
Development of a handheld liquid extraction pen for on-site mass spectrometric analysis of daily goods†
Florian
Lotz
,
Paula
Baar
,
Bernhard
Spengler
 and
Sabine
Schulz
*
and
Sabine
Schulz
*
Institute of Inorganic and Analytical Chemistry, Justus Liebig University Giessen, Germany. E-mail: Sabine.schulz@anorg.chemie.uni-giessen.de
First published on 22nd March 2021
Abstract
We present a handheld liquid extraction pen (LEP) combined with a self-sustaining electrospray ionization platform for ambient mass spectrometry within a laboratory-independent workspace. The LEP enables direct sampling from various surfaces and textures, independent of sample shape without precise sample positioning or dedicated sample preparation. The combination of liquid extraction of analytes through the pen and electrospray ionization (ESI) opens a broad field of applications. Qualitative and semi-quantitative analysis is presented for pesticides, plasticizers and drugs which were analyzed from representative consumer goods, such as fruits, toys and pills. Food authentication via metabolomic fingerprinting and multivariate statistics is demonstrated for the analysis of fish fillets and coffee. The LEP source uses a rechargeable battery to power a compressor. Ambient air is used for solvent nebulization in ESI. Through a pressure pump with integrated solvent reservoir, a solvent flow through the LEP and ESI source is generated. Measurement times of more than three hours are possible. The ion source is adaptable to any kind of mass spectrometer equipped with an atmospheric pressure interface. Measurements were performed on orbital trapping instruments and on a miniature mass spectrometer. Coupled to the miniaturized mass spectrometer, the completely portable LEP-MS instrument has dimensions of 48.4 × 27.0 × 18.0 cm (l × w × h).
Introduction
Ambient liquid extraction ionization methods are a powerful tool for fast mass spectrometric analyses. The first application appeared about three decades ago, when a liquid junction was used to connect capillary zone electrophoreses with electrospray ionization (ESI).1 In the last two decades, several liquid extraction methods were developed. All are based on the direct contact between solvent and a solid sample surface which contains the analyte of interest. Then the analyte is dissolved by the solvent, ionized and transferred into the gas phase for mass spectrometric analysis.The three main established techniques in the field of liquid extraction from solid sample materials using microjunctions are nanospray desorption electrospray ionization (nano-DESI),2 liquid extraction surface analysis (LESA),3 and the liquid micro junction surface sampling probe (LMJ-SSP).4 Each of these three approaches use ESI for ionization and vaporization. A common characteristic is that analyte sampling and analyte ionization occur spatially and temporally separated. The methods differ in the way how the solvent is directed onto the sample and towards the ESI tip after analyte extraction, qualifying each method for different applications.
In nano-DESI the solvent is pumped onto the surface of interest with a primary capillary. A secondary capillary with a length of a few centimeters is placed directly next to the primary capillary. A liquid bridge is formed between the two capillaries and the sample surface, with a liquid flow from the primary to the secondary capillary. The other end of the secondary capillary points to the inlet of a mass spectrometer and generates the nano-ESI spray. The ESI plume is forced by a high voltage applied between secondary capillary and the mass spectrometer inlet. The continuous liquid flow is self-aspirating with flow rates in the sub-microliter range.2 As one of the applications of nano-DESI is mass spectrometry imaging (MSI),5 the capillary diameters are typically chosen as small as possible to obtain a high lateral resolution.
A further methodological step was made with the development of liquid extraction surface analysis (LESA)3 which is based on a discontinuous sample extraction. A small amount of solvent (0.5–2.5 μL) is transferred to the sample and picked up again by the same probe.3 The probe is then moved to a commercially available chip with an array of nano-ESI tips where an ESI spray is generated from the analyte solution. To prevent any sample carry-over, each measurement uses a new ESI tip of the chip.6 The broad usability of LESA, especially for tissue samples, led to commercialized instruments.7,8 One LESA-like device is the MasSpec Pen which uses the handheld-pen principle for clinical applications.9 Defined volumes of solvent in the range of several microliters are directed through a channel to the tip of a handheld pen probe. A small droplet contacts the sample and desorbs analyte. Then a second channel pumps gas into the probe tip area and pushes the droplet trough a third channel to the mass spectrometer inlet where solvent evaporation and ionization takes place by solvent-assisted inlet ionization. Optional ionization methods like ESI, atmospheric pressure chemical ionization and others can be installed. The MasSpec Pen is a useful tool for rapid non-destructive tissue analysis, used for in vivo cancer diagnosis.9,10
Extending the concept of handheld probes, a tool that accomplishes DESI, easy ambient sonic-spray ionization (EASI) and low-temperature plasma (LTP) ionization was presented. This device performs both, analyte desorption and ionization on the sample surface, and ions are transported in the gas phase through up to 1 m long transfer tubing. An implemented camera module allows for interactive mass spectrometry imaging (IMSI) with a lateral resolution in the mm range.11
LMJ-SSP is based on a two-capillary setup, usually designed in a coaxial manner with one capillary inside the other. The outer capillary transfers the solvent to the sample surface. The capillary end is placed closely above the sample surface, forming a microjunction between capillary tip and sample surface. The inner capillary ends on the same level as the outer capillary or is drawn inward up to three times the inner diameter of the inner capillary at maximum.12 It transfers the solvent back from the sample surface to an ESI sprayer. Like nano-DESI, LMJ-SSP maintains a continuous flow of solvent between the two capillaries and the sample surface. LMJ-SSP was also used for the analyses of microbial colonies. The so-called flow-probe uses the coaxial capillary design in combination with an automated sampling mechanism. During sampling, the capillaries of the flow probe do not directly contact the sample but form a liquid bridge between outer capillary, sample surface and inner capillary. To maintain the small probe-to-sample-distance of 20 μm for different sample spots on heterogeneous shaped samples, a precise computer controlled x, y and z positioning is used.12,13 Later it was shown, that a liquid flow rate that exceeds the aspiration rate, maintains effective sampling but also allows a direct contact to the sample surface.14 First attempts to expand the distance between sampling and ionization were recently realized with the tethered, open-port sampling interface (TOPSI). Here the transport of analyte solution from a handheld sampling probe to the atmospheric pressure chemical ionization (APCI) source is supported by an external vacuum line.15
Due to identical analyte transition principles from solid into the solvent, the sample classes which can be analyzed are comparable for the three techniques. Nevertheless, the methods have different fields of application. Nano-DESI is designed for a minimized contact between sample and probe tip via a liquid bridge to achieve an optimized lateral resolution for MSI. The achievable spatial resolution is in the range of some ten micrometers. In this matter, tissue sections of biological materials are often the object of interest. LMJ-SSP deals with higher liquid flow rates compared to nano-DESI and its design causes bigger sampling spots.16 Typical applications are in the field of spatially resolved liquid–solid extraction from biological sample materials often combined with liquid chromatography analyses.13,17,18 Even if imaging applications were performed with LMJ-SSP, the method is typically not the method of choice because of its limited lateral resolution.13,19 A clear advantage is its robustness regarding efficiency of analyte sampling and ionization via ESI. Such continuous flow LMJ-SSP sources are already commercially available.20
Recently we developed an self-sustaining DESI platform for on-site analysis of consumer goods.21 We found that it is well suited for qualitative high-throughput analysis, but quantification and sensitivity is still limited. We figured that a flexible and handheld LMJ-SSP probe would provide better sensitivity, reproducibility, and quantitative results, especially in a portable MS system. Therefore, we developed a handheld LMJ-SSP based pen named liquid extraction pen (LEP) and integrated it in the existing self-sustaining DESI platform, then operated in ESI mode. The handheld pen device allows a flexible measurement procedure without any precise placement of the sample object. Design and function of the LEP as well as qualitative and quantitative capabilities of the portable LEP source are presented for the analysis of representative consumer goods.
Experimental
Chemicals
Di-n-octyl phthalate (DNOP) was purchased from Tokyo Chemical Industry Co., Ltd (Tokyo, Japan). Diisononyl phthalate (DINP) was obtained from Sigma Aldrich Chemie GmbH (Steinheim, Germany). Diethylhexyl phthalate (DEHP) was purchased from Bernd Kraft GmbH (Duisburg, Germany). Diisobutyl phthalte (DIBP) was purchased from Alfa Aesar GmbH & Co KG (Karlsruhe, Germany). 1,2-Cyclohexane dicarboxylic acid diisononyl ester (DINCH) was provided by Combi GmbH (Mönchengladbach, Germany). Polyvinyl chloride (PVC) powder Vinnolit P70 was provided by Vinnolit GmbH & Co. KG (Ismaning, Germany). Formic acid and pure water were purchased from Fluka (Neu Ulm, Germany) in chromatographic grade. Methanol was obtained from Merck (Darmstadt, Germany) in Uvasol quality. Flusilazole was obtained from Sigma Aldrich (Steinheim, Germany). Carbendazim was purchased from LGC Standards (Teddington, UK).Sample preparation
Plastisol pills with 0, 1, 5, 10, 20 or 40%w of DIBP, DEHP, DINP and DNOP were prepared as standards for quantitative analyses. The phthalate was mixed with PVC powder Vinnolit P70 and DINCH until a homogenous suspension was formed with a total plasticizer content of 40%w. 1 g of each suspension was then filled into an aluminum mold of 20 mm in diameter and 8 mm in depth. The mold was baked in an oven for 20 minutes at 200 °C for solidification of the pills. After cooling to room temperature, the plastisol pill was removed from the mold. Toys and other plastics were used as examples for consumer goods, containing phthalate-based plasticizers in known amounts. Samples and respective data were provided by the Bavarian State Laboratory for Health and Food Safety (Erlangen, Germany) and from State Laboratory of Rhineland-Palatinate (Koblenz, Germany). LEP-MS measurements were performed from the surface of the complete sample without any further sample preparation steps. Medical pills were purchased from a local pharmacy and used without sample preparation. Coffees, fish fillets and the vegetables were purchased from local supermarket or online shop (see ESI Table 1†). Fish fillets were stored at −20 °C in plastic bags. Prior to measurement the fillets were defrosted within the bag in warm water. After unpacking, the excess liquid was removed and the samples were cut to obtain a fresh surface of the tissue. Analyses of pesticides (carbendazim and flusilazole) were directly performed from pepper peel which was spiked with the respective substances. Prior to spiking, the pepper was thoroughly cleaned with warm water. The measurement area was defined with a red marker on the pepper peel. Each pesticide was diluted in ethanol in respective concentrations. A volume of 10 μL of each pesticide solution was distributed on a spot of about 1 cm2 size. The distribution was carried out carefully using pipet tips to prevent any carryover beyond the assigned areas. The solvent was allowed to dry off resulting in surface concentrations of 0.01 μg cm−2 to 2.00 μg cm−2.Measurement procedure
For all measurements, the solvent flow rate was individually adjusted, so that the volume pumped to the sample surface matched the volume that was consumed by the ESI spray. For the solvent mixture of MeOH/H2O (9/1), flow rates were between 30 μl min−1 and 80 μl min−1. Via the high voltage connector, a potential of 4 kV was applied to the solvent passing through the secondary capillary. The distance between the ESI sprayer and the transfer capillary of the MS was set to about 5 cm. Changes to these general parameters are mentioned specifically in the text. The spray was directed in a 0° angle to the MS inlet for all measurements except for the DESI/LEP switching experiment. Here the angle was 70°.For the analyses of plasticizers, the LEP was placed directly on the sample surface. For quantitative measurements, three calibration rows were measured consecutively, consisting of the different plastisol pills analyzed in order of increasing concentration, followed by the real samples. Each plastisol pill and real sample was analyzed at three different sampling spots, representing three technical replicates with other samples measured in between. Each plastisol pill and real sample was measured for about 20–30 s by moving the pen along the surface or from a single spot. At the miniaturized mass spectrometer, ten consecutive single spectra were averaged to a sum spectrum for each pill and sample. After sampling, the LEP tip was placed for 30 s on a glass slide to avoid carry-over. During the measurement procedure of pesticides, the LEP was moved 20 s across each sample spot with direct contact to the pepper peel. Three calibration rows were measured consecutively. Sample spots were analyzed in order of increasing concentration. Each pesticide concentration was analyzed at three different sampling spots, representing three technical replicates with other samples measured in between. After each concentration, the LEP tip was placed for 10 s on a glass slide to avoid carry-over. Medical pills were analyzed by placing the LEP for 30 s onto the surface without movement. The fish and coffee samples were measured by moving the LEP slowly over the surface until 50 spectra were recorded.
The self-sustaining DESI source,21 used in ESI mode in this work, was designed to fit to the dimensions of the prototype of the miniaturized and portable mass spectrometer Mini 11.22 The prototype has a total size of 28.4 × 20.1 × 18 cm and a weight of 8 kg without batteries. The power consumption is less than 35 W, running on 24 V DC with a power adapter for lab-based measurements or with an appropriate battery for in-field applications. The rectilinear ion trap features unit resolution and was operated in full scan (m/z 50–700) mode. For each scan cycle, the magnetic valve of the discontinuous atmospheric interface was set to an opening time of 14 ms, allowing ions to enter the analyzer. Follow-up products of this instrument are commercially available (Mini β, PURSPEC Technologies Inc., West Lafayette, Indiana, USA). One measurement cycle took about 2 seconds before the next injection step. However, many sample measurements were performed with orbital trapping mass spectrometers (Exactive and Q Exactive HF-X, Thermo Fisher Scientific GmbH, Bremen, Germany). An extended ion transfer capillary (48 cm length, 0.625 mm inner diameter) was used to connect the source to the Exactive mass spectrometer. Therefore, the standard capillary was exchanged according to the instruments manual in stand-by mode by a custom-made capillary with the same ID and length protruding into the instrument but with an extended length outside the instrument. Measurements were performed in a m/z range of 100–500 if not stated otherwise. Ion injection was controlled via automatic gain control (maximum injection time 500 ms) with a resolution of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (Exactive) and 240000 (Q Exactive HF-X) at 200 m/z. HCD fragmentation experiments were performed at the Q Exactive HF-X with an isolation window of ±0.5.
000 (Exactive) and 240000 (Q Exactive HF-X) at 200 m/z. HCD fragmentation experiments were performed at the Q Exactive HF-X with an isolation window of ±0.5.
Data analysis
For quantitative pesticide analysis with the Exactive Orbitrap instrument, signal intensities of the protonated ion and the total ion current (TIC) were extracted from all spectra corresponding to one sampling spot, using the Xcalibur software. Ion intensities were TIC normalized per spectrum and averaged for one sampling spot. For quantitative phthalate analysis with the Exactive instrument, signal intensities for the protonated phthalate ion, its sodium and potassium adduct as well as the total ion current (TIC) were extracted from 15 spectra, corresponding to one plastisol pill, using the Xcalibur software. Adduct ion intensities were summed, TIC normalized per spectrum and averaged for one pill. For quantitative phthalate analysis with the Mini 11, signal intensities for the protonated phthalate ion were extracted with an m/z window of ±5 from the sum spectrum, corresponding to one plastisol pill or real sample, using the Excel software. Ion intensities were summed for one plastisol pill or real sample. Results for three measurement cycles were averaged, highest value was set to 100% and plotted against the analyte concentration. Linear regression analysis was performed using the least-squares method to fit a linear function with intercept set to zero to the data points and to obtain a coefficient of determination (R2). Intercepts were forced through zero, since we observed larger variance in the signal intensities of the highly concentrated standards compared the other standards which would otherwise bias our result obtained in the low- and medium-concentration range. Error bars represent standard deviation. Phthalate concentrations in the real samples were calculated from the slope of calibration curves and the measured ion intensities. The limit of detection (LOD) was calculated from the analyte signal intensity of the calibration standard containing no added analyte averaged for the three technical replicates plus three times its standard deviation. The calculated intensity value was divided by the calibration curve slope to obtain LOD in %wt. LOQ was calculated the same way as LOD, but using ten times the standard deviation of the averaged phthalate signal intensity of the plastisol disc containing only DINCH and PVC.Results and discussion
