
Rapid isolation of extracellular vesicles from diverse biofluid matrices via capillary-channeled polymer fiber solid-phase extraction micropipette tips
Kaylan K.
Jackson
 a,
Rhonda R.
Powell
b,
Terri F.
Bruce
c and
R. Kenneth
Marcus
*a
a,
Rhonda R.
Powell
b,
Terri F.
Bruce
c and
R. Kenneth
Marcus
*a
aClemson University, Department of Chemistry, Clemson, SC 29634, USA. E-mail: marcusr@clemson.edu
bClemson University, Clemson Light Imaging Facility, Clemson, SC 29634, USA
cClemson University, Department of Bioengineering, Clemson, SC 29634, USA
First published on 9th June 2021
Abstract
Extracellular vesicles (EVs) play essential roles in biological systems based on their ability to carry genetic and protein cargos, intercede in cellular communication and serve as vectors in intercellular transport. As such, EVs are species of increasing focus from the points of view of fundamental biochemistry, clinical diagnostics, and therapeutics delivery. Of particular interest are 30–200 nm EVs called exosomes, which have demonstrated high potential for use in diagnostic and targeted delivery applications. The ability to collect exosomes from patient biofluid samples would allow for comprehensive yet remote diagnoses to be performed. While several exosome isolation methods are in common use, they generally produce low recoveries, whose purities are compromised by concomitant inclusion of lipoproteins, host cell proteins, and protein aggregates. Those methods often work on lengthy timescales (multiple hours) and result in very low throughput. In this study, capillary-channeled polymer (C-CP) fiber micropipette tips were employed in a hydrophobic interaction chromatography (HIC) solid-phase extraction (SPE) workflow. Demonstrated is the isolation of exosomes from human urine, saliva, cervical mucus, serum, and goat milk matrices. This method allows for quick (<15 min) and low-cost (<$1 per tip) isolations at sample volume and time scales relevant for clinical applications. The tip isolation was evaluated using absorbance (scattering) detection, nanoparticle tracking analysis (NTA), and transmission electron microscopy (TEM). Exosome purity was assessed by Bradford assay, based on the removal of free proteins. An enzyme-linked immunosorbent assay (ELISA) to the CD81 tetraspanin protein was used to confirm the presence of the known exosomal-biomarker on the vesicles.
Introduction
Extracellular vesicles (EVs) are a diverse group of cell-derived membrane vesicles, typically ranging in size from 30 nm to 1 μm in diameter.1,2 EVs are released by all cell types and contain the biomolecular characteristics of the mother cell (i.e., DNA, RNA, miRNA, mRNA, biomarker proteins).3–7 While no official EV classification system exists, three main EV subtypes have been identified based on size and mechanism of biogenesis.8,9 Microvesicles are 100 nm to 1 μm vesicles created by the outward budding of a cell membrane. Apoptotic bodies (reflective of cell death10) are 1 to 5 μm vesicles created during the programmed cell death process. Exosomes are 30 to 200 nm vesicles created through the multivesicular body (MVB) endosomal pathway. Due to their similarities in composition, overlapping size range, and characteristic cup/dimpled shape when observed by electron microscopy, the exosome and microvesicle subtypes are difficult to differentiate. For this reason, the vesicles are generically referred to as EVs.11 Not surprisingly, within the heterogeneity in EV sources, size, and content, the specific mechanisms of action and distribution of potential biomarkers varies immensely.12EVs are primary vehicles in intercellular communication, signal transduction, and local and distal transport processes.13,14 The exosome subset of EVs has become increasingly targeted both as mediums for diagnostic information and cargo transmission.15,16 The lack of understanding of EV physiochemical and biological characteristics, along with a lack of field-wide consensus, has hindered the progress of the fundamental and clinical use of exosomes. A thorough understanding of exosome biophysical attributes would allow for details of several vital cell interaction mechanisms to be revealed (i.e., immune regulation, communication, and disease progression).17,18 The analysis of EV-associated biomarker components during liquid biopsies has become a valued tool for cancer detection, allowing for the surveillance of progression and treatment with a reduced physical burden on the patient.16,19 Alternatively, the large-scale processing of exosomes has become a key goal for researchers in many areas, including in the biopharmaceutical industry. EVs from mesenchymal stem cell (MSC) origin are of particular interest, having demonstrated the ability to enhance therapeutic transport of targeted drugs,20 initiate tissue regeneration,21 and support immune response modulation.14 Nevertheless, for the full extent of EV analyses to be realized, the inefficient tools for EV retrieval must be addressed.
Due to their ubiquitous nature in terms of the cells of origin, exosomes and other EVs are found in diverse biofluids, including urine,22–24 saliva,25–27 blood (serum and plasma),28–30 cervical mucus,25,31,32 breast milk,20–22 and cerebrospinal,33,34 lymph,35,36 synovial,37 and amniotic38 fluids. As such, these media are reservoirs to derive clinical and research scale populations. EVs may also be harvested from cell culture media during the cell growth process for fundamental studies or subsequent use as biotherapeutic vectors.39 Despite the high bioavailability of EVs, the extraction of EVs from biofluids has proven to be a challenge due to sample and vesicle heterogeneity and intense matrix effects. In terms of characterizing the effectiveness of generic EV isolation processes, several metrics exist relative to the final product's quality (versus the cost/time aspects of the procedures). The first, most obvious feature is the yield; how many microvesicles can be extracted per unit volume of the primary matrix. Practical working volumes can range from tens of microliters of cerebrospinal fluid (CSF) to milliliters of urine and liters of cell culture media. The second is the purity of the isolate. In the case of EVs, the primary contaminants/co-eluates are matrix and host cell proteins. In the case of serum/plasma samples, these would typically include albumins and, most problematically, lipoproteins.40,41 Finally, the most critical aspect is the retention of biological functionality. Whether the end-use is clinical analysis, fundamental research, or production of biotherapeutic vectors, the recovered EVs’ physical and chemical integrity must remain intact. Additional metrics come into play during high-specificity isolations of targeted EV populations. In all instances, aspects regarding processing time, capital and supply costs, and operational complexity must be considered.
It has been documented the needs for the development and optimization of methods specifically for the isolation and quantification of EVs from complex biofluid samples.42 The available methods for these purposes limit the downstream characterization and application of EV recoveries due to concentration and purity concerns. The lack of efficient EV isolation methods has become the rate-limiting step towards realizing the full potential of EVs in clinical and fundamental research and prevents large-scale processing of EVs. Many EV isolation methods are available based on a wide variety of chemical/physical properties. Riekkola and co-workers have recently presented an excellent review of the topic,43 with many papers describing comparisons of the methods. At this point, it is clear that no single method can be universally applied.44,45 The employed isolation method is usually chosen based on the subsequent means of characterization and utilization of the EVs. At present, ultracentrifugation (UC) methods are most commonly used to isolate EVs.46 The UC isolation method consists of several differential centrifugation steps, potentially reaching 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g.13 UC introduces high-costs regarding time (2 hours to overnight), sample volume (10–45 mL), and capital (up to $100000 for equipment, and $3000 in running costs per year), producing low recovery/yields (5–25%) which are typically contaminated with protein/lipoprotein aggregates.46,47 Variations of this technique employing density gradients and other reagents have also been implemented but continue to present the previously-mentioned challenges.46,48 Other size/density-based methods include ultrafiltration, size-exclusion spin downs, and field flow fractional.49–51 Here again, low purity recoveries are problematic. As a final class of methods, immune-affinity and polymer precipitation “kits” are finding increased use.52,53 Still, concerns lie in the low yield and impure recoveries, skewing the downstream characterization of the vesicles. Ultimately, an isolation method with the ability to efficiently produce high-yield, high-purity EVs on practical time/cost scales is of critical importance.
000g.13 UC introduces high-costs regarding time (2 hours to overnight), sample volume (10–45 mL), and capital (up to $100000 for equipment, and $3000 in running costs per year), producing low recovery/yields (5–25%) which are typically contaminated with protein/lipoprotein aggregates.46,47 Variations of this technique employing density gradients and other reagents have also been implemented but continue to present the previously-mentioned challenges.46,48 Other size/density-based methods include ultrafiltration, size-exclusion spin downs, and field flow fractional.49–51 Here again, low purity recoveries are problematic. As a final class of methods, immune-affinity and polymer precipitation “kits” are finding increased use.52,53 Still, concerns lie in the low yield and impure recoveries, skewing the downstream characterization of the vesicles. Ultimately, an isolation method with the ability to efficiently produce high-yield, high-purity EVs on practical time/cost scales is of critical importance.
To address the aforementioned issues, researchers from the Bruce and Marcus groups have demonstrated the use of a polyester (PET) capillary-channeled polymer (C-CP) fiber stationary phase in hydrophobic interaction chromatography (HIC) workflows for EV isolation.54–60 The C-CP fibers consist of an 8-legged periphery that creates 1 to 4 μm-wide channels upon colinear packing in a column format. The relative hydrophobicity of the stationary phase and the high-salt retention of the EVs allows for the capture and elution of the vesicles based on hydrophobicity. HIC has been traditionally applied to protein separations61 due to the non-denaturing, on–off partitioning of the solute, allowing the preservation of structure/function.62–64 Taking advantage of this, the efficient and vesicle-preserving isolation of EVs from urine,54,56 blood plasma,55 and cell culture milieu54,58 have been demonstrated in a 10 min HPLC workflow enabling simultaneous EV isolation and quantification. Importantly, recent proteomics analysis of the eluates has revealed a very efficient removal of serum proteins and lipoproteins, yielding extremely high purity fractions in comparison to other methods.59 The method has been extended to a more clinically-favorable EV isolation workflow using 1 cm C-CP fiber phases attached to micropipette tips, allowing for the solid-phase extraction (SPE) of EVs to occur in a table-top centrifuge.57 Both methods have proven to be beneficial in terms of efficiency, purity, and yield, producing recoveries of EVs on clinically relevant scales of time (<15 minutes) and cost (<$1 per column per tip). Here, the versatility of the C-CP fiber spin-down tip to produce concentrated and contaminant-free EV recoveries is demonstrated for the complex matrices of urine, saliva, cervical mucus, serum, and milk. The tip recovery of exosomes was evaluated using absorbance (scattering) detection, nanoparticle tracking analysis (NTA), and transmission electron microscopy (TEM). The exosome purity was assessed by Bradford assay of free proteins. The bioactivity and identity of the recovered vesicles was confirmed with an enzyme-linked immunosorbent assay (ELISA) to the CD81 tetraspanin protein. It is believed that the methodology presented here will have relevance to both clinical and fundamental biology research settings.
