
Efficient co-delivery of microRNA 21 inhibitor and doxorubicin to cancer cells using core–shell tecto dendrimers formed via supramolecular host–guest assembly†
Cong
Song
a,
Yunchao
Xiao
a,
Zhijun
Ouyang
a,
Mingwu
Shen
 a and
Xiangyang
Shi
*ab
a and
Xiangyang
Shi
*ab
aState Key Laboratory for Modification of Chemical Fibers and Polymer Materials, International Joint Laboratory for Advanced Fiber and Low-dimension Materials, College of Chemistry, Chemical Engineering and Biotechnology, Donghua University, Shanghai 201620, P. R. China. E-mail: xshi@dhu.edu.cn
bCQM-Centro de Quimica da Madeira, Universidade da Madeira, Campus da Penteada, 9020-105 Funchal, Portugal
First published on 5th March 2020
Abstract
Development of versatile and powerful nanoplatforms for efficient therapeutic delivery represents a major topic for current nanomedicine. Herein, we present the development of core–shell tecto dendrimers (CSTDs) for co-delivery of a therapeutic gene and drug for enhanced anticancer therapy applications. In this work, CSTDs were first prepared via supramolecular recognition of β-cyclodextrin (CD)-decorated generation 5 (G5) poly(amidoamine) (PAMAM) dendrimers as cores and adamantane (Ad)-functionalized G3 PAMAM dendrimers as shell components. The formed CSTDs with each G5 dendrimer surrounded with 4.2 G3 dendrimers were evaluated as a gene vector for delivery of plasmid DNA encoding enhanced green fluorescent protein as well as microRNA 21 inhibitor (miR 21i). We show that under an appropriate N/P ratio, the CSTDs enable effective transfection of both genetic materials to cancer cells. In particular, the transfection of miR 21i led to the inhibition of cancer cell migration, decreased miR 21 gene expression, and the effective regulation of the target genes and proteins (e.g., PTEN, PDCD4, p53, and Caspase-3). Furthermore, we revealed that the CSTDs were able to co-deliver miR 21i and an anticancer drug doxorubicin, leading to enhanced therapeutic efficacy to cancer cells in vitro. Our findings imply that the developed CSTDs could be adopted as a versatile platform for effective co-delivery of different therapeutic components for enhanced anticancer therapy applications.
Introduction
As is well known, the occurrence and development of tumors is a multi-factor, multi-step and complex biological process. At present, single-mode chemotherapy has limited efficacy, and drug resistance is always a major obstacle of tumor chemotherapy.1 Therefore, finding new approaches to integrate different therapeutic elements for combination therapy is always promising. The combined drug and gene therapy strategy has been widely used for enhanced cancer therapy due to the potential synergetic effects.2–5 For example, Xiong et al.3 employed multifunctional micelles to simultaneously carry MDR-1 siRNA and doxorubicin (DOX) to overcome the multidrug resistance of breast cancer. Deng et al.4 loaded microRNA 34a and DOX into hyaluronic acid-modified chitosan nanoparticles (NPs) for co-delivery to breast cancer cells to induce synergistic tumor suppression effects. Therefore, co-delivery of gene and drug using nanocarriers can be considered as an important strategy for enhanced anticancer therapy for future clinical translations.MicroRNAs, as a class of small RNAs with regulatory gene expression, control biological process such as cell proliferation, differentiation, angiogenesis and apoptosis.6,7 The microRNAs may serve as either oncogenes or tumor suppressors under certain conditions and involve in the occurrence of many types of cancer.8 For instance, microRNA 21 (miR 21) has been noted to have a high expression in several tumors such as triple negative breast cancer, pancreatic cancer, and so on.9–11 Abnormal expression of miR 21 could facilitate the tumor growth and spread by regulating phosphatase and TENsin homolog (PTEN) expression and PTEN-associated pathways to mediate the growth, migration, and invasion of cancer cells.12 Consequently, miR 21 inhibitors (miR 21i), miR 21 antisense oligonucleotides, have been designed to specifically inhibit the proliferation of tumor cells.10,13 In our previous work, we have shown that co-delivery of gemcitabine and miR 21i to pancreatic cancer cells can be realized by dendrimer-entrapped gold NPs as a vector, and the co-delivery affords significantly enhanced cytotoxic response when compared to the single delivery of gemcitabine.10 The prior work underscores the importance to apply both microRNA inhibitor and anticancer drug for synergistic tumor therapy, in particular for a tumor subgroup still lacking an efficient and specific therapy.
Compared with low-generation poly(amidoamine) (PAMAM) dendrimers, high-generation PAMAM dendrimers have been widely investigated in the areas of gene14,15 and drug16,17 delivery due to their better performances such as perfect water solubility, non-immunogenicity, and easiness of functionalization.16,18 Unfortunately, synthesis of high generation PAMAM dendrimers is time consuming and tedious, limiting their further biomedical applications.19,20 Therefore, core–shell tecto dendrimers (CSTDs) with similar structures and performances to high-generation dendrimers have aroused considerable attention.21–23 Among them, CSTDs have been prepared through supramolecular host–guest assembly between cyclodextrin (CD)-modified generation 5 (G5) dendrimer and adamantane (Ad)-modified generation 3 (G3) dendrimer.24 Through the host–guest supramolecular assembly of CD and Ad, amine-terminated G5-CD/Ad-G3 CSTDs with G5 core and G3 shell can be formed for significantly enhanced gene delivery applications. Particularly, the amine-surfaced CSTDs were able to transfect the luciferase gene with an efficiency 20 times and 170 times higher than the single-generation G5-CD and G3-Ad dendrimers, respectively. However, the developed CSTDs have not been ever used for functional gene delivery or drug/gene co-delivery to achieve therapeutic applications.
Based on the above findings, we present here the synthesis and use of the G5-CD/Ad-G3 CSTDs for co-delivery of miR 21i and DOX for enhanced anticancer therapy applications (Fig. 1). The synthesized G5-CD/Ad-G3 CSTDs were characterized via different techniques. The CSTDs were then systematically evaluated after complexing with an enhanced green fluorescent protein (EGFP)-encoding plasmid DNA (pDNA) or microRNA 21 inhibitor (miR 21i) to form polyplexes through different methods. Besides the use of cytotoxicity assay, flow cytometry and confocal microscopy to check the cytocompatibility, cellular uptake efficiency, and gene expression, we also checked the ability of CSTDs/miR 21i transfection to inhibit the cancer cell migration through wound healing assay, and to regulate the expression of genes and proteins through real-time polymerase chain reaction (PCR) and Western blot assays. Moreover, the co-delivery of DOX/miR 21i using CSTDs was also investigated for enhanced anticancer therapy in vitro. According to our thorough literature investigation, the current study provides a very first example related to the development of CSTDs formed through supramolecular host–guest recognition for co-delivery of drug/gene for enhanced anticancer therapy applications. Compared to other nanocarriers (e.g., micelles and NPs), the prepared CSTDs have defined structure, size and composition, and should be more controllable for biomedical applications.
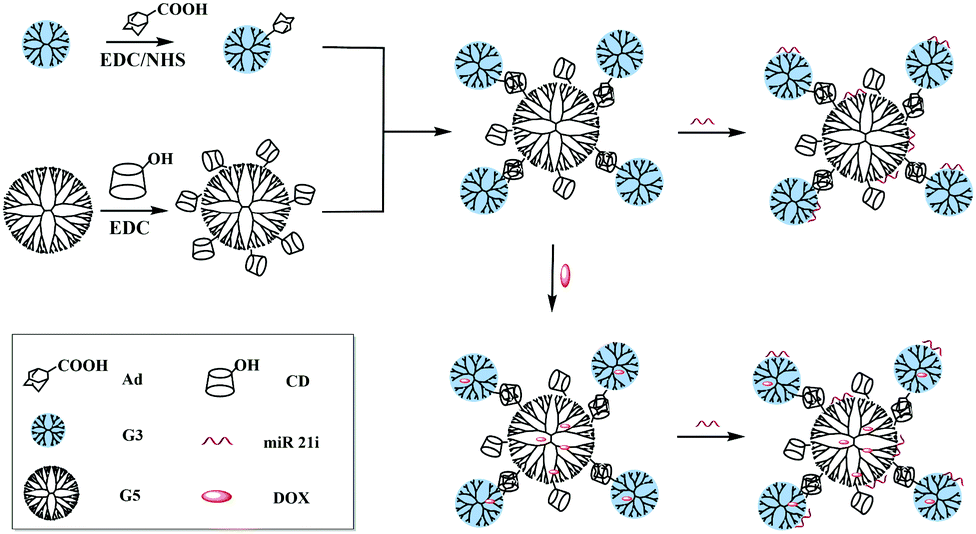 | ||
| Fig. 1 Synthesis of G5-CD/G3-Ad CSTDs for compacting miR 21i or co-load DOX and miR 21i for drug delivery applications. | ||
Experimental
Preparation and characterization of G5-CD/Ad-G3 CSTDs
The G5-CD/Ad-G3 CSTDs were prepared according to a procedure reported in our previous work.24 The CSTDs were characterized using 1H NMR, two dimensional nuclear Overhauser effect NMR spectroscopy (2D NOESY), and atomic force microscopy (AFM). 1H NMR and 2D NOESY spectra were recorded using a Bruker AV600 NMR spectrometer. Samples were dissolved in D2O before measurements. AFM was performed with 3D molecular force probe (Asylum Research, Santa Barbara, CA) to observe the morphology of the samples. Samples were dropped onto silicon wafers and air dried before measurements. The terminal primary amine groups present on the surface of the G5-CD/Ad-G3 CSTDs were quantified using Megazyme's PANOPA Assay Kit according to the manufacturer's instruction.Preparation and characterization of CSTDs/pDNA and CSTDs/miR 21i polyplexes
pDNA encoding EGFP was used as a model, and CSTDs/pDNA polyplexes were formed according to the literature.24 Similarly, CSTDs/miR 21i polyplexes were prepared under different N/P ratios using the same method. The CSTDs with different amounts were dissolved in phosphate buffered saline (PBS, pH = 7.4) and mixed with 1 μg miR 21i. The mixture was incubated at 37 °C for 30 min before further characterization or transfection.For gel retardation assay, 0.1 μg mL−1 ethidium bromide (EB) and 1.0% (w/v) agarose gel were mixed in Tris–acetate–EDTA buffer. Polyplexes were prepared using 1 μg pDNA or 1 μg miR 21i at different N/P ratios (0.125![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1–5:1), naked pDNA or miR 21i (1 μg) was used for comparison. Gel electrophoresis was performed at 80–90 V for 20–30 min. The retardation of the polyplexes was imaged using a gel image analysis system (Shanghai FURI Science & Technology, Shanghai, China). Surface potentials and hydrodynamic sizes of the polyplexes under various N/P ratios (2:1, 5:1, and 10:1, respectively) were determined via a Malvern Zetasizer Nano ZS system (Worcestershire, UK) equipped with a standard 633 nm laser. The morphology of the polyplexes was also observed by AFM.
:1–5:1), naked pDNA or miR 21i (1 μg) was used for comparison. Gel electrophoresis was performed at 80–90 V for 20–30 min. The retardation of the polyplexes was imaged using a gel image analysis system (Shanghai FURI Science & Technology, Shanghai, China). Surface potentials and hydrodynamic sizes of the polyplexes under various N/P ratios (2:1, 5:1, and 10:1, respectively) were determined via a Malvern Zetasizer Nano ZS system (Worcestershire, UK) equipped with a standard 633 nm laser. The morphology of the polyplexes was also observed by AFM.
Preparation and characterization of DOX-loaded CSTDs or CSTDs/miR 21i polyplexes
To load DOX within the CSTDs or prepare DOX-loaded CSTDs/miR 21i polyplexes, DOX was first physically encapsulated within the CSTDs according to the literature,23 and then DOX-loaded CSTDs were complexed with the miR 21i at an N/P ratio of 10:1. Firstly, DOX·HCl was dispersed in methanol, and added with triethylamine to neutralize the hydrochloride salt to form the hydrophobic DOX. The methanolic solution of DOX was added to an aqueous solution of CSTDs under stirring overnight to allow the evaporation of the methanol solvent. The solution was centrifuged to get the drug-loaded complexes in the supernatant. Meanwhile, the precipitate associated to non-encapsulated free DOX was also collected, dissolved in methanol, and quantified using a Lambda 25 UV-vis spectrophotometer (PerkinElmer, Waltham, MA). The loading of DOX within the dendrimers was quantified by subtracting the free DOX amount in the collected precipitate from the initial DOX amount according to the literature.23 To co-load DOX and miR 21i, the generated CSTDs/DOX complexes were employed to compact miR 21i at an N/P ratio of 10:1, and the mixture was incubated at 37 °C for 30 min. The DOX release kinetics from the CSTDs/DOX complexes was studied under two different pHs (pH = 7.4 and pH = 5.0) at 37 °C according to the literature.25,26 For PCR experiments, the upstream and downstream primer sequences of each gene are shown in Table S1. See more experimental details in ESI.†
Results and discussion
Synthesis and characterization of G5-CD/Ad-G3 CSTDs
In this study, G5-CD/Ad-G3 CSTDs were selected as a vector to co-deliver therapeutic gene and anticancer drug due to their sufficient stability, large internal space and high gene transfection efficiency.21–24 G5-CD/Ad-G3 CSTDs were prepared via supramolecular recognition of β-cyclodextrin (CD)-modified G5 PAMAM dendrimers as cores and adamantane (Ad)-modified G3 PAMAM dendrimers as shell components following protocols reported in our previous study.24 The synthesized G5-CD, G3-Ad, and CSTDs were characterized via1H NMR (Fig. S1, ESI†). Shown in the 1H NMR spectra of G3-Ad, G5-CD and G5-CD/G3-Ad CSTDs, the peaks at 1.5–1.9 ppm (Fig. S1a, ESI†), 3.5–4.1 ppm/5 ppm (Fig. S1b, ESI†), and 2.4–3.4 ppm (Fig. S1a–c, ESI†) can be respectively assigned to the attached Ad and β-CD moieties, and the dendrimer methylene protons. By integrating the characteristic peaks, the numbers of Ad conjugated onto each G3 dendrimer, β-CD conjugated on each G5 dendrimer, and G3-Ad assembled onto each G5-CD dendrimer were calculated to be 1.2, 7.6, and 4.2, respectively.To further demonstrate the successful formation of G5-CD/Ad-G3 CSTDs via β-CD-Ad host–guest recognition, 2D NOESY spectrum of the CSTDs was collected (Fig. S2, ESI†). The strong correlation signals of G5-CD/Ad-G3 can be observed between β-CD and Ad moieties (the protons of Ad at 1.5–1.7 ppm and the protons of β-CD at 3.6 ppm and 3.9 ppm), suggesting the success of the synthesis of CSTDs via supramolecular host–guest interaction.
Preparation and characterization of CSTDs/pDNA polyplexes for pDNA transfection
Generally speaking, pDNA encoding EGFP was usually employed to evaluated the gene transfection efficiency of the designed vectors in vitro.18,22 First, the amount of terminal primary amines of the G5-CD/Ad-G3 CSTDs was measured by Megazyme's PANOPA Assay Kit to be 80 per CSTD (Table S2, ESI†), in agreement with the literature.24 Then, the ability of G5-CD/Ad-G3 CSTDs to compact pDNA under different N/P ratios was tested by agarose gel retardation assay (Fig. S3, ESI†). Clearly, at the N/P ratio of 2 or above, the migration of pDNA can be completely retarded, implying that the pDNA can be condensed at an N/P ratio of 2 or above to create stable polyplexes. Further, the hydrodynamic sizes and surface potentials of the CSTDs/pDNA polyplexes at different N/P ratios were determined (Fig. S4, ESI†). Clearly, the hydrodynamic sizes of the CSTDs/pDNA polyplexes are in the range of 135–155 nm, while their surface potentials are in the range of 24–28 mV under the studied N/P ratios. Our data reveal that although a higher N/P ratio theoretically consumes more G5-CD/Ad-G3 CSTDs to form a larger polyplexes in a certain range of N/P ratios, the hydrodynamic sizes and positive surface potentials do not significantly change. The measured hydrodynamic sizes and surface potentials are appropriate for their further gene delivery applications.To test the CSTDs/pDNA polyplexes for gene delivery application, we first checked the cytotoxicity of the vector and the polyplexes via CCK-8 cell viability assay (Fig. S5, ESI†). Apparently, the viability of MDA-MB-231 cells after treated with both G5-CD/Ad-G3 CSTDs and CSTDs/pDNA polyplexes remain at 64% or above at the CSTD concentration up to 3000 nM. As expected, the CSTDs/pDNA polyplexes possess a lower cytotoxicity than G5-CD/Ad-G3 CSTDs due to the partial neutralization of the vector surface amines with the negatively charged pDNA. The good cytocompatibility of the polyplexes is mandatory for their safe and efficient gene transfection applications.
Then, cellular uptake of the CSTDs/pDNA polyplexes was examined via flow cytometry (Fig. S6, ESI†). Apparently, cells treated with the polyplexes show the highest Cy3-derived fluorescence intensity at an N/P ratio of 5, validating the effective cellular uptake of the polyplexes at the optimized N/P ratio. Fluorescence microscopy was adopted to investigate the effective gene transfection of the CSTDs/pDNA polyplexes at different N/P ratios for 24 h through the observation of EGFP green fluorescence (Fig. S7, ESI†). Clearly, at the N/P ratio of 5, the cells display the strongest green fluorescence signals, correlating well with the optimized cellular uptake of the polyplexes at an N/P ratio of 5 (Fig. S6, ESI†).
Formation and characterization of CSTDs/miR 21i polyplexes before transfection
With the proven gene transfection ability of the CSTDs, we next compressed miR 21i with the vector and attempted to transfect MDA-MB-231 cells for cancer therapy applications. The morphology of the vector and vector/miR 21i polyplexes was firstly observed via AFM (Fig. 2). It can be seen that both the vector and the polyplexes display a semi-spherical shape with the heights of CSTDs (Fig. 2a) and CSTDs/miR 21i polyplexes (Fig. 2b) at about 9 and 15 nm, respectively. The CSTDs possess a larger size than those of the single-generation G3-Ad (3.1 nm) and G5-CD (5.2 nm) dendrimers described in our previous report,24 suggesting that the CSTDs have been successfully synthesized via supramolecular host–guest assembly. The larger size of the CSTDs/miR 21i polyplexes than that of the CSTDs indicates the successful compaction of the miR 21i through electrostatic interaction.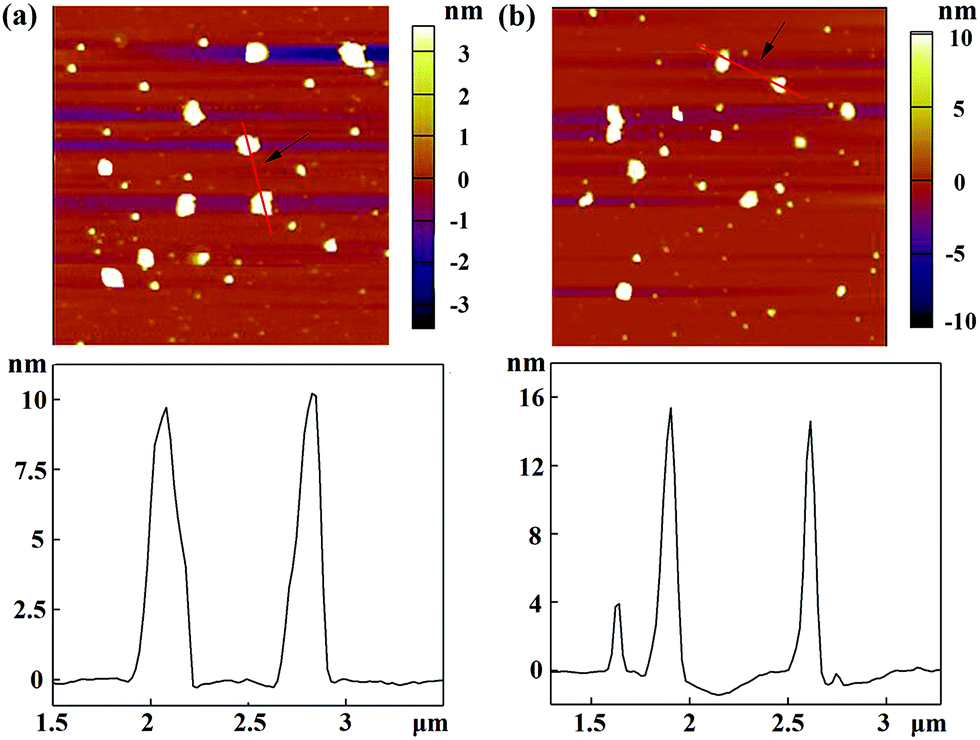 | ||
| Fig. 2 AFM images and corresponding height profiles of (a) G5-CD/Ad-G3 CSTDs and (b) CSTDs/miR 21i polyplexes (N/P ratio = 10:1) deposited onto silicon wafers. | ||
The ability of CSTDs to compact miR 21i was next assessed by gel retardation assay (Fig. 3a). Apparently, at the N/P ratio of 2 or above, the CSTDs are able to absolutely inhibit the migration of miR 21i, in agreement with the above pDNA compaction data. The hydrodynamic sizes and surface potentials of the CSTDs/miR 21i polyplexes were determined under different N/P ratios (Fig. 3b and c). The measured ranges of hydrodynamic sizes and surface potentials of the polyplexes are 200–250 nm and 28–35 mV, respectively, which are suitable for further gene transfection studies.
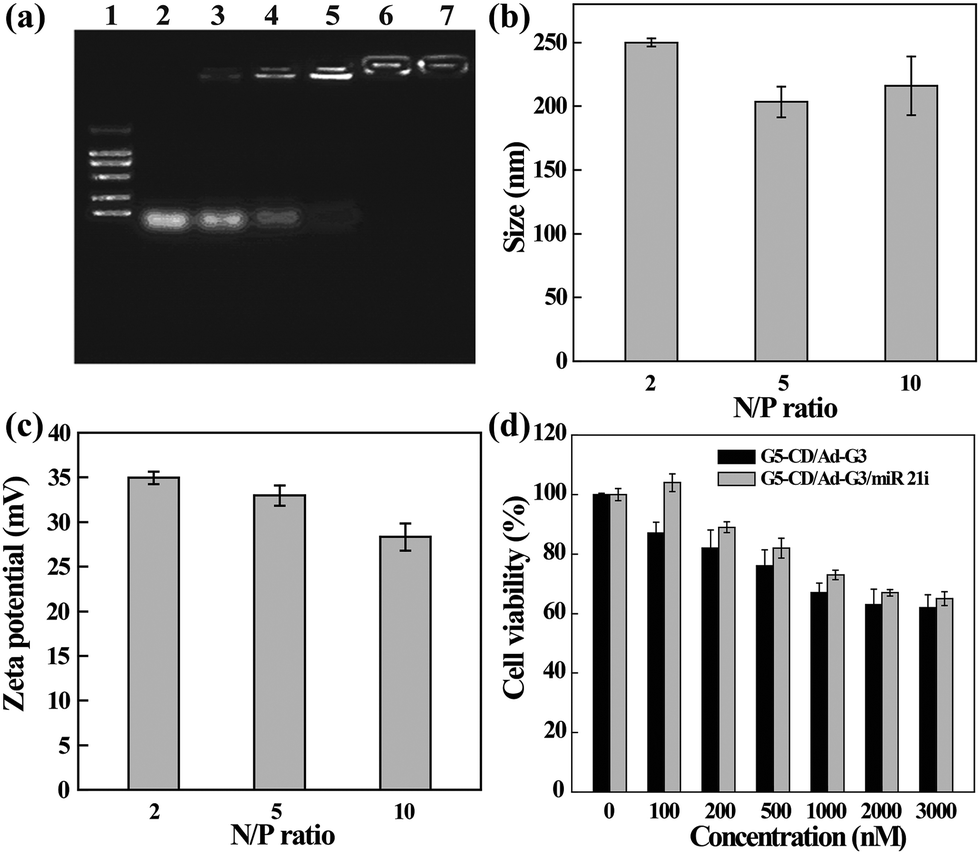 | ||
| Fig. 3 (a) Agarose gel retardation assay of miR 21i compacted with G5-CD/Ad-G3 CSTDs under different N/P ratios. Lane 1, DNA marker 2000; lane 2, N/P = 0.125:1; lane 3, N/P = 0.25:1; lane 4, N/P = 0.5:1; lane 5, N/P = 1:1; lane 6, N/P = 2:1; and lane 7, N/P = 5:1. (b) The hydrodynamic sizes and (c) zeta potentials of the CSTDs/miR 21i polyplexes under different N/P ratios (mean ± SD, n = 3). (d) The viability of MDA-MB-231 cells treated with the CSTDs and CSTDs/miR 21i polyplexes for 24 h at different concentrations. | ||
The cytotoxicity and intracellular uptake of the CSTDs/miR 21i polyplexes were next tested in vitro before transfection studies. As shown in Fig. 3d, the viability of MDA-MB-231 cells treated with the CSTDs/miR 21i polyplexes for 24 h remains at a high level (65% or above) under different concentrations, and at the same concentration, cells treated with the polyplexes have a better viability than those treated with the CSTDs due to the partial neutralization of the surface positive charge of the CSTDs. The results are consistent with those of the CSTDs/pDNA polyplexes, revealing that the CSTDs/miR 21i polyplexes possess good cytocompatibility for gene therapy applications, and single mode of gene transfection does not appreciably affect the cell viability.
In addition, the cellular uptake of G5-CD/Ad-G3/miR 21i polyplexes at different N/P ratios was also evaluated by using confocal microscopy (Fig. 4) and flow cytometry (Fig. S8, ESI†). Compared to control group, free miR 21i group does not display any red fluorescence signals, meaning that free miR 21i is unable to be taken up by cells. However, at different N/P ratios, red fluorescence signals can be found around the nucleus, suggesting that the CSTDs/Cy3-miR 21i polyplexes can be taken up by MDA-MB-231 cells. The cellular uptake efficiency is the highest at the N/P ratio of 10, which can also be quantitatively confirmed through flow cytometry assay (Fig. S8, ESI†).
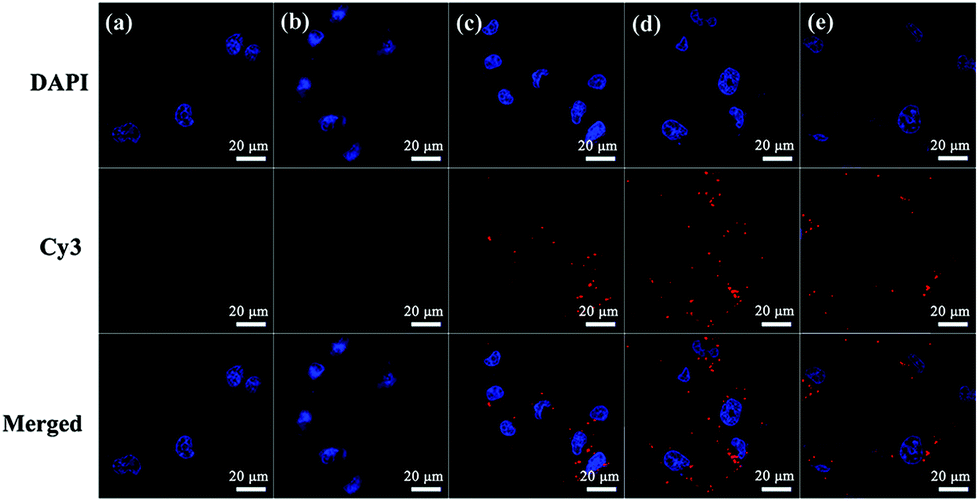 | ||
| Fig. 4 Confocal microscopic images of MDA-MB-231 cells treated with the CSTDs/Cy3-miR 21i polyplexes at different N/P ratios: cells without treatment (a), cells treated with naked Cy3-miR 21i (b), and cells treated with the CSTDs/Cy3-miR 21i polyplexes at N/P = 5:1 (c), N/P = 10:1 (d), and N/P = 15:1 (e). Red fluorescence represents Cy3-labeled miR 21i and blue fluorescence shows the DAPI-stained cell nucleus. | ||
Transfection of CSTDs/miR 21i polyplexes for cancer cell therapy
To prove the effective cancer cell therapy after the CSTDs/miR 21i polyplexes were transfected, we investigated the inhibition of cancer cell migration through wound healing assay (Fig. 5a). The images of the wound under different conditions were observed by optical microscopy and the cell migration area was quantified by Image J software. Clearly, compared to the control cells without treatment and cells treated with free miR 21i, the cells treated with the polyplexes display much more limited migration in-between the scraped wound gap at 24 and 48 h post transfection. The corresponding quantitative data of cell migration area further reveal that the cell migration area in the polyplexes group at 48 h is significantly less than those of the control and free miR 21i groups (Fig. 5b, p < 0.01). This suggests that the transfection of CSTDs/miR 21i polyplexes leads to a strong anti-migratory effect of cancer cells.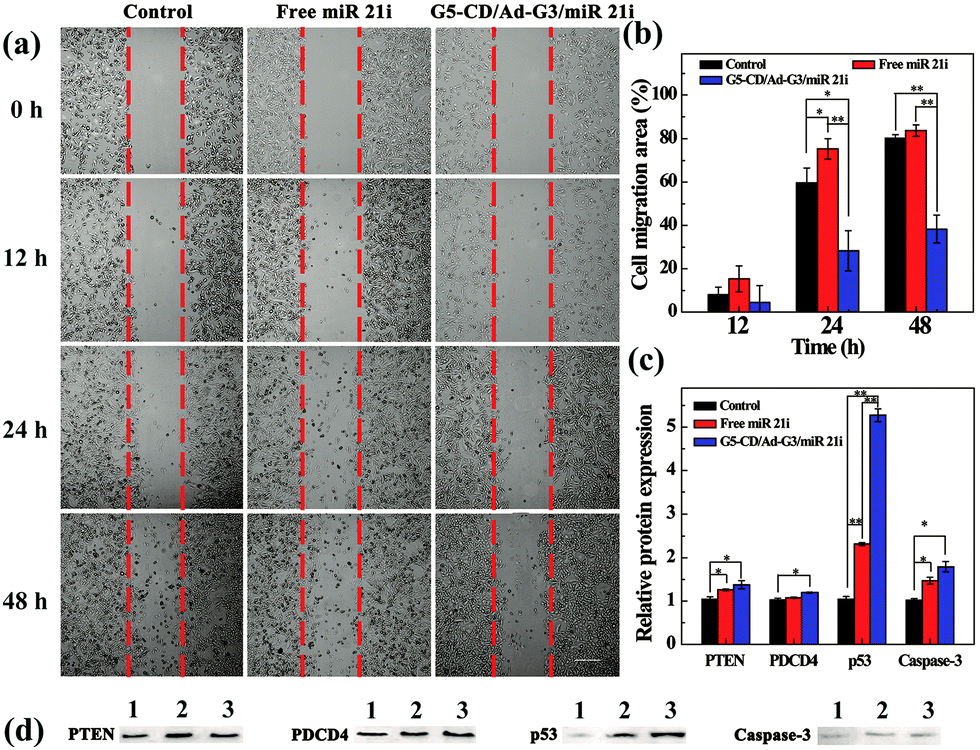 | ||
| Fig. 5 (a) Phase contrast microscopic images and (b) the corresponding cell migration area of MDA-MB-231 cells after transfection with the CSTDs/miR 21i polyplexes for 0, 12, 24 and 48 h, respectively for the wound-healing assay. (c) Quantitative analysis of protein expression based on Western blot assay. (d) Images of protein expression based on Western blot assay. Lane 1, control cells without treatment; lane 2, cells treated with free miR 21i; and lane 3, cells treated with the CSTDs/miR 21i polyplexes. | ||
The transfection of CSTDs/miR 21i polyplexes to cancer cells was further proven via PCR analysis of the expression of miR 21 and other genes (PTEN, PDCD4, p53, and Caspase-3) within the cells. CSTDs/miR 21i polyplexes at an N/P ratio of 10 were transfected to cells for 48 h (Fig. S9, ESI†). Apparently, the miR 21 gene expression level in the polyplexes group is significantly lower than those in the control and free miR 21i groups (p < 0.05). The expression levels of the corresponding target genes such as PTEN, PDCD4, p53, and Caspase-3 in the polyplexes group are significantly higher than those in the control and free miR 21i groups.
Western blot assay was finally carried out to study the effect of the CSTDs/miR 21i polyplexes on the expression of target proteins in cancer cells (Fig. 5c and d). The corresponding target protein (PTEN, PDCD4, p53, and Caspase-3) expression level in the polyplexes group are also much higher than those in the control and free miR 21i groups, especially the p53 protein expression. These data indicate that the G5-CD/Ad-G3 CSTDs are an excellent gene vector to transfect miR 21i to modulate the expression of related target genes/proteins and the activation of the cell apoptosis process.
Co-delivery of miR 21i and DOX by CSTDs for enhanced cancer cell therapy in vitro
The co-delivery of gene and drug using the vector could be considered to achieve synergistic treatment of cancer due to the weak therapeutic efficacy of the single-mode gene therapy.4 DOX, as a common chemotherapy drug, has been reported to crosslink DNA, inhibit cellular DNA, RNA and protein synthesis, and induce apoptosis of cancer cells.27,28 Therefore, we attempted to load DOX within the interior of CSTDs via physical encapsulation. By calculation, we show that the DOX encapsulation efficiency within the CSTDs is 53.9%, and there are around 5.0 DOX molecules contained in each CSTD on the average. It should be noted that the encapsulation capacity of CSTDs is much higher than that of single-generation functional G5 dendrimers,29 which can only encapsulate around one DOX molecule per G5 dendrimer. The increased DOX loading capacity should be due to the significantly enlarged internal cavity of the CSTDs. Next, the DOX release kinetics in vitro under different pHs was investigated (Fig. 6a). The DOX release rate from the CSTDs/DOX complexes at pH 5.0 is much greater than at pH 7.4. In the first 12 h, there are approximately 22% of DOX released from the CSTDs under pH 5.0, while only about 8% of the DOX was released under pH 7.4. At 48 h, there are about 57% and 34% of the DOX released under pH 5.0 and pH 7.4, respectively. Since the tumor microenvironment is slightly acidic,30 the therapeutic effect of tumor cells could be enhanced when the anticancer drugs are fast released.31 Therefore, the result of drug release kinetics assay can be considered to be beneficial for the treatment of tumors.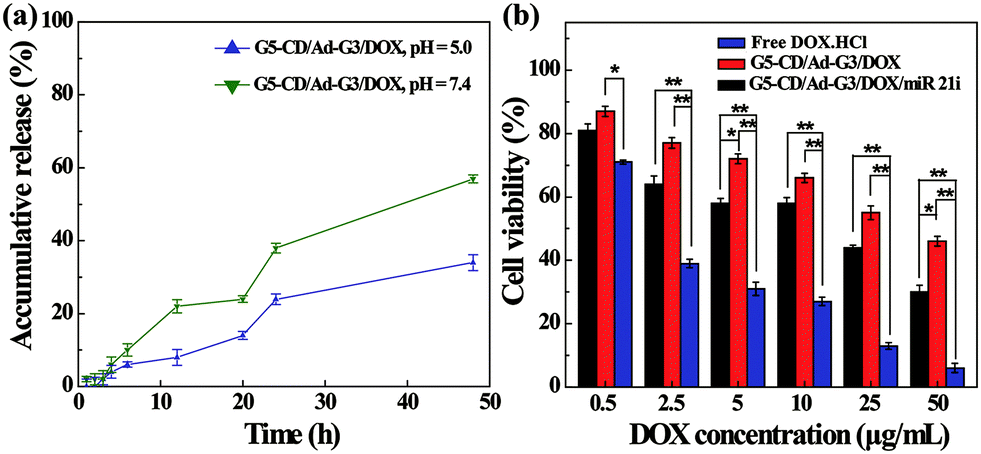 | ||
| Fig. 6 (a) DOX release profiles from the CSTDs in phosphate buffer (pH = 5.0 and pH = 7.4) at 37 °C. (b) The viability of MDA-MB-231 cells treated with the CSTDs/DOX/miR 21i polyplexes, CSTDs/DOX complexes, and free DOX·HCl under different DOX concentrations. | ||
The therapeutic efficacy of the different formulations was evaluated by CCK-8 cell viability assay. As shown in Fig. 6b, the viability of MDA-MB-231 cells decreases with the increase of DOX concentration for all groups. As expected, the cell viability of the CSTDs/DOX/miR 21i group or CSTDs/DOX group were significantly higher than that of free DOX·HCl group at each DOX concentration (Except for the DOX concentration of 0.5 μg mL−1, p < 0.01). This is because for the CSTDs/DOX/miR 21i and CSTDs/DOX groups, it takes a certain time period for the DOX to be released to exert its therapeutic functionality, and the concentration of released DOX is lower than that of free DOX at the same time point, in agreement with the literature.29,32,33 The DOX IC50 follows the order of free DOX·HCl (1.64 μg mL−1) < CSTDs/DOX/miR 21i (11.55 μg mL−1) < CSTDs/DOX (45.37 μg mL−1). Apparently, the cooperative cancer cell inhibition effect can be realized through co-delivery of miR 21i and DOX with the CSTDs.
The cellular uptake and internalization of the CSTDs/DOX/miR 21i and CSTDs/DOX formulations were checked by flow cytometry (Fig. S10, ESI†) and confocal microscopy (Fig. S11, ESI†), respectively. Clearly, the fluorescence intensity of cells treated with the CSTDs/DOX/miR 21i and CSTDs/DOX increases with the DOX concentration, implying the cellular uptake of the complexes or polyplexes. Furthermore, the internalization of the complexes or polyplexes by cancer cells was validated by confocal microscopy. With the increase of DOX concentration, the red fluorescence signals of cells that are associated to DOX increase, indicating that the complexes or polyplexes can be internalized within the cytoplasm and part of DOX can be delivered to cell nucleus for effective cancer cell treatment.
Conclusions
In summary, we report an efficient approach to co-deliver therapeutic miR 21i gene and anticancer drug DOX within CSTDs formed via supramolecular assembly for enhanced cancer therapy applications. Due to the large interior space and the excellent gene compact ability of the CSTDs, we were able to load anticancer drug DOX within their internal cavities, and compact miR 21i via electrostatic interaction. We show that the CSTDs enable effective transfection of both pDNA and miR 21i to cancer cells under a suitable N/P ratio, and the transfection efficiency of CSTDs/miR 21i is the highest at the N/P ratio of 10, leading to effective regulation of the target genes and proteins. In particular, the effective miR 21i transfection leads to the inhibition of cancer cell migration and activation of apoptosis of cancer cells, and co-delivery of miR21i and DOX using CSTDs leads to significantly improved therapeutic efficacy of cancer cells when compared to vectors loaded only with DOX. The strategy to co-load genetic materials and anticancer drugs within CSTDs may be extended to load other therapeutically active ingredients for enhanced therapy of other types of diseases.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research has been financially supported by the National Natural Science Foundation of China (81761148028 and 21773026), the Science and Technology Commission of Shanghai Municipality (19410740200, 19XD1400100, and 18520750400), the National Key R&D Program (2017YFE0196200), and the Fundamental Research Funds for the Central Universities (CUSF-DH-D-2019073). X. Shi also thanks the support by FCT-Fundação para a Ciência e a Tecnologia through the CQM Base Fund – UIDB/00674/2020, and Programmatic Fund – UIDP/00674/2020, and by ARDITI-Agência Regional para o Desenvolvimento da Investigação Tecnologia e Inovação, through the project M1420-01-0145-FEDER-000005 – Centro de Química da Madeira – CQM+ (Madeira 14-20 Program).Notes and references
- D. Hanahan and R. A. Weinberg, Cell, 2011, 144, 646–674 CrossRef PubMed.
- R. J. Youle and A. Strasser, Nat. Rev. Mol. Cell Biol., 2008, 9, 47–59 CrossRef PubMed.
- L. Zhang, Z. Lu, Q. Zhao, J. Huang, H. Shen and Z. Zhang, Small, 2011, 7, 460–464 CrossRef PubMed.
- D. Xiongwei, C. Minjun, Z. Jiakun, H. Kelei, Y. Zhaoxia, Z. Zhixiang, X. Xiangqian, Y. Yishu, S. Wang, W. Yan and Z. Yi, Biomaterials, 2014, 35, 4333–4344 CrossRef PubMed.
- X. Li, L. X. Xing, Y. Hu, Z. J. Xiong, R. Z. Wang, X. Y. Xu, L. F. Du, M. W. Shen and X. Y. Shi, Acta Biomater., 2017, 62, 273–283 CrossRef PubMed.
- F. Passetti, C. G. Ferreira and F. F. Costa, Pharmacogenomics J., 2009, 9, 1–13 CrossRef.
- M. D. Jansson and A. H. Lund, Mol. Oncol., 2012, 6, 590–610 CrossRef.
- P. E. Blower, J.-H. Chung, J. S. Verducci, S. Lin, J.-K. Park, Z. Dai, C.-G. Liu, T. D. Schmittgen, W. C. Reinhold, C. M. Croce, J. N. Weinstein and W. Sadee, Mol. Cancer Ther., 2008, 7, 1–9 CrossRef.
- M. M. Abdulhussain, N. A. Hasan and A. G. Hussain, Ind. J. Clin. Biochem., 2019, 34, 26–38 Search PubMed.
- L. Lin, Y. Fan, F. Gao, L. Jin, D. Li, W. Sun, F. Li, P. Qin, Q. Shi, X. Shi and L. Du, Theranostics, 2018, 8, 1923–1939 CrossRef.
- T. J. Lee, J. Y. Yoo, D. Shu, H. Li, J. Zhang, J.-G. Yu, A. C. Jaime-Ramirez, M. Acunzo, G. Romano, R. Cui, H.-L. Sun, Z. Luo, M. Old, B. Kaur, P. Guo and C. M. Croce, Mol. Ther., 2017, 25, 1544–1555 CrossRef.
- H. Mollaei, R. Safaralizadeh and Z. Rostami, J. Cell. Physiol., 2019, 234, 12369–12384 CrossRef.
- R. Devulapally, K. Foygel, T. V. Sekar, J. K. Willmann and R. Paulmurugan, ACS Appl. Mater. Interfaces, 2016, 8, 33412–33422 CrossRef.
- W. X. Hou, P. Wei, L. D. Kong, R. Guo, S. G. Wang and X. Y. Shi, J. Mater. Chem. B, 2016, 4, 2933–2943 RSC.
- L. D. Kong, Y. L. Wu, C. S. Alves and X. Y. Shi, Nanomedicine, 2016, 11, 3103–3115 CrossRef.
- J. Zhu and X. Shi, J. Mater. Chem. B, 2013, 1, 4199–4211 RSC.
- Z. Xiong, M. Shen and X. Shi, Sci. China Mater., 2018, 61, 1387–1403 CrossRef.
- J. Haensler and F. C. Szoka, Bioconjugate Chem., 1993, 4, 372–379 CrossRef.
- C. J. Hawker and J. M. J. Frechet, J. Am. Chem. Soc., 1990, 112, 7638–7647 CrossRef.
- D. A. Tomalia, Nanomedicine, 2012, 7, 953–956 CrossRef CAS PubMed.
- J. Li, D. R. Swanson, D. Qin, H. M. Brothers, L. T. Piehler, D. Tomalia and D. J. Meier, Langmuir, 1999, 15, 7347–7350 CrossRef CAS.
- S. Uppuluri, D. R. Swanson, L. T. Piehler, J. Li, G. L. Hagnauer and D. A. Tomalia, Adv. Mater., 2000, 12, 796–800 CrossRef CAS.
- J. Wang, D. Li, Y. Fan, M. Shi, Y. Yang, L. Wang, Y. Peng, M. Shen and X. Shi, Nanoscale, 2019, 11, 22343–22350 RSC.
- F. Chen, L. Kong, L. Wang, Y. Fan, M. Shen and X. Shi, J. Mater. Chem. B, 2017, 5, 8459–8466 RSC.
- D. Li, Y. Fan, M. Shen, I. Banyai and X. Shi, J. Mater. Chem. B, 2019, 7, 277–285 RSC.
- B. Zhou, L. Zhao, M. Shen, J. Zhao and X. Shi, J. Mater. Chem. B, 2017, 5, 1542–1550 RSC.
- S. Gorini, A. De Angelis, L. Berrino, N. Malara, G. Rosano and E. Ferraro, Oxid. Med. Cell. Longevity, 2018, 2018, 7582730 Search PubMed.
- T. A. Nissinen, J. Degerman, M. Rasanen, A. R. Poikonen, S. Koskinen, E. Mervaala, A. Pasternack, O. Ritvos, R. Kivela and J. J. Hulmi, Sci. Rep., 2016, 6, 32695 CrossRef CAS.
- Y. Wang, X. Y. Cao, R. Guo, M. W. Shen, M. G. Zhang, M. F. Zhu and X. Y. Shi, Polym. Chem., 2011, 2, 1754–1760 RSC.
- M. Martinez-Carmona, D. Lozano, A. Baeza, M. Colilla and M. Vallet-Regi, Nanoscale, 2017, 9, 15967–15973 RSC.
- L. Wang, W. Wang, Z. Rui and D. Zhou, Drug Delivery, 2016, 23, 3200–3208 CrossRef CAS PubMed.
- Y. Wang, V. Bansal, A. N. Zelikin and F. Caruso, Nano Lett., 2008, 8, 1741–1745 CrossRef CAS PubMed.
- K. Kono, C. Kojima, N. Hayashi, E. Nishisaka, K. Kiura, S. Wataral and A. Harada, Biomaterials, 2008, 29, 1664–1675 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details and results. See DOI: 10.1039/d0tb00346h |
| This journal is © The Royal Society of Chemistry 2020 |