
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ru and RuOx decorated carbon nitride for efficient ammonia photosynthesis†
Hui
Wang
 ,
Xiyi
Li
,
Qiushi
Ruan
and
Junwang
Tang
*
,
Xiyi
Li
,
Qiushi
Ruan
and
Junwang
Tang
*
Solar Energy & Advanced Materials Research Group, Department of Chemical Engineering, UCL, Torrington Place, London, WC1E 7JE, UK. E-mail: junwang.tang@ucl.ac.uk
First published on 1st May 2020
Abstract
Photocatalytic ammonia synthesis is a promising strategy for sustainable development compared to the energy-intensive industrial Haber–Bosch approach. Herein, a ternary heterostructure that consists of ruthenium species and carbon nitride (C3N4) was rationally explored for ammonia photosynthesis. Compared to the small ammonia yield from the g-C3N4 and Ru/g-C3N4 system, the Ru/RuO2/g-C3N4 system represents 6 times higher activity with excellent stability under full-spectrum irradiation. Such an enhancement is not only due to efficient transfer of electrons and holes to Ru and RuO2, respectively, facilitating both the reduction and oxidation reaction, but also taking advantage of Ru for N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N activation.
N activation.
1. Introduction
Ammonia is one of the most fundamental and essential feedstocks in the chemical industry due to its application in fertilizer production, medicaments and biological molecules.1,2 The main industrial process of ammonia production is the Haber–Bosch process, which requires extremely high temperatures and pressures, leading to an energy-intensive and highly carbon-emitting process.3 Ammonia photosynthesis is believed to be a low cost and environmentally friendly approach which could be regarded as a promising next-generation ammonia synthesis technique.4,5 However, the efficiency obtained to date has been very moderate, mainly due to the high recombination of photo-induced charges and the lack of efficient activation sites for cleavage of the NN triple bond.
Graphitic carbon nitride (g-C3N4), a thermally and chemically stable nitride semiconductor photocatalyst, is one of the most promising polymer photocatalysts for ammonia photosynthesis.6,7 It not only has good visible light absorption ability but also has enough driving force for the reduction reaction of N2. Many g-C3N4 based photocatalysts, including g-C3N4/CsxWO3,8 g-C3N4/ZrO2,9 g-C3N4/ZnMoCdS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10 and so on, have been applied in ammonia photosynthesis. However, they mainly suffered from low efficiency of charge separation, thus resulting in a low yield in photocatalytic N2 reduction. Engineering heterojunctions has been proved to be one of the most effective ways for the spatial separation of photo-induced carriers.11,12 Besides, the precise design of suitable active sites and matching of functionalities during heterojunction construction certainly aid in the specific photocatalytic reaction.13 Experimental and theoretical studies indicated that N2 could be easily adsorbed and activated on the Ru catalyst surface to form N2Hx species to facilitate the cleavage of the NN triple bonds in thermal catalysis.14,15 Recently, Ru has been used as an efficient cocatalyst for photo-assisted ammonia synthesis in aqueous solutions such as Ru/GaN and Ru/TiO2 due to the electron trapping sites of Ru.16,17 It is more efficient if a dual cocatalyst could be loaded for efficient trapping of both electrons and holes and also facilitate both reduction and oxidation reactions.
10 and so on, have been applied in ammonia photosynthesis. However, they mainly suffered from low efficiency of charge separation, thus resulting in a low yield in photocatalytic N2 reduction. Engineering heterojunctions has been proved to be one of the most effective ways for the spatial separation of photo-induced carriers.11,12 Besides, the precise design of suitable active sites and matching of functionalities during heterojunction construction certainly aid in the specific photocatalytic reaction.13 Experimental and theoretical studies indicated that N2 could be easily adsorbed and activated on the Ru catalyst surface to form N2Hx species to facilitate the cleavage of the NN triple bonds in thermal catalysis.14,15 Recently, Ru has been used as an efficient cocatalyst for photo-assisted ammonia synthesis in aqueous solutions such as Ru/GaN and Ru/TiO2 due to the electron trapping sites of Ru.16,17 It is more efficient if a dual cocatalyst could be loaded for efficient trapping of both electrons and holes and also facilitate both reduction and oxidation reactions.
Ruthenium oxide (RuO2) has been widely reported to be efficient as a hole acceptor, specifically it could extract photo-generated holes from excited photocatalysts efficiently and therefore suppresses charge recombination. Hence, it is of particular interest to rationally design the Ru/RuO2/g-C3N4 ternary heterojunction by coating RuO2 and Ru clusters on g-C3N4 nanosheets as dual co-catalysts for efficient ammonia photosynthesis. The 2D g-C3N4 nanosheets, with short bulk-to-surface diffusion length, accelerate the transfer of photo-induced carriers. Moreover, it also provides large surface area and abundant unsaturated sites (e.g. –NH2) for coordination of active components. Based on this excellent platform, the suitable choice of Ru as an electron acceptor not only takes advantage of its unique interaction and activation with NN, but also makes good use of the formed oxide species (RuO2) as a well-known hole acceptor. This ternary heterostructure Ru/RuO2/g-C3N4 exhibits a much higher NH3 generation rate (13.3 μmol g−1 h−1) than Ru/g-C3N4 (2.5 μmol g−1 h−1) and g-C3N4 (almost zero). The optimised catalyst can also remain stable for at least four cycles.