Setup and function
The LEP is based on the two-parallel-capillary principle of LMJ-SSP. In our setup the two capillaries are not fitted into each other, but placed next to each other (Fig. 1). The primary capillary delivers solvent to a solid sample surface, while a secondary capillary directs analyte-enriched solvent to an ESI source. The analyte extraction takes place via a liquid bridge between the capillary tips and the sample. Both capillaries were glued together and guided through the case of a standard ballpoint pen. The glue prevents that solvent rises into the pen case via capillary forces. The capillary tips protrude the pen case about 1 cm and bend slightly during the measurement procedure. The capillaries are fixed into a fitting which is mounted on a spring permitting some movement of the capillaries along the pen axis. This allows sensitive surface contact and minimizes the risk of damage to the sampling tip. After extraction, the solution is transferred to an ESI sprayer by self-aspiration where ionization of analytes for mass spectrometric analysis takes place. The connection between the secondary capillary and the ESI spray capillary is established via a high voltage connector. In the presented setup the primary capillary (100 μm inner diameter (ID), 193 μm outer diameter (OD)) and secondary capillary (250 μm ID, 375 μm OD) were used in a length of 50 cm. The ESI sprayer was constructed according to literature23 with a 10 cm long ESI spray capillary (100 μm ID, 193 μm OD) coupled to the high voltage connector. A battery-powered compressor pumps ambient air through a gas filter and a drier tube to remove dust particles and humidity. The pressurized air is then used in the ESI sprayer as nebulizing gas. Via a gas bypass, part of the pressurized air is directed to the pressure pump containing the 10 ml ESI solvent reservoir. In combination with a valve which allows a precise pressure regulation, the solvent flow rate through the primary capillary is controlled. The implemented rechargeable battery allows continuous runtimes of more than 3.5 hours. A more detailed description of the setup and function of the self-sustaining ESI platform as well as of the parameters of the whole supply system can be found elsewhere.21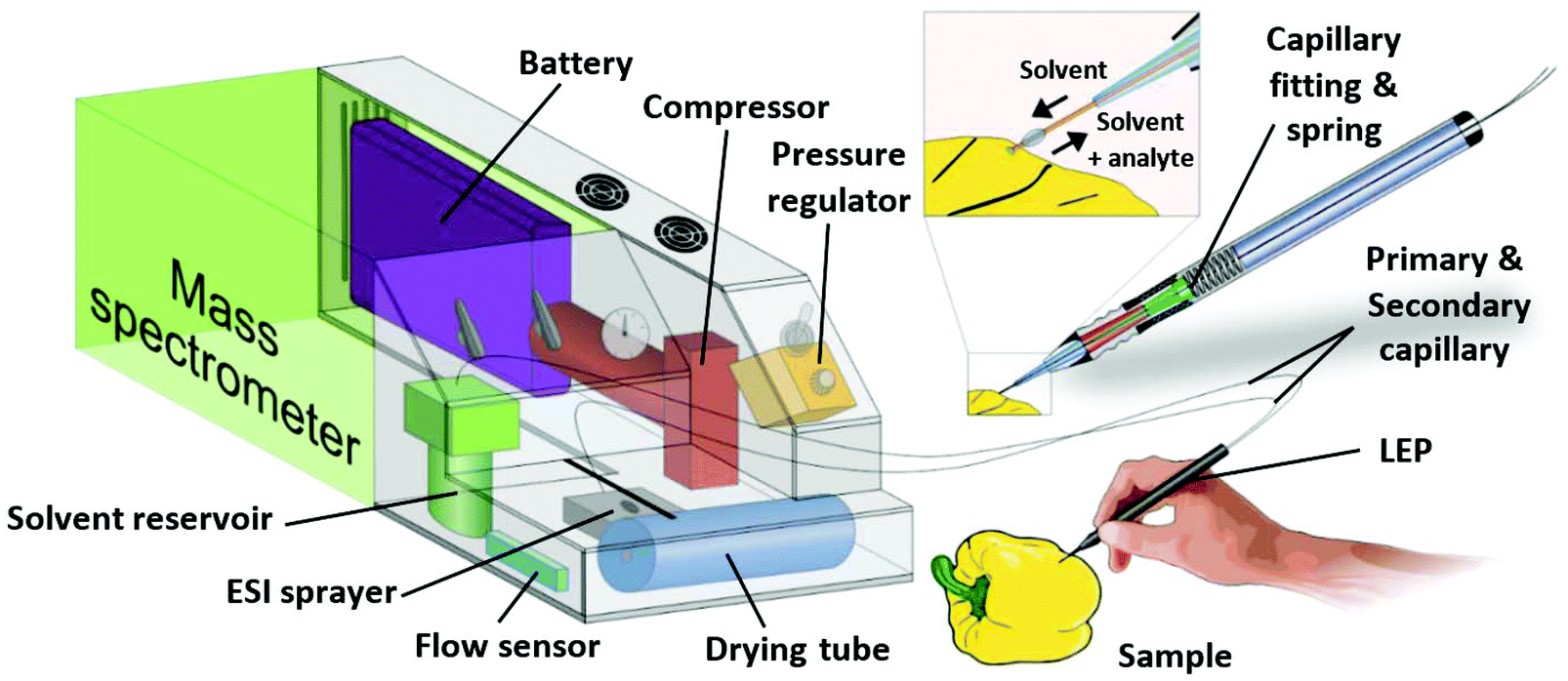 | ||
| Fig. 1 Scheme of the portable ESI source with its major components and the handheld LEP. | ||
Characterization and optimization
The lengths and IDs of primary and secondary capillaries were chosen with respect to short measurement times per sample and easy measurement procedure. A length of 50 cm of both capillaries allows flexible usage of the pen in the close environment of the source to analyze e.g. large samples which cannot be placed on the sample table. The ID of the primary capillary is optimized to provide highest flow rate precision, available with the pressure regulator installed at the pressure pump. The pressure regulator has a range of 0 to 2 bar. With 50 cm length, ID of the primary capillary was chosen in such a way that flow rates between 10 and 90 μL min−1 for methanol–water and acetonitrile–water mixtures can be controlled using the full pressure range, providing a flow rate precision of ±0.05 μL min−1 by manual adjustment. The flow rate is measured by a flow sensor which must be calibrated for each solvent mixture.21 For all measurements the solvent flow rate delivered to the sample was adjusted depending on the surface texture and absorptivity of samples, so that the volume pumped to the sample surface matched the volume aspirated via the secondary capillary and ESI spray.Aspiration of the solvent through the secondary capillary of LEP depends on the geometry of the ESI sprayer tip, the capillary length and inner diameter and the solvent viscosity. It is known,1 that a solvent flow is induced in the ESI spray capillary by the nebulizing gas flowing around it, based on the venturi effect. This effect is less intense, if the spray capillary protrudes from the surrounding gas capillary (as it is usually the case in ESI or DESI). If the spray capillary is placed back into the gas capillary, the force on the solvent is significantly higher, resulting in a higher induced flow rate. This phenomenon is exploited to transfer solvent from the sample trough the secondary capillary of the LEP to the ESI sprayer. As a result, a time delay between sampling and detection in the mass spectrometer occurs which depends on the offset of the spray capillary to the gas capillary in the ESI sprayer, the length and inner diameter of the secondary capillary and the solvent. ESI Fig. 1† shows the influence of the spray capillary offset on the flow rate for the given capillary setup and a solvent mixture of ACN/H2O (9/1) which was used as a model system. At high flow rates, analyte dilution in the solvent increases, ionization efficiency decreases and the maximum measurement time with one filling of the solvent reservoir shortens. For our setup, we found flow rates between 30 and 80 μl min−1 best suited. Within this range, detection delays were between 20 s and 50 s and in case of a 10 ml solvent reservoir the sustained measurement time was between 2 h and 5.5 h. To optimize solvent aspiration through the secondary capillary, the spray capillary was flexibly mounted in its fitting so that higher or lower desirable flow rates were adjustable by moving it manually. A longer secondary capillary resulted in longer time delays between sampling and ionization, causing longer measurement times per sample. A reduced ID of the secondary capillary increases the flow velocity, thus reduces measurement times, but also increases flow resistance and the possibility of clogging. Therefore, the inner diameter of the secondary capillary can only be reduced to a certain extend. Mixtures of water and the organic solvents methanol and acetonitrile are commonly used in ESI. The LEP setup is optimized for the mixtures ACN/H2O (9/1) and MeOH/H2O (9/1). The high organic content was chosen, since the viscosity increases with increasing portion of water, resulting in lower flow rates. In addition, quick vaporization is promoted by the high proportion of organic solvent improving ionization efficiency at the miniaturized mass spectrometer which has no heated inlet capillary. We found that MeOH/H2O (9/1) worked for all shown analytes. Change of the solvent mixture may require an adaption of the solvent leading capillaries or the pump system. ESI Fig. 2(a)† shows two consecutive measurements from a toy puppet containing the plasticizer DEHP in 25.9%w. The sampling time with the LEP was 10 s. The delay between sampling and DEHP ion signal detection at the MS was about 20 s at a flow rate of 40 μL min−1. The DEHP signal was detected for about 45 s. Due to the slowly falling signal after the time corresponding to the sampling, it is recommended to wait 30 s after each sampling to avoid carry-over. ESI Fig. 2(b)† shows two consecutive measurements of an Arabica coffee bean, naturally containing trigonelline. Due to the solvent flow rate of 30 μL min−1, an increased delay time of about 30 s was obtained. Compared to the plasticizer the carry-over of trigonelline was less pronounced. Washing intervals in the following qualitative and quantitative experiments between samples were estimated before the analysis by measuring the time till the analyte signal decreased to 10% of the signal intensity detected during sampling.
In our homebuilt ESI sprayer the spray capillary typically has unwanted contact to the inner wall of the gas capillary at some point.24 Consequently, the emerging solvent at the sprayer tip is not coaxially enclosed by gas. This can result in an uneven spray pattern in case of the common mounting. With the spray capillary ending inside the gas capillary, analyte-enriched solvent may get into contact with the inner wall of the gas capillary. Thus it was observed that slowly growing droplets formed around the gas capillary tip. To prevent such droplet formation which can disturb the ESI spray, a thermoplastic polymer coating was applied to the gas capillary as shown in ESI Fig. 3.† We did not observe any interferences caused by the polymer coating in the mass spectrometry data. With a flow rate in the range of 30–50 μl min−1 and MeOH/H2O 9:1 as solvent, a spatial resolution of about 0.5 mm in diameter was obtained with this setup for line scans and one of 1.1 mm in diameter, if individual spots were measured. Analyte consumption and achieved spatial resolution are shown in ESI Fig. 4† where a glass slide, covered with rhodamine 6G was measured.
We did not observe any influence of the LEP movement or its speed on the detected ion signal intensities if analyte was distributed homogenous in the sample volume. We assume that the analyte transfer from the solid phase into the liquid phase occurs fast, so that the movement speed along the surface have no significant influence on the analyte concentration in the liquid phase.
Analysis of daily goods with LEP coupled to a lab-based mass spectrometer
The functionality of the LEP was optimized and tested on an orbital trapping mass spectrometer till its performance was suitable for the rapid analysis of daily goods. Various daily goods were then analyzed qualitatively with LEP. Fig. 2 exemplifies which samples can be tested without any sample preparation. Fig. 2(a) is representative for rapid drug analysis as needed for example at crime scenes, at border controls or for counterfeit identification. It shows a LEP spectrum of paracetamol measured directly from a drug pill (Thomapyrin CLASSIC, 200 mg, Boehringer Ingelheim). The protonated ions of paracetamol ([M + H]+, m/z 152.071) and caffeine ([M + H]+, m/z 195.088) were identified by accurate mass with a tolerance of ±2 ppm and HCD fragmentation experiments. In addition a doxycycline pill (Doxycyclin AL 100 T, 100 mg, Aliud Pharma) was analyzed (ESI section ‘list of mass spectra’). Both pills were also analyzed with LEP at the miniaturized MS (ESI section ‘list of mass spectra’).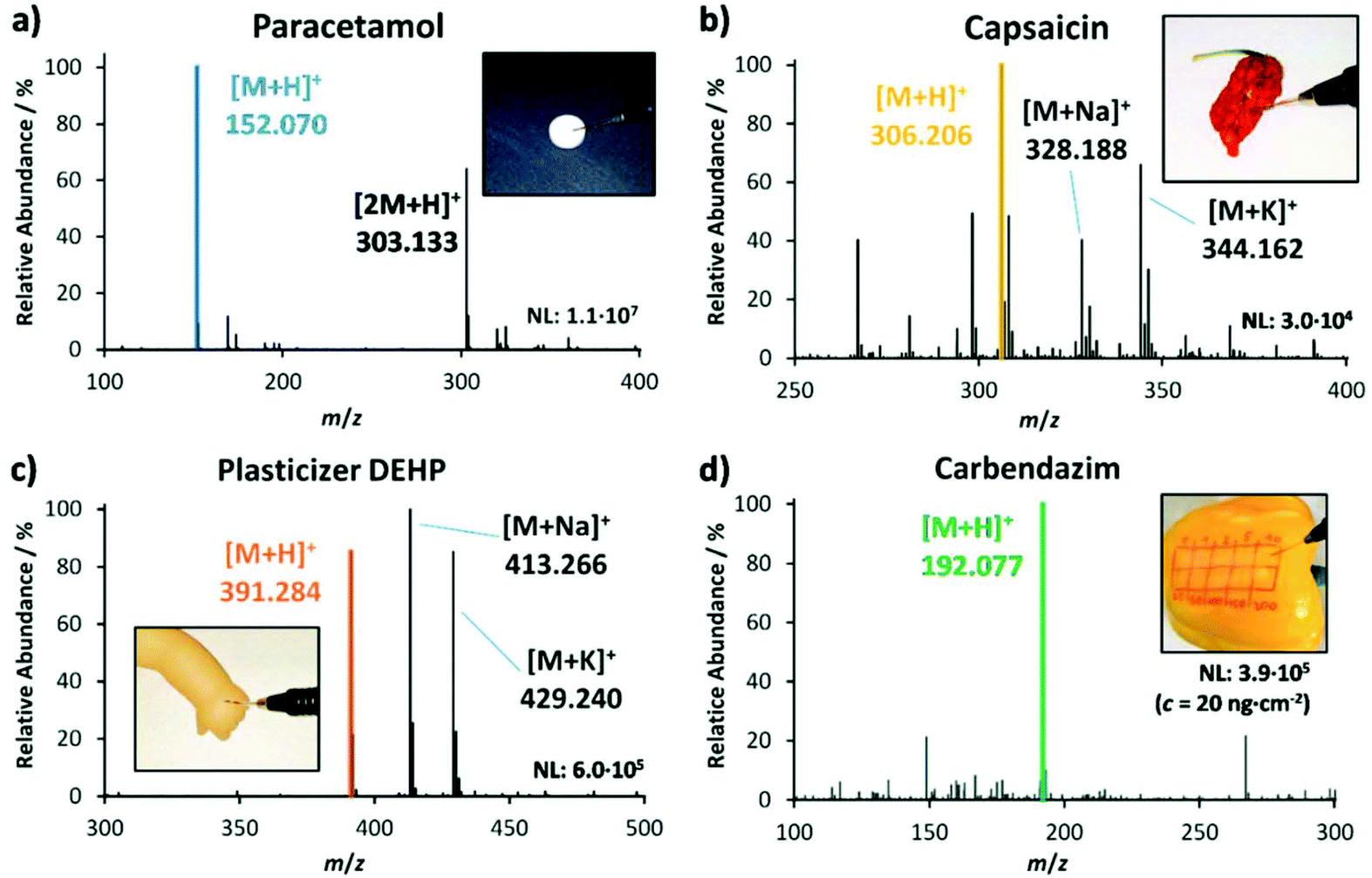 | ||
| Fig. 2 Mass spectra of various analytes measured directly from the sample object with the self-sustaining LEP source attached to an orbital-trapping mass spectrometer. (a) Paracetamol pill, (b) capsaicin from a dried chili pepper ‘Carolina Reaper’, (c) plasticizer DEHP from a toy puppet and (d) carbendazim from a fresh pepper. | ||
Fig. 2(b) stands for rapid food analysis, needed for example to identify food fraud or adulteration. Here a dried chili pepper ‘Carolina Reaper’ was analyzed, and capsaicin was detected as one of the main compounds next to dihydrocapsaicin (+2 u). Food fingerprints as shown here can be used in combination with multivariate statistical tools for food authentication25–28 (also shown below). Because of the crumpled surface of the dried pepper, it is challenging to generate good spectra from it with DESI, but with LEP we were able to generate spectra with stable ion intensities.
Fig. 2(c) is representative for the analysis of plasticizers in plastic-containing daily goods such as toys and table ware. Here a puppet, containing the phthalate-based plasticizer DEHP in 25.9%w was analyzed with the LEP. The mass spectrum shows the three adduct ions [DEHP + H]+ (m/z 391.284), [DEHP + Na]+ (m/z 413.265) and [DEHP + K]+ (m/z 429.240). The use of phthalate-based plasticizers in daily goods with oral contact is restricted in many countries due to their hormone-disrupting properties.29–31Fig. 2(d) stands for rapid pesticide detection on fruits and vegetables as needed at border controls and for customer protection. Here the pesticide carbendazim was detected from a spiked pepper. The concentration of carbendazim was 20 ng cm−2 to mimic realistic conditions. We detected its protonated ion ([M + H]+ at m/z 192.077). The corresponding HCD fragment spectra of analytes shown in Fig. 1(a)–(d) can be found in the ESI section ‘list of mass spectra’. During the measurement procedure the pen was held in hand in a slant position, similar to writing. The sampling tip was moved along the surface following the hand movement. Apart from its own weight, the pen was not forced onto the surface, so that the 1 cm long sampling tip was only slightly bent. Pushing the pen along the sample surface with the tip ahead increases the risk that small particles detach from the sample and clog the capillaries, making a pulling movement more advisable. The analysis time per sample was 20–30 seconds. Between samples, the LEP was flushed for 30 seconds to remove any residues from the previous sample from the capillary system and to prevent carry-over. In comparison to the performance of the DESI operation, the LEP source showed similar ion signal intensities for highly concentrated analytes, such as paracetamol in the pill and DEHP in the puppet. For the lowly concentrated analytes, such as the pesticide on pepper, the LEP showed two orders of magnitude higher signal intensities than DESI, providing a better sensitivity for quantification.
In addition, rapid switching between ionization methods LEP and DESI was tested at the portable platform. First results are shown in ESI Fig. 5.† Here, four different samples (a beach ball, a chili pepper, a drug pill and a toy puppet) were analyzed in seven minutes. The first sample was analyzed with DESI, the second with LEP, the third with DESI again and the fourth with LEP again. Switching between ionization methods took only 10 seconds. Details how the switching was done are given in the figure caption of ESI Fig. 5.†
Suitability of LEP-MS for food authentication and food fraud detection was tested by an experiment focused on fish species detection. The surface of the defrosted fish fillets was directly measured. The mass spectrometric fingerprints of fish fillets obtained via LEP were used for food classification. Two different fish species were analyzed: Solea solea and Pangasianodon hypophthalmus.
These species are common suspects for food fraud by miss-labeling.32 The low-cost fish Pangasianodon hypophthalmus is labeled as the higher-priced fish Solea solea. While the species are easily distinguishable if the fish is intact, it becomes quite challenging after processing (e.g. to fillets). Then a rapid analytical method for food authentication is needed as presented in the following. For each species, 450 LEP mass spectra were recorded from three biological replicates. Each biological replicate was measured at three different areas representing three technical replicates (50 spectra each). The measurements were performed in a m/z range from 50 to 500 in positive-ion mode. Data was then subjected to principal component analysis (PCA) using the in-house developed software “MS Food classifier”.27 For PCA, data was binned with a mass window of 1 u, intensity values were log10 transformed and the QR algorithm was used with 99% variance coverage. Fig. 3(a) shows the corresponding score plot. Both fish species were separated well from each other in the PCA score plot and hence could be differentiated based on the metabolomic fingerprint. Cross validation of the PCA model via the leave-10%-out method showed 100% correct classification of the fish spectra to their species, using the LDA (linear discriminant analysis) coefficient as classifier. Robustness of the PCA model is further highlighted by the fact, that the biological replicates were measured on three days within a four weeks’ time frame. Further experiments in negative-ion mode (m/z 50–500) and the higher mass range (m/z 700–900) of positive-ion mode allowed the discrimination of the fish species with very similar quality. Related PCA score plots are given in ESI Fig. 7.† Corresponding loading plots are given in ESI Fig. 8.† In another experiment, intact coffee beans of the two species Arabica and Robusta from the regions Kerala and Karnataka in India, were analyzed with LEP. Since Arabica is higher in quality, it is often blended with lower-quality Robusta coffee.33,34 For each combination of species (Robusta or Arabica) and cultivation region (Kerala or Karnataka), 450 LEP mass spectra were collected from coffee beans of three different retailers (biological replicates). For each biological replicate 150 spectra from three coffee beans (technical replicates) were acquired. Data sets were analyzed via PCA, using the same parameters as for the fish analyses. Fig. 3(b) shows a clear grouping of the respective biological replicates and good separation of the different species. In addition, data points belonging to the two cultivation regions Kerala and Karnataka of Arabica coffee separate into two groups while the data corresponding to the cultivation regions overlap for Robusta coffee. The PCA model gives 99.1% correct classification using the LDA coefficient for the differentiation of species and growing region. Corresponding loading plots are given in ESI Fig. 8.† The binning mass window of 1 u for PCA analyses simulate the unit mass resolving power of portable mass spectrometers. In fact, we found no improvement in separation of the species using smaller bin sizes. Since biomarker detection was not the goal of these experiments, we choose this bin size to save resources during PCA analysis. Nevertheless, we performed MS/MS on some high abundant metabolites (see ESI section ‘list of mass spectra’) and labeled the corresponding m/z bins in the loading plots of fish and coffee data.
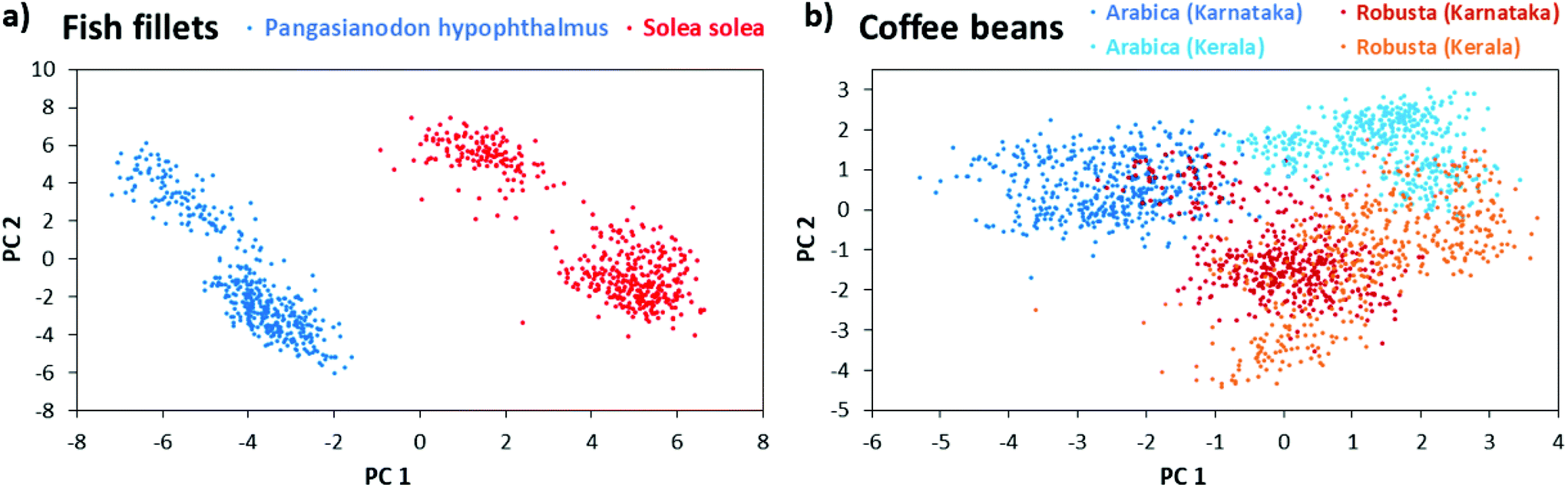 | ||
| Fig. 3 Food authentication via multivariate data analysis of LEP mass spectral metabolite fingerprints acquired in positive-ion mode (m/z range 50–500). (a) PCA score plot showing the differentiation of fish species Solea solea (N = 3) and Pangasianodon hypophthalmus (N = 3). (b) PCA score plot showing the differentiation of the coffee species Arabica (N = 6) and Robusta (N = 6) as well as their growing regions in India. | ||
The performance of LEP in quantification was evaluated for the two pesticides carbendazim and flusilazole, measured from pepper peel. Fig. 4(a) shows the measurement procedure. The peel of a pepper was marked with several sampling spots. The spots were spiked with the pesticides in increasing concentration. Each sampling spot was analyzed for 20 s by moving the LEP in the pattern shown in Fig. 4(b). Between sampling spots, the LEP was placed on a glass slide for 10 s to flush the capillary system and to prevent carry-over. Fig. 4(c) shows the ion signal intensities of the protonated carbendazim ion, recorded from the sampling spots analyzed consecutively in the order of increasing pesticide concentrations. The measurement of one calibration row (10 concentrations) took about 6 min. This is a major improvement in measurement time compared to our previously published method using conventional DESI.35 There the measurement of a single spiked sampling spot took about 6 min. Fig. 4(d) and (e) show the calibration curves obtained for carbendazim and flusilazole.
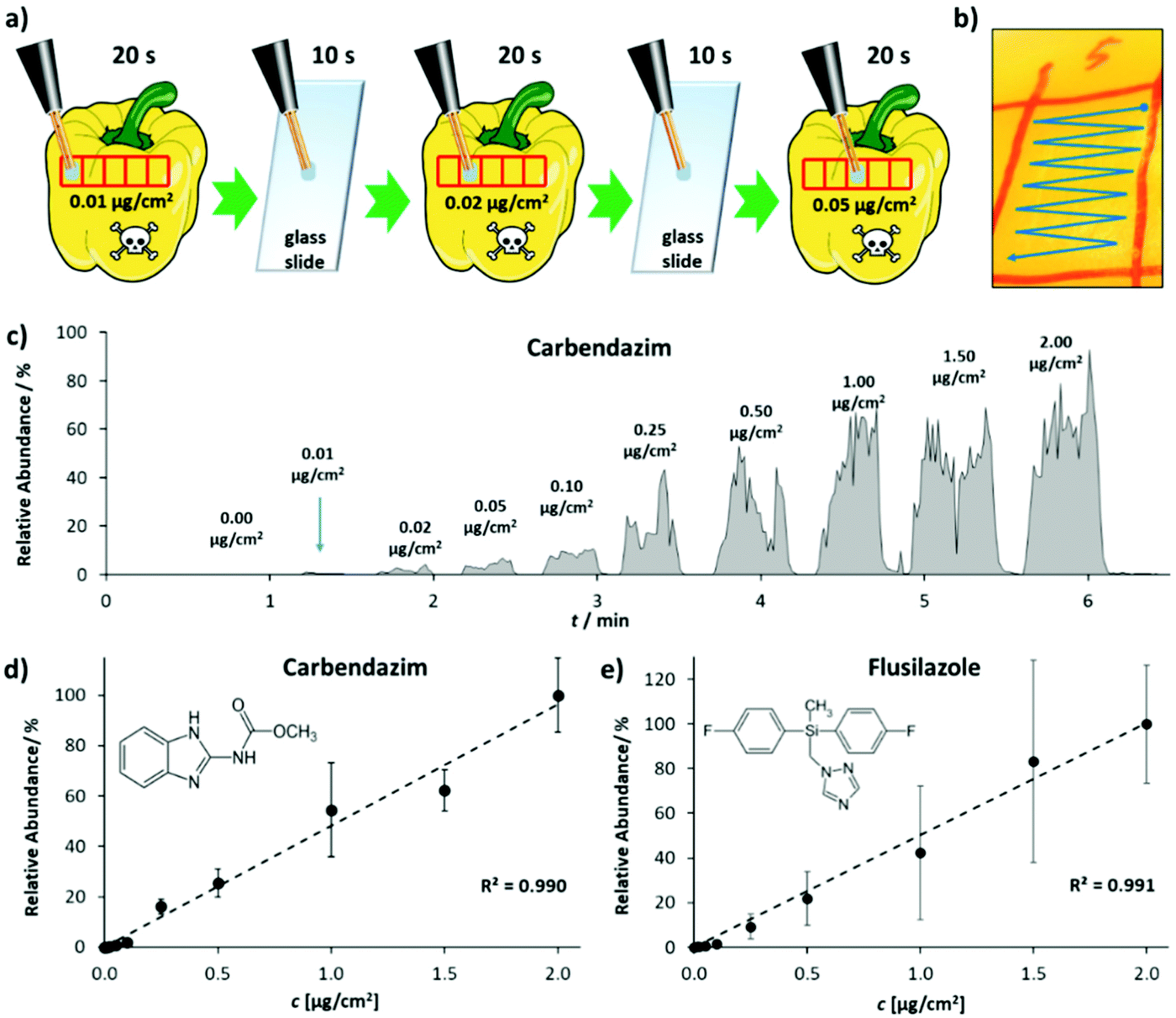 | ||
| Fig. 4 Quantitative analysis of pesticides from pepper peel, using the self-sustaining and portable LEP source attached to an orbital-trapping mass spectrometer. (a) Sampling procedure for pesticide quantification from pepper peel. (b) Sampling across the spiked sampling spot. (c) Ion intensity chromatogram for different Carbendazim concentrations, detected from pepper peel. (d) and (e) show calibration curves, obtained for the two pesticides carbendazim and flusilazole. Data points represent averages of three technical replicates. Error bars indicate standard deviation. | ||
Both calibration curves feature an R2 value of 0.99. A LOD of 0.7 ng cm−2 and a limit of quantification (LOQ) of 1.8 ng cm−2 were calculated for carbendazim. For flusilazole, the LOD was determined as 0.2 ng cm−2 and the LOQ as 0.4 ng cm−2. In comparison to conventional DESI,35 where we determined an LOD of 3.3 ng cm−2 (on apple peel, single calibration row), this corresponds to an improvement by a factor of 16.5. Reproducibility was assessed analyzing one carbendazim concentration (500 ng cm−2) on five consecutive days (5 technical replicates each day). Intra-day reproducibility ranged from 9.1 to 25.3% relative standard deviation. Inter-day reproducibility was 18.5% relative standard deviation (ESI Fig. 9†). In addition, the performance of the LEP in quantification was analyzed for phthalates. Calibration curves for DEHP, DNOP and DINP were recorded, and intra-day and inter-day reproducibility were studied (ESI Fig. 6†). The measurement of one calibration row (6 concentrations) took about 3 minutes. In comparison to DESI,21,36R2-values of the calibration curves obtained for DEHP, DNOP and DINP were all higher with the LEP. LODs for DEHP, DNOP and DINP were calculated as 0.02, 0.01 and 0.1%w for LEP. These values were by a factor of 31, 8 and 12 lower than those obtained by conventional DESI.21,36 LOQ for DEHP, DNOP and DINP were calculated as 0.02, 0.02 and 0.2%w for LEP and were by a factor of 43, 9 and 11 lower than those obtained by DESI.21,36 This illustrates that the sensitivity of the LEP is higher and it is more suited for quantification than DESI. Intra-day and inter-day reproducibility were studied on two consecutive days by analyzing a 20%w plastisol pill of DNOP on each day 10 times. On day 1, the relative standard deviation for the 10 consecutive measurements was 3.5%. The relative standard deviation for the 10 consecutive measurements on day 2 was 1.6% and inter-day was 3.6%. With DESI, the intra-day relative standard deviation was 1.8% (day 1, n = 3) and 3.5% (day 2, n = 2) and inter-day 6% (n = 5) for a 20%w plastisol pill of DEHP using a conventional source. With the self-sustaining DESI source,21 the intra-day relative standard deviation was 12% (day 1, n = 3) and 19% (day 2, n = 5) and inter-day 21% (n = 8) for a 20%w plastisol pill of DEHP. While the intra-day reproducibility was comparable for the LEP and conventional DESI, inter-day reproducibility was better with the LEP. Compared to the reproducibility of DESI with the portable DESI source, LEP reproducibility was found to be significantly better (F-Test p-value 3.3 × 10−9).
Analysis of daily goods with the portable LEP-MS instrument
After the functionality of LEP was optimized, the source was installed in front of the miniaturized ion trap mass spectrometer. With this fully self-sustaining and portable setup, several plasticizer-containing samples (Fig. 5) as well as two medical pills and the dried chili pepper ‘Carolina Reaper’ (ESI section ‘list of mass spectra’) were analyzed qualitatively. For each sample and analyte, the protonated molecular ion was clearly visible in the mass spectrum. In addition, CID was used for identification via specific fragment ions at the Mini 11 prototype. Further details regarding these MS/MS experiments can be found in the ESI section ‘list of mass spectra’. The performance of the portable LEP-MS regarding quantification was evaluated for phthalate analysis in daily goods (Fig. 6). Calibration curves were obtained for the phthalates DEHP, DNOP and DIBP (Fig. 6(a–c)). The calibration curves of DEHP and DNOP featured high R2 values (0.998 and 0.995) even with the miniaturized mass spectrometer. For DIBP, a lower R2 value (0.956) was determined. The calculated LOD for DEHP was 0.4%w, for DNOP 0.5%w and for DIBP 1.3%w. The calculated LOQ for DEHP was 0.8%w, for DNOP 1.3%w and for DIBP 3%w. For DEHP and DNOP, the LOD was by a factor of 20 and 50 higher than on the orbital trapping mass spectrometer, and the LOQ was by a factor of 40 and 65 higher, most likely due to the lower sensitivity and performance of the miniaturized mass spectrometer. The phthalate contents of seven real samples were determined using these calibration curves. Fig. 6(d) shows the values obtained with the portable LEP-MS in comparison to the values measured with confirmatory methods. Assuming that the confirmatory methods are providing the true value, six out of seven phthalate values determined by LEP-MS matched the values of the confirmatory methods within a relative deviation range of ±15%. More details regarding the confirmatory methods, the determined phthalate contents and the measurement error can be found in ESI Table 2.† For five of the samples, the value given by the confirmatory method was within the LEP-MS measurement error. In case of the toy stethoscope tube, only a small sample piece was available, used for LEP-MS and for the previously published DESI-MS measurements. This could explain the significant deviation from the value found with the confirmatory methods.21,36 For all other samples, larger surface areas were accessible, making oversampling more unlikely. Overall, a good correlation was observed between LEP-MS and confirmatory methods. Given the great difference in sample preparation and analysis between the confirmatory methods and LEP-MS, the LEP appears as an appropriate alternative with clear advantages in time exposure and simplified working procedure.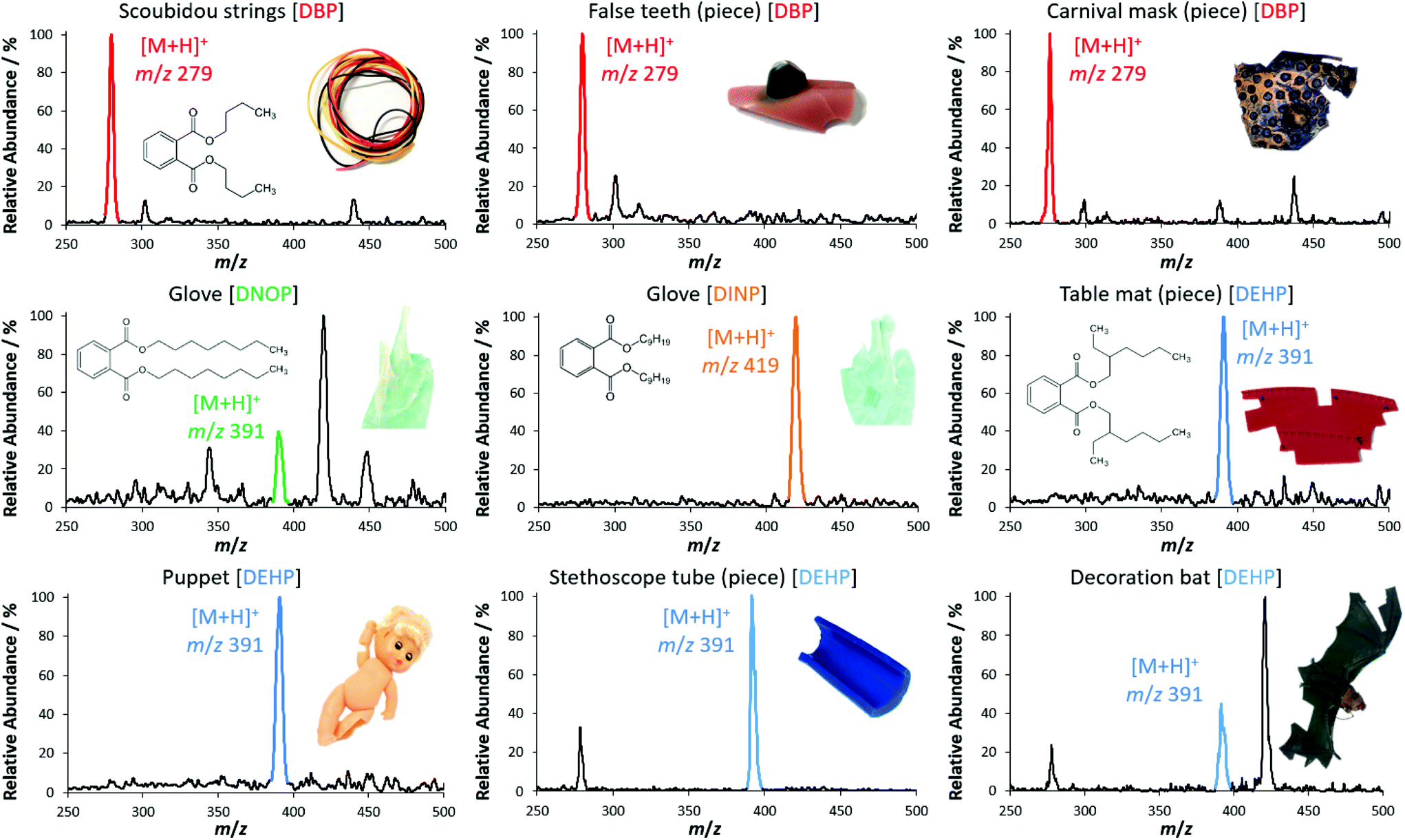 | ||
| Fig. 5 Qualitative plasticizer measurements from real sample objects. The portable LEP source was coupled to the miniaturized mass spectrometer Mini 11. For some objects, only small pieces are illustrated. The photographs of the samples are not true to scale due to distinct differences in size. Corresponding MS/MS data and analyses of other compounds using the LEP connected to the portable mass spectrometer can be found in the ESI.† | ||
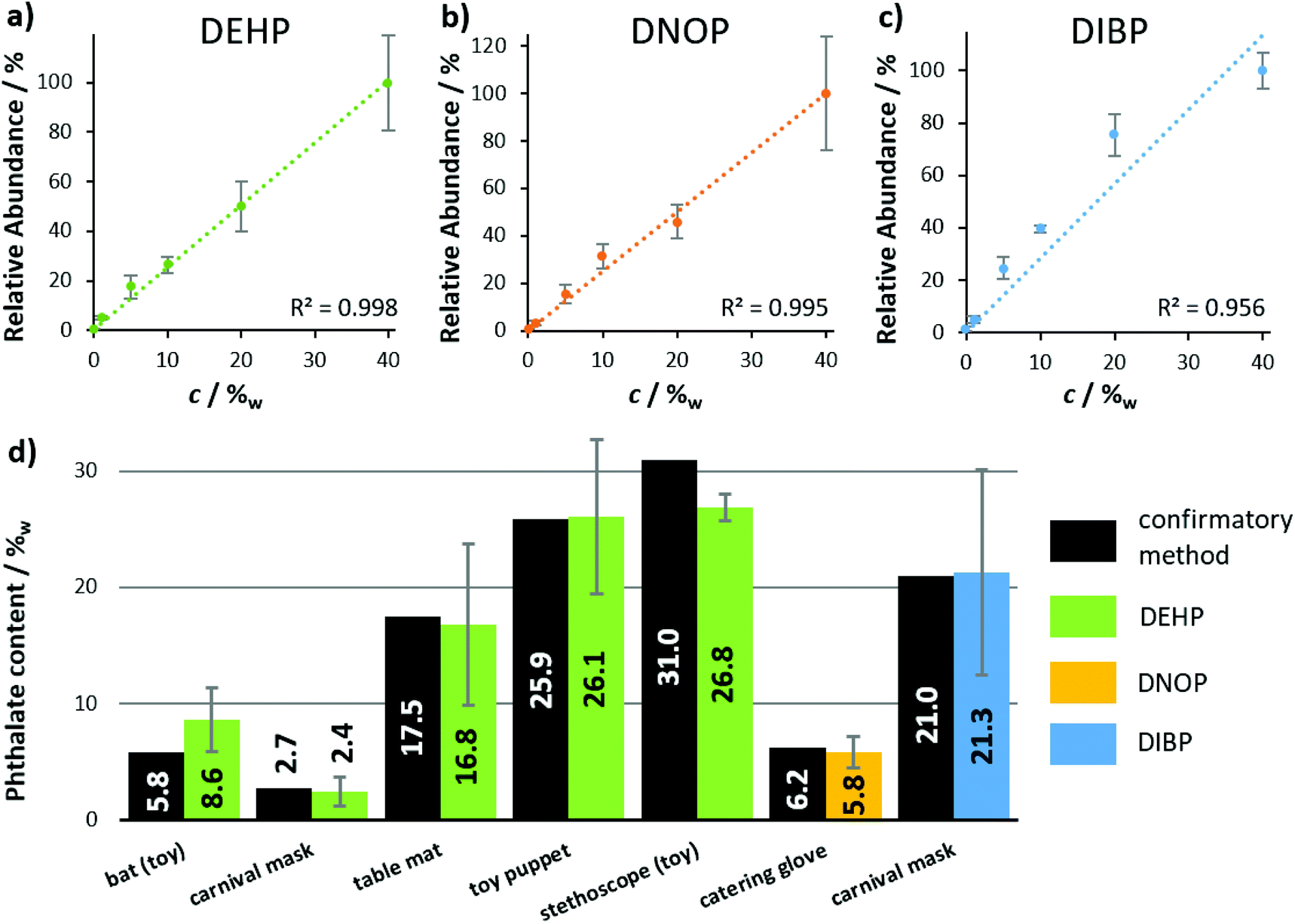 | ||
| Fig. 6 Quantitative analysis of phthalates in consumer goods, using the self-sustaining and portable LEP-MS system. Calibration curves were obtained from plastisol pills with 0–40%w of (a) DEHP, (b) DNOP and (c) DIBP. Data points represent averages of three technical replicates. Error bars indicate standard deviations. (d) Phthalate concentrations of the real samples, determined by LEP-MS, compared to concentrations obtained by confirmatory methods such as GC-FID, GC-MS and HPLC-DAD (see ESI Table 1† for more details). Error bars indicate measurement errors. | ||
Even if the sampling tip is formed by only two thin coated glass capillaries, no damage of capillaries was observed during the measurements. Unintended hard impact onto the sample is damped by the spring loading of the pen or the elasticity of the capillaries. The measurement procedure can leave traces on sample objects when the surface layers are soluble to the used solvent and no persistent analyte matrix is present (ESI Fig. 4†). This should be considered if analyses of e.g. soluble paints from paintings or similar sensitive samples are of interest. None of the presented sample objects did show any destruction due to physical contact to the LEP tip. Nevertheless, chemical and structural alterations of the sample surface and bulk material due to interactions with the solvent, cannot be excluded and must be evaluated for the individual sample. The bulk material of medical pills often consists of the soluble analyte itself. Due to the porous texture and absorbed solvent, structural degradation or liquefaction can appear during longer lasting measurements of several minutes.
In case of oily analytes like phthalates and capsaicin, a carry-over for several seconds was observed. Hence non-constant liquid extraction sampling methods rely on separately controlled washing steps after each analyte measurement, the constant liquid flow of fresh solvent through the LEP automatically rinses the secondary capillary and the spray capillary. One challenging aspect when working with the LEP is the sensitivity to clogging of the secondary capillary. It is advisable to perform measurements from particle-free surfaces to suppress any intake of solid materials into the capillary system. The low pumping efficiency of the ESI spray does not allow an implementation of a porous filter. If the capillary is clogged, a solvent droplet emerges on the sample surface. In this case, both capillaries must be flushed with fresh solvent through the high-voltage connector which can be performed in two minutes. More details on this topic are given in the ESI.†
Although delay times between sampling and ionization appear to be a disadvantage of the method, this feature of the source could be turned into an advantage, e.g. in a reactive LEP approach. If reactive additives are part of the solvent, reaction with analytes can proceed during delay time. Slow reactions which do not result in a sufficient product yield during short analyte–solvent interaction times like in DESI, become available using the LEP system. Analyte modifications are also conceivable via UV irradiation, if parts of the glass capillaries are placed in the beam path of appropriate light sources. Such a setup could be a useful tool for direct analyses of phospholipids from biological samples regarding the identification of double-bond position37 and many other applications.
The LEP was designed for manual but simple operation. The distinct difference to commercial and automated sources of using a handheld probe, further expands the range of applications of LMJ-SSP. Irregular sample topologies which are hardly accessible with present liquid-extraction methods such as nano-DESI, LESA or LMJ-SSP, predominantly rely on implementing sophisticated sample preparation steps like sectioning or sample flattening. Instead of adding complex robotic mechanics for proper positioning of the sample and the probe relative to each other, we come up with a robust tool for flexible operator-driven sampling. The method is inspired and closely related to the MassSpec pen setup but can be deployed almost everywhere in combination with a portable mass spectrometer, opening up a diverse field of new applications.
There are several portable MS with atmospheric pressure interface commercially available: Mini β (PURSPEC Technologies Inc.), which is the follow-up product of the prototype we used; MT Explorer 50 (MassTech); Portability (BaySpec, Inc.) and the Griffin AI-MS 1.2 (FLIR Systems). All use ion trap mass analysers featuring MS/MS capabilities for reliable compound identification but differ in size, weight, and performance. To our knowledge LMJ-SSP has not been used in combination with portable mass spectrometers by other researchers. Besides the portable and self-sustaining design of the LEP source the main differences to the MassSpec Pen are: (i) continuous (LEP) vs. discontinuous solvent flow (MassSpec Pen), (ii) use of an ESI source for analyte transport and ionization in case of LEP, (iii) open (LEP) vs. enclosed sampling spot (MassSpec Pen). In comparison to earlier presented results using DESI for ambient analysis of consumer goods, we found significantly better signal intensities for the tested analytes, especially for samples with low analyte concentrations, resulting in better LOD values by one to two orders of magnitude.
The implementation of a LEP, supplied by a portable solvent- and gas-delivery system makes LMJ-SSP a well-suited device for in-field analysis if connected to portable mass spectrometers, but can also allow for fast sample evaluation in a laboratory environment.
Conclusion
In this study, the capabilities of a portable and self-sustaining liquid microjunction surface sampling probe were tested. The probe was implemented in a handheld pen device, called a liquid-extraction pen (LEP), which gave maximum flexibility in sample handling. It was shown to allow rapid qualitative and quantitative analysis directly from solid sample materials, working independently of sample's topology or dimension. No sample preparation steps were needed. Profiting from the broad field of application and the robustness of ESI, the LEP can be connected to any kind of mass spectrometer, equipped with an atmospheric-pressure interface and can be applied to a variety of samples and analytes. In connection with a portable mass spectrometer and a high-performance lab-based mass spectrometer, qualitative and semi-quantitative applicational examples were presented for pesticides, plasticizers and drugs from corresponding daily consumer goods and for food fraud identification. The simple handling of the autarkic LEP source enables even untrained operators to perform LEP measurements.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by Deutsche Forschungsgemeinschaft (DFG, grant no. INST 162/500-1 FUGG) and by Justus Liebig University Giessen (research grant for junior academic staff) is gratefully acknowledged. The Bavarian State Laboratory for Health and Food safety (Erlangen, Germany) and the State Laboratory of Rhineland-Palatinate (Koblenz, Germany) are gratefully acknowledged for providing phthalate-containing samples and corresponding analytical data. Thermo Fisher Scientific Bremen GmbH is gratefully acknowledged for providing an Exactive orbital trapping mass spectrometer for this work. Vinnolit GmbH und Co. KG (Ismaning, Germany) is gratefully acknowledged for providing a sample of the PVC powder Vinnolit P 70. We obtained the biological samples (fish fillets, coffees, vegetables) from local supermarkets and online shops.Notes and references
- E. D. Lee, W. Mück and J. D. Henion, Biomed. Environ. Mass Spectrom., 1989, 844–850 Search PubMed.
- P. J. Roach, J. Laskin and A. Laskin, Analyst, 2010, 135, 2233–2236 RSC.
- V. Kertesz and G. J. van Berkel, J. Mass Spectrom., 2010, 45, 252–260 CrossRef CAS PubMed.
- G. J. van Berkel, V. Kertesz and R. C. King, Anal. Chem., 2009, 81, 7096–7101 CrossRef CAS PubMed.
- J. Laskin, B. S. Heath, P. J. Roach, L. Cazares and O. J. Semmes, Anal. Chem., 2012, 84, 141–148 CrossRef CAS PubMed.
- G. A. Schultz, T. N. Corso, S. J. Prosser and S. Zhang, Anal. Chem., 2000, 72, 4058–4063 CrossRef CAS PubMed.
- http://www.htximaging.com/htx-sepquant-dropletprobe .
- https://www.advion.com/products/triversa-nanomate/ .
- J. Zhang, J. Rector, J. Q. Lin, J. H. Young, M. Sans, N. Katta, N. Giese, W. Yu, C. Nagi, J. Suliburk, J. Liu, A. Bensussan, R. DeHoog, K. J. Garza, B. Y. Ludolph, A. G. Sorace, A. Syed, A. Zahedivash, T. E. Milner and L. S. Eberlin, Sci. Transl. Med., 2017, 1–11 Search PubMed.
- M. Sans, J. Zhang, J. Q. Lin, C. L. Feider, N. Giese, M. T. Breen, K. Sebastian, J. Liu, A. K. Sood and L. S. Eberlin, Clin. Chem., 2019, 65, 674–683 CrossRef CAS PubMed.
- C. Meisenbichler, F. Kluibenschedl and T. Müller, Anal. Chem., 2020, 92, 14314–14318 CrossRef PubMed.
- M. S. Elnaggar, C. Barbier and G. J. van Berkel, J. Am. Soc. Mass Spectrom., 2011, 22, 1157–1166 CrossRef CAS PubMed.
- C.-C. Hsu, M. S. Elnaggar, Y. Peng, J. Fang, L. M. Sanchez, S. J. Mascuch, K. A. Møller, E. K. Alazzeh, J. Pikula, R. A. Quinn, Y. Zeng, B. E. Wolfe, R. J. Dutton, L. Gerwick, L. Zhang, X. Liu, M. Månsson and P. C. Dorrestein, Anal. Chem., 2013, 85, 7014–7018 CrossRef CAS PubMed.
- G. J. van Berkel and V. Kertesz, Rapid Commun. Mass Spectrom., 2015, 29, 1749–1756 CrossRef CAS PubMed.
- C. L. Walton, V. Kertesz and J. F. Cahill, J. Am. Soc. Mass Spectrom., 2021, 32, 198–205 CrossRef CAS PubMed.
- G. J. van Berkel and V. Kertesz, Rapid Commun. Mass Spectrom., 2013, 27, 1329–1334 CrossRef CAS PubMed.
- V. Kertesz, D. Calligaris, D. R. Feldman, A. Changelian, E. R. Laws, S. Santagata, N. Y. R. Agar and G. J. van Berkel, Anal. Bioanal. Chem., 2015, 407, 5989–5998 CrossRef CAS PubMed.
- V. Kertesz, T. M. Weiskittel and G. J. van Berkel, Anal. Bioanal. Chem., 2015, 407, 2117–2125 CrossRef CAS PubMed.
- V. Kertesz, M. J. Ford and G. J. van Berkel, Anal. Chem., 2005, 77, 7183–7189 CrossRef CAS PubMed.
- https://www.advion.com/products/touch-express/ .
- F. Lotz, S. Gerbig, C. Lotze, B. Spengler and S. Schulz, Analyst, 2020, 145, 5584–5593 RSC.
- L. Gao, A. Sugiarto, J. D. Harper, R. G. Cooks and Z. Ouyang, Anal. Chem., 2008, 80, 7198–7205 CrossRef CAS PubMed.
- Z. Takáts, J. M. Wiseman, B. Gologan and R. G. Cooks, Anal. Chem., 2004, 76, 4050–4058 CrossRef PubMed.
- J. Tillner, V. Wu, E. A. Jones, S. D. Pringle, T. Karancsi, A. Dannhorn, K. Veselkov, J. S. McKenzie and Z. Takats, J. Am. Soc. Mass Spectrom., 2017, 28, 2090–2098 CrossRef CAS PubMed.
- D. I. Ellis, V. L. Brewster, W. B. Dunn, J. W. Allwood, A. P. Golovanov and R. Goodacre, Chem. Soc. Rev., 2012, 41, 5706–5727 RSC.
- K. Böhme, P. Calo-Mata, J. Barros-Velázquez and I. Ortea, TrAC, Trends Anal. Chem., 2019, 110, 221–232 CrossRef.
- S. Gerbig, S. Neese, A. Penner, B. Spengler and S. Schulz, Anal. Chem., 2017, 89, 10717–10725 CrossRef CAS PubMed.
- E. Cubero-Leon, R. Peñalver and A. Maquet, Food Res. Int., 2014, 60, 95–107 CrossRef CAS.
- S. H. Swan, Environ. Res., 2008, 108, 177–184 CrossRef CAS PubMed.
- J. M. Braun, S. Sathyanarayana and R. Hauser, Curr. Opin. Pediatr., 2013, 25, 247–254 CrossRef CAS PubMed.
- R. U. Halden, Annu. Rev. Public Health, 2010, 31, 179–194 CrossRef PubMed.
- K. Kappel and U. Schröder, Food Control, 2016, 59, 478–486 CrossRef.
- M. J. Martín, F. Pablos and G. Gonzáles, Talanta, 1998, 46, 1259–1264 CrossRef.
- P. Piotr Konieczka, M. J. Aliaño-González, M. Ferreiro-González, G. F. Barbero and M. Palma, Sensors, 2020, 20, 3123 CrossRef PubMed.
- S. Gerbig, G. Stern, H. E. Brunn, R.-A. Düring, B. Spengler and S. Schulz, Anal. Bioanal. Chem., 2017, 409, 2107–2117 CrossRef CAS PubMed.
- S. Schulz, S. Wagner, S. Gerbig, H. Wächter, D. Sielaff, D. Bohn and B. Spengler, Analyst, 2015, 140, 3484–3491 RSC.
- F. Wäldchen, S. Becher, P. Esch, M. Kompauer and S. Heiles, Analyst, 2017, 142, 4744–4755 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional photos of the ESI emitter tip and the influence of the primary capillary offset on the solvent flow rate. The time delay between LEP sampling and electrospray ionization is shown for two analytes. Analyte consumption and spatial resolution of LEP are visualized on a dye covered glass slides. Fast switching between LEP-MS and DESI-MS mode is demonstrated. Quantitative plasticizer analysis with the LEP source coupled to an orbital-trapping mass spectrometer and inter-day reproducibility data are shown for plasticizer and pesticides. For the food authentication PCA results are given for fish in the low and high mass range in positive-ion mode and low mass range in negative-ion mode. Further information on the origin of the biological samples. MS and MS/MS data for all presented and additional analytes, collected with an orbital trapping mass spectrometer and a miniaturized instrument. See DOI: 10.1039/d0an02281k |
| This journal is © The Royal Society of Chemistry 2021 |