
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In cell Gd3+-based site-directed spin labeling and EPR spectroscopy of eGFP†
Svetlana
Kucher‡
a,
Sergej
Korneev
b,
Johann P.
Klare
 *a,
Daniel
Klose
*c and
Heinz-Jürgen
Steinhoff
*a
*a,
Daniel
Klose
*c and
Heinz-Jürgen
Steinhoff
*a
aDepartment of Physics, Osnabrück University, Barbarastr. 7, Osnabrück, Germany. E-mail: jklare@uni-osnabrueck.de; hsteinho@uni-osnabrueck.de
bDepartment of Biology, Osnabrück University, Barbarastr. 11, Osnabrück, Germany
cDepartment of Chemistry and Applied Biosciences, ETH Zurich, Vladimir-Prelog-Weg 2, Zurich, Switzerland. E-mail: daniel.klose@phys.chem.ethz.ch
First published on 1st June 2020
Abstract
Label-based functional studies of biomolecules in their native environment require labeling reactions inside living cells. In cell spin labeling using alkyne–azide click chemistry with a Gd3+-DOTAM-azide complex is shown to provide high spin label stability and narrow EPR lines for EPR spectroscopic detection of a spin labeled protein in living cells at ambient temperatures.
Information about the structure and dynamics of biomolecules is a prerequisite to understand their physiological function. The vast majority of structural data present to date has been obtained under highly artificial conditions, i.e. in crystals or in highly concentrated solutions and purified from “contaminants”. However, the complex cellular environment can have strong influence on the biomolecules’ behavior,1 which raises the requirement to develop techniques that allow obtaining structural data in cell. Techniques such as X-ray crystallography and cryoEM, which provide atomic resolution, meet their limitations in such complex environments and in cell NMR is currently growing, but still a very challenging tool.2,3 EPR spectroscopy together with site-directed spin-labeling (SDSL) is an alternative approach, which can give site-specific information on structure and dynamics of complex biomolecular systems independent of their size and homogeneity of the environment.4 (Time-resolved) continuous wave (cw) EPR measurements of the spin label dynamics can be performed at physiological temperatures, yielding insights into local structure and dynamics.5,6 Moreover, double-electron–electron resonance (DEER) spectroscopy has become a powerful tool for measurements of inter- or intramolecular spin distance distributions within a range of 1.5–10 nm, providing structural data with near-atomic resolution.7 Paramagnetic labeling has also found a broad application in NMR spectroscopy, offering a significant sensitivity enhancement.8
First attempts to study paramagnetically labeled biological systems in a cellular context have successfully been made by incorporation of spin labeled proteins into living cells,9 using either micro-injection into oocytes or by membrane penetration (via electroporation, hypotonic swelling or incubation).10–15 However, most of these applications are based on in vitro labeling, usually with cysteine specific labels followed by incorporation of the protein mostly into a different cell line and in higher than physiological concentrations. Alternatively, membrane proteins were labeled on the outer cell surface or recombinant proteins were directly expressed with spin-labeled unnatural amino acids (UAA).16,17