2. Experimental section
g-C3N4 was synthesized using urea as the precursor. In detail, 10 g urea powder was calcined in a muffle furnace (Carbolite, CWF 1300) at 550 °C for 4 h with a rate of 5 °C min−1. The obtained yellow product was washed with water, 0.1 M HCl, 0.1 M NaOH and water three times, respectively, then dried in an oven at 70 °C overnight. RuO2 and/or Ru loaded carbon nitride was synthesized via wet impregnation. Typically, 200 mg bulk C3N4 and 45 wt% RuCl3·H2O were suspended in 40 ml water followed by ultra-sonication (Fisherbrand) for 30 min. After that, the suspension was dried at 100 °C and calcined at 300 °C for 2 h in air and under a 10% H2/Ar atmosphere, and were labelled as Ru/RuO2/g-C3N4 and Ru/g-C3N4, respectively. Ru loaded on g-C3N4 by photodeposition was labelled as Ru/g-C3N4-p. The obtained materials were washed several times with water to remove RuCl3 residues. A series of Ru/RuO2/g-C3N4 samples were labelled as Ru/RuO2/g-C3N4-5% x% (x = 0.5–7.5), where x is the Ru loading weight percentage (nominal ratio).The powder X-ray diffraction (XRD) was carried out using a Bruker D4 diffractometer equipped with a Cu-Kα source (Kα1 = 1.540562 Å and Kα2 = 1.544398 Å). Transmission electron microscopy (TEM) (JEOL2010) was used to analyse the morphology and composition of the sample. The surface elemental composition analyses was characterized by X-ray Photoelectron Spectroscopy (XPS) (Thermoscientific XPS K-alpha). UV-vis-NIR diffuse reflectance spectra (DRS) were recorded using an Agilent Carry 3500 UV-Vis-NIR spectrophotometer equipped with a diffuse reflectance unit. Steady-state photoluminescence (PL) and Raman spectra were examined using a Renishaw inVia Raman microscope. Photocurrent testing was carried out using a closable three-electrode electrolytic cell, using an electrochemical workstation (IVIUM) control voltage with 0.1 mol L−1 Na2SO4 solution as the electrolyte, where the FTO electrode coated with the catalyst was used as the working electrode, the platinum plate was used as the counter electrode, and the Ag/AgCl electrode was used as the reference electrode.
In a typical photocatalytic N2 reduction reaction, a certain amount of photocatalyst was well dispersed in 30 ml of aqueous solution containing a sacrificial electron donor and then transferred to a 100 ml reactor. An in-house-built 100 mL two-necked flat-bottomed quartz container was used as the reactor, 50 mL of deionized water was used as the solvent and proton source for the reaction, and 0.03 g of the obtained photocatalyst was added into the solution. The reactor was sealed, purged with N2 gas for 20 min and then irradiated. The N2 and Ar control experiment was irradiated under LED irradiation (365 nm). Other control and cycling experiments were irradiated under full arc using a 300 W xenon lamp. The solution was taken out at regular intervals to detect ammonia concentration using ion chromatography.
Photocurrent testing was carried out using a three-electrode electrolytic cell, using an electrochemical workstation (IVIUM) control voltage. 0.1 M Na2SO4 solution was used as the electrolyte, the FTO electrode coated with the catalyst was used as the working electrode, the platinum plate served as the counter electrode, and the Ag/AgCl electrode acted as the reference electrode. A 150 W xenon lamp was used as the full arc light source. The photocurrent test was carried out using a potentiostatic technique with a switching period of 5 s, which mainly measured the photocurrent density of the photocatalysts. The alternating current impedance spectrum was measured using a three-electrode system with the following parameters: at −0.4 V vs. Ag/AgCl, high frequency 10 kHz, low-frequency 1 Hz, amplitude 10 mV. The test was carried out under irradiation with 100 mL of 0.1 M Na2SO4 solution as the electrolyte.
3. Results and discussion
The crystal structure of the obtained samples along with pure carbon nitride was firstly examined by powder X-ray diffraction (Fig. 1a). The characteristic diffraction peak at 27.4° assigned to the interlayer stacking of g-C3N4 in all three samples remains unchanged, excluding the insertion of Ru species at the interlayer.18 The Ru/g-C3N4-5% sample shows a series of peaks at 38.0°, 41.9°, 43.6°, 57.8°, 68.7° and 77.7° which are ascribed to the (1 0 −1 0), (0 0 0 2), (1 0 −1 1), (1 0 −1 2), (2 –1 –1 0) and (1 0 −1 3) lattice planes of the hexagonal Ru crystal structure, which corresponds to the commercial Ru. In contrast, no peaks attributed to RuOx species can be observed over the Ru/RuO2/g-C3N4-5% sample which may be due to the low amount or high dispersion of Ru species. FTIR and Raman spectroscopy were used to characterize the stable chemical structure of the obtained samples. For FTIR spectra (Fig. 1b), the typical signal at 810 cm−1 is assigned to the out-of-plane bending vibration characteristic of heptazine. The multiple peaks at 1200–1600 cm−1 represent the stretching vibration of tri-s-triazine heterocyclic stretches.19 Moreover, the absorption band in the range of 3000–3300 cm−1 is ascribed to the –NH2 stretching.18 Raman spectroscopy was carried out to examine the fine structure of g-C3N4 and Ru/RuO2/g-C3N4-5% in detail (Fig. 1c). In the range of 1200–1700 cm−1, a series of peaks are attributed to C–N stretching vibrations, specifically “G” and “D” band profiles of structurally disordered graphitic carbon and other carbon/nitrogen layered compounds.20,21 The peak at 980 cm−1 could be assigned to the symmetric N-breathing mode of heptazine, whilst the peak at 690 cm−1 corresponds to the in-plane bending vibrations of the tri/heptazine C–N–C linkages.22 All the above characterizations confirm the preservation of carbon nitride's structure after loading Ru species, while the specific peaks assigned to Ru species are not observed, again due to the highly dispersed and low amount of Ru loaded.23,24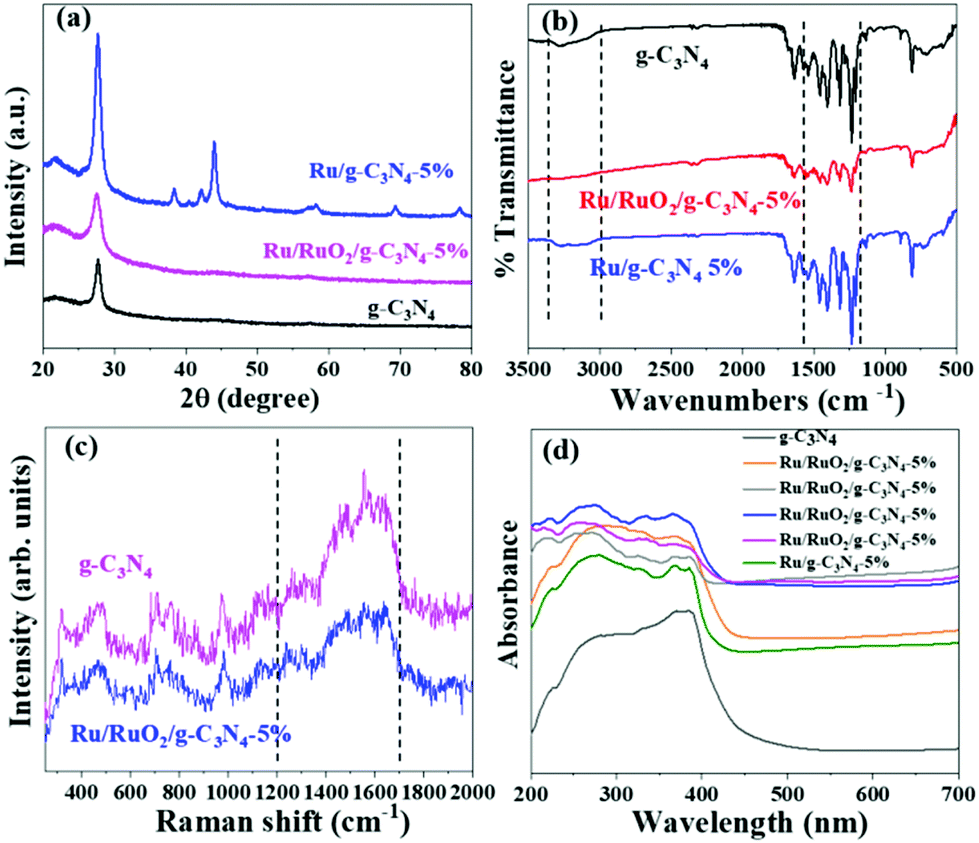 | ||
