
Gradient copolymer prepared from alternating ring-opening metathesis of three monomers†
Francis O.
Boadi
 and
Nicole S.
Sampson
*
and
Nicole S.
Sampson
*
Department of Chemistry, Stony Brook University, Stony Brook, New York 11794-3400, USA. E-mail: Nicole.sampson@stonybrook.edu
First published on 13th September 2021
Abstract
Bicyclo[4.2.0]oct-6-ene-7-carboxamide is a simple but highly strained olefin monomer which forms an alternating copolymer with cyclohexene in the presence of N-heterocyclic carbene-ruthenium catalyst. [4.2.0] moieties with a bulky substituent on C7 that chelate with the ruthenium center of the catalyst propagate more slowly than monomers that cannot chelate. Accordingly, the reactivity ratio of N-propylbicyclo[4.2.0]oct-6-ene-7-carboxamide with cyclohexene is significantly higher than that of N-(2-(2-ethoxyethoxy)ethan)-bicyclo[4.2.0]oct-6-ene-7-carboxamide with cyclohexene. A copolymerization involving the three monomers in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2 (propyl:ethylene glycol:cyclohexene) molar ratio formed a gradient copolymer in a one-pot reaction. Surface hydrophobicity, topology, and thermal properties of the gradient copolymer were similar to those of a copolymer comprised of six microblocks prepared through multistep synthesis by alternately employing the same two bicyclo[4.2.0]oct-6-ene-7-carboxamides in each microblock. The properties of the gradient copolymer were distinct from a copolymer comprised of two larger blocks based on the same bicyclo[4.2.0]oct-6-ene-7-carboxamides.
:1:2 (propyl:ethylene glycol:cyclohexene) molar ratio formed a gradient copolymer in a one-pot reaction. Surface hydrophobicity, topology, and thermal properties of the gradient copolymer were similar to those of a copolymer comprised of six microblocks prepared through multistep synthesis by alternately employing the same two bicyclo[4.2.0]oct-6-ene-7-carboxamides in each microblock. The properties of the gradient copolymer were distinct from a copolymer comprised of two larger blocks based on the same bicyclo[4.2.0]oct-6-ene-7-carboxamides.
Introduction
Polymers containing more than one monomer (thus copolymers) are useful because their properties and functions may be tuned by varying comonomer compositions and sequence. Compositional variation of monomers, whether continuous (gradient) or discontinuous (block), along polymer chains, can influence the thermal,1 mechanical,2 surface,3 and interfacial behaviors of copolymers.4 Almost all gradient copolymers (GCPs) synthesized to date are two-monomer component systems. Even with only two monomers, gradient copolymers are useful and sometimes have properties superior to block copolymers. For example, when used as an additive in a polymer blend, gradient copolymers tend to decrease domain sizes; thus GCPs are suitable as reinforcing agents and blend compatibilizers.5 In addition, GCPs usually have broad glass transition temperatures2,6 and are attractive for applications such as pressure sensitive adhesives.Numerous non-templated “living” polymerization techniques such as reversible addition–fragmentation chain transfer (RAFT),7 atom transfer radical polymerization (ATRP),8 nitroxide-mediated radical polymerization (NMRP)5 and ring opening polymerization (ROP)9 are popularly used in gradient polymer synthesis. Although ring opening metathesis polymerization (ROMP) possesses a living character10 and allows for tolerance of a wide range of functional groups,11 its use in gradient copolymer synthesis is underexploited. The first ROMP gradient copolymer was reported in 2004 by Nguyen et al.12 To control reactivities, they used two norbornene monomers whose substituents differed by their steric bulk. Since then, only a few other reports on GCP preparation have been issued, with all copolymers based on norbornene (Fig. 1).13–16 Our laboratory has shown that three cyclic olefin monomers can be used to prepare AB alternating copolymers via alternating ring-opening polymerization (AROMP) with specific sequences of different A-type monomers and provided evidence that sequence control modulates behavior.3 Alternating copolymers were made by stepwise addition of the relevant number of equivalents of each monomer, a labor intensive and iterative process. Thus, we were motivated to develop gradient copolymerization with AROMP. Gradient copolymerization relies on different reactivities of monomer species.12,14,17 In developing sequence controlled alternating copolymers, we had observed that bicyclo[4.2.0]oct-6-ene-7-carboxamide moieties with propyl side chain (1a) have marginally faster reactivity with cyclohexene (2) than glycine 7-substituted carboxamides.3 Thus, the reactivities were insufficiently different and their copolymerization would result in a predominantly random copolymer. However, olefins substituted with chelating ligands can coordinate with ruthenium catalyst and impose steric constraints on an approaching monomer,13 thus rendering them slower to propagate.
 | ||
| Fig. 1 (A) typical ROMP gradient copolymer bearing cyclopentanyl backbone with a 2-carbon spacer, (B) AROMP gradient copolymer bearing cyclohexanyl backbone with a 6-carbon spacer (black line). | ||
We reasoned that the reactivity of an “ethylene glycol” side chain would have further reduced reactivity compared to a glycine side chain because of the longer side chain and possibly strong chelating property of the glycol side chain (1b, Chart 1). Therefore, we hypothesized that the combination of 1a-alt-2, 1b, and 2 would yield a gradient copolymer in which the compositions 1a-alt-2 and 1b-alt-2 vary continuously along the chains. Herein, we employed bicyclo[4.2.0]oct-6-ene-7-carboxamides 1a and 1b and examined their reactivities with cyclohexene 2. We prepared uniform, diblock, hexablock, and gradient copolymers (Fig. 2). Uniform copolymers are the copolymers consisting of only 1a-alt-2 (P1) or 1b-alt-2 (P2) strands while diblock, hexablock and gradient copolymers are made up of both 1a-alt-2 and 1b-alt-2 strands. Topological and thermal analyses of the copolymers revealed the dependence of functional properties on the length and location of (1a-alt-2) and (1b-alt-2) sequences within the copolymer chains. To the best of our knowledge, this is the first report of a ROMP gradient copolymer which does not rely on norbornene monomers. Importantly, the inclusion of monomer 2 introduces a larger spacer between the side chains (Fig. 1) offering entry to a more flexible copolymer.