In order to label a specific protein at desired positions inside the cell with a spin label of interest, an orthogonal labeling approach is required due to the broad range of chemical functionalities present inside cells. Orthogonal labeling with the genetically encoded p-acetyl-L-phenylalanine was applied using a hydroxylamine reagent to generate a nitroxide side chain in vitro.18 However, reaction conditions do not allow in cell applications. Click chemistry – here an alkyne–azide cycloaddition (AAC) – is a highly selective labeling approach, which has been applied for labeling of proteins in, e.g., fluorescence studies.19,20 Both types of click reactions, the strain-promoted (SPAAC)21 and the copper-catalyzed (CuAAC)22,23 variant, were shown to be successfully applicable for SDSL with nitroxides in vitro.24,25 Recently, we demonstrated that the green fluorescent protein eGFP can be spin labeled with nitroxides inside living E. coli cells via click reaction with genetically encoded UAAs.26 However, in cell EPR detection is limited by the fast reduction of nitroxides.27
Gd3+ complexes, long known as MRI contrast agents, were introduced for SDSL of proteins and show an outstanding stability in a reducing environment.13,14 Successful ligation of a Gd3+ tag to p-azido-L-phenylalanine introduced into proteins using CuAAC has been shown, but so far only in vitro.9,28 Moreover, Gd3+ labels have been applied for distance measurements using pulse EPR at high magnetic fields.29,30
In this communication we show the applicability of in vitro and in cell click labeling using a newly synthesized Gd3+-based DOTAM-azide spin label and its direct in cell detection with continuous wave EPR at conventional X-band frequency (9–10 GHz) with high sensitivity at room temperature (RT). We use eGFP with an amber mutation at position 39 with strained-cyclic octanyl (SCO, Scheme 1A) because it has successfully been applied for the labeling with azido-functionalized nitroxides and FRET labels and allows precise concentration determination.26,31 The incorporation of UAAs containing azide groups for the labeling with alkyne-functionalized markers is possible as well, however, this is less efficient for in cell labeling due to azide reduction to amines.32–34 However, a lower degree of degradation was reported for mammalian cells than for bacteria making them promising for further applications.35
 | ||
| Scheme 1 (A) Structure of strained-cyclic octanyl (SCO). (B) Synthesis of DOTAM-azide. Reaction conditions: (a) NaN3, H2O, 70 °C, 3 days; (b) ClCOCH2Br, Et3N, CH2Cl2, 0 °C → RT, 15 h; (c) K2CO3, MeOH, 55 °C, 24 h. (C) The side chain of SCO after binding with Gd3+-DOTAM-azide (GDMA). | ||
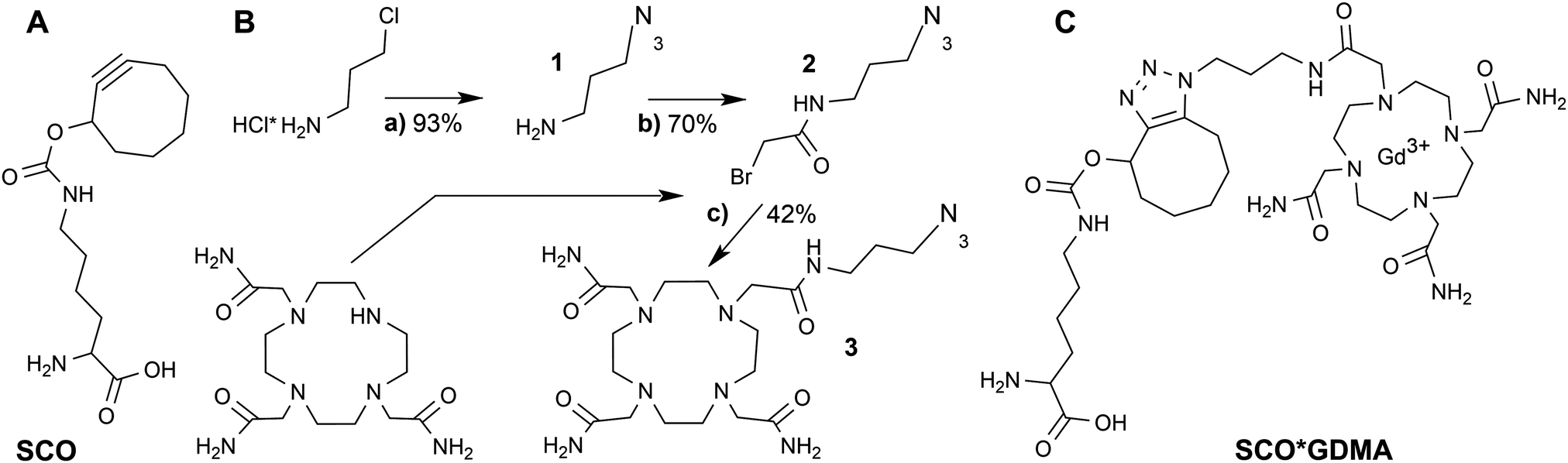
Gd3+ ions are high spin systems (S = 7/2) that exhibit large zero-field splitting (ZFS) leading to broad linewidths at conventional X-band frequencies (∼9–10 GHz), which become sharper with increasing frequency. However, the ZFS strongly depends on the chelator structure and symmetry.36 So far one of the smallest ZFS for Gd3+ complexes from commercially available chelators was reported for Gd3+-DOTAM (GDM) (∼560 MHz) exhibiting a narrow spectrum with a linewidth Γ of ∼2 mT at X-band (Fig. 1A) compared to ∼10 mT of the commonly used Gd3+-DOTA complex.36–38 This effect was explained by the only structural differences between the DOTA and DOTAM tags – substitution of the Gd-coordinating carboxyl groups by the less electronegative acetamides.37 The linewidth of protein bound Gd3+-DOTA derivatives has been previously shown to be correlated with dynamics at X-band in a similar way to nitroxides.39 However, its cw EPR detection is limited to the sub-millimolar concentration range due to the large ZFS. In contrast, the narrow linewidth of the GDM complex allows for cw EPR detection at X-band in the low μM concentration range providing a sensitivity comparable to nitroxides, thereby making this complex promising for in vitro and in cell EPR spectroscopic studies. Due to the lack of DOTAM functionalized as a labeling tag, we synthesized a DOTAM-azide derivative as described in the following, suitable for subsequent labeling of alkyne groups.
 | ||
| Fig. 1 (A) X-band EPR spectra (T = 293 K) of GDM (grey), GDMA free in solution (black) and bound to eGFP-Y39SCO (green) with Lorentzian fits (dotted lines). (B) Viscosity dependence of the inverse linewidth for GDM (grey) and GDMA (black). | ||
DOTAM-azide was synthesized in three steps from commercially available educts according to Scheme 1B. First, 3-chloropropylamine salt was converted into 3-azidopropylamine (1) in 93% yield by the exchange reaction with sodium azide as described.40 Next, the azide 1 was coupled to bromoacetyl chloride in the presence of Et3N to give N-(3-azidopropyl)-2-bromoacetamide (2) in 70% yield. Final condensation of the bromide 2 with 1,4,7,10-tetraazacyclododecane-1,4,7-triacetamide in the presence of K2CO3 at elevated temperature resulted in the formation of DOTAM-azide 3 that was isolated in 42% yield. Its structure was confirmed by 1H and 13C NMR spectroscopy and electrospray-ionization mass spectrometry (ESI MS, see Fig. S1, ESI†). A detailed description of the synthesis together with spectral data is provided in the SI1 (ESI†).
The room temperature cw EPR spectrum of DOTAM-azide (3 in Scheme 1B) after complexation with Gd3+ (GDMA) exhibits a peak-to-peak linewidth of 2.8 mT, which is only slightly broader than for the symmetric Gd3+-DOTAM (Fig. 1). EPR spectra of both, GDM and GDMA, recorded at different viscosities (see SI4, Fig. S3, ESI†) show that a decreasing motional averaging of the ZFS anisotropy is reflected in a considerable increase of the linewidth (Fig. 1B) proving its sensitivity to reorientational dynamics.
The EPR spectrum of the spin labeled eGFP-Y39SCO*GDMA (Fig. 1A) reveals a pronounced increase of the linewidth to 4.4 mT, as obtained from single Lorentzian fits, compared to the spectrum of the unbound label (2.8 mT) due to the spin label immobilization. This allows discrimination between bound and unbound spin labels and makes this spin label a promising candidate to probe reorientational motion (Fig. 1A, B and Fig. S3, ESI†) at ambient temperatures.