| Fig. 1 XRD patterns (a), FTIR spectra (b), Raman spectra (c), and UV-Vis spectra (d) of g-C3N4, Ru/RuO2/g-C3N4-5% and Ru/g-C3N4-5%. | ||
TEM was applied to analyse the microstructures of photocatalysts after loading Ru and RuO2 (Fig. 2). The thin g-C3N4 nanosheets are firmly assembled with small nanoparticles, constructing stable 0D/2D heterojunctions. The size of Ru species is around 2 nm, as shown in Fig. S1.†Fig. 2b shows the high-resolution image along with the fast Fourier transform (FFT) image. The transformed lattice fringe with a spacing of 0.207 nm matches well with the (101) plane of the metallic Ru, while the spacing of 0.18 nm corresponds to the (211) lattice plane of RuO2.25,26 Ru and RuO2 formation was believed to be due to calcination of the Ru precursor on carbon nitride at 300 °C in air. Similarly, the formation of Ru and RuO2 by thermal decomposition in air was reported, where a mixture of RuO2 as the dominant phase and Ru was found in the sample to be calcined between 200 °C and 400 °C.27 The reason is likely due to the organic substrate working as a reductant.28 The EDS mapping (Fig. 2c) shows the homogeneous distribution of the corresponding elements (C, N, O and Ru), indicating the high dispersion of RuOx on the g-C3N4. The high dispersion of Ru species can be attributed to two reasons: (i) long duration of sonication can help disperse the Ru precursor during uniform precipitation on the carbon nitride; (ii) carbon nitride possesses rich NH2– groups, which are widely reported to coordinate and anchor transition metals, avoiding the aggregation. Furthermore, Ru and O are ubiquitously dispersed in the selected region of Ru/RuO2/g-C3N4-5%, indicating successful formation of RuOx.
 | ||
| Fig. 2 TEM (a), the diffraction pattern of the selected area (b) and the corresponding elemental mapping images (c) of Ru/RuO2/g-C3N4-5% nanosheets. The inset in (b) is the corresponding fast Fourier transform image. | ||
To further investigate the chemical states of Ru species in the as-prepared sample, X-ray photoelectron spectroscopy (XPS) measurement was carried out (Fig. 3). The C 1s spectra overlap with Ru 3d spectra (Fig. 3a), illustrating three main peaks at 288.0 eV, 286.2 eV and 284.6 eV corresponding to the sp2-bonded carbon (N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), C–O and adventitious C–C bond, respectively.29 The signals at 280.2 eV and 281.7 eV are ascribed to Ru0 and RuO2, respectively.30 The peaks in N 1s spectra (Fig. 3b) at 398.5 eV and 400.0 eV are assigned to heptazine C–N–C and N–(C)3, while the signal centred at 401.2 eV corresponds to the C–N–H bond. In order to unravel the coexistence of Ru and RuO2, we further analysed the Ru species by XPS (Fig. 3c). The peaks at 461.9 eV and 484.1 eV are assigned to metallic Ru, while those at 463.7 eV and 485.9 eV are correlated to RuO2.31,32 The average ratio of Ru0/RuO2 in Ru/RuO2/g-C3N4 is calculated to be 0.18 by the curve fit of Ru 3p spectra. The peaks at 531.2 eV and 532.3 eV in O 1s spectra are related to Ru–O–Ru and –C–O, respectively. The binding energy at 534 eV is assigned to the adsorbed H2O (Fig. 3d).33 In addition, the XPS Ru 3p spectra of Ru/RuO2/g-C3N4-5% and Ru/g-C3N4-5% are shown in Fig. S2.† Compared with the Ru 3p spectra of Ru/RuO2/g-C3N4-5%, Ru/g-C3N4-5% shows peaks at 461.9 eV and 484.2 eV, corresponding to Ru 3p3/2 and Ru 3p1/2 of metallic Ru. The absence of RuO2 in Ru/g-C3N4-5% is due to the reduction condition of H2. Combining the TEM and XPS analysis of the Ru/RuO2/g-C3N4-5% sample, Ru species exist as Ru and RuO2 clusters on carbon nitride nanosheets.
N), C–O and adventitious C–C bond, respectively.29 The signals at 280.2 eV and 281.7 eV are ascribed to Ru0 and RuO2, respectively.30 The peaks in N 1s spectra (Fig. 3b) at 398.5 eV and 400.0 eV are assigned to heptazine C–N–C and N–(C)3, while the signal centred at 401.2 eV corresponds to the C–N–H bond. In order to unravel the coexistence of Ru and RuO2, we further analysed the Ru species by XPS (Fig. 3c). The peaks at 461.9 eV and 484.1 eV are assigned to metallic Ru, while those at 463.7 eV and 485.9 eV are correlated to RuO2.31,32 The average ratio of Ru0/RuO2 in Ru/RuO2/g-C3N4 is calculated to be 0.18 by the curve fit of Ru 3p spectra. The peaks at 531.2 eV and 532.3 eV in O 1s spectra are related to Ru–O–Ru and –C–O, respectively. The binding energy at 534 eV is assigned to the adsorbed H2O (Fig. 3d).33 In addition, the XPS Ru 3p spectra of Ru/RuO2/g-C3N4-5% and Ru/g-C3N4-5% are shown in Fig. S2.† Compared with the Ru 3p spectra of Ru/RuO2/g-C3N4-5%, Ru/g-C3N4-5% shows peaks at 461.9 eV and 484.2 eV, corresponding to Ru 3p3/2 and Ru 3p1/2 of metallic Ru. The absence of RuO2 in Ru/g-C3N4-5% is due to the reduction condition of H2. Combining the TEM and XPS analysis of the Ru/RuO2/g-C3N4-5% sample, Ru species exist as Ru and RuO2 clusters on carbon nitride nanosheets.
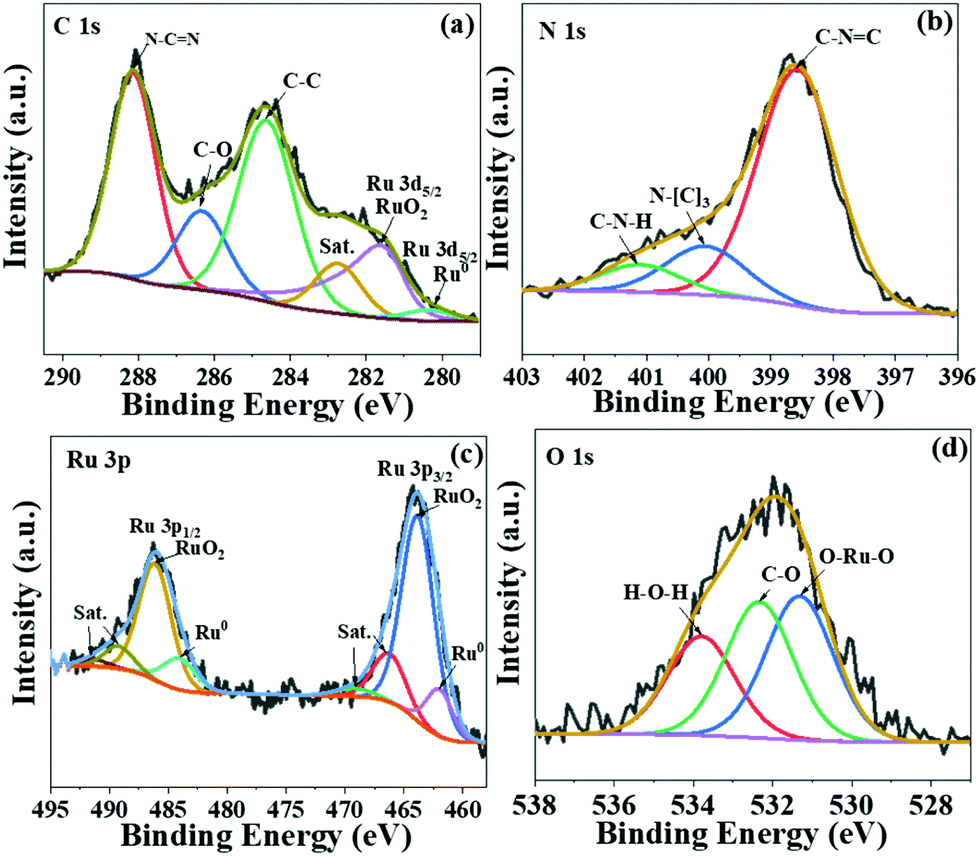 | ||
| Fig. 3 The C 1s (a), N 1s (b), Ru 3p (c), and O 1s (d) XPS spectra of Ru/RuO2/g-C3N4-5%. | ||
The photocatalytic activity for ammonia synthesis is shown in Fig. 4. Control experiments were conducted and are shown in Fig. 4a. No ammonia is detected from the pure g-C3N4 and in the absence of illumination, indicating that the Ru species and light irradiation are both indispensable to the photocatalytic ammonia photosynthesis. A small concentration of 15 μmol g−1 NH4+ was initially observed in the Ru/RuO2/g-C3N4-5% sample under an Ar atmosphere while no further increase of NH4+ can be observed in the following 5 hours. The production of NH4+ in the first hour is likely due to the remaining NHx species adsorbed on g-C3N4 during sample preparation rather than photosynthesis of N2. In the absence of the Ru cocatalyst, this adsorbed NHx cannot be easily desorbed, while with the loading of the Ru cocatalyst, this small amount of adsorbed NHx is readily removed within one hour. This is an interesting note for ammonia synthesis if there were NHx groups produced during photocatalyst synthesis. When changing to a N2 atmosphere, the ammonia yield on the Ru/RuO2/g-C3N4-5% sample linearly increases, on average 13.3 μmol g−1 h−1, indicating that N2 gas is the main source of ammonia photosynthesis. Then, the Ru cocatalyst amount was investigated between 0.5 wt% and 7.5 wt% to optimise its loading. All samples present enhanced activity compared with pure C3N4. The optimum amount of the loaded co-catalyst has been determined to be 5%, showing the highest N2 fixation activity with an NH4+ concentration of 13.3 μmol g−1 h−1 under full arc irradiation. The ammonia generation rate is comparable to that of the reported Ru system photocatalysts which have been list in Table S1.†34,35 A further increase in the amount of loaded co-catalysts results in decreased activity, because excess co-catalyst may block the light absorption of g-C3N4.36 In order to elucidate the important role of RuO2 in ternary heterostructures, other methods including photodeposition and H2 reduction have been used to load Ru species on g-C3N4 (denoted Ru/g-C3N4-5%-p and Ru/g-C3N4-5%) for comparison (Fig. 4c). All samples exhibited a linear increase in ammonia production with increasing time. Ru/g-C3N4-5% and Ru/g-C3N4-5%-p present an ammonia yield of less than 10 μmol g−1 after 3 h full arc irradiation, only one-fourth of that achieved by Ru/RuO2/g-C3N4-5%. On the former samples, only Ru metallic species were observed as mentioned above, which can act as an electron acceptor and activation sites for N2 reduction.12 The extraordinary high activity of Ru/RuO2/g-C3N4-5% can be ascribed to the synergistic effect between metallic Ru and RuO2 on g-C3N4 for both electron and hole extraction as well as for N2 activation as discussed below. In addition, a little change is observed from cycling experiments in Fig. 3d, which is believed to be due to the loss of a photocatalyst amount as we filtered the sample from the solution for cycling. Furthermore, the XRD and XPS of Ru/RuO2/g-C3N4-5% before and after the reaction are presented in Fig. S3 and Fig. S4.† No obvious differences in the XRD peaks and Ru 3p XPS spectra of the used photocatalyst are observed compared with that of the fresh photocatalyst. The ratio of the Ru metal and RuO2 before and after the reaction has been compared to further confirm the stability of Ru/RuO2/g-C3N4-5%. The Ru/RuO2 ratio after the reaction is estimated to be 0.19, which is similar to that before the reaction (0.20). All these results prove that the two cocatalysts remain very stable.
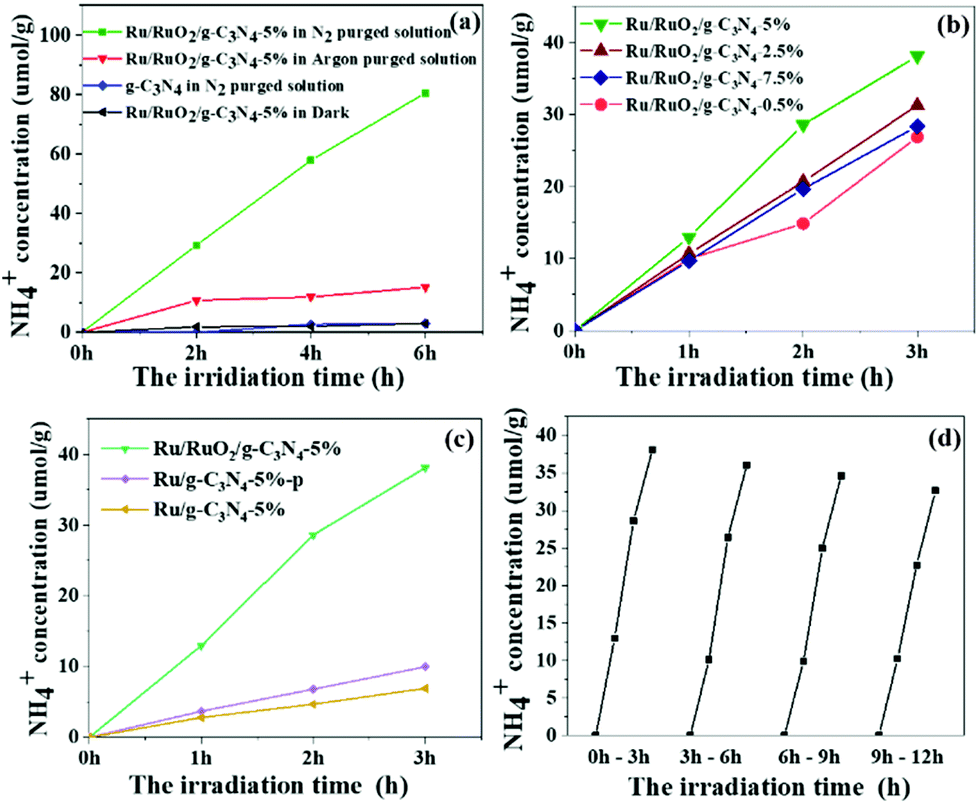 | ||
| Fig. 4 The photocatalytic N2 reduction and control experiments by Ru/RuO2/g-C3N4-5% under 365 nm LED illumination (a), photocatalytic ammonia yield of Ru/RuO2/g-CN4 with co-catalyst loading (0.5 wt%–7.5 wt%) in N2 purged water under full arc irradiation (b), photocatalytic activity of Ru/RuO2/g-C3N4-5%, Ru/g-C3N4-5%-p and Ru/g-C3N4-5% (c), and photocatalytic cycling tests for Ru/RuO2/g-C3N4-5% in N2 purged water under full arc irradiation (d). All experiments were carried out under neutral conditions with a pH value of 7. | ||
UV-Vis DRS analysis was implemented to investigate the light absorption of all samples (Fig. 1d). g-C3N4 exhibits light absorption from the UV to visible range, and the sharp edge suggests that the visible light absorption is due to the band gap transition. After loading Ru species, there is no obvious difference in the absorption edge compared with the pristine g-C3N4, indicating that loading Ru and RuO2 does not have a great influence on the band structure of g-C3N4. In the visible light region, the light absorption intensities of Ru/g-C3N4-5% and Ru/RuO2/g-C3N4-5% samples are strengthened gradually as Ru species increase, which may be due to the light scattering of the Ru species. Photoluminescence (PL) spectra of the samples were recorded to study the behaviour of photogenerated charge carriers, shown in Fig. 5a. The g-C3N4 exhibits broad and robust doublet peaks at 440 nm and 500 nm. The emission centre at around 440 nm is ascribed to π–π* transitions, which are usually observed in conjugated ring systems including heterocyclic aromatic compounds. The peak at around 500 nm is explained as the n–π* transition,37 which involves lone pairs on the N atom on the edge of the triazine/heptazine ring. After introducing Ru species, Ru/g-C3N4-5% exhibits a similar emission peak profile to that of pure g-C3N4 with decreased intensity. Ru/RuO2/g-C3N4-5% also shows the same trend but the lowest intensity compared with Ru/g-C3N4 and g-C3N4, indicating the least charge carrier recombination in the Ru/RuO2/g-C3N4 sample.
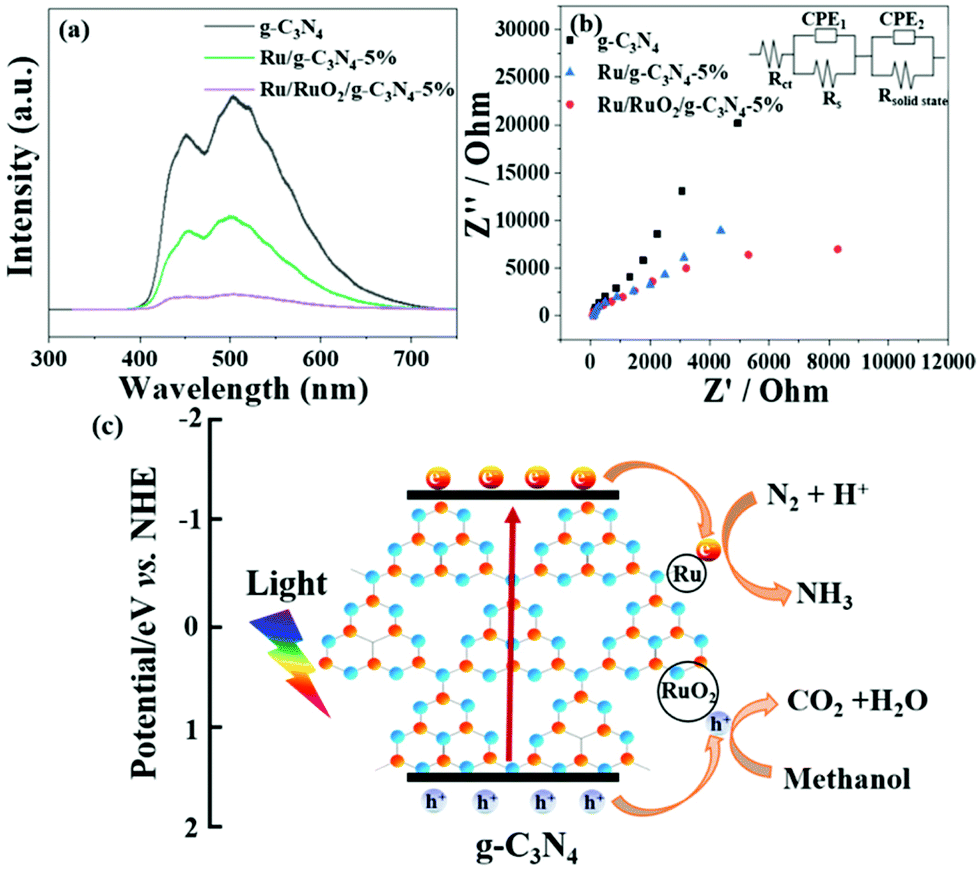 | ||
| Fig. 5 The photoluminescence (a) and EIS (b) of Ru/RuO2/g-C3N4-5% and g-C3N4 (inset is the interface charge transfer by fitting the impedance plots). A potential mechanism for the ammonia photosynthesis activity of Ru/RuO2/g-C3N4-5% (c). | ||
To investigate charge transfer kinetics among different samples, in situ photoelectrochemical and electrochemical impedance spectroscopy measurements were performed on all three samples in N2 purged solution. From Fig. S5,† Ru/RuO2/g-C3N4-5% exhibits higher reduction photocurrent density (e.g. 2 μA cm−2) compared to Ru/g-C3N4-5% (0.6 μA cm−2). It is consistent with the enhanced NH3 evolution rate on Ru/RuO2/g-C3N4-5% compared with that on Ru/g-C3N4-5%. Pure g-C3N4 has shown negligible photocurrent density, which also maintains consistency with its negligible NH3 synthesis performance. The interface charge transfer of all three samples was described by fitting the impedance scan plots to the equivalent circuit shown in the inset of Fig. 5b. The equivalent circuit model consists of two components: the electrode/electrolyte interface impedance (Rct and CPE1) and solid-state interface impedance (Rsolid-state and CPE2). Rs is the system resistance, Rct is the electrode/electrolyte interface charge transfer resistance and Rsolid-state is the solid-state interface (e.g. g-C3N4/RuO2 and g-C3N4/Ru) charge transfer resistance, respectively. The results summarized in Table 1 show that all three samples have a similar system resistance around 40 Ω cm2. However, charge transfer resistance (Rct) dramatically differs among different samples. Obviously, co-catalyst (Ru/RuO2 or Ru) loading has essentially reduced the electrode/electrolyte interface charge transfer resistance, as Rct has decreased from 2.46 × 106 Ω cm2 of g-C3N4 to 9.8 × 103 Ω cm2 of Ru/RuO2/g-C3N4-5% and 3.38 × 104 Ω cm2 of Ru/g-C3N4-5%. The reduced charge transfer resistance or enhanced charge transfer is attributed to the presence of metallic Ru and RuO2. In consequence, g-C3N4 has shown negligible NH3 production and photocurrent in part due to a high charge transfer resistance, while Ru/RuO2/g-C3N4-5% and Ru/g-C3N4-5% exhibit significant NH3 synthesis activities of 13.3 μmol g−1 h−1 and 2.5 μmol g−1 h−1, respectively.
| g-C3N4 | Ru/g-C3N4-5% | Ru/RuO2/g-C3N4-5% | |
|---|---|---|---|
| R s (Ω cm2) | 42.5 | 45.9 | 36.8 |
| R ct (Ω cm2) | 2.46 × 106 | 9.8 × 103 | 3.38 × 104 |
| CPE1 (Sn Ω−1 cm−2) | 1.02 × 10−4 | 1.63 × 10−4 | 4.02 × 10−4 |
| R solid-state (Ω cm2) | 2.21 × 102 | 1.82 × 103 | 1.74 × 103 |
| CPE2 (Sn Ω−1 cm−2) | 2.12 × 10−4 | 1.63 × 10−4 | 3.25 × 10−4 |
Apart from the solid-solution interface, charge transfer at the solid-state interface is also imperative in deciding the photocatalytic activity. To be precise, the solid-state interface charge transfer resistances (Rsolid-state) are 2.21 × 102 Ω cm2 on g-C3N4, 1.74 × 103 Ω cm2 on Ru/RuO2/g-C3N4 5% and 1.82 × 104 Ω cm2 on Ru/g-C3N4-5%. As Rsolid-state represents the resistance between g-C3N4/g-C3N4, RuO2/g-C3N4 or Ru/g-C3N4 particle boundaries, their smaller values compared to the solid-solution interface resistance confirmed over 5 times faster solid–solid charge transfer than the solid-solution charge transfer. One can see the increased Rsolid-state value on Ru/RuO2/g-C3N4-5%, indicating that the enhanced activity is in part due to the improved charge transfer between the catalyst and solution instead of between C3N4 particles.
Based on the above characterization and experimental results, a possible ammonia synthesis mechanism is proposed on the best sample of Ru/RuO2/g-C3N4-5% (Fig. 5c). When irradiated, electrons can be excited from the valence band to the conduction band of g-C3N4, which are further trapped by metallic Ru as the electrons sink. Then, these electrons are transferred from the Ru to the π antibonding orbital of N2, facilitating the cleavage of the NN triple bonds, thus activating N2.12,38 The activated N2 on the Ru catalyst surface further reacts with H+ in water to form NH3, and finally forms NH4+ in water. Meanwhile, the left holes are transported to RuO2 and scavenged by methanol, leading to a decrease of the electron–hole recombination.
4. Conclusions
In summary, the ternary heterostructure Ru/RuO2/g-C3N4 system was explored and it exhibited excellent photocatalytic N2 reduction activity and stability under full arc irradiation. It was found that g-C3N4 has shown negligible activity, while Ru-g-C3N4 and Ru/RuO2-g-C3N4 yielded 2.5 μmol g−1 h−1 and 13.3 μmol−1 g h−1 ammonia under full arc irradiation, respectively. The resulting structure was characterized by XRD, Raman, FTIR and TEM. The electron transfer mechanism was confirmed by spectroscopy including PL and PEC. The higher activity of Ru/RuO2/g-C3N4 over Ru-g-C3N4 illustrated the imperative role of RuO2 in enhancing the N2 fixation efficiency of g-C3N4. More importantly, a large amount of electrons trapped on Ru enhanced the electron transfer to the antibonding orbital of N2, leading to N2 activation. Hence, the synthetic strategy to incorporate both Ru and RuO2 to form the ternary system, where Ru acts as N2 adsorption and activation sites for the reduction reaction and RuO2 serves as fast hole extraction sites for the oxidation reaction, leads to the enhanced photocatalytic activity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
H. W. acknowledges the support from the UCL Dean's prize and China CSC scholarship. We all gratefully acknowledge the financial support provided by the UK EPSRC (EP/N009533/1), the Royal Society-Newton Advanced Fellowship grant (NA170422) and the Leverhulme Trust (RPG-2017-122).Notes and references
- S. Vaclav, Detonator of the population explosion, Nature, 1999, 400, 415 CrossRef.
- R. Raja, G. Sankar and J. M. Thomas, Bifunctional Molecular Sieve Catalysts for the Benign Ammoximation of Cyclohexanone: One-Step, Solvent-Free Production of Oxime and ε-Caprolactam with a Mixture of Air and Ammonia, J. Am. Chem. Soc., 2001, 123, 8153–8154 CrossRef CAS PubMed.
- S. Licht, S. B. Cui, B. Wang, F. Li, J. Lau and S. Liu, Ammonia synthesis by N2 and steam electrolysis in molten hydroxide suspensions of nanoscale Fe2O3, Science, 2014, 345, 637–640 CrossRef CAS PubMed.
- N. Zhang, A. Jalil, D. Wu, S. Chen, S. Chen, Y. i. Liu, C. Gao, W. Ye, Z. Qi, H. Ju, C. Wang, X. Wu, L. Song, J. Zhu and Y. Xiong, Refining Defect States in W18O49 by Mo Doping: A Strategy for Tuning N2 Activation towards Solar-Driven Nitrogen Fixation, J. Am. Chem. Soc., 2018, 140, 9434–9443 CrossRef CAS PubMed.
- H. Hirakawa, M. Hashimoto, Y. Shiraishi and T. Hirai, Photocatalytic Conversion of Nitrogen to Ammonia with Water on Surface Oxygen Vacancies of Titanium Dioxide, J. Am. Chem. Soc., 2017, 139, 10929–10936 CrossRef CAS PubMed.
- H. Liu, P. Wu, H. Lia, Z. Chen, L. Wang, X. Zeng, Y. Zhu, Y. Jiang, X. Liao, B. S. Haynes, J. Yee, C. Stampfl and J. Huang, Unravelling the effects of layered supports on Ru nanoparticles for enhancing N2 reduction in photocatalytic ammonia synthesis, Appl. Catal., B, 2019, 259, 118026 CrossRef CAS.
- Y. Shiraishi, S. Shiota, Y. Kofuji, M. Hashimoto, K. Chishiro, H. Hirakawa, S. Tanaka, S. Ichikawa and T. Hirai, Nitrogen fixation with water on carbon-nitride-based metal-free photocatalysts with 0.1% solar-to-ammonia Energy Conversion Efficiency, ACS Appl. Energy Mater., 2018, 1, 4169–4177 CrossRef CAS.
- A. Shi, H. Li, S. Yin, Z. Hou, J. Rong, J. Zhang and Y. Wang, Photocatalytic NH3 versus H2 evolution over g-C3N4/CsxWO3: O2 and methanol tipping the scale, Appl. Catal., B, 2018, 235, 197–206 CrossRef CAS.
- H. Mou, J. Wang, D. Yu, D. Zhang, W. Chen, Y. Wang, D. Wang and T. Mu, Fabricating Amorphous g-C3N4/ZrO2 Photocatalysts by One-Step Pyrolysis for Solar-Driven Ambient Ammonia Synthesis, ACS Appl. Mater. Interfaces, 2019, 11, 44360–44365 CrossRef CAS PubMed.
- Q. Zhang, S. Hu, Z. Fan, D. Liu, Y. Zhao, H. Ma and F. Li, , Preparation of g-C3N4/ZnMoCdS hybrid heterojunction catalyst with outstanding nitrogen photofixation performance under visible light via hydrothermal post-treatment, Dalton Trans., 2016, 45, 3497–3505 RSC.
- R. Marschall, Semiconductor composites: Strategies for enhancing charge carrier separation to improve photocatalytic activity, Adv. Funct. Mater., 2014, 24, 2421–2440 CrossRef CAS.
- X. Wang, C. Liow, A. Bisht, X. Liu, T. C. Sum, X. Chen and S. Li, Engineering Interfacial Photo-Induced Charge Transfer Based on Nanobamboo Array Architecture for Efficient Solar-to-Chemical Energy Conversion, Adv. Mater., 2015, 27, 2207–2214 CrossRef CAS PubMed.
- H. Wang, L. Zhang, Z. Chen, J. Hu, S. Li, Z. Wang, J. Liu and X. Wang, Semiconductor heterojunction photocatalysts: Design, construction, and photocatalytic performances, Chem. Soc. Rev., 2014, 43, 5234–5244 RSC.
- H. Duan, J. Liu, M. Xu, Y. Zhao, X. Ma, J. Dong, X. Zheng, J. Zheng, C. S. Allen, M. Danaie, Y. Peng, T. Issariyakul, D. Chen, A. I. Kirkland, J. Buffet, J. Li, S. C. E. Tsang and D. O'Hare, Molecular nitrogen promotes catalytic hydrodeoxygenation, Nat. Catal., 2019, 2, 1078–1087 CrossRef CAS.
- K. Honkala, A. Hellman, I. N. Remediakis, A. Logadottir, A. Carlsson, S. Dahl, C. H. Christensen and J. K. Nørskov, Ammonia Synthesis from First-Principles Calculations, Science, 2005, 307, 555–558 CrossRef CAS PubMed.
- L. Li, et al., Nitrogen Photofixation over III-Nitride Nanowires Assisted by Ruthenium Clusters of Low Atomicity, Angew. Chem., Int. Ed., 2017, 56, 8701–8705 CrossRef CAS PubMed.