 | ||
| Chart 1 Monomers used for gradient copolymer synthesis. 1 and 2 react to form alternating strand poly(1-alt-2). All metathesis polymerization reactions were afforded by catalyst 3. | ||
 | ||
| Fig. 2 Synthesis of (A) uniform AROMP copolymers from monomers 1 and 2 to form P1, poly(1a-alt-2)20 or, P2, poly(1b-alt-2)20 and, (B) sequence-defined AROMP copolymers from 1a, 1b, and 2 in (i) multiple steps to from diblock copolymer P3 or hexablock copolymer P4, (ii) one-pot to yield gradient copolymer P5 poly(1-alt-2)10-g-poly(1b-alt-2)10. | ||
Results and discussion
N-Propylbicyclo[4.2.0]oct-6-ene-7-carboxamide 1a was synthesized by coupling bicyclo[4.2.0]oct-6-ene-7-carboxylic acid and N-propylamine according to literature procedure (Fig. S1†)18 and the ethylene glycol bicyclo[4.2.0]oct-6-ene-7-carboxamide monomer 1b was similarly prepared by coupling bicyclo[4.2.0]oct-6-ene-7-carboxylic acid and N-2-(2-ethoxyethoxy)ethan-1-amine (Fig. S2†).To demonstrate the impact of copolymer sequence on material properties such as topological and thermal behaviors, we synthesized uniform copolymers (P1 and P2) whereby P1 is made up of polymer chains containing only 1a-alt-2 units, P2 consists solely of 1b-alt-2 units, and diblock (P3), hexablock (P4), and gradient (P5) are copolymers of 1a, 1b, and 2. The reactivity ratios of 1a & 1b require that P3 and P4 be synthesized by multi-step AROMP (Fig. 2B). In contrast, P5 was prepared in one step. All the polymers synthesized, P1–P5, were characterized by 1H-NMR spectroscopy (Fig. S3–S7†). Their molecular weights (MWs) and degree of polymerization (DP) were determined by gel permeation chromatography (GPC) and 1H-NMR spectroscopy, respectively. As determined by GPC, the polymers exhibited a unimodal distribution (Fig. S8†) and MW dispersities (ĐM) were narrow with ĐM = 1.2–1.5 (Table 1). The theoretical molecular weights of the polymers match the molecular weights determined by DP analysis. However, molecular weights determined by polystyrene standard calibration were different from the theoretical MW, most likely due to the difference in hydrodynamic volumes of the polymers described here compared with the polystyrene standards.19 With polymer P1, 100% monomer conversion was achieved in less than 1.5 hours due to the higher reactivity of propyl[4.2.0]-based monomer with cyclohexene. In contrast for polymer P2, the ethylene glycol[4.2.0]-based monomer required twice the reaction time. The diblock copolymer P3 was prepared in two steps by sequential addition. First, (1a-alt-2)10 block was prepared using a twofold molar excess of cyclohexene. The second block (1b-alt-2)10 was successfully installed by adding 1b monomer to the reaction mixture containing poly(1a-alt-2)10-with a living Ru-carbene and excess cyclohexene.
| Polymer | Type | [1a]:[1b]:[2]:[3] |
Time (min) | DPb |
1b-alt-2c |
M
n,theod (kDa) |
M
nf (kDa) |
Đ
Me |
|---|---|---|---|---|---|---|---|---|
| a Polymers were prepared with catalyst 3 (10 mM) at 40 °C in CH2Cl2. b DP refers to the total number of alternating -AB-pairs in the polymer; DP was determined by 1H NMR spectroscopy integrating polymer against the end group phenyl (2H). c Mole fraction of 1b-alt-2 strand in the copolymers determined by 1H NMR spectroscopy. d Theoretical molecular weight calculated based on total molar feed. e Molecular weight dispersity determined by GPC with polystyrene calibration. f Number average molecular weight (Mn) determined based on DP and percent composition of 1a-alt-2 and 1b-alt-2 strands in the copolymers. g Synthesis of P3 & P4 involved multiple steps and an excess of 2 was used to increase the reaction rate. | ||||||||
| P1 | Uniform | 20:0:20:1 |
80 | 20 | 0.0 | 5.5 | 5.5 | 1.2 |
| P2 | Uniform | 0:20:20:1 |
180 | 19 | 1.0 | 7.0 | 6.6 | 1.3 |
| P3 | 2 Blocks | 10:10:25g:1 |
180 | 19 | 0.5 | 6.3 | 6.0 | 1.5 |
| P4 | 6 Blocks | 10:10:30g:1 |
180 | 16 | 0.4 | 6.3 | 4.9 | 1.3 |
| P5 | Gradient | 10:10:20:1 |
180 | 20 | 0.5 | 6.3 | 6.3 | 1.4 |
Full consumption of the [4.2.0]-based monomers 1a and 1b was confirmed by disappearance of protons between δ 2.6–3.0 ppm in the 1H NMR spectra (Fig. 3A). Successful installation of the two blocks is also confirmed by GPC (Fig. 3B). The preparation of hexablock copolymer P4, was afforded by six sequential addition steps. Microblocks (1a-alt-2)4-b-(1b-alt-2)4-b-(1a-alt-2)3 were fully installed in steps 1 through 3. However due to the slower reactivity of 1b, steps 4–6 suffered from partial incorporation (Fig. S9†) which may result in reduced “blockiness” of the second half of the hexablock copolymers. Approximately 80% overall conversion was achieved for steps 1–6. Hence, a (1-alt-2) 16-mer with less than 50% incorporation of the 1b-alt-2 repeat (Table 1) was obtained. For gradient copolymer P5, full incorporation of both monomers 1a and 1b was achieved.
