
Recent advances in perovskite oxides as electrode materials for supercapacitors
Yang
Cao†
ab,
Jie
Liang†
b,
Xue
Li
a,
Luchao
Yue
b,
Qian
Liu
 b,
Siyu
Lu
c,
Abdullah M.
Asiri
d,
Jianming
Hu
*a,
Yonglan
Luo
*b and
Xuping
Sun
*b
b,
Siyu
Lu
c,
Abdullah M.
Asiri
d,
Jianming
Hu
*a,
Yonglan
Luo
*b and
Xuping
Sun
*b
aSchool of Physics and Electrical Engineering, Chongqing Normal University, Chongqing 401331, China. E-mail: hujianming@cqnu.edu.cn
bInstitute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China, Chengdu 610054, Sichuan, China. E-mail: luoylcwnu@hotmail.com; xpsun@uestc.edu.cn
cGreen Catalysis Center, and College of Chemistry, Zhengzhou University, Zhengzhou 450001, Henan, China
dChemistry Department, Faculty of Science & Center of Excellence for Advanced Materials Research, King Abdulaziz University, P.O. Box 80203, Jeddah 21589, Saudi Arabia
First published on 29th January 2021
Abstract
Owing to the high power density and ultralong cycle life, supercapacitors represent an alternative to electrochemical batteries in energy storage applications. However, the relatively low energy density is the main challenge for supercapacitors in the current drive to push the entire technology forward to meet the benchmark requirements for commercialization. To effectively solve this issue, it is crucial to develop electrode materials with excellent electrochemical performance since the electrode used is closely related to the specific capacitance and energy density of supercapacitors. With the unique structure, compositional flexibility, and inherent oxygen vacancy, perovskite oxides have attracted wide attention as promising electrode materials for supercapacitors. In this review, we summarize the recent advances in perovskite oxides as electrode materials for supercapacitors. Firstly, the structures and compositions of perovskite oxides are critically reviewed. Following this, the progress in various perovskite oxides, including single perovskite and derivative perovskite oxides, is depicted, focusing on their electrochemical performance. Furthermore, several optimization strategies (i.e., modulating the stoichiometry of the anion or cation, A-site doping, B-site doping, and constructing composites) to improve their electrochemical performance are also discussed. Finally, the significant challenges facing the advancement of perovskite oxide electrodes for supercapacitor applications and future outlook are proposed.
 Xuping Sun | Xuping Sun received his PhD degree from the Changchun Institute of Applied Chemistry (CIAC), Chinese Academy of Sciences, in 2006. During 2006–2009, he carried out postdoctoral research at Konstanz University, University of Toronto, and Purdue University. In 2010, he started his independent research career as a full Professor at CIAC and then moved to Sichuan University in 2015. In 2018, he joined the University of Electronic Science and Technology of China, where he found the Research Center of Nanocatalysis & Sensing. He was recognized as a highly cited researcher (2018–2020) in both areas of chemistry and materials science by Clarivate Analytics. He has published over 490 papers with total citations over 42 |
1. Introduction
The energy paradigm has changed from an era of fossil fuel resources to sustainable energy sources and new technologies associated with energy conversion and storage to address the rapid depletion of fossil fuels. Currently, energy storage and conversion technologies mainly include batteries, fuel cells, and supercapacitors (SCs).1–3 Among them, SCs have aroused considerable interest due to their high power density (>10 K W kg−1), long cycle life (>105 cycles), and bridging function of the power/energy gap between traditional dielectric capacitors with high power output and batteries/fuel cells with high energy storage (Fig. 1).4–7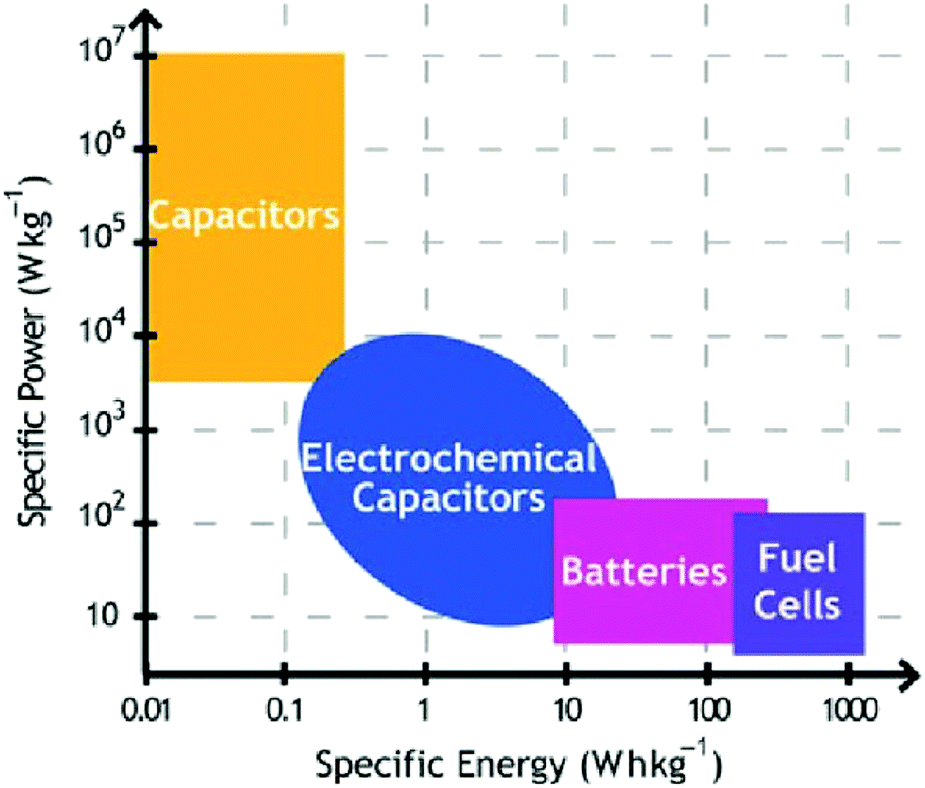 | ||
| Fig. 1 Comparison of specific power vs. specific energy of capacitors and batteries. Reproduced from ref. 6 with permission from Materials Research Society, copyright 2011. | ||
Based on the charge storage mechanism, SCs can be classified into two categories:8 electrochemical double layer capacitors (EDLCs) and pseudocapacitors (PCs). EDLCs store charge through charge accumulation at the interface between the electrolyte and the electrode's surface. This process is a physical process without any electrochemical reaction (non-faradaic), guaranteeing fast energy uptake and delivery and avoiding the electrode materials' swelling. As such, EDLCs have high power density (>15 K W kg−1) and long cycle life (>105 cycles).9,10 Because of their high specific surface area (1000–3000 m2 g−1) and desirable conductivity, carbon-based materials (such as carbon nanotubes, activated carbon, and graphene) are the most attractive to date for EDLCs.11 However, experimentally, owing to the finite conductivity and unavailability of all of the active sites, the practical specific capacitance of pure carbon-based EDLCs achieved has usually been limited to ∼100–350 F g−1.12–14 In comparison to EDLCs, PCs store charge via reversible redox reaction (faradaic reaction) between the electrode materials and the electrolyte. Since the electrochemical reactions occur both on the surface and in the bulk near the solid electrode surface, PCs often show far higher capacitance values and energy density (by a factor of 10 or higher) than EDLCs.15 Metal oxides and conductive polymers typically work as electrode materials in PCs due to their fast reversible redox reactions, cost-effectiveness as well as easy processability.16,17 Although PCs have a high energy density, they are not satisfactory in terms of power density and stability due to their poor electrical conductivity and framework swelling during cycling.18 Therefore, achieving high energy density while maintaining the high power density of SCs has become a challenging task.
As per the calculation formula (E = 1/2CV2), the energy capability of SCs is in proportion to the specific capacitance (C) and the square of voltage windows (V). Hence, the energy density can be improved by enhancing either the potential windows or the specific capacitance value. For the measurement of electrochemical response, electrochemical cells can be designed in either a half cell or a full cell configuration. The half cell is composed of three electrodes (working electrode, counter electrode, and reference electrode), while the full cell is composed of two electrodes (positive and negative electrodes).19 Generally, the cell potential can be increased by assembling asymmetric electrodes (two different electrode materials for the anode and the cathode) or by using organic electrolytes.19,20 Typically, in an aqueous-based symmetric system, the working voltage is limited to less than 1.2 V (due to the thermodynamic breakdown potential of water molecules).21 Based on the same active material, when it is assembled into an asymmetric supercapacitor (ASC), the voltage window will be expanded to 2.5 V, while when using organic electrolytes, the potential window will be expanded to 3 V.22,23 On the other hand, finding sustainable electrode materials is an effective strategy to improve the specific capacitance. Among the many candidate materials, metal oxides have attracted significant interest because they can provide higher energy density than conventional carbon materials and have better cycle lives than polymer materials.24 With a wide potential window (about 1.2 V), excellent electronic conductivity (3 × 102 S cm−1), and high theoretical specific capacitance (1358 F g−1), RuO2 is the most active electrode material.25 Nonetheless, high cost and limited abundance restrict its widespread applications. In this context, non-noble metal oxides such as MnO2, NiO, and Fe3O4 have been widely explored.26–28 Nevertheless, some inherent shortcomings, such as low electrical conductivity and poor cycling stability, limit their practical applications.
As star materials, perovskite oxides have rapidly drawn intensive attention owing to their low cost, robust skeleton structure, high tap density, and inherent nature of containing oxygen vacancies and distinctive chemical tailoring.29,30 Over the past few decades, perovskite oxides have traditionally been used as functional materials in energy-related fields. In 2014, a breakthrough work on the oxygen anion intercalation mechanism for nanostructured lanthanum-based perovskite oxides was reported by Meffold et al.31 The mechanism is as follows: in the first step, the diffusion of oxygen is in the form of OH− from the electrolyte. Subsequently, the oxygen vacancies are filled by OH− groups and they diffuse along the octahedral edges through the crystal concomitant with the oxidation of two Mn2+ centers to two Mn3+ and yielding water as a product. In the next step of the reaction, excess oxygen is intercalated at the surface by diffusion of manganese to the surface and oxidation of two Mn3+ centers to two Mn4+ (Fig. 2).31,32 As mentioned above, the bulk of perovskite oxide is involved in the storage of charge, which means that perovskite oxide does not require a high surface area to achieve high energy storage. There are metal oxides (such as TiO2-B, T-Nb2O5, and α-MoO3) with similar intercalation behavior to perovskite oxides but are based on positive ions (such as Li+ or Na+) as the charge carrier.33–35 In principle, O2− can carry two negative charges per unit, which means that the intercalation pseudocapacitance of O2− can store twice charges in one charge/discharge cycle than Li+ intercalation. Therefore, perovskite oxides have been regarded as tremendously promising candidate materials for SCs.
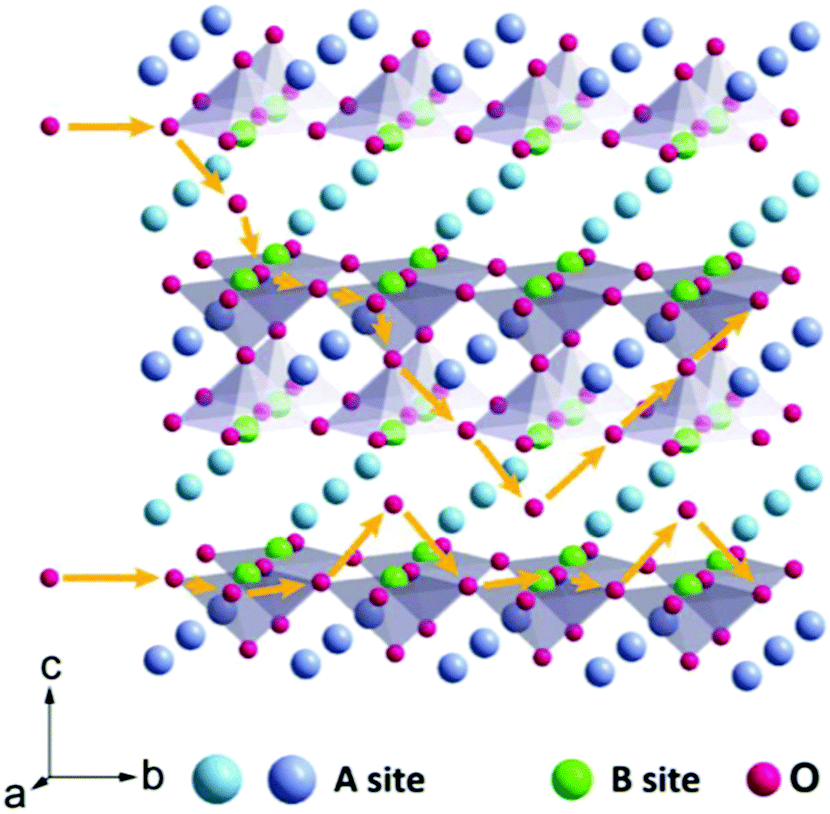 | ||
| Fig. 2 Mechanism of oxygen intercalation into perovskite oxides. Reproduced from ref. 32 with permission from Elsevier B.V., copyright 2020. | ||
To date, some high-quality reviews on perovskite oxides for energy conversion and storage applications have been published.36–39 However, there is still a lack of application and development of perovskite oxides in SCs. In this review, we first review the structure and composition of perovskite oxides. Secondly, perovskite oxides as electrode materials for SCs are introduced. Thirdly, the optimization strategies and applications of perovskite oxides in SCs are discussed. Finally, the application opportunities and challenges of perovskite oxides in SCs are presented.