Experimental
Chemicals and reagents
Deionized water (DI-H2O, 18.2 MΩ cm) was obtained from a Milli-Q water purification system (Millipore Sigma, Merck, Darmstadt, Germany). Biotechnology-grade glycerol and ammonium sulfate were purchased from VWR (Sokon, OH, USA). Phosphate buffered saline (PBS, pH = 7.4), bovine serum albumin (BSA), and Pierce™ Coomassie Plus (Bradford) Assay Reagent were purchased from ThermoFisher Scientific (Waltham, MA, USA).Instrumentation
A NanoVue Plus UV-Vis spectrophotometer (GE Healthcare, Chicago, IL, USA) was used to measure the absorbance/scattering (203 nm) of the EV fractions. A Synergy H1 Hybrid Multi-Mode Plate Reader (BioTek, Winooski, VT, USA) was used to measure the UV-Vis absorbance (595 nm) of samples in the 96 cell-well format during the Bradford assay of protein content, employing the colorimetric Pierce™ Coomassie Plus (Bradford) Assay Reagent. The plate reader was also used in the chemiluminescent detection of the Pierce™ ECL Substrate during the enzyme-linked immunosorbent (ELISA) assay. A Hitachi HT7830 transmission electron microscope (Chiyoda City, Tokyo, Japan) was used for TEM imaging to determine the structural integrity, size, and purity of the EVs in the C-CP tip recoveries from various biofluids. A Malvern Panalytical NanoSight NS300 nanoparticle tracking analysis (NTA) system (Malvern, Worcestershire, United Kingdom) was used to determine the concentration and size distribution of isolated vesicles.Extracellular vesicles
Commercial lyophilized “exosome standards” from the urine of healthy donors were obtained from Galen Laboratories Supplies (Craigavon, Northern Ireland). To be clear, the material has not been certified as a reference standard. No information regarding purity or classification was supplied from the manufacturer. However, the product is a means of preparing EV solutions of known concentration (2.7 × 1012 particles per mL), though vesicles exceeding typical exosome diameter, lipoproteins, and other protein contaminants have been previously identified in the material.65 Despite the potential of systematic error (impurities) introduced by these standards, they have proven useful for order-of-magnitude estimation of recovered EV concentrations.Fresh-morning urine, saliva, and cervical mucus (collected using a cotton swab and dissolved in PBS) were obtained from consenting, anonymous donors. After sample collection, the cervical mucus samples were stored at −80 °C until thawed for EV processing. Corning™ Human AB Blood Serum was obtained from ThermoFisher Scientific (Waltham, MA, USA). The frozen human serum was thawed and aliquoted before use. Unpasteurized raw goat milk (serving as a surrogate for human breast milk) was obtained from Split Creek Farm (Anderson, SC, USA). All biofluid samples were filtered using a sterile syringe filter of 0.22 μm pore size (Frogga Bio, Toronto, Canada) prior to processing.
C-CP fiber tip creation and methodology
The C-CP fiber micropipette tips were assembled as previously reported,57 with the same HIC isolation workflow employed. Briefly, the 1 cm long C-CP fiber tips were cut from 30 cm long, 0.8 mm inner diameter fluorinated ethylene–propylene (FEP) C-CP packed columns consisting of ∼450 PET C-CP fibers. The C-CP tips had an interstitial fraction of ∼0.6, with ∼3 μL of bed volume, which was press-fit to 200 μL low-retention micropipette tips and secured with a small amount of superglue, as depicted in Fig. 1. The EV isolation methodology for the various biofluids was initiated by mixing 100 μL of the raw biofluid with 100 μL of ammonium sulfate (2 M final concentration) to induce the hydrophobic interaction between the biofluid components and the fibers. The total volume was vortexed, then deposited inside the sample reservoir of the C-CP tip assembly. The apparatus was then placed inside a 15 mL conical, table-top centrifuge tube and spun-down at 300g for 1 minute. (Due to the high viscosity of the saliva matrix, the tip containing the saliva sample was centrifuged at 500g for 10 minutes.) Next, the fiber-bound vesicles were washed with 200 μL of DI-water (300g, 1 min) before inducing the elution of free proteins (including lipoproteins59,60) using 200 μL of 25% glycerol with 1 M ammonium sulfate in PBS (300g, 1 min). For the protein-rich serum and milk matrices, two protein elution steps were employed to minimize protein carryover in the EV elution. Finally, the elution of the EVs was induced using 50 μL of 50% glycerol in PBS (300g, 1 min) and the final fraction collected. Based on the respective sample/elution volumes, a 2× concentration factor is realized.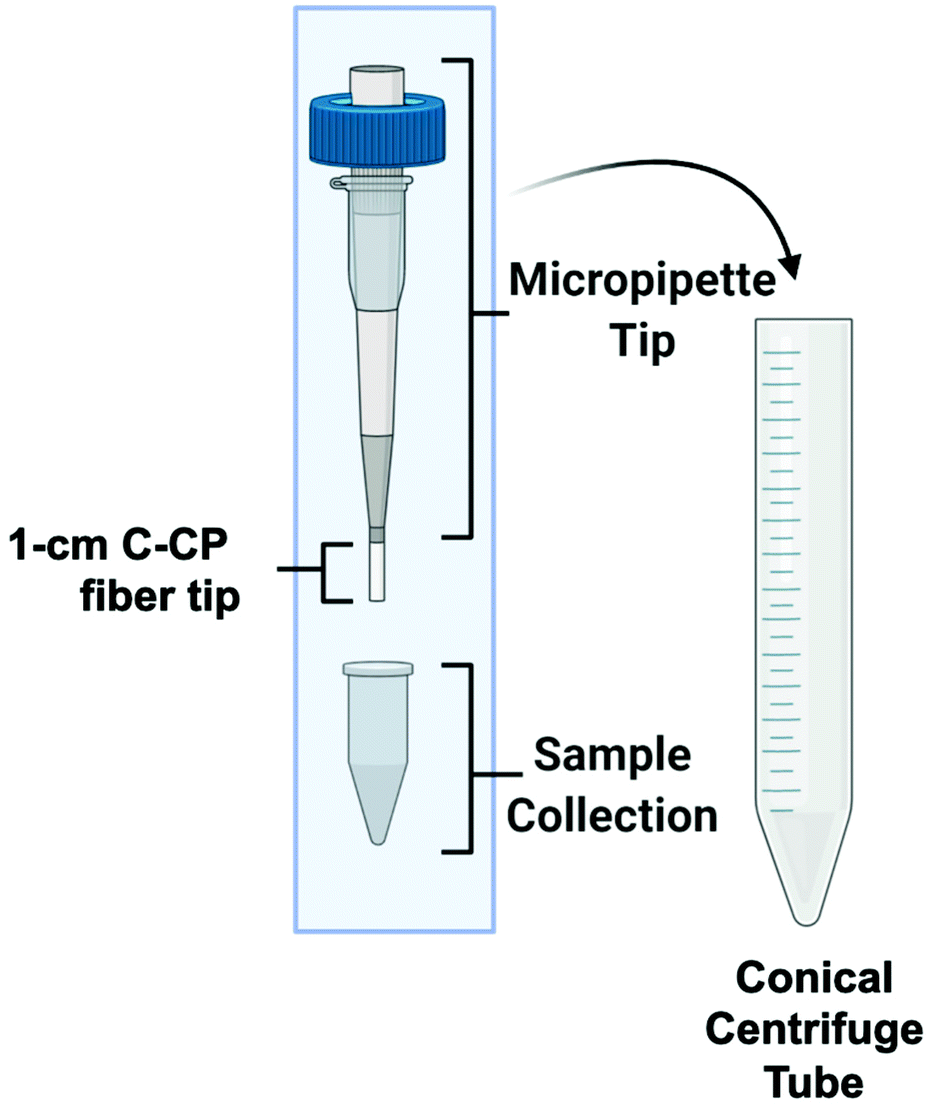 | ||
| Fig. 1 The capillary-channeled polymer (C-CP) fiber solid-phase extraction up setup for the isolation of EVs from complex biofluids in a tabletop centrifuge. | ||
Quantification and characterization of EV recoveries
Previous reports have demonstrated the validity of using standard absorbance (scattering) measurements as a means of quantifying isolated exosomes.54–56 In those efforts, quantification was achieved by generating linear response curves based on serial dilutions of the commercial exosome standards in the elution solvent. Given the high complexity and presence of matrix-associated components in the diverse biofluid matrices, the method of standard addition was also used to more accurately quantify the EVs. For the method, 10 μL of the unknown sample (S0) was spiked once (S1), twice (S2), and three times (S3) with 10 μL of EV standards of known concentration (2.7 × 1010 particles per mL), with the total sample volumes adjusted to 50 μL using DI-water. The absorbance of each sample was measured at 203 nm (n = 5). The optical absorbance of the raw and spiked samples (S0–4) and the known added concentrations of exosome standards were used to create a standard addition response curve for EV quantification. The resulting linear regression was extrapolated to determine the concentration of EVs in the unknown sample.The structure, size, and concentration of the recovered EVs were evaluated using TEM and NTA. The sample preparation for TEM imaging was performed as previously reported.57 The size distribution of the eluted EVs was determined using the NanoSight NS300 NTA system, equipped with a 532 nm laser. Throughout NTA experimentation, five replicates were collected for each sample in 60-second intervals, with a minimum of 200 valid tracks recorded per video and a minimum of 1000 valid tracks recorded per sample. The focal plane for each sample was manually adjusted using the focus knob to achieve the best optical field of view. The syringe pump for sample introduction was set to a constant flow rate of 50 μL per minute. The camera level was set to 14, and the detection threshold was set to 3, as optimized by Vestad et al.66 To clarify, the concentration values based on the NTA data are not the direct concentration values of the EV recoveries. Instead, the recovered EVs were diluted to be compatible with the NTA system's working concentration range (107–109 particles per mL).
Protein components of the biofluids and EV recoveries were evaluated using a Bradford assay and an indirect enzyme-linked immunosorbent assay (ELISA). The Bradford assay was used to determine the total protein concentration of both the whole samples and EV elution fractions. For the total protein determinations, 250 μL of Bradford reagent was added to 25 μL of each sample and allowed to incubate at room temperature for 20 minutes before detecting the absorbance response at 595 nm using the Synergy H1 Plate Reader. The absorbance responses were compared to a standard curve using BSA standards. All samples and standards were applied to the cell well plate in triplicate.
The presence of EVs in biofluids is commonly confirmed using antibodies to the CD81, CD63, and CD9 tetraspanin proteins, which are incorporated in the transmembrane space of EVs during biogenesis.67 Despite their wide use as marker proteins, tetraspanins are in fact not universally expressed in EVs, and the overall expression is also heterogeneous among singular EV populations.68 Therefore, the presence of EVs may be confirmed by the detection of these proteins, but their absence does not preclude the presence of EVs. Prior to chemical processing for the CD81 ELISA assay, the tip-isolated EVs were applied to a 100 kDa filter unit to remove latent glycerol, as high concentrations of glycerol are known to interfere with antibody binding.69,70 The EVs isolated from the target biofluids were first diluted in 1:1 ELISA coating buffer (0.05 M carbonate–bicarbonate in PBS) and then incubated on a shaker overnight at 4 °C to coat the cell well plate with the analytes. An exosome standard positive control and negative controls of PBS, protein elution buffer, and EV elution buffer were also applied to the cell well plate. All samples and controls were applied to the cell well plate in triplicate. After incubation, the cell well plates were washed with sterile PBS (200 μL per well, 30 min, 6 buffer changes) and then blocked with 5% BSA in PBS at room temperature for 30 min. The wells were incubated overnight with 50 μL of a mouse monoclonal antibody to the CD81 protein (1 μg mL−1) on an orbital shaker (4 °C). The washing and blocking steps were repeated before applying 200 μL of the goat anti-mouse HRP conjugated secondary antibody (1 μg mL−1, 200 μL, RT, 2 hours). The cell well plate was washed using 200 μL of PBS per well and 6 buffer changes. Finally, the Pierce ECL Substrate was applied and incubated at room temperature for 30 minutes before detection. The Synergy H1 microplate reader was used to measure the chemiluminescent response resulting from the HRP catalyzed oxidation of the substrate, correlating to the concentration of species containing the CD81 antigen.
Results and discussion
EV quantification via standard addition
Concentrated EV recoveries with high purity, preserved morphology, viability, and stability are essential for the most efficient use of EVs derived via any isolation method. Given the complexity and diversity of the biofluids (and culture media), removing matrix contaminants is of utmost importance. Carryover of matrix species with the target EV isolates, including proteins and genetic material, hinders the implementation of downstream characterization techniques (i.e., MS proteomics or RNA-Seq), their use in clinical analysis schemes, and use as vectors in gene therapy applications. In this regard, the use of optical absorbance as an EV quantification tool is particularly susceptible to interferences due to the presence of low concentrations of matrix species. However, the quantification of isolated EVs by absorbance has been previously demonstrated using simple optical absorbance measurements at 203 nm.54–59,61 To be clear, the absorbance response observed at this wavelength is not credited to the common electronic transitions typical of biomolecules in solution. Instead, the “absorbance” response is caused by light scattering due to the presence of the nanobodies, which is conveniently proportional to the EV concentration. A cause for concern with this method for quantifying EVs is that matrix proteins and nucleic acids will skew the absorbance detection, especially at the 216 and 280 nm wavelengths traditionally used for determinations of proteins. These effects are lessened at 203 nm, where a higher absorbance (light scattering) response is observed at shorter wavelengths.48 In fact, absorbance spectra obtained for EV solutions follow the anticipated responses (exponentially decreasing with wavelength) for particles of ∼150 nm, based on Mie scattering theory.The method of standard addition is widely used for the quantification of analytes whose responses (regardless of the methodology) are subjected to significant matrix interferences.71 The method has not been previously employed for the quantification of EVs in biofluids, but could prove useful in this application as diverse matrices are being evaluated. A proof of concept for this method is illustrated in Fig. 2, where the method of standard addition was used to quantify EVs in aqueous solution using the commercial exosome stock. The method was first applied to test the “unknown”, which was the initial exosome stock solution of 7.0 × 1010 particles per mL. The test unknown (S0) was spiked once (S1), twice (S2), and three times (S3) with aqueous aliquots of the EV standard, increasing the theoretical concentrations by 1.1 × 1010, 2.2 × 1010, and 3.2 × 1010 particles per mL, respectively. As shown in Fig. 2 (red line), the absorbance responses for the un-spiked (S0) and spiked (S1, S2, S3) EV stock aliquots in DI-water are well behaved, yielding a correlation coefficient (R2) of >0.999. Based on the linear regression, the “unknown” concentration was determined to be 7.4 × 1010 particles per mL, a 5% error. As a point of reference, the concentration of the same solution determined by a standard calibration curve (R2-value = 0.998) yielded a concentration of 6.3 × 1010 particles per mL, a 10% error (accuracy that would be considered outstanding by virtually any other EV assay method).
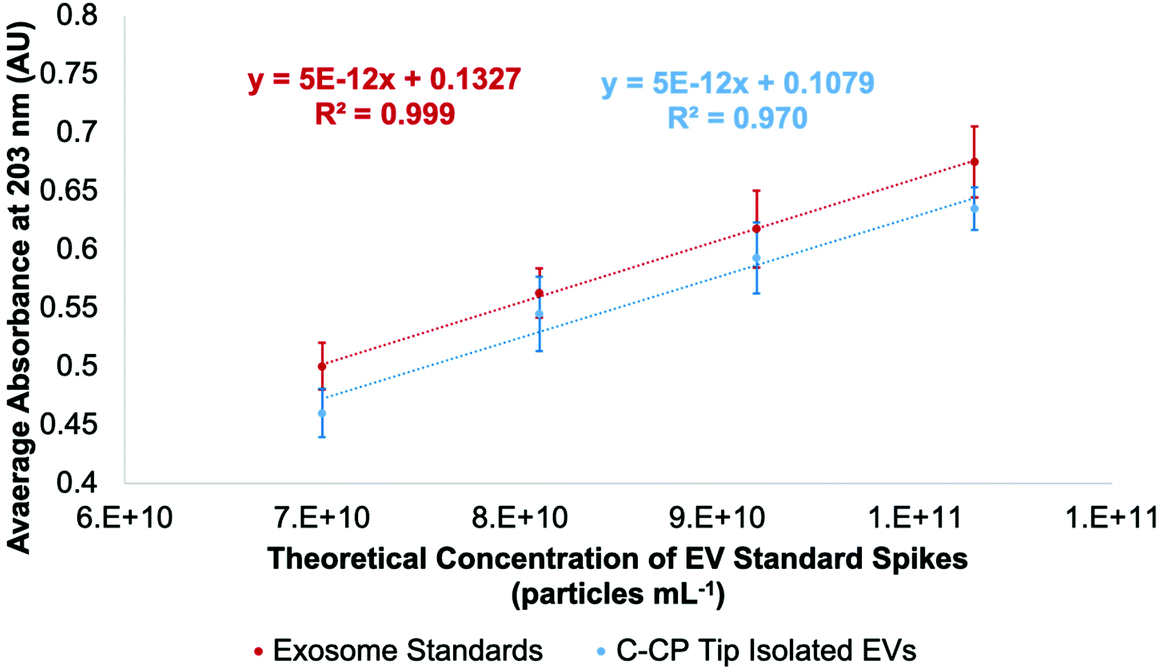 | ||
| Fig. 2 Standard addition curve using a commercial exosome standard stock of 1.1 × 1010 particles per mL concentration based on absorbance measurement at 203 nm (red). Quantification of EVs based on absorbance detection after employing aqueous EV solutions of known concentration to the C-CP tip (blue). | ||
As mentioned previously, the stock exosome material is known to contain undetermined amounts of proteinaceous material and other vesicular bodies. As a further test of the use of the standard addition quantification method, the “unknown” sample and the equivalent spike samples were put through the spin-down protocol. As seen in the response curve (blue line), proportional recoveries are indeed maintained, reflecting a lack of any sort of overloading of the fiber phase. Indeed, the recoveries are quite high versus the EVs in the stock aqueous solution, ranging from 96–102% (concentration of recovered EVs /raw stock), with the lower y-intercept being attributed to the removal of the latent proteins in the original stock material. Also of relevance, the average variation for the bulk measurements was 4%RSD, while for the full extraction process the variability averaged 5%RSD. There is some level of degraded quantitative performance (scatter) in the tip recoveries, as seen in the lessened goodness-of-fit (R2 = 0.970).
EV recoveries from diverse matrices
After confirming the ability of the standard addition method to determine the concentration of EVs and the C-CP tip's ability to produce quantitative EV recoveries, the experimental protocol was applied to the raw biofluid matrices. The urine, saliva, cervical mucus, serum, and milk biofluids samples were spiked as described above, followed by tip isolation. The raw biofluids were spiked once, twice, and three times with EV stock solutions of increasing concentration (1.1 × 1010 particles per mL per spike), then diluted to 200 μL with ammonium sulfate (2 M final concentration) before applying to the C-CP spin-down tips for the isolation process (load, protein wash, EV elution). The absorbance response of the EV eluates was measured at 203 nm. The relative absorbance responses presented in Fig. 3 reflect the fact that the C-CP tip does produce EV recoveries of proportionally increasing concentrations, despite the biofluid sample complexity. The respective regressions of each have an R2 correlation coefficient of >0.98. The determined values for each of the biofluids are provided in each case, with respective values each falling in line with expectations based on literature values.72–76 The relative precision of the determined values (n = 3) is excellent, with an average value of ∼7%RSD across the matrices.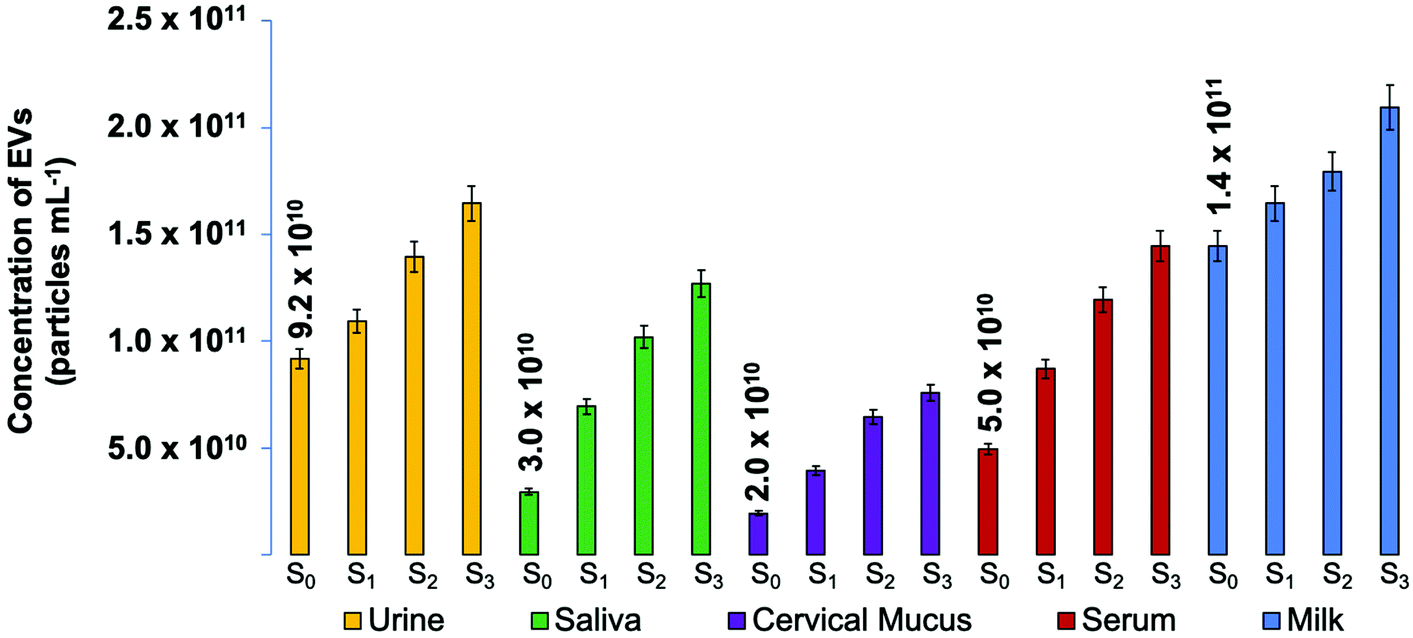 | ||
| Fig. 3 Determined EV particle concentrations (n = 3) for human urine, saliva, cervical mucus, blood serum and goat milk biofluid unknown samples spiked once, twice, and three times with a commercial exosome standard stock of 1.1 × 1010 particle per mL before EV isolation using the C-CP tip workflow. The biofluid-originating EV recoveries were quantified based on absorbance at 203 nm and compared to a response curve of linear response. | ||
The relative responses for the spikes across the different matrices are fundamentally instructive. In theory, consecutive increases of 1.1 × 1010 particles per mL EV concentration were applied. Therefore, given a homogenous and ideal biofluid sample, the difference between the determined concentrations of the Sx and Sx+n samples should be 1.1 × 1010 particles per mL. While the responses here are proportional within each matrix type, there is a definitive difference in the slopes; i.e. the method of standard addition reveals the existence of matrix effects. That said, given the vast physico-chemical differences among these biofluids, the extent of the effects, based on the slopes of the response curves, are less than a factor of 2×. As such, the use of a single absorbance calibration function would deliver that level of accuracy, with higher levels achieved with the use of matrix-matched standards. Analysis across multiple matrices would benefit most using the standard addition method.
Physical characterization of EV isolates
To confirm that the C-CP tip elution fractions do indeed contain EVs in the correct size range and consist of the expected characteristic shape, NTA and TEM imaging were performed. Fig. 4 presents both the size distributions observed via NTA and electron micrographs of the intact vesicles following isolation. The eluted EVs presented average diameters from 121.7–160.3 nm across the matrices. Based on the NTA data, the populations of EVs recovered from the urine, saliva, and milk samples presented the most “gaussian-like” size distributions, though with minor subsets of vesicles detected at larger sizes (as is typical). On the other hand, the EVs isolated from the cervical mucus and blood serum samples were far less homogeneous, with several distinct subpopulations.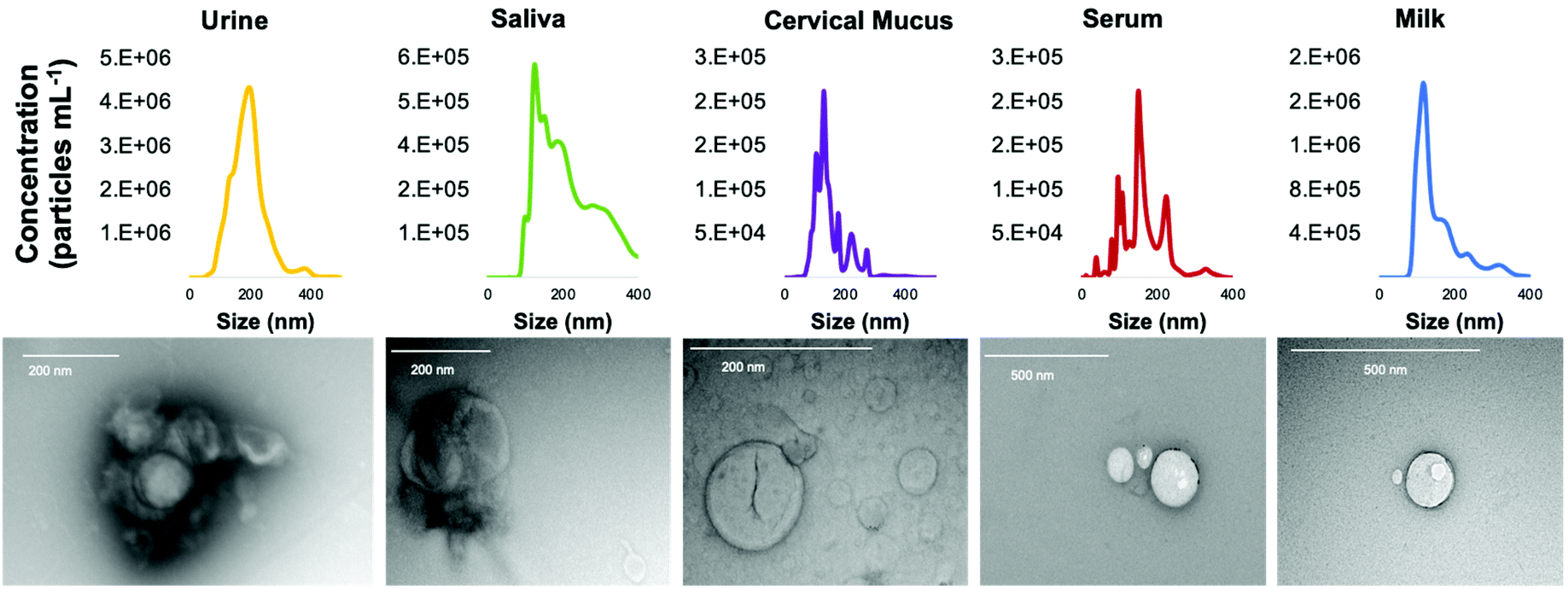 | ||
| Fig. 4 Size distribution of vesicles in the EV recoveries resulting from the C-CP fiber tip isolation from human urine, saliva, cervical mucus, blood serum, and goat milk, measured using the Nanosight NS300 nanoparticle tracking analysis system. TEM micrographs of EVs isolated from biofluids using the C-CP fiber tip, taken using the Hitachi HT7830. | ||
Visualization via transmission electron microscopy (TEM) is another benchmark method to identify extracellular vesicles. The TEM micrographs presented in Fig. 4 confirm that EVs were isolated from the biofluids using the C-CP spin-down tips. In each of the images, either cup-shaped, donut-shaped, or spherical-shaped vesicles with a dark halo can be observed. The EVs observed in the TEM micrographs fall within the exosome size range. One key aspect to emphasize is that, even in the potentially lipoprotein-heavy biofluids (cervical mucus, serum, milk), no vesicles are observed that would correspond to the anticipated lipoprotein size range (∼20 nm) characteristic of LDLs. The isolation of EVs from lipoproteins is a fundamental challenge due to the similarities of the vesicles’ size, structure, composition, and biological interactions.40,41 High purity recovery of EVs (i.e., the lack of matrix proteins/lipoproteins) using the fiber isolation methodology has been demonstrated in recent mass spectrometric proteomics analyses,59,60 and is a significant advantage of the C-CP tip isolation technique. This point is further demonstrated in the following section. The TEM images show that the HIC-based C-CP tip isolation preserves the characteristic vesicular shape with no visual contamination.
Beyond the size distribution, NTA can also be used as a semi-quantitative means of determining nanoparticle densities. As presented in Fig. 5a, the particle densities determined for the raw biofluids via NTA can be appreciably higher than the corresponding values generated by absorbance measurements. Not surprisingly, this is particularly true for the most proteinaceous matrices (where agglomeration would likely occur). In these cases, the densities determined by NTA can exceed those of absorbance by as much as an order of magnitude, with the measurement variability also highest for those samples. Importantly, the same analyses performed on the spin-down isolates (Fig. 5b) yield values in far better agreement between the two quantification methods, with much-improved measurement precision realized for the NTA. It is noteworthy that the relative concentrations across the matrices parallel each other between the two independent measurement methods, with the values not differing by more than 2×. This level of agreement is seen as validation of the efficacy of the C-CP fiber spin-down tip methodology.