Surprisingly, while SPAAC of eGFP-Y39SCO with GDMA resulted in a high labeling efficiency of ∼85% (4 h at 37 °C, see ESI†), comparable with the efficiency previously achieved for nitroxides,26 the same did not hold true for CuAAC. CuAAC of eGFP-Y39PrK (PrK is abbreviation for propargyl-L-lysine) with GDMA did not result in fast and sufficient labeling as it has been reported for nitroxides and FRET labels (see SI3, Fig. S2, ESI†).26,41 Such a retardation of the usually faster click reaction variant might be caused by the interference of the positively charged spin label with the copper catalyst under the present experimental conditions. This makes CuAAC not well suited for in cell applications with Gd3+ labels and shows SPAAC to be the more efficient strategy even for a strained UAA with a low reaction rate.20
Since distance measurements are of general interest in EPR-aided structural biology, the applicability of the new side chain Y39SCO*GDMA for DEER is addressed in the following. For this purpose, eGFP-Y39SCO*GDMA was additionally labeled in vitro with the S-(1-oxyl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)methyl methanesulfonothionate spin label (MTSSL) at the outer cysteine position 48 and at the inner, less accessible, cysteine position 70, yielding side chains C48R1 and C70R1 (see Fig. 2A, for labeling details see SI3, ESI†) in a similar way as has been shown before.26
 | ||
| Fig. 2 eGFP-Y39SCO*GDMA-C48R1-C70R1 (A) Structure (PDB: 4EUL) with calculated rotamers of GDMA, position 39 (99.5% probability envelope in gray), and of R1, positions 48 and 70. (B) Q-band DEER data: experimental (black, grey) and calculated (RL, blue and MD, green) distance distributions for Y39SCO*GDMA-C48R1 sites. | ||
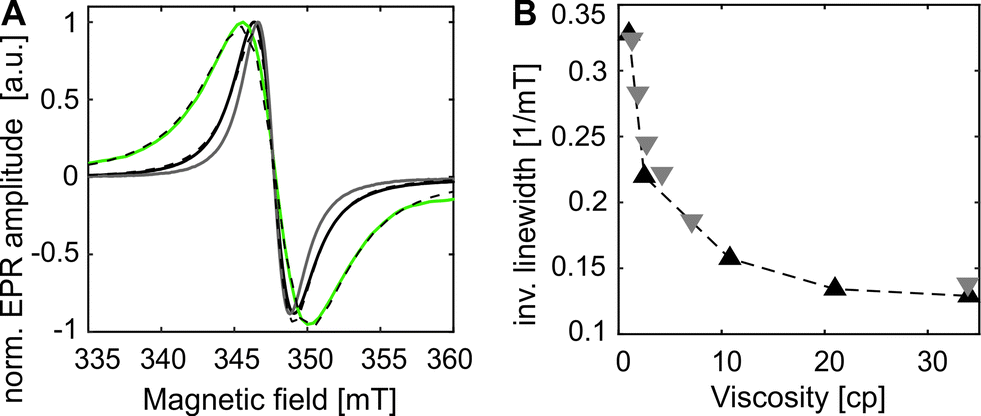
The distance distribution obtained from a Q-band DEER trace by a “user-unbiased” Neural network approach in DeerAnalysis201942,43 (Fig. 2B) shows agreement with the distribution obtained by Tikhonov regularization (Fig. S6 and S8, ESI†), both revealing a broad distance distribution with two main peaks at 3.5 and 4.3 nm and a smaller contribution at 2.3 nm. To compare the experimental results to the protein structure, we prepared a rotamer library (RL) for the SCO*GDMA side chain. 8192 rotamers were generated by hierarchical clustering of a 250k-membered Monte Carlo ensemble using a forgive factor of 0.8. The library is implemented in MMM44 versions 2019.1 and later as the spin label with code GM1 (see SI5 for details, ESI†). The distance distribution between C48R1 and Y39SCO*GDMA using the default MTSSL library of MMM2019.1 and the newly calculated one, respectively, fits well to the middle part of experimental distribution and the main peak at 3.5 nm (Fig. 2B, blue). Only one populated rotamer was obtained for both C48R1 and C70R1 (see Fig. 2A), which is in agreement with the observed low modulation depth in DEER as well as with the low experimental labeling efficiency for C70R1 (see SI3, ESI†). Thus, the large distribution width is mainly caused by the conformational flexibility of the long SCO*GDMA side chain. The presence of larger distances found in the experimental distribution can be attributed to the possible dynamics of the outer loop where C48R1 is located. To verify this assumption, MD simulations on eGFP were performed revealing additional possible conformations (see SI5 for details, ESI†). This led to additional rotamers for C48R1 resulting in the distance distribution shown in green in Fig. 2B, which covers the experimentally observed long distances. The small contribution at 2.3 nm in the experimental distribution likely arises from a less probable dipolar interaction between C70R1 and Y39SCO*GDMA (see Fig. S6, ESI†).26
For application of the SPAAC for labeling of eGFP inside E. coli cells we tested the biocompatibility of required reagents. Exposing E. coli cells to the Gd3+-DOTAM complexes revealed no detectable toxic effects at a wide range of concentrations (see SI7, ESI†). Furthermore, Gd3+ complexes were able to penetrate the cell membrane of E. coli cells during cell growth at 37 °C in a concentration and incubation time dependent manner without the need of applying external stress or additional procedures (see SI7, Fig. S10, ESI†). External millimolar spin label concentrations resulted in micromolar in cell concentrations. Moreover, no reduction in E. coli cell lysate was found for Gd3+-DOTAM complex after 10 h of incubation (see SI7, Fig. S11, ESI†), which is in line with the previous reports for other chelators.13–15
To test the label specificity, E. coli BL21 cells were incubated with GDMA spin label. Afterwards the medium was exchanged to remove extracellular labels and the cells were collected (see SI8 for details, ESI†). As shown in Fig. 3A the cells without eGFP overexpression exhibited a broadened signal originating from penetrated GDMA as expected due to the higher viscosity in cells (see Fig. 1B). The lysate of these control cells did not show any EPR signal after washing and concentrating using 10 kDa cutoff concentrators (Fig. 3B), which proves that the EPR signal of the control present in Fig. 3A originates from unbound GDMA only.
 | ||
| Fig. 3 (A) Amplitude-normalized EPR spectra of E. coli BL21 cells after incubation with GDMA at 37 °C without (grey) and with (green) overexpressed eGFP-Y39SCO. (B) The lysates of the samples shown in A were washed to remove free GDMA. Lorentzian fits are shown in dotted lines. | ||
For in cell labeling, E. coli BL21 cells expressing eGFP-Y39SCO were incubated with GDMA for 2 h at 37 °C (see SI8 for details, ESI†). The EPR spectrum of the cells shows a line broadening which is increased compared to the control (Fig. 3A). This is the evidence that labeling of eGFP-Y39SCO occurred inside the E. coli cells because bound GDMA exhibits a broader line (Fig. 1A) than free GDMA in both, buffer and cells. To further prove in cell spin labeling, the cells were lyzed and the lysate washed under conditions that restrain further binding reactions (see SI8, ESI†). Only for cells that had expressed eGFP, the resulting EPR spectrum was similar to that observed for eGFP-Y39SCO*GDMA obtained in vitro (Fig. 3B and 1A). The spin concentration of the bound spin label inside cells was calculated to be ∼9 μM, which is about 50% of the total GDMA concentration (∼18 μM) determined for intact cells before purification. This means that half of the GDMA present in the cells reacted with eGFP-Y39SCO. This is in reasonable agreement with the result of a fitting of two Lorentzians with fixed linewidths for free and bound GDMA to the EPR spectrum of E. coli cells with eGFP-Y39SCO incubated with GDMA, yielding an amplitude ratio for free![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :bound of 1.9:1.
:bound of 1.9:1.
Prolongation of the incubation time from 2 to 6 hours increased the labeling efficiency from 5 to ∼22% limited by the GDMA to protein ratio of 1:5 inside cells. For native protein concentrations below 20 μM the GDMA concentration present in the cell is not a limiting factor, and the achievable labeling efficiencies are expected to be higher. The observed reaction yields agree with previous reports for SCO as a strained UAA with a low reaction rate in SPAAC, whereas future utilization of other variants can accelerate the reaction rate by several orders of magnitudes.20
In summary, the SPAAC with a Gd3+-DOTAM-azide spin label is shown to proceed in living E. coli cells, thus demonstrating the feasibility of this reaction for in cell labeling of UAA-modified proteins using Gd3+-based spin labels. The newly synthesized Gd3+ spin label allowed in cell EPR detection, overcoming previous limits for in cell cw EPR due to reduction of nitroxides by the high chemical stability of the new Gd3+ spin label and its small line width. The observed increase in the (ambient temperature) cw EPR line width upon binding of the label to the protein opens the possibility for studying protein dynamics in cell. The method is shown to be applicable for (low temperature) inter spin distance measurements in vitro. Utilization of artificial amino acids functionalized with strained alkenes providing higher click reaction rates and short chain lengths will further enhance this applicability.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
H. J. S. & D. K. acknowledge funding from the DFG priority program SPP1601 (STE 640/12). The authors thank Prof. Edward A. Lemke for providing the cell strains for the expression of eGFP with unnatural amino acids and Prof. Gunnar Jeschke for support in designing the rotamer library as well as critical reading of the manuscript.References
- G. Rivas and A. P. Minton, Trends Biochem. Sci., 2016, 41, 970–981 CrossRef CAS PubMed.
- Z. Serber and V. Dötsch, Biochemistry, 2001, 40, 14317–14323 CrossRef CAS PubMed.
- C. Vénien-Bryan, Z. Li, L. Vuillard and J. A. Boutin, Acta Crystallogr., Sect. F: Struct. Biol. Commun., 2017, 73, 174–183 CrossRef PubMed.
- W. L. Hubbell, C. J. Lopez, C. Altenbach and Z. Yang, Curr. Opin. Struct. Biol., 2013, 23, 725–733 CrossRef CAS PubMed.
- M. Drescher, in EPR Spectroscopy: Applications in Chemistry and Biology, ed. M. Drescher, G. Jeschke and E. Bordignon, Springer, Heidelberg, 2012, pp. 91–119 Search PubMed.
- E. Bordignon, S. Kucher and Y. Polyhach, in Lipid-Protein Interactions, ed. J. H. Kleinschmidt, Springer, New York, 2019, vol. 2003, pp. 493–528 Search PubMed.
- G. Jeschke, Annu. Rev. Phys. Chem., 2012, 63, 419–446 CrossRef CAS PubMed.
- A. J. Pell, G. Pintacuda and C. P. Grey, Prog. Nucl. Magn. Reson. Spectrosc., 2019, 111, 1–271 CrossRef CAS PubMed.
- P. Roser, M. J. Schmidt, M. Drescher and D. Summerer, Org. Biomol. Chem., 2016, 14, 5468–5476 RSC.
- M. Azarkh, V. Singh, O. Okle, I. T. Seemann, D. R. Dietrich, J. S. Hartig and M. Drescher, Nat. Protoc., 2013, 8, 131–147 CrossRef CAS PubMed.
- S. Dunkel, L. P. Pulagam, H. J. Steinhoff and J. P. Klare, Phys. Chem. Chem. Phys., 2015, 17, 4875–4878 RSC.
- I. Krstic, R. Hansel, O. Romainczyk, J. W. Engels, V. Dotsch and T. F. Prisner, Angew. Chem., Int. Ed., 2011, 50, 5070–5074 CrossRef CAS PubMed.
- A. Martorana, G. Bellapadrona, A. Feintuch, E. Di Gregorio, S. Aime and D. Goldfarb, J. Am. Chem. Soc., 2014, 136, 13458–13465 CrossRef CAS PubMed.
- M. Qi, A. Gross, G. Jeschke, A. Godt and M. Drescher, J. Am. Chem. Soc., 2014, 136, 15366–15378 CrossRef CAS PubMed.
- F. X. Theillet, A. Binolfi, B. Bekei, A. Martorana, H. M. Rose, M. Stuiver, S. Verzini, D. Lorenz, M. van Rossum, D. Goldfarb and P. Selenko, Nature, 2016, 530, 45–50 CrossRef CAS PubMed.
- B. Joseph, A. Sikora, E. Bordignon, G. Jeschke, D. S. Cafiso and T. F. Prisner, Angew. Chem., Int. Ed., 2015, 54, 6196–6199 CrossRef CAS PubMed.
- M. J. Schmidt, J. Borbas, M. Drescher and D. Summerer, J. Am. Chem. Soc., 2014, 136, 1238–1241 CrossRef CAS PubMed.
- M. R. Fleissner, E. M. Brustad, T. Kalai, C. Altenbach, D. Cascio, F. B. Peters, K. Hideg, S. Peuker, P. G. Schultz and W. L. Hubbell, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21637–21642 CrossRef CAS PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- C. P. Ramil and Q. Lin, Bioorthogonal chemistry: strategies and recent developments, Chem. Commun., 2013, 49, 11007–11022 RSC.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- C. W. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef CAS PubMed.
- L. Garbuio, E. Bordignon, E. K. Brooks, W. L. Hubbell, G. Jeschke and M. Yulikov, J. Phys. Chem. B, 2013, 117, 3145–3153 CrossRef CAS PubMed.
- P. Widder, F. Berner, D. Summerer and M. Drescher, ACS Chem. Biol., 2019, 14, 839–844 CrossRef CAS PubMed.
- S. Kucher, S. Korneev, S. Tyagi, R. Apfelbaum, D. Grohmann, E. A. Lemke, J. P. Klare, H. J. Steinhoff and D. Klose, J. Magn. Reson., 2017, 275, 38–45 CrossRef CAS PubMed.
- J. T. Paletta, M. Pink, B. Foley, S. Rajca and A. Rajca, Org. Lett., 2012, 14, 5322–5325 CrossRef CAS PubMed.
- E. H. Abdelkader, A. Feintuch, X. Yao, L. A. Adams, L. Aurelio, B. Graham, D. Goldfarb and G. Otting, Chem. Commun., 2015, 51, 15898–15901 RSC.
- E. Matalon, T. Huber, G. Hagelueken, B. Graham, V. Frydman, A. Feintuch, G. Otting and D. Goldfarb, Angew. Chem., Int. Ed., 2013, 52, 11831–11834 CrossRef CAS PubMed.
- A. M. Raitsimring, C. Gunanathan, A. Potapov, I. Efremenko, J. M. Martin, D. Milstein and D. Goldfarb, J. Am. Chem. Soc., 2007, 129, 14138–14139 CrossRef CAS PubMed.
- T. Plass, S. Milles, C. Koehler, C. Schultz and E. A. Lemke, Angew. Chem., Int. Ed., 2011, 50, 3878–3881 CrossRef CAS PubMed.
- K. L. Kiick, E. Saxon, D. A. Tirrell and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 19–24 CrossRef CAS PubMed.
- S. Milles, S. Tyagi, N. Banterle, C. Koehler, V. VanDelinder, T. Plass, A. P. Neal and E. A. Lemke, J. Am. Chem. Soc., 2012, 134, 5187–5195 CrossRef CAS PubMed.
- J. V. Staros, H. Bayley, D. N. Standring and J. R. Knowles, Biochem. Biophys. Res. Commun., 1978, 80, 568–572 CrossRef CAS PubMed.
- L. Jakob, A. Gust and D. Grohmann, Biochem. Biophys. Rep., 2019, 17, 1–9 Search PubMed.
- M. Benmelouka, A. Borel, L. Moriggi, L. Helm and A. E. Merbach, J. Phys. Chem. B, 2007, 111, 832–840 CrossRef CAS PubMed.
- A. Borel, H. Kang, C. Gateau, M. Mazzanti, R. B. Clarkson and R. L. Belford, J. Phys. Chem. A, 2006, 110, 12434–12438 CrossRef CAS PubMed.
- L. Garbuio, K. Zimmermann, D. Haussinger and M. Yulikov, J. Magn. Reson., 2015, 259, 163–173 CrossRef CAS PubMed.
- J. O. Lagerstedt, J. Petrlova, S. Hilt, A. Marek, Y. Chung, R. Sriram, M. S. Budamagunta, J. F. Desreux, D. Thonon, T. Jue, A. I. Smirnov and J. C. Voss, Contrast Media Mol. Imaging, 2013, 8, 252–264 CrossRef CAS PubMed.
- J. Hannant, J. H. Hedley, J. Pate, A. Walli, S. A. Farha Al-Said, M. A. Galindo, B. A. Connolly, B. R. Horrocks, A. Houlton and A. R. Pike, Chem. Commun., 2010, 46, 5870–5872 RSC.
- S. Tyagi and E. A. Lemke, Method. Cell Biol., Elsevier, 2013, vol. 113, pp. 169–187 Search PubMed.
- G. Jeschke, V. Chechik, P. Ionita, A. Godt, H. Zimmermann, J. Banham, C. R. Timmel, D. Hilger and H. Jung, Appl. Magn. Reson., 2006, 30, 473–498 CrossRef CAS.
- S. G. Worswick, J. A. Spencer, G. Jeschke and I. Kuprov, Sci. Adv., 2018, 4, eaat5218 CrossRef CAS PubMed.
- G. Jeschke, Protein Sci., 2018, 27, 76–85 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cp01930e |
| ‡ Present address: Faculty of Chemistry and Biochemistry, Ruhr University Bochum, Universitaetsstr. 150, Bochum, Germany. |
| This journal is © the Owner Societies 2020 |