2. Structure and composition of perovskite oxides
Perovskite oxides with the general formula of ABO3, originating from CaTiO3, are a family of oxides.40 An ideal structure of perovskite oxides is shown in Fig. 3a. The A cation is located at the cube's body center, the B cation is at each of the eight corners, and O2− is at each of the centers of the 12 edges. In this structure, twice the B–O bond distance is equal to a (a is the cubic unit cell parameter), and the A–O bond distance is equal to a/21/2.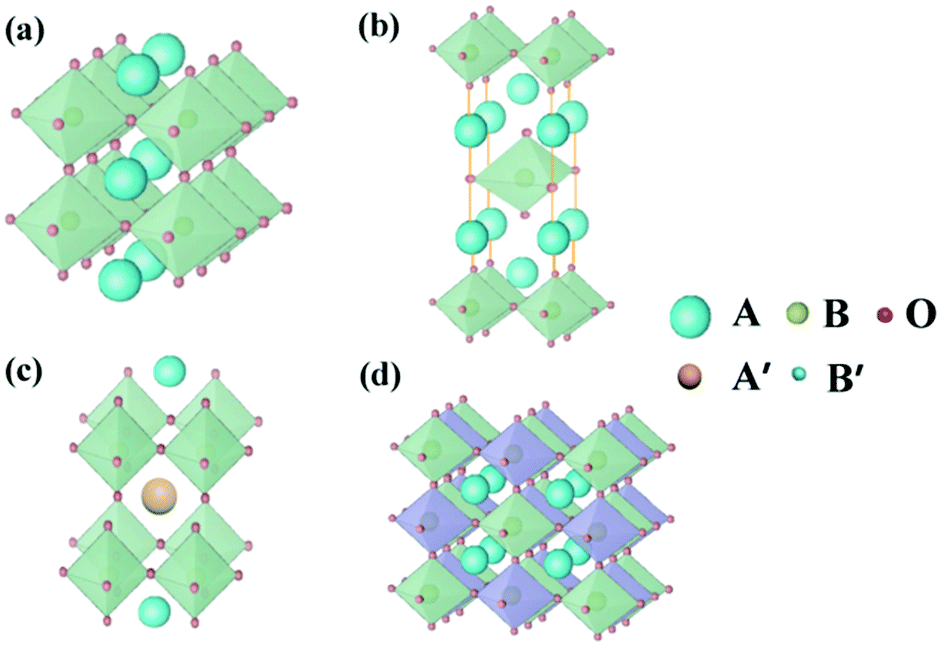 | ||
| Fig. 3 Crystal structure of (a) the ideal ABO3-type perovskite oxides, (b) Ruddlesden–Popper perovskite oxides ((AO)(ABO3)n), (c) A-site ordered double perovskite oxides (AA′B2O6) and (d) B-site ordered double perovskite oxides (A2BB′O6). | ||
In reality, it was found that the cubic structure was still retained in ABO3 compounds, even though the bond length relationship between A–O and B–O does not strictly meet the above rule. To measure the ideal deviation, Goldschmidt introduced a tolerance factor (t), defined by the equation41
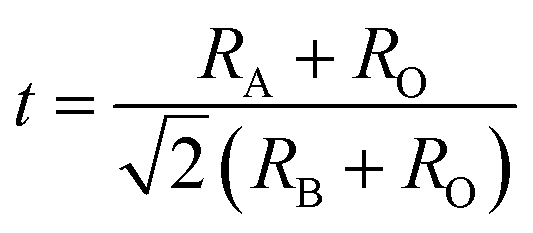
In addition to the ABO3-type perovskite, other oxides are also associated with perovskites, such as Ruddlesden–Popper (RP) perovskites ((AO)(ABO3)n, Fig. 3b),47 A-site ordered double perovskite oxides (AA′B2O6, Fig. 3c),48 and B-site ordered double perovskite oxides (A2BB′O6, Fig. 3d).49 From the composition, about 90% of the metal elements in the periodic table can be the cations in ABO3-type perovskite oxides.50 The common elements are shown in Fig. 4. Generally, A-site metal's significant role is to elevate the thermodynamic stability, and the B-site metal regulates the electrochemical reactions. For the electrode material applications in SCs, alkaline-earth (such as Sr and Ba) and rare-earth metals (such as La and Sm) are the principal A-site cations, and the B-site cation is usually a transition metal (such as Mn, Co, and Ni).
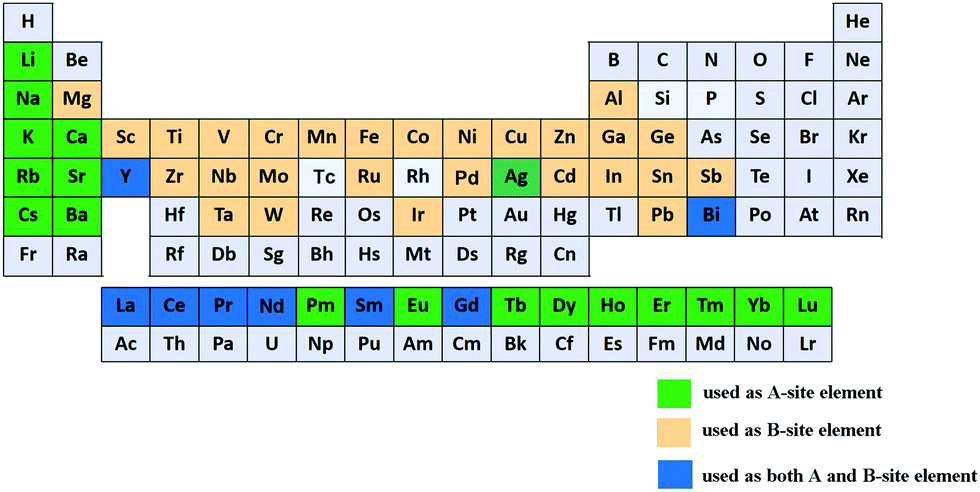 | ||
| Fig. 4 Common elements in ABO3-type perovskite oxides. | ||
3. Perovskite oxides as electrode materials for SCs
3.1 The single perovskite oxides
ABO3 holds great promise as an active material for SCs due to its high tap density and oxygen vacancy concentration. In this section, perovskite oxides are divided into four categories, lanthanum-based, strontium-based, cerium-based, and other perovskite oxides, to review their application.LaMnO3 is the first lanthanum-based perovskite oxide to be applied in SCs. Because of the defective cation-deficient lattice and the presence of manganese in two oxidation states (Mn 3p/Mn 4p), LaMnO3 shows a relatively stable and constant oxygen excess.52 In 2014, a specific capacitance of about 609.8 F g−1 for LaMnO3 was reported by Mefford et al.31 LaNiO3 is reported to exhibit a similar charge storage mechanism to LaMnO3 but with lower electrical resistivity (about 10−4 Ω).53–58 Shao et al.59 developed a template-free solvothermal approach to fabricate a hollow spherical structure of LaNiO3, showing a high specific capacitance (422 F g−1 at 1 A g−1) and good cycle stability (83.3% capacitance retention after 5000 cycles).
Compared with LaNiO3, LaFeO3 is more stable because Fe3+ has a stable electronic configuration of 3d5.60 Zhang et al.61 reported mesoporous LaFeO3 nanoparticles as the electrode material for SCs (Fig. 5a). A two-electrode symmetric SC cell (SSC) based on this material displayed a high energy density of 34 W h kg−1 at a power density of 900 W kg−1 with 92.2% capacitance retention after 5000 cycles (Fig. 5b and c). In Harikrishnan's work,62 LaCoO3 nanoparticles were prepared by the co-precipitation method. Because cobalt atoms have many oxidation states (2+, 3+, and 4+), LaCoO3 exhibited good electrochemical redox properties with a specific capacitance of 299.64 F g−1 at 10 A g−1. Recently, Hussain's group used the sol–gel method to synthesise a hierarchical mesoporous nanostructure of LaCrO3. The obtained sample revealed superior specific capacitance (1268 F g−1 at 2 A g−1) and good stability (91.5% capacitance retention after 5000 cycles).63
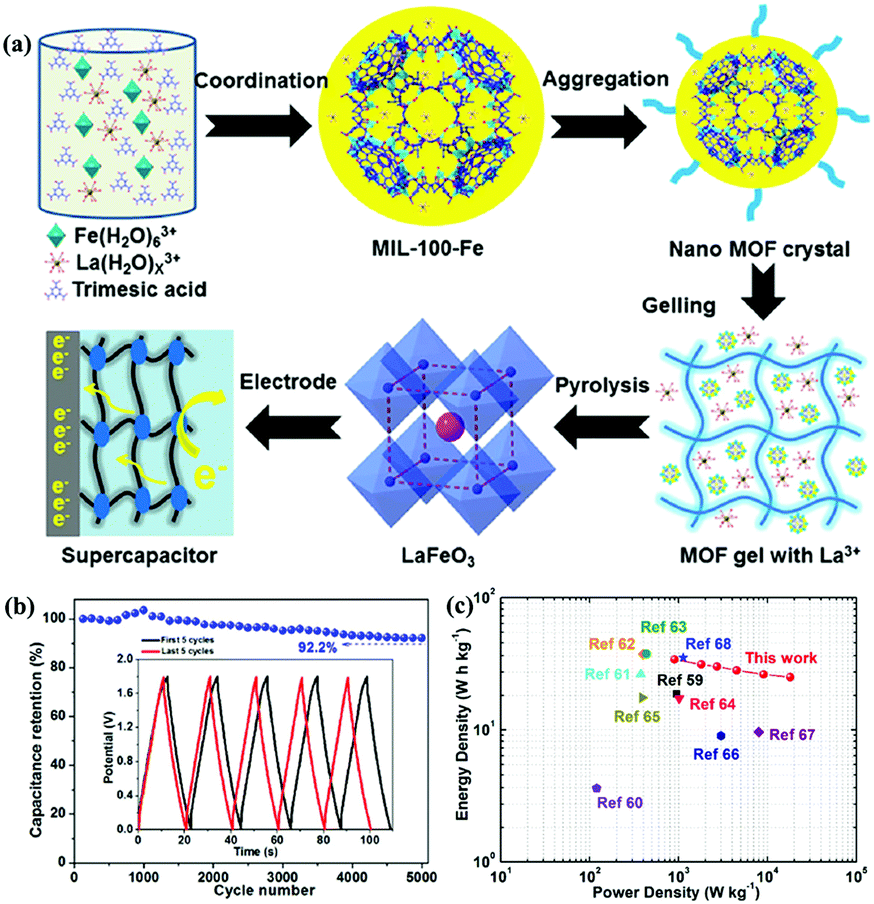 | ||
| Fig. 5 (a) Schematic illustrations showing the synthesis of perovskite LaFeO3. (b) Cycling stability versus cycle number graph, inset: the first and last five GCD cycles. (c) Ragone plots (energy density vs. power density) of this work and other devices. Reproduced from ref. 61 with permission from Elsevier B.V., copyright 2020. | ||
The earliest application of strontium-based perovskite oxides in the field of SCs can be traced back to 1999.65 Garche et al.66 reported SrRuO3 with a capacitance of 270 F g−1. Other strontium-based perovskite oxides (such as SrCoO2.5, SrMnO3, and SrTiO3) also show potential for SCs. Xiao et al.67 used a conventional solid-state reaction route to synthesize the orthorhombic structure of SrCoO2.5, and the resultant SrCoO2.5 displayed a specific capacitance of 168.5 F g−1. George et al.68 fabricated SrMnO3 nanofibers using sol–gel assisted electrospinning, which was followed by calcination at different temperatures (600 °C, 700 °C, and 800 °C). It was found that the nanofibers synthesized at 700 °C with small grains comprising a porous structure performed (321.7 F g−1 at 0.5 A g−1) better than other samples. The high porosity and surface area are likely to improve the SC performance with more ion diffusion and adsorption in the charge–discharge process. Additionally, with high porosity, sufficient electrochemically active sites are exposed, which can hold a large quantity of the electrolyte, providing more electrode–electrolyte accessibility.69 Similarly, Sharma et al.70 demonstrated the charge storage properties of mesoporous SrTiO3 (STO) with a cubic crystal structure. In their study, the Barrett-Joyner-Halenda (BJH) average pore diameter and pore volume of STO were tested as 15.18 nm and 0.249 cm3 g−1, respectively. Such pores provide a large number of active sites and low charge transfer resistance.71 Consequently, STO displayed a high capacitance of 592 F g−1 at 5 mV s−1. Interestingly, based on this material, a SSC was constructed, which exhibited maximum energy (27.8 W h kg−1) and power density (1921 W kg−1) with excellent cycling stability (99% capacitance retention after 5000 cycles).