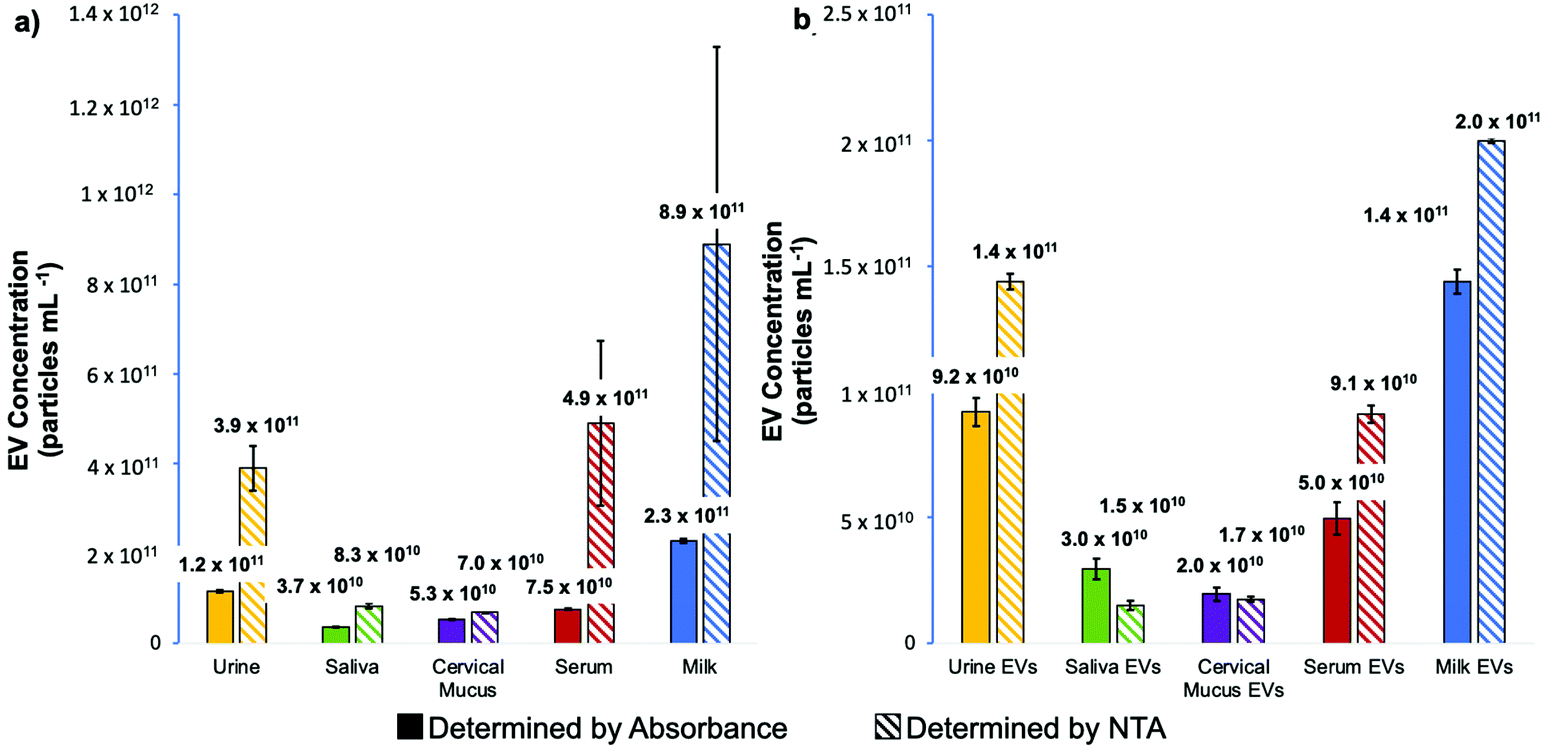 | ||
| Fig. 5 Comparison of the determined concentration of (a) EVs in each bulk blofluid sample and (b) EVs recovered from each biofluid using the C-CP tip method as determined using the method of standard addition by absorbance at 203 nm and by nanoparticle tracking analysis. | ||
Characterization of EV purity
To investigate the purity of EVs (based on the removal of matrix proteins) isolated using the C-CP tip method, a Bradford assay was performed. The total protein concentrations of the whole biofluid samples and the EVs eluted from those biofluids using the C-CP tip isolation method were determined. To be clear, a Bradford assay reflects the total proteinaceous material present in a sample. As such, in the ideal case of perfect isolation of EVs, a positive response will still result due to the interaction between the Bradford reagent and surface proteins and externally exposed basic and aromatic amino acid residues. The Bradford assay results for the raw matrix materials and the EV isolates are presented in Fig. 6. As would be anticipated, the goat milk and human blood serum matrices were the most protein-dense, with the human urine matrix having the lowest amount of protein present. After conducting the C-CP fiber spin-down tip EV isolation workflow, most of the contaminating proteins were removed while leaving behind the EVs, which contribute to only a small fraction of the total protein response for the protein-rich matrices. Here, the 67–89% removal of “total protein” was demonstrated for the saliva, cervical mucus, serum, and milk biofluid samples. A much lower (17%) removal of proteins was observed from the human urine sample, as expected given the much lower relative concentration of free protein in healthy urine samples. The EV recoveries present a low (346–412 μg mL−1) total protein concentration based on the Bradford assay. While not perceivable on this scale, the relative amounts of determined protein for the isolates are a very close reflection of their relative EV densities determined via the standard addition and NTA methods (Fig. 3 and 5), suggesting the efficacy of the method to yield high-purity EVs. The C-CP tip method demonstrates here the ability to remove up to 89% of protein contaminant species. The efficiency of the method is demonstrated by the absence of proteinaceous aggregates in the TEM micrographs of EVs after the tip isolation process. Perhaps most definitive, recent MS proteomic analysis work has confirmed the removal of common contaminant lipoprotein species from serum samples using this method, based on the virtual absence (<0.3% of total proteins) of the Apo-B100 content in the EV isolates.60 The depletion of the lipid marker protein was confirmed by ELISA analysis, as well.60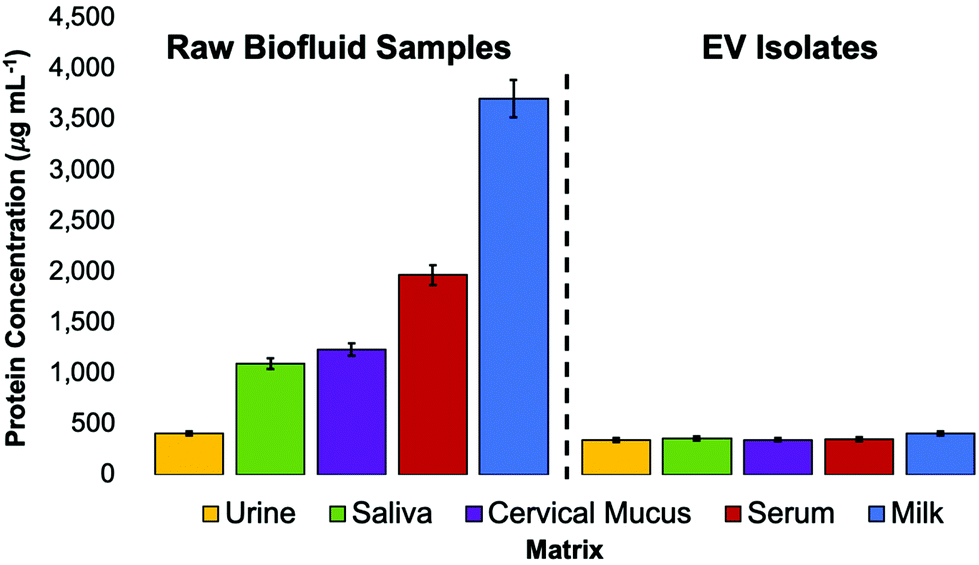 | ||
| Fig. 6 Bradford assay of raw biofluid matrices and concentrated EV recoveries after isolation with the C-CP tip. The total protein concentration was determined using the absorbance measurement of Bradford reagent at 595 nm, as compared to a BSA standard curve of linear response. n = 3. | ||
Verification of EV identity
While no universally expressed EV/exosome marker exists, the CD81 tetraspanin protein has been identified in high concentrations in many exosome populations.55 (The CD63 and CD9 tetraspanins have been used as identifiers in previous works from this laboratory,57 but CD81 generally exists in higher concentrations.) As such, the marker has been accepted as a general marker for the presence of EVs, with the acknowledgment that it is expressed to different extents even within the same EV population, and in some cases not at all. To confirm the presence of EVs in the C-CP tip eluates and assess the recovered vesicles’ bioactivity, a semi-quantitative ELISA using an antibody to the CD81 tetraspanin protein was employed. As shown in Fig. 7, serial dilutions of the commercial exosome standard stock were used to create a standard curve for the ELISA response quantification. With this standard curve of linear response (R2 = 0.985), the concentration of recovered EVs containing the CD81 tetraspanin protein was estimated. When the concentration of EV standards presenting a CD81 response was compared to the EV concentration as determined by absorbance detection (Fig. 3), the relative concentrations show the same general trends among the matrices. Even so, the quantitative numbers for the exosome concentrations reflect recoveries of 53–91% across the matrix types versus the absorbance-determined concentrations (Fig. 3). This level of agreement between the highly generic (absorbance) and highly specific (ELISA) means of quantification is quite remarkable. Based solely on the CD81 ELISA, the highest percentage of recovery for EVs containing CD81 was found for the blood serum sample (91%), followed by the saliva (70%), urine (59%), cervical mucus (58%), and the goat milk (53%). This level of variation is not at all surprising because CD81 is not universally expressed and is upregulated/downregulated in EVs of different origins.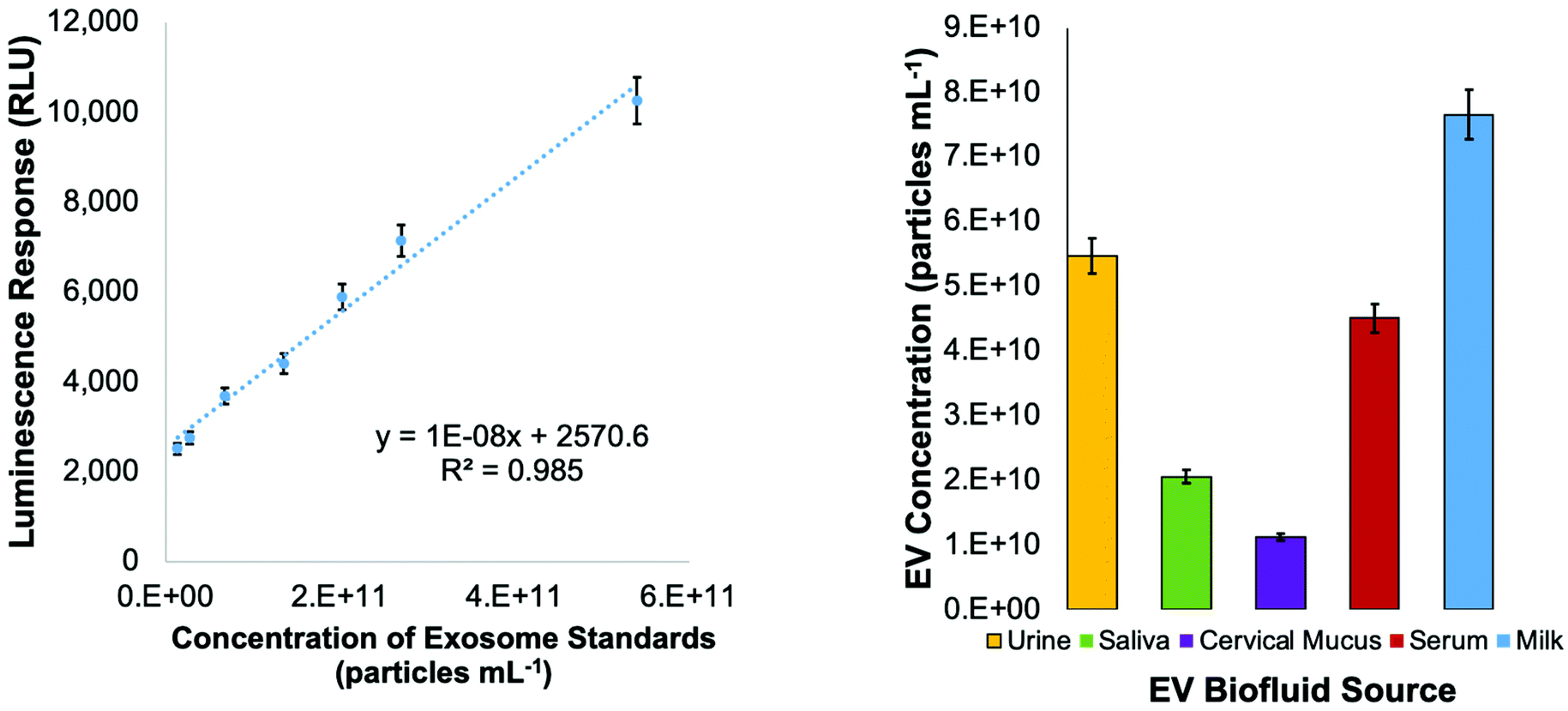 | ||
| Fig. 7 Indirect ELISA standard curve employing an antibody to the CD81 tetraspanin protein using serial dilutions of a commercial exosome standard (2.7 × 1012 particles per mL), and the CD81 responses of the C-CP tip isolated EVs from biofluid samples. | ||
Conclusions
The C-CP fiber tip isolation method has proven to be an efficient means of EV isolation, with the ability to withstand potentially complex matrix effects from human urine, saliva, cervical mucus, blood serum, and goat milk. The HIC-based EV isolation technique presents significant benefits regarding time, cost, and ease of use. The C-CP spin-down tip workflow enables the processing of multiple samples simultaneously in 15 min, limited only by the table-top centrifuge capacity. The method of standard addition employing a commercial exosome standard stock was demonstrated as an accurate means to determine the concentration of EVs, regardless of the matrix type. That said, the respective responses showed very little difference in sensitivity (i.e., minimal matrix effects). NTA analysis provided the determination of particle size distributions and overall particle densities for the different matrices. TEM analysis confirms that the EVs isolated from all biofluids retained the characteristic cup or donut-shaped morphology after the isolation process. The purity of the EV isolates was confirmed through Bradford assays, revealing total protein content before and after isolation, with up to 89% of biofluid-originating proteins being removed. The efficacy of the method to isolate bioactive EVs was demonstrated through an ELISA assay for the CD81 tetraspanin marker protein. Overall, there was a self-consistency in the relative (and absolute) amounts of EVs isolated from the different matrices based on the multiple, independent measurement approaches. This agreement serves to validate the quantitative aspects of the isolation process.The bench-top C-CP spin-down tip protocol introduces a relatively facile means of EV isolation. The C-CP tip HIC isolation method's capabilities make it an ideal candidate for use in laboratory settings. The ability to work with microliter volumes while achieving high EV yields and purity lends itself to both clinical and fundamental EV research applications. For example, the ability to alleviate the complicating aspects of serum/lipoproteins is an essential element in performing high-fidelity proteomics analysis. Likewise, the same factors are key in developing bioassays based on the presence of targeted surface marker proteins. Finally, while likely requiring the use of preparative scale columns, the characteristics demonstrated here are essential in the development of EVs as gene therapy vectors.
Author contributions
Kaylan K. Jackson: Methodology, data curation, visualization, writing – original draft preparation. Rhonda R. Powell: Methodolog. Terri F. Bruce: Conceptualization, supervision. R. Kenneth Marcus: Conceptualization, supervision, writing – reviewing and editing.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
Financial support for the chromatography development efforts came from the National Science Foundation, Division of Chemistry under grant CHE-1608663. Financial support for the EV and exosome isolation efforts came from the Eppley Foundation for Scientific Research and The Gibson Foundation. Dr Larry Puls (Prisma Health), the Prisma Health ITOR Biorepository, and Prisma Health are gratefully acknowledged for patient identification and sample collection. Special thanks to George Wetzel, Clemson University Electron Microscopy Facility for assistance with EM. The content of this material and any opinions, findings, conclusions, or recommendations expressed in this material are solely the responsibility of the author(s) and do not necessarily represent the official views of the National Science Foundation. The graphical abstract presented here was created with BioRender.com.References
- F. Momen-Heravi, L. Balaj, S. Alian, P.-Y. Mantel, A. E. Halleck, A. J. Trachtenberg, C. E. Soria, S. Oquin, C. M. Bonebreak and E. Saracoglu, Biol. Chem., 2013, 394, 1253–1262 CAS.
- F. M. Heravi, S. Bala, K. Kodys and G. Szabo, Sci. Rep., 2015, 5, 9991 CrossRef PubMed.
- Y. Ouyang, A. Bayer, T. Chu, V. A. Tyurin, V. E. Kagan, A. E. Morelli, C. B. Coyne and Y. Sadovsky, Placenta, 2016, 47, 86–95 CrossRef CAS PubMed.
- S. Keerthikumar, D. Chisanga, D. Ariyaratne, H. Al Saffar, S. Anand, K. Zhao, M. Samuel, M. Pathan, M. Jois, N. Chilamkurti, L. Gangoda and S. Mathivanan, J. Mol. Biol., 2016, 428, 688–692 CrossRef CAS PubMed.
- S. Pant, H. Hilton and M. E. Burczynski, Biochem. Pharmacol., 2012, 83, 1484–1494 CrossRef CAS PubMed.
- Z. J. Smith, C. Lee, T. Rojalin, R. P. Carney, S. Hazari, A. Knudson, K. Lam, H. Saari, E. L. Ibanez, T. Viitala, T. Laaksonen, M. Yliperttula and S. Wachsmann-Hogiu, J. Extracell. Vesicles, 2015, 4, 28533 CrossRef PubMed.
- K. L. Schey, J. M. Luther and K. L. Rose, Methods, 2015, 87, 75–82 CrossRef CAS PubMed.
- M. Colombo, C. Moita, G. van Niel, J. Kowal, J. Vigneron, P. Benaroch, N. Manel, L. F. Moita, C. Thery and G. Raposo, J. Cell Sci., 2013, 126, 5553–5565 CAS.
- J. Kowal, M. Tkach and C. Théry, Curr. Opin. Cell Biol., 2014, 29, 116–125 CrossRef CAS PubMed.
- M. Kanada, M. H. Bachmann and C. H. Contag, Trends Cancer, 2016, 2, 84–94 CrossRef PubMed.
- V. Sokolova, A. K. Ludwig, S. Hornung, O. Rotan, P. A. Horn, M. Epple and B. Giebel, Colloids Surf., B, 2011, 87, 146–150 CrossRef CAS PubMed.
- E. Willms, H. J. Johansson, I. Mager, Y. Lee, K. E. M. Blomberg, M. Sadik, A. Alaarg, C. I. E. Smith, J. Lehtio, S. E. L. Andaloussi, M. J. A. Wood and P. Vader, Sci. Rep., 2016, 6, 12 CrossRef PubMed.
- A. Bobrie, M. Colombo, S. Krumeich, G. Raposo and C. Thery, J. Extracell. Vesicles, 2012, 1, 18397 CrossRef CAS PubMed.
- A. Bobrie, M. Colombo, G. Raposo and C. Thery, Traffic, 2011, 12, 1659–1668 CrossRef CAS PubMed.
- A. V. Vlassov, S. Magdaleno, R. Setterquist and R. Conrad, Biochim. Biophys. Acta, 2012, 1820, 940–948 CrossRef CAS PubMed.
- S. Roy, F. H. Hochberg and P. S. Jones, J. Extracell. Vesicles, 2018, 7, 1438720 CrossRef PubMed.
- D. M. Pegtel and S. J. Gould, Annu. Rev. Biochem., 2019, 88, 487–514 CrossRef CAS PubMed.
- L. L. Yu, J. Zhu, J. X. Liu, F. Jiang, W. K. Ni, L. S. Qu, R. Z. Ni, C. H. Lu and M. B. Xiao, BioMed. Res. Int., 2018, 2018, 3634563 Search PubMed.
- S. Halvaei, S. Daryani, Z. Eslami-S, T. Samadi, N. Jafarbeik-Iravani, T. O. Bakhshayesh, K. Majidzadeh-A and R. Esmaeili, Mol. Ther. – Nucleic Acids, 2018, 10, 131–141 CrossRef CAS PubMed.
- K. B. Johnsen, J. M. Gudbergsson, M. N. Skov, L. Pilgaard, T. Moos and M. Duroux, Biochim. Biophys. Acta, 2014, 1846, 75–87 CAS.
- R. C. Lai, F. Arslan, M. M. Lee, N. S. K. Sze, A. Choo, T. S. Chen, M. Salto-Tellez, L. Timmers, C. N. Lee, R. M. El Oakley, G. Pasterkamp, D. P. V. de Kleijn and S. K. Lim, Stem Cell Res., 2010, 4, 214–222 CrossRef CAS PubMed.
- X. Yang, Z. Weng, D. L. Mendrick and Q. Shi, Toxicol. Lett., 2014, 225, 401–406 CrossRef CAS PubMed.
- M. L. Merchant, I. M. Rood, J. K. J. Deegens and J. B. Klein, Nat. Rev. Nephrol., 2017, 13, 731–749 CrossRef CAS PubMed.
- J. Q. Gerlach, A. Kruger, S. Gallogly, S. A. Hanley, M. C. Hogan, C. J. Ward, L. Joshi and M. D. Griffin, PLoS One, 2013, 8, e74801 CrossRef CAS PubMed.
- C. Lasser, V. S. Alikhani, K. Ekstrom, M. Eldh, P. T. Paredes, A. Bossios, M. Sjostrand, S. Gabrielsson, J. Lotvall and H. Valadi, J. Transl. Med., 2011, 9, 9 CrossRef PubMed.
- L. A. Aqrawi, H. K. Galtung, B. Vestad, R. Øvstebø, B. Thiede, S. Rusthen, A. Young, E. M. Guerreiro, T. P. Utheim and X. Chen, Arthritis Res. Ther., 2017, 19, 14 CrossRef PubMed.
- F. V. Winck, A. C. Prado Ribeiro, R. R. Domingues, L. Y. Ling, D. M. Riano-Pachon, C. Rivera, T. B. Brandao, A. F. Gouvea, A. R. Santos-Silva, R. D. Coletta and A. F. Paes Leme, Sci. Rep., 2015, 5, 16305 CrossRef CAS PubMed.
- B. Pang, Y. Zhu, J. Ni, J. Ruan, J. Thompson, D. Malouf, J. Bucci, P. Graham and Y. Li, Int. J. Nanomed., 2020, 15, 10241–10256 CrossRef CAS PubMed.
- T. Shtam, S. Naryzhny, A. Kopylov, E. Petrenko, R. Samsonov, R. Kamyshinsky, Y. Zabrodskaya, D. Nikitin, M. Sorokin, A. Buzdin and A. Malek, J. Hematol., 2018, 7, 149–153 CrossRef CAS PubMed.
- X. Zhao, Y. Wu, J. Duan, Y. Ma, Z. Shen, L. Wei, X. Cui, J. Zhang, Y. Xie and J. Liu, J. Proteome Res., 2014, 13, 5391–5402 CrossRef CAS PubMed.
- K. Blans, M. S. Hansen, L. V. Sorensen, M. L. Hvam, K. A. Howard, A. Moller, L. Wiking, L. B. Larsen and J. T. Rasmussen, J. Extracell. Vesicles, 2017, 6, 1294340 CrossRef PubMed.
- M. I. Zonneveld, A. R. Brisson, M. J. van Herwijnen, S. Tan, C. H. van de Lest, F. A. Redegeld, J. Garssen, M. H. Wauben and E. N. Nolte-'t Hoen, J. Extracell. Vesicles, 2014, 3, 24215 CrossRef PubMed.
- Y. K. Yoo, J. Lee, H. Kim, K. S. Hwang, D. S. Yoon and J. H. Lee, Micromachines, 2018, 9, 634 CrossRef PubMed.
- D. Chiasserini, J. R. van Weering, S. R. Piersma, T. V. Pham, A. Malekzadeh, C. E. Teunissen, H. de Wit and C. R. Jimenez, J. Proteomics, 2014, 106, 191–204 CrossRef CAS PubMed.
- A. Milasan, N. Tessandier, S. Tan, A. Brisson, E. Boilard and C. Martel, J. Extracell. Vesicles, 2016, 5, 31427 CrossRef PubMed.
- N. Tessandier, I. Melki, N. Cloutier, I. Allaeys, A. Miszta, S. Tan, A. Milasan, S. Michel, A. Benmoussa, T. Levesque, F. Cote, S. E. McKenzie, C. Gilbert, P. Provost, A. R. Brisson, A. S. Wolberg, P. R. Fortin, C. Martel and E. Boilard, Arterioscler. Thromb. Vasc. Biol., 2020, 40, 929–942 CrossRef CAS PubMed.
- E. I. Buzas, B. Gyorgy, G. Nagy, A. Falus and S. Gay, Nat. Rev. Rheumatol., 2014, 10, 356–364 CrossRef CAS PubMed.
- C. Balbi, M. Piccoli, L. Barile, A. Papait, A. Armirotti, E. Principi, D. Reverberi, L. Pascucci, P. Becherini, L. Varesio, M. Mogni, D. Coviello, T. Bandiera, M. Pozzobon, R. Cancedda and S. Bollini, Stem Cells Transl. Med., 2017, 6, 1340–1355 CrossRef CAS PubMed.
- C. Gardiner, D. Di Vizio, S. Sahoo, C. Thery, K. W. Witwer, M. Wauben and A. F. Hill, J. Extracell. Vesicles, 2016, 5, 32945 CrossRef PubMed.
- J. B. Simonsen, Circ. Res., 2017, 121, 920–922 CrossRef CAS PubMed.
- B. W. Sódar, Á. Kittel, K. Pálóczi, K. V. Vukman, X. Osteikoetxea, K. Szabó-Taylor, A. Németh, B. Sperlágh, T. Baranyai and Z. Giricz, Sci. Rep., 2016, 6, 24316–24327 CrossRef PubMed.
- C. Thery, K. W. Witwer, E. Aikawa, M. J. Alcaraz, J. D. Anderson, R. Andriantsitohaina, A. Antoniou, T. Arab, F. Archer, G. K. Atkin-Smith, D. C. Ayre, J. M. Bach, D. Bachurski, H. Baharvand, L. Balaj, S. Baldacchino, N. N. Bauer, A. A. Baxter, M. Bebawy, C. Beckham, A. B. Zavec, A. Benmoussa, A. C. Berardi, P. Bergese, E. Bielska, C. Blenkiron, S. Bobis-Wozowicz, E. Boilard, W. Boireau, A. Bongiovanni, F. E. Borras, S. Bosch, C. M. Boulanger, X. Breakefield, A. M. Breglio, M. A. Brennan, D. R. Brigstock, A. Brisson, M. L. D. Broekman, J. F. Bromberg, P. Bryl-Gorecka, S. Buch, A. H. Buck, D. Burger, S. Busatto, D. Buschmann, B. Bussolati, E. I. Buzas, J. B. Byrd, G. Camussi, D. R. F. Carter, S. Caruso, L. W. Chamley, Y. T. Chang, C. C. Chen, S. Chen, L. Cheng, A. R. Chin, A. Clayton, S. P. Clerici, A. Cocks, E. Cocucci, R. J. Coffey, A. Cordeiro-da-Silva, Y. Couch, F. A. W. Coumans, B. Coyle, R. Crescitelli, M. F. Criado, C. D'Souza-Schorey, S. Das, A. D. Chaudhuri, P. de Candia, E. F. De Santana, O. De Wever, H. A. del Portillo, T. Demaret, S. Deville, A. Devitt, B. Dhondt, D. Di Vizio, L. C. Dieterich, V. Dolo, A. P. D. Rubio, M. Dominici, M. R. Dourado, T. A. P. Driedonks, F. V. Duarte, H. M. Duncan, R. M. Eichenberger, K. Ekstrom, S. E. L. Andaloussi, C. Elie-Caille, U. Erdbrugger, J. M. Falcon-Perez, F. Fatima, J. E. Fish, M. Flores-Bellver, A. Forsonits, A. Frelet-Barrand, F. Fricke, G. Fuhrmann, S. Gabrielsson, A. Gamez-Valero, C. Gardiner, K. Gartner, R. Gaudin, Y. S. Gho, B. Giebel, C. Gilbert, M. Gimona, I. Giusti, D. C. I. Goberdhan, A. Gorgens, S. M. Gorski, D. W. Greening, J. C. Gross, A. Gualerzi, G. N. Gupta, D. Gustafson, A. Handberg, R. A. Haraszti, P. Harrison, H. Hegyesi, A. Hendrix, A. F. Hill, F. H. Hochberg, K. F. Hoffmann, B. Holder, H. Holthofer, B. Hosseinkhani, G. K. Hu, Y. Y. Huang, V. Huber, S. Hunt, A. G. E. Ibrahim, T. Ikezu, J. M. Inal, M. Isin, A. Ivanova, H. K. Jackson, S. Jacobsen, S. M. Jay, M. Jayachandran, G. Jenster, L. Z. Jiang, S. M. Johnson, J. C. Jones, A. Jong, T. Jovanovic-Talisman, S. Jung, R. Kalluri, S. Kano, S. Kaur, Y. Kawamura, E. T. Keller, D. Khamari, E. Khomyakova, A. Khvorova, P. Kierulf, K. P. Kim, T. Kislinger, M. Klingeborn, D. J. Klinke, M. Kornek, M. M. Kosanovic, A. F. Kovacs, E. M. Kramer-Albers, S. Krasemann, M. Krause, I. V. Kurochkin, G. D. Kusuma, S. Kuypers, S. Laitinen, S. M. Langevin, L. R. Languino, J. Lannigan, C. Lasser, L. C. Laurent, G. Lavieu, E. Lazaro-Ibanez, S. Le Lay, M. S. Lee, Y. X. F. Lee, D. S. Lemos, M. Lenassi, A. Leszczynska, I. T. S. Li, K. Liao, S. F. Libregts, E. Ligeti, R. Lim, S. K. Lim, A. Line, K. Linnemannstons, A. Llorente, C. A. Lombard, M. J. Lorenowicz, A. M. Lorincz, J. Lotvall, J. Lovett, M. C. Lowry, X. Loyer, Q. Lu, B. Lukomska, T. R. Lunavat, S. L. N. Maas, H. Malhi, A. Marcilla, J. Mariani, J. Mariscal, E. S. Martens-Uzunova, L. Martin-Jaular, M. C. Martinez, V. R. Martins, M. Mathieu, S. Mathivanan, M. Maugeri, L. K. McGinnis, M. J. McVey, D. G. Meckes, K. L. Meehan, I. Mertens, V. R. Minciacchi, A. Moller, M. M. Jorgensen, A. Morales-Kastresana, J. Morhayim, F. Mullier, M. Muraca, L. Musante, V. Mussack, D. C. Muth, K. H. Myburgh, T. Najrana, M. Nawaz, I. Nazarenko, P. Nejsum, C. Neri, T. Neri, R. Nieuwland, L. Nimrichter, J. P. Nolan, E. N. M. Nolte-'t Hoen, N. Noren Hooten, L. O'Driscoll, T. O'Grady, A. O'Loghlen, T. Ochiya, M. Olivier, A. Ortiz, L. A. Ortiz, X. Osteikoetxea, O. Ostegaard, M. Ostrowski, J. Park, D. M. Pegtel, H. Peinado, F. Perut, M. W. Pfaffl, D. G. Phinney, B. C. H. Pieters, R. C. Pink, D. S. Pisetsky, E. P. von Strandmann, I. Polakovicova, I. K. H. Poon, B. H. Powell, I. Prada, L. Pulliam, P. Quesenberry, A. Radeghieri, R. L. Raffai, S. Raimondo, J. Rak, M. I. Ramirez, G. Raposo, M. S. Rayyan, N. Regev-Rudzki, F. L. Ricklefs, P. D. Robbins, D. D. Roberts, S. C. Rodrigues, E. Rohde, S. Rome, K. M. A. Rouschop, A. Rughetti, A. E. Russell, P. Saa, S. Sahoo, E. Salas-Huenuleo, C. Sanchez, J. A. Saugstad, M. J. Saul, R. M. Schiffelers, R. Schneider, T. H. Schoyen, A. Scott, E. Shahaj, S. Sharma, O. Shatnyeva, F. Shekari, G. V. Shelke, A. K. Shetty, K. Shiba, P. R. M. Siljander, A. M. Silva, A. Skowronek, O. L. Snyder, R. P. Soares, B. W. Sodar, C. Soekmadji, J. Sotillo, P. D. Stahl, W. Stoorvogel, S. L. Stott, E. F. Strasser, S. Swift, H. Tahara, M. Tewari, K. Timms, S. Tiwari, R. Tixeira, M. Tkach, W. S. Toh, R. Tomasini, A. C. Torrecilhas, J. P. Tosar, V. Toxavidis, L. Urbanelli, P. Vader, B. W. M. van Balkom, S. G. van der Grein, J. Van Deun, M. J. C. van Herwijnen, K. Van Keuren-Jensen, G. van Niel, M. E. van Royen, A. J. van Wijnen, M. H. Vasconcelos, I. J. Vechetti, T. D. Veit, L. J. Vella, E. Velot, F. J. Verweij, B. Vestad, J. L. Vinas, T. Visnovitz, K. V. Vukman, J. Wahlgren, D. C. Watson, M. H. M. Wauben, A. Weaver, J. P. Webber, V. Weber, A. M. Wehman, D. J. Weiss, J. A. Welsh, S. Wendt, A. M. Wheelock, Z. Wiener, L. Witte, J. Wolfram, A. Xagorari, P. Xander, J. Xu, X. M. Yan, M. Yanez-Mo, H. Yin, Y. Yuana, V. Zappulli, J. Zarubova, V. Zekas, J. Y. Zhang, Z. Z. Zhao, L. Zheng, A. R. Zheutlin, A. M. Zickler, P. Zimmermann, A. M. Zivkovic, D. Zocco and E. K. Zuba-Surma, J. Extracell. Vesicles, 2018, 7, 43 Search PubMed.
- T. Liangsupree, E. Multia and M.-L. Riekkola, J. Chromatogr. A, 2021, 1636, 461773 CrossRef CAS PubMed.
- K. W. Witwer, C. Soekmadji, A. F. Hill, M. H. Wauben, E. I. Buzás, D. Di Vizio, J. M. Falcon-Perez, C. Gardiner, F. Hochberg, I. V. Kurochkin, J. Lötvall, S. Mathivanan, R. Nieuwland, S. Sahoo, H. Tahara, A. C. Torrecilhas, A. M. Weaver, H. Yin, L. Zheng, Y. S. Gho, P. Quesenberry and C. Théry, J. Extracell. Vesicles, 2017, 6, 1396823 CrossRef PubMed.
- M. Macias, V. Rebmann, B. Mateos, N. Varo, J. L. Perez-Gracia, E. Alegre and A. Gonzalez, Clin. Chem. Lab. Med., 2019, 57, 1539–1545 CrossRef CAS PubMed.
- Y. Yuana, J. Levels, A. Grootemaat, A. Sturk and R. Nieuwland, J. Extracell. Vesicles, 2014, 3, 23262 CrossRef PubMed.
- B. J. Tauro, D. W. Greening, R. A. Mathias, H. Ji, S. Mathivanan, A. M. Scott and R. J. Simpson, Methods, 2012, 56, 293–304 CrossRef CAS PubMed.
- D. Freitas, M. Balmaña, J. Poças, D. Campos, H. Osório, A. Konstantinidi, S. Y. Vakhrushev, A. Magalhães and C. A. Reis, J. Extracell. Vesicles, 2019, 8, 1621131 CrossRef CAS PubMed.
- G. Vergauwen, B. Dhondt, J. Van Deun, E. De Smedt, G. Berx, E. Timmerman, K. Gevaert, I. Miinalainen, V. Cocquyt, G. Braems, R. Van den Broecke, H. Denys, O. De Wever and A. Hendrix, Sci. Rep., 2017, 7, 2704 CrossRef PubMed.
- A. Gámez-Valero, M. Monguió-Tortajada, L. Carreras-Planella, M. L. Franquesa, K. Beyer and F. E. Borràs, Sci. Rep., 2016, 6, 33641 CrossRef PubMed.
- H. Zhang, D. Freitas, H. S. Kim, K. Fabijanic, Z. Li, H. Chen, M. T. Mark, H. Molina, A. B. Martin, L. Bojmar, J. Fang, S. Rampersaud, A. Hoshino, I. Matei, C. M. Kenific, M. Nakajima, A. P. Mutvei, P. Sansone, W. Buehring, H. Wang, J. P. Jimenez, L. Cohen-Gould, N. Paknejad, M. Brendel, K. Manova-Todorova, A. Magalhães, J. A. Ferreira, H. Osório, A. M. Silva, A. Massey, J. R. Cubillos-Ruiz, G. Galletti, P. Giannakakou, A. M. Cuervo, J. Blenis, R. Schwartz, M. S. Brady, H. Peinado, J. Bromberg, H. Matsui, C. A. Reis and D. Lyden, Nat. Cell Biol., 2018, 20, 332–343 CrossRef CAS PubMed.
- R. Stranska, L. Gysbrechts, J. Wouters, P. Vermeersch, K. Bloch, D. Dierickx, G. Andrei and R. Snoeck, J. Transl. Med., 2018, 16, 1 CrossRef CAS PubMed.
- M. Macías, V. Rebmann, B. Mateos, N. Varo, J. L. Perez-Gracia, E. Alegre and Á. González, Clin. Chem. Lab. Med., 2019, 57, 1539–1545 CrossRef PubMed.
- T. F. Bruce, T. J. Slonecki, L. Wang, S. Huang, R. R. Powell and R. K. Marcus, Electrophoresis, 2019, 40, 571–581 CrossRef CAS PubMed.
- L. Wang, T. F. Bruce, S. Huang and R. K. Marcus, Anal. Chim. Acta, 2019, 1082, 186–193 CrossRef CAS PubMed.
- S. Huang, L. Wang, T. F. Bruce and R. K. Marcus, Anal. Bioanal. Chem., 2019, 411, 6591–6601 CrossRef CAS PubMed.
- K. K. Jackson, R. R. Powell, T. F. Bruce and R. K. Marcus, Anal. Bioanal. Chem., 2020, 412, 4713–4724 CrossRef CAS PubMed.
- S. Huang, L. Wang, T. F. Bruce and R. K. Marcus, Biotechnol. Prog., 2020, e2998 CAS.
- X. Ji, S. Huang, J. Zhang, T. F. Bruce, Z. Tan, D. Wang, J. Zhu, R. K. Marcus and D. M. Lubman, Electrophoresis, 2021, 42, 245–256 CrossRef CAS PubMed.
- S. Huang, X. Ji, K. K. Jackson, T. F. Bruce, D. M. Lubman and R. K. Marcus, Anal. Chim. Acta, 2021, 1167, 338578 CrossRef CAS PubMed.
- L. Wang and R. K. Marcus, J. Chromatogr. A, 2019, 1585, 161–171 CrossRef CAS PubMed.
- J. Fausnaugh and F. Regnier, J. Chromatogr. A, 1986, 359, 131–146 CrossRef CAS PubMed.
- A. Jungbauer, C. Machold and R. Hahn, J. Chromatogr. A, 2005, 1079, 221–228 CrossRef CAS PubMed.
- J. A. Queiroz, C. T. Tomaz and J. M. Cabral, J. Biotechnol., 2001, 87, 143–159 CrossRef CAS PubMed.
- S. Sitar, A. Kejžar, D. Pahovnik, K. Kogej, M. Tušek-Žnidarič, M. Lenassi and E. Žagar, Anal. Chem., 2015, 87, 9225–9233 CrossRef CAS PubMed.
- B. Vestad, A. Llorente, A. Neurauter, S. Phuyal, B. Kierulf, P. Kierulf, T. Skotland, K. Sandvig, K. B. F. Haug and R. Øvstebø, J. Extracell. Vesicles, 2017, 6, 1344087 CrossRef PubMed.
- Z. Andreu and M. Yanez-Mo, Front. Immunol., 2014, 5, 442 Search PubMed.
- E. Willms, C. Cabanas, I. Mager, M. J. A. Wood and P. Vader, Front. Immunol., 2018, 9, 738 CrossRef PubMed.
- V. Vagenende, A. X. Han, M. Mueller and B. L. Trout, Chem. Biol., 2013, 8, 416–422 CAS.
- V. Vagenende, M. G. Yap and B. L. Trout, Biochemistry, 2009, 48, 11084–11096 CrossRef CAS PubMed.
- B. E. Saxberg and B. R. Kowalski, Anal. Chem., 1979, 51, 1031–1038 CrossRef CAS.
- K. Vaswani, M. D. Mitchell, O. J. Holland, Y. Q. Koh, R. J. Hill, T. Harb, P. S. W. Davies and H. Peiris, J. Nutr. Metab., 2019, 2019, 5764740 Search PubMed.
- L. Musante, S. V. Bontha, S. La Salvia, A. Fernandez-Piñeros, J. Lannigan, T. H. Le, V. Mas and U. Erdbrügger, Sci. Rep., 2020, 10, 3701 CrossRef CAS.
- S. R. Kumar, E. T. Kimchi, Y. Manjunath, S. Gajagowni, A. J. Stuckel and J. T. Kaifi, Sci. Rep., 2020, 10, 2800 CrossRef CAS PubMed.
- P. J. Wermuth, S. Piera-Velazquez and S. A. Jimenez, Clin. Exp. Rheumatol., 2017, 35(Suppl. 106), 21–30 Search PubMed.
- C. Conzelmann, R. Groß, M. Zou, F. Krüger, A. Görgens, M. O. Gustafsson, S. El Andaloussi, J. Münch and J. A. Müller, J. Extracell. Vesicles, 2020, 9, 1808281 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |