
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStimulus-responsive surface-enhanced Raman scattering: a “Trojan horse” strategy for precision molecular diagnosis of cancer†
Cai
Zhang
a,
Xiaoyu
Cui
 a,
Jie
Yang
a,
Xueguang
Shao
a,
Yuying
Zhang
b and
Dingbin
Liu
*a
a,
Jie
Yang
a,
Xueguang
Shao
a,
Yuying
Zhang
b and
Dingbin
Liu
*a
aCollege of Chemistry, Research Center for Analytical Sciences, State Key Laboratory of Medicinal Chemical Biology, Tianjin Key Laboratory of Molecular Recognition and Biosensing, Collaborative Innovation Center of Chemical Science and Engineering, Nankai University, Tianjin 300071, China. E-mail: liudb@nankai.edu.cn
bSchool of Medicine, Nankai University, Tianjin 300071, China
First published on 19th May 2020
Abstract
Molecular diagnosis has played an increasingly important role in cancer detection. However, it remains challenging to develop an in situ analytical method capable of profiling the molecular phenotype of tumors for precision cancer diagnosis. A “Trojan horse” strategy based on stimulus-responsive surface-enhanced Raman scattering (SR-SERS) is reported here for selectively recording the comprehensive molecular information of tumors in situ, without resorting to destructive sample preparation and complex data analysis. This technique is employed to delineate the margin between tumors and normal tissues with high accuracy, and to further discriminate the molecular fingerprints of tumors in the early and late stages. Based on molecular profiling, we discovered that the signal ratios of fatty acid-to-phenylalanine could serve as promising indicators for identifying the primary tumors in different stages. This simple SR-SERS technique also provides a potential useful means for identifying tumor classifications or distinguishing primary and metastatic tumors.
Introduction
The tumor microenvironment (TME) is recognized as the “soil” of cancer that plays a critical role in tumor growth, metastasis, and prognosis.1,2 The TME constitutes a complex network of biospecies whose abnormal functions are highly associated with cancer development.3 Gaining access to the comprehensive molecular information of the TME could revolutionize molecular diagnosis and help physicians make accurate and timely decisions for clinical intervention.4 A number of analytical methods such as metabolomics,5 proteomics,6 gene sequencing,7 immunoassays,8 and mass spectrometry9 have been established for collecting the molecular information of tumors. However, all these techniques can only provide fragmented molecular information of the TME, which is unable to reflect the overall molecular changes. At present, obtaining the comprehensive molecular details of biological tissues relies on the coupling of multiple analytical methods, which are complicated, time-consuming, and costly. Moreover, these techniques often rely on the destructive preparation of target tissues into solutions, causing the loss of tissue's morphological information.10 A non-destructive analytical method that can provide in situ comprehensive molecular signatures of the TME is highly required for the precision diagnosis of cancer.Raman spectroscopy represents a biocompatible detection tool that provides unique vibrational bands for target molecules.11 However, the extremely weak Raman signals of native biomolecules make this technique difficult for direct profiling of the TME molecular vibrations in situ. Surface-enhanced Raman scattering (SERS) has been proven to be a powerful strategy for enhancing the Raman scattering of a molecule when it is brought in close proximity to a metallic nanoparticle surface.12–25 The detection sensitivity of SERS can be down to single-molecule levels.26,27 Thus, SERS has been utilized to analyze the molecular vibrations of different tissue types by coating the tissue surfaces with metal nanoparticles.28 However, Raman signals on these tissues are “always-on”. As a result, it is rather difficult to directly discriminate the molecular vibrations between cancer and healthy tissues since they possess similar chemical compositions. There is lacking a strategy that can selectively amplify the biomolecular vibrations of tumor tissues in situ for precision cancer diagnosis.
Herein, we report a stimulus-responsive SERS (referred to as SR-SERS) probe, which functions like a Trojan horse, to specifically augment the vibrational signals of the TME for precision molecular diagnosis of cancer. This TME-specific SERS probe is designed based on a manganese dioxide-encapsulated gold nanoparticle (Au@MnO2 NP, abbreviated as AM NP) core–shell structure. The AM NPs were assembled using a polyethylene glycol (PEG) layer to minimize non-specific protein adsorption in physiological environments. When the PEGylated AM NPs are introduced into the blood circulation, the PEG and MnO2 shells isolate biomolecules from interacting with the Au NP core, thus turning off the SERS signals of the probe (Fig. 1a). After accumulation into the tumors by the enhanced permeability and retention (EPR) effect,29 the AM NPs can be decomposed into the naked Au NPs and Mn2+ in the presence of acidic pH/H2O2 species that are abundant in the TME. The freshly-exposed Au NPs are adsorbed by the native biomolecules, thus amplifying their Raman signals remarkably in situ. At the same time, the released Mn2+ offers intense T1-weighted magnetic resonance (MR) signals to report the location and morphological information of the TME. The proposed SR-SERS probes were applied to determining the margin between tumor and normal tissues with high accuracy. Further, we employed the SR-SERS technique to differentiate the overall molecular fingerprints of tumors in different growth stages.