3.2 Optimization strategy of perovskite oxides
Because the oxygen vacancies can provide charge-storage sites, increasing the number of oxygen vacancies is an effective strategy to enhance capacitance. For perovskite oxides, the number of oxygen vacancies is highly dependent on their structure and composition. Therefore, well-designed perovskite oxides are critical for achieving high electrode performance. Moreover, composite materials generally have better properties than the individual components. In the following sections, we discuss two aspects to optimize the SC of perovskite oxides: creating oxygen vacancies and compositing with other materials.3.2.1.1 Modulating the stoichiometry of the anion or cation. The non-stoichiometry phenomenon in perovskite oxides is widespread, arising from either anion deficiency (ABO3−δ) or anion excess (ABO3+δ) or cation deficiency in the A/B-site.82 Modulating the stoichiometry of the anion or cation to enhance electrochemical performance is crucial because it generates oxygen vacancies.37
Due to stability issues, there have been few studies on oxygen-excess perovskite ABO3+δ with interstitial oxygen atoms.83 However, oxygen-deficient perovskite oxides have been studied extensively. Typically, ABO3−δ is prepared by post-processing methods, such as, thermal treatment of perovskite oxides at elevated temperatures under a low-oxygen partial pressure or inert or reducing atmosphere (such as nitrogen, argon, hydrogen, or their mixture, or a vacuum).84–88 One typical example is that of Che et al.,89 who fabricated two kinds of nonstoichiometric LaNiO3−δ through the sol–gel method and further calcination at different temperatures (800 and 850 °C), which were named LNO and LNO-HT, respectively. In their experiment, the oxygen non-stoichiometry δ of LNO and LNO-HT was 0.37 and 0.27, respectively. They found that the specific capacitance of LNO increased by 75 F g−1 compared with LNO-TH. Interestingly, LNO showed cycling stability (94.5% capacitance retention after 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles) superior to that of hollow spherical LaNiO3.56 LaMnO3 suffers from similar limitations as other oxides, such as low conductivity and short cycle stability.90 Elsiddig et al.91 synthesized a series of LaMn1±xO3 by controlling Mn/La molar ratios (0.90, 0.95, 1, 1.05, and 1.1). Compared with the stoichiometric LaMnO3 sample, significant differences were observed regarding Mn4+ ions and oxygen vacancies, which improve their conductivity and provide additional sites to accept more ions from the electrolyte and hence improve the electrochemical performance (Fig. 6a and b). In particular, LaMn1.1O3−δ showed the lowest equivalent series resistance (Rs = 3.5 Ω) and the best specific capacity (727.6 C g−1 at 1 A g−1).
000 cycles) superior to that of hollow spherical LaNiO3.56 LaMnO3 suffers from similar limitations as other oxides, such as low conductivity and short cycle stability.90 Elsiddig et al.91 synthesized a series of LaMn1±xO3 by controlling Mn/La molar ratios (0.90, 0.95, 1, 1.05, and 1.1). Compared with the stoichiometric LaMnO3 sample, significant differences were observed regarding Mn4+ ions and oxygen vacancies, which improve their conductivity and provide additional sites to accept more ions from the electrolyte and hence improve the electrochemical performance (Fig. 6a and b). In particular, LaMn1.1O3−δ showed the lowest equivalent series resistance (Rs = 3.5 Ω) and the best specific capacity (727.6 C g−1 at 1 A g−1).
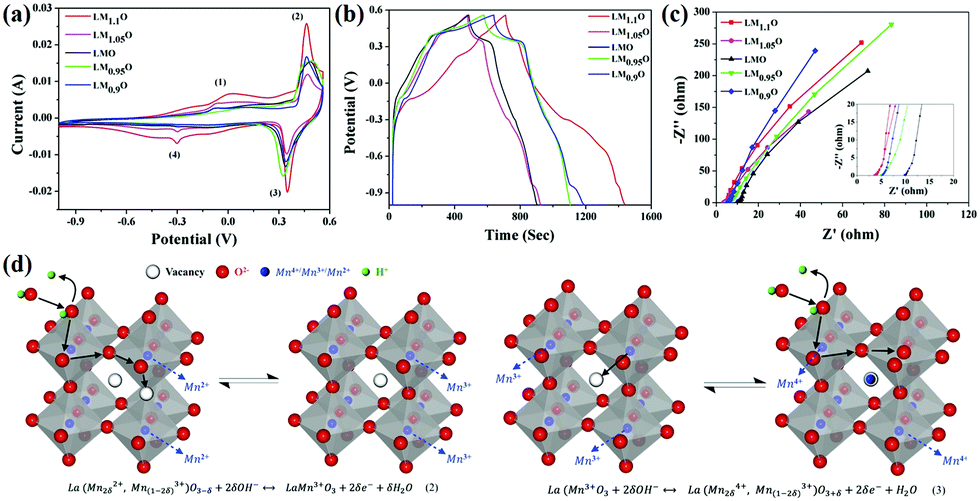 | ||
| Fig. 6 (a) Cyclic performance of the LM1±xO3 samples at a scan rate of 0.01 V s−1. (b) Charge–discharge curves in a potential window of −1 to 0.56 V at 1 A g−1. (C) EIS of the LM1±xO3 samples with the inset of enlarged Rs at high frequencies. (d) Charge storage mechanism in LM1±xO3 perovskite oxide materials. Reproduced from ref. 91 with permission from Elsevier B.V., copyright 2017. | ||
3.2.1.2 A-site doping. In general, the SC performance of perovskite oxides is closely related to the B-site transition metal element. The A-site element (such as lanthanide and alkaline earth element) is inert for the redox reaction directly. However, the A-site cation may affect the electronic structure and coordination. Partial substitution of the A-site by a low-valence cation (such as Sr2+ and Ca2+) generates addition oxygen vacancies and shifts a large proportion of the B-site transition-metal ions to unstable oxidation states (Bm+/B(m+1)+ redox couple).92 As a result, electronic conductivity is increased, and electrochemical performance is enhanced.
Since the alkaline earth metals and rare earth metals share similar atomic radius, it is energetically favorable for the former to substitute the latter.38 In this direction, Mo et al.93 prepared Ca-doped perovskite lanthanum manganates (La0.5Ca0.5MnO3) by a sol–gel method. The specific capacitance of La0.5Ca0.5MnO3 was 2.4 times that of pure LaMnO3 (72 F g−1). Similarly, Wang et al.94 doped LaMnO3 with Sr and reported that its capacitance was slightly increased from 187 to 198 F g−1 at 0.5 A g−1, and the cycle life was enhanced from 40% to 80% after 1000 cycles. To gain more insight into the influence of the substitution degree on electrochemical performance, Tian's group synthesized La1−xSrxMnO3 (x = 0, 0.15, 0.3, 0.5) by the sol–gel method.95 It was observed that the aggregation degree of nanoparticles, specific capacitance, and charge transfer resistance are affected by the value of x. In particular, when x = 0.15 (La0.85Sr0.15MnO3), the lowest Rct (1.6 Ω) and the best specific capacitance (102 F g−1) were observed. Furthermore, the ASCs employing La0.85Sr0.15MnO3 as the anode material and AC as the cathode material reached an energy density of 3.6 W h kg−1 at a low power density of 120 W kg−1. Recently, Alexander and coworkers synthesized a series of perovskite oxides with the composition La1−xSrxBO3−δ (x = 0–1; B = Fe, Mn, Co) to study anion-based PCs systematically.96 They found that more significant oxygen vacancy content upon systematic incorporation of Sr2+ linearly increases the surface-normalized capacity with a slope controlled by the B-site element. Moreover, La0.2Sr0.8MnO2.7 exhibited the best specific capacitance (492 A g−1) relative to the Fe and Co oxides. The formation energy of oxygen vacancies due to aliovalent substitution depends on the alkaline-earth metals.97 The influence of Ba and Ca doping on the SC of SrMnO3 nanofibers was studied by Luo et al.68 Doping 20 mol% Ba to the SrMnO3 matrix significantly enhanced the specific capacitance from 321.7 F g−1 to up to 446.8 F g−1. Moreover, the ASC fabricated using Sr0.8Ba0.2MnO3 showed an energy density of 37.3 W h kg−1 at a power density of 400 W kg−1 and a superior capacitance retention of 87% after 5000 cycles.
3.2.1.3 B-site doping. The charge storage mechanism embraces electrochemical reactions. Incorporating oxygen ions into the vacancies requires a simultaneous change in the B cation's oxidation state. B-site doping is another effective measure to enhance electrical performance because it can produce oxygen vacancies and improve the crystal structure's stability.98
Cobalt-based perovskite oxides are superior to Mn-based ones due to their larger oxygen vacancy concentration and higher oxygen-ion mobility.99 However, some cobalt-based perovskite oxides (such as cubic phase SrCoO3−δ) are not stable at room temperature. To widely implement cobalt-based perovskite oxides application in SCs, Sharma and coworkers doped Mo to substitute Co in SrCoO3−δ partially (Fig. 7a).100 It was found that the oxygen vacancies of SrCo0.9Mo0.1O3 (SCM) were 2.1 times greater than that of SrCoO3−δ. Additionally, SCM's specific capacitance (1223.34 F g−1 at 1 A g−1) was increased by 1.36 times that of SrCoO3−δ (Fig. 7b). Notably, the ASC constructed by using lacey reduced graphene oxide nanoribbon (LRGONR) as a negative electrode showed long-term stability (165.1% activated specific capacitance retention after 10000 cycles) (Fig. 7c). Most importantly, it also achieved a specific energy density of 74.8 W h kg−1 at a power density of 734.5 W kg−1 (Fig. 7d). Similarly, Nb-doped SrCoO3−δ with a gravimetric capacitance of ca. 773.6 F g−1 (2034.6 F cm−3) and great cycling stability (95.7% capacitance retention after 3000 cycles) was reported by Shao et al.101 Furthermore, an ASC was assembled using SrCo0.9Nb0.1O3−δ (SCN) and AC as the cathode and anode. The device possessed an energy density of 37.6 W h kg−1 at a power density of 433.9 W kg−1, and it still maintained an energy density of 32.9 W h kg−1 at a higher power density of 9864.2 W kg−1. Also, it displayed long-duration cycling performance (about 1.7% loss after 5000 cycles). The B-site doping can also affect the potential window of perovskite oxides. Garche's team studied the influence of B site element doping on the stability window of SrRuO3. Interestingly, 20 mol% Mg doping was found to increase the specific capacitance of SrRuO3 without changing its stability window.66 In contrast, Fe or Co substitution could lead to a decreased stability window.
