
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStabilization effects in binary colloidal Cu and Ag nanoparticle electrodes under electrochemical CO2 reduction conditions†
Longfei
Wu
 a,
Kees E.
Kolmeijer
a,
Yue
Zhang
b,
Hongyu
An
a,
Sven
Arnouts
cd,
Sara
Bals
c,
Thomas
Altantzis
d,
Jan P.
Hofmann
e,
Marta
Costa Figueiredo
b,
Emiel J. M.
Hensen
b,
Bert M.
Weckhuysen
a and
Ward
van der Stam
*a
a,
Kees E.
Kolmeijer
a,
Yue
Zhang
b,
Hongyu
An
a,
Sven
Arnouts
cd,
Sara
Bals
c,
Thomas
Altantzis
d,
Jan P.
Hofmann
e,
Marta
Costa Figueiredo
b,
Emiel J. M.
Hensen
b,
Bert M.
Weckhuysen
a and
Ward
van der Stam
*a
aInorganic Chemistry and Catalysis group, Institute for Sustainable and Circular Chemistry, Utrecht University, 3584 CG Utrecht, The Netherlands. E-mail: w.vanderstam@uu.nl
bLaboratory for Inorganic Materials and Catalysis, Department of Chemical Engineering and Chemistry, Eindhoven University of Technology, 5600 MB Eindhoven, The Netherlands
cElectron Microscopy for Materials Research (EMAT), University of Antwerp, 2020 Antwerp, Belgium
dApplied Electrochemistry & Catalysis (ELCAT), University of Antwerp, 2610 Wilrijk, Belgium
eSurface Science Laboratory, Department of Materials and Earth Sciences, Technical University of Darmstadt, 64287 Darmstadt, Germany
First published on 22nd February 2021
Abstract
Nanoparticle modified electrodes constitute an attractive way to tailor-make efficient carbon dioxide (CO2) reduction catalysts. However, the restructuring and sintering processes of nanoparticles under electrochemical reaction conditions not only impedes the widespread application of nanoparticle catalysts, but also misleads the interpretation of the selectivity of the nanocatalysts. Here, we colloidally synthesized metallic copper (Cu) and silver (Ag) nanoparticles with a narrow size distribution (<10%) and utilized them in electrochemical CO2 reduction reactions. Monometallic Cu and Ag nanoparticle electrodes showed severe nanoparticle sintering already at low overpotential of −0.8 V vs. RHE, as evidenced by ex situ SEM investigations, and potential-dependent variations in product selectivity that resemble bulk Cu (14% for ethylene at −1.3 V vs. RHE) and Ag (69% for carbon monoxide at −1.0 V vs. RHE). However, by co-deposition of Cu and Ag nanoparticles, a nanoparticle stabilization effect was observed between Cu and Ag, and the sintering process was greatly suppressed at CO2 reducing potentials (−0.8 V vs. RHE). Furthermore, by varying the Cu/Ag nanoparticle ratio, the CO2 reduction reaction (CO2RR) selectivity towards methane (maximum of 20.6% for dense Cu2.5–Ag1 electrodes) and C2 products (maximum of 15.7% for dense Cu1–Ag1 electrodes) can be tuned, which is attributed to a synergistic effect between neighbouring Ag and Cu nanoparticles. We attribute the stabilization of the nanoparticles to the positive enthalpies of Cu–Ag solid solutions, which prevents the dissolution-redeposition induced particle growth under CO2RR conditions. The observed nanoparticle stabilization effect enables the design and fabrication of active CO2 reduction nanocatalysts with high durability.
Introduction
Converting carbon dioxide (CO2) into value-added chemicals or fuels with electricity has attracted extensive attention as it provides an alternative route to store intermittent renewable energy, such as solar and wind energy.1 Ever since the pioneering work of electrochemical CO2 reduction (CO2RR) on metal surfaces by Hori et al.,2 tremendous efforts have been devoted to optimizing CO2RR electrocatalysts with high selectivity, activity and stability.3 Among the various materials being investigated, copper (Cu) stands out as appealing catalyst as it produces a variety of products ranging from C1 (e.g. CH4, CO, formate) to C2+ hydrocarbons and alcohols (e.g. C2H4, C2H5OH, C3H7OH).4,5 The selectivity of Cu catalysts for specific products can be tuned by a variety of approaches, such as nanostructuring to increase the active surface area, shape or facet control for selective exposure of Cu(100) or Cu(111), and alloying with other elements to induce synergistic effects, among others.6–13 Although significant progress has been made, the challenge of finding stable catalysts with high selectivity and efficiency still remains.14,15Colloidally synthesized nanoparticles (NPs) act as ideal material platform to study the effect of size, shape (or exposed facet) and composition on the activity and selectivity towards electrochemical CO2RR.16 For instance, Strasser et al. studied the size effect of colloidal Cu NPs (diameter: 2 to 15 nm) during CO2RR.17 A dramatic increase of activity and selectivity towards H2 and CO with decreasing Cu particle size was observed at an applied potential of −1.1 V vs. RHE. Furthermore, tailor-made colloidal nanomaterials with exposed facets allow studying the facet-dependent structural selectivity of Cu NPs.5,8,18 Buonsanti et al. synthesized Cu nano-octahedra (with predominantly Cu(111) facets exposed) and nanocubes (with prevalent Cu(100) facets), which showed high selectivity to CH4 and C2 products (ethylene), respectively.19,20 It was also suggested that the stability of the colloidal Cu nanocubes and nano-octahedra was due to the large nanocrystal size (stable nanocrystals for diameter >40 nm), whereas smaller nanoparticles displayed severe nanoparticle sintering.19,20 In a later study, the colloidal spherical, cuboidal and octahedral Cu NPs were deposited on gas-diffusion electrodes (GDE) and tested at industrially viable current densities (100–300 mA cm−2).8 Cu octahedra and nanocubes were reported to reach a selectivity of up to 51% methane and 57% ethylene, respectively, which highlights the applicability of colloidal NPs in industrial-scale CO2 electrolyzers.8 However, the lifetime of the catalysts was only improved to several hours by changing the electrochemical H-cell to a flow cell, associated with the aforementioned stability issues of colloidal nanocatalysts, and many challenges remain to stabilize nanoparticles <25 nm under CO2RR conditions.8
Although colloidal chemistry enables selectivity and activity tuning of catalysts at the nanoscale, investigating and solving the stability issue is of the utmost importance for large-scale application of nanostructured catalysts. The degradation phenomenon of nanostructured electrocatalysts (size <25 nm) has been widely reported, and it is found that larger NPs appear during reaction, which is often attributed to sintering of the initial smaller nanoparticles, or to a dissolution-redeposition mechanism.21,22 For example, under electrochemical CO2RR conditions, NPs with an initial diameter of ∼7 nm have been reported to evolve into sizes above 20 nm within 10 min, and the authors ascribe such a morphology change to nanoparticle coalescence and a dissolution-redeposition mechanism.23 By following the morphology evolution of Cu nanocubes during CO2RR with ex situ TEM, it was found that the potential driven nanoclustering at −1.1 V vs. RHE is the predominant degradation pathway.24 In addition, densely packed Cu NP ensembles were reported to undergo structural transformation to electrocatalytically active nanocubes during CO2RR, which are selective for multicarbon products (ethylene, ethanol and n-propanol).25,26 Therefore, the active sites of NPs might be misinterpreted without correlating the selectivity to the dynamic structure of nanostructured electrocatalysts. This points to the importance of stabilizing NPs during catalysis, which not only facilitates deeper understanding of the catalyst active sites but also ensures long-term tests under industrially relevant operating conditions. Compared to the vast majority of efforts devoted in optimizing the catalyst's activity and selectivity, fewer attention has been paid to mitigation strategies towards enhanced durability.6,22
Here, we report the stabilization of colloidal binary Cu and Ag NP electrodes under CO2RR conditions (−0.8 V vs. RHE), which allows us to assess the selectivity of the nanostructured electrocatalysts at this overpotential. We developed synthesis protocols that result in colloidal Cu (9.4 ± 0.7 nm) and Ag (6.5 ± 0.6 nm) NPs with a narrow size distribution. Furthermore, structural analysis revealed that the nanoparticles are polyhedral Cu and Ag nanoparticles, with predominant fcc(111) facets. When the monometallic electrodes were used in CO2RR, severe restructuring was observed by ex situ SEM, even at moderate overpotential of −0.8 V vs. RHE. However, when Cu and Ag nanoparticles are evenly distributed on the glassy carbon substrates through electrophoresis or drop-casting of pre-mixed solutions, we find that the neighbouring Cu and Ag NPs retain their initial size and shape after CO2RR. We attribute the increased stability of the Cu and Ag NPs to the immiscibility of Cu and Ag that inhibits the dissolution-redeposition process of NPs.27–30 In other words, the Cu–Ag solid solutions have positive enthalpies and do not tend to form alloys under moderate CO2RR conditions.27–30 At higher overpotentials, alloyed clusters are observed in ex situ TEM measurements. We correlate the nanoparticle morphology evolution with the CO2RR selectivity, and find that the binary colloidal Cu and Ag nanoparticle electrodes have a maximum selectivity of 15.7% C2 products and 20.6% methane at moderate overpotential (−0.8 V vs. RHE). Furthermore, the selectivity can be easily tuned by the Cu/Ag nanoparticle ratio, which is attributed to a synergistic effect between the Ag and Cu NPs. The observed stabilization effect between binary colloidal Cu and Ag NPs provides an alternative way to improve the stability of CO2RR electrocatalysts.
Results and discussion
Cu NPs capped with tetradecylphosphonic acid (TDPA) and oleic acid were synthesized via a colloidal method adopted from a previous report.23 As shown in Fig. 1a, the NPs have a uniform size distribution of 9.4 ± 0.7 nm (see ESI Fig. S1† for the size histogram). The colloidal Cu NPs were dropcasted on ITO substrates inside a N2 filled glovebox, loaded into a dome to avoid oxidation of the NPs, and were further characterized by X-ray diffraction (XRD). The XRD pattern (Fig. S2†) indicates that the NPs have an fcc Cu lattice. Electron tomography series were acquired from a number of Cu NPs using high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM), in order to investigate their morphology. From the reconstructed volume, shown in Fig. S3,† it was revealed that the shape of the particles is close to spherical, although some faceting appears to be present.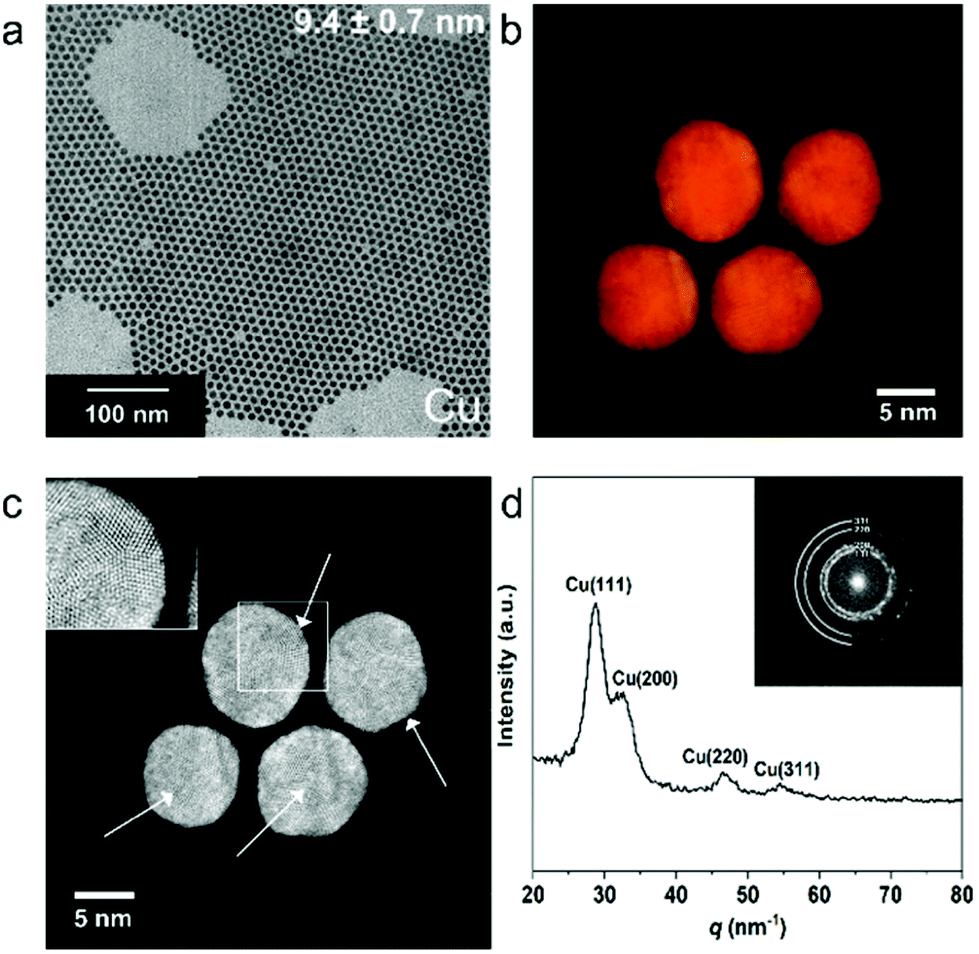 | ||
| Fig. 1 (a) Transmission electron microscopy (TEM) image of colloidal Cu NPs. (b) Visualization of the 3D atomic resolution reconstructions of four Cu NPs. (c) Atomic resolution HAADF-STEM image of four Cu NPs, showing the pentatwinned domains, indicated by white arrows. A magnified image of a region with a pentatwinned domain, indicated by the white rectangle, is shown as inset. (d) Fourier transform (FT) pattern (inset) obtained from the Cu NPs shown in (c) and its radially integrated 1D powder electron diffraction (PED) pattern. | ||
In order to investigate the faceting of the nanoparticles in more detail, High-Angle Annular Dark Field Scanning Transmission Electron Microscopy (HAADF-STEM) measurements were performed. From these high resolution HAADF-STEM images (Fig. S3†), it is clear that the NPs have several crystalline domains, of which the majority is pentatwinned in projection. These pentatwinned crystalline domains suggest a polyhedral shape with a dominant presence of Cu(111) surface facets. In order to verify this observation, atomic resolution electron tomography was performed for four Cu NPs.31,32 Visualization of the 3D reconstructions are shown in Fig. 1b and c respectively, and an animated version is presented in ESI Movie 1.† From these results it is clear that pentatwinned domains (inset of Fig. 1c) are indeed present in all the investigated single NPs, confirming that the (111) facets are dominant. The radially-integrated Fourier transform (FT) pattern (Fig. 1d) is in close agreement with the XRD pattern (Fig. S2†), confirming the Cu fcc lattice. From the HAADF-STEM images obtained for Cu nanoparticles loaded in a TEM holder inside a N2 filled glovebox, no (surface) copper oxide shell is observed, which is confirmed by energy dispersive X-rays spectroscopy (EDXS) mapping (Fig. S3†). However, HAADF-STEM images of Cu NPs after air exposure for 3 hours show severe oxidation on the surface (Fig. S4†), indicating that the NPs are prone to structural changes due to oxidation.
The colloidal Ag NPs were prepared in a similar way, ensuring that the decomposition of silver nitrate was performed at much lower temperature (120 °C for Ag compared to 180 °C for Cu, see ESI Fig. S5†). The as-synthesized colloidal Ag NPs had a narrow size distribution with an average size of 6.5 ± 0.6 nm (Fig. 2a, see ESI Fig. S6† for the size histogram). The Ag NP-glassy carbon electrodes were characterized by surface sensitive Grazing Incidence Diffraction (GID, angle of incidence 0.5°), which revealed an fcc Ag lattice and exposed Ag (111), (200) and (220) facets (Fig. S7†). The polycrystalline fcc lattice was confirmed by HAADF-STEM imaging (Fig. 2c and Fig. S8†) and the corresponding FT pattern (Fig. 2d), which revealed that the Ag NPs are less prone to surface oxidation compared to Cu NPs, since no crystalline surface oxide species are observed even after air exposure during sample transfer. This is further corroborated by EDXS mapping (Fig. S8†), where no oxide is observed. Furthermore, the Ag NPs also have several crystalline domains, with most of them being pentatwinned in projection as revealed by the high resolution HAADF-STEM images (Fig. 2c). However, in this case the acquisition of atomic resolution HAADF-STEM series at high magnification was not possible due to the sensitivity of the Ag NPs under prolonged electron beam irradiation.
 | ||
| Fig. 2 (a) TEM image of colloidal Ag NPs. (b) 3D visualization of the reconstructed volume of a number of Ag NPs. (c) High resolution HAADF-STEM image of Ag NPs, showing the polycrystallinity of the NPs and the presence of pentatwinned domains. A magnified image of a region with a pentatwinned domain is shown as inset. (d) Fourier transform (FT) pattern (inset) of Ag NPs and the radially integrated 1D Powder Electron Diffraction (PED) pattern. | ||
The colloidal NPs were deposited on glassy carbon substrates via a versatile electrophoresis deposition method (EPD).33 Due to the ligands (TPDA and oleic acid) on the surface of the NPs, the suspended colloidal NPs have an electric surface charge. As shown in Fig. 3a, by applying an electric field, the negatively charged NPs (due to the negatively charged ligands) could be easily deposited on the anode and positively charged NPs (due to partial ligand removal) on the cathode. Densely packed NP films on glassy carbon electrodes with varying composition and film thickness were obtained by EPD through optimization of the NP concentration, applied voltage, deposition time, and solvent.34
 | ||
| Fig. 3 (a) Schematic illustration of the film preparation via electrophoresis. The arrows indicate the moving direction of NPs under applied potential. (b and c) SEM images of the anode Cu (b) and Cu2.5–Ag1 (c) NPs film prepared at 100 V for 60 min. (d and e) Cu 2p (d) and Ag 3d (e) XPS spectra of a Cu2.5–Ag1 NPs film prepared by electrophoresis at 100 V for 60 min. | ||
A typical anode Cu NP film prepared by EPD is shown in the SEM images in Fig. 3b and Fig. S9,† which indicate that the NPs were uniformly distributed on the substrate. X-ray photoelectron spectroscopy (XPS) was performed on both the anode and cathode Cu NP films after EPD (Fig. S10†). We find that the NPs deposited on the cathode had a much higher oxide composition (Fig. S10†). This is explained by the partial removal of the negatively charged surface ligands at the cathode, which made the NPs more susceptible to surface oxidation. Therefore, the anode films were used for electrochemical CO2RR in the remainder of this work. In a similar approach, mixed Cu–Ag NP films were also prepared with the EPD method. Fig. 3c–e display the SEM image (Fig. 3c) and XPS spectra (Fig. 3d and e) of the binary Cu–Ag electrode, prepared by mixing Cu and Ag colloidal solutions in a 1 to 1 volume ratio. The XPS spectra indicate the presence of some copper oxide (13.6 at% oxygen) on the surface of Cu NPs, while only Ag0 is observed for the Ag NPs, consistent with the FT patterns above (Fig. 2d). Based on the ICP-OES analysis of the NP solutions (ESI,† Experimental details), the concentration of Cu and Ag NPs in the stock solution is determined to be 0.064 and 0.026 mol L−1, respectively. Therefore, the Cu to Ag atomic ratio of the mixed colloidal solution is 2.5. Finally, SEM-EDX analysis (Fig. S11†) indicates that the Cu and Ag NPs are well distributed across the film, and no phase separation is observed.