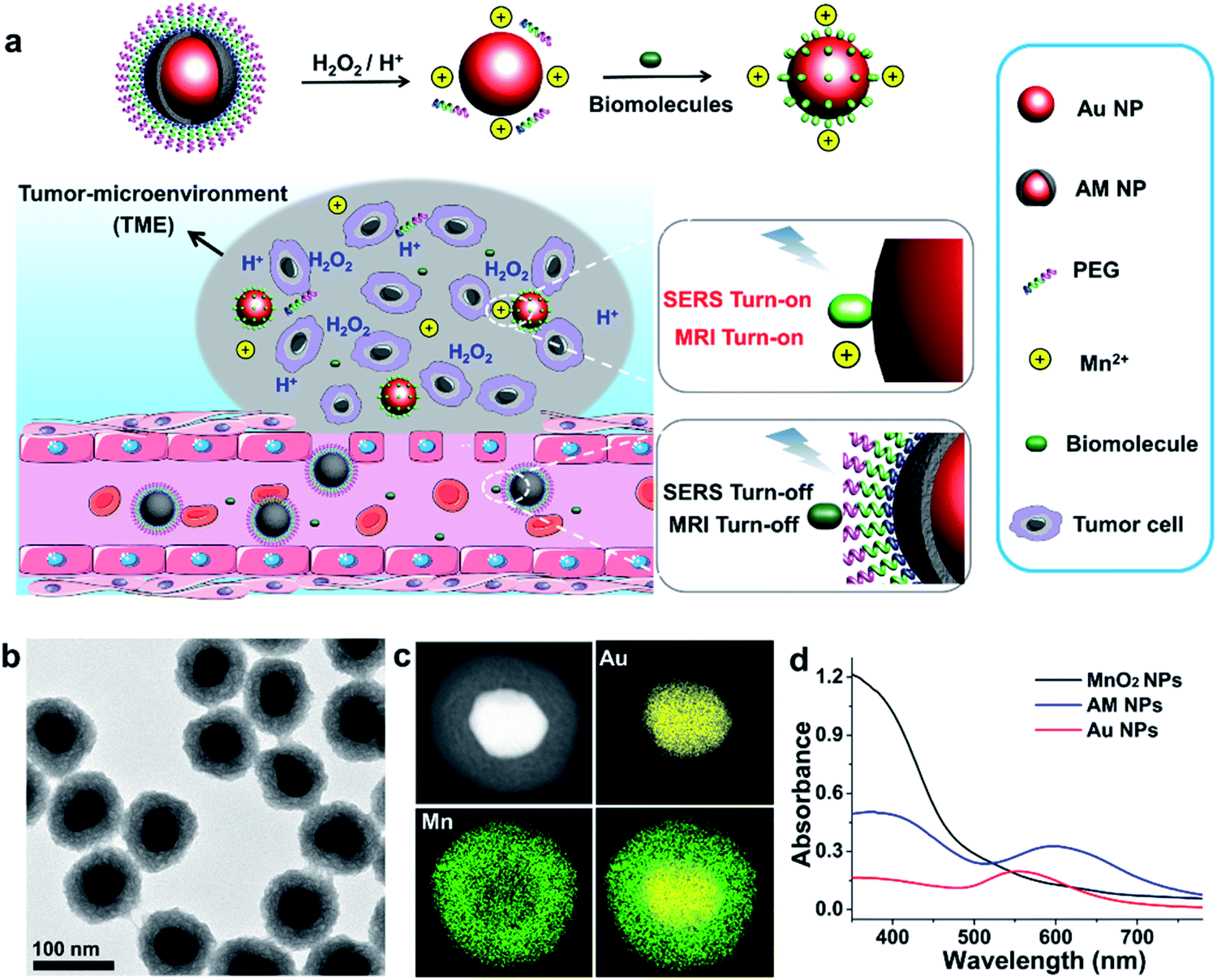 | ||
| Fig. 1 Schematic illustration of the SR-SERS strategy and characterization of the AM NPs. (a) The design of the “Trojan horse” strategy based on SR-SERS for molecular diagnosis of cancer. The abundant H2O2/H+ species in the TME trigger the release of Mn2+ and the naked Au NPs, which serve as T1 contrast agents for MRI and SERS substrates to amplify the vibrational fingerprints of the native biomolecules in the tumor, respectively. (b) HRTEM image of AM NPs. (c) HAADF-STEM image of AM NPs and the EDX elemental mapping of the Au core, MnO2 shell, and the merged image. (d) UV-vis spectra of Au NPs, MnO2 NPs, and the AM NPs. | ||
Results and discussion
Synthesis and characterization
The AM NPs were synthesized with a simple in situ growth method, in which Au NPs were prepared as the core, followed by depositing MnO2 shells. KMnO4 and poly(allylamine hydrochloride) (PAH) act as the manganese source and reducing agent, respectively.30 The dosages of KMnO4 and PAH were optimized to obtain a core–shell spherical structure (Fig. S1†).We next characterized the as-obtained AM NPs with different analytical tools. The morphology of the AM NPs was first characterized by high-resolution transmission electron microscopy (HRTEM). Fig. 1b reveals that the AM NPs possess uniform core–shell structures with an Au core (54 ± 5 nm in diameter) wrapped by a MnO2 shell with a thickness of 24 ± 2 nm. The core–shell structure of AM NPs was confirmed by energy-dispersive X-ray spectroscopy (EDS) mapping (Fig. 1c). The UV-vis spectra of the Au NPs, MnO2 NPs, and AM NPs were recorded subsequently. The two typical absorption peaks of AM NPs at 380 and 595 nm are overlapped with the surface plasmon bands of MnO2 and Au NP cores, respectively (Fig. 1d), validating the formation of AM NPs. X-ray photoelectron spectroscopy (XPS) was further employed to analyze the chemical composition of AM NPs. As shown in Fig. S2,† the O 1s spectrum has two peaks centered at 529.7 and 531.5 eV, which correspond to O 1s binding energies in the anhydrous compounds (Mn–O–Mn) and hydrated manganese (Mn–O–H) respectively. The binding energies for Mn 2p1/2 and Mn 2p3/2 are calculated to be 653.9 and 642.6 eV, respectively; while those for Au 4f7/2 and Au 4f5/2 are 83.9 and 87.6 eV, respectively.31 The loss of the SERS effect of AM NPs was assessed by incubating with Raman dyes and compared to that of the naked Au NPs. When the AM NPs were incubated with rhodamine 6G (R6G, 0.2 μM), no Raman signals can be detected; while the naked Au NPs (∼55 nm in diameter) can enhance the intensity of the same concentration of R6G for more than 4 orders of magnitude (Fig. S3†). This result implies that the MnO2 shell can inhibit the SERS effect of the Au NP core on the free dyes.
To enhance the stability and biocompatibility of the yielded AM NPs, the NP surfaces were wrapped with PEG chains via a layer-by-layer coating strategy (Scheme S1†). The PEG coating was characterized by Fourier-transform infrared spectroscopy, which shows the characteristic C–O–C bands (1179 cm−1) of PEG in the spectrum (Fig. S4†). The zeta potential of the AM NPs was determined to be 5.1 mV due to the presence of amino groups in PAH, and the NPs became negatively charged after functionalization with polyacrylic acid (PAA) and NH2-PEG (Fig. S5†). DLS analysis indicates that, before and after modification with PEGs, the average hydrodynamic diameters of AM NPs were measured to be 106 and 140 nm, respectively (Fig. S6†). All the characterization experiments confirmed the formation of AM NPs as well as those coated with PEGs.
Acidic pH/H2O2-dependent AM NP decomposition
The specific decomposition of the MnO2 shells was visualized by the naked eye, accompanied by the change of UV-vis absorption spectra in different media. It is well known that the TME is characteristic of acidic pH (6.2–6.9) and high-level H2O2 (∼100 μM),32 which possess high reactivity with MnO2 to release Mn2+. As shown in Fig. 2a, when the AM NPs were immersed in acidic pH/H2O2 solution (pH 6.5, H2O2: 100 μM), the color of the solution changed from dark-green to red in 5 min. In parallel, the absorption peak at 380 nm disappeared, along with the appearance of an absorption peak at around 550 nm that is assigned to the 55 nm naked Au NPs (Fig. S7†). The changes of both solution color and corresponding UV-vis spectra indicate the rapid decomposition of the MnO2 shells and the production of the naked Au NPs. In contrast, the UV-vis absorption spectra of AM NPs showed a trivial change in either H2O2 solution or pH 6.5 buffer solution in 30 min (Fig. S8†). The TEM images further demonstrated the entire decomposition of the MnO2 shells after treatment with acidic pH/H2O2 solution for 30 min (Fig. 2b). However, the core–shell structures of AM NPs still kept intact after treatment with acidic solution alone or neutral pH/H2O2 solution. These results demonstrate that the MnO2 shells of AM NPs can be specifically decomposed by the synergistic effect of H+ and H2O2. The reaction between MnO2 and H2O2 produces the intermediate Mn-oxo-hydroxide (MnOOH), which reacts with H+ to generate Mn2+ and O2.33–38 The following reactions illustrate the acidic pH/H2O2-triggered hydrolysis of the MnO2 shell:| 2MnO2 + H2O2 → 2MnOOH + O2 | (1) |