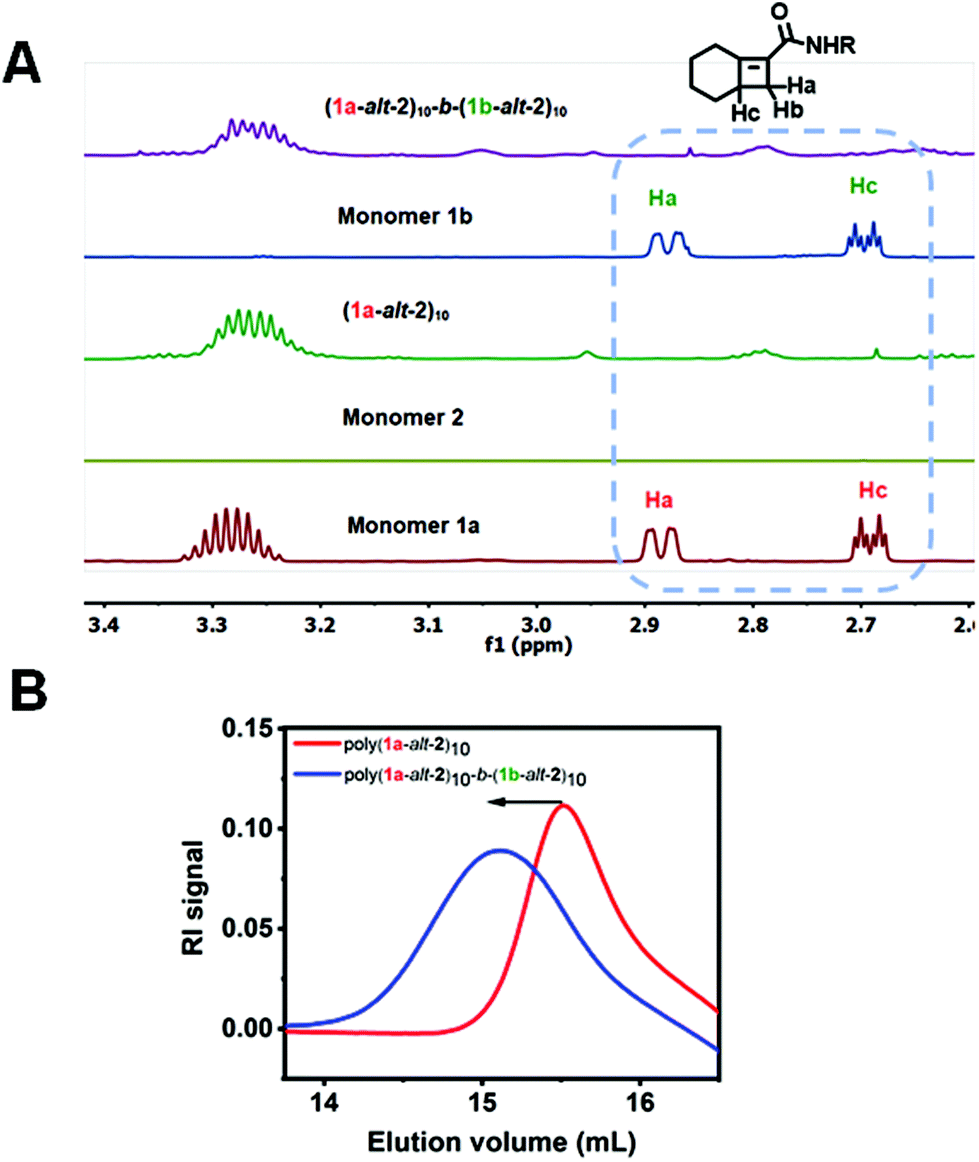 | ||
| Fig. 3 Diblock copolymer P3 was successfully prepared in two steps. In step 1, monomer 1a was fully incorporated to form the first block, and monomer 1b was fully incorporated at step 2 to install the second block as shown by both the 1H-NMR overlay (A) and GPC chromatogram (B). | ||
Ring opening kinetics
To determine the reactivity ratios of 1a-alt-2 and 1b-alt-2, we examined ring opening metathesis (ROM) of 1a and 1b (Fig. 4A, Fig. S10–13†). Either 1a or 1b solution was treated with a equimolar amount of 3. Evidence of ROM was confirmed by the disappearance of Ru-styrenylidene proton (1H NMR δ 19.17) (Fig. S12 & S13†). ROM of both 1a and 1b followed second-order reaction kinetics and 1a reacted approximately three-fold faster than 1b with catalyst 3 (t1/2 = 21 min and 60 min respectively [1]0 = [3]0 = 0.15 M). The slower reactivity of 1b with [Ru]-alkylidene is most likely due to the sterically bulky side chain.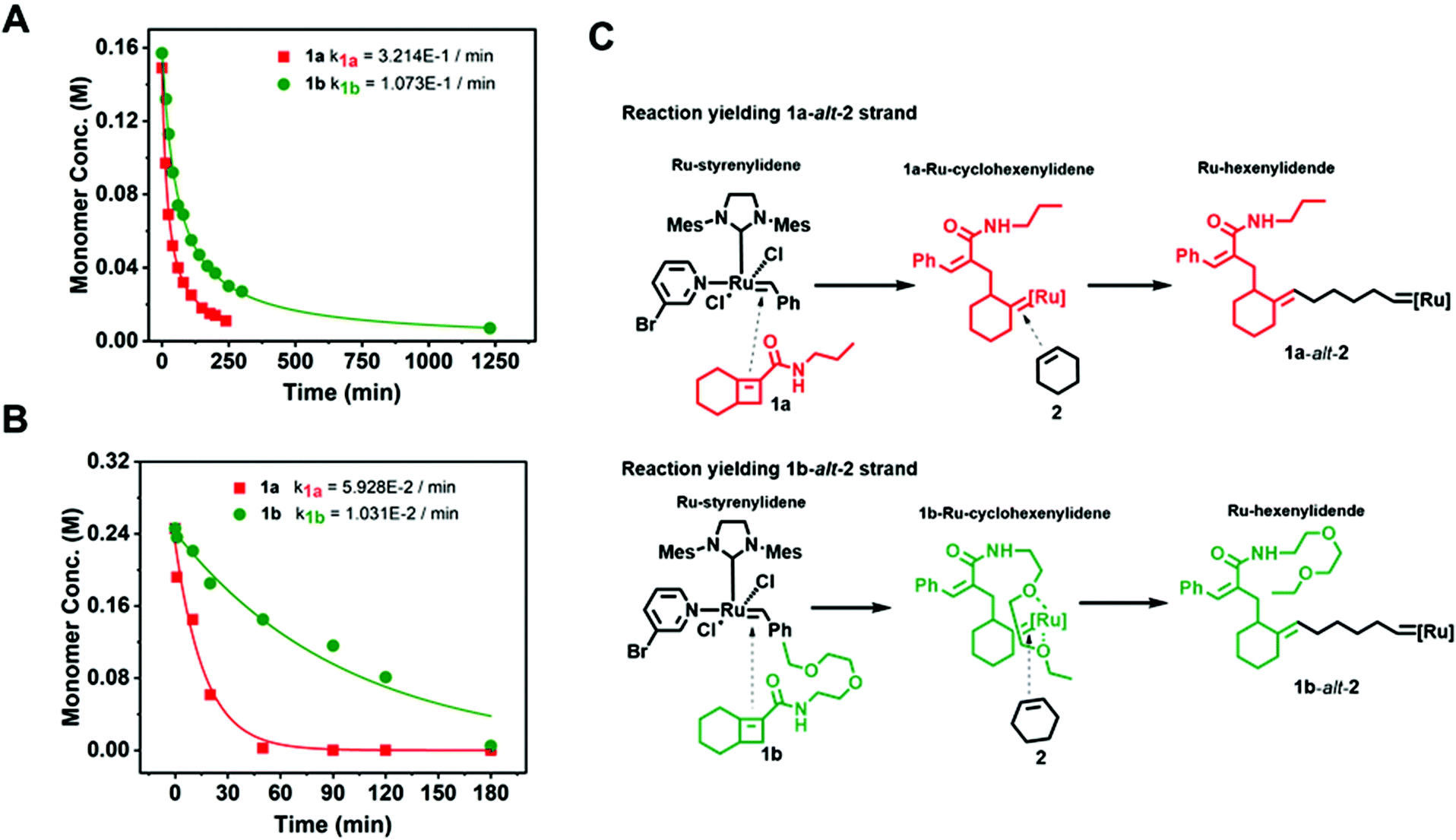 | ||
| Fig. 4 The substituent on monomer 1 influences the rate of reaction of single monomer ring-opening metathesis (ROM) and of gradient copolymerization. (A) ROM reactions run with a single monomer 1: [1a] = [3] = 0.15 M, or [1b] = [3] = 0.155 M. (B) Rate of disappearance of each 1 monomer in a gradient copolymerization of a mixture of 1a, 1b, and 2 at [1a] = 0.25 M, [1b] = 0.25 M, [2] = 0.49 M, [3] = 0.025 M. (C) ROM and AROM reactions of 1a and 1b with 2 and proposed orientation of alkylidenes and monomers that contribute to rate differences. | ||
Gradient copolymer kinetics
The incorporation of 1a with respect to 2 and 1b with respect to 2, was assessed by 1H-NMR spectroscopy at regular time intervals for the copolymerization of 1a:2:1b (1:2:1 molar ratio) (Fig. 4B). The reaction kinetics of 1a or 1b disappearance in the gradient copolymerization follow first order kinetics. In the gradient copolymerization, the rate of incorporation of 1a is about 6 times faster than 1b (half-lives t1/2 (1a), t1/2 (1b) ≈ 12 and 67 min respectively). The difference in rate of disappearance of 1a and 1b in the gradient copolymerization is larger than in the ROM reaction which suggests longer range effects on the addition to Ru-species occur. We also determined the relative reactivity ratios (r1a&2, r1b&2) for 1a & 2 and 1b & 2 respectively. Reactivity ratio is defined as the tendency for the propagating species to react with the same monomer over the other. To determine the reactivity ratios, we used the extended Kelen–Tüdös model (Fig. 5)20 by analyzing the reaction mixture at moderate conversion (∼40%) with feed ratios of monomers 1 varying from 0.1–0.9 equivalents (for 1a), 0.9–0.1 equivalents (for 1b), with one equivalent of monomer 2 (Tables S1 & S2†). The calculated relative reactivity ratios were r1a&2 (2.17) and r1b&2 (0.36). Data when fitted with the Jaacks model also gave similar reactivity ratios (Fig. S14†). The product of the reactivity ratios, r1a&2.r1b&2 = 0.78 and indicates a nonideal copolymerization character, that is not exclusive or random, and thus a gradient copolymerization. The reactivity ratios suggest a potential medium gradient copolymer formation.21
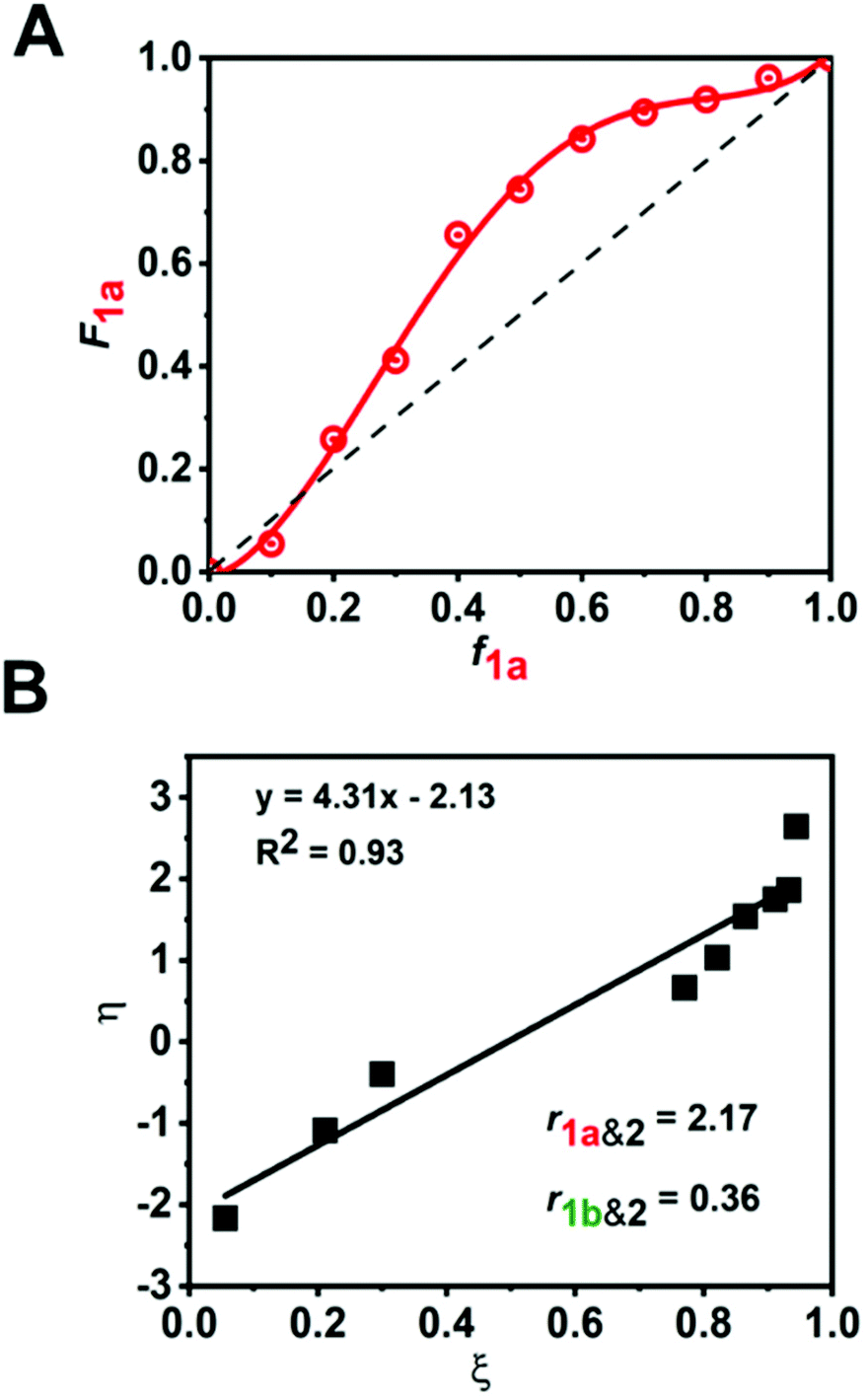 | ||
Fig. 5 (A) Lewis–Mayo plot for composition (F) of 1a in the copolymer vs. feed ratio (f) of 1a. The dashed line is an imaginary line which represents ideal copolymerization, (B) extended Kelen–Tüdös plot, given by the equation  . . | ||
Copolymer microstructure
The probabilities of finding certain sequences (N(x)) where x is the length of the sequence and μ is the mean sequence length are absolutely critical to make a more accurate prediction of compositional gradient heterogeneity in a copolymer.22 To confirm that the molar combination of monomers 1a, 2, and 1b (1:2:1) yields copolymer with gradient microstructure, N(x) and μ were calculated (see ESI Table S3†). N1a-alt-2 (1) = 0.32 and N1b-alt-2(1) = 0.74 are the probabilities of finding a sequence of a unit length, for (1a-alt-2)1 and (1b-alt-2)1; whereas the probabilities of finding a sequence of 3 unit lengths, (1a-alt-2)3 and (1b-alt-2)3, are N1a-alt-2(3) = 0.15, N1b-alt-2(3) = 0.05 respectively (Table S3, Fig. S15†). These imply there is a greater propensity of forming longer units of (1a-alt-2)n≥3 at early stages of polymerization. Longer sequences of (1b-alt-2)n≥3 are incorporated when 1a monomer is significantly depleted (thus at later stages of polymerization). In addition, the mean sequence lengths were 3 and 1 for 1a-alt-2 and 1b-alt-2 respectively. Moreover, following the kinetics of polymerization to form P5 with a molar feed ratio of 1:1:2 (1a, 1b, 2) by 1H NMR spectroscopy (Fig. S16†) revealed formation of gradient copolymer. In this experiment, aliquots of polymer were taken from the reaction vial at different time points (10, 20, 50, 90, 120, and 180 min), quenched with ethyl vinyl ether, precipitated with diethyl ether, and dried under reduced pressure before 1H NMR analysis. To determine the degree of polymerization (DP), the polymer protons were integrated against the phenyl end group proton at δ: 7.38 ppm (2H) (see ESI Fig. S17–21†). Other protons of the phenyl end group are obscured under the solvent peak (chloroform at δ 7.28 ppm). Unfortunately at 10 min, the short oligomers formed did not precipitate with diethyl ether (Fig. S22†) and analysis of the crude mixture was required. At this time point, a DP of 5 was attained and incorporation of 1b-alt-2 was only 19%. As shown in Fig. S16,† the fraction of 1b-alt-2 strand increases gradually along the copolymer chain as the molar fraction of 1a monomer is depleted. Combined, this evidence confirms gradient copolymer (P5) formation.
Effects of sequence on material properties
The thermal1 and topological3 properties of copolymers are highly dependent on microblock size or monomer sequence but not necessarily on their bulk composition.23 The molar compositions and the molecular weights of all the copolymers prepared were similar. So, differences in functional properties are attributed to comonomer sequence.Thermal properties
Material processability is dependent on the thermal properties of a copolymer. Using differential scanning calorimetry (DSC), the glass transition temperatures (Tgs) for the copolymers in bulk were determined (Fig. 6 and S23†). As expected, the uniform copolymer with propyl sidechain (P1) has the highest Tg whereas the polymer with an ethylene glycol sidechain (P2) has the lowest Tg. There was an obvious difference in Tg between the gradient copolymer (P5) and the diblock copolymer (P3). P3 has two glass transition temperatures (Tg1 = −13.3 and Tg2 = 26.5 °C) which suggests that the two blocks, (1a-alt-2)10 and (1b-alt-2)10, act independently as two different phases. Consistent with their smaller block size, their glass transitions temperatures are lower than the measured Tg values for P1 and P2 of −6.9 °C and 63.0 °C, respectively. The Tg of the parent block of 1a in P1 is much higher than in the block copolymer P3. Although this partly due to reduction of chain length from 20-AB- units to 10; however, with 1b chains having a Tg well below room temperature, it could have a plasticizing effect on the harder 1a block and therefore lowering its Tg.24 In contrast, P5 has only one glass transition temperature (Tg = 22.7 °C) which may be attributed to the absence of interfacial separation between the multiple 1a-alt-2 and 1b-alt-2 units. Like P5, P4 also has only one Tg. The single transition suggests that short block sizes of 3–4 repeating 1-alt-2 units do not induce microphase separation. Other thermal events such as melting temperature (Tm) and crystallization temperature (Tc) can be seen in thermograms of P4 and P5 but not present in P3. Thermal degradation of the polymers in bulk was assessed using thermogravimetric analysis (TGA) and derivative thermogravimetry (DTG) (Fig. S24†). None of the copolymers showed detectable weight loss up to 275 °C. These copolymers are quite stable. The least stable polymer among the five was P1 which exhibited a maximum degradation at ∼370 °C. The other copolymers, P2–5 exhibited maximum degradation at ∼400 °C. Since the diblock copolymer is made up of two phases we expected to see two degradation maxima. The presence of a single maximum is most likely due to the close proximity of the P1 ((1a-alt-2)20) and P2 ((1b-alt-2)20) thermal degradation temperatures. | ||
| Fig. 6 (A) Differential scanning calorimetry (DSC), (B) thermogravimetric analysis (TGA) thermograms for the sequence-controlled copolymers, P3–5. | ||
Topological properties
Degradation and interaction of polymeric material with other molecules such as solvents and atmospheric gases are influenced by topological features including surface wetability.25 Here, we measured the water contact angles (WCA) for each polymer (Fig. 7 and S25†). Silicon wafers were dip-coated with 1.0% (w/w) polymer solution in chloroform and allowed to dry at ambient temperature and pressure for 48 hours. Each polymer solution was coated on three separate silicon wafers. A precise volume of water (2.5 μL) was delivered onto the polymer coated silicon wafer at a delivery speed of 1.0 μL s−1. The measured contact angles of the polymer-coated substrates were consistent between replicates with only small deviations (±0.8–2.5) an indication of the reproducibility of coating. Contact angles measured for three different synthetic batches of polymers gave similar results. The contact angles on films of P1 and P2 were measured to establish a reference. P1 with propyl amide side chain (1a) was relatively hydrophilic (CA = 79°). To our surprise, P2 with an ethylene glycol amide side chain (1b) was less hydrophilic (CA = 88°) than P1. We believe that on a silicon matrix P2 exposes the hydrophobic of the polymer to the air interface and therefore behaves more like a hydrophobic film. | ||
| Fig. 7 Surface hydrophobicity is dependent on the sequence of 1a-alt-2 and 1b-alt-2 in the copolymer chains. Water contact angle for each polymer, from three different synthetic batches, was measured; the error bars represent the standard deviations of three measurements of the representative batch. P1: poly(1a-alt-2)20; P2: poly(1b-alt-2)20 shown in red and green respectively are used as reference. The contact angle for silicon wafer without polymer coat is 50° ± 2.2. | ||
Copolymers P3–P5 displayed hydrophobicities that fall between the uniform copolymers P1 and P2. Interestingly, as the number of 1b-alt-2 chain blocks within the copolymer increases, the hydrophobicity increases, as seen for hexablock P4 and gradient P5 copolymers. This suggests that P3 containing only two chain blocks phase segregates more efficiently than P4 and P5, exposing a hydrophilic shell to the air interface.26
To confirm that the surface hydrophobicity was indeed influenced by the polymer-type, we examined the surface of the polymer-coated silicon substrate by scanning electron microscopy (SEM, Fig. 8 and S26†). Measurements were done on one representative sample batch.
 | ||
| Fig. 8 SEM micrographs of films of copolymers P1–P5 prepared in, (A) chloroform, (B) 2-propanol. Scale bars 200 nm. | ||
Copolymer films were prepared from chloroform or 2-propanol solutions, cast on silicon wafer substrates, and dried at ambient temperature for 48 hours. Polymer films prepared from 2-propanol solutions of P3 exhibited distinctly hollow topological features. In contrast, films prepared from P4 or P5 solutions were similarly smooth. We believe that the hollow airy features seen in P3 are dominated by the 1a-alt-2 block. The diblock copolymer most likely phase-separates efficiently as its 1b-alt-2 block strongly adheres to the silicon surface due to intermolecular H-bond interactions and exposes the 1a-alt-2 block to the air interface. SEM images of P1 cast from 2-propanol (Fig. S26B†) also form films that trap air-bubbles at the air interface.
Using dynamic light scattering (DLS), we explored the particle size of the copolymers in chloroform and 2-propanol (Fig. S27 and 28,†Table 2). The particle size was dependent on polymer type as well as the solvent. In general, copolymers formed smaller particles in the more polar solvent (2-propanol) than in the less polar solvent (chloroform). The size of the P2 particle with ethylene glycol side chain is bigger in 2-propanol than in chloroform. In contrast, P1 with propyl side chain is bigger in chloroform than in 2-propanol. Due to the presence of the ethylene glycol system in P2, intermolecular interaction through hydrogen-bonding with 2-propanol can result in higher solvation, and therefore swelling. The particle sizes of the sequence-defined copolymers increase or decrease based on the distribution 1b-alt-2 strand in the copolymer chains. For instance, particle size of diblock copolymer P3 is larger than hexablock P4 and gradient copolymer P5 in 2-propanol but smaller in chloroform. This solvent dependent particle size behavior of the sequence-defined copolymers suggests that the diblock copolymer strongly phase segregates than the hexablock and gradient copolymers.
| Entry | Type | T g (°C) | Contact angle (°) | D H, chloroform (nm) | D H, 2-propanol (nm) |
|---|---|---|---|---|---|
| P1 | Uniform | 63.0 | 78.6 ± 2.5 | 45.1 | 14.6 |
| P2 | Uniform | −6.9 | 87.6 ± 0.6 | 14.1 | 17.8 |
| P3 | 2 Blocks | −13.3, 26.5 | 80.9 ± 0.8 | 21.4 | 12.2 |
| P4 | 6 Blocks | 21.0 | 81.8 ± 0.8 | 35.0 | 10.8 |
| P5 | Gradient | 22.7 | 84.7 ± 1.2 | 48.7 | 9.2 |
Conclusion
In this work, we demonstrate that AROMP is a versatile method to prepare a myriad of polymeric materials with molecularly defined structures whose surface and thermal properties can be tuned. A three-monomer system 1a/1b/2 was utilized to prepare a gradient copolymer which had thermal and morphological properties similar to copolymer with hexablock units but different than that of the diblock copolymer. This work in addition to current works17,27,28 suggest that with the choice of monomers with suitable reactivities, laborious multi-step synthetic routes could be bypassed while still achieving copolymers with multiblock segments in a one-pot approach.Therefore, gradient copolymer preparation with AROMP presents an opportunity to synthesize variable sequence copolymers with a wider variety of possibilities than traditional norbornene-ROMP.
There are additional prospects to tune copolymer flexibility by varying spacer length,29 and to prepare degradable copolymers18 through appropriate selection of the unstrained cyclic alkene in place of monomer 2.
Experimental section
Materials and general methods
All air-sensitive and metathesis reactions were performed under N2 by means of standard Schlenk or glove box techniques. Polystyrene standards and Cl2(H2IMes)(PCy3)Ru![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHPh (Grubbs 2nd generation catalyst) were purchased from Aldrich. Grubbs 3rd generation catalyst (3-BrPyr)2Cl2(H2IMes)RuCHPh, 3, was prepared from 2nd generation catalyst and 3-bromopyridine.30 Dry, oxygen-free CH2Cl2, was obtained with a Pure Process Technology solvent purification system. Mallinckrodt silica gel-60 (230–400 mesh) was used for column chromatography. Analytical thin-layer chromatography was performed on precoated silica gel plates (60F254), and Combi-Flash chromatography on RediSep normal-phase silica columns (silica gel-60, 230–400 mesh). Bruker Nanobay 400, AVANCE III 500 MHz, and AVANCE III 700 MHz instruments were used for NMR spectroscopy.
CHPh (Grubbs 2nd generation catalyst) were purchased from Aldrich. Grubbs 3rd generation catalyst (3-BrPyr)2Cl2(H2IMes)RuCHPh, 3, was prepared from 2nd generation catalyst and 3-bromopyridine.30 Dry, oxygen-free CH2Cl2, was obtained with a Pure Process Technology solvent purification system. Mallinckrodt silica gel-60 (230–400 mesh) was used for column chromatography. Analytical thin-layer chromatography was performed on precoated silica gel plates (60F254), and Combi-Flash chromatography on RediSep normal-phase silica columns (silica gel-60, 230–400 mesh). Bruker Nanobay 400, AVANCE III 500 MHz, and AVANCE III 700 MHz instruments were used for NMR spectroscopy.
Gel permeation chromatography/size exclusion chromatography
Analyses were performed on a system composed of Shimadzu SCL-10A controller, a Shimadzu LC-20AT pump, and a Shimadzu CTO-10AS column oven equipped with combined Phenogel columns: 5 μm linear (2) (300 × 7.8 mm, 100–10000k) and 5 μm 50 Å (300 × 7.8 mm, 100–3k), and coupled with a Brookhaven Instruments BI-DNDC refractometer. The mobile phase used was HPLC-grade tetrahydrofuran (filtered with 0.2 μL PTFE filter) at 30 °C and 0.7 mL min−1. High resolution mass spectra were recorded on an Agilent 1260 LC/G6224 qTOF instrument.
Water contact angle
1% (w/w) polymer solutions were prepared in chloroform. Silicon wafers were dip-coated with the polymer solutions and dried at ambient temperature and pressure for 48 h. The water contact angles were measured with Data Physics instrument's optical contact angle goniometers and drop shape analysis systems and SCA 20 software in sessile drop mode. Each measurement was performed on three dip-coated wafers for each polymer sample; the average and standard deviations were taken.Thermal analysis
Solid polymer samples were used. The experiments were performed on a TA Instruments Q2000 differential scanning calorimeter with Wizard software (TA Instruments Q Series software). Heat flow (Tzero) calibration was performed with sapphire standards, and the temperature (cell constant) was calibrated using indium at a scan rate of 10 °C min−1. All differential scanning calorimetry was performed with a DSC Q2000 V24.11 Build 124 Module DSC Standard Cell RC under a N2 flow (50 mL min−1) using S5 Tzero aluminum pans and hermetic lids. The sample weights ranged from 5 to 8 mg. The samples were first equilibrated at −50 °C and then heated to 200 °C at 5 °C min−1. Thermogravimetric analysis was performed with a TGA Q50 V20.13 Build 39 equipped with a platinum pan. The sample sizes ranged from 6 to 12 mg. Weight loss data were collected from 30 to 600 °C at 5 °C min−1 under a N2 flow (60 mL min−1).Scanning electron microscopy (SEM)
Samples were analyzed using the SEISS Crossbeam 340 focus ion beam-scanning electron microscope (FIB-SEM) at 30 kV. Polymer films were prepared by dip-coating a silicon substrate with 1% (w/w) polymer solution in chloroform or 2-propanol and dried under vacuum. Samples were analyzed without staining.Dynamic light scattering (DLS)
Samples were analyzed on a 90 Plus Particle Size Analyzer (Brookhaven Instruments), a 35 mW red diode laser with a wavelength of 659.0 nm, scattering angle 90, 25 °C. Each signal was recorded for 60 s, and the signals for 5 consecutive runs were averaged. 1% (w/w) polymer solutions were prepared in the desired solvent, filtered using 0.2 μm PTFE membrane, and 200 μL solution transferred into a cuvette.Monomer and polymer preparation
1b: 500 mg bicyclo[4.2.0]oct-6-ene-7-carboxylic acid (3.29 mmol, 1 eq.) and 633 mg EDC·HCl (3.3 mmol, 1 eq.) were dissolved in 15 ml CH2Cl2. Diisopropylethylamine (1.15 ml, 6.58 mmol, 2 eq.) and N-2-(2-ethoxyethoxy)ethan-1-amine (0.48 ml, 3.29 mmol, 1 eq.) were added to the mixture. The reaction was stirred for 3 h at 23 °C. The mixture was diluted with 100 mL CH2Cl2 and then sequentially washed with 1 N HCl (25 mL), sat. NaHCO3 (25 mL), DI H2O (25 mL). Flash chromatography on silica gel using 20% acetone in CH2Cl2 (Rf ≈ 0.45) afforded the desired product as colorless viscous oil in 52% (459 mg) isolated yield. 1H NMR (500 MHz, CDCl3) δ ppm 5.93 (s, 1H), 3.64–3.61 (m, 2H), 3.61–3.58 (m, 4H), 3.54 (q, 2H, J = 6.95 Hz), 3.54–3.50 (m, 2H), 2.88 (d, 1H, J = 14.0 Hz), 2.69 (dt, 1H, J = 12.0, 3.71 Hz), 2.35 (m, 1H), 2.20 (d, 1H, J = 12.3 Hz), 2.13 = 2.05 (m, 2H), 1.91 (m, 1H), 1.75 (m, 1H), 1.32 (m, 2H), 1.23 (t, 3H, J = 6.93 Hz), 1.12 (m, 1H). 13C NMR (500 MHz, CDCl3, δ) 164.4, 161.5, 126.8, 70.5, 70.2, 69.9, 66.9, 38.7, 38.0, 34.1, 33.1, 27.4, 26.9, 24.9, 15.4. HRMS: m/z calc 268.1907; found 268.1906 [M + H]+.P1: poly(1a-alt-2)20: A solution of 1a (90 mg, 0.47 mmol) in dichloromethane (1000 μL) and a solution of 3 (20.5 mg, 0.023 mmol) in dichloromethane (500 μL) were mixed in a septum capped vial previously sparged with nitrogen. The mixture was incubated at 40 °C for 15 min. Monomer 2 (94 μL, 76.1 mg, 0.927 mmol) was added and after 80 min, the reaction was terminated with ethyl vinyl ether (100 μL). Polymer was precipitated in diethyl ether and dried under vacuum to yield the copolymer as off-white sponge-like solid (75% isolated yield).
P2: poly(1b-alt-2)20: A solution of 1b (100 mg, 0.37 mmol) in dichloromethane (1100 μL) and a solution of 3 (16.5 mg, 0.019 mmol) in dichloromethane (400 μL) were mixed in a septum capped vial previously sparged with nitrogen. The mixture was incubated at 40 °C for 15 min. Monomer 2 (93 μL, 75.6 mg, 0.92 mmol) was added and after 180 min, reaction was terminated with ethyl vinyl ether (100 μL). Polymer was precipitated in diethyl ether and dried under vacuum to yield the copolymer as off-white very viscous liquid-solid-like material (66% isolated yield).
P3: diblock, poly(1a-alt-2)10-b-(1b-alt-2)10: A solution of 3 (23 mg, 0.026 mmol) in dichloromethane (550 μL) and a solution of 1a (50 mg, 0.26 mmol, 10 eq.) in dichloromethane (600 μL) were mixed in a septum capped vial previously sparged with nitrogen. The mixture was incubated at 40 °C for 15 min. Monomer 2 (66 μL, 53.2 mg, 0.65 mmol, 25 eq.) was added in excess. The reaction was allowed to proceed for an additional 40 min and then a solution of 1b (69.2 mg, 0.26 mmol) in dichloromethane (700 μL) was added. After an additional 140 min, the reaction was terminated with ethyl vinyl ether (100 μL). Polymer was precipitated in diethyl ether and dried under vacuum to yield the copolymer as off-white solid (74% isolated yield).
P4: hexablock, poly[(1a-alt-2)-b-(1b-alt-2)]n=4, 3, 3: A solution of 3 (23 mg, 0.026 mmol) in dichloromethane (400 μL) and a solution of 1a (20 mg, 0.103 mmol, 4 eq.) in dichloromethane (200 μL) were mixed in a septum capped vial previously sparged with nitrogen. Step 1: after the mixture had been incubated at 40 °C for 15 min, monomer 2 was added in excess (79 μL, 63.8 mg, 0.78 mmol, 30 eq.) and the reaction allowed to proceed for an additional 25 min. Step 2: a solution of 1b (27.5 mg, 0.103 mmol, 4 eq.) in dichloromethane (250 μL) was added to the mixture from step 1 and the reaction allowed to continue for an additional 35 min. Step 3: a solution of 1a (15 mg, 0.078 mmol, 3 eq.) in dichloromethane (151 μL) was added to the step 2 mixture and the reaction allowed to continue for an additional 25 min. Step 4: a solution of 1b (20.6 mg, 0.077 mmol, 3 eq.) in dichloromethane (187 μL) was added to the step 3 mixture and the reaction allowed to continue for an additional 35 min. Steps 3 and 4 were repeated in sequence and the reaction was terminated with ethyl vinyl ether (500 μL). Polymer was precipitated in diethyl ether and dried under vacuum to yield the copolymer as off-white solid (78% isolated yield).
P5: gradient, poly(1a-alt-2)10-grad-(1b-alt-2)10: A solution of 3 (22.9 mg, 0.026 mmol) in dichloromethane (550 μL) and a solution of 1a (50 mg, 0.26 mmol) in dichloromethane (600 μL) were mixed. The mixture was incubated at 40 °C for 15 min, followed by addition of a solution of 1b (69.2 mg, 0.26 mmol) in dichloromethane (700 μL) and 2 (53 μL, 42.6 mg, 0.52 mmol). After 180 min, the reaction was terminated with ethyl vinyl ether (100 μL). Polymer was precipitated in diethyl ether and dried under vacuum to yield the copolymer as off-white solid (65% isolated yield).
