 29460 CrossRef PubMed.
- H.-Y. Chang, Y.-J. Sheng and H.-K. Tsao, Soft Matter, 2014, 10, 6373–6381 RSC.
- V. Ortiz, S. O. Nielsen, D. E. Discher, M. L. Klein, R. Lipowsky and J. Shillcock, J. Phys. Chem. B, 2005, 109, 17708–17714 CrossRef CAS PubMed.
- L. Pourtau, H. Oliveira, J. Thevenot, Y. Wan, A. R. Brisson, O. Sandre, S. Miraux, E. Thiaudiere and S. Lecommandoux, Adv. Healthcare Mater., 2013, 2, 1420–1424 CrossRef CAS PubMed.
- J. Chen, Q. Liu, J. Xiao and J. Du, Biomacromolecules, 2015, 16, 1695–1705 CrossRef CAS PubMed.
- P. Kulkarni, M. K. Haldar, F. Karandish, M. Confeld, R. Hossain, P. Borowicz, K. Gange, L. Xia, K. Sarkar and S. Mallik, Chem. – Eur. J., 2018, 24, 12490–12494 CrossRef CAS PubMed.
- P. Yao, Y. Zhang, H. Meng, H. Sun and Z. Zhong, Biomacromolecules, 2019, 20, 184–191 CrossRef CAS PubMed.
- Y. Zou, M. Zheng, W. Yang, F. Meng, K. Miyata, H. J. Kim, K. Kataoka and Z. Zhong, Adv. Mater., 2017, 29, 1703285 CrossRef PubMed.
- F. Karandish, J. Froberg, P. Borowicz, J. C. Wilkinson, Y. Choi and S. Mallik, Colloids Surf., B, 2018, 163, 225–235 CrossRef CAS PubMed.
- L. Simón-Gracia, P. Scodeller, S. S. Fuentes, V. G. Vallejo, X. Ríos, E. S. Sebastián, D. D. Silvio, M. Suck, F. D. Lorenzi, L. Y. Rizzo, S. V. Stillfried, K. Kilk, T. Lammers, S. E. Moya and T. Teesalu, Oncotarget, 2018, 9, 18682–18697 CrossRef PubMed.
- Y. Fang, W. Yang, L. Cheng, F. Meng, J. Zhang and Z. Zhong, Acta Biomater., 2017, 64, 323–333 CrossRef CAS PubMed.
- T. Anajafi, J. Yu, A. Sedigh, M. K. Haldar, W. W. Muhonen, S. Oberlander, H. Wasness, J. Froberg, M. S. Molla, K. S. Katti, Y. Choi, J. B. Shabb, D. K. Srivastava and S. Mallik, Mol. Pharmaceutics, 2017, 14, 1916–1928 CrossRef CAS PubMed.
- T. Jia, Z. Sun, Y. Lu, J. Gao, H. Zou, F. Xie, G. Zhang, H. Xu, D. Sun, Y. Yu and Y. Zhong, Int. J. Nanomed., 2016, 11, 3765–3775 CrossRef CAS PubMed.
- Y. Jiang, J. Zhang, F. Meng and Z. Zhong, ACS Nano, 2018, 12, 11070–11079 CrossRef CAS PubMed.
- C. Zelmer, L. P. Zweifel, L. E. Kapinos, I. Craciun, Z. P. Güven, C. G. Palivan and R. Y. H. Lim, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 2770–2778 CrossRef CAS PubMed.
- W. Yang, Y. Xia, Y. Fang, F. Meng, J. Zhang, R. Cheng, C. Deng and Z. Zhong, Adv. Healthcare Mater., 2018, 7, 1701135 CrossRef PubMed.
- Y. Wei, X. Gu, L. Cheng, F. Meng, G. Storm and Z. Zhong, Acta Biomater., 2019, 92, 196–204 CrossRef CAS PubMed.
- Y. Yu, X. Jiang, S. Gong, L. Feng, Y. Zhong and Z. Pang, Nanoscale, 2014, 6, 3250–3258 RSC.
- Y. Xiao, H. Sun and J. Du, J. Am. Chem. Soc., 2017, 139, 7640–7647 CrossRef CAS PubMed.
- L. Klermund, S. T. Poschenrieder and K. Castiglione, J. Nanobiotechnol., 2016, 14, 48 CrossRef PubMed.
- M. Alibolandi, M. Ramezani, K. Abnous and F. Hadizadeh, J. Pharm. Sci., 2016, 105, 1741–1750 CrossRef CAS PubMed.
- X. Li, X. Zhu and L. Qiu, Acta Biomater., 2016, 35, 269–279 CrossRef CAS PubMed.
- H. Lomas, J. Du, I. Canton, J. Madsen, N. Warren, S. P. Armes, A. L. Lewis and G. Battaglia, Macromol. Biosci., 2010, 10, 513–530 CrossRef CAS PubMed.
- F. Wang, J. Gao, J. Xiao and J. Du, Nano Lett., 2018, 18, 5562–5568 CrossRef CAS PubMed.
- A. Belluati, I. Craciun, C. E. Meyer, S. Rigo and C. G. Palivan, Curr. Opin. Biotechnol., 2019, 60, 53–62 CrossRef CAS PubMed.
- F. Axthelm, O. Casse, W. H. Koppenol, T. Nauser, W. Meier and C. G. Palivan, J. Phys. Chem. B, 2008, 112, 8211–8217 CrossRef CAS PubMed.
- D. Dobrunz, A. C. Toma, P. Tanner, T. Pfohl and C. G. Palivan, Langmuir, 2012, 28, 15889–15899 CrossRef CAS PubMed.
- A. Belluati, I. Craciun, J. Liu and C. G. Palivan, Biomacromolecules, 2018, 19, 4023–4033 CrossRef CAS PubMed.
- D. Hvasanov, J. R. Peterson and P. Thordarson, Chem. Sci., 2013, 4, 3833–3838 RSC.
- P. Tanner, V. Balasubramanian and C. G. Palivan, Nano Lett., 2013, 13, 2875–2883 CrossRef CAS PubMed.
- K. Langowska, J. Kowal, C. G. Palivan and W. Meier, J. Mater. Chem. B, 2014, 2, 4684–4693 RSC.
- C. G. Palivan, R. Goers, A. Najer, X. Zhang, A. Car and W. Meier, Chem. Soc. Rev., 2016, 45, 377–411 RSC.
- S. F. M. van Dongen, M. Nallani, J. J. L. M. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, Chem. – Eur. J., 2009, 15, 1107–1114 CrossRef CAS PubMed.
- T. Nishimura, Y. Sasaki and K. Akiyoshi, Adv. Mater., 2017, 29, 1702406 CrossRef PubMed.
- Q. Chen, H. Schönherr and G. J. Vancso, Small, 2009, 5, 1436–1445 CrossRef CAS PubMed.
- W. Siti, H.-P. M. de Hoog, O. Fischer, W. Y. Shan, N. Tomczak, M. Nallani and B. Liedberg, J. Mater. Chem. B, 2014, 2, 2733–2737 RSC.
- P. Baumann, M. Spulber, O. Fischer, A. Car and W. Meier, Small, 2017, 13, 1603943 CrossRef PubMed.
- H.-O. Kim, J.-W. Lim, J. Choi, H. Lee, H. Y. Son, J. Kim, G. Park, H. Chun, D. Song, Y.-M. Huh and S. Haam, J. Mater. Chem. B, 2017, 5, 9571–9578 RSC.
- Z. Liu, C. Dong, X. Wang, H. Wang, W. Li, J. Tan and J. Chang, ACS Appl. Mater. Interfaces, 2014, 6, 2393–2400 CrossRef CAS PubMed.
- X. Liu and W. Gao, ACS Appl. Mater. Interfaces, 2017, 9, 2023–2028 CrossRef CAS PubMed.
- I. C. Reynhout, J. J. L. M. Cornelissen and R. J. M. Nolte, J. Am. Chem. Soc., 2007, 129, 2327–2332 CrossRef CAS PubMed.
- T. Einfalt, R. Goers, I. A. Dinu, A. Najer, M. Spulber, O. Onaca-Fischer and C. G. Palivan, Nano Lett., 2015, 15, 7596–7603 CrossRef CAS PubMed.
- C. Edlinger, T. Einfalt, M. Spulber, A. Car, W. Meier and C. G. Palivan, Nano Lett., 2017, 17, 5790–5798 CrossRef CAS PubMed.
- R. Stoenescu, A. Graff and W. Meier, Macromol. Biosci., 2004, 4, 930–935 CrossRef CAS PubMed.
- M. Lomora, G. Gunkel-Grabole, S. Mantri and C. G. Palivan, Chem. Commun., 2017, 53, 10148–10151 RSC.
- H.-J. Choi, J. Germain and C. D. Montemagno, Nanotechnology, 2006, 17, 1825–1830 CrossRef CAS.
- M. Kumar, M. Grzelakowski, J. Zilles, M. Clark and W. Meier, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20719–20724 CrossRef CAS PubMed.
- R. Goers, J. Thoma, N. Ritzmann, A. Di Silvestro, C. Alter, G. Gunkel-Grabole, D. Fotiadis, D. J. Müller and W. Meier, Commun. Chem., 2018, 1, 35 CrossRef.
- L. You and H. Schlaad, J. Am. Chem. Soc., 2006, 128, 13336–13337 CrossRef CAS PubMed.
- G. Pasparakis and C. Alexander, Angew. Chem., Int. Ed., 2008, 47, 4847–4850 CrossRef CAS PubMed.
- S. Haas, N. Hain, M. Raoufi, S. Handschuh-Wang, T. Wang, X. Jiang and H. Schönherr, Biomacromolecules, 2015, 16, 832–841 CrossRef CAS PubMed.
- L. Su, Y. Zhao, G. Chen and M. Jiang, Polym. Chem., 2012, 3, 1560–1566 RSC.
- K. Panneerselvam, M. E. Lynge, C. F. Riber, S. Mena-Hernando, A. A. A. Smith, K. N. Goldie, A. N. Zelikin and B. Städler, Biomicrofluidics, 2015, 9, 052610 CrossRef PubMed.
- W. Zong, Y. Hu, Y. Su, N. Luo, X. Zhang, Q. Li and X. Han, J. Microencapsulation, 2016, 33, 257–262 CrossRef CAS PubMed.
- W. Zong, B. Thingholm, F. Itel, P. S. Schattling, E. Brodszkij, D. Mayer, S. Stenger, K. N. Goldie, X. Han and B. Städler, Langmuir, 2018, 34, 6874–6886 CrossRef CAS PubMed.
- J. Nam, T. Kyle Vanderlick and P. A. Beales, Soft Matter, 2012, 8, 7982–7988 RSC.
- M. Schulz and W. H. Binder, Macromol. Rapid Commun., 2015, 36, 2031–2041 CrossRef CAS PubMed.
- M. Chemin, P.-M. Brun, S. Lecommandoux, O. Sandre and J.-F. L. Meins, Soft Matter, 2012, 8, 2867–2874 RSC.
- S. K. Lim, H.-P. De Hoog, A. N. Parikh, M. Nallani and B. Liedberg, Polymers, 2013, 5, 1102–1114 CrossRef.
- S. Khan, J. McCabe, K. Hill and P. A. Beales, J. Colloid Interface Sci., 2020, 562, 418–428 CrossRef CAS PubMed.
- W. F. Paxton, P. T. McAninch, K. E. Achyuthan, S. H. R. Shin and H. L. Monteith, Colloids Surf., B, 2017, 159, 268–276 CrossRef CAS PubMed.
- S. Khan, M. Li, S. P. Muench, L. J. C. Jeuken and P. A. Beales, Chem. Commun., 2016, 52, 11020–11023 RSC.
- Z. Cheng, D. R. Elias, N. P. Kamat, E. D. Johnston, A. Poloukhtine, V. Popik, D. A. Hammer and A. Tsourkas, Bioconjugate Chem., 2011, 22, 2021–2029 CrossRef CAS PubMed.
- N. Pippa, D. Stellas, A. Skandalis, S. Pispas, C. Demetzos, M. Libera, A. Marcinkowski and B. Trzebicka, Eur. J. Pharm. Biopharm., 2016, 107, 295–309 CrossRef CAS PubMed.
- W. Kawamura, Y. Miura, D. Kokuryo, K. Toh, N. Yamada, T. Nomoto, Y. Matsumoto, D. Sueyoshi, X. Liu, I. Aoki, M. R. Kano, N. Nishiyama, T. Saga, A. Kishimura and K. Kataoka, Sci. Technol. Adv. Mater., 2015, 16, 035004 CrossRef PubMed.
- D. Sueyoshi, Y. Anraku, T. Komatsu, Y. Urano and K. Kataoka, Biomacromolecules, 2017, 18, 1189–1196 CrossRef CAS PubMed.
- J. Richardson, M. Choy, J. Guo, K. Liang, K. Alt, Y. Ping, J. Cui, L. S. Law, C. Hagemeyer and F. Caruso, Adv. Mater., 2016, 28, 7703–7707 CrossRef CAS PubMed.
- H. He, Y. Lu, J. Qi, Q. Zhu, Z. Chen and W. Wu, Acta Pharm. Sin. B, 2019, 9, 36–48 CrossRef PubMed.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS PubMed.
- T. P. T. Dao, F. Fernandes, M. Fauquignon, E. Ibarboure, M. Prieto and J. F. Le Meins, Soft Matter, 2018, 14, 6476–6484 RSC.
- J. Y. Kang, I. Choi, M. Seo, J. Y. Lee, S. Hong, G. Gong, S. S. Shin, Y. Lee and J. W. Kim, J. Colloid Interface Sci., 2020, 561, 318–326 CrossRef CAS PubMed.
- M. Fauquignon, E. Ibarboure, S. Carlotti, A. Brulet, M. Schmutz and J. F. Le Meins, Polymers, 2019, 11, 2013 CrossRef CAS PubMed.
- T. P. Dao, F. Fernandes, E. Ibarboure, K. Ferji, M. Prieto, O. Sandre and J. F. Le Meins, Soft Matter, 2017, 13, 627–637 RSC.
- J. F. Le Meins, C. Schatz, S. Lecommandoux and O. Sandre, Mater. Today, 2013, 16, 397–402 CrossRef CAS.
- S.-W. Hu, C.-Y. Huang, H.-K. Tsao and Y.-J. Sheng, Phys. Rev. E, 2019, 99, 012403 CrossRef CAS PubMed.
- C. Magnani, C. Montis, G. Mangiapia, A. F. Mingotaud, C. Mingotaud, C. Roux, P. Joseph, D. Berti and B. Lonetti, Colloids Surf., B, 2018, 168, 18–28 CrossRef CAS PubMed.
- R. J. Linhardt and T. Toida, Acc. Chem. Res., 2004, 37, 431–438 CrossRef CAS PubMed.
- S. Thamboo, A. Najer, A. Belluati, C. von Planta, D. Wu, I. Craciun, W. Meier and C. G. Palivan, Adv. Funct. Mater., 2019, 29, 1904267 CrossRef.
- L. Martin, P. Gurnani, J. Zhang, M. Hartlieb, N. R. Cameron, A. M. Eissa and S. Perrier, Biomacromolecules, 2019, 20, 1297–1307 CrossRef CAS PubMed.
- A. Kubilis, A. Abdulkarim, A. M. Eissa and N. R. Cameron, Sci. Rep., 2016, 6, 32414 CrossRef CAS PubMed.
- A. Najer, D. Wu, A. Bieri, F. Brand, C. G. Palivan, H.-P. Beck and W. Meier, ACS Nano, 2014, 8, 12560–12571 CrossRef CAS PubMed.
- B. Pandey, J. Mahato, K. B. Cotta, S. Das, D. K. Sharma, S. Sen Gupta and A. Chowdhury, ACS Omega, 2016, 1, 600–612 CrossRef CAS PubMed.
- P. A. Beales, S. Khan, S. P. Muench and L. J. Jeuken, Biochem. Soc. Trans., 2017, 45, 15–26 CrossRef CAS PubMed.
- M. Sauer, T. Haefele, A. Graff, C. Nardin and W. Meier, Chem. Commun., 2001, 2452–2453, 10.1039/B107833J.
- S. Semrau and T. Schmidt, Soft Matter, 2009, 5, 3174–3186 RSC.
- A. Belluati, V. Mikhalevich, S. Yorulmaz Avsar, D. Daubian, I. Craciun, M. Chami, W. P. Meier and C. G. Palivan, Biomacromolecules, 2020, 21, 701–715 CrossRef CAS PubMed.
- M. Bieligmeyer, F. Artukovic, S. Nussberger, T. Hirth, T. Schiestel and M. Müller, Beilstein J. Nanotechnol., 2016, 7, 881–892 CrossRef CAS PubMed.
- M. Garni, T. Einfalt, R. Goers, C. G. Palivan and W. Meier, ACS Synth. Biol., 2018, 7, 2116–2125 CrossRef CAS PubMed.
- A. Kishimura, A. Koide, K. Osada, Y. Yamasaki and K. Kataoka, Angew. Chem., Int. Ed., 2007, 46, 6085–6088 CrossRef CAS PubMed.
- W.-S. Jang, S. C. Park, E. H. Reed, K. P. Dooley, S. F. Wheeler, D. Lee and D. A. Hammer, Soft Matter, 2016, 12, 1014–1020 RSC.
- C. Martino, S.-H. Kim, L. Horsfall, A. Abbaspourrad, S. J. Rosser, J. Cooper and D. A. Weitz, Angew. Chem., Int. Ed., 2012, 51, 6416–6420 CrossRef CAS PubMed.
- J. Gaitzsch, X. Huang and B. Voit, Chem. Rev., 2016, 116, 1053–1093 CrossRef CAS PubMed.
- X. Huang, M. Li, D. C. Green, D. S. Williams, A. J. Patil and S. Mann, Nat. Commun., 2013, 4, 2239 CrossRef PubMed.
- X. Huang, A. J. Patil, M. Li and S. Mann, J. Am. Chem. Soc., 2014, 136, 9225–9234 CrossRef CAS PubMed.
- R. Kurapati, T. W. Groth and A. M. Raichur, ACS Appl. Bio Mater., 2019, 2, 5512–5527 CrossRef CAS.
- A. Larrañaga, M. Lomora, J. R. Sarasua, C. G. Palivan and A. Pandit, Prog. Mater. Sci., 2017, 90, 325–357 CrossRef.
- X. Liu, D. Appelhans and B. Voit, J. Am. Chem. Soc., 2018, 140, 16106–16114 CrossRef CAS PubMed.
- X. Huang, D. Appelhans, P. Formanek, F. Simon and B. Voit, ACS Nano, 2012, 6, 9718–9726 CrossRef CAS PubMed.
- O. S. Sakr and G. Borchard, Biomacromolecules, 2013, 14, 2117–2135 CrossRef CAS PubMed.
- E. T. Kisak, B. Coldren and J. A. Zasadzinski, Langmuir, 2002, 18, 284–288 CrossRef CAS.
- M. J. York-Duran, M. Godoy-Gallardo, C. Labay, A. J. Urquhart, T. L. Andresen and L. Hosta-Rigau, Colloids Surf., B, 2017, 152, 199–213 CrossRef CAS PubMed.
- T. Einfalt, M. Garni, D. Witzigmann, S. Sieber, N. Baltisberger, J. Huwyler, W. Meier and C. G. Palivan, Adv. Sci., 2020, 7, 1901923 CrossRef CAS PubMed.
- O. Kreft, M. Prevot, H. Mohwald and G. B. Sukhorukov, Angew. Chem., Int. Ed., 2007, 46, 5605–5608 CrossRef CAS PubMed.
- R. Chandrawati, L. Hosta-Rigau, D. Vanderstraaten, S. A. Lokuliyana, B. Städler, F. Albericio and F. Caruso, ACS Nano, 2010, 4, 1351–1361 CrossRef CAS PubMed.
- H. Lomas, A. P. Johnston, G. K. Such, Z. Zhu, K. Liang, M. P. van Koeverden, S. Alongkornchotikul and F. Caruso, Small, 2011, 7, 2109–2119 CrossRef CAS PubMed.
- B. Stadler, R. Chandrawati, A. D. Price, S. F. Chong, K. Breheney, A. Postma, L. A. Connal, A. N. Zelikin and F. Caruso, Angew. Chem., Int. Ed., 2009, 48, 4359–4362 CrossRef CAS PubMed.
- L. Hosta-Rigau, S. F. Chung, A. Postma, R. Chandrawati, B. Stadler and F. Caruso, Adv. Mater., 2011, 23, 4082–4087 CrossRef CAS PubMed.
- P. Schattling, C. Dreier and B. Stadler, Soft Matter, 2015, 11, 5327–5335 RSC.
- L. Hosta-Rigau, B. Städler, Y. Yan, E. C. Nice, J. K. Heath, F. Albericio and F. Caruso, Adv. Funct. Mater., 2010, 20, 59–66 CrossRef CAS.
- R. Chandrawati, P. D. Odermatt, S. F. Chong, A. D. Price, B. Stadler and F. Caruso, Nano Lett., 2011, 11, 4958–4963 CrossRef CAS PubMed.
- L. Hosta-Rigau, M. J. York-Duran, T. S. Kang and B. Städler, Adv. Funct. Mater., 2015, 25, 3860–3869 CrossRef CAS.
- M. Godoy-Gallardo, C. Labay and L. Hosta-Rigau, ACS Appl. Mater. Interfaces, 2019, 11, 5862–5876 CrossRef CAS PubMed.
- B. Thingholm, P. Schattling, Y. Zhang and B. Stadler, Small, 2016, 12, 1806–1814 CrossRef CAS PubMed.
- M. Godoy-Gallardo, C. Labay, V. D. Trikalitis, P. J. Kempen, J. B. Larsen, T. L. Andresen and L. Hosta-Rigau, ACS Appl. Mater. Interfaces, 2017, 9, 15907–15921 CrossRef CAS PubMed.
- M. Godoy-Gallardo, C. Labay, M. M. Jansman, P. K. Ek and L. Hosta-Rigau, Adv. Healthcare Mater., 2017, 6, 1601190 CrossRef PubMed.
- Z. Fu, M. A. Ochsner, H. P. de Hoog, N. Tomczak and M. Nallani, Chem. Commun., 2011, 47, 2862–2864 RSC.
- D. McPhail, L. Tetley, C. Dufes and I. F. Uchegbu, Int. J. Pharm., 2000, 200, 73–86 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © the Owner Societies 2020 |