- K. T. Ranjit and B. Viswanathan, Photocatalytic reduction of dinitrogen to ammonia, Indian J. Chem., 1996, 35, 443–453 Search PubMed.
- X. Wang, X. Chen, A. Thomas, X. Fu and M. Antonietti, Metal-containing carbon nitride compounds: a new functional organic-metal hybrid material, Adv. Mater., 2009, 21, 1609–1612 CrossRef CAS.
- J. Oh, J. Lee, J. C. Koo, H. R. Choi, Y. Lee, T. Kim, N. D. Luong and J. Nam, Graphene oxide porous paper from amine-functionalized poly(glycidyl methacrylate)/graphene oxide core-shell microspheres, J. Mater. Chem., 2010, 20, 9200–9204 RSC.
- A. C. Ferrari and J. Robertson, Interpretation of Raman spectra of disordered and amorphous carbon, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095–14107 CrossRef CAS.
- A. B. Jorge, D. J. Martin, M. T. S. Dhanoa, A. S. Rahman, N. Makwana, J. Tang, A. Sella, F. Corà, S. Firth, J. A. Darr and P. F. McMillan, H2 and O2 Evolution from Water Half-Splitting Reactions by Graphitic Carbon Nitride Materials, J. Phys. Chem. C, 2013, 117, 7178–7185 CrossRef CAS.
- P. J. Larkin, M. P. Makowski and N. B. Colthup, The form of the normal modes of s-triazine: Infrared and Raman spectral analysis and ab initio force field calculations, Spectrochim. Acta, Part A, 1999, 55, 1011–1020 CrossRef.
- M. Xiao, L. Gao, Y. Wang, X. Wang, J. Zhu, Z. Jin, C. Liu, H. Chen, G. Li, J. Ge, Q. He, Z. Wu, Z. Chen and W. Xing, Engineering energy level of metal center: Ru single-atom site for efficient and durable oxygen reduction catalysis, J. Am. Chem. Soc., 2020, 141, 19800–19806 CrossRef PubMed.
- X. Wang, X. Peng, W. Chen, Gu. Liu, A. Zheng, L. Zheng, J. Ni, C. Au and L. Jiang, The Insight into dynamic and steady-state active sites for nitrogen activation to ammonia by cobalt-based catalyst, Nat. Commun., 2020, 11, 1–10 CAS.
- H. Li, X. Li, J. Liang and Y. Chen, Hydrous RuO2-decorated MXene coordinating with silver nanowire inks enabling fully printed micro-supercapacitors with extraordinary volumetric performance, Adv. Energy Mater., 2019, 9, 1–13 Search PubMed.
- P. Jiang, Y. Yang, R. Shi, G. Xia, J. Chen, J. Su and Q. Chen, Pt-like electrocatalytic behavior of Ru-MoO2 nanocomposites for the hydrogen evolution reaction, J. Mater. Chem. A, 2017, 5, 5475–5485 RSC.
- S. Musić, S. Popović, M. Maljković, K. Furić and A. Gajović, Formation of RuO2 and Ru by thermal decomposition of ruthenium(III)-acetylacetonate, J. Mater. Sci. Lett., 2002, 21, 1131–1134 CrossRef.
- M. L. Green, M. E. Gross, L. E. Papa, K. J. Schnoes and D. Brasen, Chemical Vapor Deposition of Ruthenium and Ruthenium Dioxide Films, J. Electrochem. Soc., 1985, 132, 2677 CrossRef CAS.
- D. J. Martin, K. Qiu, S. A. Shevlin, A. D. Handoko, X. Chen, Z. Guo and J. Tang, Highly efficient photocatalytic H2 evolution from water using visible light and structure-controlled graphitic carbon nitride, Angew. Chem., Int. Ed., 2014, 53, 9240–9245 CrossRef CAS PubMed.
- Z. Ma, S. Zhao, X. Pei, X. Xiong and B. Hu, New insights into the support morphology-dependent ammonia synthesis activity of Ru/CeO2 catalysts, Catal. Sci. Technol., 2017, 7, 191–199 RSC.
- B. Feng, C. Chen, H. Yang, X. Zhao, L. Hua, Y. Yu, T. Cao, Y. Shi and Z. Hou, Ionic liquid-promoted oxidant-free dehydrogenation of alcohols with water-soluble ruthenium nanoparticles in aqueous phase, Adv. Synth. Catal., 2012, 354, 1559–1565 CrossRef CAS.
- D. J. Morgan, Resolving ruthenium: XPS studies of common ruthenium materials, Surf. Interface Anal., 2015, 47, 1072–1079 CrossRef CAS.
- D. Briggs and G. Beamson, XPS studies of the oxygen 1s and 2s levels in a wide range of functional polymers, Anal. Chem., 1993, 65, 1517–1525 CrossRef CAS.
- A. Awati, H. Maimaiti, S. Wang and B. Xu, Photo-derived fixation of dinitrogen into ammonia at ambient condition with water on ruthenium/coal-based carbon nanosheets, Sci. Total Environ., 2019, 695, 133865 CrossRef CAS PubMed.
- Y. Liao, J. Lin, B. Cui, G. Xie and S. Hu, Well-dispersed ultrasmall ruthenium on TiO2 (P25) for effective photocatalytic N2 fixation in ambient condition, J. Photochem. Photobiol., A, 2010, 387, 112100 CrossRef.
- J. Ran, J. M. aroniec and S. Z. Qiao, Cocatalysts in semiconductor-based Photocatalytic CO2 Reduction: achievements, challenges, and cpportunities, Adv. Mater., 2018, 30, 1–31 Search PubMed.
- A. B. Jorge, D. J. Martin, M. T. S. Dhanoa, A. S. Rahman, N. Makwana, J. Tang, A. Sella, F. Corà, S. Firth, J. A. Darr and P. F. McMillan, H2 and O2 evolution from water half-splitting reactions by graphitic carbon nitride materials, J. Phys. Chem. C, 2013, 117, 7178–7185 CrossRef CAS.
- M. Kitano, Y. Inoue, Y. Yamazaki, F. Hayashi, S. Kanbara, S. Matsuishi, T. Yokoyama, S. Kim, M. Hara and H. Hosono, Ammonia synthesis using a stable electride as an electron donor and reversible hydrogen store, Nat. Chem., 2012, 4, 934–940 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr02527e |
| This journal is © The Royal Society of Chemistry 2020 |