The EPD nanoparticle films served as working electrodes in a three-electrode system using Ag/AgCl and Pt foil as reference and counter electrodes (Fig. S12†), respectively.35 CO2RR selectivity measurements of pure Cu and mixed Cu–Ag films were performed at −0.8 V vs. RHE in CO2-saturated 0.1 M KHCO3 (pH = 6.8) aqueous electrolyte solution. Two monometallic Cu nanoparticle films were prepared by EPD at two different applied potentials (100 V and 200 V), hereafter denoted as Cu-thin (100 V, Fig. 4c) and Cu-thick (200 V, Fig. 4e), and one Cu–Ag NP electrode (Cu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ag ratio 2.5, 100 V, see Fig. 4g). As shown in Fig. 4a, the current density of the films over the course of an hour gradually decays, which is due to possible particle detachment or dissolution from the substrate.36,37 The initial drop in current density in the first 100 seconds after cathodic bias onset (Fig. S13†) is attributed to surface oxide removal.38,39 The FE reported in Fig. 4b suggests a clear difference among the tested films. All films display a large FE contribution of H2 (70–78%, Fig. S14†), which could be explained by the small NP size that is typically more favourable for H2 production.17 We find that for monometallic Cu NP films, methane FE is increased from 0 to 7.5% by increasing the EPD deposition potential, and hence the film thickness, while CO production is almost constant (∼6%). In addition, the production of formic acid is also suppressed from 12.5% to 8.7% for increasing film thickness. Both CO and formic acid are 2e− reduction products, but they follow distinct and separate reaction pathways.3 Unlike CO, formic acid is not typically involved in subsequent reduction reactions, which means that formic acid is the dead end for this particular CO2 reduction pathway.3 In other words, if the reduction pathway towards formic acid is suppressed, pathways towards CO and methane are increased, as observed here.3,40,41 Upon addition of Ag NPs, methane and CO production are further enhanced to 10.3 and 8.8% at −0.8 V vs. RHE, respectively, while FE of formic acid was further suppressed to 5.2%. We note that the overall FE for CO2RR products is increased from 19.2% (for Cu-thin) to 24.3% (for Cu–Ag). To gain insights into the FE selectivity of EPD films and its relation to possible restructuring and sintering of the nanoparticle electrodes, ex situ SEM measurements of the EPD films were performed prior to and after CO2RR. As indicated by red circles in Fig. 4d and f, a substantial part of the Cu NPs on the electrode surface have merged and grown into larger NPs due to sintering after CO2RR at −0.8 V vs. RHE was performed. Compared to the Cu-thin sample, Cu-thick has a much denser NP loading and the sintering process is enhanced after CO2RR (Fig. 4f). This suggests that NP sintering is more notable for NP electrodes with a higher density of nanoparticles.22 The severe sintering process in thick Cu NP electrodes might explain the FE increase of methane compared to Cu-thin, since the increase of Cu NP size was found to promote methane production.17 However, SEM images of the Cu–Ag NP electrode before and after CO2RR (Fig. 4h) displayed no apparent NP sintering, which indicates that the addition of Ag NPs mitigated the sintering process of Cu NPs. This is also confirmed by high resolution SEM and EDXS mapping measurements (Fig. S15 and S16†), which indicated that only a minor fraction of the nanoparticles in the binary electrodes increased in diameter, whereas a substantial part of the nanoparticles in the monometallic electrode displayed a drastic increase in size. The increase in CO FE of the Cu2.5–Ag1 electrode (8.8%) compared to the monometallic Cu electrode (5.2%) is attributed to the presence of stable Ag NPs, since Ag is known to be very selective toward CO production.42 At the same time, the FE for methane increased to 10.3%, which we attribute to the synergistic effect between Cu and Ag NPs, where CO2 is converted to CO over the Ag NPs, and subsequently further reduced to methane over the Cu NPs.43–48
:Ag ratio 2.5, 100 V, see Fig. 4g). As shown in Fig. 4a, the current density of the films over the course of an hour gradually decays, which is due to possible particle detachment or dissolution from the substrate.36,37 The initial drop in current density in the first 100 seconds after cathodic bias onset (Fig. S13†) is attributed to surface oxide removal.38,39 The FE reported in Fig. 4b suggests a clear difference among the tested films. All films display a large FE contribution of H2 (70–78%, Fig. S14†), which could be explained by the small NP size that is typically more favourable for H2 production.17 We find that for monometallic Cu NP films, methane FE is increased from 0 to 7.5% by increasing the EPD deposition potential, and hence the film thickness, while CO production is almost constant (∼6%). In addition, the production of formic acid is also suppressed from 12.5% to 8.7% for increasing film thickness. Both CO and formic acid are 2e− reduction products, but they follow distinct and separate reaction pathways.3 Unlike CO, formic acid is not typically involved in subsequent reduction reactions, which means that formic acid is the dead end for this particular CO2 reduction pathway.3 In other words, if the reduction pathway towards formic acid is suppressed, pathways towards CO and methane are increased, as observed here.3,40,41 Upon addition of Ag NPs, methane and CO production are further enhanced to 10.3 and 8.8% at −0.8 V vs. RHE, respectively, while FE of formic acid was further suppressed to 5.2%. We note that the overall FE for CO2RR products is increased from 19.2% (for Cu-thin) to 24.3% (for Cu–Ag). To gain insights into the FE selectivity of EPD films and its relation to possible restructuring and sintering of the nanoparticle electrodes, ex situ SEM measurements of the EPD films were performed prior to and after CO2RR. As indicated by red circles in Fig. 4d and f, a substantial part of the Cu NPs on the electrode surface have merged and grown into larger NPs due to sintering after CO2RR at −0.8 V vs. RHE was performed. Compared to the Cu-thin sample, Cu-thick has a much denser NP loading and the sintering process is enhanced after CO2RR (Fig. 4f). This suggests that NP sintering is more notable for NP electrodes with a higher density of nanoparticles.22 The severe sintering process in thick Cu NP electrodes might explain the FE increase of methane compared to Cu-thin, since the increase of Cu NP size was found to promote methane production.17 However, SEM images of the Cu–Ag NP electrode before and after CO2RR (Fig. 4h) displayed no apparent NP sintering, which indicates that the addition of Ag NPs mitigated the sintering process of Cu NPs. This is also confirmed by high resolution SEM and EDXS mapping measurements (Fig. S15 and S16†), which indicated that only a minor fraction of the nanoparticles in the binary electrodes increased in diameter, whereas a substantial part of the nanoparticles in the monometallic electrode displayed a drastic increase in size. The increase in CO FE of the Cu2.5–Ag1 electrode (8.8%) compared to the monometallic Cu electrode (5.2%) is attributed to the presence of stable Ag NPs, since Ag is known to be very selective toward CO production.42 At the same time, the FE for methane increased to 10.3%, which we attribute to the synergistic effect between Cu and Ag NPs, where CO2 is converted to CO over the Ag NPs, and subsequently further reduced to methane over the Cu NPs.43–48
 | ||
| Fig. 4 (a) Chronoamperometry curves of Cu NP and Cu2.5–Ag1 NP films recorded at −0.8 V vs. RHE in CO2 saturated 0.1 M KHCO3 aqueous electrolyte solution. (b) Faradaic efficiency of Cu NP films for CO2RR at −0.8 V vs. RHE in CO2 saturated 0.1 M KHCO3. (c–h) SEM images of Cu-thin (c and d), Cu-thick (e and f) and Cu2.5–Ag1 (g and h) before (c, e and g) and after (d, f and h) CO2RR at −0.8 V vs. RHE for 1 h. Red circles highlight the sintered nanoparticles after CO2RR. | ||
The overall FE of carbon-containing products for CO2RR on Cu-thin, Cu-thick and Cu2.5–Ag1 electrodes are 19.2, 21.4 and 24.3%, respectively. We suggest that the Ag NPs facilitated tandem catalysis at neighbouring Cu NPs (CO spillover or sequential adsorption on Ag and subsequent CO conversion to methane on Cu), which results in higher methane selectivity for the binary Cu–Ag electrodes compared to the monometallic electrodes.3,12,45,49 Furthermore, it has been demonstrated that copper-silver binary systems exhibit positive enthalpy of mixing in both solid and liquid states, which means that copper and silver are immiscible with each other in the absence of external stimuli.27 Cu–Ag alloyed phases, either in the form of bulk films or nanoparticles, are experimentally found to show phase separation during the preparation procedure,28–30 suggesting that fcc Cu and Ag are indeed immiscible phases and do not form solid solutions. Based on the CO2RR activity and ex situ SEM measurements of the different EPD films, we suggest that there is a stabilization effect between Cu and Ag NPs that mitigates the sintering process of small monometallic NPs (diameter <10 nm) during electrocatalytic reduction reactions. Moreover, by tuning the NP composition of the binary electrodes, the CO2RR selectivity can be adjusted accordingly.
To further explore the synergistic and stabilization effect between Cu and Ag NPs towards CO2RR, we have prepared a series of dense Cu–Ag NP films with varying Cu/Ag ratio by electrophoresis at 200 V for 60 min, namely Cu2.5–Ag1_thick, Cu1–Ag1_thick and Cu1–Ag2.5_thick. As shown in Fig. 5a, the methane production is improved to ∼20% for the dense Cu2.5–Ag1_thick sample. However, the methane FE decreases to ∼9% with higher Ag content (Cu1–Ag2.5_thick). In addition, the FE for C2 products for the Cu1–Ag1_thick sample (15.7% FE) is slightly higher than for both the Cu2.5–Ag1_thick (13.3% FE) and Cu1–Ag2.5_thick (12.0% FE) sample. These results show that the selectivity of the binary nanoparticle electrodes can be easily tuned, and that methane production is favoured with higher Cu/Ag ratios, while a Cu/Ag ratio of 1 results in enhanced C2 products formation. Furthermore, the stabilization effect is well pronounced for the NP electrodes with different Cu/Ag ratios, although there is more notable sintering in Cu2.5–Ag1_thick compared to other samples (Fig. 5c–h and Fig. S17†). The slight increase in sintering for the Cu2.5–Ag1_thick electrode compared to the binary electrode displayed in Fig. 3h can be ascribed to the NP density, since dense NP films are more prone to sintering (compare Cu_thin to Cu_thick, Fig. 3d and f).
 | ||
| Fig. 5 (a and b) Faradaic efficiency of dense Cu–Ag NP films with varying Cu/Ag ratios prepared by electrophoresis deposition. CO2RR experiments were performed at −0.8 V vs. RHE in CO2 saturated 0.1 M KHCO3 electrolyte solution (pH = 6.8). (c–h) SEM images of Cu2.5–Ag1_thick (c and d), Cu1–Ag1_thick (e and f) and Cu1–Ag2.5_thick (g and h) before (c, e and g) and after (d, f and h) CO2RR at −0.8 V vs. RHE for 1 h. Red circles highlight the sintered nanoparticles after CO2RR. | ||
As discussed above, the EPD film preparation method provides a versatile approach to prepare homogeneous NP electrodes with varying thickness, nanoparticle density and composition, resulting in stable binary Cu–Ag NP electrodes at −0.8 V vs. RHE. Furthermore, we have shown that the immiscibility of Cu and Ag results in stable NP electrodes, which suggests that Cu and Ag need to be homogeneously mixed in order to utilize the stabilization effect.
To study the stabilization of binary Cu–Ag electrodes further, we prepared mixed electrodes via another deposition method (drop-casting) and performed CO2RR. Drop-casting typically results in less homogeneous films, which allows us to verify whether indeed neighbouring Cu and Ag NPs are prone to sintering, whereas homogeneous mixing of Cu and Ag NPs results in stable NP electrodes. Fig. 6a presents the FE of drop-casted Cu NP films tested at various potentials. At relatively low overpotentials (−0.8, −1.0 V vs. RHE), the majority (>90%) product is H2 and the FE of CO is around 1–2%. Ex situ SEM images reveal a clear NP coalescence phenomenon at low overpotentials of −0.8 and −1.0 V vs. RHE (Fig. 6c and d), confirming the instability of monometallic Cu NPs under CO2RR conditions as observed for the EPD samples. The production of hydrocarbons (C2H4, C2H6) on monometallic Cu NP electrodes has an onset potential of −1.1 V vs. RHE, and the FE of ethylene reaches a maximum of 14% at −1.3 V vs. RHE, similar to bulk Cu foil.50,51 By comparing the morphology of a pristine Cu NP film (Fig. 6c) and that tested at −1.2 V vs. RHE (Fig. S18†), we observed severe particle migration, aggregation/sintering and detachment from the substrate, in line with the dissolution-redeposition mechanism.27 Therefore, we attribute the production of hydrocarbons at high overpotentials to the coalescence of Cu NPs, which thus not reflects the selectivity of colloidal Cu NPs. As comparison, we also looked into the selectivity (Fig. S19†) and stability (Fig. S20 and S21†) of Ag NPs at different applied potentials. As shown in Fig. 6e and f, Ag NPs evolved into dendritic structures52 already at applied potentials of −0.8 V vs. RHE, with a CO FE of ∼50% (Fig. S19†). At higher applied potentials (−1.0 and −1.2 V vs. RHE), the CO FE even increases to ∼70%. In contrast, the sintering of NPs is not observed for a binary Cu–Ag (molar ratio Cu/Ag = 1) electrode at −0.8 V vs. RHE (Fig. 6g), and SEM-EDX mapping displayed a relatively uniform distribution of Cu and Ag (Fig. S22†). The stabilization of binary Cu–Ag NP electrodes and the sintering of monometallic Cu and Ag NP electrodes is further corroborated by Electrochemical Surface Area (ECSA) measurements, which revealed a much higher ECSA for stabilized NP electrodes after CO2RR compared to the sintered NP electrodes (Fig. S23†).36 It should be noted that the ECSA of the sample after CO2RR increased compared to the pristine electrodes, which is ascribed to the fact that the hydrophobic ligands on the nanoparticle surface electro-desorbed under applied negative potentials (CO2RR conditions).16,53 However, phase segregation of Cu and Ag nanoparticles on the nanoparticle electrodes occurred at higher applied potential (−1.0 V vs. RHE), and it was clearly visible that the Cu-rich region underwent severe coalescence (Fig. S22†). It could be possible that the small amount of ethylene (1.3%) at −1.0 V vs. RHE (Fig. 6b) originates from the sintered Cu-rich region on the binary Cu–Ag electrodes.26 After further increasing the overpotential to −1.2 V vs. RHE, coalescence happens across the entire binary Cu1.0Ag1.0 film (Fig. S22†). From these experiments we conclude that drop-casted Cu–Ag binary electrodes still result in Cu-rich and Ag-rich regions, resulting in dissolution-redeposition and growth of larger nanostructures under CO2RR conditions. Therefore, the stabilization and synergistic effect between Cu and Ag NPs are more pronounced in samples prepared by EPD, which resulted in a homogeneous distribution of Cu and Ag NPs, thereby explaining the observed selectivity difference among samples prepared by the two methods.
 | ||
| Fig. 6 (a and b) Faradaic efficiency of drop-casted Cu (a) and Cu1.0_Ag1.0 (b) NP electrodes for CO2RR at various potentials for 1 h in CO2 saturated 0.1 M KHCO3. (c and d) SEM images of drop-casted Cu NP films after CO2RR at −0.8 V (c) and −1.0 V (d) vs. RHE for 1 h. (e and f) SEM images of drop-casted Ag NP films after CO2RR at −0.8 V (e) and −1.0 V (f) vs. RHE for 1 h. (g and h) SEM images of Cu1.0_Ag1.0 films after CO2RR at −0.8 V (g) and −1.0 V (h) vs. RHE for 1 h. | ||
To gain further insights into the nanostructures formed under different CO2RR conditions, the drop-casted NP films after CO2RR are scratched and deposited on ultrathin carbon coated Au grids for ex situ TEM characterization. Fig. 7a–c show the HAADF-STEM and EDXS maps of a binary Cu1.0_Ag1.0 film after CO2RR at −0.8 V vs. RHE for 1 h, which indicates a homogeneous distribution of Cu and Ag in the mixture under this mild overpotential. HAADF-STEM images of another region (Fig. S25†) show that individual NPs could be observed all over the grid, confirming the NP stabilization effect for binary Cu–Ag nanoparticle electrodes. When the cathodic bias is increased to – 1.0 V vs. RHE, a clear separation of Cu- and Ag-rich regions can be observed (Fig. 7d–g). High magnification HAADF-STEM images of individual particles after CO2RR at −1.0 V vs. RHE for 1 h (Fig. 7h, i and Fig. S26†) show a thick surface oxidation layer on Cu NPs (Fig. 7h), while Ag NPs are preserved in the metallic state (Fig. 7i). The resistance of Ag toward surface oxidation after CO2RR can be explained by the Ag-H2O Pourbaix diagram, which shows that metallic Ag is the dominant phase over a wide potential-pH range.54 For the oxide layer on Cu NPs, it has been reported that Cu NPs undergo a dissolution-redeposition process during CO2RR and the first step of this process is the spontaneous oxidation of NP surface upon contact with the electrolyte.55 Such a dissolution-redeposition process leads to the growth of Cu NPs during cell operation at sufficiently cathodic bias.55 However, we find that the growth of Cu NPs according to this dissolution-redeposition mechanism is alleviated by mixing with Ag NPs. A possible explanation is that under mild CO2RR conditions, deposition of dissolved copper species on Ag NPs is much more difficult than deposition on Cu NPs, because of the immiscibility between Cu and Ag.27,56,57 Therefore, redeposition of dissolved species likely results in similar Cu and Ag NP morphologies as in the pristine binary Cu–Ag electrodes. We also characterized the Cu–Ag binary electrode after CO2RR at high cathodic potential (−1.2 V vs. RHE, Fig. S27†), and a clear phase separation of Cu- and Ag-rich regions is observed in the ex situ STEM-EDX measurements. Furthermore, the NPs have merged into larger clusters, in which metallic Ag and oxidized Cu domains can be discerned. The Ag-rich region has a similar dendritic structure to that of monometallic Ag NP electrodes (Fig. 6e and f), which indicates that the high cathodic potential drives the phase separation and results in destabilization of the binary Cu–Ag system.
 | ||
| Fig. 7 (a) HAADF-STEM image and the corresponding EDXS elemental maps of (b) Cu and (c) Ag for Cu–Ag NP electrodes after CO2RR at −0.8 V vs. RHE for 1 h. (d) HAADF-STEM image and the corresponding EDXS elemental maps of (e) Cu, (f) Ag and (g) Cu and Ag for Cu–Ag NP electrodes after CO2RR at −1.0 V vs. RHE for 1 h. (h) High magnification HAADF-STEM image of (h) individual Cu@copper oxide and (i) Ag particles of Cu–Ag NP electrodes after CO2RR at −1.0 V vs. RHE for 1 h. | ||
As evidenced by the selectivity and stability measurements for the nanoparticle electrodes prepared by both electrophoresis and drop-casting, homogeneous co-deposition of Cu and Ag NPs results in stabilization of nanoparticles under moderate electrochemical CO2RR conditions. Fig. 8 presents a schematic summary of the structural evolution of nanoparticle electrodes under mild CO2RR conditions (−0.8 V vs. RHE). First, monometallic Cu NPs undergo severe sintering and result in larger nanoparticle sizes after CO2RR (Fig. 8a), favouring ethylene production at larger overpotentials (<−1.0 V vs. RHE). Second, we have found that dendritic assemblies of Ag NPs are formed during electrocatalytic CO2RR at moderate overpotential (−0.8 V vs. RHE). These dendritic electrodes are selective for CO, as often observed for Ag electrodes in CO2RR. Third, by homogeneously mixing Cu and Ag NPs through electrophoresis, the NPs are stable under CO2RR conditions (up to −0.8 V vs. RHE), resulting in a synergistic effect between Cu and Ag NPs. We attribute the stabilization of the NPs to the immiscibility of fcc Cu and Ag. Although they both have an fcc structure, their lattice constants differ substantially (3.6 and 4.1 Å for Cu and Ag, respectively), which would result in severe strain for Cu–Ag solid solutions. Furthermore, both experimental and theoretical studies have shown that copper–silver mixtures have positive enthalpies and are therefore immiscible.30,58 Additionally, by introducing Ag NPs into the Cu NP electrodes prepared by electrophoresis, the CO2RR selectivity towards methane and C2 products can be adjusted, which allowed us to evaluate the synergistic effect between Cu and Ag NPs (maximum FE of 20.6% methane and 15.7% C2 products, respectively). The stabilization effect as well as the tunable selectivity of Cu–Ag mixed nanoparticle electrodes provides an alternative way of preparing stable nanocatalysts towards electrochemical production of valuable base chemicals.59
 | ||
| Fig. 8 Schematic illustration of the structural evolution of (a) Cu, (b) Ag and (c) Cu–Ag NP electrodes under electrochemical CO2RR conditions (−0.8 V vs. RHE), and the observed stabilization effect in binary Cu–Ag nanoparticle electrodes. | ||
Conclusions
In this work, synthesis protocols for colloidal Cu and Ag nanoparticles with narrow size distributions were developed, and these nanoparticles were applied in electrochemical CO2 reduction reaction (CO2RR). HAADF-STEM characterization revealed that the Cu and Ag nanoparticles have pentatwinned domains, which shows that the nanoparticles are polyhedral, and predominantly consist of fcc(111) facets. A versatile electrophoresis deposition (EPD) method was utilized to fabricate homogeneous mono- and bimetallic nanoparticle films. Monometallic Cu and Ag nanoparticle electrodes were found to be very unstable, even at moderate overpotential of −0.8 V vs. RHE, resulting in larger Cu and Ag structures with activities similar to bulk Cu and Ag electrodes. When Cu and Ag nanoparticles were homogeneously mixed, a stabilization effect was observed, and the Cu and Ag nanoparticles retained their shape at −0.8 V vs. RHE applied potential. The stabilization of the binary Cu and Ag NP electrodes under CO2RR conditions allowed us to tune the CO2RR selectivity, resulting in a maximum of 20% FE for methane and 15.7% FE for C2 products at −0.8 V vs. RHE. The stabilization effect in binary Cu–Ag nanoparticle electrodes was further confirmed for drop-casted nanoparticle films. We attribute the stabilization effect to the immiscibility of the Cu and Ag lattices, which hinders the dissolution-redeposition nanoparticle growth process of Cu during CO2RR. The observed stabilization effect between the Cu and Ag nanoparticles provides an efficient approach to prepare CO2RR catalysts with high durability and study the synergistic effect of colloidal nanoparticles.Author contributions
L. W. and W. v. d. S. conceived the idea, L. W. performed the SEM analysis, XRD characterization and activity measurements, and wrote the manuscript under the supervision of W. v. d. S., K. E. K. synthesized the nanoparticles and prepared the electrodes via dropcasting. L. W. and Y. Z. prepared the electrophoresis deposition electrodes, and Y. Z. performed XPS characterization under the supervision of J. P. H., M. C. F. and E. J. M. H. H. A. performed the activity measurements together with L. W., under the supervision of B. M. W. and W. v. d. S., S. A. performed the ex situ HAADF-STEM measurements, under the supervision of S. B. and T. A. All authors have given feedback on the manuscript and agree on its contents.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is funded by the Strategic UU-TU/e Alliance project ‘Joint Centre for Chemergy Research’ (budget holder B. M. W.). S. B. acknowledges support from the European Research Council (ERC Consolidator Grant #815128 REALNANO). S. A. and T. A. acknowledge funding from the University of Antwerp Research fund (BOF). We thank Eric Hellebrand (Faculty of Geosciences, Utrecht University) for the assistance in SEM measurements. Dr Ramon Oord (ARC Chemical Building Blocks Consortium, Faculty of Science, Utrecht University) is acknowledged for assisting with the grazing incidence XRD measurements. We thank Shuang Yang (Inorganic Chemistry and Catalysis group, Utrecht University) for performing ICP analysis and NMR measurements.References
- M. G. Kibria, J. P. Edwards, C. M. Gabardo, C. T. Dinh, A. Seifitokaldani, D. Sinton and E. H. Sargent, Adv. Mater., 2019, 31, 1807166 CrossRef.
- Y. Hori, K. Kikuchi and S. Suzuki, Chem. Lett., 1985, 14, 1695–1698 CrossRef.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Norskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS.
- F. P. G. de Arquer, C.-T. Dinh, A. Ozden, J. Wicks, C. McCallum, A. R. Kirmani, D.-H. Nam, C. Gabardo, A. Seifitokaldani, X. Wang, Y. C. Li, F. Li, J. Edwards, L. J. Richter, S. J. Thorpe, D. Sinton and E. H. Sargent, Science, 2020, 367, 661–666 CrossRef.
- Y. Wang, Z. Wang, C.-T. Dinh, J. Li, A. Ozden, M. G. Kibria, A. Seifitokaldani, C.-S. Tan, C. M. Gabardo, M. Luo, H. Zhou, F. Li, Y. Lum, C. McCallum, Y. Xu, M. Liu, A. Proppe, A. Johnston, P. Todorovic, T.-T. Zhuang, D. Sinton, S. O. Kelley and E. H. Sargent, Nat. Catal., 2019, 3, 98–106 CrossRef.
- S. Popovic, M. Smiljanic, P. Jovanovic, J. Vavra, R. Buonsanti and N. Hodnik, Angew. Chem., 2020, 59, 14736–14746 CrossRef CAS.
- R. Kas, R. Kortlever, A. Milbrat, M. T. Koper, G. Mul and J. Baltrusaitis, Phys. Chem. Chem. Phys., 2014, 16, 12194–12201 RSC.
- G. L. De Gregorio, T. Burdyny, A. Loiudice, P. Iyengar, W. A. Smith and R. Buonsanti, ACS Catal., 2020, 10, 4854–4862 CrossRef CAS.
- D. Gao, I. Zegkinoglou, N. J. Divins, F. Scholten, I. Sinev, P. Grosse and B. Roldan Cuenya, ACS Nano, 2017, 11, 4825–4831 CrossRef CAS.
- M. B. Ross, C. T. Dinh, Y. Li, D. Kim, P. De Luna, E. H. Sargent and P. Yang, J. Am. Chem. Soc., 2017, 139, 9359–9363 CrossRef CAS.
- P. Wang, M. Qiao, Q. Shao, Y. Pi, X. Zhu, Y. Li and X. Huang, Nat. Commun., 2018, 9, 4933 CrossRef.
- D. Kim, J. Resasco, Y. Yu, A. M. Asiri and P. Yang, Nat. Commun., 2014, 5, 4948 CrossRef CAS.
- D. Kim, C. Xie, N. Becknell, Y. Yu, M. Karamad, K. Chan, E. J. Crumlin, J. K. Norskov and P. Yang, J. Am. Chem. Soc., 2017, 139, 8329–8336 CrossRef CAS.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- D. Gao, R. M. Arán-Ais, H. S. Jeon and B. Roldan Cuenya, Nat. Catal., 2019, 2, 198–210 CrossRef CAS.
- J. Huang and R. Buonsanti, Chem. Mater., 2018, 31, 13–25 CrossRef.
- R. Reske, H. Mistry, F. Behafarid, B. Roldan Cuenya and P. Strasser, J. Am. Chem. Soc., 2014, 136, 6978–6986 CrossRef CAS.
- W. Luc, X. Fu, J. Shi, J.-J. Lv, M. Jouny, B. H. Ko, Y. Xu, Q. Tu, X. Hu, J. Wu, Q. Yue, Y. Liu, F. Jiao and Y. Kang, Nat. Catal., 2019, 2, 423–430 CrossRef CAS.
- P. Iyengar, J. Huang, G. L. De Gregorio, C. Gadiyar and R. Buonsanti, Chem. Commun., 2019, 55, 8796–8799 RSC.
- A. Loiudice, P. Lobaccaro, E. A. Kamali, T. Thao, B. H. Huang, J. W. Ager and R. Buonsanti, Angew. Chem., Int. Ed., 2016, 55, 5789–5792 CrossRef CAS.
- C. Spori, J. T. H. Kwan, A. Bonakdarpour, D. P. Wilkinson and P. Strasser, Angew. Chem., Int. Ed., 2017, 56, 5994–6021 CrossRef.
- E. D. Goodman, J. A. Schwalbe and M. Cargnello, ACS Catal., 2017, 7, 7156–7173 CrossRef CAS.
- K. Manthiram, B. J. Beberwyck and A. P. Alivisatos, J. Am. Chem. Soc., 2014, 136, 13319–13325 CrossRef CAS.
- J. Huang, N. Hormann, E. Oveisi, A. Loiudice, G. L. De Gregorio, O. Andreussi, N. Marzari and R. Buonsanti, Nat. Commun., 2018, 9, 3117 CrossRef.
- D. Kim, C. S. Kley, Y. Li and P. Yang, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 10560–10565 CrossRef CAS.
- Y. Li, D. Kim, S. Louisia, C. Xie, Q. Kong, S. Yu, T. Lin, S. Aloni, S. C. Fakra and P. Yang, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 9194–9201 CrossRef CAS.
- P. R. Subramanian and J. H. Perepezko, J. Phase Equilib., 1993, 14, 62–75 CrossRef CAS.
- G. Radnóczi, E. Bokányi, Z. Erdélyi and F. Misják, Acta Mater., 2017, 123, 82–89 CrossRef.
- V. Elofsson, G. A. Almyras, B. Lü, R. D. Boyd and K. Sarakinos, Acta Mater., 2016, 110, 114–121 CrossRef CAS.
- S. Nag, K. C. Mahdak, A. Devaraj, S. Gohil, P. Ayyub and R. Banerjee, J. Mater. Sci., 2009, 44, 3393–3401 CrossRef CAS.
- T. Milagres de Oliveira, W. Albrecht, G. Gonzalez-Rubio, T. Altantzis, I. P. Lobato Hoyos, A. Beche, S. Van Aert, A. Guerrero-Martinez, L. M. Liz-Marzan and S. Bals, ACS Nano, 2020, 14, 12558–12570 CrossRef CAS.
- T. Altantzis, I. Lobato, A. De Backer, A. Beche, Y. Zhang, S. Basak, M. Porcu, Q. Xu, A. Sanchez-Iglesias, L. M. Liz-Marzan, G. Van Tendeloo, S. Van Aert and S. Bals, Nano Lett., 2019, 19, 477–481 CrossRef CAS.
- S. Whan Lee, D. Zhang and I. P. Herman, Appl. Phys. Lett., 2014, 104, 053113 CrossRef.
- L. Ye, K. Wen, Z. Zhang, F. Yang, Y. Liang, W. Lv, Y. Lin, J. Gu, J. H. Dickerson and W. He, Adv. Energy Mater., 2016, 6, 1502018 CrossRef.
- M. Ma, K. Djanashvili and W. A. Smith, Angew. Chem., Int. Ed., 2016, 55, 6680–6684 CrossRef CAS.
- R. Kas, K. Yang, D. Bohra, R. Kortlever, T. Burdyny and W. A. Smith, Chem. Sci., 2020, 11, 1738–1749 RSC.
- H. Yang, Q. Lin, C. Zhang, X. Yu, Z. Cheng, G. Li, Q. Hu, X. Ren, Q. Zhang, J. Liu and C. He, Nat. Commun., 2020, 11, 593 CrossRef CAS.
- Q. Zhu, X. Sun, D. Yang, J. Ma, X. Kang, L. Zheng, J. Zhang, Z. Wu and B. Han, Nat. Commun., 2019, 10, 3851 CrossRef.
- L. Mandal, K. R. Yang, M. R. Motapothula, D. Ren, P. Lobaccaro, A. Patra, M. Sherburne, V. S. Batista, B. S. Yeo, J. W. Ager, J. Martin and T. Venkatesan, ACS Appl. Mater. Interfaces, 2018, 10, 8574–8584 CrossRef CAS.
- R. Kortlever, J. Shen, K. J. Schouten, F. Calle-Vallejo and M. T. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS.
- K. J. P. Schouten, Y. Kwon, C. J. M. van der Ham, Z. Qin and M. T. M. Koper, Chem. Sci., 2011, 2, 1902–1909 RSC.
- M. Ma, B. J. Trzesniewski, J. Xie and W. A. Smith, Angew. Chem., Int. Ed., 2016, 55, 9748–9752 CrossRef CAS.
- E. L. Clark, C. Hahn, T. F. Jaramillo and A. T. Bell, J. Am. Chem. Soc., 2017, 139, 15848–15857 CrossRef CAS.
- P. B. O'Mara, P. Wilde, T. M. Benedetti, C. Andronescu, S. Cheong, J. J. Gooding, R. D. Tilley and W. Schuhmann, J. Am. Chem. Soc., 2019, 141, 14093–14097 CrossRef.
- Gurudayal, D. Perone, S. Malani, Y. Lum, S. Haussener and J. W. Ager, ACS Appl. Energy Mater., 2019, 2, 4551–4559 CrossRef CAS.
- S. Lee, G. Park and J. Lee, ACS Catal., 2017, 7, 8594–8604 CrossRef CAS.
- H. S. Jeon, J. Timoshenko, F. Scholten, I. Sinev, A. Herzog, F. T. Haase and B. Roldan Cuenya, J. Am. Chem. Soc., 2019, 141, 19879–19887 CrossRef CAS.
- K. Liu, M. Ma, L. Wu, M. Valenti, D. Cardenas-Morcoso, J. P. Hofmann, J. Bisquert, S. Gimenez and W. A. Smith, ACS Appl. Mater. Interfaces, 2019, 11, 16546–16555 CrossRef CAS.
- J. Huang, M. Mensi, E. Oveisi, V. Mantella and R. Buonsanti, J. Am. Chem. Soc., 2019, 141, 2490–2499 CrossRef CAS.
- Y. Hori, in Modern Aspects of Electrochemistry, ed. C. Vayenas, R. White and M. Gamboa-Aldeco, Springer, New York, 2008, vol. 42, pp. 89–189 Search PubMed.
- Y. Hori, A. Murata and R. Takahashi, J. Chem. Soc., Faraday Trans., 1989, 85, 2309–2326 RSC.
- K. Manthiram, Y. Surendranath and A. P. Alivisatos, J. Am. Chem. Soc., 2014, 136, 7237–7240 CrossRef CAS.
- D. Li, C. Wang, D. Tripkovic, S. Sun, N. M. Markovic and V. R. Stamenkovic, ACS Catal., 2012, 2, 1358–1362 CrossRef CAS.
- W. Wei, X. Mao, L. A. Ortiz and D. R. Sadoway, J. Mater. Chem., 2011, 21, 432–438 RSC.
- J. Vavra, T. H. Shen, D. Stoian, V. Tileli and R. Buonsanti, Angew. Chem., 2021, 60, 1347–1354 CrossRef CAS.
- N. Okamoto, F. Wang and T. Watanabe, Mater. Trans., 2004, 45, 3330–3333 CrossRef CAS.
- E. Ma, Prog. Mater. Sci., 2005, 50, 413–509 CrossRef CAS.
- J. L. Murray, Metall. Trans. A, 1984, 15, 261–268 CrossRef.
- F. Sastre, M. J. Muñoz-Batista, A. Kubacka, M. Fernández-García, W. A. Smith, F. Kapteijn, M. Makkee and J. Gascon, ChemElectroChem, 2016, 3, 1497–1502 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr09040a |
| This journal is © The Royal Society of Chemistry 2021 |