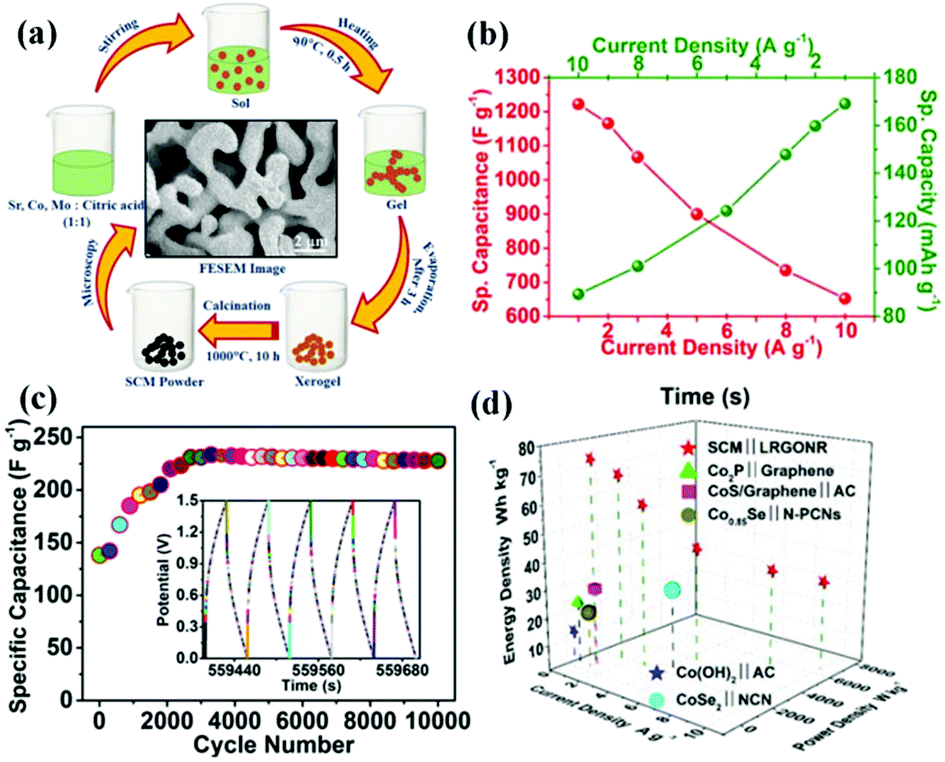 | ||
| Fig. 7 (a) Schematic illustration of the synthesis of SCM. (b) Electrochemical performance of the SCM electrode evaluated through GCD at different current densities. (c) Cycling stability of the ASC at 10 A g−1. The inset shows the last five GCD cycles. (d) Ragone plot of a hybrid SCM cell compared to literature reports. Reproduced from ref. 100 with permission from WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, copyright 2018. | ||
3.2.2.1 Perovskite oxides/noble metals. Noble metals, including gold (Au) and platinum (Pt) group metals with good electric conductivity, can facilitate the transport of electrons that arise from the oxidation/reduction of PCs to the current collectors.102 Nevertheless, owing to the scarcity and high cost of noble metals, their integration with other sustainable and cheap materials is considered one of the most attractive strategies to minimize their consumption. Perovskite oxides possess characteristics of natural abundance and low cost. Unfortunately, the electrical conductivity of perovskite oxides is still not satisfactory. Therefore, combining perovskite oxides with noble metals is expected to improve their SC performance by creating synergistic effects.
Ag has the highest conductivity among the noble metals. Besides, it also has the advantage of reasonable cost and has an acceptable activity.103 Lang et al.104 reported that Ag nanoparticle decorated La0.85Sr0.15MnO3 was designed and used as an electrode for SCs. Since Ag has much higher electrical conductivity than that of carbon materials, it could build electron transfer channels. Additionally, the redox reaction between Ag and Ag2O in an alkaline electrolyte solution could contribute a small amount of pseudocapacitance. As a result, the Ag@LSM15 composite delivered a high specific capacitance (186 F g−1 at 1 A g−1) and a long cycle life (100% capacitance retention after 1000 cycles). Moreover, based on this material, the ASC was constructed, showing a maximum energy density of 20.6 W h kg−1 at a power density of 1700 W kg−1. In another study, Ag nanoparticles were grown directly on a porous perovskite La0.7Sr0.3CoO3−δ (LSC) substrate (Ag/LSC) (Fig. 8a).105 The effect of different mass loading of Ag (10.61, 30.60, and 51.31 mg) on performance was studied. The porous structure of LSC was preserved, and the surface became rough when the content of Ag was 30 mg (30 Ag/LSC) or less. This favorable structure can provide more active surface sites and facilitate rapid mass transport. Besides, the Ag/LSC electrode with 30 mg Ag loading also displayed lower Rs (1.28 Ω cm2) and Rct (0.61 Ω cm2), giving rise to the best performance (14.8 F cm−2) (Fig. 8b). Besides, an ASC based on 30 Ag/LSC as the negative electrode and carbon cloth (CC) as the positive electrode was constructed, displaying an energy density of 21.9 mW h cm−3 and a power density of 90.1 mW cm−3 at 5 mA cm−2 (Fig. 8c). Visibly, two ASCs in series were used to light an LED blue for 15 min after charged for 0.5 min at 50 mA cm−2 (Fig. 8c).
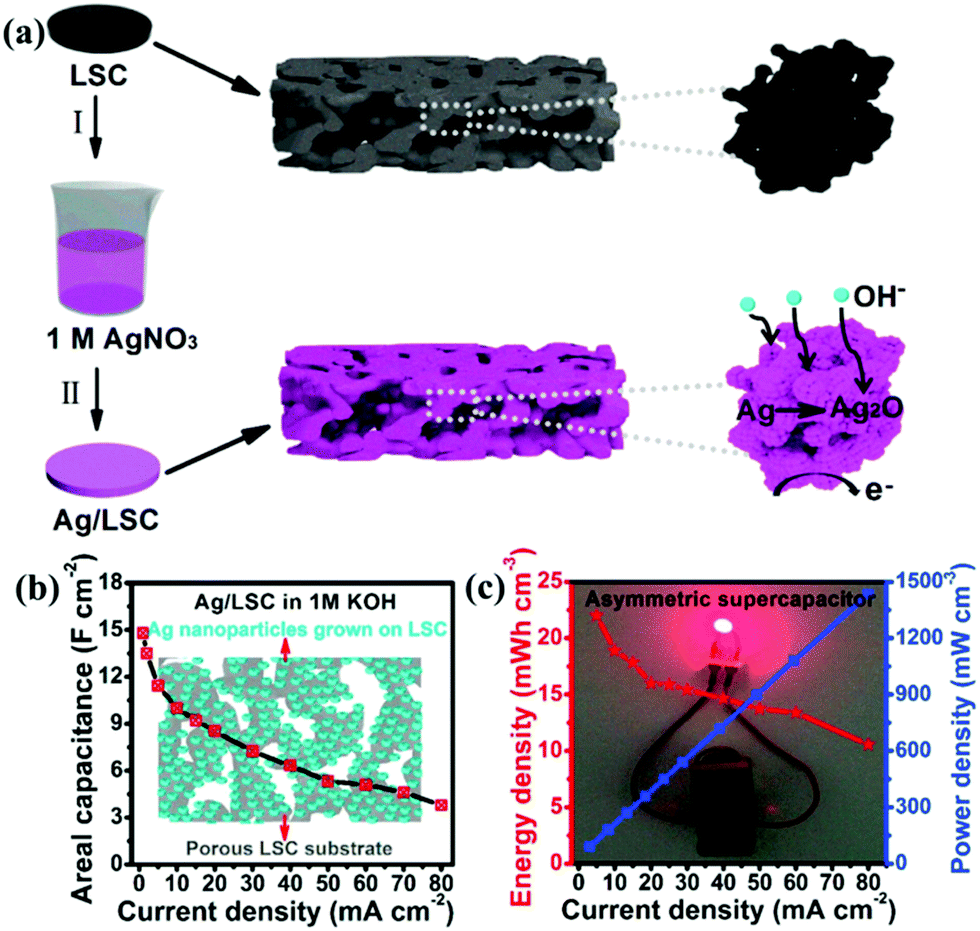 | ||
| Fig. 8 (a) Schematic illustration of the preparation of Ag nanoparticles grown on a LSC substrate. (b) The areal capacitance of 30 Ag/LSC. (c) Electrochemical measurements of the asymmetric supercapacitor: Ragone plot at different current densities (inset: an LED bulb lighted by two asymmetric cells in series). Reproduced from ref. 105 with permission from Elsevier B.V., copyright 2017. | ||
3.2.2.2 Perovskite oxides/metal oxides. Metal oxides have received more and more attention because their multiple numbers of oxidation states allow for up to an order of magnitude more generous energy storage compared to carbon-based EDLCs.12 However, most metal oxides suffer from low conductivity, poor rate capability, and poor durability during charge/discharge processes. In contrast, perovskite oxides possess a stable structure and can produce much higher oxygen ion/electron conductivity and better surface oxygen exchange kinetics.106 Hence, the composites of perovskite oxides and metal oxides are attractive alternatives because of their low cost in addition to the possibility to achieve high specific capacitance/capacity values and superb stability.
Among the metal oxides, MnO2 stands out due to its high theoretical specific capacitance (about 1370 F g−1), low cost, and natural abundance.107,108 Lv et al.109 synthesized (La0.75Sr0.25)0.95MnO3−δ ((LSM)/MnO2) composite as an electrode for SCs via a hydrothermal method. The resultant sample exhibited a larger specific capacitance (437.2 F g−1 at 2 mV s−1). Compared with MnO2, CeO2 has low theoretical capacitance, but it has unique chemical properties. That is, it can be easily oxidized and reduced during the oxidation–reduction process.110,111 Nagamuthu and co-workers reported LaMnO3 mixed CeO2 (CeO2/LaMnO3) nanocomposites with a higher specific capacitance (262 F g−1 at 1 A g−1) in 1 M Na2SO4 solution.112 Interestingly, they found that the CeO2/LaMnO3 nanocomposite was more suited for ASC negative electrodes. More specifically, an ASC device was assembled using the CeO2/LaMnO3 nanocomposites as the negative electrode and AC as the positive electrode, revealing an energy density of 17.2 W h kg−1 at a power density of 1015 W kg−1. It is well established that the energy density (E) is proportional to the potential window (V). Thus, another way to elevate the energy density is to enlarge the potential window. A La0.8Sr0.15MnO3@NiCo2O4 (LSM15@NC) core–shell nanoflower structure directly grown on Ni foam (Fig. 9a) was constructed by Lang et al.113 LSM15@NC showed a wide window and the coexistence of PC as well as EDLC behavior (Fig. 9b). Benefitting from the unique structure acting as a buffer for the volume change during the long-term charge–discharge tests, the specific capacitance of LSM15@NC was twice as much as that of the first cycle after 10000 cycles (Fig. 9c). Moreover, the ASC using AC as the negative electrode and the LSM15@NC composite as the positive electrode delivered an energy density of 63.5 W h kg−1 at a power density of 900 W kg−1 (Fig. 9d).
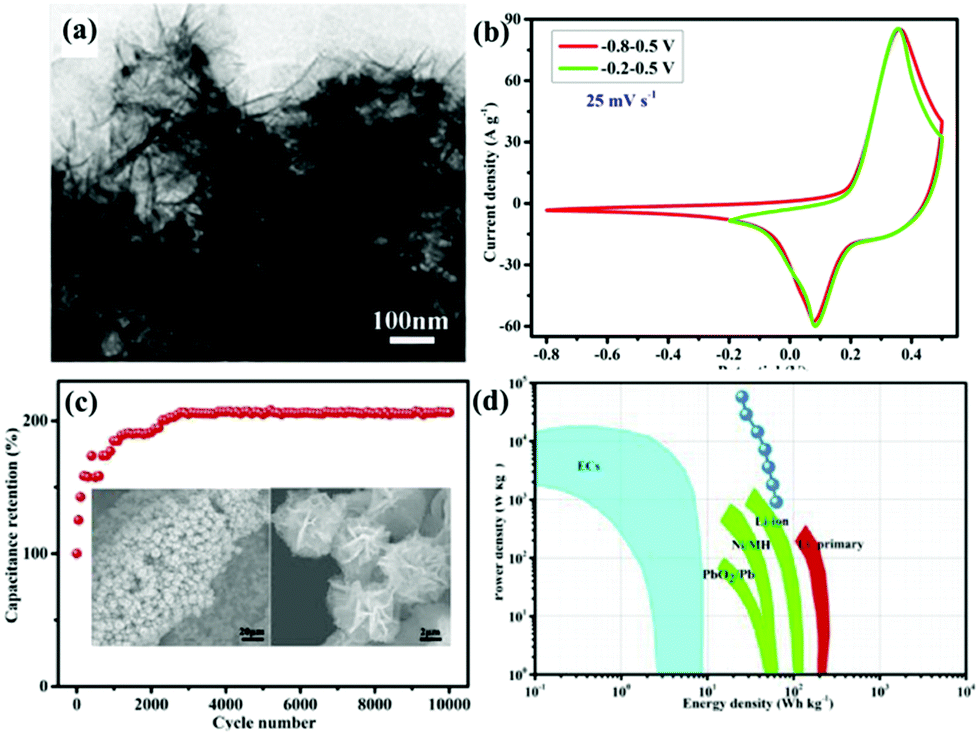 | ||
| Fig. 9 (a) TEM images of LSM15@NC scratched down from the Ni foam. (b) Comparative CV curves at a scan rate of 25 mV s−1 within different potential windows of −0.8 to 0.5 V and −0.2 to 0.5 V. (c) Cycling performance (the insets show the morphology of LSM15@NC after 10000 cycles). (d) Ragone plot. Reproduced from ref. 113 with permission from Elsevier B.V., copyright 2018. | ||
3.2.2.3 Perovskite oxides/carbon materials. It is well known that carbonaceous materials (such as AC, graphene, graphene oxide, and reduced graphene oxide) with large specific surface area, good electronic conductivity, and high chemical stability have been widely applied in SCs.114 The low intrinsic conductivity of perovskite oxides is the main drawback that limits their further application in SCs. The addition of carbonaceous materials to form perovskite oxides/carbon composites is an effective strategy to remedy this deficiency.