| 2MnOOH + 4H+ + H2O2 → 2Mn2+ + 4H2O + O2 | (2) |
| 2MnOOH + 2H+ → MnO2 + Mn2+ + 2H2O | (3) |
| MnO2 + H2O2 + 2H+ → O2 + Mn2+ + 2H2O | (4) |
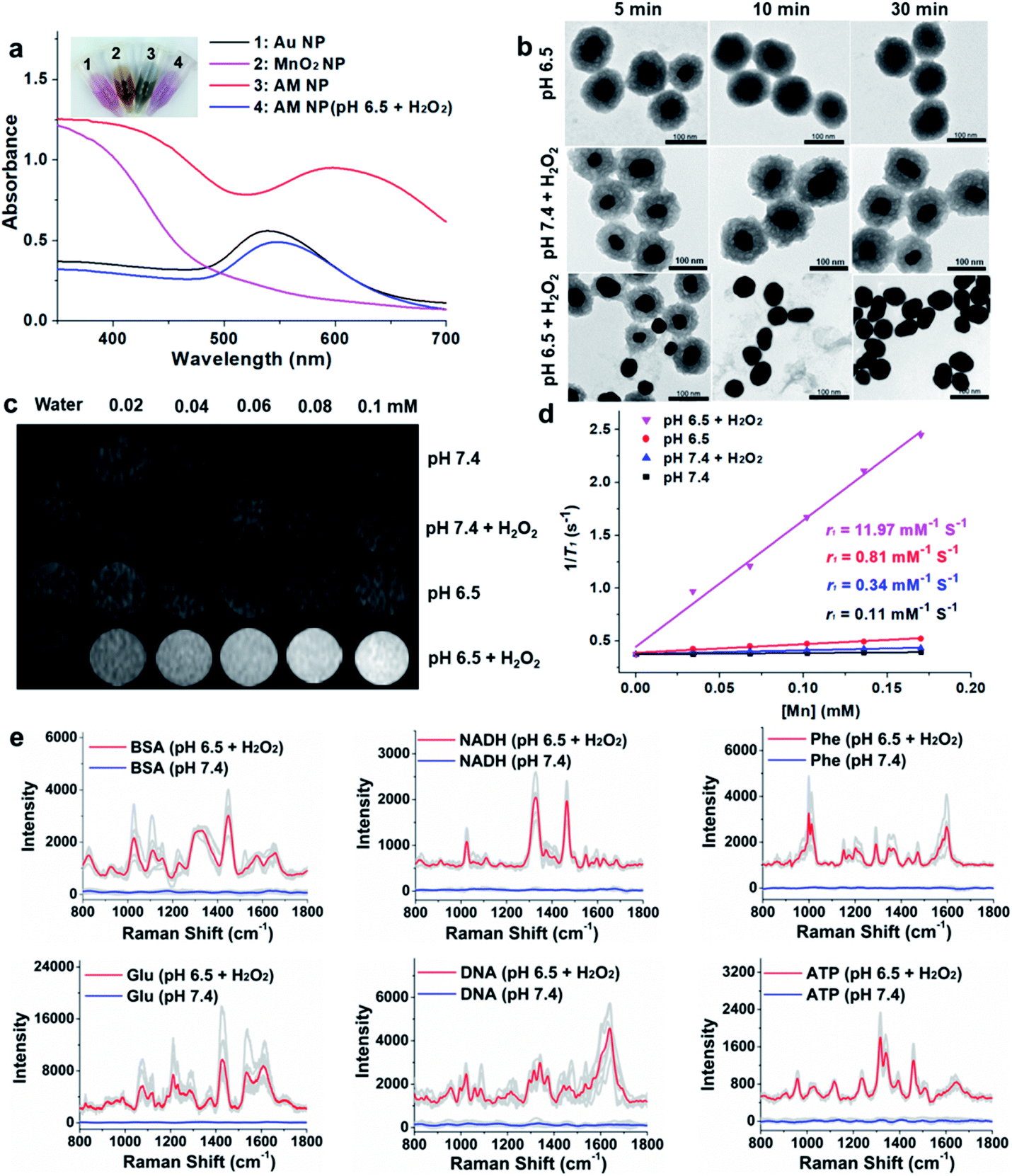 | ||
| Fig. 2 Characterization of the acidic pH/H2O2-responsive AM NPs and SERS/MR turn on ability. (a) UV-vis spectra and corresponding color of the solutions containing Au NPs, MnO2 NPs, AM NPs, and the AM NPs decomposed by the acidic pH/H2O2 solution (pH 6.5, H2O2: 100 μM). (b) TEM images of AM NPs before and after treatment with acidic buffer solution (pH 6.5), neutral pH/H2O2 solution (pH 7.4, H2O2: 100 μM) or acidic pH/H2O2 solution (pH 6.5, H2O2: 100 μM) for 5, 10, and 30 min. (c) T1-Weighted MR imaging and the corresponding T1 relaxation rates (r1) (d) of varying concentrations of AM NPs in solutions with different pH values (pH 6.5 and 7.4) in the absence or presence of H2O2 (0.5 mM). (e) Raman spectra of various biomolecules in AM NP solutions (0.6 nM) that were pretreated with an acidic pH/H2O2 mixture and compared to those pretreated with pH 7.4 buffer. All the spectra were collected with a confocal Raman spectrometer using 633 nm (3 mW) laser excitation. Data acquirement time, 10 s. 7 different sets of SERS spectra were recorded for each kind of biomolecule (in gray), while the average spectra for the activated and silent signals are displayed in red and blue, respectively. | ||
Acidic pH/H2O2-activated turn-on MRI of AM NPs
Since Mn2+ is an excellent T1 MR contrast agent,39 we observed the concentration-dependent brightening effect of AM NPs for T1-weighted MR imaging in the acidic pH/H2O2 solution, whereas the MR contrast effect under the other three conditions appeared to be much weaker (Fig. 2c). The enhanced T1 signal intensities of these samples were quantified by calculating the T1 relaxivity (r1). Fig. 2d shows that the r1 of the AM NPs was as high as 11.97 mM−1 s−1 in the acidic pH/H2O2 solution owing to the decomposition of MnO2 shells into the paramagnetic Mn2+. In contrast, the r1 values of the AM NPs under the other conditions were much smaller (0.11–0.81 mM−1 s−1). These results indicated the superior acidic pH/H2O2-activated MR imaging ability of the AM NPs.Acidic pH/H2O2-activated Raman enhancement of AM NPs
To verify the SR-SERS effect of AM NPs in vitro, the AM NPs (0.02 nM) were pretreated with acidic pH/H2O2 solution or neutral buffer solution (as control) for 30 min and centrifuged to 0.6 nM (6000 rpm, 2 min) and then incubated with several kinds of common biomolecules including bovine serum albumin (BSA, 12 mg mL−1), nicotinamide adenine dinucleotide (NADH, 1 mg mL−1), phenylalanine (3 mg mL−1), glucose (12 mg mL−1), nucleic acid (TCCATGACGTTCCTGACGTT, 0.3 mg mL−1), and ATP (5 mg mL−1) respectively at their optimized concentrations. The Raman fingerprint signals of the biomolecules were dramatically enhanced after incubation with AM NPs which had been pretreated with acidic pH/H2O2 solution (Fig. 2e). Under the same spectral recording conditions, all the biomolecules remained silent when they were incubated with AM NPs pretreated with pH 7.4 buffer alone. The results imply that the AM NPs are able to report the comprehensive Raman fingerprints of biomolecules after decomposition of the MnO2 shells to release the naked Au NPs in the TME.TME-activated MR imaging and SERS detection in vivo
Encouraged by the acidic pH/H2O2-responsive MR and SERS effects in vitro, we then evaluated the feasibility of PEGylated AM NPs for TME-activated MR imaging and SERS detection in vivo. After intratumoral injection of the PEGylated AM NPs (100 μL, 1 mg mL−1), the MR signals of the tumors became intense (Fig. S9a–c†). As a comparison, when the same amount of PEGylated AM NPs was injected into normal subcutaneous tissues, the MR signals had no noticeable change. This observation suggests that the AM NPs possess outstanding contrast effects resulting from the rapid, specific decomposition of MnO2 shells into Mn2+ in the tumor sites. Besides, the Raman intensity of tumor tissues was much higher than that of normal tissues after treatment with the PEGylated AM NPs, which was attributed to the exposure of the naked Au NP cores to the TME that dramatically amplified the Raman signals of the native biomolecules (Fig. S9d†). However, the Raman signals in the normal tissue were virtually undetectable, most likely due to the inertness of the AM NPs in the neutral environments. These results indicate that the PEGylated AM NPs may allow tumor-specific turn-on MR imaging and label-free SERS detection.The superior TME responsiveness of the dual turn-on MRI/SERS probes via intratumoral injection motivated us to explore the MRI and SERS performance in vivo. 200 μL of 1 mg mL−1 PEGylated AM NP solution was intravenously injected into a tumor-bearing mouse, and then the specific MR images were collected using a 3.0 T clinical MRI scanner. T1 contrast enhancement of the tumor was observed gradually after intravenous injection (Fig. 3a and b). The highest enhancement of MRI signals was found at 4 h post-injection. These results demonstrated the excellent activated MR imaging capability of the PEGylated AM NPs in tumors. The TME-activated MRI holds great promise for monitoring the location and morphology of tumors with high spatial resolution.
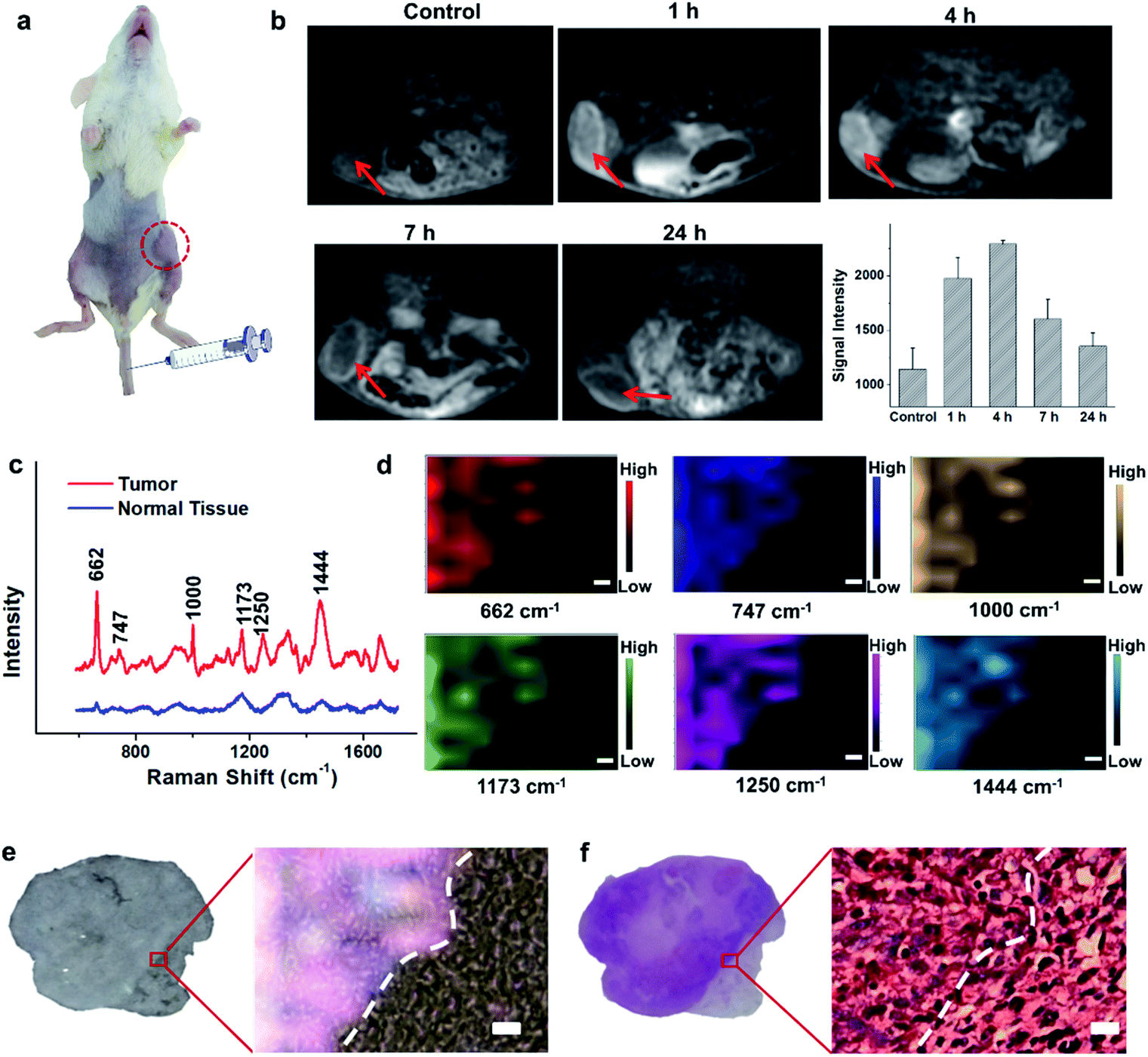 | ||
| Fig. 3 In vivo MRI and ex vivo imaging of the TME using the SR-SERS strategy. (a) The photo of a tumor-bearing mouse that was intravenously injected with PEGylated AM NPs (200 μL, 1 mg mL−1). (b) MR imaging of the tumor model after intravenous injection with PEGylated AM NPs (200 μL, 1 mg mL−1) at different time points and corresponding quantitative T1-weighted MR signals of the tumors derived from six different mice (error bars). (c) Raman spectra of the native biomolecules in the tumor and the surrounding normal tissues (n = 35) after intravenous injection with the PEGylated AM NPs. The spectra were recorded using 633 nm (3 mW) laser excitation (acquirement time 8 s), while the mean spectra for the tumor and normal signals are displayed in red and blue, respectively. (d) Raman mapping images that were recorded via 6 channels, which correspond to the bands at 662, 747, 1000, 1173, 1250, and 1444 cm−1 respectively. (e) Bright-field image of the tissue and that merged with the multiple Raman mapping channels in (d). (f) H&E-staining image of (e) scale bars: 10 μm. | ||
TME-activated SERS detection ex vivo
To study the comprehensive SERS fingerprinting signatures of the native biomolecules in the tumor sites, the probe-administered mice were sacrificed at 4 h post-injection. Subsequently, the tumors and surrounding normal tissues were cut out, then sliced and fixed on glass slides. The Raman spectra and mapping images of the tissue slices were recorded using a confocal Raman microscope in a non-invasive fashion. Fig. 3c shows that the Raman signals of the native biomolecules in the tumor tissues were much higher than those in the surrounding normal tissues. Based on this observation, we attempted to determine the boundary between the tumor and normal tissues by means of Raman mapping. Accurate delineation of the margin between tumors and normal tissues is extremely important for tumor resection in the clinic.40,41 Through mapping the tissues with multiple channels that are assigned to various native biospecies in the tumors (Fig. 3d), strong Raman signals can be observed in the tumor site but are undetectable in the neighboring normal tissues, providing a very clear boundary between the tumor and normal tissues (Fig. 3e). Impressively, the boundary delineated by Raman mapping correlated well with that determined by hematoxylin and eosin (H&E) staining (Fig. 3f). In contrast, if no probes were administered, the Raman intensities of the native biomolecules in both tumor and the surrounding normal tissues appeared to be quite weak, and no boundary was observed between the two types of tissues (Fig. S10†). These results verified the specific Raman enhancement effect of the PEGylated AM NPs towards tumors. The TME-specific comprehensive molecular profiling ability of the SR-SERS probe makes it promising for precision cancer diagnosis. Additionally, the TEM images in Fig. S11† show that the AM NPs were decomposed into naked Au NPs in the tumor tissues, confirming the TME-triggered SERS of the native biomolecules.We further wanted to apply the PEGylated AM NPs to discriminating the Raman fingerprints of primary tumors with different growth periods, where the early growth group was less than 4 days and the late growth group was more than 14 days after transplantation of tumor cells. Fig. 4a shows that the T1-weighted tumors at different growth periods exhibit spectral changes in specific bands (Fig. 4b and c), indicating the diverse expression and distribution of the native biomolecules in tumors at different growth periods. Note that the mean spectra for each tumor at different growth periods were obtained from 420 spectra randomly chosen in different tumor slices. In detail, the Raman peaks at 747, 855, 941, 1173, 1250, 1300, 1365, 1396, 1444, 1579 and 1657 cm−1 showed distinctive changes in spectral intensity (Table 1).42–46 Among them, the peaks at 855 and 941 cm−1 are assigned to carbohydrates (red bars);46,47 the peaks at 1300, 1444 and 1657 cm−1 belong to fatty acids (yellow bars);45 and the peaks at 747, 1173, and 1365 cm−1 are related to lactic acids, urea, and creatinine, respectively (green bars).48–51 After comparing the Raman spectra of tumors in the two groups, we found that the intensity ratios of the peaks at 1444 and 1000 cm−1 have a significant difference. In detail, the intensity ratio in the early growth stage was much higher than that in the late growth stage (p < 0.001), which may be linked to the differences in the energy metabolism of the tumors in different growth stages. As shown in Fig. S12,† the SERS spectra of phenylalanine and two types of fatty acids (oleic acid and stearic acid) were determined; the spectra further demonstrated the Raman peaks at 1000 and 1444 cm−1 related to phenylalanine and fatty acids respectively.
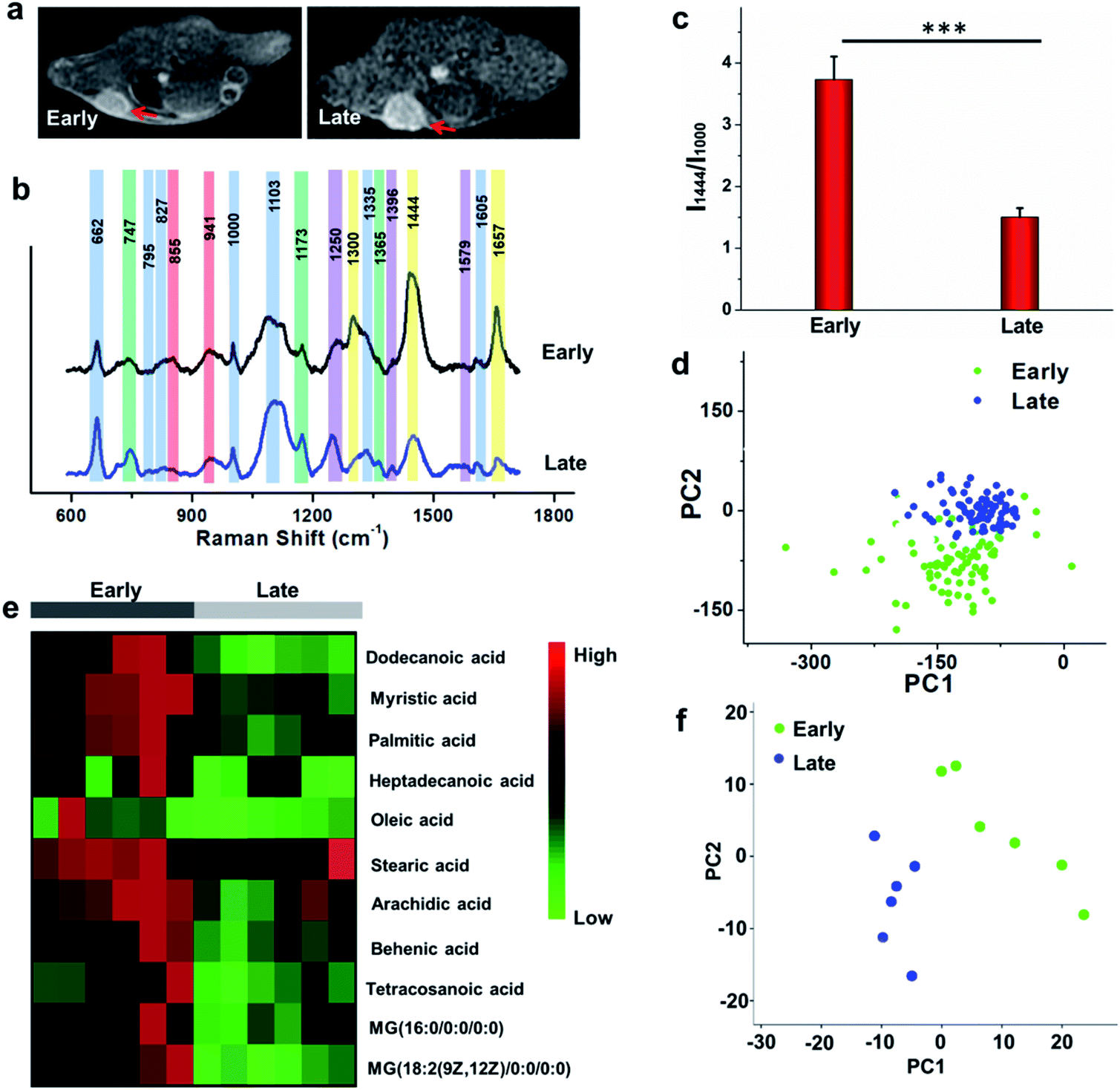 | ||
| Fig. 4 MR imaging, Raman spectroscopic detection, and metabolomics study of primary tumors in different growth stages. (a) MR images of tumors in different growth stages after intravenous injection with PEGylated AM NPs (200 μL, 1 mg mL−1) for 4 h. (b) The mean Raman spectra of the tumors in different growth stages. The spectra for each tumor at different growth periods were recorded from seven parallel tumor-bearing mice. Each tumor was cut into multiple layers of slices with a thickness of 10 μm, where the Raman spectra from three slices collected near the middle of tumors were recorded. The average spectra of 20 different single spectra were randomly recorded from each tumor slice using 633 nm (3 mW) laser excitation (acquirement time 8 s). Thus, the mean spectra for each tumor at different growth periods were collected from 420 different spectra. Blue, amino acids or proteins; red, carbohydrates; yellow, fatty acids; purple, nucleic acids or nucleotide; and green, metabolites. (c) SERS intensity ratios of peaks at 1444 and 1000 cm−1 for the tumor tissues in different growth periods, which correspond to the expression ratios of fatty acids and phenylalanine (p < 0.001). (d) PCA of the Raman spectral distribution of the tumor tissues at different growth periods in PC1–PC2. (e) Heat map for the ratios of fatty acid-to-phenylalanine for the tumors in different growth stages that were detected by gas chromatography-time-of-flight mass spectrum (GC-TOF/MS). The MS data were collected from six parallel samples of the tumors in the two groups. (f) PCA analysis of metabolites of the primary tumor tissues in different growth periods. | ||
| Shift (cm−1) | Component | SERS band assignment |
|---|---|---|
| 662 | Protein | C–S stretching mode of cysteine |
| 747 | Metabolite | Lactic acid |
| 795 | Protein | L-Serine |
| 827 | Protein | Tyrosine |
| 855 | Carbohydrates | Monosaccharides and disaccharides |
| 941 | Carbohydrates | C–O–C |
| 1000 | Protein | Phenylalanine |
| 1103 | Protein | Amide III and other groups |
| 1173 | Metabolite | Urea |
| 1250 | Nucleic acid | Guanine, cytosine (NH2) |
| 1300 | Fatty acid | CH2 twisting modes |
| 1335 | Protein | CH3CH2 wagging |
| 1365 | Metabolite | Creatinine |
| 1396 | Nucleic acid | Inosine |
| 1444 | Fatty acid | CH2 bending mode |
| 1579 | Nucleic acid | Ring breathing modes in the DNA bases |
| 1605 | Protein | Phenylalanine |
| 1657 | Fatty acid |
ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) C) |
We further made use of principal component analysis (PCA) to classify the Raman spectra of the tumors in different growth stages. PCA is a statistical tool that compresses complex multivariate data into fewer dimensions, by which an overview of data is illustrated in a two- or three-dimensional plane. The distribution of the Raman spectra among the tumors in different groups is illustrated in the PC1–PC2 subspace (Fig. 4d). The results show that the most Raman spectra of the tumors in the two groups could be distinguished. These observations suggest that the new SR-SERS technique has the potential to differentiate the molecular fingerprints of tumors at different growth periods.
To validate the Raman intensity ratio changes of tumor tissues analyzed by the SR-SERS probes, a metabolomics study was carried out with the same set of samples by gas chromatography-time-of-flight mass spectrum (GC-TOF/MS) (Fig. 4e). The results of fatty acid/phenylalanine changes obtained from the metabolomics were consistent with the semi-quantitative SR-SERS results. Therefore, the ratio of fatty acid-to-phenylalanine has great potential to serve as an indicator for growth stages of primary tumors particularly in the early growth stages. Furthermore, the PCA analysis of the metabolites in the tumors at different growth stages (Fig. 4f) indicated tremendous differences, which was similar to the Raman profiling results.
Biodistribution of PEGylated AM NPs in vivo
To study the biodistribution of the PEGylated AM NPs in vivo, the tumor and major organs (heart, liver, spleen, kidney and brain) of the mice (n = 7) that had been treated with PEGylated AM NPs were collected at different time points (4 h, 1 day and 7 days) post-injection. After digestion in aqua regia, the tissue samples were analyzed by ICP-OES. The results in Fig. S13† showed that the majority of the Au elements accumulated in the liver and spleen within 4 h and 1 day. The elemental Au was also enriched in the tumor, heart, and kidney at 4 h post-injection, while no detectable Au elements were taken up by the brain. After 7 days, the Au amounts distributed in the organs approached that of the control. On the other hand, the Mn uptake in the kidney and liver was significantly high at 4 h post-injection, and a considerable amount of Mn was also accumulated in the tumor, heart, and spleen, while there was still no Mn uptake in the brain. One day later, most of the Mn elements were cleared out from the organs except for the kidney, which may correspond to the renal metabolism of the Mn ions. After 7 day post-injection, the Mn levels in all the organs declined to that of the control, indicating that the Mn elements had been cleared out completely. These results implied that the PEGylated AM NPs can be metabolized and cleared out from the body in one week.Assessment of in vivo toxicity
We finally evaluated the chemical stability and biocompatibility of PEGylated AM NPs. The TEM images show that the core–shell NPs kept intact after incubation in the freshly-collected mouse serum for 4 h (Fig. S14†). The biocompatibility of the NPs was estimated by measuring the body weight, H&E analysis of major organs and biochemical analysis of blood of the mice that were intravenously injected with the PEGylated AM NPs (200 μL, 1 mg mL−1). The body weight of the mice increased gradually with the growth of the mice, showing no discernible difference with the control group in 30 days (Fig. S15†). Besides, H&E analysis results indicated that there was no visible tissue damage in the major organs (heart, liver, spleen, lung, kidney, brain, and thymus) of the mice after the treatment with the nanoparticle probes (Fig. 5a). The biochemical analysis results show that most of the parameters in the AM NP-treated group had similar levels to those of the control group after one-day post-injection, except for AST and ALT whose levels were elevated (Fig. 5b). However, the levels of AST and ALT declined to that of the control group on the 7th day, while other parameters in the experimental group remained persistent during the NP treatment.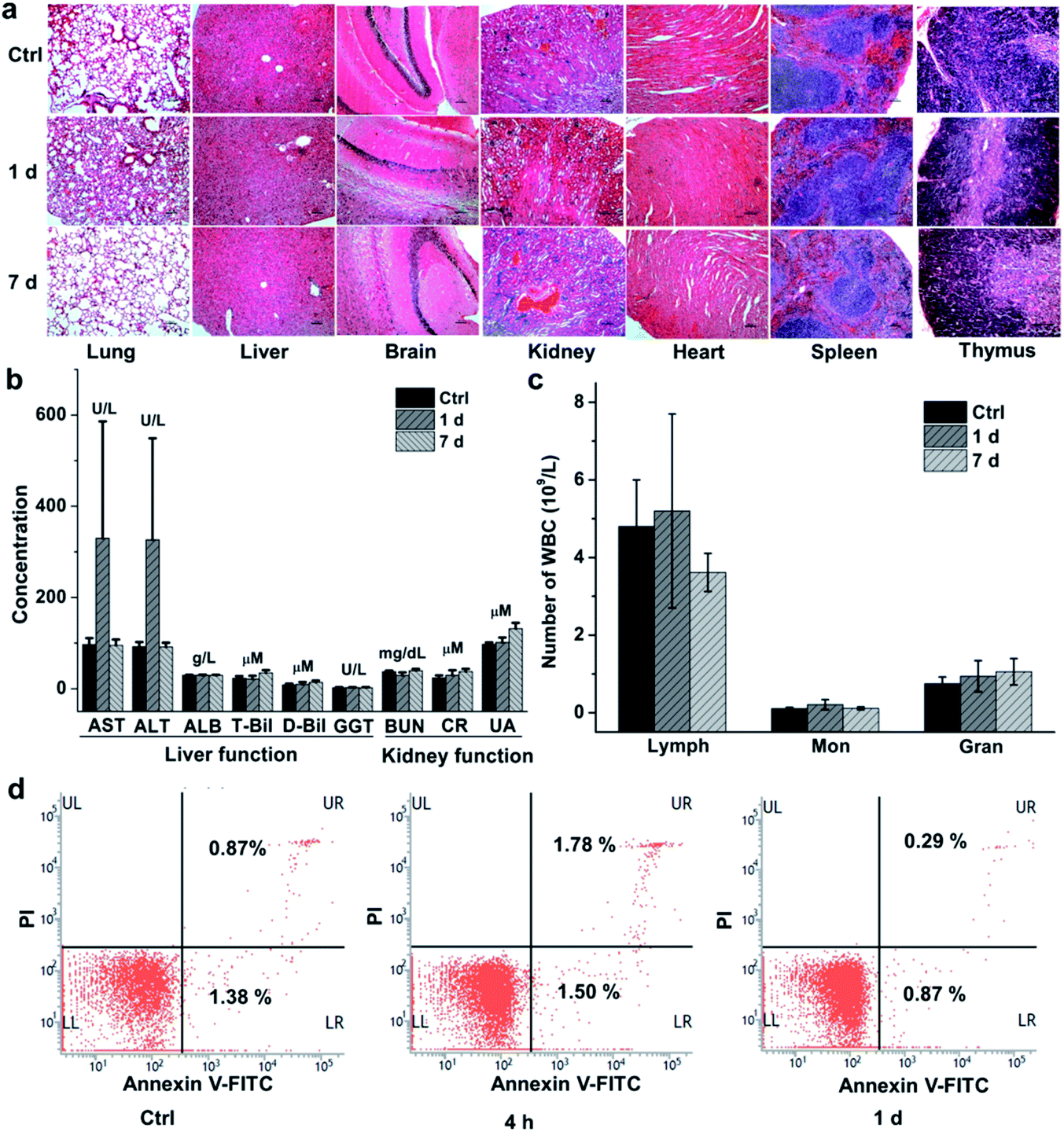 | ||
| Fig. 5 In vivo toxicity evaluation of the PEGylated AM NPs. (a) Histopathological results of different organs in the mice after they were intravenously injected with PEGylated AM NPs (200 μL, 1 mg mL−1) for 1 and 7 days and that without any treatment. Scale bar, 100 μm. (b) The blood levels of the liver function markers (AST, ALT, ALB, T-Bil, D-Bil, GGT) and the kidney function markers (BUN, CR, UA) in the mice treated with and without PEGylated AM NPs (200 μL, 1 mg mL−1) for 1 and 7 days. (c) Cell counting of lymphocytes (Lymph), monocytes (Mon) and neutrophil granulocytes (Gran) from the peripheral blood of mice intravenously injected with PEGylated AM NPs (200 μL, 1 mg mL−1) for 1 and 7 days and that without any treatment was set as control. Error bars represent the standard deviations between seven parallel mice. (d) Flow cytometry analysis for measuring the apoptotic peripheral blood mononuclear cells collected from the mice exposed to the PEGylated AM NPs (200 μL, 1 mg mL−1) for 4 h and 1 day. The peripheral blood mononuclear cells of the mice without any treatment were set as control. | ||
Since the cytotoxic effect of the AM NPs on the immune system can be reflected by the change of the immune cells in the blood, we counted the immune cells after intravenous injection of the NPs. In detail, after injection of the PEGylated AM NPs, the blood of the mice (n = 7) was collected at different time points for counting the immune cells including lymphocytes, monocytes, and neutrophil granulocytes. As shown in Fig. 5c, the numbers of the three types of cells on the 1st and 7th day post-injection were approximate to those of the control group. Besides, the peripheral mononuclear cells including lymphocytes and monocytes were isolated from the blood for apoptosis analysis by fluorescent-activated cell sorting (FACS). Fig. 5d shows that only a few numbers of cells in the early apoptosis (FITC-labeled) or necrosis (PI-labeled) were detected within both 4 h and 1 day, which are comparable to that of the control without any treatment. These results revealed that the PEGylated AM NPs had no apparent adverse effect on the immune cells.
To further verify the neurotoxicity of the PEGylated AM NPs, brain sections from the mice treated with the NPs were analyzed with a TUNEL (TdT-mediated dUTP nick end labeling) staining kit (Roche). The fragmented DNA could be stained through labeling the 3′-hydroxyl termini. Thus, apoptotic cells in the brain sections would be labeled in green. Fig. S16† reveals that no obvious TUNEL-positive cells appeared in the brain tissues of the mice intravenously injected with PEGylated AM NPs for 1 and 7 days, which was similar to the control group. The result demonstrates that the PEGylated AM NPs possess negligible neurotoxicity. All of the preliminary in vivo toxicity results revealed that the PEGylated AM NPs have excellent biocompatibility, which is crucially important to their biological and biomedical applications.
Conclusions
In summary, we have developed a powerful yet biocompatible “Trojan horse” SR-SERS nanoprobe to augment the vibrational fingerprints of tumors in situ for precision molecular diagnosis of cancer. The proposed SR-SERS strategy is, to our knowledge, the first approach that is capable of profiling the molecular fingerprints of tumor tissues selectively without destruction of the samples. The “Trojan horse” SR-SERS approach shows at least two advantages over traditional molecular diagnostic tools. First, the SR-SERS probes can only be activated by the TME while remained silent in other tissues, thus showing extremely high specificity to tumors. Second, the TME-triggered exposure of the naked Au NPs to native biomolecules could significantly amplify their overall Raman fingerprints to offer comprehensive molecular information, rather than to provide fragmented information as traditional molecular profiling approaches do. The high specificity of the SR-SERS probes in the TME made this new technique extremely potent in delineating the margin between tumors and healthy tissues, which may greatly facilitate precision tumor resection. Furthermore, the TME-responsive SERS spectra of tumors in different growth stages provided useful information that may reveal molecular changes of energy metabolism and genetic mutants in the process of tumor development, showing great potential in studying tumor growth, differentiation, and migration especially in combination with the Mn2+-based MRI. Impressively, the molecular profiling results showed that fatty acid/phenylalanine is a promising indicator of primary tumors in different growth stages.To achieve diagnostic recommendations in clinical settings, long-term biosafety of the PEGylated AM NPs should be demonstrated with large data. To demonstrate the easy generalizability of the “Trojan horse” strategy as a cancer diagnostic tool, we plan to bring this technique into profiling molecular signatures in tumor classifications as well as those under different treatments. To minimize the amount of SR-SERS probes used for cancer diagnosis, we will focus on the enhancement of SERS sensitivity by using anisotropic nanoparticles such as nanocubes, nanorods, nanostars, nanotriangles, and core/satellite nanoparticles, to name a few, because such nanostructures show much more intense field enhancements for SERS than single spherical nanoparticles.52–57
Ethical statement
All animal studies were performed in compliance with the institutional guidelines made by the Tianjin Committee of Use and Care of Laboratory Animals and the overall regulations approved by the Animal Ethics Committee of Nankai University.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the National Key R&D Program of China (2019YFA0210103), the National Natural Science Foundation of China (21775075 and 21977053), and the Fundamental Research Funds for Central Universities (China). We are grateful to Professor Xiaobing Zhang from Hunan University for his enthusiastic discussion about the data.Notes and references
- B. Mlecnik, G. Bindea, A. Kirilovsky, H. K. Angell, A. C. Obenauf, M. Tosolini, S. E. Church, P. Maby, A. Vasaturo, M. Angelova, T. Fredriksen, S. Mauger, M. Waldner, A. Berger, M. R. Speicher, F. Pages, V. Valge-Archer and J. Galon, Sci. Transl. Med., 2016, 8, 327ra26 CrossRef PubMed.
- X. Huang, J. Song, B. C. Yung, X. Huang, Y. Xiong and X. Chen, Chem. Soc. Rev., 2018, 47, 2873–2920 RSC.
- A. Sharma, M.-G. Lee, H. Shi, M. Won, J. F. Arambula, J. L. Sessler, J. Y. Lee, S.-G. Chi and J. S. Kim, Chem, 2018, 4, 2370–2383 CAS.
- X. H. Fang and W. H. Tan, Acc. Chem. Res., 2010, 43, 48–57 CrossRef CAS PubMed.
- Z. Wang, B. Cui, F. Zhang, Y. Yang, X. Shen, Z. Li, W. Zhao, Y. Zhang, K. Deng, Z. Rong, K. Yang, X. Yu, K. Li, P. Han and Z.-J. Zhu, Anal. Chem., 2019, 91, 2401–2408 CrossRef CAS PubMed.
- G. Yanovich, H. Agmon, M. Harel, A. Sonnenblick, T. Peretz and T. Geiger, Cancer Res., 2018, 78, 6001–6010 CAS.
- H.-M. Lu, S. Li, M. H. Black, S. Lee, R. Hoiness, S. Wu, W. Mu, R. Huether, J. Chen, S. Sridhar, Y. Tian, R. McFarland, J. Dolinsky, B. T. Davis, S. Mexal, C. Dunlop and A. Elliott, JAMA Oncol., 2019, 5, 51–57 CrossRef PubMed.
- J. Shan and Z. Ma, Microchim. Acta, 2017, 184, 969–979 CrossRef CAS.
- P. Saudemont, J. Quanico, Y.-M. Robin, A. Baud, J. Balog, B. Fatou, D. Tierny, Q. Pascal, K. Minier, M. Pottier, C. Focsa, M. Ziskind, Z. Takats, M. Salzet and I. Fournier, Cancer Cell, 2018, 34, 840–851 CrossRef CAS PubMed.
- M. V. Yezhelyev, A. Al-Hajj, C. Morris, A. I. Marcus, T. Liu, M. Lewis, C. Cohen, P. Zrazhevskiy, J. W. Simons, A. Rogatko, S. Nie, X. Gao and R. M. O'Regan, Adv. Mater., 2007, 19, 3146–3151 CrossRef CAS.
- S. Schluecker, Angew. Chem., Int. Ed., 2014, 53, 4756–4795 CrossRef CAS PubMed.
- S. Kim, T. G. Kim, S. H. Lee, W. Kim, A. Bang, S. W. Moon, J. Song, J. H. Shin, J. S. Yu and S. Choi, ACS Appl. Mater. Interfaces, 2020, 12, 7897–7904 CrossRef CAS PubMed.
- Z. Cheng, N. Choi, R. Wang, S. Lee, K. C. Moon, S. Y. Yoon, L. X. Chen and J. Choo, ACS Nano, 2017, 11, 4926–4933 CrossRef CAS PubMed.
- J. Wang, D. W. Liang, J. Feng and X. J. Tang, Anal. Chem., 2019, 91, 11045–11054 CrossRef CAS PubMed.
- Y. Q. Wang, B. Yan and L. X. Chen, Chem. Rev., 2013, 113, 1391–1428 CrossRef CAS PubMed.
- C. Zong, M. X. Xu, L. J. Xu, T. Wei, X. Ma, X. S. Zheng, R. Hu and B. Ren, Chem. Rev., 2018, 118, 4946–4980 CrossRef CAS PubMed.
- Y. X. Zou, S. Q. Huang, Y. X. Liao, X. P. Zhu, Y. Q. Chen, L. Chen, F. Liu, X. X. Hu, H. J. Tu, L. Zhang, Z. K. Liu, Z. Chen and W. H. Tan, Chem. Sci., 2018, 9, 2842–2849 RSC.
- H. W. Wang, Y. F. Cui, J. F. Wang, S. Liu, X. Zhang, X. L. Song, Y. Wang, J. D. Huang and J. H. Yu, Sens. Actuators, B, 2019, 287, 535–543 CrossRef CAS.
- H. Karabeber, R. Huang, P. Iacono, J. M. Samii, K. Pitter, E. C. Holland and M. F. Kircher, ACS Nano, 2014, 8, 9755–9766 CrossRef CAS PubMed.
- A. Oseledchyk, C. Andreou, M. A. Wall and M. F. Kircher, ACS Nano, 2017, 11, 1488–1497 CrossRef CAS PubMed.
- L. A. Lane, X. Qian and S. Nie, Chem. Rev., 2015, 115, 10489–10529 CrossRef CAS PubMed.
- S. Harmsen, M. A. Bedics, M. A. Wall, R. Huang, M. R. Detty and M. F. Kircher, Nat. Commun., 2015, 6, 6570 CrossRef CAS PubMed.
- S. Pal, A. Ray, C. Andreou, Y. Zhou, T. Rakshit, M. Wlodarczyk, M. Maeda, R. Toledo-Crow, N. Berisha, J. Yang, H.-T. Hsu, A. Oseledchyk, J. Mondal, S. Zou and M. F. Kircher, Nat. Commun., 2019, 10, 1926 CrossRef PubMed.
- M. F. Kircher, A. de la Zerda, J. V. Jokerst, C. L. Zavaleta, P. J. Kempen, E. Mittra, K. Pitter, R. Huang, C. Campos, F. Habte, R. Sinclair, C. W. Brennan, I. K. Mellinghoff, E. C. Holland and S. S. Gambhir, Nat. Med., 2012, 18, 829–834 CrossRef CAS PubMed.
- Y. Zhang, Y. Gu, J. He, B. D. Thackray and J. Ye, Nat. Commun., 2019, 10, 1926 CrossRef PubMed.
- S. Nie and S. R. Emory, Science, 1997, 275, 1102–1106 CrossRef CAS PubMed.
- K. Kneipp, Y. Wang, H. Kneipp, L. T. Perelman, I. Itzkan, R. R. Dasari and M. S. Feld, Phys. Rev. Lett., 1997, 78, 1667–1670 CrossRef CAS.
- D. Cialla-May, X. S. Zheng, K. Weber and J. Popp, Chem. Soc. Rev., 2017, 46, 3945–3961 RSC.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271–284 CrossRef CAS PubMed.
- X. Yi, L. Chen, X. Zhong, R. Gao, Y. Qian, F. Wu, G. Song, Z. Chai, Z. Liu and K. Yang, Nano Res., 2016, 9, 3267–3278 CrossRef CAS.
- Y. Ling, D. Zhang, X. Cui, M. Wei, T. Zhang, J. Wang, L. Xiao and Y. Xia, Angew. Chem., Int. Ed., 2019, 58, 1–6 CrossRef.
- F. Gong, N. Yang, X. Wang, Q. Zhao, Q. Chen, Z. Liu and L. Cheng, Nano Today, 2020, 32, 100851 CrossRef CAS.
- Q. Chen, L. Feng, J. Liu, W. Zhu, Z. Dong, Y. Wu and Z. Liu, Adv. Mater., 2016, 28, 7129–7136 CrossRef CAS PubMed.
- B. Li, Z. Gu, N. Kurniawan, W. Chen and Z. P. Xu, Adv. Mater., 2017, 29, 1700373 CrossRef PubMed.
- C.-C. Huang, W.-T. Chia, M.-F. Chung, K.-J. Lin, C.-W. Hsiao, C. Jin, W.-H. Lim, C.-C. Chen and H.-W. Sung, J. Am. Chem. Soc., 2016, 138, 5222–5225 CrossRef CAS PubMed.
- G. Yang, L. Xu, Y. Chao, J. Xu, X. Sun, Y. Wu, R. Peng and Z. Liu, Nat. Commun., 2017, 8, 902 CrossRef PubMed.
- Z. L. Zhao, H. H. Fan, G. F. Zhou, H. R. Bai, H. Liang, R. W. Wang, X. B. Zhang and W. H. Tan, J. Am. Chem. Soc., 2014, 136, 11220–11223 CrossRef CAS PubMed.
- M. Song, T. Liu, C. Shi, X. Zhang and X. Chen, ACS Nano, 2016, 10, 633–647 CrossRef CAS PubMed.
- P. Mi, D. Kokuryo, H. Cabral, H. Wu, Y. Terada, T. Saga, I. Aoki, N. Nishiyama and K. Kataoka, Nat. Nanotechnol., 2016, 11, 724–730 CrossRef CAS PubMed.
- S. Harmsen, R. Huang, M. A. Wall, H. Karabeber, J. M. Samii, M. Spaliviero, J. R. White, S. Monette, R. O'Connor, K. L. Pitter, S. A. Sastra, M. Saborowski, E. C. Holland, S. Singer, K. P. Olive, S. W. Lowe, R. G. Blasberg and M. F. Kircher, Sci. Transl. Med., 2015, 7, 271ra77 Search PubMed.
- Y. Qiu, Y. Zhang, M. Li, G. Chen, C. Fan, K. Cui, J.-B. Wan, A. Han, J. Ye and Z. Xiao, ACS Nano, 2018, 12, 7974–7985 CrossRef CAS PubMed.
- J. K. Lim and S.-W. Joo, J. Electroanal. Chem., 2007, 605, 68–72 CrossRef CAS.
- U. K. Sarkar, J. Mol. Struct., 2013, 1045, 42–46 CrossRef CAS.
- G. Kister, G. Cassanas and M. Vert, Polymer, 1998, 39, 267–273 CrossRef CAS.
- K. Kochan, H. Peng, E. S. H. Gwee, E. Izgorodina, V. Haritos and B. R. Wood, Analyst, 2019, 144, 901–912 RSC.
- A. C. S. Talari, Z. Movasaghi, S. Rehman and I. U. Rehman, Appl. Spectrosc. Rev., 2015, 50, 46–111 CrossRef CAS.
- M. Dudek, G. Zajac, E. Szafraniec, E. Wiercigroch, S. Tott, K. Malek, A. Kaczor and M. Baranska, Spectrochim. Acta, Part A, 2019, 206, 597–612 CrossRef CAS.
- A. Liang, H. Wang, D. Yao and Z. Jiang, Food Chem., 2019, 271, 39–46 CrossRef CAS PubMed.
- H. Zhang, G. Li, S. Li, L. Xu, Y. Tian, A. Jiao, X. Liu, F. Chen and M. Chen, Appl. Surf. Sci., 2018, 457, 684–694 CrossRef CAS.
- Y. Lu, C. Wu, R. You, Y. Wu, H. Shen, L. Zhu and S. Feng, Biomed. Opt. Express, 2018, 9, 4988–4997 CrossRef CAS PubMed.
- C. J. Saatkamp, M. L. de Almeida, J. A. Martins Bispo, A. L. Barbosa Pinheiro, A. B. Fernandes and L. Silveira Jr, J. Biomed. Opt., 2016, 21, 037001 CrossRef PubMed.
- D. M. Solis, J. M. Taboada, F. Obelleiro, L. M. Liz-Marzan and F. Javier Garcia de Abajo, ACS Photonics, 2017, 4, 329–337 CrossRef CAS PubMed.
- T. Vi, B. Walkenfort, M. Koenig, M. Salehi and S. Schluecker, Angew. Chem., Int. Ed., 2019, 58, 442–446 CrossRef PubMed.
- A. Kumar, S. Kim and J.-M. Nam, J. Am. Chem. Soc., 2016, 138, 14509–14525 CrossRef CAS PubMed.
- J.-E. Park, Y. Lee and J.-M. Nam, Nano Lett., 2018, 18, 7419 CrossRef CAS PubMed.
- D.-K. Lim, K.-S. Jeon, H. M. Kim, J.-M. Nam and Y. D. Suh, Nat. Mater., 2010, 9, 60–67 CrossRef CAS PubMed.
- D.-K. Lim, K.-S. Jeon, J.-H. Hwang, H. Kim, S. Kwon, Y. D. Suh and J.-M. Nam, Nat. Nanotechnol., 2011, 6, 452–460 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc01649g |
| This journal is © The Royal Society of Chemistry 2020 |