Author contributions
Francis Boadi: conceptualization, validation, investigation, writing-original draft, writing-review and editing. Nicole S. Sampson: conceptualization, writing-review and editing, resources, supervision, project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is funded by NSF CHE1609494 (N. S. S.), NIH R01GM097971 (N. S. S.), and NIH T32GM092714 (F. O. B.). We thank Dr Chung-Chueh Chang (Simon) of the AERTC facility at Stony Brook for acquisition of TGA, DSC, and SEM data. We also thank Gyusaang Youn (Stony Brook University) for proofreading and editing manuscript, Madani Khan (Stony Brook University) for helping in acquisition of water contact angle data, and Dr Béla Ruzsicska (Stony Brook) for HRMS data.Notes and references
- E. A. Hoff, G. X. De Hoe, C. M. Mulvaney, M. A. Hillmyer and C. A. Alabi, J. Am. Chem. Soc., 2020, 142, 6729–6736 CrossRef CAS PubMed.
- W. Jakubowski, A. Juhari, A. Best, K. Koynov, T. Pakula and K. Matyjaszewski, Polymer, 2008, 49, 1567–1578 CrossRef CAS.
- G. Li and N. S. Sampson, Macromolecules, 2018, 51, 3932–3940 CrossRef CAS PubMed.
- W. Yuan, M. M. Mok, J. Kim, C. L. H. Wong, C. M. Dettmer, S. T. Nguyen, J. M. Torkelson and K. R. Shull, Langmuir, 2010, 26, 3261–3267 CrossRef CAS PubMed.
- J. Kim, H. Zhou, S. T. Nguyen and J. M. Torkelson, Polymer, 2006, 47, 5799–5809 CrossRef CAS.
- J. Kim, M. M. Mok, R. W. Sandoval, D. J. Woo and J. M. Torkelson, Macromolecules, 2006, 39, 6152–6160 CrossRef CAS.
- X. Guo, T. Zhang, Y. Wu, W. Shi, B. Choi, A. Feng and S. H. Thang, Polym. Chem., 2020, 11, 6794–6802 RSC.
- Z. Wang, T. Liu, Y. Zhao, J. Lee, Q. Wei, J. Yan, S. Li, M. Olszewski, R. Yin, Y. Zhai, M. R. Bockstaller and K. Matyjaszewski, Macromolecules, 2019, 52, 9466–9475 CrossRef CAS.
- O. Sedlacek, K. Lava, B. Verbraeken, S. Kasmi, B. G. De Geest and R. Hoogenboom, J. Am. Chem. Soc., 2019, 141, 9617–9622 CrossRef CAS PubMed.
- K. Matyjaszewski, Macromolecules, 1993, 26, 1787–1788 CrossRef CAS.
- C. W. Bielawski and R. H. Grubbs, Prog. Polym. Sci., 2007, 32, 1–29 CrossRef CAS.
- C. M. Dettmer, M. K. Gray, J. M. Torkelson and S. T. Nguyen, Macromolecules, 2004, 37, 5504–5512 CrossRef CAS.
- A. B. Chang, T.-P. Lin, N. B. Thompson, S.-X. Luo, A. L. Liberman-Martin, H.-Y. Chen, B. Lee and R. H. Grubbs, J. Am. Chem. Soc., 2017, 139, 17683–17693 CrossRef CAS PubMed.
- T.-P. Lin, A. B. Chang, H.-Y. Chen, A. L. Liberman-Martin, C. M. Bates, M. J. Voegtle, C. A. Bauer and R. H. Grubbs, J. Am. Chem. Soc., 2017, 139, 3896–3903 CrossRef CAS PubMed.
- L. Jiang, D. Nykypanchuk, A. E. Ribbe and J. Rzayev, ACS Macro Lett., 2018, 7, 619–623 CrossRef CAS.
- H. Yuan, T. Kida, R. Tanaka, Z. Cai, Y. Nakayama and T. Shiono, Polym. Chem., 2021, 12, 189–195 RSC.
- S. Pal, M. Alizadeh, P. Kong and A. F. M. Kilbinger, Chem. Sci., 2021, 12, 6705–6711 RSC.
- F. O. Boadi, J. Zhang, X. Yu, S. R. Bhatia and N. S. Sampson, Macromolecules, 2020, 53, 5857–5868 CrossRef CAS PubMed.
- J. Engelke, J. Brandt, C. Barner-Kowollik and A. Lederer, Polym. Chem., 2019, 10, 3410–3425 RSC.
- F. Tüdos, T. Kelen, T. Földes-berezsnich and B. Turcsányi, J. Macromol. Sci., Part A, 1976, 10, 1513–1540 Search PubMed.
- T. Gleede, J. C. Markwart, N. Huber, E. Rieger and F. R. Wurm, Macromolecules, 2019, 52, 9703–9714 CrossRef CAS.
- A. S. Brar, Sunita and C. V. V. Satyanarayana, Polym. J., 1992, 24, 879–887 CrossRef CAS.
- K. Matyjaszewski, Science, 2011, 333, 1104–1105 CrossRef CAS PubMed.
- A. B. Kutikov and J. Song, ACS Biomater. Sci. Eng., 2015, 1, 463–480 CrossRef CAS PubMed.
- N. J. Shirtcliffe, G. McHale and M. I. Newton, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 1203–1217 CrossRef CAS.
- S. K. Filippov, B. Verbraeken, P. V. Konarev, D. I. Svergun, B. Angelov, N. S. Vishnevetskaya, C. M. Papadakis, S. Rogers, A. Radulescu, T. Courtin, J. C. Martins, L. Starovoytova, M. Hruby, P. Stepanek, V. S. Kravchenko, I. I. Potemkin and R. Hoogenboom, J. Phys. Chem. Lett., 2017, 8, 3800–3804 CrossRef CAS PubMed.
- M. Yasir, P. Liu, J. C. Markwart, O. Suraeva, F. R. Wurm, J. Smart, M. Lattuada and A. F. M. Kilbinger, Angew. Chem., 2020, 59, 13597–13601 CrossRef CAS PubMed.
- C. Wahlen, J. Blankenburg, P. von Tiedemann, J. Ewald, P. Sajkiewicz, A. H. E. Müller, G. Floudas and H. Frey, Macromolecules, 2020, 53, 10397–10408 CrossRef CAS.
- J. Zhang, G. Li and N. S. Sampson, ACS Macro Lett., 2018, 7, 1068–1072 CrossRef CAS PubMed.
- J. A. Love, J. P. Morgan, T. M. Trnka and R. H. Grubbs, Angew. Chem., Int. Ed., 2002, 41, 4035–4037 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectral characterization data and kinetic data. See DOI: 10.1039/d1py00690h |
| This journal is © The Royal Society of Chemistry 2021 |