Due to the extra-large theoretical specific surface area (2630 m2 g−1) and superior electrical conductivity (6000 S cm−1), graphene is usually added into perovskite oxides to enhance SC performance.114,115 Graphene can effectively restrain the agglomeration of perovskite oxides. Meanwhile, it can provide a high-speed electron transport channel.116 In return, the loading perovskite oxide nanoparticles are also available to maintain the structural nature of monolayer graphene for their function of increasing the distance between the graphene sheets.116Fig. 10a shows BiFeO3 (BFO) nanowires immobilized on nanometer-thin RGO.117 Based on this material, the specific capacitance (368.28 F g−1) and charge transfer resistance are found to be superior to those of BFO and RGO (Fig. 10b and c). Notably, the electrolyte also has a more significant impact on the performance of BFO–RGO (Fig. 10d). To investigate the capacitive behavior of graphene–perovskite oxide compound materials in aqueous electrolytes of various acidity or basicity, Ataa et al.118 tested reduced graphene performance sheets decorated SrRuO3 (SRGO) in different electrolytes (1.0 M NaNO3, 1.0 M H3PO4, and 1.0 M KOH). The SRGO exhibited the largest capacitance (160 F g−1) in 1.0 M KOH.
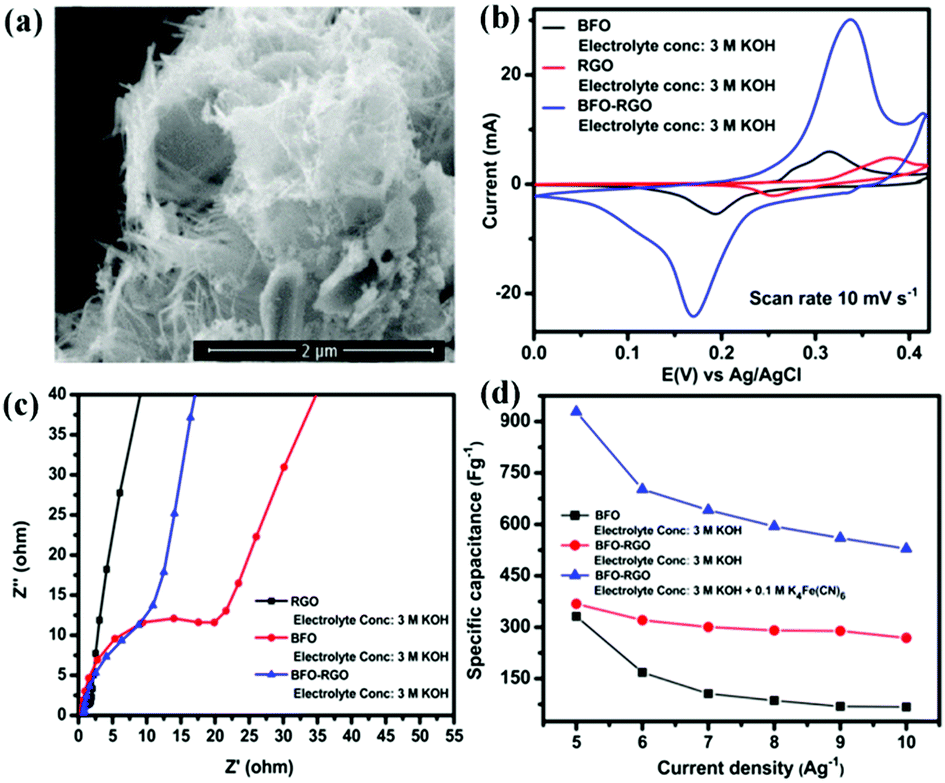 | ||
| Fig. 10 FESEM micrographs of (a) BFO. (b) Cyclic voltammetry curves of BFO, RGO, and BFO–RGO electrodes at a scan rate of 10 mV s−1 in a 3 M KOH electrolyte. (c) Electrochemical impedance spectra of RGO, BFO, and BFO–RGO electrodes in a 3 M KOH electrolyte. (d) Change of the specific capacitance of BFO and BFO–RGO electrodes with changing current density from 5 to 10 A g−1. Reproduced from ref. 117 with permission from American Chemical Society, copyright 2018. | ||
As stated above, the electrochemical performance is enhanced compared with pure perovskite oxides after carbon introduction. The redox reaction of oxygenated groups on carbon nanostructures' surface contribute to pseudocapacitance.119 In this regard, introducing surface functional groups or heteroatoms on the surface of carbon materials appears to be an effective way of improving the composite electrode's capacitance.120,121 Elsiddig et al.122 introduced a heteroatom to reduced graphene oxide (rGO) by substituting the hydroxyl groups with the nitrogen atoms. Then, the as-prepared nitrogen-doped graphene (N-rGO) sheets can be directly integrated with LaMnO3 (LMO) through electrostatic interactions to form a three-dimensional network (LMO/N-rGO). Compared with pristine graphene and LMO, the obtained nanocomposites displayed the best specific capacitance (687 F g−1 at 5 mV s−1) and stability (79% capacitance retention after 2000 cycles at 10 A g−1). In another study, Shafi et al.123 synthesized a composite material containing LaMnO3, RGO, and polyaniline (PANI) by in situ chemical polymerization. Since the RGO support and PANI coating again provided good structural stability and good electrical conductivity, the synthesized ternary composite exhibited a specific capacitance of 802 F g−1 at 1 A g−1. More importantly, the ASC with RGO as the negative electrode and ternary nanocomposite as the positive electrode exhibited a maximum energy density of 50 W h kg−1 at a power density of 2.25 kW kg−1, with admirable long-term stability (117% capacitance retention after 100000 cycles).
4. Derivative perovskite oxides
Some derivative perovskite oxides were reported to possess fast oxygen ion diffusion channels, in which the oxygen diffusion exhibited shallow activation energy.124,125 Additionally, the oxygen-ion conductivity of some derivative perovskite oxides is even higher than that of many benchmark ABO3.45,126 Moreover, many derivative perovskite oxides also can present high concentrations of variable oxygen vacancies. In this context, derivative perovskite oxides have some potential for application in SCs, as shown in Table 1.| Perovskite oxides | Methods | Structure | Electrolyte | Potential window | Capacitance | Ref. |
|---|---|---|---|---|---|---|
| LaMnO2.97 | Reverse-phase hydrolysis approach | Nanoparticles | 1 M KOH | −1.2 to 0.0 V | 609.8 F g−1 at 2 mV s−1 | 31 |
| LaMn1.1O3 | Sol–gel method | Mesoporous | 1 M KOH | −1.0 to 0.56 V | 727.6 C g−1 at 1 A g−1 | 90 |
| LaNiO3 | Template-free solvothermal method | Hollow spheres | 6 M KOH | 0.0–0.45 V | 422 F g−1 at 1 A g−1 | 59 |
| LaNiO2.63 | Sol–gel method | Nanoparticles | 1 M KOH | 0.2–0.6 V | 478.7 F g−1 at 0.1 mV s−1 | 89 |
| LaCoO3 | Co-precipitation method | Nanoparticles | 3 M KOH | 0.0–0.5 V | 299.64 F g−1 at 10 A g−1 | 62 |
| LaCrO3 | Sol–gel method | Hierarchical mesoporous | 1 M LiCl | 0.0–1.0 V | 1268 F g−1 at 2 A g−1 | 63 |
| BiFeO3 | Hydrothermal method | Nanoplates | 1 M NaOH | 0.0–0.5 V | 254.6 F g−1 at 1 mV s−1 | 77 |
| LaFeO3 | The MOF gel method | Mesoporous | 1 M Na2SO4 | −1.0 to 0.0 V | 241.3 F g−1 at 1 A g−1 | 61 |
| SrRuO3 | Pyrolysis route | Porous | 6 M KOH | −0.9 to 0.3 V | 270 F g−1 at 20 mV s−1 | 66 |
| SrTiO3 | Sol–gel method | Mesoporous | 3 M KOH | −0.2 to 0.6 V | 592 F g−1 at 5 mV s−1 | 70 |
| CeMnO3 | Electrospinning | Nanofibers | 6 M KOH | −0.1 to 0.6 V | 155 F g−1 at 1 A g−1 | 74 |
| CeCoO3 | Electrospinning | Nanofibers | 6 M KOH | −0.1 to 0.6 V | 128 F g−1 at 0.5 A g−1 | 75 |
| CeNiO3 | Electrospinning | Nanofibers | 6 M KOH | −0.1 to 0.65 V | 189 F g−1 at 0.5 A g−1 | 75 |
| CeCuO3 | Electrospinning | Nanofibers | 6 M KOH | −0.1 to 0.7 V | 117 F g−1 at 0.5 A g−1 | 75 |
| NiTiO3 | Sol–gel method | Rod-like | 2 M KOH | 0.0–0.6 V | 542.26 F g−1 at 1 A g−1 | 80 |
| NiMnO3 | Hydrothermal method | Three-dimensional hierarchical | 6 M KOH | 0.0–0.9 V | 99.03 F g−1 at 100 mV s−1 | 79 |
| CoTiO3 | Sol–gel method | Mesoporous microrods | 2 M KOH | 0.0–0.6 V | 608.4 F g−1 at 5 mV s−1 | 81 |
| La0.85Sr0.15MnO3 | Sol–gel method | Sphere | 1 M KOH | −0.96 to 0.65 V | 198 F g−1 at 0.5 A g−1 | 94 |
| La0.5Ca0.5MnO3 | Sol–gel method | Nanoparticles | 1 M KOH | −1.0 to 0.6 V | 170 F g−1 at 1 A g−1 | 93 |
| Sr0.8Ba0.2MnO3 | Sol–gel and electrospinning | Nanofibers | 1 M Na2SO4 | 0.0–0.8 V | 446.8 F g−1 at 0.5 A g−1 | 68 |
| SrCo0.9Mo0.1O3 | Sol–gel method | Nanoparticles | 6 M KOH | −0.1 to 0.5 V | 1223.34 F g−1 at 1 A g−1 | 100 |
| SrCo0.9Nb0.1O3 | Solid-state | Nanoparticles | 1 M KOH | 0.0–0.5 V | 773.6 F g−1 at 0.5 A g−1 | 101 |
| La2ZnMnO6 | Hydrothermal route | Nanoflakes | 2 M KOH | 0.0–0.6 V | 718.6 F g−1 at 1 mV s−1 | 128 |
| La2CuMnO6 | Hydrothermal route | Nanoparticles | 2 M KOH | 0.0–0.45 V | 205.5 F g−1 at 0.5 A g−1 | 129 |
| Gd2NiMnO6 | Wet chemical route | Nanoparticles | 4 M KOH | 0.2–0.6 V | 400.46 F g−1 at 1 A g−1 | 130 |
| La2CoMnO6 | Template impregnation method | Hollow spheres | 1 M Na2SO4 | −0.1 to 1.0 V | 376 F g−1 at 1 A g−1 | 131 |
| Y2NiMnO6 | Hydrothermal route | Nanowires | 0.5 M KOH | 0.2–0.5 V | 77.76 F g−1 at 30 mA g−1 | 132 |
| PrBaMn2O6−δ | Sol–gel method | Nanoparticles | 6 M KOH | 0.0–0.5 V | 1034.8 F g−1 at 1 A g−1 | 133 |
| PrBaCo2O5+δ | Sol–gel method | Nanoparticles | 1 M KOH | −0.1 to 0.55 V | 428.2 C g−1 at 1 mV s−1 | 136 |
| Ba2Bi0.1Sc0.2Co1.7O6−δ | Sol–gel method | Nanoparticles | 6 M KOH | 0.0–0.6 V | 796.7 F g−1 at 1 A g−1 | 137 |
| Sr2CoMoO6−δ | Sol–gel method | Spherical particles | 6 M KOH | −0.1 to 0.4 V | 747 F g−1 at 1 A g−1 | 138 |
| La2NiO4+δ | Citrate method | Porous | 3 M KOH | 0.0–0.6 V | 657.4 F g−1 at 2 mV s−1 | 141 |
4.1 Double perovskite oxides
In 1951, double perovskite oxides were first discovered by Steward and Rooksby, expressed by the general formula of AA'BB'O6, a derivative of ABO3, extending the pristine framework to a super-lattice.127 To date, double perovskite oxides have been widely investigated, because they have high oxygen surface exchange kinetics and fast oxygen ion diffusion rates, and good mixed ionic and electronic conductivity at elevated temperature. Compared with element-doped perovskite oxides, AA′BB′O6 holds a more orderly structure arrangement, thereby preventing lattice distortion and improving cycling stability.In general, cations are disordered and distributed homogeneously inside the AA′BB′O6 lattice. Nevertheless, in some situations, cation ordering could occur (such as AA′B2O6 and A2BB′O6). It is worth mentioning that cation ordering could display better physical and chemical properties. Kumar's team reported the electrochemical performance of R2MMnO6 (R = La, Gd; M = Zn, Cu, Ni) perovskite oxides including La2ZnMnO6, La2CuMnO6, and Gd2NiMnO6, which showed a specific capacitance of 718.6 F g−1, 205.5 F g−1, 400.46 F g−1, respectively, at different current densities.128–130 To improve the SC properties, increasing the specific surface area and improving the nanoscale charge transport are useful measures. Meng and coworkers fabricated a hollow spherical porous structure of La2CoMnO6 by template impregnation (HS-LCMO).131 For comparison, La2CoMnO6 was also prepared by the sol–gel method (SG-LCMO). The specific surface area of HS-LCMO and SG-LCMO was estimated to be 22.14 and 10.36 m2 g−1, respectively. Owing to its large specific surface area, HS-LCMO displayed a specific capacitance (376 F g−1) which was 4 times that of SG-LCMO (94 F g−1). Similarly, Y2NiMnO6 nanowires (NWs) were prepared by a hydrothermal route. The resultant Y2NiMnO6 NWs exhibited a specific capacitance (77.76 F g−1) which was higher than that of the counterparts prepared by the sol–gel method.132 The crystal structure of perovskite oxides is closely related to the calcination temperature. Shao's team demonstrated that the reduction of PrBaMn2O6−δ (r-PBM) in a hydrogen atmosphere at 850 °C led to a phase transition from a mixed hexagonal and cubic phase to a cubic phase PBM (Fig. 11a and b).133 Based on density functional theory (DFT) calculations, it is somewhat easier to form an oxygen vacancy in cubic PBM than in the hexagonal phase. Correspondingly, the formation of a layered double perovskite oxide structure after hydrogen reduction (800 °C for 45 min) significantly increased the oxygen vacancy concentration and oxygen anion diffusion rate, thus contributing to the capacitance (Fig. 11c). Notably, the r-PBM with a low specific surface area (7.83 m2 g−1) exhibited a gravimetric capacitance of 1034.8 F g−1 (Fig. 11d).
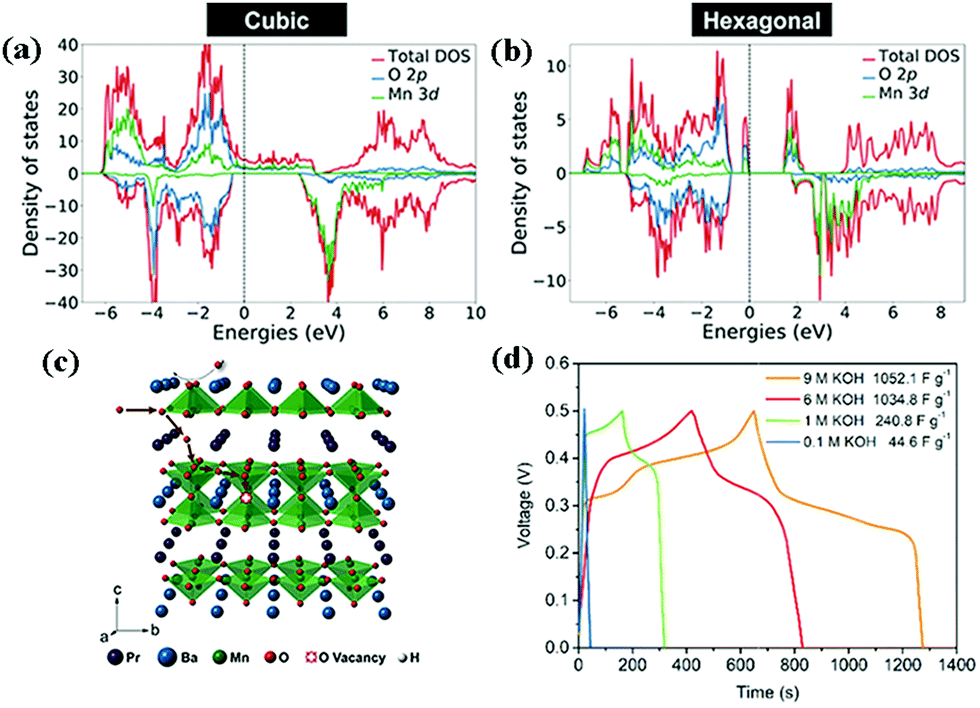 | ||
| Fig. 11 DFT+U calculated density of states (DOS) for (a) cubic and (b) hexagonal PBM. (c) A schematic diagram of oxygen intercalation into r-PBM during the energy storage process. (d) Galvanostatic charge–discharge profiles of r-PBM. Reproduced from ref. 133 with permission from WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, copyright 2018. | ||
To further explore the effect of cation ordering on the performance of SCs, more effort has been made.134,135 Wang et al.136 synthesized A-site cation-ordered double perovskite oxide PrBaCo2O5+δ (PBCO) by the sol–gel method. They found that oxygen atoms located in the Pr3+ planes can be partially or even entirely removed, producing numerous oxygen vacancies in the perovskite oxide structure. Accordingly, the synthesized PBCO showed a good specific capacity (428.2 C g−1). Notably, PBCO displayed cycling stability (93% capacitance retention after 2000 cycles at 10 A g−1) superior to that of some single perovskite oxides (such as BiFeO3 and LaMnO3).77,91 Beyond that, Xu et al.137 reported that B-site cation-ordered double perovskite oxide Ba2Bi0.1Sc0.2Co1.7O6−δ (BBSC) was a good anion-intercalation-type electrode for SCs. For comparison, the oxygen vacancy-disordered Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF) electrode was also studied. It was found that the oxygen vacancy density of BBSC is larger than that of BSCF, and the simultaneous leaching of Ba from the A site and Bi from the B site of BBSC could minimize the cation deficiency in BBSC. As a result, BBSC delivered a specific capacitance (1050 F g−1) higher than that of BSCF. Hence, the ASC fabricated using BBSC displayed an energy density of 70 W h kg−1 at a power density of 787 W h kg−1. Sr2CoMoO6−δ (DP-SCM) as an intercalation PC was reported by Tomar et al.138 In their study, B site-ordering was based on the difference between oxidation states of Co and Mo (greater than 2) with a large ionic radii difference. It was found that the synergy of Co/Mo has good redox ability, further facilitating high oxygen mobility, resulting in DP-SCM achieving a specific capacitance of 747 F g−1 at 1 A g−1 with a rate capability of 56% up to 10 A g−1.
4.2 Ruddlesden–Popper oxides
RP perovskite oxides were first studied in 1958 by Ruddlesden and Popper.139 Alternating perovskite (ABO3)− and rock-salt (AO)+ layers in the homologous series of titanates (Srn+1TinO3n+1) were discovered.140 The unique layered structure allows two sorts of oxygen species (oxygen vacancy, interstitial oxygen). The oxygen vacancy exists in the perovskite layer, while the interstitial oxygen is found in the rock-salt layer. Compared with ABO3-type, RP perovskite oxides show superior mixed ionic and electronic conductivity. Sang et al.141 prepared RP-type perovskite oxide La2NiO4+δvia a citrate method and used it as the active material for SCs. It was found that there are two ways of oxygen extrusion and only one way of oxygen insertion in La2NiO4+δ. Interestingly, La2NiO4+δ showed a high specific capacitance of 657.4 F g−1 at a scan rate of 2 mV s−1, which was better than the capacitance of LaMnO3 (609.8 F g−1).31 Recently, to optimize the surface properties of RP particles, Wei and coworkers synthesized La2NiO4+δ coated with Ag (La2NiO4+δ@Ag).142 Because of the unique layered structure of La2NiO4+δ and the quick reversible redox process of Ag/AgO2, the La2NiO4+δ@Ag electrode showed better anion intercalation performance compared to La2NiO4+δ, such as large capacity (466.4 C g−1 at 1 mV s−1) and good cycling stability (average efficiency approximated 93% in 10000 cycles). Furthermore, a hybrid SC device assembled using La2NiO4+δ@Ag and AC as the cathode and the anode showed a high energy density of 44.7 W h kg−1 at a power density of 800 W h kg−1.
5. Conclusions and outlooks
With the unique structure, compositional flexibility, and inherent oxygen vacancy, perovskite oxides have recently been widely employed as electrode materials for SCs. Notably, as an active material of intercalation-type capacitors, perovskite oxides are characterized by high oxygen vacancy concentrations and do not require high surface area to achieve a high energy storage capacity. This article mainly summarizes the recent advances in perovskite oxides (i.e., single, double, and RP perovskite oxides) for SCs. Further improvement of perovskite oxides' electrochemical properties by increasing oxygen vacancy concentration and constructing composites is discussed. Despite many efforts in this field, several aspects still need to be considered in the design of perovskite oxide electrode materials in the future.(1) During the charge storage process, the valence state change of the B-site cation with a low energy barrier in perovskite oxides is preferred.137 To date, Mn, Ni, Fe, and Co have been studied as B site elements. Some early transition metals, such as Ti, Zr, V, and Nb, share similar ionization energies and ionization energy differences with Mn,143 which is expected to be the B site element of perovskite oxide electrodes.
(2) B-site substitution by a metal element has been proven as a feasible approach to create oxygen vacancies in perovskite oxides, thereby boosting their performance as SC electrode materials.100,101 In contrast, non-metal doping at the B-site was shown to improve the phase stability, electrical conductivity, and oxygen vacancies of perovskite oxides.144–146 Therefore, more research work on non-metal doping at the B-site is expected in this field.
(3) The electronic conductivity of RP perovskite oxides is anticipated to increase as their n value increases.147 However, most of the reports are on RP perovskite oxides with n = 1. RP perovskite oxides with higher n values as advanced SC electrode materials are therefore expected.
(4) The morphology of perovskite oxides plays a crucial role in boosting electrochemical performance. However, perovskite oxides generally are synthesized at high temperatures above 600 °C, leading to limited low surface areas (<10 m2 g−1) and less exposure to active sites.148 Therefore, controllable synthesis of nanostructures (such as hollow, core–shell, and nanoarray) with large specific surface area and high redox activity is vital for achieving high energy/power densities of perovskite oxide SC electrodes.
(5) Apart from the architecture and electronic configurations, electrolytes also optimize perovskite oxides' energy-storage performance. Aqueous electrolytes like KOH with a narrow working voltage range of ∼1.23 V and Na2SO4 (∼2 V) are by far the most investigated media. The utilization of organic (2.5–3.5 V) or ionic liquid electrolytes (2–6 V) offers a wide voltage window, further improving the energy density. Notably, the introduction of additional redox-active compounds such as CuCl2, KMnO4, KI, KBr, Na2MoO4, and K3Fe(CN)6, i.e., the formation of a redox electrolyte, can further improve the electrode materials' electrochemical performance. For instance, an additional 0.1 M K4[Fe(CN)6] in 3 M KOH causes a nearly two-fold increase in the BFO–RGO electrode's specific capacitance.117 Therefore, the use of a redox electrolyte may be promising to boost the electrochemical performance of SCs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22072015).References
- Z. Guo, S. Zhao, T. Li, D. Su, S. Guo and G. Wang, Adv. Energy Mater., 2020, 10, 1903591 CrossRef CAS.
- L. Zu, W. Zhang, L. Qu, L. Liu, W. Li, A. Yu and D. Zhao, Adv. Energy Mater., 2020, 10, 2002152 CrossRef CAS.
- Y. Huang, M. Zhu, Y. Huang, Z. Pei, H. Li, Z. Wang, Q. Xue and C. Zhi, Adv. Mater., 2016, 28, 8344–8364 CrossRef CAS.
- J. Liang, B. Tian, S. Li, C. Jiang and W. Wu, Adv. Energy Mater., 2020, 10, 2000022 CrossRef CAS.
- J. Libich, J. Máca, J. Vondrák, O. Čech and M. Sedlaříková, J. Energy Storage, 2018, 17, 224–227 CrossRef.
- J. W. Long, D. Bélanger, T. Brousse, W. Sugimoto, M. B. Sassin and O. Crosnier, MRS Bull., 2011, 36, 513–522 CrossRef CAS.
- J. Liang, H. Zhao, L. Yue, G. Fan, T. Li, S. Lu, G. Chen, S. Gao, A. M. Asiri and X. Sun, J. Mater. Chem. A, 2020, 8, 16747–16789 RSC.
- J. Yan, S. Li, B. Lan, Y. Wu and P. S. Lee, Adv. Funct. Mater., 2020, 30, 1902564 CrossRef CAS.
- X. Lu, C. Wang, F. Favier and N. Pinna, Adv. Energy Mater., 2017, 7, 1601301 CrossRef.
- Z. Zhai, W. Yan, L. Dong, J. Wang, C. Chen, J. Lian, X. Wang, D. Xia and J. Zhang, Nano Energy, 2020, 78, 105193 CrossRef CAS.
- Y. Wang, L. Zhang, H. Hou, W. Xu, G. Duan, S. He, K. Liu and S. Jiang, J. Mater. Sci., 2021, 56, 173–200 CrossRef CAS.
- Y. Zhang, H. Feng, X. Wu, L. Wang, A. Zhang, T. Xia, H. Dong, X. Li and L. Zhang, Int. J. Hydrogen Energy, 2009, 34, 4889 CrossRef CAS.
- D. Qu, J. Power Sources, 2002, 109, 403 CrossRef CAS.
- R. S. Kate, S. A. Khalate and R. J. Deokate, J. Alloys Compd., 2018, 734, 89–111 CrossRef CAS.
- Y. Liu, Z. Wang, Y. Zhong, X. Xu, J.-P. M. Veder, M. R. Rowles, M. Saunders, R. Ran and Z. Shao, Chem. Eng. J., 2020, 390, 124645 CrossRef CAS.
- N. Choudhary, C. Li, J. Moore, N. Nagaiah, L. Zhai, Y. Jung and J. Thomas, Adv. Mater., 2017, 29, 1605336 CrossRef.
- G. Wang, L. Zhang and J. Zhang, Chem. Soc. Rev., 2012, 41, 797–828 RSC.
- Y. Zhai, Y. Dou, D. Zhao, P. F. Fulvio, R. T. Mayes and S. Dai, Adv. Mater., 2011, 23, 4828–4850 CrossRef CAS.
- H. Lv, Q. Pan, Y. Song, X.-X. Liu and T. Liu, Nano-Micro Lett., 2020, 12, 118 CrossRef CAS.
- M. Yu, Y. Lu, H. Zheng and X. Lu, Chem. – Eur. J., 2018, 24, 3639–3649 CrossRef CAS.
- Y. Shao, M. F. El-Kady, J. Sun, Y. Li, Q. Zhang, M. Zhu, H. Wang, B. Dunn and R. B. Kaner, Chem. Rev., 2018, 118, 9233–9280 CrossRef CAS.
- X. Lu, M. Yu, T. Zhai, G. Wang, S. Xie, T. Liu, C. Liang, Y. Tong and Y. Li, Nano Lett., 2013, 13, 2628–2633 CrossRef CAS.
- N. Choudhary, C. Li, J. Moore, N. Nagaiah, L. Zhai, Y. Jung and J. Thomas, Adv. Mater., 2017, 29, 1605336 CrossRef.
- V. Augustyn, P. Simon and B. Dunn, Energy Environ. Sci., 2014, 7, 1597–1614 RSC.
- W. Zhao, M. Jiang, W. Wang, S. Liu, W. Huang and Q. Zhao, Adv. Funct. Mater., 2020, 2009136 CrossRef.
- Z.-H. Huang, Y. Song, D.-Y. Feng, Z. Sun, X. Sun and X.-X. Liu, ACS Nano, 2018, 12, 3557–3567 CrossRef CAS.
- G. Meng, Q. Yang, X. Wu, P. Wan, Y. Li, X. Lei, X. Sun and J. Liu, Nano Energy, 2016, 30, 831–839 CrossRef CAS.
- V. D. Nithya and N. Sabari Arul, J. Mater. Chem. A, 2016, 4, 10767–10778 RSC.
- P. Tan, M. Liu, Z. Shao and M. Ni, Adv. Energy Mater., 2017, 7, 1602674 CrossRef.
- X. Xu, W. Wang, W. Zhou and Z. Shao, Small Methods, 2018, 2, 1800071 CrossRef.
- J. T. Mefford, W. G. Hardin, S. Dai, K. P. Johnston and K. J. Stevenson, Nat. Mater., 2014, 13, 726–732 CrossRef CAS.
- Y. Liu, S. P. Jiang and Z. Shao, Mater. Today Adv., 2020, 7, 100072 CrossRef.
- C. Chen, Y. Mei, Y. Huang and X. Hu, Mater. Res. Bull., 2017, 96, 365–371 CrossRef CAS.
- K. Brezesinski, J. Wang, J. Haetge, C. Reitz, S. O. Steinmueller, S. H. Tolbert, B. M. Smarsly, B. Dunn and T. Brezesinski, J. Am. Chem. Soc., 2010, 132, 6982–6990 CrossRef CAS.
- B. Mendoza-Sánchez and P. S. Grant, Electrochim. Acta, 2013, 98, 294–302 CrossRef.
- W. J. Yin, B. Weng, J. Ge, Q. Sun, Z. Li and Y. Yan, Energy Environ. Sci., 2019, 12, 442–462 RSC.
- A. Kumar, A. Kumar and V. Krishnan, ACS Catal., 2020, 10, 10253–10315 CrossRef CAS.
- Q. Ji, L. Bi, J. Zhang, H. Cao and X. S. Zhao, Energy Environ. Sci., 2020, 13, 1408–1428 RSC.
- J. Hwang, R. R. Rao, L. Giordano, Y. Katayama, Y. Yu and Y. Shao-Horn, Science, 2017, 358, 751–756 CrossRef CAS.
- J. P. Attfield, P. Lightfoot and R. E. Morris, Dalton Trans., 2015, 44, 10541–10542 RSC.
- V. M. Goldschmidt, Naturwissenschaften, 1926, 14, 477–485 CrossRef CAS.
- R. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- M. A. Peña and J. L. G. Fierro, Chem. Rev., 2001, 101, 1981–2018 CrossRef.
- T. Jia, Z. Zeng, X. Zhang, P. Ohodnicki, B. Chorpening, G. Hackett, J. Lekse and Y. Duan, Phys. Chem. Chem. Phys., 2019, 21, 20454–20462 RSC.
- M. Saiful Islam, J. Mater. Chem., 2000, 10, 1027–1038 RSC.
- X. Huang, G. Zhao, G. Wang and J. T. S. Irvine, Chem. Sci., 2018, 9, 3623–3637 RSC.
- S. Yoo, A. Jun, Y. W. Ju, D. Odkhuu, J. Hyodo, H. Y. Jeong, N. Park, J. Shin, T. Ishihara and G. Kim, Angew. Chem., Int. Ed., 2014, 53, 13064–13067 CrossRef CAS.
- D. Chen, C. Chen, Z. M. Baiyee, Z. Shao and F. Ciucci, Chem. Rev., 2015, 115, 9869–9921 CrossRef CAS.
- G. Gou, N. Charles, J. Shi and J. M. Rondinelli, Inorg. Chem., 2017, 56, 11854–11861 CrossRef CAS.
- R. Ceravola, J. Oró Solé, A. P. Black, C. Ritter, I. Puente Orench, I. Mata, E. Molins, C. Frontera and A. Fuertes, Dalton Trans., 2017, 46, 5128–5132 RSC.
- A. A. Emery and C. Wolverton, Sci. Data, 2017, 4, 170153 CrossRef CAS.
- G. Jácome-Acatitla, M. Álvarez-Lemus, R. López-González, C. García-Mendoza, A. Sánchez-López and D. Hernández-Acosta, J. Photochem. Photobiol., A, 2020, 390, 112330 CrossRef.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. Shao Horn, Nat. Chem., 2011, 3, 546–550 CrossRef CAS.
- M. Retuerto, A. G. Pereira, F. J. Pérez-Alonso, M. A. Peña, J. L. G. Fierro, J. A. Alonso, M. T. Fernández-Díaz, L. Pascual and S. Rojas, Appl. Catal., B, 2017, 203, 363–371 CrossRef CAS.
- Z. Du, P. Yang, L. Wang, Y. Lu, J. B. Goodenough, J. Zhang and D. Zhang, J. Power Sources, 2014, 265, 91–96 CrossRef CAS.
- T. Shao, H. You, Z. Zhai, T. Liu, M. Li and L. Zhang, Mater. Lett., 2017, 201, 122–124 CrossRef CAS.
- Z. Li, W. Zhang, H. Wang and B. Yang, Electrochim. Acta, 2017, 258, 561–570 CrossRef CAS.
- K. H. Ho and J. Wang, J. Am. Ceram. Soc., 2017, 100, 4629–4637 CrossRef CAS.
- T. Shao, H. You, Z. Zhai, T. Liu, M. Li and L. Zhang, Mater. Lett., 2017, 201, 122–124 CrossRef CAS.
- C. Sun, R. Hui and J. Roller, J. Solid State Electrochem., 2010, 14, 1125–1144 CrossRef CAS.
- Y. Zhang, J. Ding, W. Xu, M. Wang, R. Shao, Y. Sun and B. Lin, Chem. Eng. J., 2020, 386, 124030 CrossRef CAS.
- M. P. Harikrishnan and A. C. Bose, AIP Conf. Proc., 2019, 2082, 060001 CrossRef.
- S. Hussain, M. S. Javed, N. Ullah, A. Shaheen, N. Aslam, I. Ashraf, Y. Abbas, M. Wang, G. Liu and G. Qiao, Ceram. Int., 2019, 45, 15164–15170 CrossRef CAS.
- N. K. Patel, R. G. Utter, D. Das and M. Pecht, J. Power Sources, 2019, 438, 227040 CrossRef CAS.
- P. M. Wilde, T. J. Guther, R. Oesten and J. Garche, J. Electroanal. Chem., 1999, 461, 154–160 CrossRef CAS.
- M. Wohlfahrt-Mehrens, J. Schenk, P. M. Wilde, E. Abdelmula, P. Axmann and J. Garche, J. Power Sources, 2002, 105, 182–188 CrossRef CAS.
- F. Xiao, X. Zhang, F. Hu and J. Zhang, Mater. Chem. Phys., 2005, 94, 221–225 CrossRef CAS.
- G. George, S. L. Jackson, C. Q. Luo, D. Fang, D. Luo, D. Hu, J. Wen and Z. Luo, Ceram. Int., 2018, 44, 21982–21992 CrossRef CAS.
- R. B. Rakhi, W. Chen, D. Cha and H. N. Alshareef, Mater. Renewable Sustainable Energy, 2013, 2, 17 CrossRef.
- A. K. Tomar, G. Singh and R. K. Sharma, J. Power Sources, 2019, 426, 223–232 CrossRef CAS.
- Y. Zhu, X. Ji, Z. Wu, W. Song, H. Hou, Z. Wu, X. He, Q. Chen and C. E. Banks, J. Power Sources, 2014, 267, 888–900 CrossRef CAS.
- Q. Wang, Z. Li, M. A. Bañares, L. T. Weng, Q. Gu, J. Price, W. Han and K. L. Yeung, Small, 2019, 15, 1903525 CrossRef CAS.
- J. Chen, Z. He, G. Li, T. An, H. Shi and Y. Li, Appl. Catal., B, 2017, 209, 146–154 CrossRef CAS.
- Q. Hu, B. Yue, F. Yang, H. Shao, J. Wang, L. Ji, Y. Jia, Y. Wang and J. Liu, ChemistrySelect, 2019, 4, 11903–11912 CrossRef CAS.
- Q. Hu, B. Yue, H. Shao, F. Yang, J. Wang, Y. Wang and J. Liu, J. Mater. Sci., 2020, 55, 8421–8434 CrossRef CAS.
- Q. Pan, C. Yang, Q. Jia, W. Qi, H. Wei, M. Wang, S. Yang and B. Cao, Chem. Eng. J., 2020, 397, 125524 CrossRef CAS.
- S. Yin, Y. Wu, J. Chen, Z. Chen, H. Hou, Q. Liu, Y. Wang and W. Zhang, Funct. Mater. Lett., 2018, 11, 1850013 CrossRef CAS.
- C. D. Lokhande, T. P. Gujar, V. R. Shinde, R. S. Mane and S.-H. Han, Electrochem. Commun., 2007, 9, 1805–1809 CrossRef CAS.
- H.-Y. Kim, J. Shin, I.-C. Jang and Y.-W. Ju, Energies, 2020, 13, 36 CrossRef.
- N. Kitchamsetti, Y. R. Ma, P. M. Shirage and R. S. Devan, J. Alloys Compd., 2020, 833, 155134 CrossRef CAS.
- N. Kitchamsetti, R. J. Choudhary, D. M. Phase and R. S. Devan, RSC Adv., 2020, 10, 23446–23456 RSC.
- M. A. Peña and J. L. G. Fierro, Chem. Rev., 2001, 101, 1981–2018 CrossRef.
- B. C. Tofield and W. R. Scott, J. Solid State Chem., 1974, 10, 183–194 CrossRef CAS.
- T. Arakawa, N. Ohara and J. Shiokawa, J. Mater. Sci., 1986, 21, 1824–1827 CrossRef CAS.
- H. Wang, C. Tablet, A. Feldhoff and J. Caro, Adv. Mater., 2005, 17, 1785–1788 CrossRef CAS.
- Y. Teraoka, M. Yoshimatsu, N. Yamazoe and T. Seiyama, Chem. Lett., 1984, 893–896 CrossRef CAS.
- J. Mizusaki, S. Yamauchi, K. Fueki and A. Ishikawa, Solid State Ionics, 1984, 12, 119–124 CrossRef CAS.
- A. J. Hauser, R. E. Williams, R. A. Ricciardo, A. Genc, M. Dixit, J. M. Lucy, P. M. Woodward, H. L. Fraser and F. Yang, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 014407 CrossRef.
- W. Che, M. Wei, Z. Sang, Y. Ou, Y. Liu and J. Liu, J. Alloys Compd., 2018, 731, 381–388 CrossRef CAS.
- Y. Cao, B. Lin, Y. Sun, H. Yang and X. Zhang, J. Alloys Compd., 2015, 638, 204–213 CrossRef CAS.
- Z. A. Elsiddig, H. Xu, D. Wang, W. Zhang, X. Guo, Y. Zhang, Z. Sun and J. Chen, Electrochim. Acta, 2017, 253, 422–429 CrossRef CAS.
- J. T. Mefford, X. Rong, A. M. Abakumov, W. G. Hardin, S. Dai, A. M. Kolpak, K. P. Johnston and K. J. Stevenson, Nat. Commun., 2016, 7, 11053 CrossRef CAS.
- H. Mo, H. Nan, X. Lang, S. Liu, L. Qiao, X. Hu and H. Tian, Ceram. Int., 2018, 44, 9733–9741 CrossRef CAS.
- X. W. Wang, Q. Q. Zhu, X. E. Wang, H. C. Zhang, J. J. Zhang and L. F. Wang, J. Alloys Compd., 2016, 675, 195–200 CrossRef CAS.
- X. Lang, H. Mo, X. Hu and H. Tian, Dalton Trans., 2017, 46, 13720–13730 RSC.
- C. T. Alexander, J. T. Mefford, J. Saunders, R. P. Forslund, K. P. Johnston and K. J. Stevenson, ACS Appl. Mater. Interfaces, 2019, 11, 5084–5094 CrossRef CAS.
- P. P. Ma, Q. L. Lu, N. Lei, Y. K. Liu, B. Yu, J. M. Dai, S. H. Li and G. H. Jiang, Electrochim. Acta, 2020, 332, 135489 CrossRef CAS.
- A. Aguadero, D. Pérez-Coll, J. A. Alonso, S. J. Skinner and J. Kilner, Chem. Mater., 2012, 24, 2655–2663 CrossRef CAS.
- M. A. Salguero Salas, J. M. De Paoli, O. E. Linarez Pérez, N. Bajales and V. C. Fuertes, Microporous Mesoporous Mater., 2020, 293, 109797 CrossRef CAS.
- A. K. Tomar, G. Singh and R. K. Sharma, ChemSusChem, 2018, 11, 4123–4130 CrossRef CAS.
- L. Zhu, Y. Liu, C. Su, W. Zhou, M. Liu and Z. Shao, Angew. Chem., Int. Ed., 2016, 55, 9576–9579 CrossRef CAS.
- Y. Yan, T. Wang, X. Li, H. Pang and H. Xue, Inorg. Chem. Front., 2017, 4, 33–51 RSC.
- H. Xia, C. Hong, X. Shi, B. Li, G. Yuan, Q. Yao and J. Xie, J. Mater. Chem. A, 2015, 3, 1216–1221 RSC.
- X. Lang, X. Sun, Z. Liu, H. Nan, C. Li, X. Hu and H. Tian, Mater. Lett., 2019, 243, 34–37 CrossRef CAS.
- P. Liu, J. Liu, S. Cheng, W. Cai, F. Yu, Y. Zhang, P. Wu and M. Liu, Chem. Eng. J., 2017, 328, 1–10 CrossRef CAS.
- Y. Cao, B. Lin, Y. Sun, H. Yang and X. Zhang, Electrochim. Acta, 2015, 178, 398–406 CrossRef CAS.
- Z. S. Wu, W. Ren, D. W. Wang, F. Li, B. Liu and H. M. Cheng, ACS Nano, 2010, 4, 5835–5842 CrossRef CAS.
- H. Jiang, Y. Dai, Y. Hu, W. Chen and C. Li, ACS Sustainable Chem. Eng., 2014, 2, 70–74 CrossRef CAS.
- J. Lv, Y. Zhang, Z. Lv, X. Huang, Z. Wang, X. Zhu and B. Wei, Electrochim. Acta, 2016, 222, 1585–1591 CrossRef CAS.
- K. Chen and D. Xue, J. Colloid Interface Sci., 2015, 446, 77–83 CrossRef CAS.
- N. Maheswari and G. Muralidharan, Dalton Trans., 2016, 45, 14352–14362 RSC.
- S. Nagamuthu, S. Vijayakumar and K. S. Ryu, Mater. Chem. Phys., 2017, 199, 543–551 CrossRef CAS.
- X. Lang, H. Zhang, X. Xue, C. Li, X. Sun, Z. Liu, H. Nan, X. Hu and H. Tian, J. Power Sources, 2018, 402, 213–220 CrossRef CAS.
- M. D. Stoller, S. Park, Y. Zhu, J. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498–3502 CrossRef CAS.
- Y. Sun, Q. Wu and G. Shi, Energy Environ. Sci., 2011, 4, 1113–1132 RSC.
- J. Hu, L. Wang, L. Shi and H. Huang, J. Power Sources, 2014, 269, 144–151 CrossRef CAS.
- D. Moitra, C. Anand, B. K. Ghosh, M. Chandel and N. N. Ghosh, ACS Appl. Energy Mater., 2018, 1, 464–474 CrossRef CAS.
- A. Galal, H. K. Hassan, T. Jacob and N. F. Atta, Electrochim. Acta, 2018, 260, 738–747 CrossRef CAS.
- H. Pan, C. K. Poh, Y. P. Feng and J. Lin, Chem. Mater., 2007, 19, 6120–6125 CrossRef CAS.
- D. Hulicova, M. Kodama and H. Hatori, Chem. Mater., 2006, 18, 2318–2326 CrossRef CAS.
- A. Rezanezhad, E. Rezaie, L. S. Ghadimi, A. Hajalilou, E. Abouzari-Lotf and N. Arsalani, Electrochim. Acta, 2020, 335, 135699 CrossRef CAS.
- Z. A. Elsiddig, D. Wang, H. Xu, W. Zhang, T. Zhang, P. Zhang, W. Tian, Z. Sun and J. Chen, J. Alloys Compd., 2018, 740, 148–155 CrossRef CAS.
- P. M. Shafi, V. Ganesh and A. C. Bose, ACS Appl. Energy Mater., 2018, 1, 2802–2812 CrossRef CAS.
- S. Choi, S. Yoo, J. Kim, S. Park, A. Jun, S. Sengodan, J. Kim, J. Shin, H. Y. Jeong, Y. Choi, G. Kim and M. Liu, Sci. Rep., 2013, 3, 2426 CrossRef.
- A. Chroneos, B. Yildiz, A. Tarancón, D. Parfitt and J. A. Kilner, Energy Environ. Sci., 2011, 4, 2774–2789 RSC.
- B. P. Uberuaga and G. Pilania, Chem. Mater., 2015, 27, 5020–5026 CrossRef CAS.
- E. G. Steward and H. P. Rooksby, Acta Crystallogr., 1951, 4, 503–507 CrossRef CAS.
- J. Singh, A. Kumar and U. K. Goutam, Appl. Phys. A: Mater. Sci. Process., 2020, 126, 11 CrossRef CAS.
- J. Singh, U. K. Goutam and A. Kumar, Solid State Sci., 2019, 95, 105927 CrossRef CAS.
- A. Kumar, A. Kumar and A. Kumar, Solid State Sci., 2020, 105, 106252 CrossRef CAS.
- Z. Meng, J. Xu, P. Yu, X. Hu, Y. Wu, Q. Zhang, Y. Li, L. Qiao, Y. Zeng and H. Tian, Chem. Eng. J., 2020, 400, 15966 CrossRef.
- M. Alam, K. Karmakar, M. Pal and K. Mandal, RSC Adv., 2016, 6, 114722–114726 RSC.
- Y. Liu, Z. Wang, J. P. M. Veder, Z. Xu, Y. Zhong, W. Zhou, M. O. Tade, S. Wang and Z. Shao, Adv. Energy Mater., 2018, 8, 1702604 CrossRef.
- A. A. Taskin, A. N. Lavrov and Y. Ando, Appl. Phys. Lett., 2005, 86, 091910 CrossRef.
- C. Bernuy-Lopez, K. Høydalsvik, M. A. Einarsrud and T. Grande, Materials, 2016, 9, 154 CrossRef.
- Z. Wang, Y. Liu, Y. Chen, L. Yang, Y. Wang and M. Wei, J. Alloys Compd., 2019, 810, 151830 CrossRef CAS.
- Z. Xu, Y. Liu, W. Zhou, M. O. Tade and Z. Shao, ACS Appl. Mater. Interfaces, 2018, 10, 9415–9423 CrossRef CAS.
- A. K. Tomar, A. Joshi, S. Atri, G. Singh and R. K. Sharma, ACS Appl. Mater. Interfaces, 2020, 12, 15128–15137 CrossRef CAS.
- P. L. Wise, I. M. Reaney, W. E. Lee, T. J. Price, D. M. Iddles and D. S. Cannell, J. Eur. Ceram. Soc., 2001, 21, 2629–2632 CrossRef CAS.
- P. L. Wise, I. M. Reaney, W. E. Lee, T. J. Price, D. M. Iddles and D. S. Cannell, J. Eur. Ceram. Soc., 2001, 21, 1723–1726 CrossRef CAS.
- Z. Sang, W. Che, S. Yang and Y. Liu, Mater. Lett., 2018, 217, 23–26 CrossRef CAS.
- M. Wei, W. Che, H. Li, Z. Wang, F. Yan, Y. Liu and J. Liu, Appl. Surf. Sci., 2019, 484, 551–559 CrossRef CAS.
- H.-s. Nan, X.-y. Hu and H.-w. Tian, Mater. Sci. Semicond. Process., 2019, 94, 35–50 CrossRef CAS.
- C. A. Hancock, J. M. Porras-Vazquez, P. J. Keenan and P. R. Slater, Dalton Trans., 2015, 44, 10559–10569 RSC.
- J. F. Shin, A. Orera, D. C. Apperley and P. R. Slater, J. Mater. Chem., 2011, 21, 874–879 RSC.
- J. Ran, T. Wang, J. Zhang, Y. Liu, C. Xu, S. Xi and D. Gao, Chem. Mater., 2020, 32, 3439–3446 CrossRef CAS.
- X. Xu, Y. Pan, Y. Zhong, R. Ran and Z. Shao, Mater. Horiz., 2020, 7, 2519–2565 RSC.
- L. Zhu, R. Ran, M. Tadé, W. Wang and Z. Shao, Asia-Pac. J. Chem. Eng., 2016, 11, 338–369 CrossRef CAS.
Footnote |
| † Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |