
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSite-selective modification of peptide backbones
Alicia
Boto
 *a,
Concepción C.
González
*a,
Dácil
Hernández
a,
Iván
Romero-Estudillo
bc and
Carlos J.
Saavedra
ad
*a,
Concepción C.
González
*a,
Dácil
Hernández
a,
Iván
Romero-Estudillo
bc and
Carlos J.
Saavedra
ad
aInstituto de Productos Naturales y Agrobiología del CSIC, Avda. Astrofísico Francisco Sánchez 3, 38206-La Laguna, Tenerife, Spain. E-mail: alicia@ipna.csic.es; ccgm@ipna.csic.es
bCentro de Investigaciones Químicas-IICBA, Universidad Autónoma del Estado de Morelos. Av. Universidad 1001, Cuernavaca, Morelos 62209, Mexico
cCatedrático CONACYT-CIQ-UAEM, Mexico
dPrograma Agustín de Betancourt, Universidad de la Laguna, 38200 Tenerife, Spain
First published on 6th September 2021
Abstract
The site-selective modification of peptide backbones allows an outstanding fine-tuning of peptide conformation, folding ability, and physico-chemical and biological properties. However, to achieve selectivity in the core of these biopolymers is challenging. In the last few years, many advances towards this goal have been developed. This review addresses the selective modification of Cα- and N-positions, from the use of “customizable units” to the residue-directed introduction of substituents. The site-selective modification of the peptide bond, i.e. the formation of thioamides or heterocycles, which alters backbone rigidity and ability to form hydrogen bonds or recognition by enzymes, is also described. Moreover, not only the modifications in internal backbone positions, but also in the N- and C-termini are discussed. In addition to chemical methodologies, the review addresses some reactions, catalyzed by natural or engineered enzymes, that afford unprecedent regio- and stereoselectivity in backbone modifications.
 Alicia Boto | Dr Alicia Boto is Research Scientist at the Spanish Research Council (CSIC) in IPNA-CSIC (Tenerife-Spain). She works on the development of synthetic methodologies and their application to drug synthesis, particularly antimicrobial and antitumoral agents. She has recently participated as an Expert in the preparation of CSIC White Report on the ‘Biomedicine and Health’ Challenges. She has also been Coordinator of Scientific Outreach (2007–2019) and Deputy Director of IPNA-CSIC (2013–2020). Her 80 publications include several Featured Articles/Covers in the selective modification of peptides. Part of the results have been transferred to chemical and agrotech companies, and some are commercialized internationally. |
 Concepción C. González | Dr Concepción González graduated in Chemistry from the University of La Laguna. She carried out her PhD work at IPNA-CSIC, obtaining her PhD in 1997 (University of La Laguna-ULL). From 1998–2001, she worked as a post-doctoral fellow at the University of Strathclyde (Glasgow) under the direction of Prof. John. A. Murphy. Since 2001 she has been a Senior Scientist at IPNA – CSIC. One of her research lines is based on the study of the different reactivity of O- and N-radicals, obtained from hypervalent iodine reagents, for the selective modification of carbohydrates and peptides. |
 Dácil Hernández | Dr Dácil Hernández received her PhD in Chemistry at the University of La Laguna (Spain) in 2007 and was awarded one of the PhD University Prizes for her work on the modification of carbohydrates and amino acids to give nucleoside analogs and alkaloids. After a two-year postdoctoral stay at Aarhus University (Denmark) with Prof. Skrydstrup, working on sila-amino acids and peptides, she returned to CSIC, where she holds a postdoctoral research contract (Transaludagro project). She has published more than 20 papers and has a licensed patent. She has codirected an industrial PhD and supervised other PhD and master work on peptide modification. |
 Iván Romero-Estudillo | Dr Iván Romero carried out his PhD studies on Chemical Sciences at the Universidad de La Laguna (Spain), working under the supervisión of Dr Alicia Boto in IPNA-CSIC. His work on the selective modification of amino acids and peptides (introducing the Hyp ‘customizable unit’) was awarded one of the University PhD Prizes, in the area of Experimental Sciences. He has several Articles and licensed patents on the subject. Currently he is Professor (Profesor-Investigador Cátedra-CONACYT) at the University of Morelos (Mexico), and has directed a Project on the development of antitumoral peptides. |
 Carlos J. Saavedra | Dr Carlos Javier Saavedra obtained his BchD in Chemistry at the University of Sevilla in 2003. He completed his Ph.D. (ULL-2013) working on the selective modification of peptides at IPNA-CSIC. He introduced the glutamic acid unit and worked with serine, threonine and C-terminal units. In 2014, he moved to University of Zaragoza for a postdoctoral contract with Dr Fraile to work on heterogeneous catalysts. Later he got a contract “Torres Quevedo” in the company Biosigma S.L. He is currently working on an “Agustín de Betancourt” project (University of La Laguna) for the development and commercialization of new agents for plant protection. |
1. Introduction
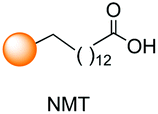
The modification of peptides and proteins is a hot area in bioorganic, medicinal and synthetic chemistry, allowing the generation of peptide drugs, antibody–drug conjugates, and probes for molecular imaging, among others.1 However, due to the similar reactivity of amino acids, the selective modification of certain positions has been troublesome until recently. Important advances have been made in the field, as shown by the reviews published recently.2–4 The ability to introduce residues with functional groups (tags) that can be replaced or transformed into other ones (tag-and-modify approach) has made possible the modification of both peptides and proteins.4 For example, residues with azido or propargyl units in certain positions have allowed click chemistry and the introduction of new chains.5 Residues with alkenyl side-chains can be modified by olefin metathesis6 or the addition of different group, such as alkyl thiol chains.7 Enzymes have been used to transform certain residues or to recognize amino acid sequences to be modified.8The progresses made in this field have been described in different reviews, and an excellent summary up to 2016–17 has been compiled in the review by Baran et al.2
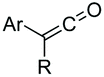
While most of the modifications have been carried out in the lateral chains, relatively few have been developed for the modification of the peptide backbone,2 in spite that backbone modifications can deeply affect the conformation, solubility, aggregation properties and therefore, the biological activity of the peptide. This review will address the advances made in the field, including not only the modifications in internal backbone positions, but also in N- and C-termini. In addition, the impact on the secondary structure and the biological activity will be highlighted.
The backbone modifications using “customizable units” will be discussed first. The most common ones use glycine residues, which are transformed into α-alkyl, aryl- or α-allyl glycines.9,10 The advances made in the last years with photoredox reactions are remarkable, since the use of chiral catalysts has allowed the attachment of lateral-chains to the carbon backbone with promising stereoselectivity and good yields.10
Other strategies such as scission reactions of serine, threonine, hydroxyproline, glutamic acid or C-terminal residues, followed in situ by the addition of new chains, have also been reported, providing from α,β- α,γ- or α,δ-peptide hybrids to peptides with a variety of N-substituents.11 The oxidation of proline with iron catalysts affords open-chain counterparts.12 The transformation of rigid, cyclic residues into acyclic ones or viceversa can have a strong impact on peptide conformation.
Some particular customizable units are dehydroalanine and related dehydroamino acids (dhAAs).13 The double bond presents both electron-withdrawing and electro-donating substituents, resulting in a capto-dative effect, which allows the addition of nucleophiles and radicals, cycloadditions and many other reactions.14 The formation of dehydroamino acids increases the rigidity of the system and favours turns, as will be commented later, while the conversion of dhAAs into acyclic residues increases backbone flexibility and affects peptide conformation.15
The formation of heterocyclic rings in the backbone also affects the overall conformation and physico-chemical properties. Nature uses this strategy frequently to create bioactive compounds, such as antimicrobial or cytotoxic oxazole and thiazole peptides, whose increased rigidity favours interaction with the biological targets.16 This strategy has also been used in the laboratories to generate a variety of heterocycles (triazoles, oxazoles, thiazoles, oxazolidines, etc.) with applications as fluorescent probes and drug candidates, among others.17
The selective introduction of N-substituents is also commented. This modification can decrease the number of hydrogen bonds available, and thus destabilize some secondary structures such as helices or β-sheets, or affect the formation of turns.18 These effects on the secondary structure have been used in peptide synthesis to increase their solubility and decrease aggregation, particularly when difficult sequences are involved. Some outstanding achievements in the solubilization of amyloid peptides, polyalanine sequences and other aggregation-prone peptides have been described.19
The site-selective introduction of N-substituents in already-formed peptides has been carried out by different methodologies, from the scission of cyclic customizable units20 to the histidine-directed N-alkenylation or arylation of the preceding residues on treatment with boronic acids.21 In other cases, units with removable N-substituents are introduced at key positions during peptide synthesis, and once it is completed, the N-substituents are cleaved under appropriate conditions (light, pH changes, etc.).22
The modification of C- and N-terminal positions will also be commented. The replacement of carboxyl groups by other functions can introduce amino acid analogues in the peptide backbone.23 The selective modification of N-terminal positions often exploits the differences in pKa between the N-terminus and the lateral chains of lysine, ornithine or arginine to perform selective N-alkylations or acylations.24 These chains can also be oxidized to glyoxalate units and further transformed.25 Their reaction with in situ formed electrophiles and residue-selective reagents (for cysteine, serine, proline) will also be summarized.26
The review will also describe the exquisite regio- and stereoselectivity achieved with some enzymes, that recognize particular motifs and transform these positions.27 Such enzymes can catalyze transamination, oxidation, (macro)cyclization and cycloaddition reactions. Their use for ligation of peptides is beyond the scope of this review,28 but some particular cases are nevertheless illustrated. Remarkably, although these enzymes are precise in motif recognition, they are however quite promiscuous, accepting different substrates containing the appropriate “key” motif.29 The rapid advances made in the field of protein engineering will undoubtly expand the catalog of enzymes for new applications.
In summary, the advances in the site-selective backbone modification is a rapidly expanding area, and each day new methodologies and synthetic tools are incorporated. The fine-tuning of backbone modification would play an important role in the understanding of peptide conformation, physicochemical properties, and biological action. In turn, this would allow the design of tailor-made peptides for a certain application in a variety of fields, from biomedicine to materials science.
2. Backbone modifications at C-α positions
2.1. Modifications at C-α using customizable units
The site-selective modification of C-α positions often uses “customizable units” which can be differentiated from the other amino acid residues.11 Among them, glycine is the most frequently employed. Other units are serine, threonine, glutamic acid, proline and hydroxyproline.11,12 The special case of dehydroamino acid units will be discussed as well.13The first significative advance was reported by Seebach et al., who generated glycine enolates at low temperatures, and trapped them with electrophiles, while the other amino acids in the peptide remained unaffected. However, the starting units were N-protected glycines, which involved extra protection and deprotection steps.30 Later on, Seebach used an aminomalonate unit as a “customizable” residue to carry out complete lateral chain replacement (conversion 1 → 2, Scheme 1).31 The alkylation in basic media was followed by benzyl ester deprotection and in situ decarboxylation, to give an aromatic residue. With this method, both D- and L-epimers could be obtained for biological studies.
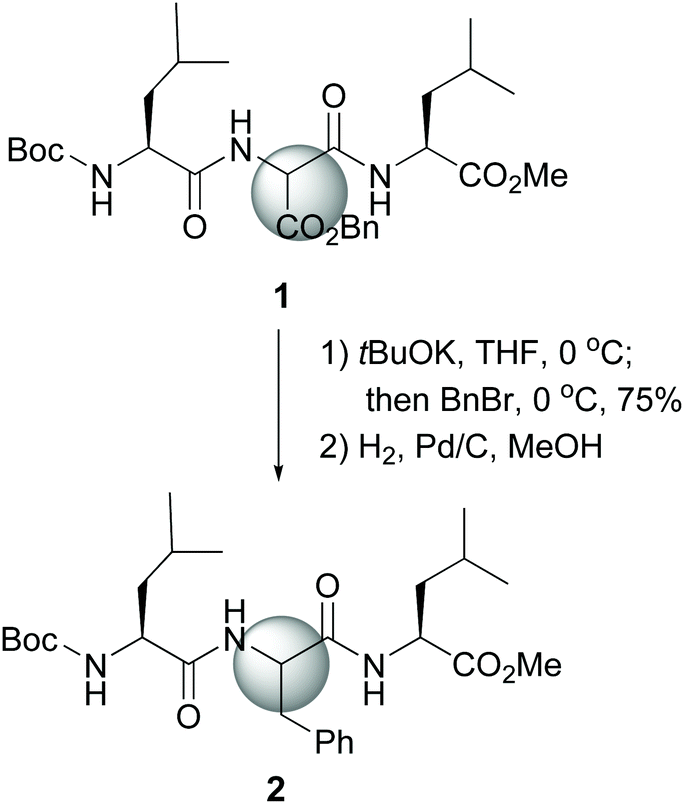 | ||
| Scheme 1 Selective modification of an aminomalonate unit by Seebach et al.31 | ||
Steglich used glycine units in a different site-selective approach32 (conversion 3 → 4/5, Scheme 2). He had shown that derivatives of α-bromohippuric esters could react with nickelocenes, cyclopentadienylthallium and other organometallics to give 2-cyclopentadienylglycines.32a He also reported that a dipeptide 3 with an alkylthio moiety was treated with thionyl chloride and then with cyclopentadienyltrimethylsilane and ZnCl2 to give the isomeric cyclopentadienyl glycines 4 and 5.32a Since the phenylalanine unit acted as a chiral auxiliary, a moderate diastereoselectivity was observed (about 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 dr).
:4 dr).
 | ||
| Scheme 2 Selective modification of glycine units by Steglich et al.32a | ||
Afterwards, Skrydstrup further improved this methodology. He carried out the selective bromination of glycine residues in peptides, and then treated them with SmI2.33 The resulting enolates reacted with electrophiles to give α-alkylglycine derivatives. This method did not require protection of the glycine units, but involved several reaction steps.33a
Another advance was reported by Kazmaier et al. working with small peptides.34 An efficient conversion of glycine residues 6 into their enolates 7 was achieved, followed by an alkylation or allylation catalyzed by palladium complexes (conversion 7 + 8 → 9, Scheme 3). Remarkably, an excellent regio- and diastereoselectivity was observed. In the examples studied, an adjacent (S)-amino acid resulted in the formation of a C-terminal (R)-amino acid, and viceversa. A N,N,O-tridentate chelate 7 is probably formed, where the lateral chain of the adjacent amino acid hinders the approach of the electrophile 8 from that face, giving the unusual (S,R)-product.
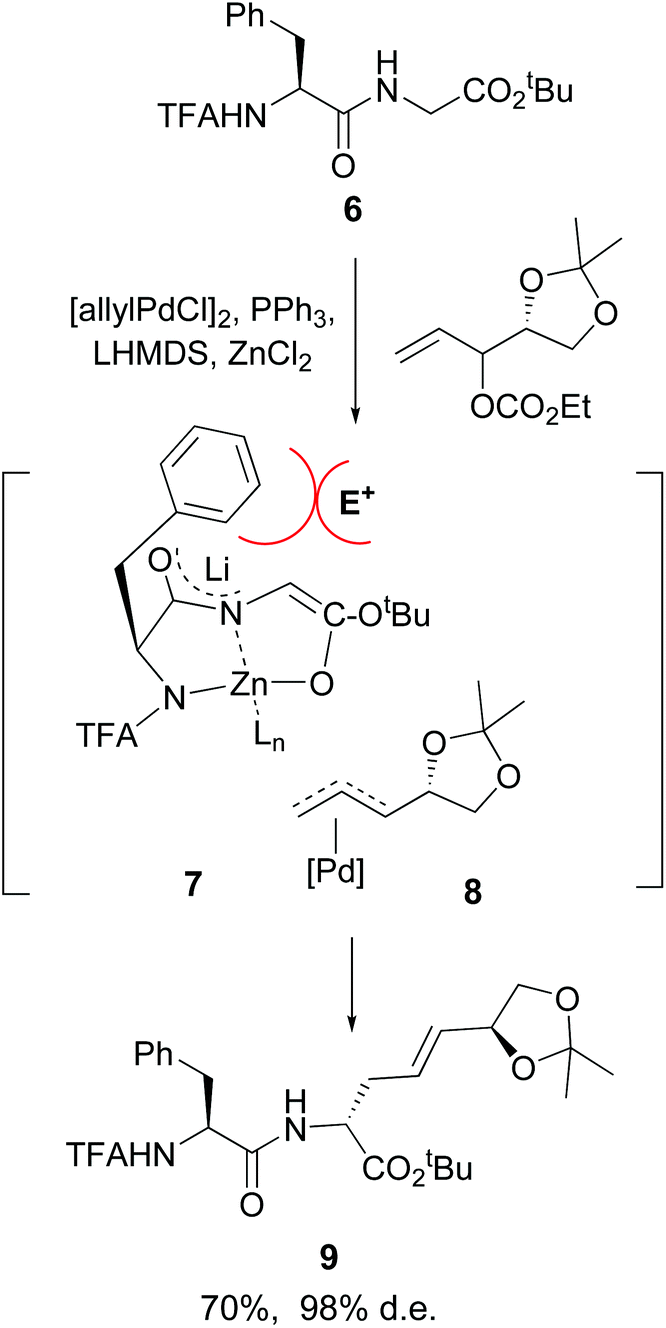 | ||
| Scheme 3 Selective modification of glycine units by Kazmaier et al.34a | ||
In another promising approach, Li et al. reported the site-specific functionalization of glycine residues in peptides using an aryl, alkenyl or alkynyl boronic acid, tert-butylhydroperoxide (TBHP) and copper catalysts (conversion 10 → 13–16, Scheme 4).35a,b The CuBr/TBHP system promotes a dehydrogenative oxidation of the substrate 10, probably through a single electron transfer process (SET) which produces a radical cation 11.35c,d The latter reacts with Cu(II) species to give the imine 12. A tautomerization follows, and the resulting iminol coordinates with the boronic acid to afford the intermediate 13.35b Then the aryl group is transferred to the imine bond, to give the final products. The proposed mechanism was based on two observations: if a tertiary amide was used and the tautomerization was not possible, the reaction did not take place. In addition, when the imine 12 was preformed and treated with the boronic acid in the absence of the copper catalyst, the arylation took place in high yield.
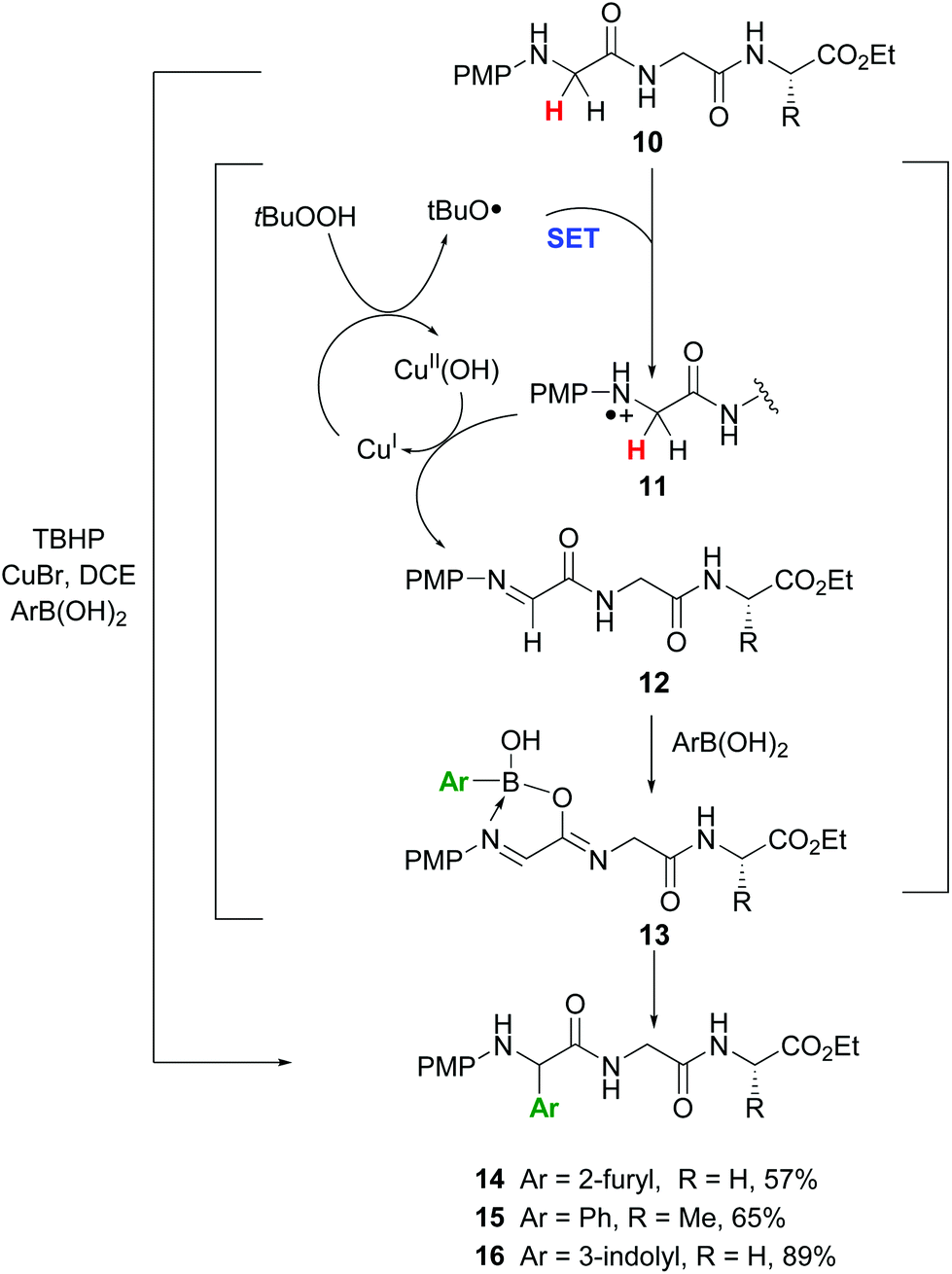 | ||
| Scheme 4 Selective modification of glycine units by Li et al.35a | ||
The reaction was completely regioselective: the PMP group makes that only the methylene group in the N-terminal position can be functionalized. No racemization of the other stereocenters was observed.
Recently, photocatalytic methods were used to introduce substituents on N-aryl glycine residues in peptides (conversion 17 → 20–24, Scheme 5).10 The reaction mechanism involves a radical–radical coupling. Thus, the copper(I)-promoted reductive decarboxylation of hydroxyftalimides under blue LED irradiation led to an alkyl radical (R˙). On the other hand, the resulting Cu(II) complex oxidized the N-terminal glycine in substrate 17 to give a cation radical 18. This intermediate underwent a H-shift in the presence of base, affording a glycyl radical 19. The coupling of radicals 19 and R yielded the final products 20–24.36 The mild conditions allowed good functional group tolerance. Although 1:1 mixtures of diastereomers were obtained, probably fine-tuning of the catalyst would improve the ratio. In any case, obtaining both isomers could be interesting for structure–activity relationship (SAR) studies. The authors illustrated the utility of this methodology for drug synthesis with the production of two enkephalin fragments and an analogue of collagen tripeptide.
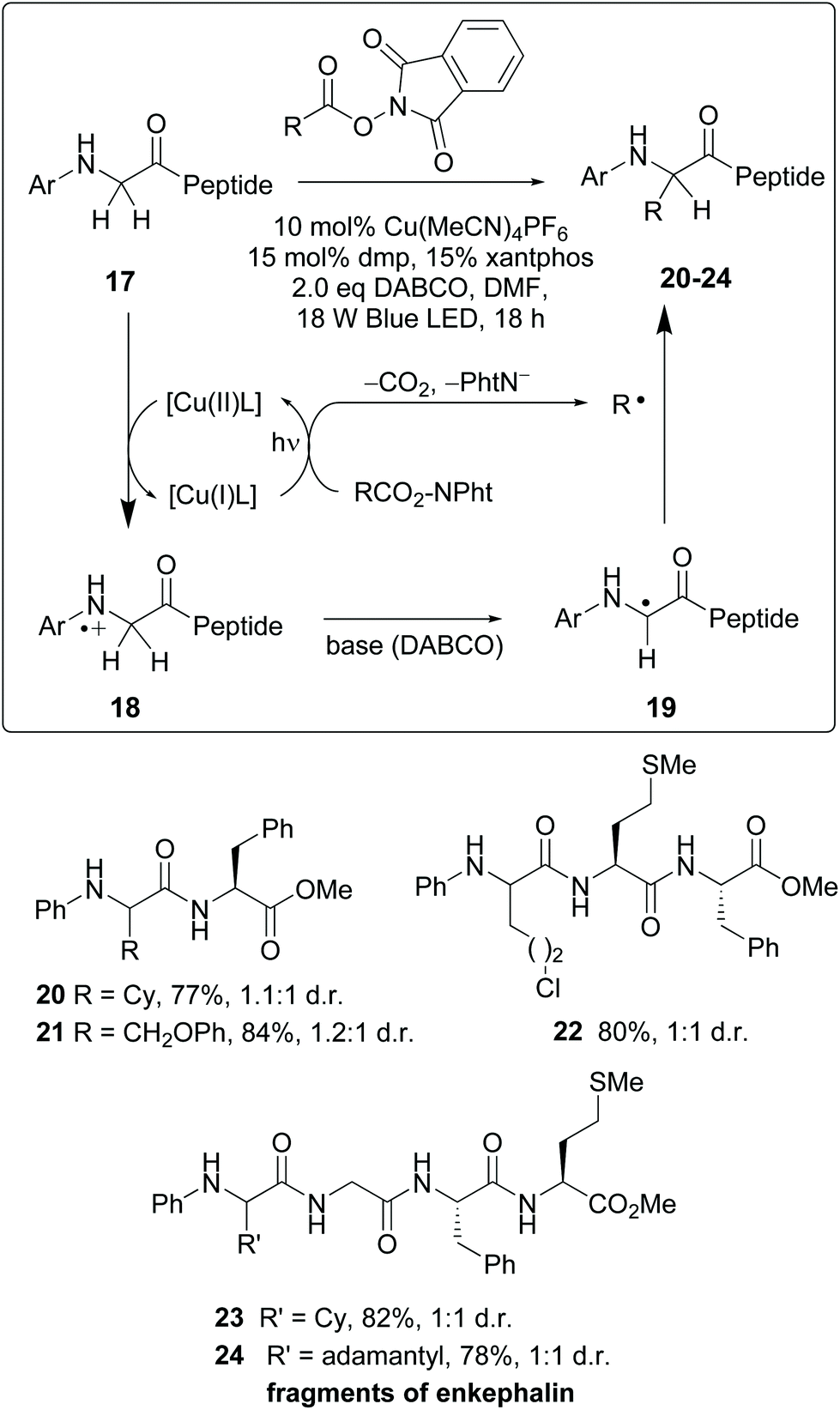 | ||
| Scheme 5 Selective modification of glycine units by Wang, Xu et al.36 | ||
Xu et al. reported the alkylation of modified N-terminal N-aryl glycine residues using Katritzky salt.37 The mechanism is similar to the previous one (conversion 25 → 28, Scheme 6).10 Upon irradiation, single electron transfer (SET) took place from the glycine amino group to the pyridinium salt, generating the intermediate 26. The dihydropyridine radical underwent rearomatization, releasing a radical (in the example, R˙ = Cy˙), while the glycyl radical cation underwent H-shift in the presence of base to give a glycyl α-radical (conversion 26 → 27). Both radicals coupled to give the final product (compound 28).
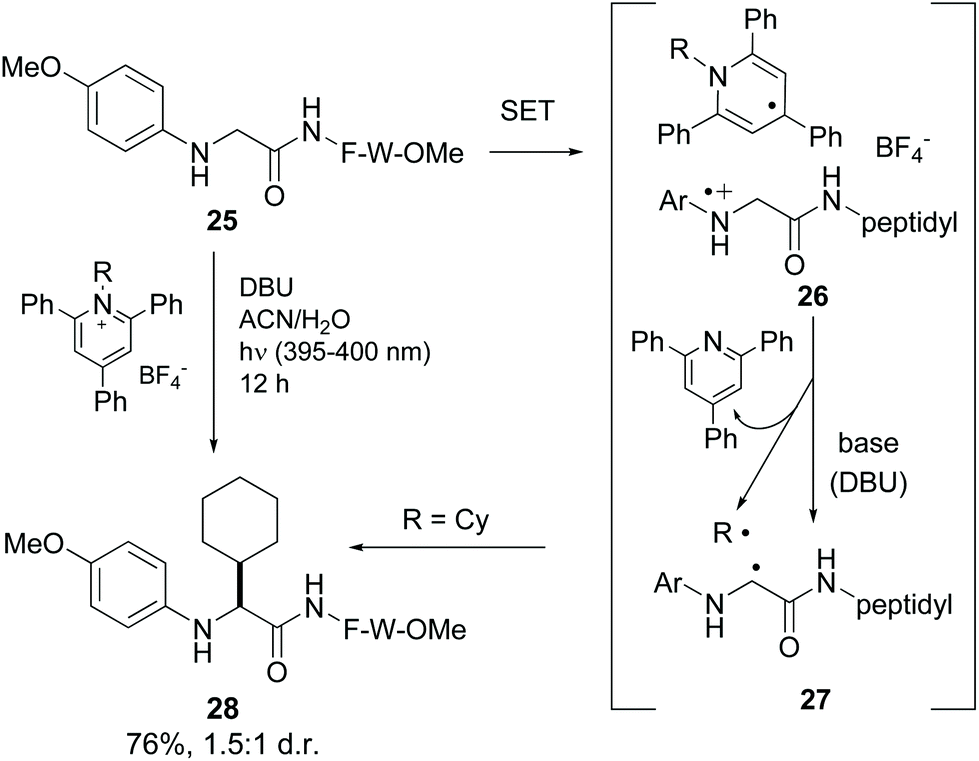 | ||
| Scheme 6 Selective modification of glycine units by Xu et al.37 | ||
Lately Xu, Chang et al. reported the transition-metal free alkylation of N-arylated glycine units in peptides, and its application to the synthesis of new Leu5-enkephalin analogues (conversion 29 → 30, Scheme 7).38 They used either alkyl chlorides or iodides, under irradiation of about 254 nm. In the case of chlorides, 50 mol% of TBAI was added to facilitate the reaction. Since a variety of alkyl chlorides and iodides are commercially available, this method could be useful to expand the range of α-alkyl glycines for SAR studies.
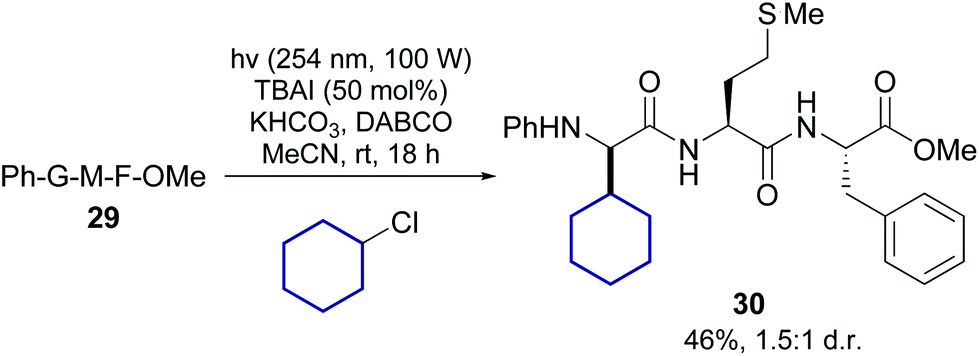 | ||
| Scheme 7 Metal-free promotion of peptide modification.38 | ||
Other type of “customizable units” use cyclic amino acids that can be opened by the addition of nucleophiles, decreasing backbone rigidity. For instance, Garner coupled amino acids and peptides 31 to derivatives of aziridin carboxylic acids (Azy units) 32 (Scheme 8). The resulting peptides were hydrolyzed under aqueous acidic conditions to give di- to hexapeptides 33 with a Xaa-Thr moiety.39 These peptides presented a more flexible backbone, and thus this transformation can be used to modulate ring rigidity and conformation. The aziridine-ring opening reaction can be used with other nucleophiles, as shown in the following examples.
 | ||
| Scheme 8 Modification of aziridin carboxylic acid units by Garner.39 | ||
Van der Donk has studied the cleavage of aziridin carboxylic acids in tri- to heptapeptides with S-nucleophiles,40b to provide valuable glycopeptides in high global yield (conversion 34 → 36, Scheme 9, 92% average yield per step in the example shown).
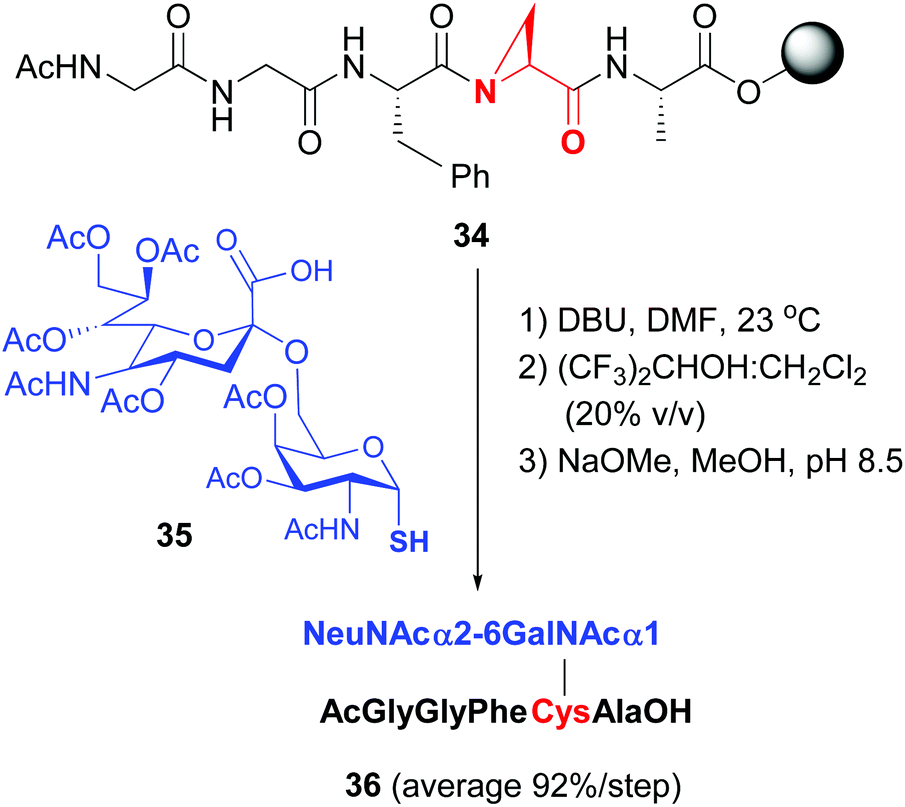 | ||
| Scheme 9 Modification of Azy units by van der Donk, Gin et al.40b | ||
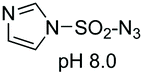
On the other hand, Cohen and Halcomb introduced sulfamidates derived from serine and allo-L-threonine for peptide modification (Scheme 10). The customizable units were introduced as their p-methoxybenzyl derivatives for solid phase synthesis. Cleavage of the protecting group in peptide 37 and treatment with the 1-thiosugar 38 at pH = 8 resulted in sulfamidate ring opening to afford a glycosyl cysteine derivative 39. Standard resin cleavage afforded peptide 40 with excellent purity.41
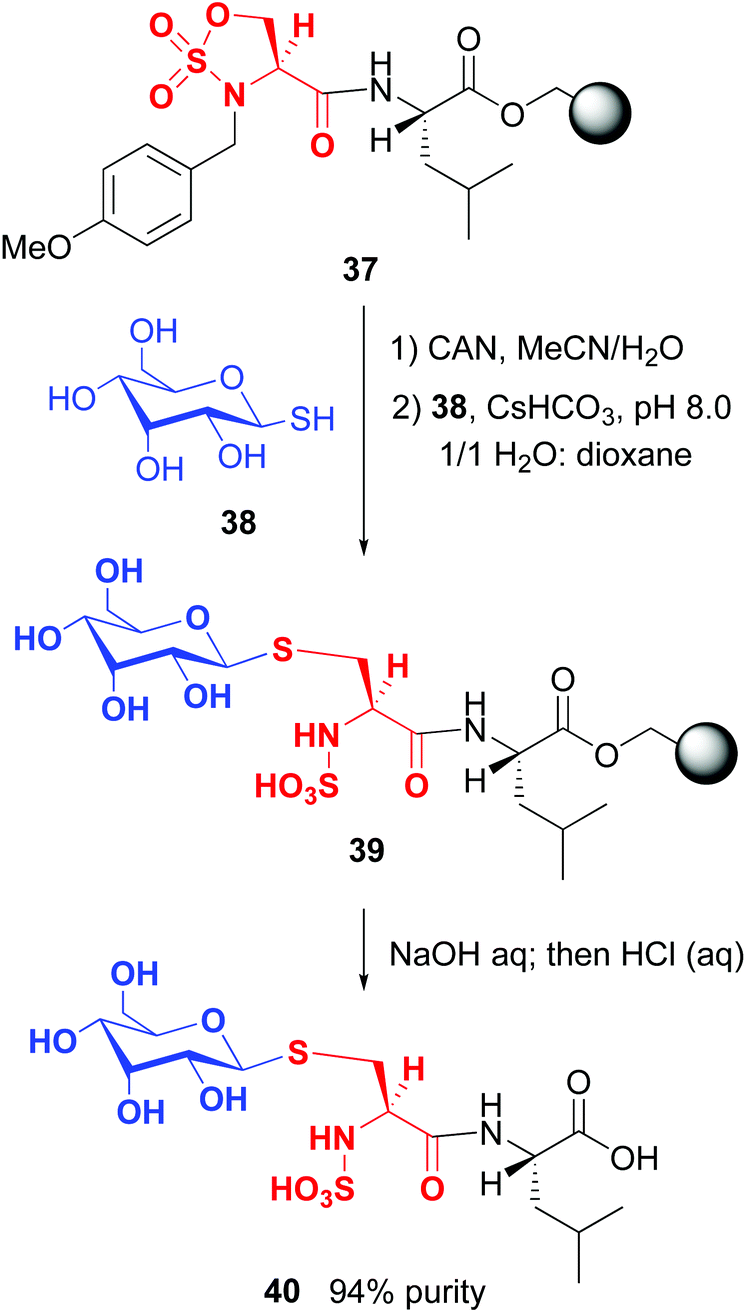 | ||
| Scheme 10 Modification of sulfamidates by Cohen and Halcomb.41 | ||
The sulfamidates were incorporated into peptides for solution and solid-phase synthesis, in both cases affording S-linked glycopeptides in good yield and excellent purity. Lubell has developed a related strategy to introduce α- and β-amino γ-lactams at the N-terminus during solid phase synthesis.42 The lactams were incorporated into analogues of the growth hormone secretagogue GHRP-6, restricting their conformational mobility. It was found that the selectivity for two receptors (GHS-R1a and CD36) could be fine-tuned by lactam introduction.42a
Busto and Peregrina have reported a related transformation of the peptide backbone, but using a 2-methylisoserine sulfamidate.43 Upon treatment with nucleophiles, a ring-opening of the cyclic sulfamidate in peptide 41 took place, producing a quaternary center with total inversion of configuration (Scheme 11). Remarkably, when sodium azide was used as a nucleophile, quaternary azides 42 were obtained, which underwent click chemistry to give substituted triazoles at C-α (conversion 42 → 43).43b Moreover, the authors described on-resin sulfamidate ring cleavage with thiocoumarin dyes. Other nucleophiles such as pyridines and thiocarbohydrates were also tried. The latter afforded thioglyco-α/β-conjugates, generally in good yields.
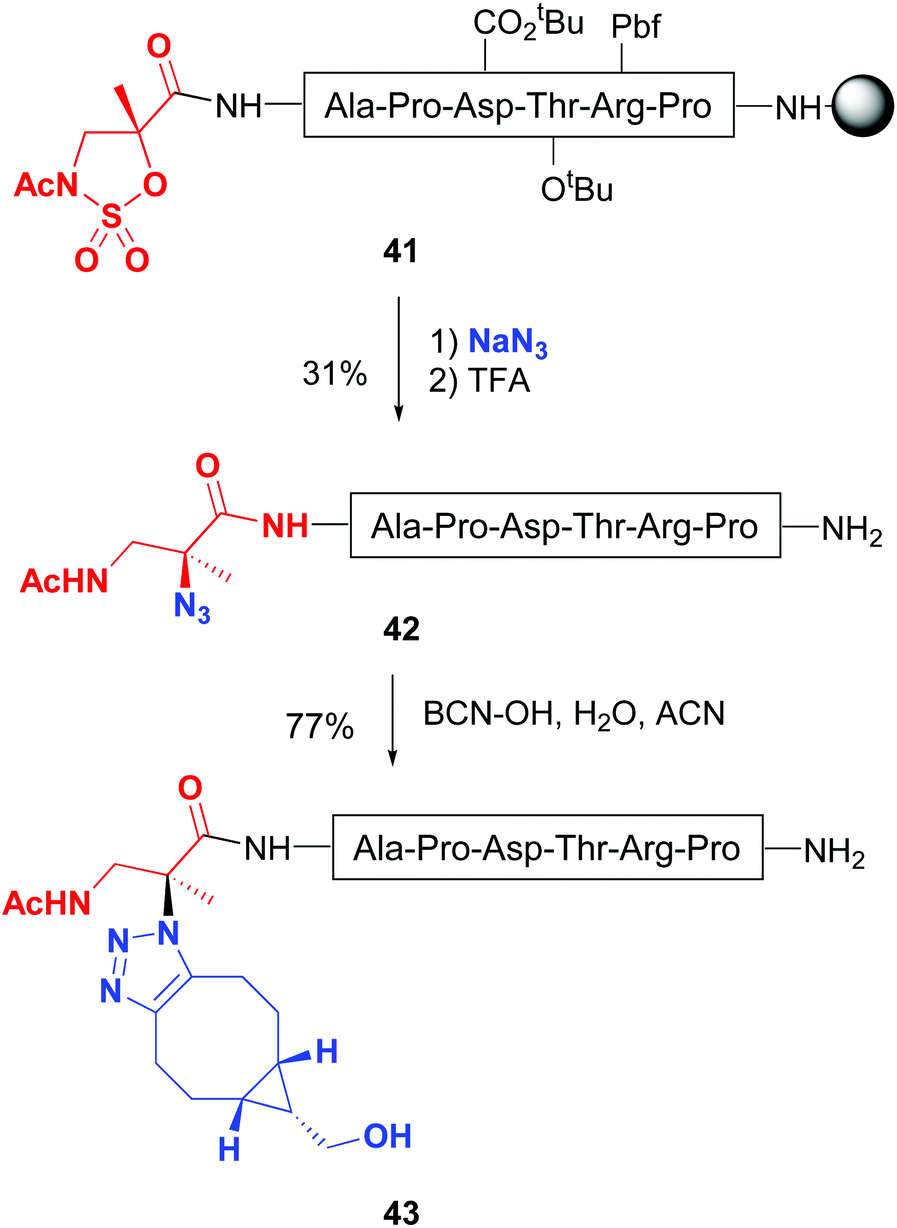 | ||
| Scheme 11 Modification of sulfamidates by Jiménez-Osés et al.43b | ||
In addition to the generation of non proteinogenic α-amino acids in peptides, the selective transformation can allow the generation of β- and γ-units, in order to provide α,β- or α,γ-peptide hybrids.44 Many β- or γ-peptides or their hybrids have the ability to form new turns, helices or fibrils, allowing interesting applications in materials science and biomedicine.45
Thus, Boto et al. used glutamic acid residues as customizable units, for the generation of a variety of γ-amino acids in the peptide backbone.46 Since the carboxyl groups of different glutamic units can have orthogonal protecting groups, it is possible to transform selectively one of the Glu units without affecting the others. An example is shown in the conversions 44 → 49a,b and 44 → 50a,b in Scheme 12, where the oxidative radical decarboxylation of substrate 44 afforded a C-radical 45 which reacted with the reagents (iodine or diacetoxyiodobenzene [DIB]) to afford an unstable halogenated intermediate 46. Substitution of the halo group by acetate from DIB gave a N,O-acetal 47, which was in equilibrium with the iminium ion 48. In the presence of a Lewis acid, the equilibrium was shifted to the formation of intermediate 48, which underwent the addition of C-nucleophiles, to afford hybrid α,γ-peptides 49a,b and 50a,b with good overall yields.
 | ||
| Scheme 12 Generation and modification of glutamic acid units.46 | ||
Most transformations commented up to now have a drawback: when several customizable units are introduced in the peptide, it is generally very difficult to differentiate them. Thus, when a transformation is carried out, all the tunable units will undergo the reaction. For instance, if the peptide contains several glycine residues, it would be challenging to achieve the selective enolization of just one of them.
A solution to this problem is to use, as customizable units, amino acids whose lateral chains can be protected with orthogonal groups. In this way, it is possible to carry out the selective transformation of one unprotected residue, while the other (protected) units remain unchanged.
Among the precedents of this strategy, Steglich pioneered the cleavage of serine residues in peptides with lead tetraacetate (LTA) to give α-hydroxy and acetoxy glycine derivatives.47 Skrydstrup carried out a variation of this methodology, where a radical oxidative scission with LTA transformed the serine unit in peptide 51 (Scheme 13) into an α-acetoxy glycine.33 The resulting compound 52 was reacted with 2-thiopyridine to afford product 53. Subsequent treatment with SmI2 generated an anion which was trapped by electrophiles, for instance carbonyl compounds, affording unnatural α-alkylglycines in the peptides (conversion 53 → 54).33a
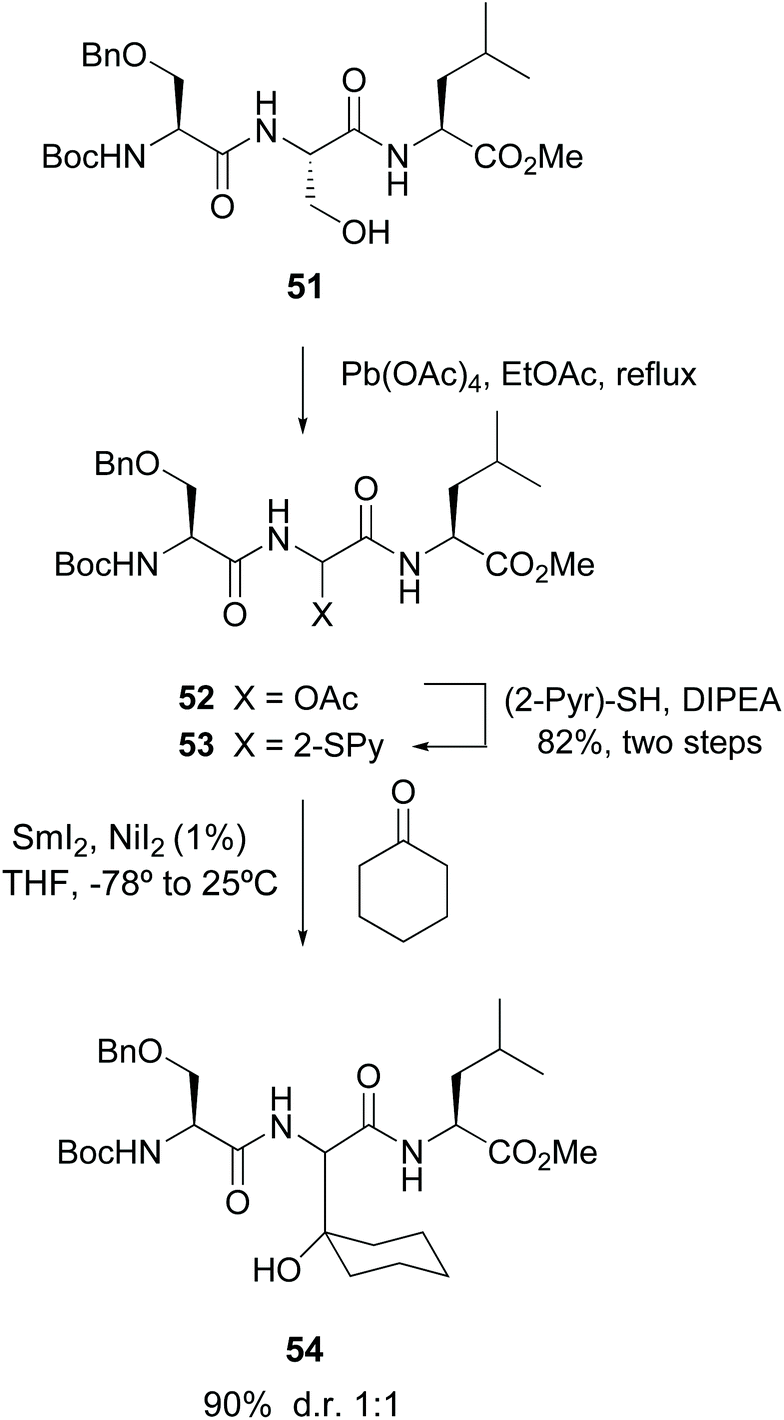 | ||
| Scheme 13 Modification of serine units by Skrydstrup et al.33a | ||
In an alternative strategy, Boto et al. used the oxidative radical scission of serine units to generate an α-acetoxyglycine which was not isolated, but treated with a Lewis acid to provide an acyliminium intermediate which was trapped by different nucleophiles.48 The adjacent amino acids served as chiral auxiliaries and low to moderate stereoselectivities were obtained. This one-pot conversion was a shorter route to a variety of α-alkyl, aryl or allyl glycines.48b However, the authors noted that stereoselectivity during nucleophile addition should still be improved, for instance by using chiral catalysts.
On the contrary, excellent stereoselectivities were obtained in the synthesis of β-substituted dehydroamino acids, which are commonly used to increase peptide rigidity, and the modulation of its conformation and biological activity.49 In the conversion 55 → 57 shown in Scheme 14 the initial step is the radical scission–oxidation of the threonine residue, which was followed by the addition of phosphorous nucleophiles.49a The resulting aminophosphonates (e.g. compound 56, 1:1 dr) underwent a Horner–Wadsworth–Emmons reaction to give β-substituted dehydroamino acids such as product 57 with excellent Z stereoselectivity. Both hydrophobic and cationic residues can be introduced, which was used for the creation of a library of potential antimicrobials.
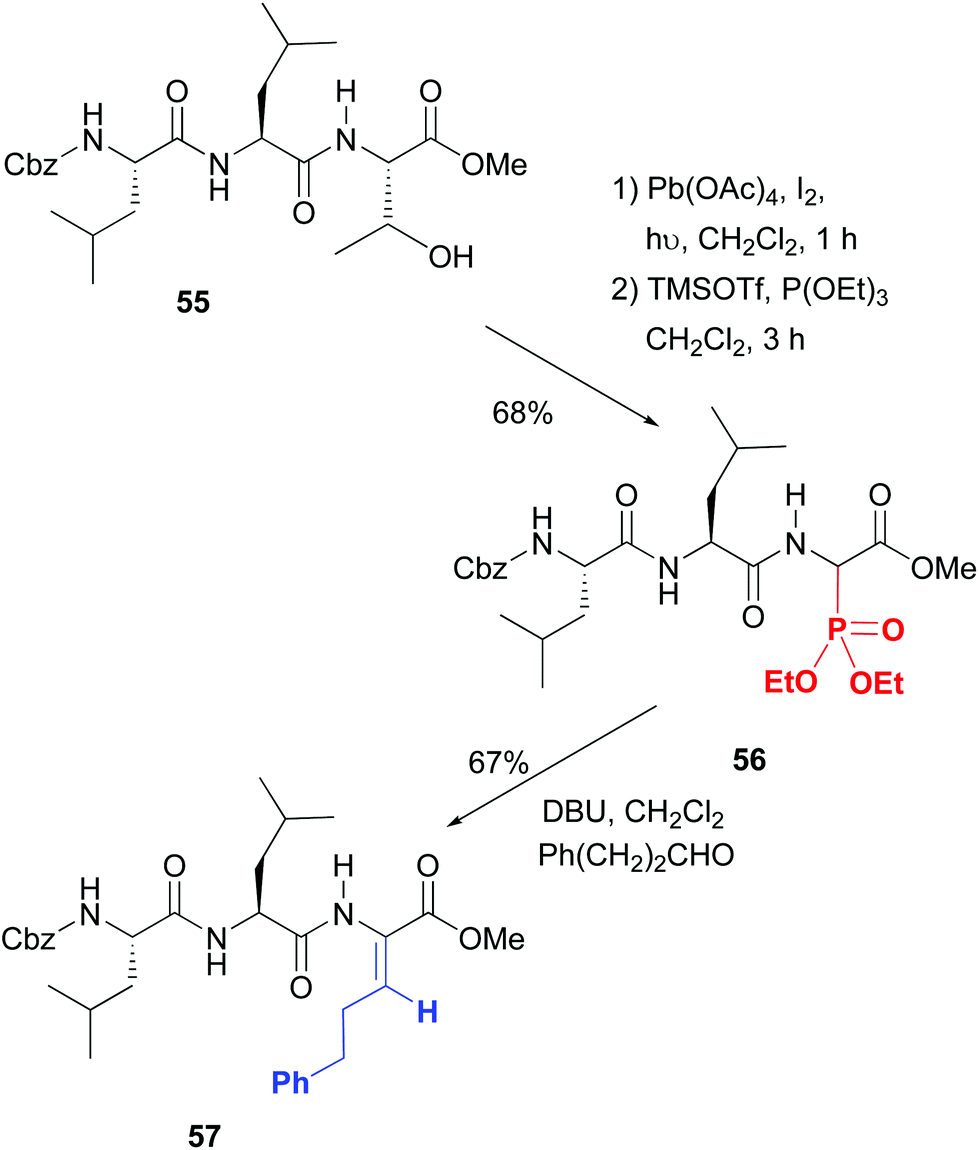 | ||
| Scheme 14 Scission of threonine and generation of dehydroamino acids by Boto et al.49a | ||
The drawback of many of these procedures is the moderate stereocontrol. Therefore, a significative advance was obtained with the development of customizable units, such as hydroxyproline by Boto et al.11,50 and proline by White et al.,12 that allowed the generation of new residues retaining a high optical purity.
The introduction of hydroxyproline units whose pyrrolidine ring could be cleaved at the C4–C5 bond allowed the conversion of a rigid, cyclic unit into a new acyclic unit with an N-substituent (Scheme 15). Both the C-α and the N-chains could be manipulated independently, which afforded a high diversity in the product libraries. In the example shown (conversion 58 → 61), an internal Hyp unit was functionalized selectively in the presence of another customizable unit. The terminal Hyp moiety, with a protected reactive group, did not undergo the oxidative radical scission.11,50a
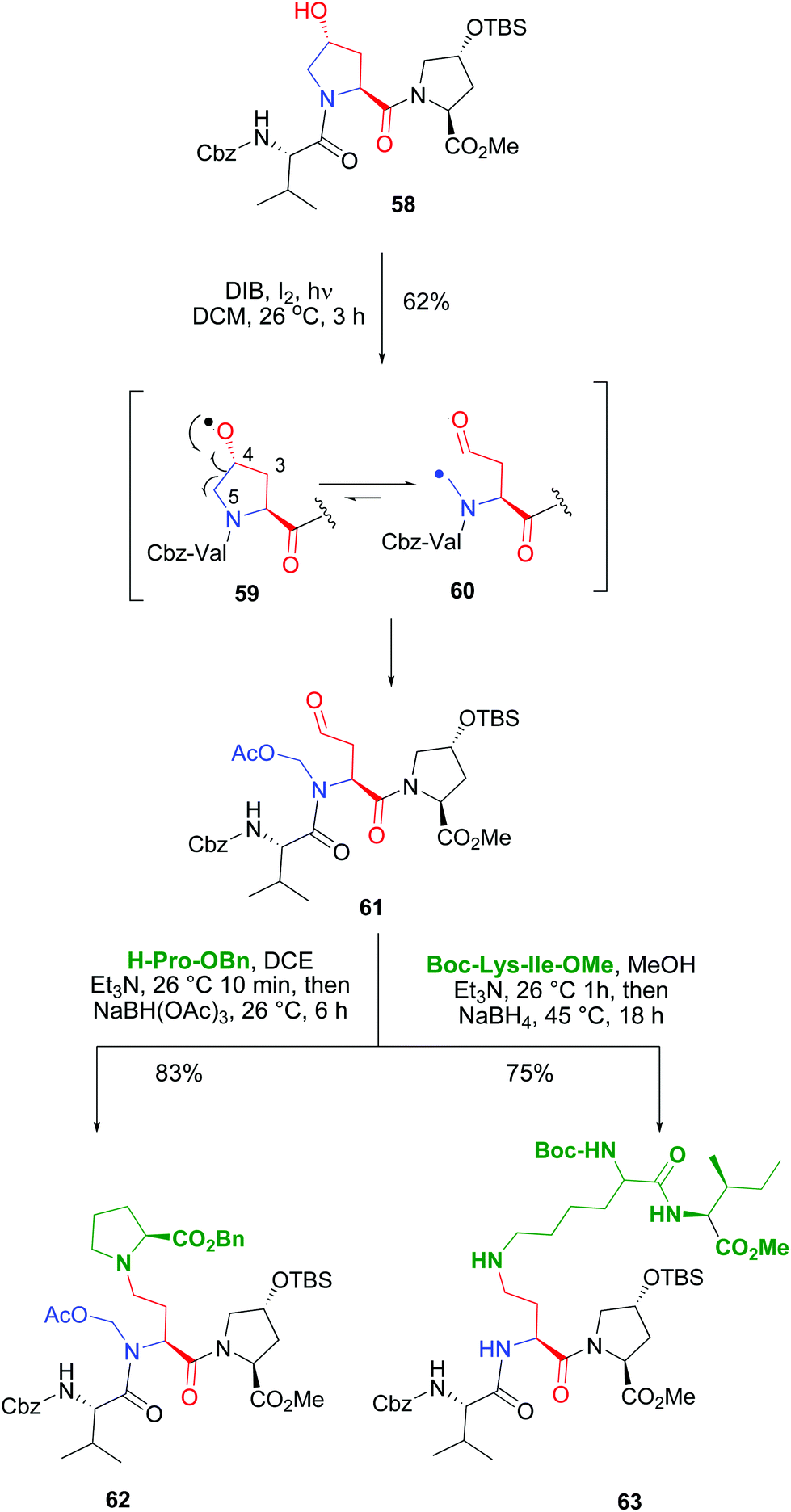 | ||
| Scheme 15 Use of Hyp units for selective peptide modification.50 | ||
The scission of the initially formed O-radical 59 was regioselective, involving only the C4–C5 bond. The alternative C3–C4 bond scission was not observed. This is due to the different stability of the resulting C-radicals. In both fragmentations a primary radical would be formed, but in the C4–C5 scission, the C-radical 60 would be stabilized by the adjacent nitrogen. Then the C-radical reacted with iodine or DIB to give an unstable halogenated intermediate (as commented before in Scheme 12), that evolved to give an N-acetoxymethyl group. The resulting fragmentation product 61 presented an α-chain with a terminal carbonyl group, and an N-acetoxymethyl substituent. Both chains could be transformed independently as shown in the conversion of compound 61 into the branched peptide 62 using a reductive amination. Interestingly, this reaction can also be used to ligate two peptides, as shown by conversion of tripeptide 61 into the pentapeptide 63. In this case, using different reductive amination conditions, the N,O-acetal was cleaved, and a more flexible backbone was achieved.
The scission-reductive amination was then used to ligate two peptides, and at the same time, to generate a rigid α-amino-γ-lactam unit to rigidify the backbone (conversion 64 → 66, Scheme 16).11,50b Other applications of this methodology have allowed the preparation of diketopiperazine scaffolds with four different substituents,51 and the production of a library of cyclic antitumoral peptides, as sansalvamide A analogues.52
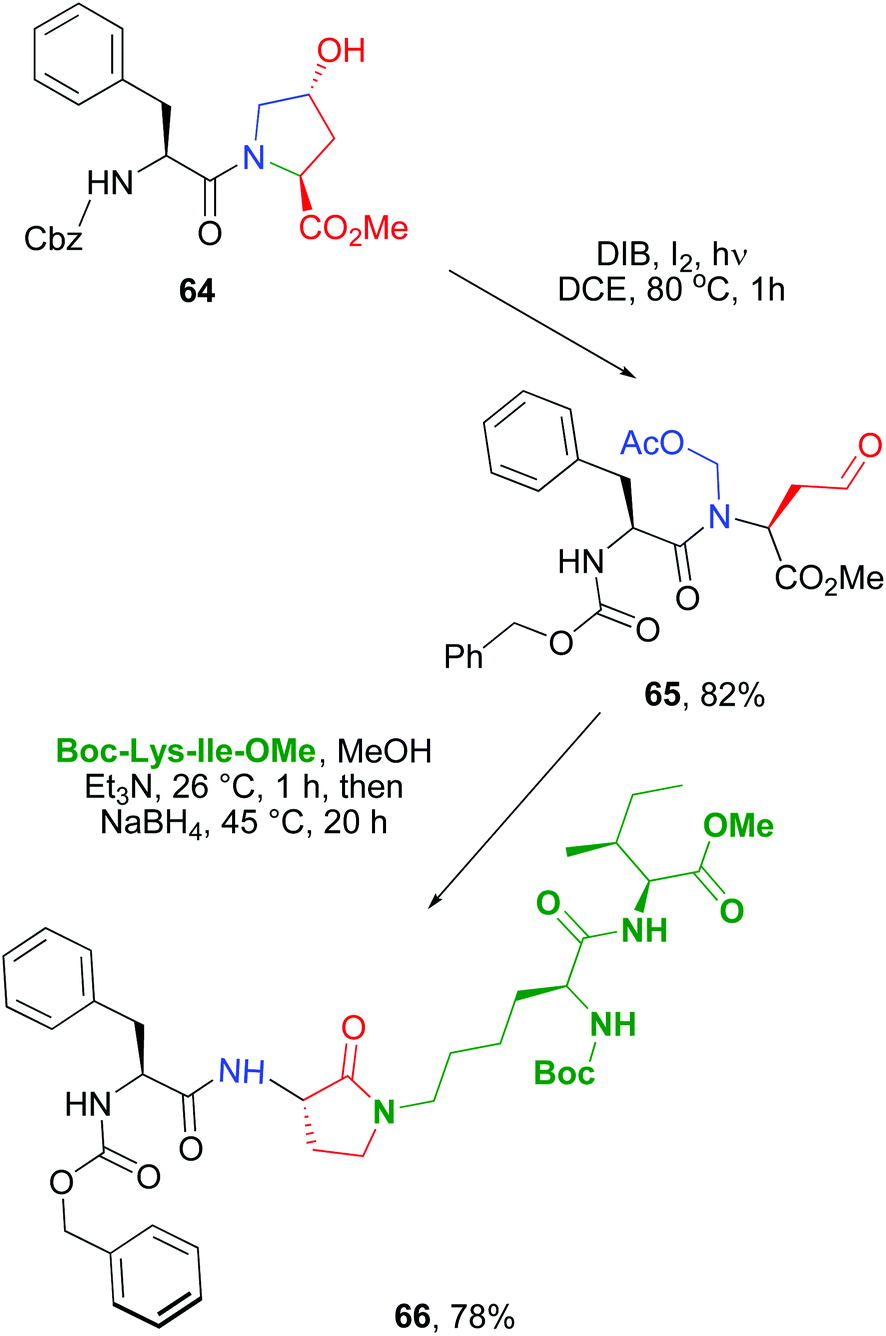 | ||
| Scheme 16 Use of Hyp units to ligate peptides with rigid Agl units.50b | ||
Recently the use of Hyp as a doubly customizable unit was reported by the same group (Scheme 17).11 A one-pot radical decarboxylation–oxidation–addition of C-nucleophiles allowed the modification of the C-terminal position of the peptide 67 and replacement of the carboxyl group by a variety of other chains, with high stereoselectivity due to the presence of an stereogenic center at C-4. Deprotection of the hydroxyl group activated the unit 68 for C4–C5 cleavage of the pyrrolidine ring, affording a β-amino aldehyde that could be converted into other derivatives as seen before. In this way, products 69 with three tailored chains and high R or S purity can be obtained.
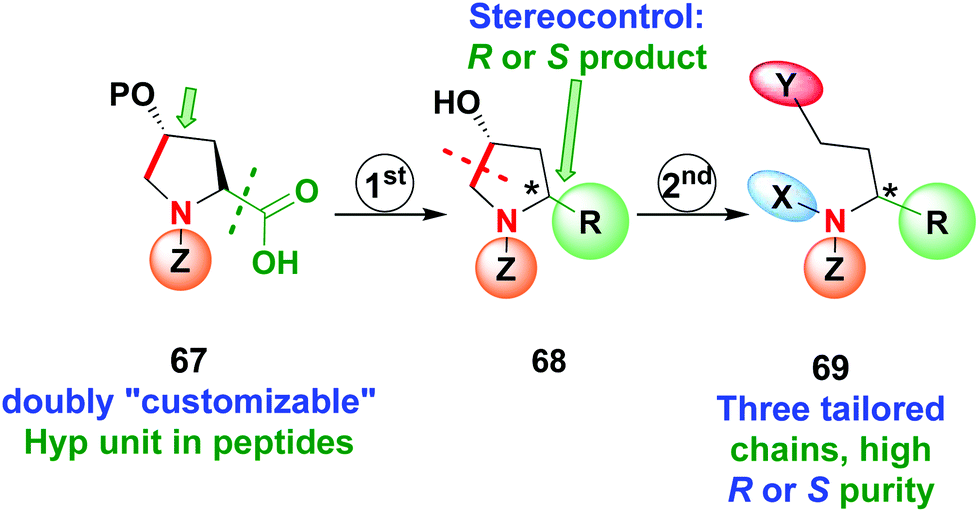 | ||
| Scheme 17 Use of doubly customizable Hyp units by Boto et al.11 | ||
In 2016, a remarkable work by White, Osberger, Rogness et al. introduced proline as a customizable unit,12 using an iron-catalyzed oxidation inspired by non-ribosomal peptide synthetases. The cleavage of proline at the C5–N1 positions proceeded with no loss of chirality (Scheme 18, conversions 70 → 71, 70 → 72 and 73 → 74). Interestingly, the chemoselectivity could be tuned. Thus, Ns-protected N-terminal prolines underwent oxidation to an aminol, while those at the C-terminus or internal position underwent oxidation either to glutaraldehyde or to glutamic acid. The intermediate aminals or aldehydes can be further modified, for instance by reductive amination (compound 71), the addition of C-nucleophiles (compound 72) or oxidation (compound 74).
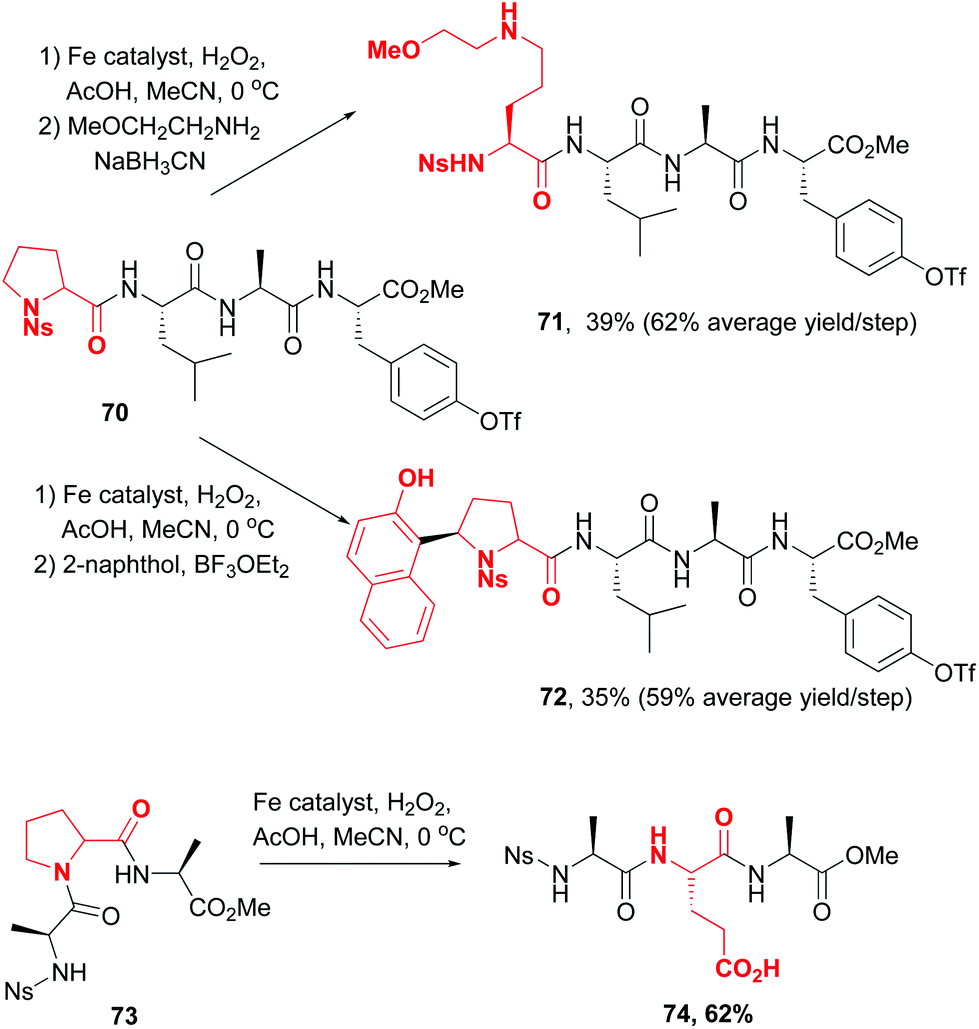 | ||
| Scheme 18 Use of proline customizable units by White et al.12 | ||
2.2. Modifications at C-α using dehydroamino acids
Dehydroalanine (Dha) and other dehydroamino acids (dhAAs) are special cases of “customizable units”. The transformation of alicyclic amino acids into dehydroamino acids and viceversa can have a profound impact on the backbone and general peptide conformation, and thus on its biological activity.13 The dhAAs present a planar conformation with restricted conformational flexibility, and a strong preference for trans peptide bonds. Therefore, they are valuable turn inducers and increase peptide rigidity, in many cases favoring interactions with the target.15Dha can be formed from serine or cysteine, by using an elimination reaction (conversion 75 → 76, Scheme 19).13,14 The advantage of using cysteine is its relative scarcity, which in many cases allows the creation of just one Dha residue (or one surface-exposed unit).53 There are different methods to prepare Dha residues from cysteine, but the most used is oxidative elimination,54–56 by treatment with o-mesitylenesulfonyl hydroxylamine (MSH)6b,54,55 or with dibromobisamides.56 For instance, mutant protein S156C SBL underwent oxidative elimination with MSH of its single surface-exposed cysteine (Cys156) to afford a derivative with one Dha residue.54 In a similar way, histone mutant H3 was treated with a dibromobisamide, affording a derivative with a single Dha unit (Dha-9).56 Other examples can be found for GFP mutants, AurA kinase domain, and antibodies, among others.3f,55a,57
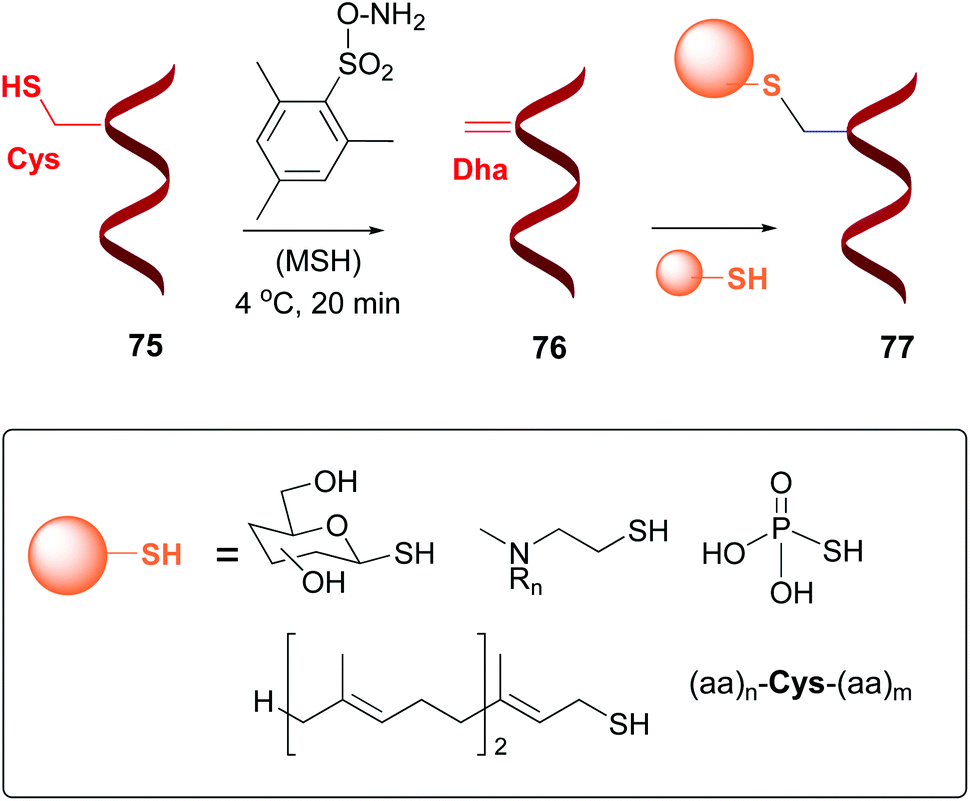 | ||
| Scheme 19 Generation and modification of Dha units.4a,55b | ||
The resulting Dha residue presents an “umpolung” reactivity with respect to the parent cysteine. Since the double bond has electron-withdrawing and electron-donor substituents, which create a push–pull effect, Dha easily undergoes the addition of different nucleophiles, as well as radical additions, cycloadditions and metal-promoted cross-coupling reactions.14 A comprehensive review by Bowers et al. was recently published13 but herein we will summarize the transformations affecting the peptidic backbone.
Davis et al. reported the transformation of cysteine into dehydroalanine residues as shown in Scheme 19, and the subsequent addition of sulfur nucleophiles to give peptides 77 (conversion 76 → 77, Scheme 19).54a,55b,56 The peptides were thus glycosidated, aminoalkylated, phosphorylated, or prenylated, among many conversions.56,58
Davis applied this method for the functionalization of subtilisin, giving more than 95% conversion in 90 min in a phosphate buffer.54a Van der Donk used a similar strategy for peptide functionalization.59
Brik et al. have reported the use of dibromobisamides to transform cysteine into Dha in ubiquitin probes.60 These probes were constituted by an ubiquitin fragment (Ub 1–75) attached to a TAMRA fluorophore (carboxytetramethylrhodamine) through a PEG linker. The dehydroamino acid “trap” units were used to capture different enzymes by their catalytic cysteines, for instance USP2 (belonging to the universal stress protein family), forming adducts with an intense fluorescence.
In a similar way, Pearson created a library of mutant N-acetylneuraminic acid lyases (NAL) by using precursors with dhAA units and treating them with different thiol nucleophiles. One NAL mutant was able to process erythrose, that was poorly accepted by the native enzyme, with high efficiency.61
Davis et al. prepared a library of pantothenate synthetases with histidine-like residues at the active site, using precursors bearing dhAAs at the active site, by treating them with azoles.62 These transformations could be expanded in a future, when reactions developed for dehydroamino acids are optimized for peptides, from the addition of different carbon nucleophiles to Friedel–Crafts reactions.63–67
Other promising strategies use selenocysteine derivatives to generate the dehydroalanine unit. For instance, Schultz et al. performed genetic incorporation of phenylselenocysteine into histone 78 (Scheme 20).58a Treatment with hydrogen peroxide afforded the H3-Dha9 derivative 79. Thiols added to the Dha-residue in a Michael reaction, giving methyl and acetyllysine analogues (compounds 80a–c). Similarly to the natural proteins, compound 80a was recognized by the enzymatic HDAC3/NCOR1 complex, which performed a selective deacetylation. Moreover, the resultant product 81 was accepted by Aurora B kinase, which phosphorylated the adjacent serine residue (S10).
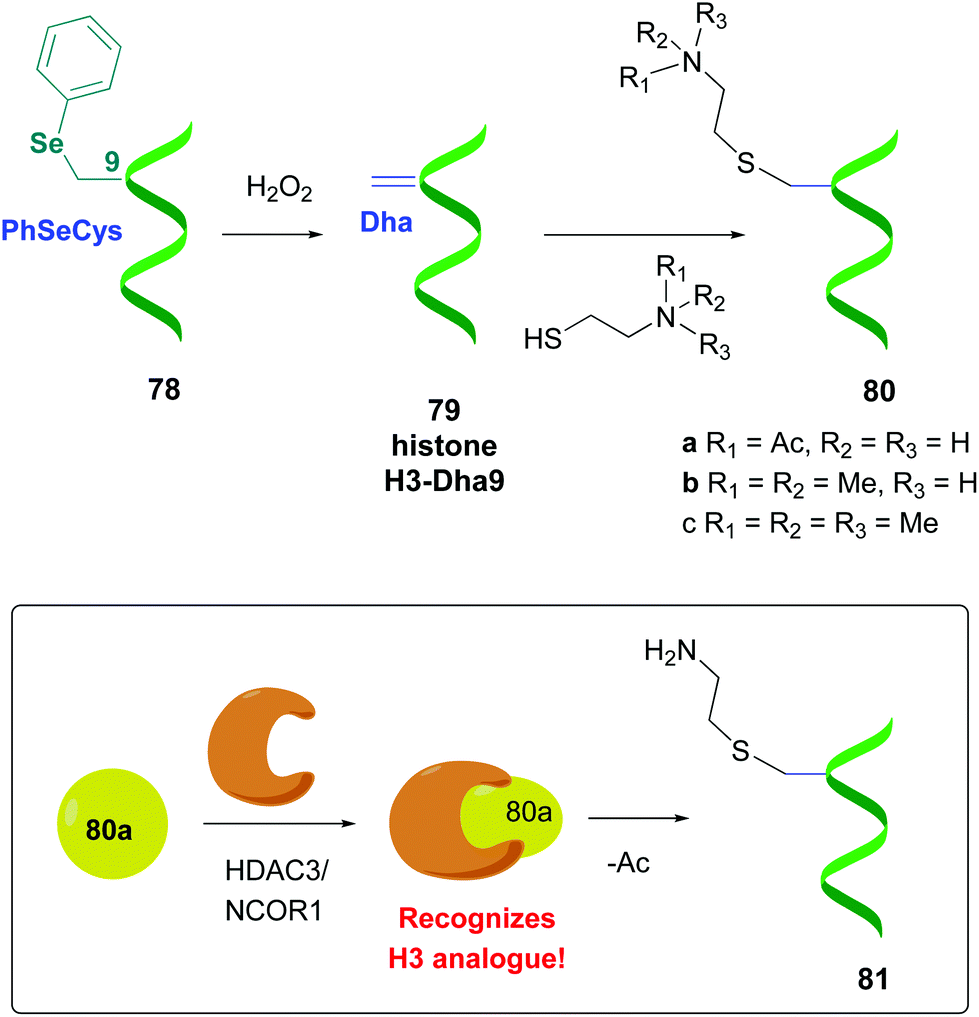 | ||
| Scheme 20 Protein modification using selenocysteine derivatives by Schultz et al., in order to study histone function.58a | ||
Davis described a reversible protein labeling method using allylselenocysteine analogues (Seac).54d,68 They used as substrate a histone H3 with a Dha residue at position 9 instead of the usual lysine (K9). When the substrate was treated with allylselenide, a conjugate addition to Dha took place, providing the allylated selenocysteine.68 Olefin metathesis then gave a mimic of K9Ac side-chain (write step). The resultant histone could be “read” (recognized) by anti-LysAc antibodies. The label could then be removed (“erased”) via selenoxide elimination to regenerate the substrate H3-9Dha. In this way, many different labels can be installed in order to study their impact on protein function.
The addition to Dha usually produces an epimeric mixture, but as Davis pointed out, the resulting products are still valuable for biological studies. For instance, Davis et al. prepared different protein substrates in order to study their interaction with endoglycosidase A (Endo A) in the synthesis of glycoproteins. Dha residues were incorporated into proteins and then GlncNAc cysteine residues were created by addition of sugar thiols.54e Apparently, both epimers were accepted by the enzyme. In another example, Davis prepared modified histones (H3) with several K9 mimic units, which were recognized by antibodies for the original histone.54a Besides, a related phosphoserine mimic which presented diastereomeric mixtures was recognized by a chromatin reader protein. In fact, even if the activities were different for epimers, comparison of both epimers and their more rigid Dha-containing precursor could be valuable in structure–activity relationship (SAR) studies.
Although the method is useful even when stereocontrol is not achieved, some promising reports on stereoselective conjugate additions to dehydroamino acids have appeared, using organocatalysts69 or chiral auxiliaries.70 Similar methods will likely be optimized for peptides.
Interestingly, Naidu et al. reported that the conjugate addition of thiols and amines to dehydroamino acids can be accelerated if the reaction was carried out in water.71 Therefore, this chemistry is quite useful for protein and peptide modification.72
Moreover, C–C bonds can be created by addition of organic radicals to the β-position of the Dha double bond, using modifications of the Luche methodology.73 Thus, these radicals are formed by treatment of organic halides with Cu(II) and Zn(0) usually under acidic conditions (pH 4.5).74,75 Baran and Blackmond have described that the zinc-catalyzed radical cleavage of an activated ester provided radicals that added to dhAAS.75 Park applied this reaction to generate C-radicals from alkyl iodides, and thus introduced different chains onto histone H3, ubiquitin, and green fluorescent protein with high site-specificity. Remarkably, in histone H3, methylated analogues of Lys79 were thus produced. Since methylated histones stimulate transcription, this methodology allowed a better understanding of this key biological process.76
In another seminal work, Davis et al. found that radicals could be generated from alkyl iodide and sodium borohydride in aqueous solutions.77a The generation of radicals was observed by electron paramagnetic resonance (EPR); besides, the reaction was inhibited by TEMPO or acrylamides. With NaBH4, less alkyl iodide equivalents were needed for reaction completion, and high-quality products were obtained. Many functionalized chains were tolerated, such as phosphate, phosphonate, ammonium, guanidino groups and sugar residues. It also served to create methylated Lys analogues. Davis functionalized many proteins, including annexin V, Npβ, SBL, histone H3, H4, p38α, cAbLys, and AcrA. In Scheme 21 the functionalization of Npβ-Dha61 (conversion 82 → 83) and H3-Dha9 (conversion 84 → 85) is shown.
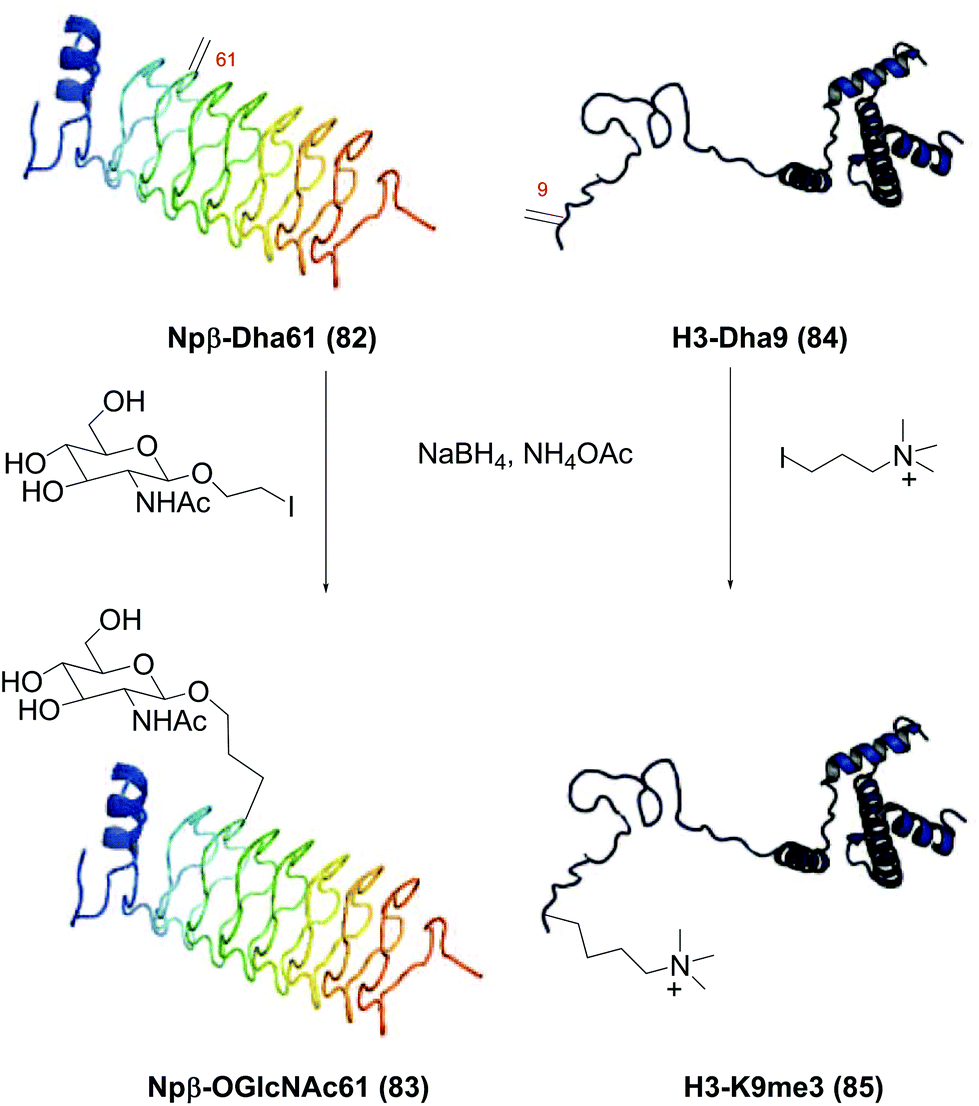 | ||
| Scheme 21 Protein functionalization with C-radicals by Davis et al.77a | ||
Dixon and Jui have generated radicals at room temperature using iridium photoredox catalysts and blue light. Ruthenium and copper catalysts can also be used in such reactions, and the use of different ligands can modulate the redox potential of the metal complex.78 Thus, Jui used an iridium catalyst and alkyl amines or 3-bromopyridines to generate either α-amino or pyridyl radicals.78,79 In this way, dimethylaniline, dimethyladenosine, and complex molecules such as strychnine and the drug diltiazem, and a cholic acid derivative, were attached to peptides (Scheme 22, conversion 86 → 87–89). Using chiral auxiliaries with amino acid substrates provided d.e. up to 95%.79 Later, Dixon generated α-aminoradicals from electrophilic imines, and their addition to dhAAs generated 1,3-diamines.80
 | ||
| Scheme 22 Peptide functionalization with C-radicals by Jui et al.79 | ||
In a remarkable work, Roelfes achieved the functionalization of complex antimicrobial peptides such as thiostrepton and nisin by addition of C-radicals to dehydroalanine (Dha) and dehydrobutyrine (Dhb), using a photocatalytic activation of organoborates under irradiation with visible light.81 The reaction used the iridium complex Ir(dF(CF3)ppy)2(dtbbpy)PF6 as photoredox catalyst. Scheme 23 illustrates the conversion of thiostrepton 90 into the addition derivatives 91. The ratio of mono-, di-, tri- or tetrasubstituted products depended on reaction time and the substrate itself. Thus, thisostrepton needed irradiation with blue LEDS for several hours to give tri- or tetrasubstituted products, but with the more hydrophylic nisin, almost complete addition to the three double bonds had been achieved after 1 h. Although the “customizable units” were not differentiated among them, the reaction was selective for the dehydroamino acids; the other residues were not affected.
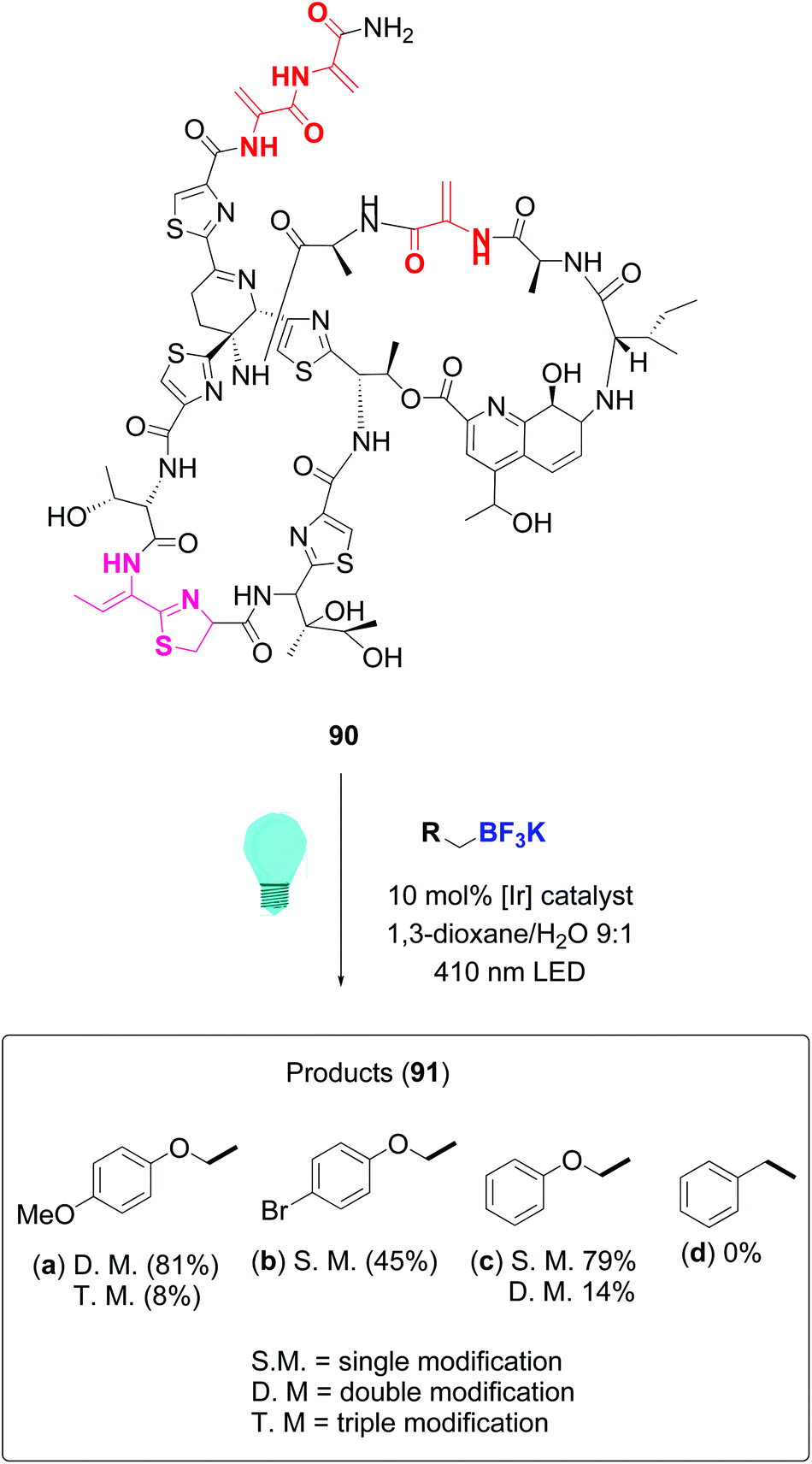 | ||
| Scheme 23 Thiostrepton functionalization by Roelfes et al.81 | ||
Other recent methods reported for dehydroamino acids will likely find applicability in peptide chemistry. Thus, Molander reported that alkyltrifluoroborate derivatives added to Dha derivatives to provide α-fluoro-α-amino acids.82a Fluoro groups have been used as bioisosters of H and other moieties and also to tune the biological properties of peptides and drugs.82a In another example, Leonori has reported that alkyl halides, in particular iodides, can be suitable radical precursors.82b Due to the commercial availability of alkyl halides, this reaction could be quite interesting to obtain peptide diversity.
Cycloaddition reactions have been less studied, but Raines reported that the lantibiotic nisin was successfully labeled in a crude protein mixture, by treatment with electron-rich diazetoamides in water/acetonitrile.83a Roelfes has recently reported the Diels–Alder reaction in peptides such as thiostrepton, nisin and nosiheptide. In the case of thiopeptide nosiheptide, only one dhAA unit reacted, in 75% yield (Scheme 24, conversion 92 → 93).83b
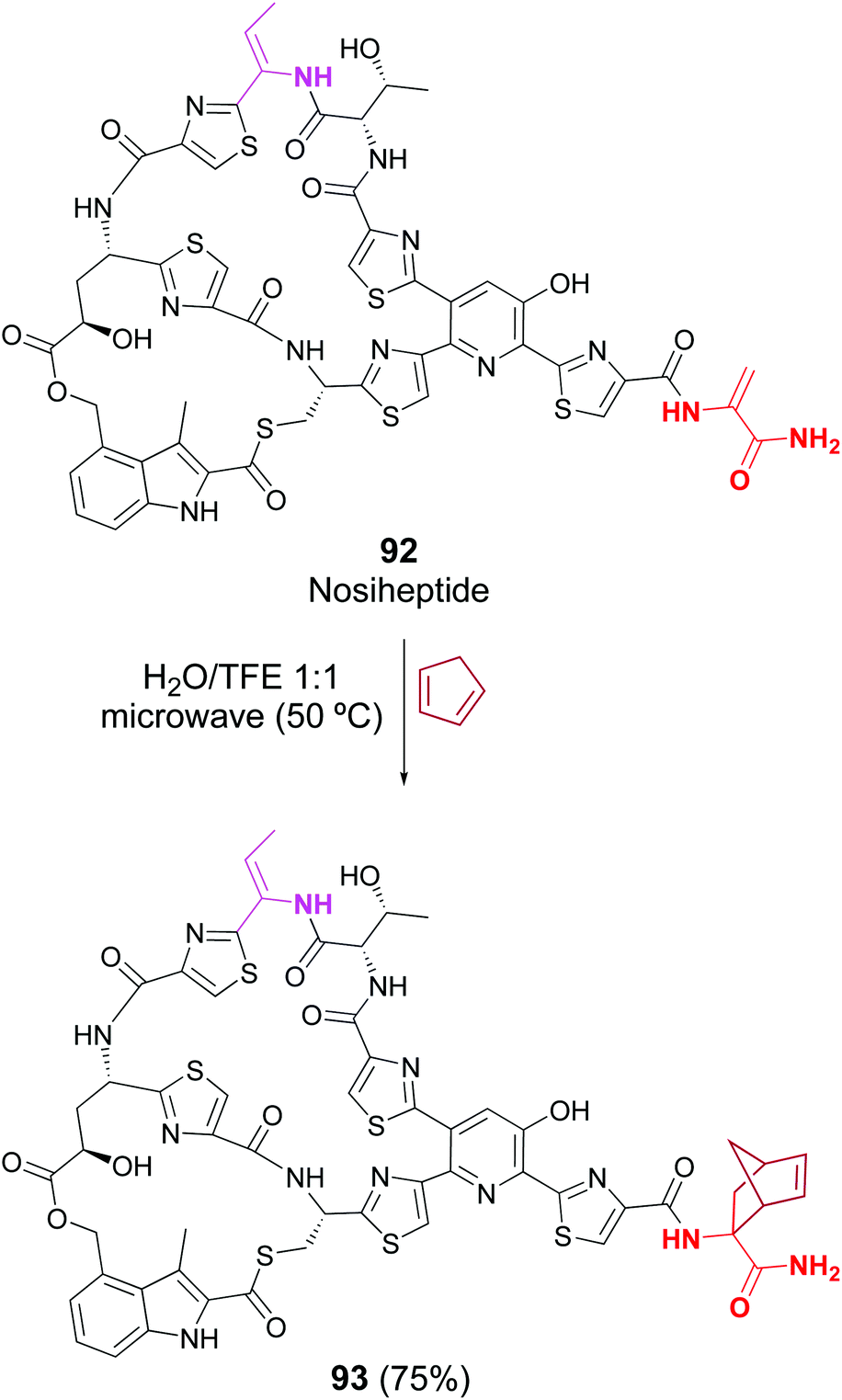 | ||
| Scheme 24 Selective [2 + 4]-cycloaddition in nosiheptide by Roelfes et al.83b | ||
Sun and Wang have reported 1,3-dipolar cycloaddition (DC) between Dha units and C,N-cyclic azomethine imines, where the protecting group (phthalimido or amido) determined predominance of normal- and inverse-electron demand 1,3-DC (conversions 94 → 95 and 96 → 97, Scheme 25).83c
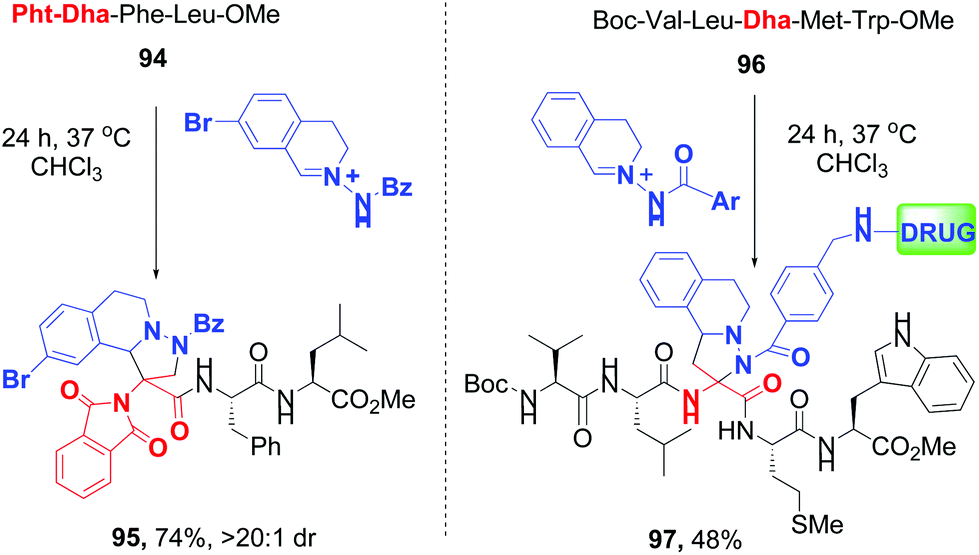 | ||
| Scheme 25 Selective cycloaddition reactions by Sun, Wang et al.83c | ||
Cycloadditions can also be catalyzed by enzymes. Different pyridine synthases from the thiopeptide biosynthesis catalyze aza-[4 + 2] cycloaddition to give a substituted pyridine.84 Other enzymes were engineered to carry out the cyclopropanation of dehydroamino acids in thiopeptides with high regio- and stereoselectivity. The cycloadditions introduce a rigid ring in the peptide backbone that restricts its conformational flexibility.
With respect to transition-metal catalyzed reactions, different from the cited above, a classical example is the reduction of dehydroamino acids using rhodium-catalyzed hydrogenation.85 Dong et al. have applied hydrogenation to the stereoselective reduction of four dehydrophenylalanines in a cyclopentapeptide in protic solvent.86 The dpp ligand gave alternating D- and L-centers, but other ligands could provide different stereochemistries. It must be said that, although the last example was not really site-selective, in other cases the method could serve as such, due to the scarcity of dhAAs in complex peptides.
In another seminal example, the stereoselective creation of C–C bonds using Dha-containing small peptides was carried out by Frost et al.87 Thus, the Dha unit was arylated using a rhodium catalyst, with moderate to good yields (30–86%). For some substrates, excellent d.e. were obtained (conversion 98 → 99, Scheme 26). In other cases, the use of a chiral ligand allowed some stereocontrol. The reaction was applied to the synthesis of urotensin analogues.87 Other examples by Willis and Frost have shown the potential of this methodology.88
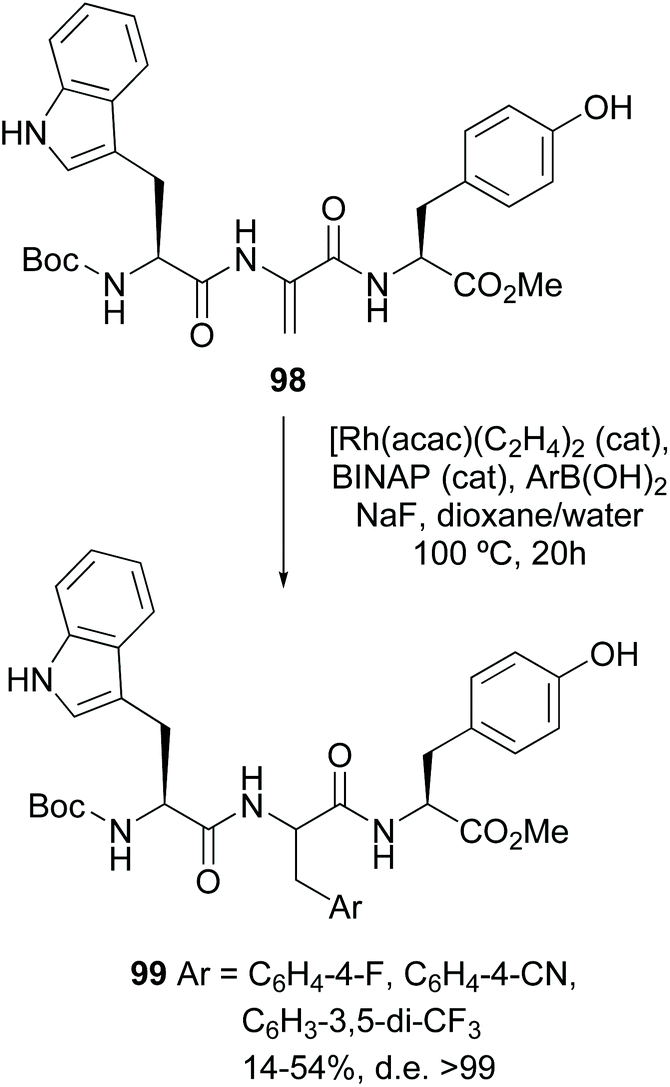 | ||
| Scheme 26 Arylation of Dha units by Frost et al.87 | ||
Miller reported milder conditions (room temperature, by increasing catalyst load) for the site-selective modification of thiostrepton. Although thiostrepton possesses four dhAAs, only Dha16 was modified, and with good stereoselectivity.89 In a related approach, Roelfes reported the use of EDTA-Pd complexes to couple arylboronic acids with dhAAs in peptides and proteins, such as nisin and a small version of SUMO protein. Remarkably, a good tolerance of an aqueous buffer was demostrated.90
Enzymes can also modify dehydroamino acids with exquisite site- and stereoselectivity. An example for their use in cycloadditions has been commented before. Other well known example is the role of lanthipeptide cyclase to create the lanthionine link by addition of a cysteine to a Dha (conversion 100 → 101, Scheme 27), and the labionin unit.55b,91
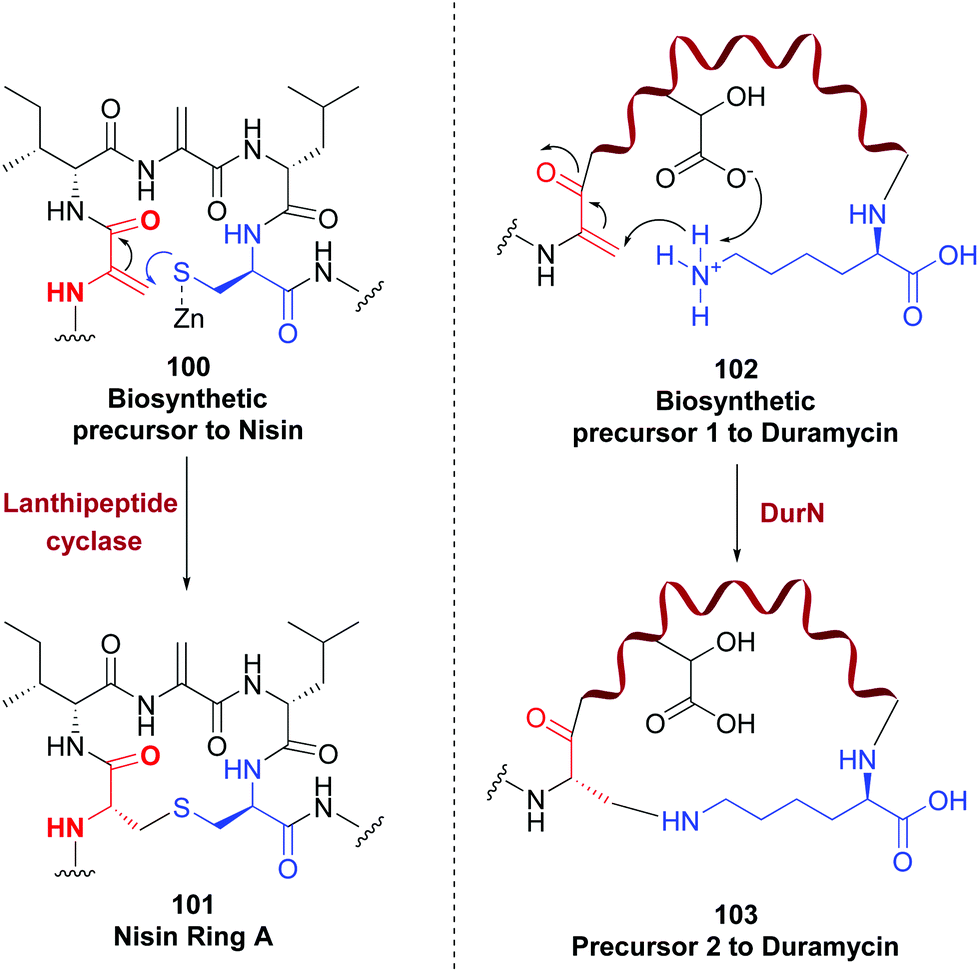 | ||
| Scheme 27 Generation and modification of Dha units.55b,94 | ||
NisC, a lanthipeptide cyclase, catalyzes the creation of five thioether bridges in nisin with total regio- and stereocontrol.92 But these enzymes show some substrate promiscuity, that makes them useful for providing modified proteins and peptides with high stereoselectivity, as shown for the cyclase ProcM.93 In a similar example, the enzyme DurN creates a lysinoalanine in duramycin biosynthesis, by addition of the ε-amine in lysine to a Dha unit (conversion 102 → 103, Scheme 27).94 The stereoselective reduction of dhAAs (in many cases to D-amino acids) is also catalyzed by enzymes such as SacJ, LtnJ, PenN, LasJ, Bsj and CrnJ, among others.95 No doubt that with the ongoing discovery of new enzymes, the range of possible reactions will increase in the next years.
3. Selective formation of heterocyclic units and other modifications of the peptidic bond
Many peptides present oxazole and oxazolidine or thiazole and thiazolidine units in their backbones. These heterocyclic units confer a high rigidity to the biomolecule, thus enhancing interaction with their biological targets.96 While oxazole and oxazolidine rings derive from serine units, by intramolecular dehydrocyclization, the thiazole and thiazolines usually derive from cysteine. There are many methodologies reported for these cyclizations.97 Since the cysteine units are relatively scarce in proteins, their selective functionalization with respect to other amino acid units is in many cases feasible.53 In case that other Cys units are present, the selectivity can be achieved by protecting the different residues with orthogonal groups, and deprotecting each Cys unit selectively before transforming that position.97 Other than this, enzymes can provide exquisite selectivity, but their substrate range is not usually very large.27For the lector interested in the formation of different heterocyclic rings in peptides, it is recommended the comprehensive review by Balalaie et al.17a In most cases, the heterocycles are formed before the unit is inserted into the peptide, and thus these syntheses escape the scope of this review. However, some site-selective examples are highlighted below.
An example of the use of orthogonal protecting groups to differentiate tunable units is shown in the synthesis of telomestatin by Pattenden et al. (Scheme 28), in which an oxazole and a thiazolidine ring were formed sequentially.98 The precursor for the oxazole ring, a serine unit, was protected as an isopropylidene acetal before macrocyclization (substrate 104). Deprotection of serine and the adjacent acid and subsequent cyclization provided the macrocycle 105. The formation of the oxazole ring then took place by dehydratation to a Dha unit and intramolecular halocyclization. The resulting N,O-acetal underwent methanol elimination under acid treatment to provide the oxazole ring in product 106. Then the cysteine residue was deprotected and transformed into the thiazolidine moiety, thus completing the synthesis of telomestatin (107) in good overall yield.
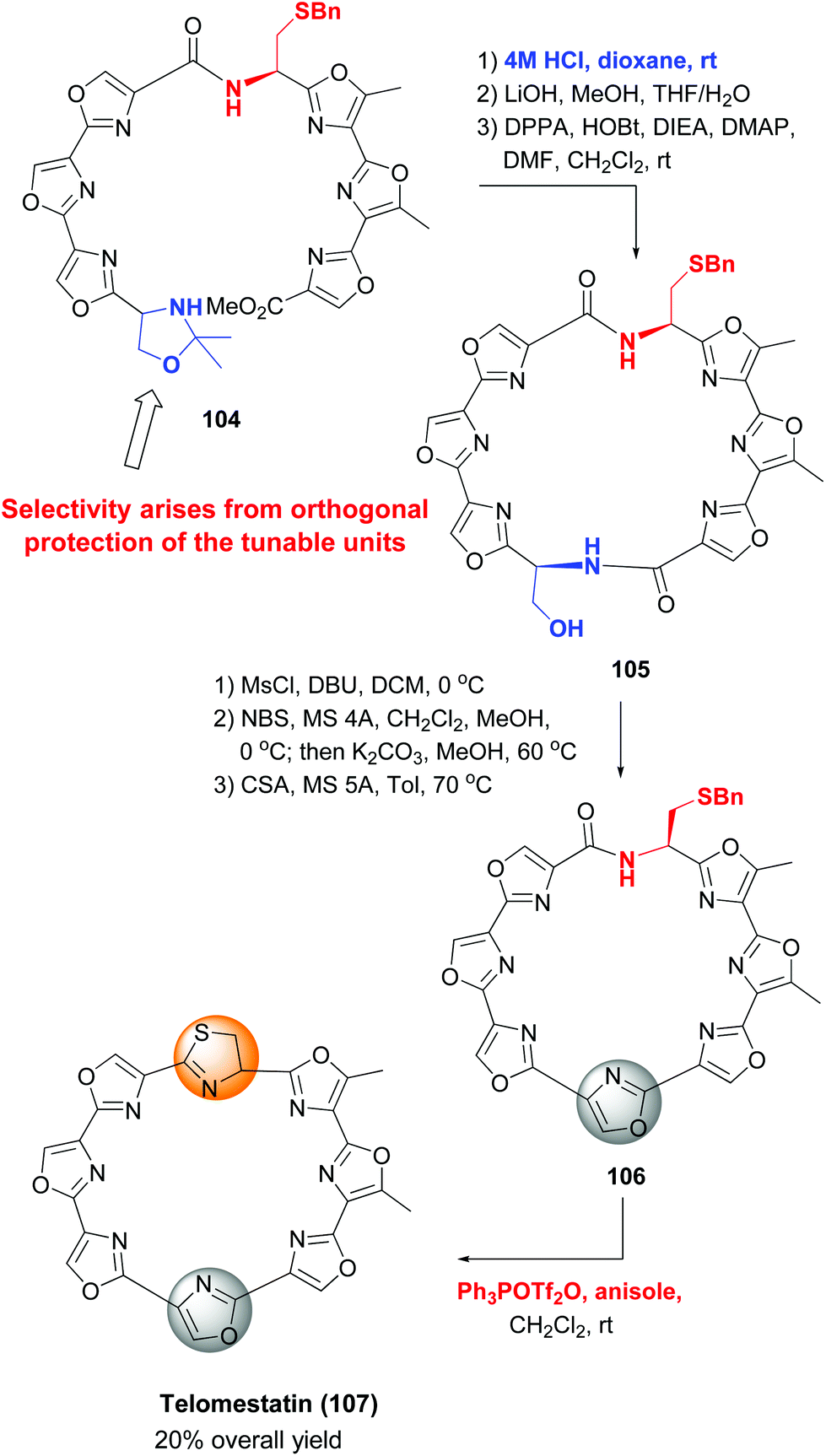 | ||
| Scheme 28 Formation of backbone heterocycles, Pattenden et al.98 | ||
In another example, the synthesis of Trunkamide A by the same group (Scheme 29), a remarkable selective conversion of just one amide of precursor 108 into a thioamide 109 was achieved, by treating the peptide with DAST and then hydrogen sulfide.99 Once installed, the thioamide underwent an intramolecular cyclization with the adjacent serine residue on treatment with DAST, to afford the final thiazolidine ring (conversion 109 → 110).
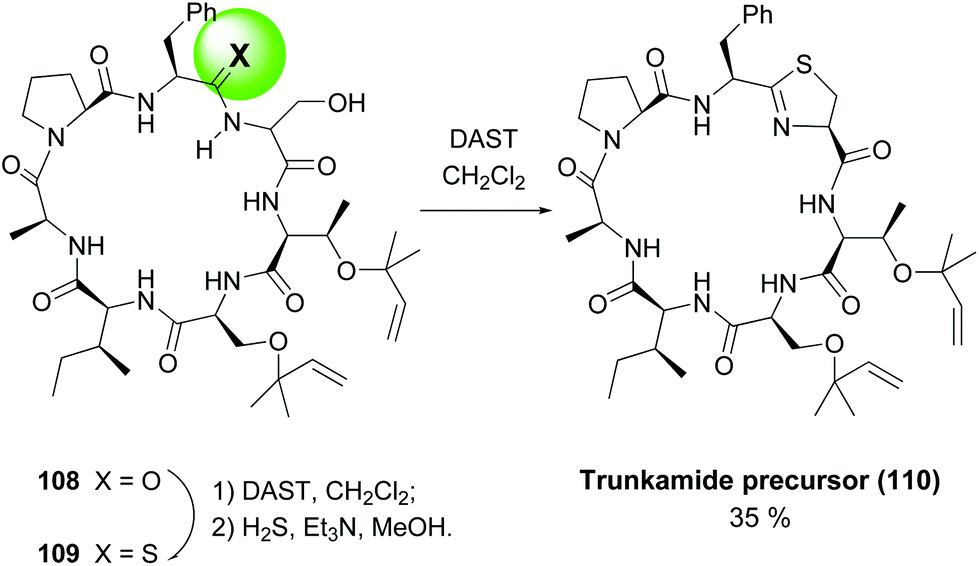 | ||
| Scheme 29 Generation of thioamides as thiazolidine precursors.99 | ||
It must be said that thioamides are another useful modification of the peptide backbone, and although they are considered bioisosters of the carbonyl group, their reactivity could differ notably. Their different ability to form hydrogen bonds and stabilize/destabilize certain conformations, can translate into different biological activities, as shown in the review by Mitchell et al.100
A method for the formation of oxazole rings that does not involve serine or hydroxylated units, and that can be applied to any residue in a terminal C-position, is shown in Scheme 30 (conversions 111 → 115 and 116 → 117).101 Thus, decarboxylation of substrate 111 on treatment with a (diacyliodo)benzene and iodine generated an iminium ion 112 which isomerized to an enamide. The latter underwent an intramolecular halocyclization involving an adjacent amide carbonyl group (113 → 114). Aromatization of the system, by elimination of the leaving group X, provided the oxazole 115. This one pot oxidative radical decarboxylation–isomerization–iodination–cyclization took place in good global yields, and proceeded with different acyl substituents.
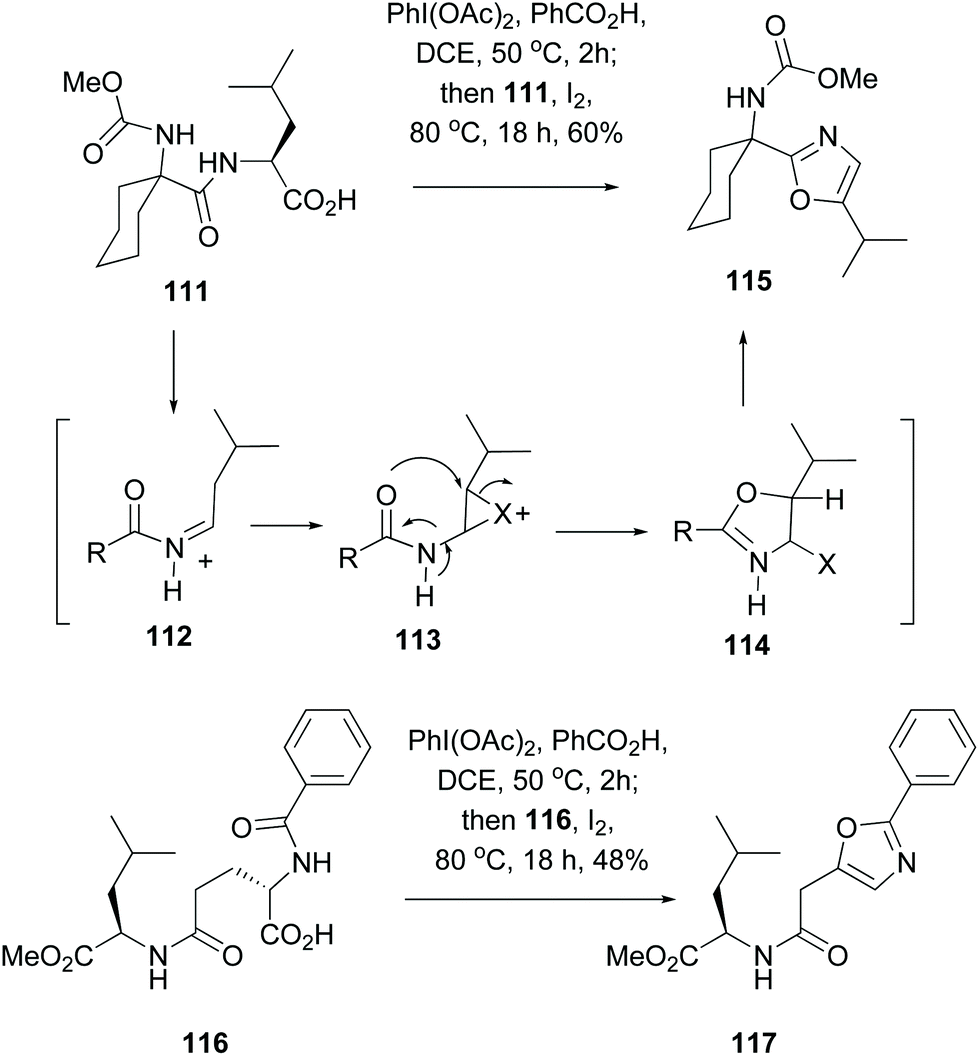 | ||
| Scheme 30 One-pot generation of oxazole units.101 | ||
The previous methodology was only reported for small-size peptides, but recently Qvortrup et al. described another site-selective oxazole synthesis that used tryptophan as a “tunable” unit, and that could be applied to large-size peptides.102 As shown in Scheme 31, treatment of a tryptophan-containing peptide 118 with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) produced the oxidation of the lateral chain to give an α-indolylketone 119. Cyclodehydration followed, to provide a conjugated indolyl-oxazole moiety 120 that could serve as a promising fluorescent label, with an intense emission at 425 nm. Interestingly, this band does not overlap with those produced by natural residues. Moreover, since tryptophan is a low-frequency amino acid in natural proteins (1.1%), many proteins of interest will present only one or a few units, facilitating the modification of selected positions.103 This was shown in the selective fluorescent labeling of GLP-1 hormone (121), which took place in 85% yield.102
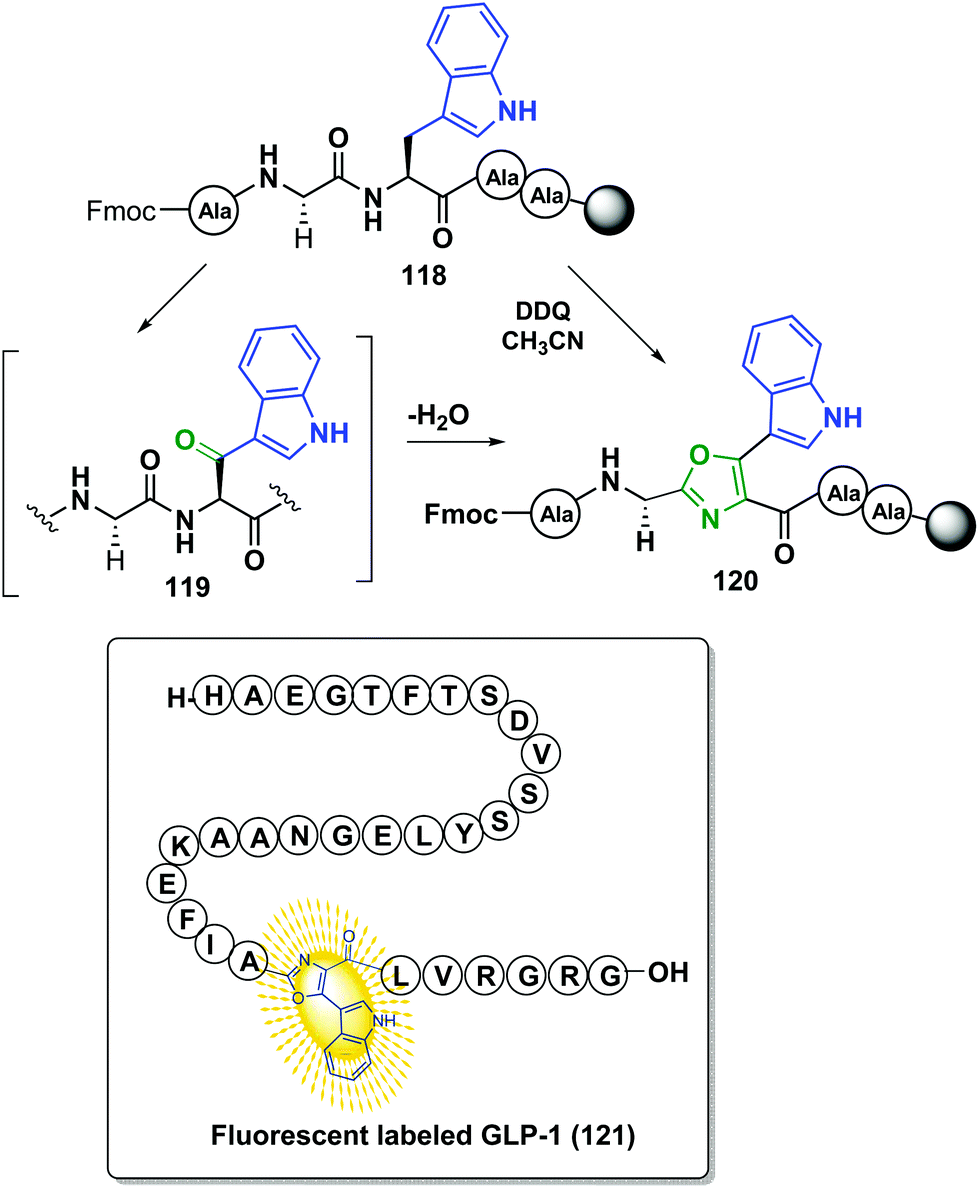 | ||
| Scheme 31 Site-selective formation of oxazole-based fluorophores by Qvortrup et al.102 | ||
Other tunable residues are pseudoprolines, which are cyclic, protected derivatives of serine and other hydroxyamino acids.104 The conformation and bioactivity of the peptide containing this unit can be different from the acyclic counterparts. Most pseudoprolines are acetal derivatives that can be deprotected using acid treatment, or other standard treatment. The promising sulfamidate derivatives have been previously commented in section 2.1.41–43
The N-terminal position in peptides offers more possibilities to form heterocyclic units. In a recent paper, van der Eycken reported the modification of a N-terminal benzamide with formation of a isoquinolone. The reaction was regiospecific and racemization-free. It provided fluorescent peptides, peptides conjugated to drugs (carriers) and other bioactive products.105 No doubt that the selective formation of different heterocycles in peptides will offer many applications for biomedicine and materials science in a future.
4. Backbone conversions: modification of the N-substituents
4.1. N-Modifications using customizable units
The introduction and variation of N-substituents is another way to create diversity in the peptide library. However, the selective modification of these substituents is difficult. The traditional way to introduce N-alkylated amino acids in a peptide is troublesome, with poor reaction rates and many side-reactions (Scheme 32, conversion 122 → 123).106–108 An exception is the coupling of cyclic amino acids, such as proline and hydroxyproline. Therefore, Boto and all have suggested an alternative route (Scheme 32, conversions 124 → 125 → 123) where hydroxyproline is introduced first in the desired position, and then its scission and functionalization would readily give the desired N-alkylated peptides.20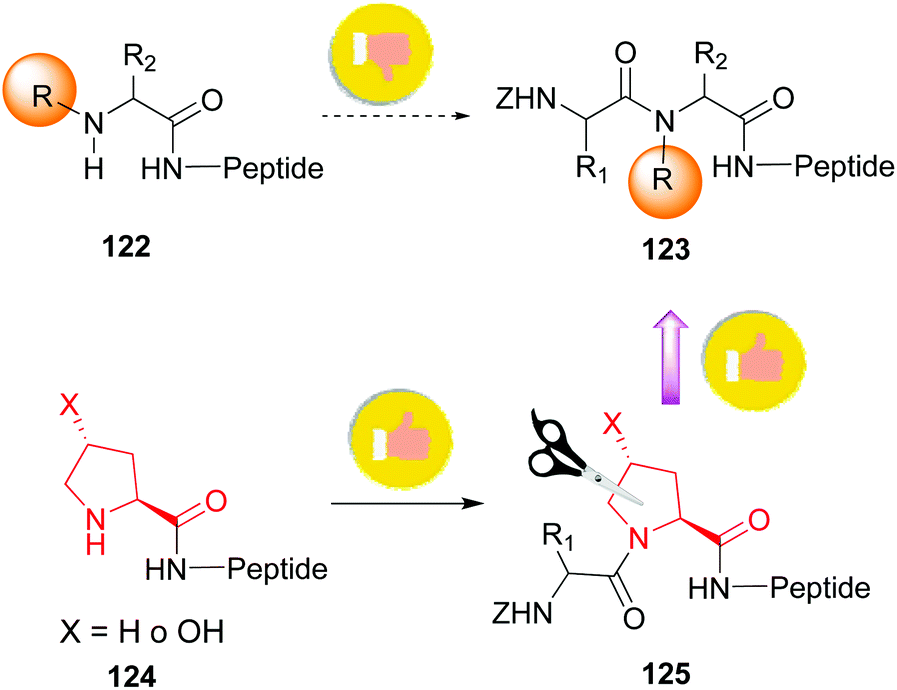 | ||
| Scheme 32 Generation of N-substituted peptides. | ||
According to this, the addition of different nucleophiles was studied (Scheme 33, conversions 126 → 127 → 128–131), and the N,O-acetals were transformed in good yields in a N-methyl group (by reduction of the N,O-acetal, compound 128) and other N-alkyl groups (by addition of C-nucleophiles, compounds 129–131).20 Encouragingly, even very bulky N-substituents (as in 130 and 131) could be introduced in good yields, and mild conditions. No side reactions were observed. The introduction of the homoallyl chain in 131 is remarkable, since even larger substituents could be now generated by olefin metathesis.
 | ||
| Scheme 33 Conversion of N,O-acetals into N-substituents.20 | ||
An application of this methodology to the preparation of tetrasubstituted diketopiperazine scaffolds was also reported (Scheme 34, conversion 132 → 136).51 This conversion is usually carried out by alkylation of unsubstituted N-diketopiperazines with strong bases and very reactive electrohiles, and usually heating or other activation source is required.109 These harsh conditions were avoided using the methodology shown in Scheme 34. In this case, the first scission process was followed by a Horner–Wadsworth–Emmons reaction to create a hydrophobic residue (conversion 132 → 133). In the final step (conversion 134 → 135, the two N,O-acetals were allylated, to give a diolefinic compound 135 that could be the base of dimers or oligomers 136.51
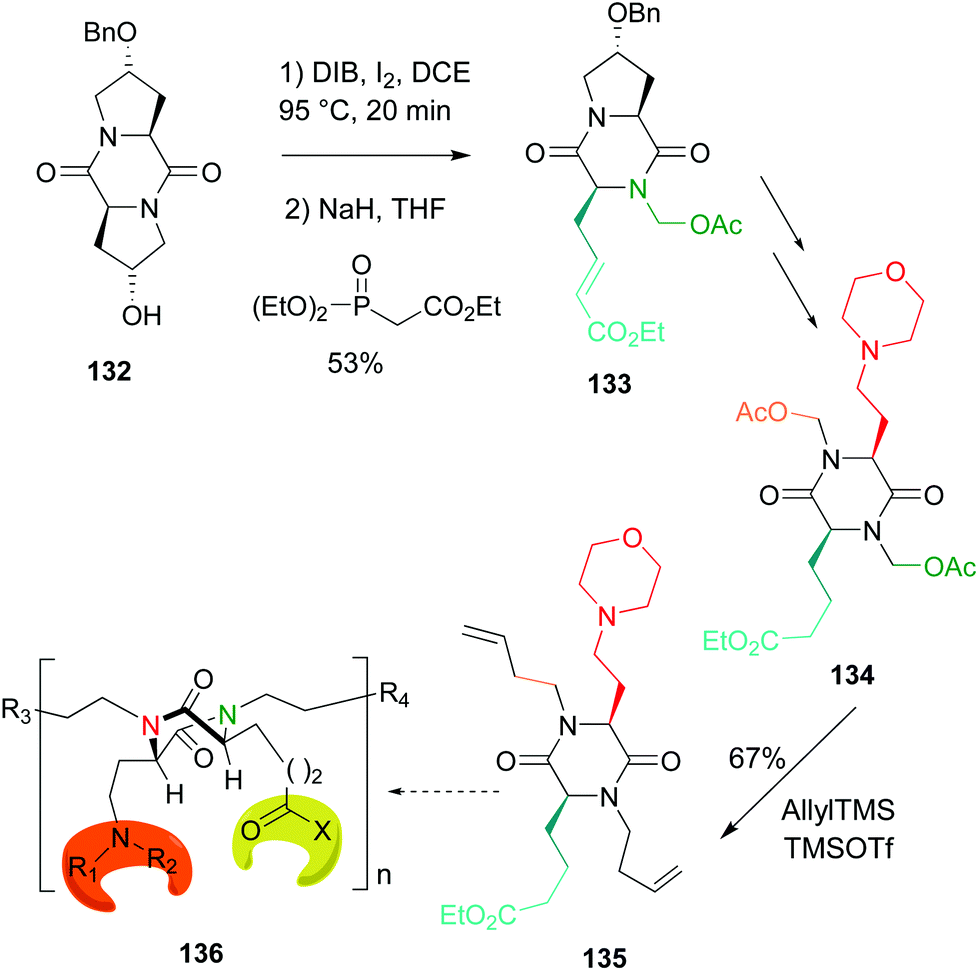 | ||
| Scheme 34 DKPs as scaffolds for oligomeric materials.51 | ||
4.2. N-Modifications using removable protecting groups
The introduction of amino acids with removable N-substituents can be useful to obtain peptides with improved solubility, less aggregation, or other useful properties.110 These N-substituted residues can be considered as “units for tunable physicochemical properties”.N-Protected glycine and alanine residues are often used as such “tunable units”. Other residues are less frequent, since the steric hindrance created by both the lateral chain and the N-substituents hinder peptide coupling.106,107 The use of backbone protecting groups to enhance solubility was originally put forward with the dimethoxybenzyl (Dmb) group (Fig. 1, structure 137).111 The introduction of Dmb disrupted the hydrogen bonding that generated scarcely-soluble secondary structures, such as β-sheets, and inhibited interchain aggregation. Even polyalanine chains could be solubilized with Dmb.111 Other protecting groups followed, such as Hmb, Hnb, Hmsb and pseudoprolines (138–142, Fig. 1).112 All of them favour open-chain, disordered and hence more soluble structures. They are widely used to deal with difficult sequences in solid phase peptide synthesis (SPPS), avoid aggregation and prevent aspartimide formation during Fmoc SPPS.112c
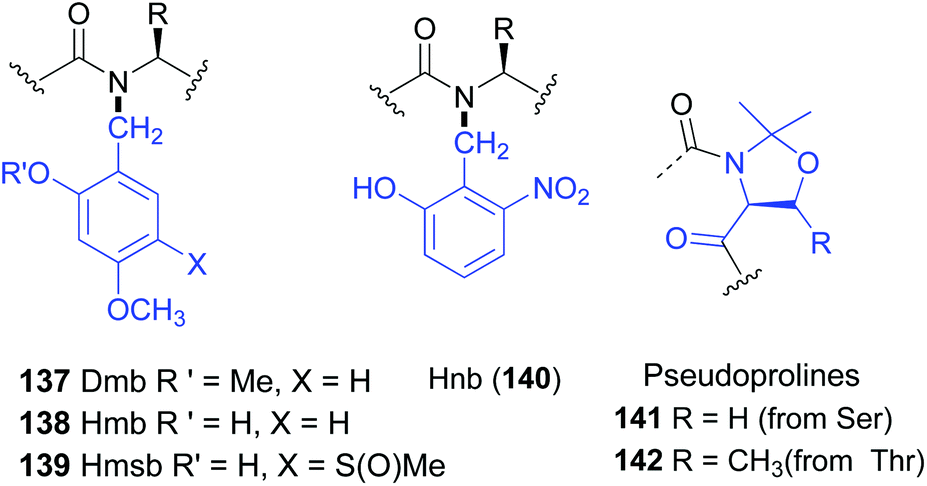 | ||
| Fig. 1 N-Removable protecting groups. | ||
In a representative example, Meredith et al. used an N-(o-nitrobenzyl) glycine to modify the bend region of amyloid-β (Ab), a peptide involved in Alzheimer's disease (Fig. 2, compound 143).113 The replacement of Gly29 at the bend region of the amiloid by the “physicochemical tunable unit” gave a peptide that was able to form oligomers but not fibrils at physiological pH and temperature. The reasons for this were not clear, but the authors presented three possible explanations. In the first, the introduction of a protecting group removed hydrogen bond/s that usually stabilize the bend. In the second, the steric bulk of the protecting group would alter the folding of the bend section, and/or approximation of the N-and C-terminal β-sheets prior to fibril formation. In the third, the cis/trans isomerization of the glycine and thus the bend conformation would change. Such variations would prevent the structural requirements for fibril formation, but oligomerization would still be possible through hydrophobic collapse of the hydrophobic sequences in the peptide. Cleavage of the protecting group by photolysis could restore the original amyloid properties.
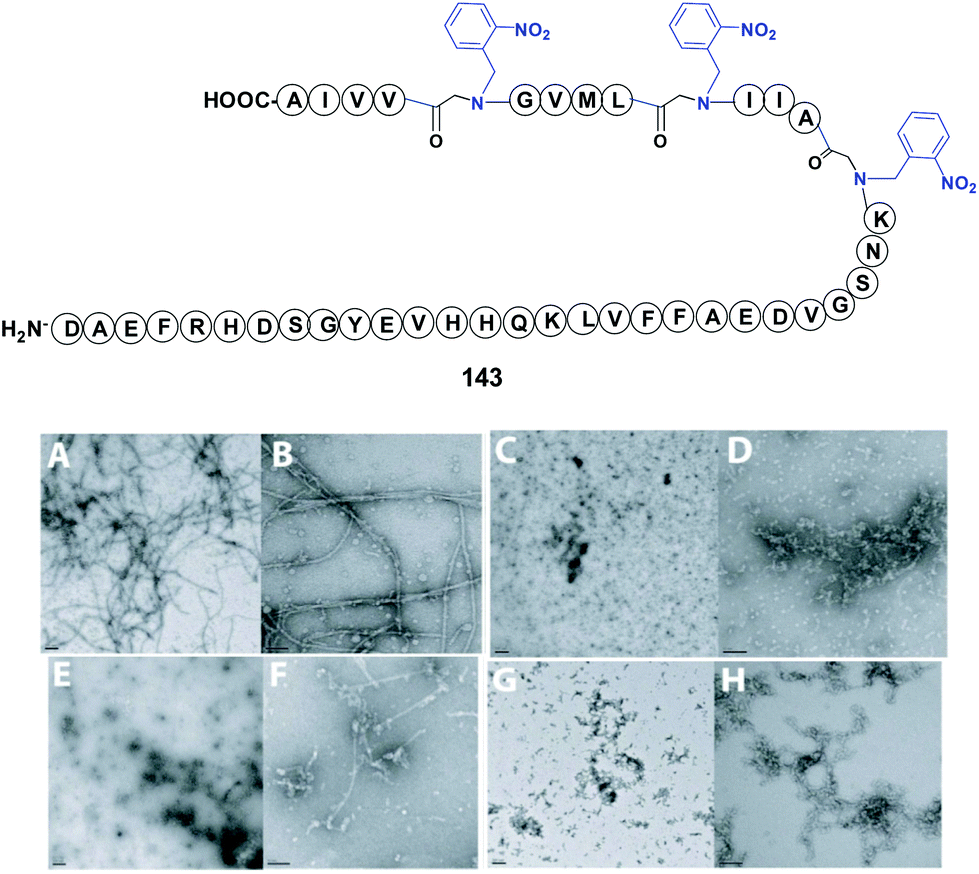 | ||
| Fig. 2 N-Removable protecting groups in amyloid synthesis. Photolabile (2-nitrobenzyl)glycine units are used in key peptide positions. (A and B) Transmission electron microscopy (TEM) of non-protected Aβ1–42 at 24 h showed formation of long fibrils. (C and D) TEM at 24 h of Aβ1–42 with backbone modification at Gly29 using the 2-nitrobenzylgroup (NB). The introduction of NB inhibited fibril generation, but smaller oligomer aggregation could be observed. (E and F) TEM at 24 h of Aβ1–42 with protecting groups in Gly29, Gly33 and Gly38. The hydrophobicity increased and also the aggregation of oligomers. (G and H) In Aβ1–42 modified at Gly33 and Gly38, fibrils formed but were shorter and thinner than those in the native Aβ1–42 sequence. TEM photos are originals from Meredith et al., reproduced by license of Wiley-VCH.113 | ||
Offer et al. used an alanine residue with an Hmnb group for the synthesis of difficult sequences, including aggregation-prone polyalanine sequences.114a The automated synthesis of ACP (65–74) with Hmnb-alanine is shown in Scheme 35. The tunable unit is formed during SPPS, by reaction of peptide 144 with the Hmnb-aldehyde, followed by reduction of the imine intermediate with sodium borohydride, to give Ala(Hmnb)-containing peptide 145. The synthesis of the peptide continued without incidents using standard SPPS. It must be said that if the protecting group had not been introduced at this point, the synthesis would probably have failed, due to peptide solubility and aggregation problems.
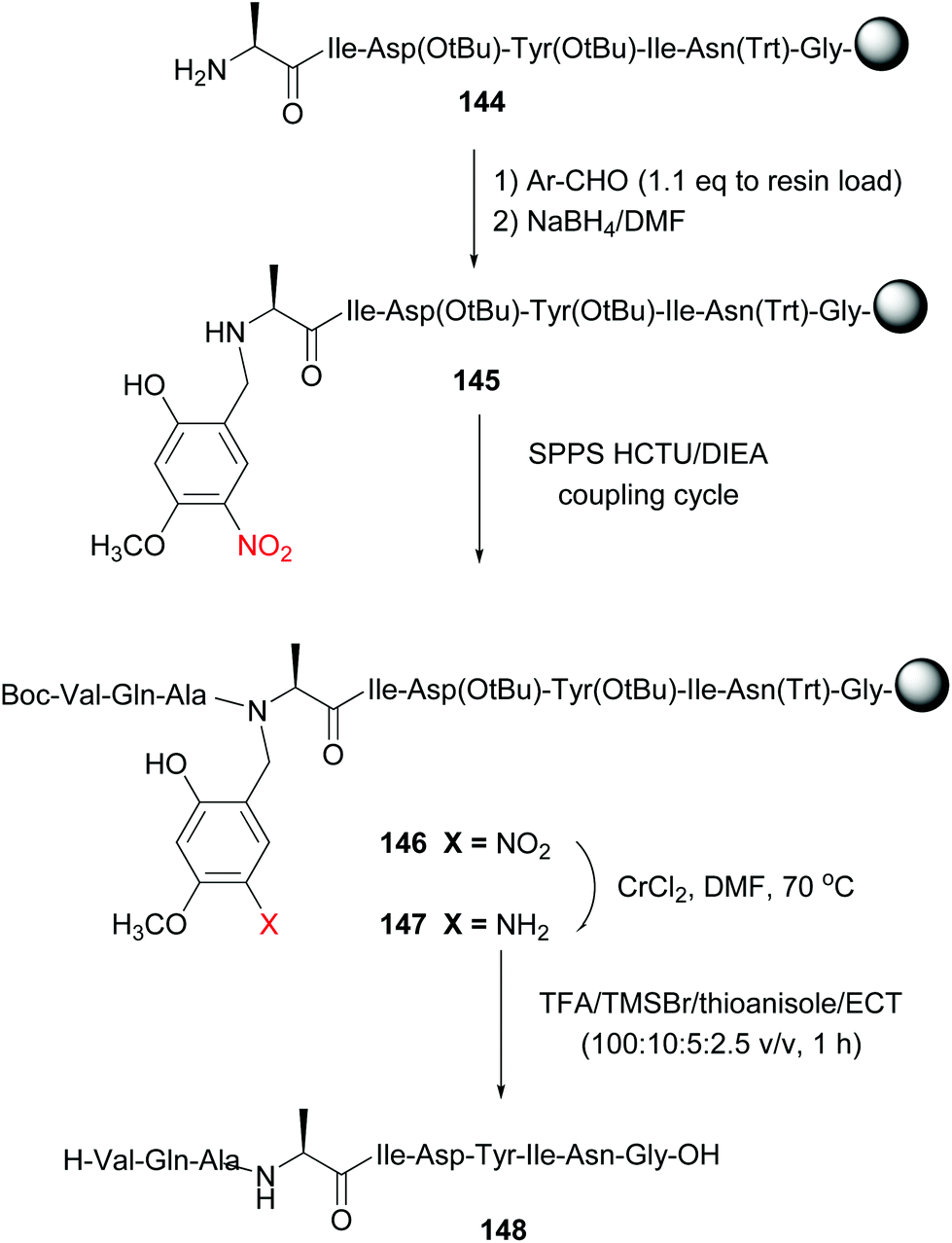 | ||
| Scheme 35 Alanine unit with a removable N-Hmnb protecting group in the synthesis of ACP (65–74) by Offer et al.114a | ||
Once the total sequence was ready (compound 146), the protecting group was removed. In a first step, the phenylnitro group was reduced under mild conditions (CrCl2/DMF system). The resulting aniline 147 was cleaved with TFA and TMSBr/thioanisole mixtures, conditions that also allowed cleavage of the remaining protecting groups and the resine linker. The free peptide 148 was obtained with high purity. Recently, Chiba has reported the use of a N-(substituted benzyl)glycine unit in the central position of peptides to favour a conformation that facilitates head to tail cyclization on solid support (a “molecular claw”).114b
The tunable units can be quite complex and can be attached through the removable link to peptidic chains (Scheme 36). Thus, Zheng et al. synthesized longer amyloid beta-peptides (Ab, from 43 to 49 amino acids) to study their role in Alzheimer's disease.115 These peptides are usually quite difficult to prepare, due to solubility and self-assembly problems. The authors introduced a glycine unit with a removable RMB group, consisting of a substituted benzyl derivative attached to a Arg4-Gly peptide chain (Scheme 36). This RMB motif solubilized the developing peptide, facilitated the ligation step (conversion 149/150 → 151), and allowed the synthesis of the 42-amino acid Aβ-peptide to proceed successfully. In the final step, a TFA cocktail cleaved the benzylic bond and thus removed the Arg4Gly tag, releasing the desired peptide 152 for biological studies.
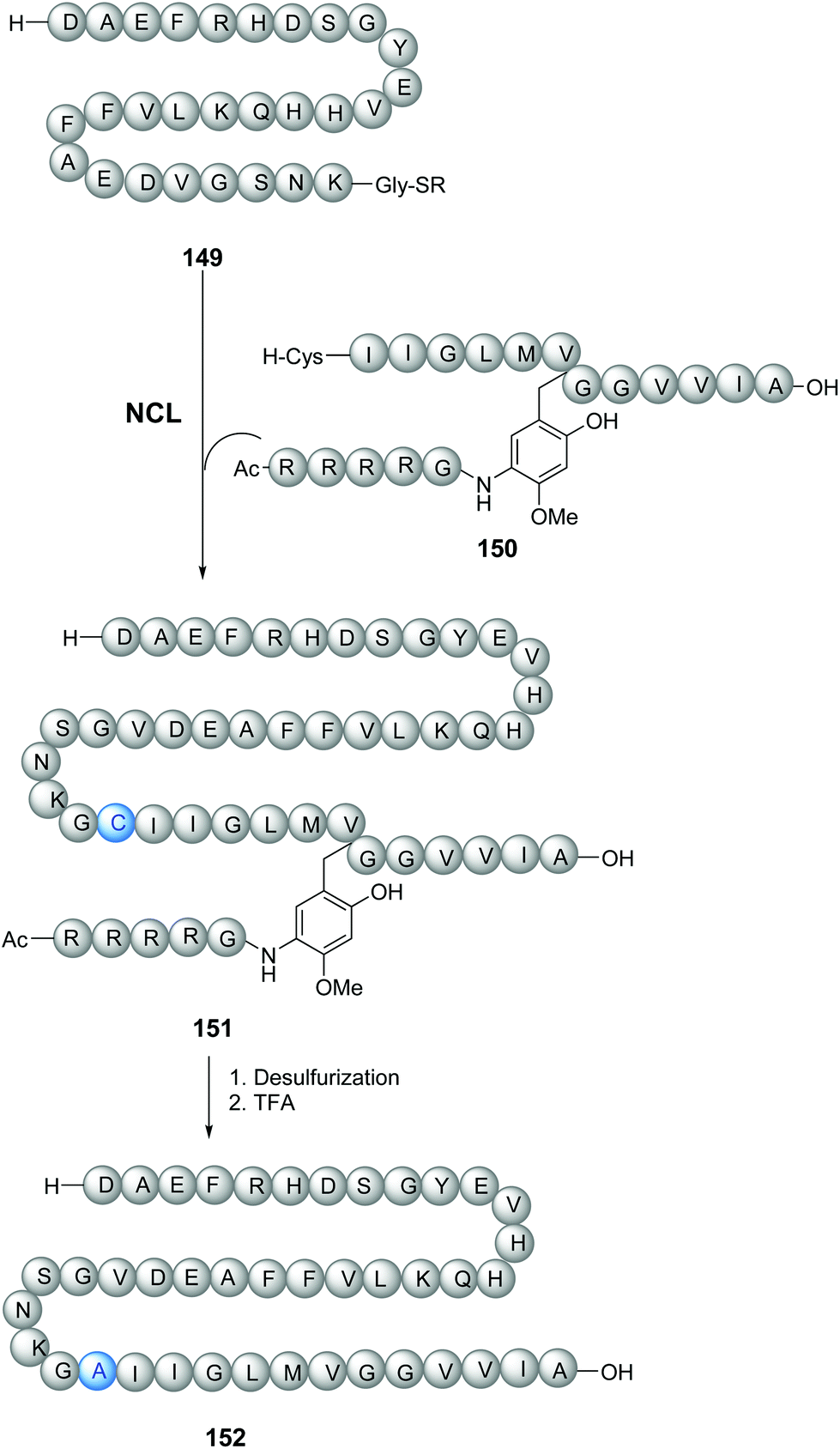 | ||
| Scheme 36 N-Removable protecting groups to avoid aggregation in the synthesis of amyloid beta-peptides by Zheng et al.115 | ||
In a similar way, Brik reported the backbone modification of synthetic ubiquitin(Ub)-aldehyde, using a Thz linker to attach a cell-penetrating peptide (CPP).116,117 The CPP allowed the cell internalization of ubiquitin, and once inside, the linker was cleaved by an external stimulus (addition of palladium). The released ubiquitin aldehyde was a potent inhibitor of cellular deubiquitinating enzyme USP2a.117 In another example (Scheme 37, conversion 153 → 156), Brik incorporated propargylated amides during the synthesis of the NEDD8 protein. This backbone modification improved solubility and avoided aggregation during SPPS. The propargyl group can be readily removed using Au(I) catalysts.118 The removal probably involves hydratation of the alkyne through Au(I) activation, to give intermediate 154. Tautomerization to the carbonyl compound 155 followed by β-N elimination would give the unprotected peptide 156.
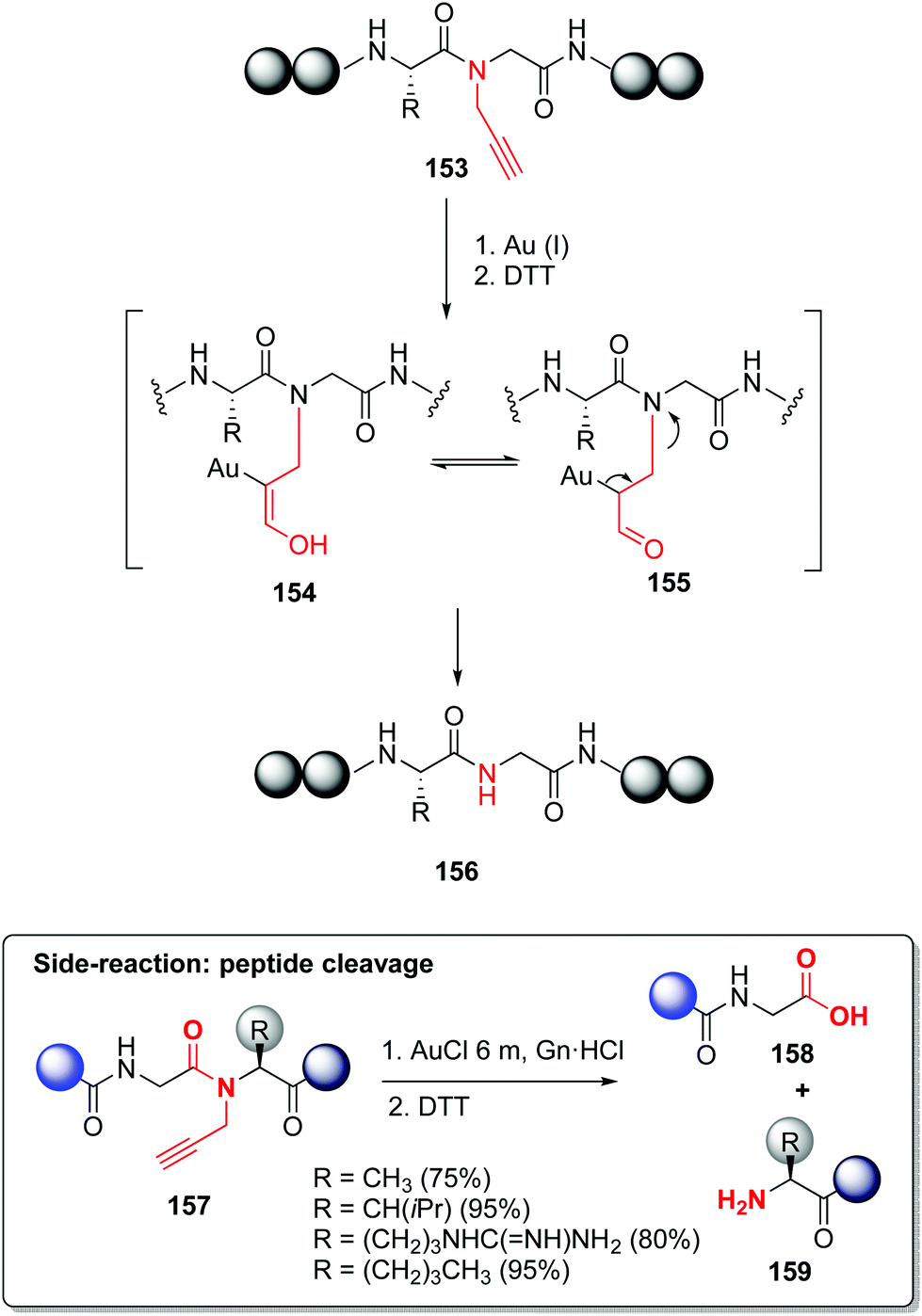 | ||
| Scheme 37 N-Removable protecting groups by Brik et al.118 | ||
The authors added a warning note: for certain Gly-Aa couples, cleavage of the peptidic backbone occurred (conversion 157 → 158/159), so the propargyl-Gly unit should be avoided in these junctions. Palladium can also be used for propargyl group removal, with even superior yields.118
4.3. Residue-directed selective N-modifications
Based on the Cham-Lam coupling (a metal-catalyzed oxidative reaction where X–H bonds are coupled with an organoboron reagent),119 Ball and others have developed a selective N-alkenylation and N-arylation of the peptide backbone.21a,b,120 The transformation takes place in the amino acid preceding a histidine residue. The histidine directs the incorporation of an N-substituent into the adjacent residue after treatment with an aryl or alkenyl boronic acid and a Cu(II) salt. A copper complex is formed involving the histidine NH and imidazole groups, and the NH group of the preceding amino acid (conversion 160 → 163, Scheme 38). The intermediate 161 resembles natural ATCUN motifs (amino-terminal Cu2+ and Ni2+ binding motifs) that occur in proteins.121,122 However, while ATCUN motifs are tetradentate complexes, the non-terminal sequences form a tridentated complex A. The free site allows the Cu(II) complex to react with the boronic acid in a transmetalation reaction, generating the Cu(III) intermediate 162, which transfers the aryl or alkenyl residue to the backbone nitrogen to give the N-arylated product 163.121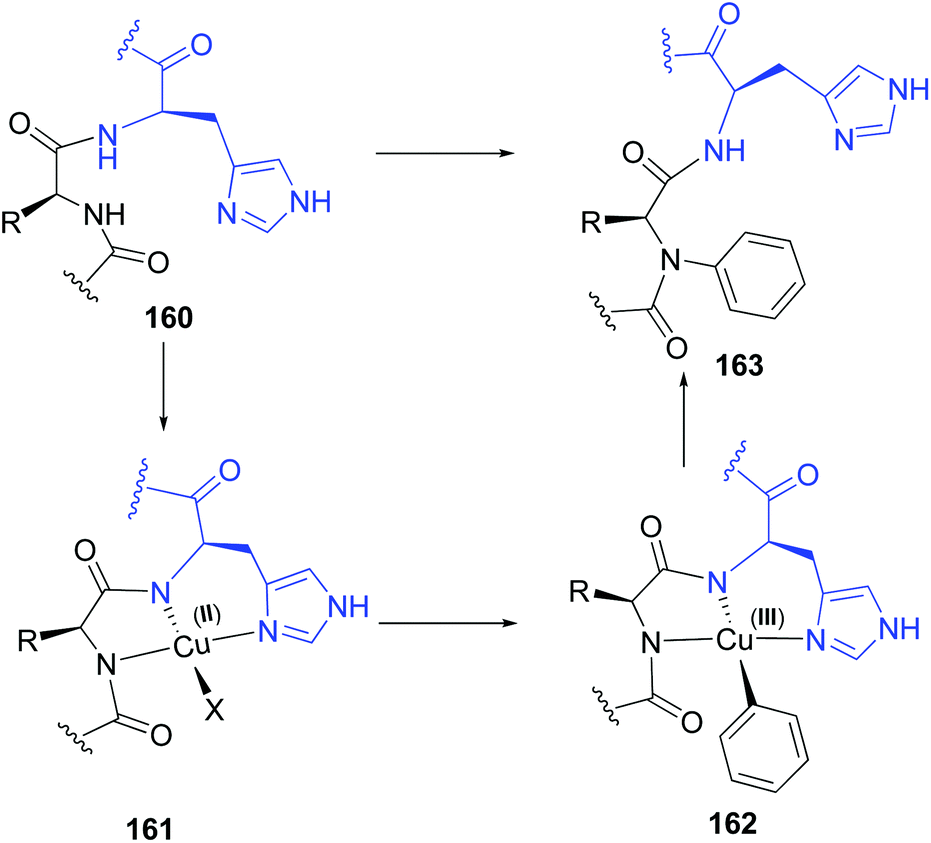 | ||
| Scheme 38 Histidin-directed backbone arylation by Ball et al.21a,b | ||
The reaction is sensitive to steric hindrance, and while trans-alkenylboronic acids give usually good yields, the ortho-substituted arylboronic acids are unreactive.
The reaction has been applied to different peptides and proteins. As shown in Scheme 39, neuromedin B 164 is only alkenylated on the glycine residue preceding histidine, to give peptide 165.21a Other residues such as tryptophan or asparagine are not involved in the transformation.
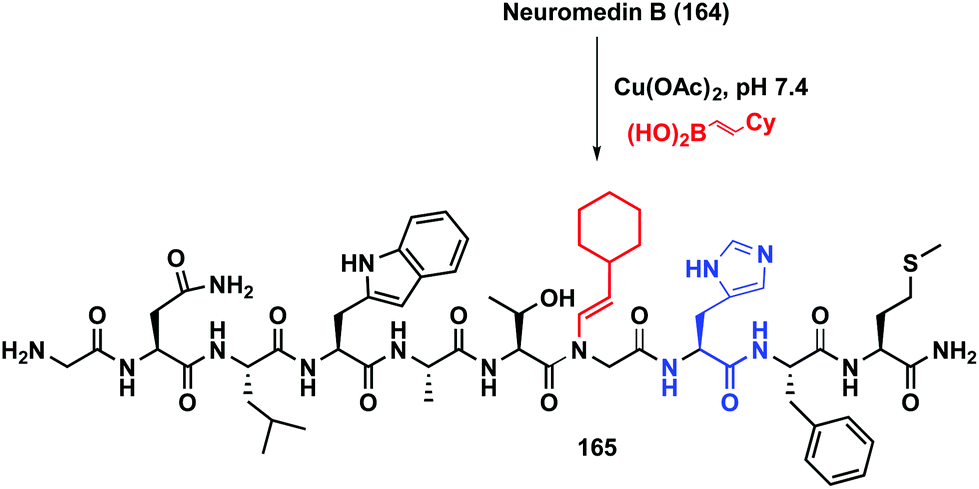 | ||
| Scheme 39 Histidin-directed neuromedin arylation by Ball et al.21a | ||
It must be said that histidine is a relatively rare amino acid in peptides and proteins, and thus this reaction can be very useful to incorporate backbone N-substituents at just one (or a few) positions.21b,120 Lysozyme (PDB:1HEL) was modified at Arg14 preceding the only histidine residue.3c,123
This reaction can be very interesting to control protein and peptide structure and function. The N–H substitution disrupts the hydrogen bonding that allow the formation of helices, β-sheets and other secondary structures.110 Conversely, the N-dealkylation would restore the folding ability of the peptide. In the photocaging system shown in Scheme 40 (conversion 166/167), the alkenylation of the protein or the removal of the alkenyl group under an external stimuli determine the protein structure.120 Thus, a collagen-resembling triple helix peptide was N-alkylated, and the resulting peptide could no longer fold in solution. However, when the modified peptide was irradiated with UV light, the N-substituent was removed, and the peptide folded again into a triple helix. The authors also showed that backbone N–H modification could also prevent protease degradation. When uncaging took place by photoirradiation, the peptide became sensitive to protease action.
 | ||
| Scheme 40 Photocaging systems by Ball et al.120 | ||
A particularly reactive sequence is pyroglutamate-histidine, which can be labeled selectively even in the complex cell lysate.21a While most backbone N–H modification took hours, Pyr-His sequences reacted in an average 5 minutes. Pyroglutamate can be formed from glutamine using the enzyme glutaminyl cyclase (conversion 168 → 169, Scheme 41). Once formed, the Pyr-His motif can undergo selective labeling (conversion 169 → 170). Since the Pyr-His unit is not frequent in natural proteins, monolabeled peptides can be obtained selectively.
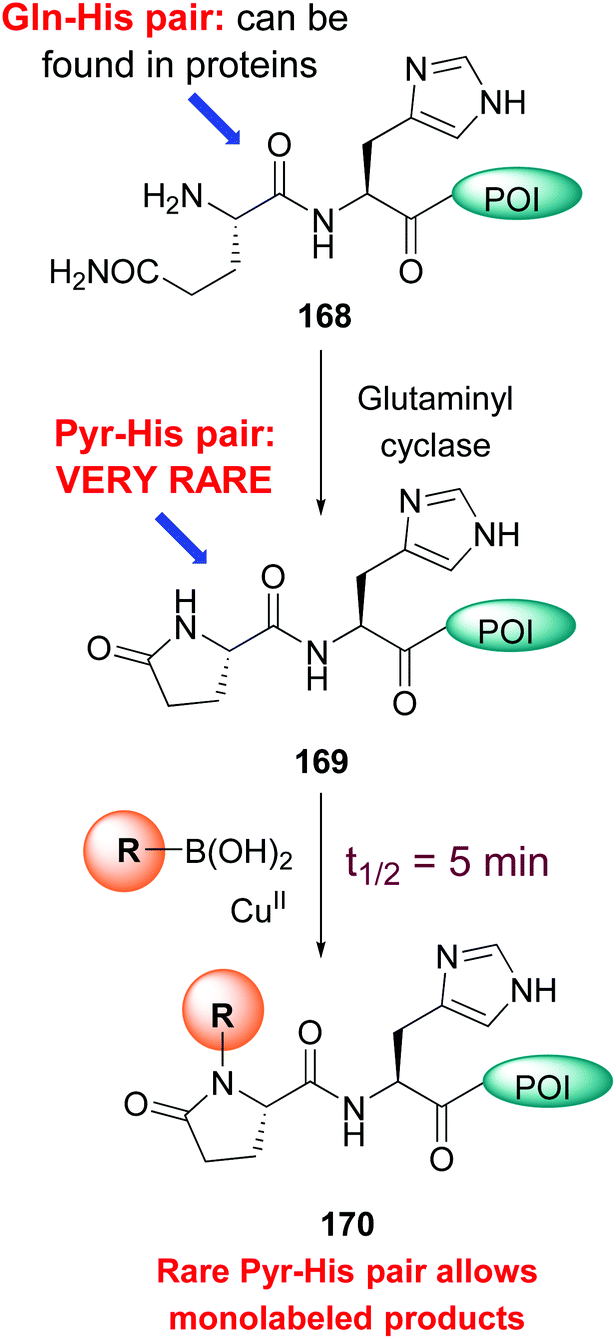 | ||
| Scheme 41 Pyr-His pair for monolabeling of proteins.21a | ||
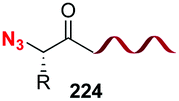
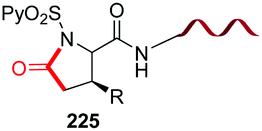
Recently, Ball et al. have reported red-shifted backbone N–H photocaging systems (Scheme 42).124 The use of near-IR light has elicited much interest due to its superior tissue penetration and since it does not overlap with the absorption of natural biomolecules, thus allowing improved precision for uncaging. The Scheme shows the preparation of a boronic acid with the cromophore (conversion 171 → 172) and its His-directed introduction at the terminal Pyr position (conversion 173 → 174). The peptide is released under uncaging conditions (irradiation at 450 nm).
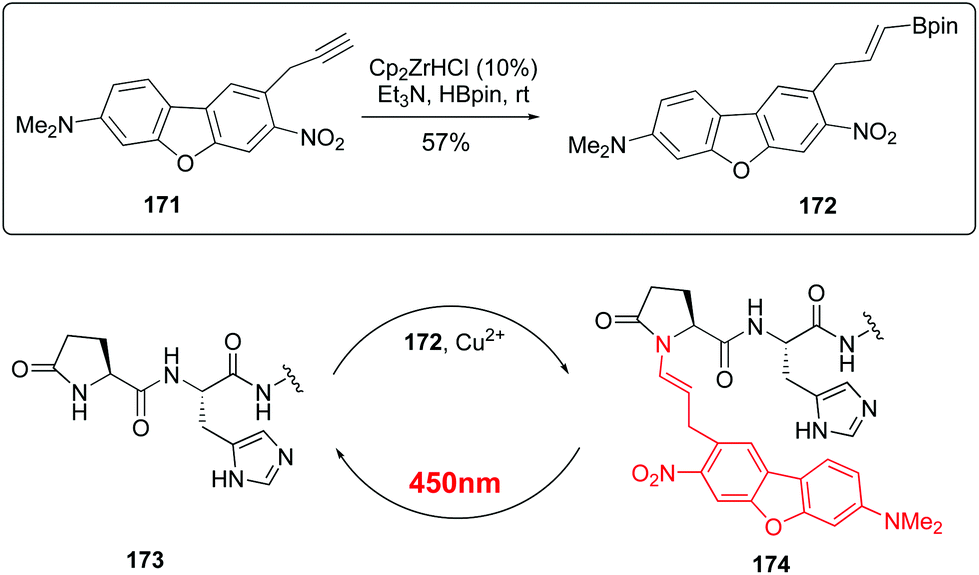 | ||
| Scheme 42 Red-shift backbone photocaging systems.124 | ||
Palladium can also form ATCUN-like complexes (Scheme 43) but the reaction outcome can be quite different. In 2002, Kostić et al. reported that on treatment of the peptide with [Pd(en)(H2O)2]2+, the histidines in interior positions directed the formation of palladium complexes which resulted in regioselective cleavage of the second peptide bond upstream and the first peptide bond downstream (as shown for angiotensine complex 175, Scheme 43).125 A first cleavage downstream produces the tetrapeptide 176, which is in equilibrium with the ATCUN-like complex 177. Ligand exchange with water produced the intermediate 178, where the second cleavage (upstream) releases a H-Val-Phe-OH dipeptide and the complex 179.
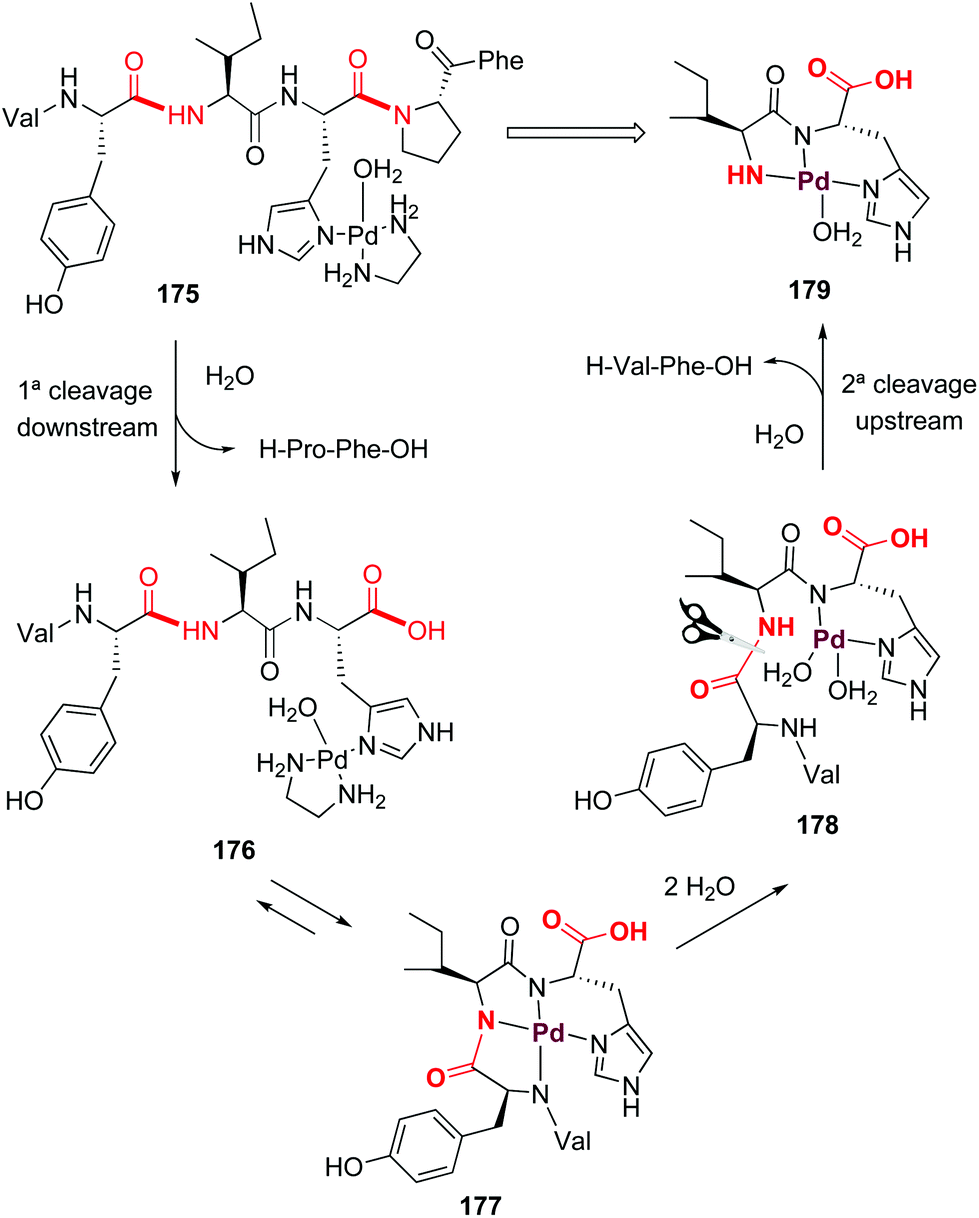 | ||
| Scheme 43 Palladium complexes promote peptide cleavage (Kostić et al.).125 | ||
5. Selective conversion of C-terminal units
The particular reactivity of the C-terminal position allows its site-selective modification. In a classical example, Ooi et al. reported the site-directed asymmetric quaternization of a peptide backbone at the C-terminal position by formation of an azlactone, alkylation of its α-position, and opening of the azlactone with a peptide (Scheme 44, conversion 180 → 182).126 Remarkably, using the chiral catalyst PTC (tetraaminophosphonium chloride), the alkylation proceeded with high diastereoselectivity and good to excellent yields, using a variety of alkyl halides. This methodology was used to create a tetrapeptide 182 that was coupled to a tetraleucine residue, affording the final octapeptide 183.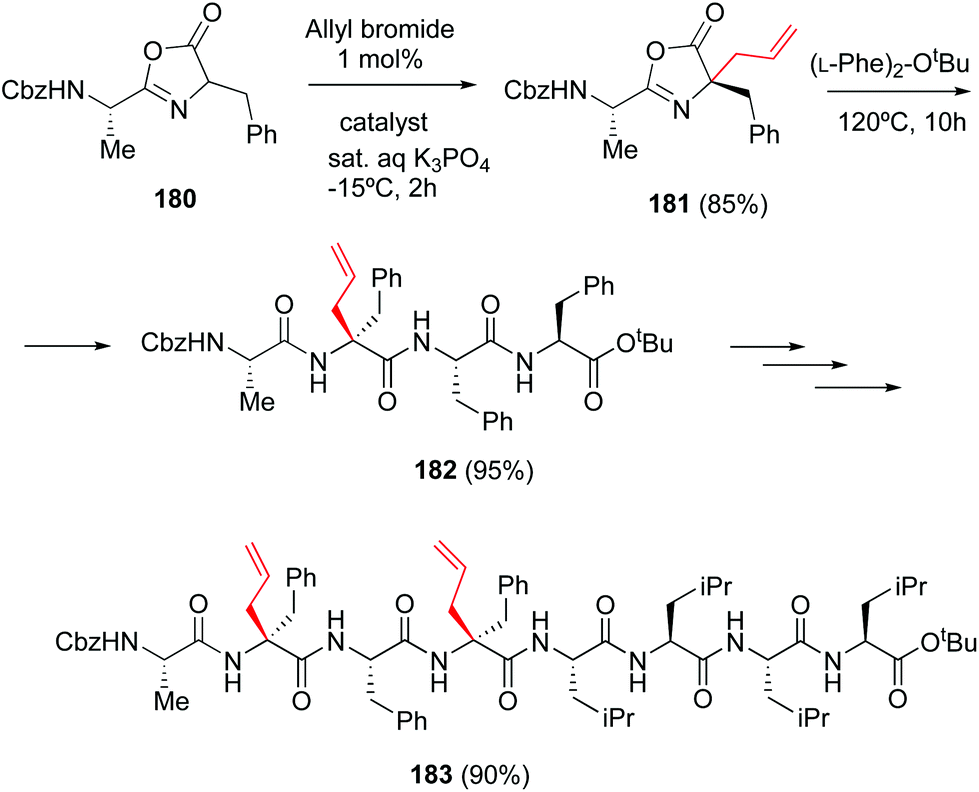 | ||
| Scheme 44 Selective C-terminal modification by Ooi et al.126 | ||
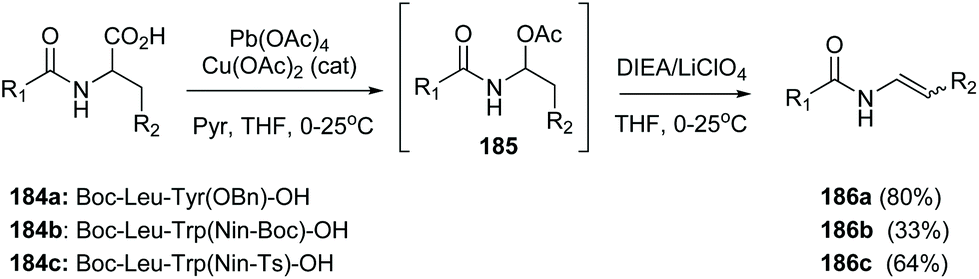
In another example, Porco carried out an oxidative radical decarboxylation of the C-terminal residue in several peptides 184a–c (Scheme 45) generating an intermediate α-acetoxyamine 185, that under base treatment yielded a peptide enamide 186a–c. The methodology was later used to synthesize chondriamides A and C.127
 | ||
| Scheme 45 Formation of C-terminal enamides by Porco et al.127 | ||
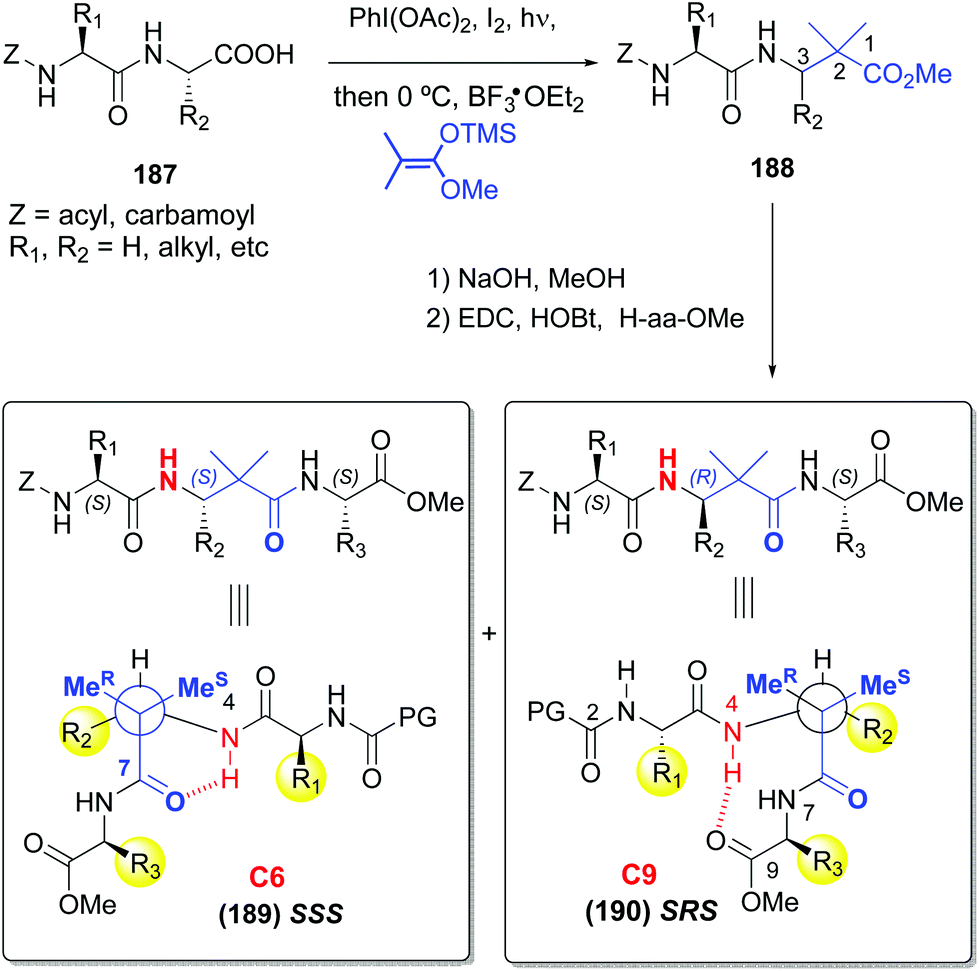
A related decarboxylation strategy was used to obtain peptide hybrids, such as α,β-peptides, which can present new folding patterns, depending on the α:β ratio and distribution.128,129 In addition, they usually display superior resistance to proteases and hence, in vivo stability. Boto et al. described the synthesis of α,β,α-tripeptides where the central unit was a α,α-disustituted β-amino acid (conversion 187 → 189/190, Scheme 46).130 These hybrids formed unusual turns such as deltaturns or extended beta-turns in solid phase. The tripeptides were formed from α,β-dipeptides 188, generated by modification of the C-terminal residue of α-peptides 187. In the conversion 187 → 188 the major diastereomer presented the “natural” (S) configuration, since the N-terminal residue acted as a chiral auxiliary. Moreover, in this work a procedure was developed which used catalytic amounts of a Lewis acid (0.1 equivalents of Cu(OTf)2), affording moderate to good yields of the final products (40–70%), which could allow in a future an increase of the stereoselectivity by using chiral catalysts.
 | ||
| Scheme 46 Generation and modification of C-terminal positions.131 | ||
Interestingly, in a subsequent work by Aizpurua et al.,131 the SSS and SRS tripeptides showed different conformations in CDCl3 or DMSO solution (Scheme 46). Thus, while the SSS peptides 189 displayed a turn by hydrogen bonding of the carbonyl and NH groups of the central β-residue, the SRS isomers 190 presented an extended delta-turn. This duality can be of interest to design drugs or peptide catalysts.
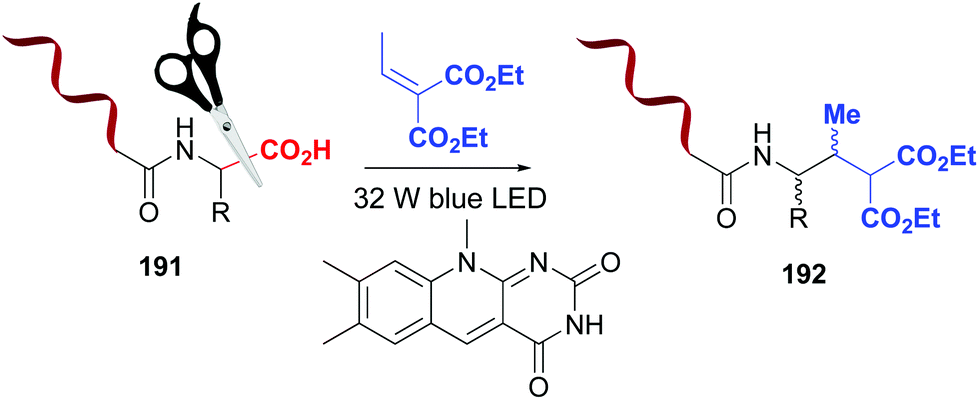
Photocatalytic methods also allow selective C-terminal modifications of short peptides, by replacing the carboxyl group by other functions.10 These reactions are regioselective, due to the difference in reduction/oxidation potentials between the C-terminal carboxyl group and those present in the lateral chains of aspartic and glutamic acid residues.10,132,133 MacMillan et al. used this difference in oxidation potentials to carry out a flavin-catalyzed decarboxylation of the C-terminus in proteins (Scheme 47, conversion 191 → 192).133 The resulting α-amino radical underwent a Michael addition to give the final product 192. Using a variant of this reaction, the alkynylation on the C-terminus of insuline A chain was achieved in 40% yield.132
 | ||
| Scheme 47 Photocatalytic modification of C-terminal positions by MacMillan et al.133 | ||
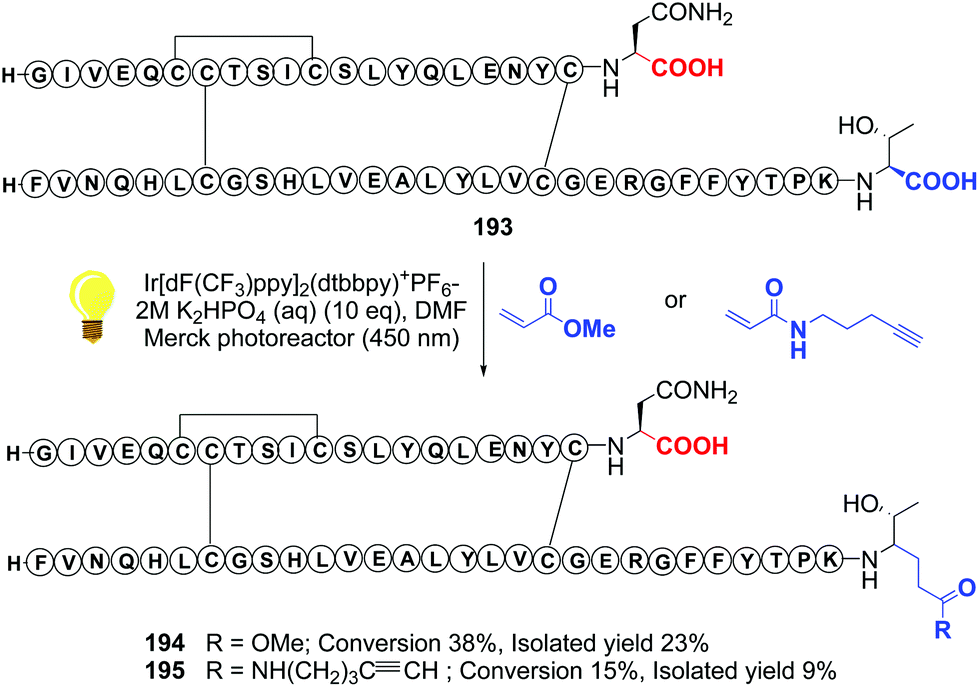
Recently, Shi reported a photocatalytic decarboxylation–conjugate addition to acrylates or acrylamides, using iridium complexes, and applied it to the C-terminal modification of insulin (conversions 193 → 194 and 193 → 195, Scheme 48).134 The authors warned that this decarboxylation was only useful when tryptophan was not present in the molecule, since in its lateral chain reacts preferentially. In proteins such as insulin, however, the reaction can be very useful to install C-terminal functionalities and create γ-amino acid units.134,135
 | ||
| Scheme 48 Photocatalytic modification of insuline by Shi et al.134 | ||
A related decarboxylation–alkynylation was developed by Waser et al.136a The method used a DACA (donor–acceptor cyanoarene) catalyst and EBX (ethynyl benzidioxolone) to achieve the modification of di- to hexapeptides, in low to excellent yields, depending on the substrate. However, aromatic amino acids such as tyrosine, histidine or tryptophan can give side reactions that decrease the yields. Recently, Waser has reported a promising decarboxylation to give N,O-acetals that on treatment with aromatic nucleophiles (indoles, phenols) provided C-aryl substituted peptides.136b
Leonori has reported the decarboxylation–azidation reaction of peptides to afford C-terminal azides, catalyzed by Rhodamine 6G (conversion 196 → 197, Scheme 49).137 These azides can be very useful to expand the chain or ligate peptides using click chemistry. The reader is directed to the review by Spring et al. to find other interesting examples of photocatalysis out of the scope of this review.10
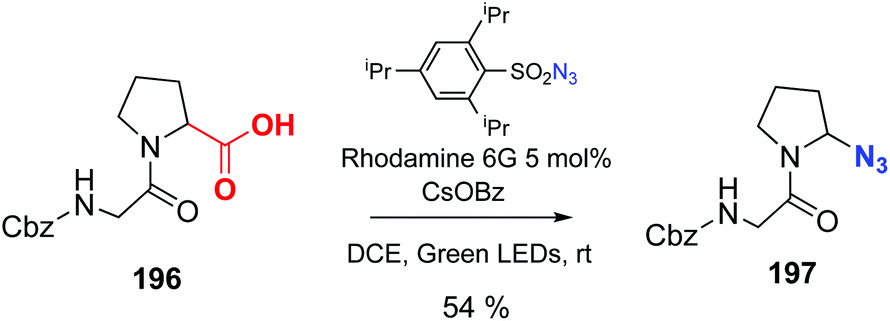 | ||
| Scheme 49 Photocatalytic decarboxylation–azidation by Leonori et al.137 | ||
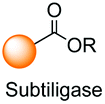
A brief comment should also be devoted to the modification of C-terminal positions with enzymes. Most of these modifications are ligation reactions, which are not covered by this review. For instance, Tubulin Tyrosine Ligase (TTL) recognizes the VDSVEGEGEEEGEE motif (Tub-Tag) in the C-terminus and attaches tyrosine derivatives, including modified tyrosines with extra functionalities.138 Subtiligase ligates the C-terminal ester of a peptide and the N-terminal amine of other peptide, without requiring a recognition motif.139 However, some enzymes not only attach fragments but also remove sections of “signal” motifs.
This is the case of Sortase A and Butelase 1 (Scheme 50). Sortase A is a transpeptidase from Staphylococcus aureus, which recognizes the LPXTG sequence on a protein, and cleaves the T-G union to form a reactive thioester between the threonine residue and a cystein unit from the SrtA active pocket.140 Then the thioester is cleaved by addition of a glycine-derived nucleophile, for instance a GGG-peptide2 chain, generating a new ligated protein (e.g. protein1-LPXT-GGG-peptide2) (conversion 198 → 199, Scheme 50). It must be said that SrtA is able to recognize the LPXTG motif both in the C- and N-terminus of a protein.
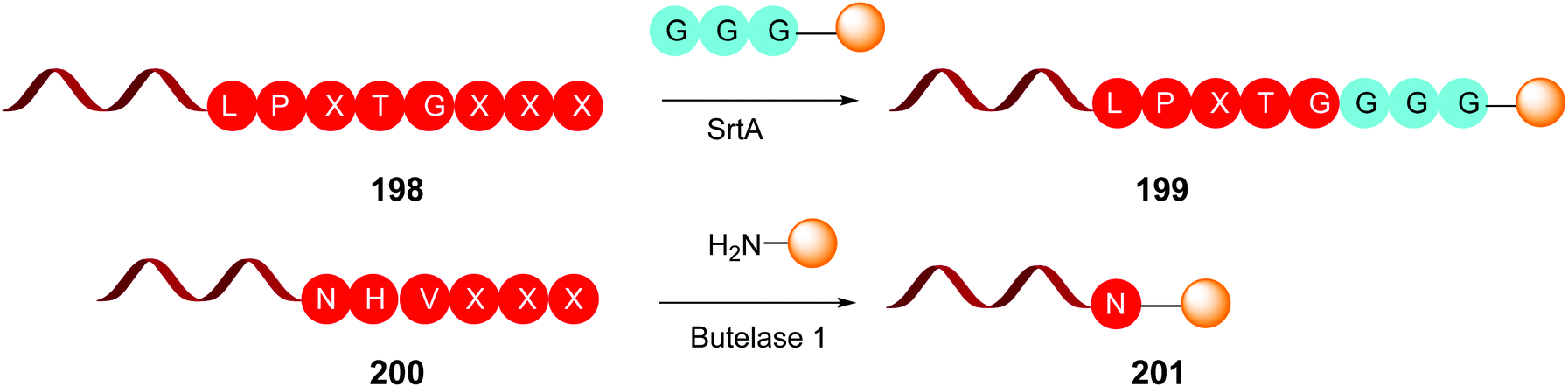 | ||
| Scheme 50 Some site-selective modifications catalyzed by enzymes at C-terminal positions. | ||
Butelase 1 was isolated from the medicinal plant Clitora ternatea and is an Asx peptide ligase that attaches C-terminal NHV or DHV sequences to the N-terminal domains of other proteins (conversion 200 → 201, Scheme 50). Although specific for the C-terminal residue, it accepts most N-terminal residues.141 Butelase is highly active (around 20000 times faster than SrtA) and has been used for protein cyclization and peptide ligation.141c
Many applications have been developed;141–144 for instance, SortA-catalyzed PEGylation has been used to attach proteins or protein conjugates to the surface of cells.143 Tam et al. have used butelase to label ubiquitin and green fluorescent protein in more than 95% yield.142
In a recent example, Ploegh et al. have reported the one-pot dual labeling of the immunoglobulin IgG1 with two different groups, based on the orthogonal specificity of butelase1 and sortase A (Scheme 51, conversion 202 → 203).145 The same group ligated two proteins through a linker 204, which had one of its end prepared to react with sortase, and the other prepared to react with butelase (Scheme 52, conversion 205/206 → 207 using linker 204). The nature of such linkers can influence the properties of the peptide/peptidomimetic backbone.145 More examples will likely appear in the next future.
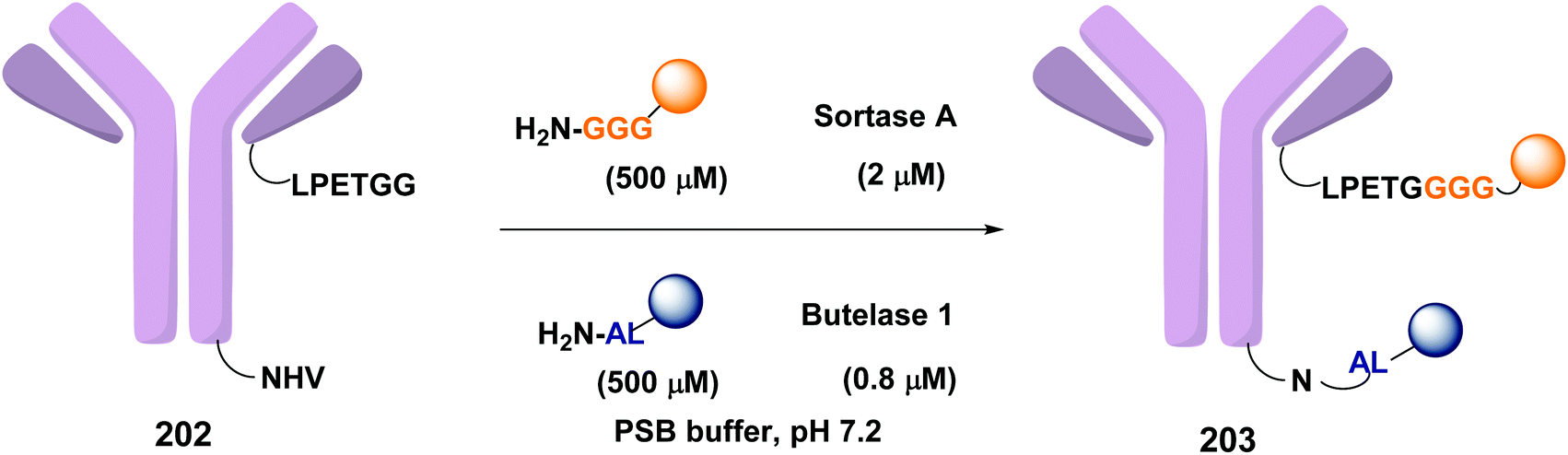 | ||
| Scheme 51 Selective labeling of IgG1 with butelase and sortase by Ploegh et al.145 | ||
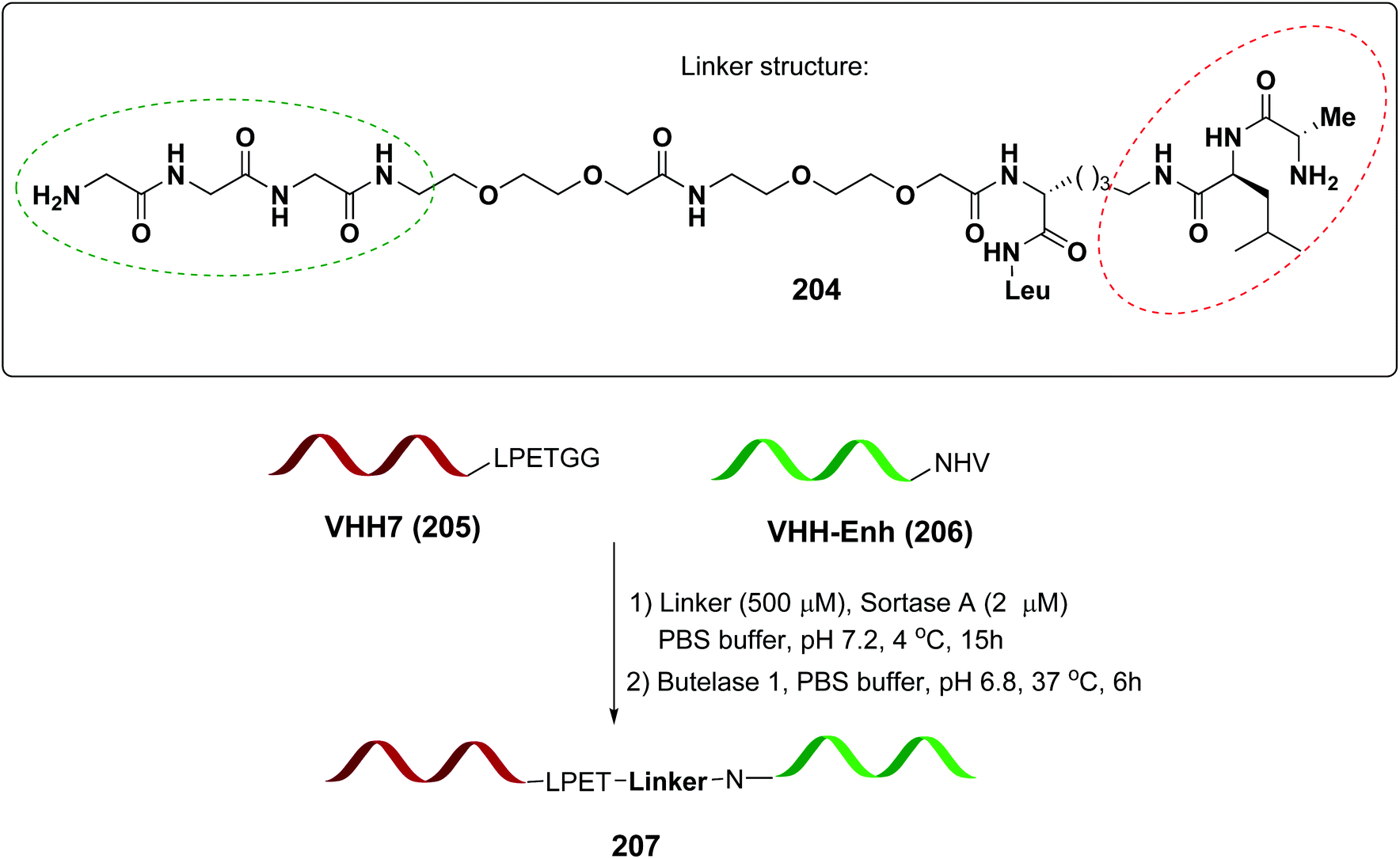 | ||
| Scheme 52 Selective protein coupling using an adaptable linker by Ploegh et al.145 | ||
6. Selective conversion of N-terminal units
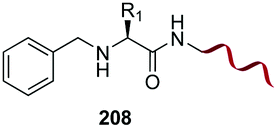
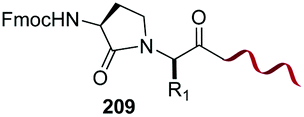
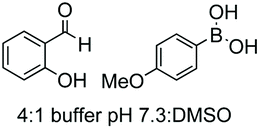
Unlike the site-selective transformation of backbone internal positions and C-terminal units, the selective conversion of N-terminal units has been covered by several reviews, and the reader will find a comprehensive treatment of the subject therein.1a,2,3a,e,g,146 Nevertheless, in this section a selection of methods has been compiled in Table 1, and commented below.| Entry | N-terminal residue | Reaction conditions | Product/s (%) | Ref. |
|---|---|---|---|---|
| 1 | Any | PhCHO NaCNBH3 |
|
148 |
| 2 | Any |
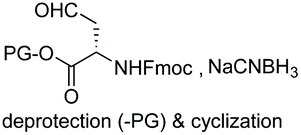
|
|
149 |
| 3 | Any |
|
|
150 |
| 4 | Any |
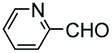
|
|
151 |
| 5 | Cys | R2-CHO |
|
152 |
| 6 | Any except Pro, Hyp, Pip |
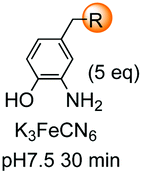
|
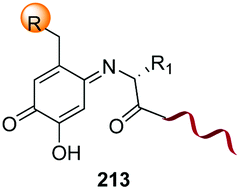
|
3i and 153 |
| 7 | Pro, Hyp |
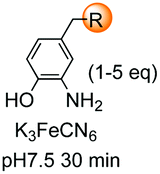
|
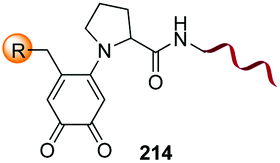
|
3i and 26a |
| 8 | Pro, others |
|

|
154 |
| 9 | Any |

|
|
25a |
| 10 | Gly |
|
|
155 |
| 11 | Any |
|
|
24b |
| 12 | Cys |
|

|
158 |
| 13 | Cys |
|
|
26b |
| 14 | Any |
|
|
160 |
| 15 | Any |
|
161–163 | |
| 16 | Any |
|
|
25b and 164 |
| 17 | Ser, Thr |
|
|
165b |
| 18 | Any |
|
|
167 |
| 19 | Val, other acyclic unit | Pd(OAc)2 (10 mol%) Mo(CO)6 (0.33 eq.) BQ (2.0 eq.), AgOAc (1.5 eq.), HFIP, 110 °C |
|
168 and 169 |
| 20 |
|
|

|
170 |
| 21 | G-N-E-A-S-Y-P-L |
|

|
171–173 |
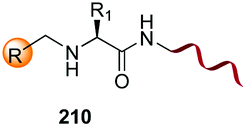
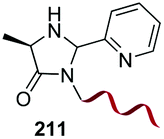
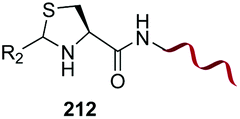
Electrophiles such as aldehydes react preferentially with the N-terminal residue over other basic units (such as lysine), as shown in entries 1–5. This selectivity is due to the fact that the N-terminal amine presents an α-electron-withdrawing carboxyl group, which lowers its pK value.147 Therefore, by controlling the pH (at slighty acidic values) it is possible to differentiate the amino group of the N-terminal residue (pKaca. 8) and those of lysine, ornithine (pKaca. 10) and arginine (ca. 12). Thus, Chou et al. selectively labeled the N-terminus of several proteins using a reductive amination, by treatment with aldehydes and sodium cyanoborohydride at pH ca. 6.1 (entry 1).148 Under this slighty acidic pH, the Nε-amino group in lysine was protonated and was unable to form the imine intermediate. Using reductive amination is preferable to N-alkylation with alkyl halides or similar electrophiles, since it avoids side reactions such as overalkylation.
Kodadek has reported an interesting reductive amination and cyclization, which generates a cycloalanine unit (CyAla) at the N-terminal position (entry 2). This modification is an efficient alternative to N-terminal methylation in order to obtain more protease-stable and cell-permeable peptides.149
In a variation of this method, Francis used an iridium complex to catalyse a reductive amination where sodium formate was used as the hydride source (entry 3).150
In another example, the group reported the use of 2-pyridine carboxyaldehydes (entry 4) to generate an imine that underwent intramolecular addition of the adjacent amide nitrogen, affording an aminal.151 In this case, the pyridine ring acted as a base that favored the cyclization reaction. Neri showed that when cysteine occupies the N-terminal position, a thiazolidine is formed (entry 5).152
A recent labeling of N-terminal residues uses the oxidation of an aminophenol by ferricyanide, which gives an o-aminoquinone intermediate (entries 6 and 7).3i,153 This reactive Michael acceptor undergoes addition of the N-terminal amino group. The reaction is fast (about 0.5 h) and tolerates many N-terminal units, with proline being the most reactive residue. The products are different for reaction with primary (entry 6) and secondary amines (entry 7).26a Ferricyanide offers advantages with respect to other oxidants (such as sodium periodate) since it avoids side-reactions (e.g. oxidation of glycans) and offers superior selectivity and yields.3i,26a Francis has used this strategy to label many proteins, as commented later.
Boronic acids are also useful to modify the N-terminal position, as shown in the example by Raj et al. where a terminal secondary amine such as proline underwent a Petasis bioconjugation to give a site-selective labeled peptide (entry 8).154 Angiotensin, bradykinin, insuline chain A, hyperthrehalosemic neuropeptide and creatine kinase were thus successfully labeled.
In another example, Ball et al. formed a heterocyclic aminal on treatment of the peptide with the boronic acid in the presence of ascorbic acid (entry 9).25a The reaction needs oxygen to take place, which suggests an “umpolung” reaction where an in situ formed dehydroascorbate acts as an oxidizing agent.
When glycine occupies the N-terminal position, treatment with aromatic aldehydes without reductive conditions leads to C-α alkylation, giving an aminoalcohol (entry 10).155 The method can differentiate N-terminal glycine even in cell lysates. Rai et al. used the method to prepare pure tagged proteins.
The selective acylation of the N-terminal position has also received much attention. Since Reid showed that the growth hormone could be selectively acetylated at the N-terminus by using pH control and a small amount of acetic anhydride,156 other authors have use different acylating agents (N-hydroxysuccinimide esters, N-hydroxyphthalimide, selenobenzaldehyde esters).157 Che et al. used an isolated alkyne-functionalized ketene (entry 11) which was generated from the acid in two steps: by forming a mixed anhydride with oxalyl chloride, and then by reaction with a base.24b Che also showed this selective labeling for lysozyme, insulin, RnaseA, BcArg and other peptides.24b
An interesting application of N-terminal acylation is to generate derivatives with better solubility and less prone to aggregation. Thus, Tolbert et al. attached betaine, which possesses a quaternary ammonium unit, to the N-terminal residue of two peptides, using native chemical ligation (entry 12).158
When cysteine occupies the N-terminal position, selective conjugation to a variety of tags can be achieved, as shown with the reaction with a cyanobenzothiazole (entry 13).26b N-Terminal cysteine can also be used for native chemical ligation, but ligation methods are beyond the scope of this review.28
The N-terminal position can also be modified by transamination promoted by metal ions such as Cu(II) and Ni(II). Inspired by previous work by other authors,159 Dixon and Moret160 used glyoxal to generate an imine that coordinated the metal (entry 14). The acidity of such imine favoured proton removal from C-α and isomerization to a second imine which was hydrolized to generate a terminal α-ketoacid.
The reaction was subsequently optimized, and Francis et al. reported a variation where pyridoxal-5-phosphate (PLP, vitamin B6) promoted the reaction (entry 15).161 Francis found that the reaction was completed in 2 h at 65 °C and 24 h at 25 °C for most amino acids,161 although some residues pose problems due to their low reactivity (Pro) or because side-reactions take place (Lys, His, Trp). Other alternative to PLP is its analog FHMDP, where the phosphate has been removed, and the pyridine is forming an N-methyl hydrochloride salt. FHMDP is much more efficient than PLP when bulky amino acids (Val, Leu, Ile) occupy the N-terminal position.162 The ketoproteins thus obtained are stable and can be readily manipulated. Francis and others have used this reaction to label different proteins, as commented later.25d,161–163 Francis has also reported other alternatives to PLP, such as Rapoport salt (N-methylpyridinium-4-carboxaldehyde-benzene sulfonate salt), which is particularly efficient for glutamate-rich sections (entry 16).25b,164 The method was applied to transamination of bacterially expressed proteins, such as cellulase mutants EES-, AEE- and AKT-EGPh in high yields.164
A particular case is when serine or threonine occupy the N-terminal position (entry 17). Fields and Dixon,165a and later Geoghegan and Stroh,165b reported that treatment with sodium periodate produced the oxidative cleavage of the hydroxymethyl group and the formation of a glyoxalate that can be trapped by amines, hydrazides, and other nucleophiles. The methodology was applied to the synthesis of modified peptides.165b A review by El Mahdi and Melnyk comments the glyoxolyl transformations to obtain bioconjugates.166
Other procedures transform the N-terminal amino group into other functions. For instance, van Hest et al. converted the terminal amino moiety into an azido group, by treatment with 1-sulfonyl azides (entry 18).167
Other selective modifications of the N-terminus use palladium-catalyzed C(sp3)–H activation.168,169 Carretero et al. converted acyclic residues into cyclic ones using this methodology (entry 19).168
Finally, enzymes can also be used to functionalize the N-terminus. Entries 20 and 21 illustrate sequence-specific conversions catalysed by the enzymes subtiligase and NMT.
Subtiligase conjugates C-terminal activated esters, such as thioesters, with the N-terminus of peptides and proteins (entry 20).170 Its promiscuity makes it ideal to conjugate a wide range of peptides and proteins. N-Myristoyltransferase (NMT) attachs a fatty acid to the N-terminal position of a protein expressing the sequence GNEASYPL, which facilitates its anchoring to the cell membrane.171 NMT can accept many different fatty acids, incorporating carbonyls, halogens, azides or alkynes for further protein functionalization.
It should be said that N-functionalization can deeply affect the stability, solubility, aggregation and other properties of the peptide. For instance, NMT was used to convert an elastin-related polypeptide into a myristoyl derivative, which then assembled in liposomes. The nanostructures were temperature-responsive and were used to encapsulate drugs such as doxorubicin and paclitaxel.172 The myristoylation has also been used to generate valuable fatty-acid-modified elastin-like polypeptides (FAMEs) which assembled into different nanostructures depending on the temperature.173
Finally, Fig. 3 presents some selected examples of protein and peptide functionalization using methodologies described in Table 1. More examples are given in the commented reviews. For instance, Francis et al. reported the modification of the N-terminus of native RNase (228) with 2-pyridine carboxaldehyde (PCA) derivatives (entry 5).151
 | ||
| Fig. 3 Application of site-selective modifications at N-terminal positions to protein functionalization. The 3D structure of GFP is authored by Francis,3i,26a of Lysozyme by Che24b (reproduced with ACS permission), and that of nanowires by Meldal175 (WILEY-VCH permission). Those of Rnase and interleukin 1a were created by Vossman176a and Boghog,176b respectively (public domain). | ||
The same group used the oxidation of aminophenols with ferricyanide (entries 6 and 7 in the Table 1) to label many different proteins, such as wild type lysozyme MS2, Lysozyme, RNase A, myoglobin, chymotrypsinogen and wild type Green Fluorecent Protein (GFP).3i,26a The N-terminal residues in these proteins were Ala (0% and 38% modification in wtMS2 and Lysozyme, respectively), Lys (30% for RNase), Gly (34% for myoglobin), Cys (42% for chymotrypsinogen) and Met (36% for wt GFP). Interestingly, when a proline was introduced into the N-terminal position of Green Fluorescent protein (compound 229) the levels of modification increased notably.3i,26a The oxidation was also employed to pegylate the enzyme creatine kinase by using PEG-derivatised aminophenols (50–60% modification).174
Ball et al. used the treatment with a boronate in the presence of ascorbic acid (entry 9)25a to label the blood-pressure regulating peptide angiotensin with a fluorophore (compound 230).
Che et al. labeled lysozyme (pdb: 2LYZ) in the N-terminal unit, in the presence of five lysines (compound 231) using ketenes (entry 11 in Table 1).24b This selective labeling was also used with insulin, RnaseA, BcArg and other peptides.24b
Tolbert et al. attached betaine to the N-terminal residue of bacterial xanthine-guanine phosphoribosyltransferase CG-GPRT (compound 232, entry 12 in Table 1), and antiHIV peptide CG-T20.158 The two peptides have a high propensity to aggregation, but once modified with betaine, their solubility was greatly increased without loss of activity. Interestingly, the modified CG-T20 is an inhibitor of the aggregation of native CG-T20.
Geoghegan and Stroh used the oxidation of serine with sodium periodate (entry 17) to label recombinant murine interleukin 1a (pdb: 2KKI) with a Lucifer yellow dye (compound 233).165b Other applications of this reaction include PEGylation, protein conjugates, protein dendrimers, and ligation of protein fragments.3f,57 Finally, Meldal used the method reported by van Hest (entry 18) to create nanowires (compound 234). Thus, the newly created azide was transformed using click chemistry.175 The fast progress in this area will likely provide more exciting examples in the next years.
7. Conclusions
The site-selective modification of peptides has achieved many advances in the last years, and although most of them are reported for side-chain manipulation, promising methodologies have also appeared to modify the backbone and thus modulate the conformation, folding ability, physico-chemical and biological properties of the peptide. This review has addressed the advances in the modification of Cα- and N-positions, from the use of “customizable units” to the residue-directed introduction of substituents. The site-selective modification of the peptide bond, i.e. the formation of thioamides or heterocycles, which alters backbone rigidity and ability to form hydrogen bonds or recognition by enzymes, was also described. In addition, not only the modifications in internal backbone positions, but also in the N- and C-termini were discussed.The use of “customizable amino acid units”, which undergo cleavage of a certain C–X bond (X![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) H, C) and its replacement by a variety of other functions (generating new C–C, C–N, C–O, C–P, C–S bonds, etc.) allow the efficient modification of the C-α. Thus, glycine has been used by different groups to introduce an alkyl, allyl or aryl chain, in some cases with excellent stereoselectivity.
H, C) and its replacement by a variety of other functions (generating new C–C, C–N, C–O, C–P, C–S bonds, etc.) allow the efficient modification of the C-α. Thus, glycine has been used by different groups to introduce an alkyl, allyl or aryl chain, in some cases with excellent stereoselectivity.
Photocatalytic methods have recently shown their promise to obtain a variety of such units with high stereoselectivity.
Glutamic acid has allowed the one-pot transformation of peptides with glutamic units into α,γ hybrids. The scission of serine and threonine, followed by the addition of new chains, has generated interesting α-alkyl and α-arylglycines. It has also afforded β-substituted dehydroamino acids with excellent stereoselectivity. Since these amino acids can be reduced with chiral catalysts, this is an interesting way to study the role of normal L-units, and compare them with D-units and rigid dehydroamino acid residues.
Serine, threonine and alloserine have been used to create cyclic sulfamidates that were transformed into α,α-disubstituted amino acids. Then the rigid sulfamidate ring could be opened with nucleophiles.
The introduction of L- or D-hydroxyproline units (Hyp) which underwent scission of the C4–C5 bond under very mild conditions, generated acyclic amino acids with a variety of α-chains which retained the stereochemistry of the starting unit. Moreover, an N,O-acetal was generated, which could be manipulated independently from the α-chain. The addition of nucleophiles under mild conditions afforded a variety of N-substituents, even bulky ones. Since peptide coupling of N-alkylated residues is challenging and plagued by side reactions and low reaction rate, this route could offer a promising solution to introduce bulky N-alkyl residues at any desired position. A “doubly customizable” Hyp unit, that can be decarboxylated and cleaved at the C4–C5 bond sequentially, with introduction of new chains/function at these positions, has recently been reported. Proline has also been developed as customizable unit, but in this case, the C5–N1 bond is cleaved by oxidation with iron catalysts. Although these customizable units have been studied mainly in small peptides, the application of these methodologies to larger molecules should be possible, and is an open line for the future.
Dehydroamino acids are special types of “customizable units”. Due to the capto-dative character of their substituted double bonds, their reactivity is quite different from that of the other amino acids. They can undergo the addition of nucleophiles, radicals, dienes, and azacompounds. The addition of thiol derivatives, for instance, has generated a variety of labeled peptides and proteins. Dha can be readily generated from serine, threonine and cysteine. The latter is particularly useful, since it is relatively scarce in peptides and proteins, and thus biomolecules with just one Dha unit in accesible (“open”) positions are feasible in many cases. The development of write-read-erase systems is an application of Dha with important biological applications.
Dha is a scarce amino acid, and as a result, it has been used to selectively label certain positions. However, if there are several units in a peptide, it can be difficult to differentiate between them. A possible solution is to have “masked” Dha units as phenyl- or alkylselenocysteine and/or cysteines using orthogonal protecting groups. These units could be activated sequentially, by varying the reaction conditions.
Another important challenge is the introduction of N-substituents, since it allows modulation of the number of hydrogen bonds, and also creates steric interactions that can strongly influence the secondary structure of the peptide, its solubility and aggregation properties. In addition to the “customizable units” commented before, glycine and alanine units with removable N-substituents have been used to disrupt structures that were aggregation-prone or presented other undesired properties. After the peptide was completed, the substituent was cleaved and the desired challenging peptide was released in high purity. Difficult β-amyloid and polyalanine sequences were completed this way.
An outstanding advance in this area was the His-directed N-alkylation of adjacent units. Since histidine is a relatively scarce amino acid, a peptide can often be monolabeled accurately. The development of Pyr-His “tags”, which are extremely rare in natural proteins and highly reactive, also affords promise for selective protein labeling. Moreover, the recent development of Red-shift backbone photocaging systems will allow the “marking” and “unmarking” of proteins and the modulation of their functions (active-unactive).
The selective labeling of C- and N-terminal positions is quite useful. C-Terminal positions have different pKa with respect to glutamic and aspartic units, which allows differentiation of these residues. Something similar happens with the N-terminal units and lysine, arginine and ornithine. Therefore, a variety of methods have been used to differentiate them. The selective oxidation of N-terminal units to glyoxalate followed by amination or other functionalizations is also remarkable. The oxidation of aminophenols with ferrycianide offers a mild, effective method to label large proteins at the N-terminus.
The decarboxylation of C-terminal units using photocatalytic methods has also found application in complex protein functionalization. With smaller peptides, the decarboxylation–alkylation of C-terminal “customizable units” has allowed the formation of terminal amino acid analogues, and thus α,β- and α,γ-hybrids.
The site-selective modification of the peptidic bond, for instance by formation of thioamides or heterocyclic rings has also been highlighted. The use of tryptophan to obtain oxazole–indole systems displaying high fluorescence is an example of the previous functionalization of the lateral chain to produce then a cyclization involving the peptide backbone. These remarkable transformations would be probably expanded in a future.
The high regio- and stereoselectivity achieved by enzymes is commented along the review. Many are quite selective, and their exquisite selectivity is at the cost of substrate limitation. But some enzymes have been reported that are promiscuous with respect to the whole substrate, recognizing a certain motif in different molecules. This is the case of enzymes such as sortase or butelase1 that recognize specific sequences in the peptide or protein and make modifications at these points. Since the discovery and engineering of new enzymes advances at a rapid pace the range of possible reactions will likely increase in the next future.
In conclusion, the site-selective backbone modification is a rapidly developing topic, with continuous promising incorporations to the synthetic toolbox. The ability to selectively modify the peptide backbone will be crucial to understand peptide conformation, folding and other physicochemical properties, and their biological action. This understanding will facilitate the design of tailor-made peptides for use in a variety of fields, such as biomedicine and materials science.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was mainly financed by project ProID2020010134 (Subvenciones a la Realización I + D Mª Carmen Betancourt y Molina, ACIISI-Gobierno de Canarias, with European Funds for Regional Development-FEDER) with contributions from project APOGEO (MAC/1.1.b./226, Cooperation Program INTERREG-MAC 2014–20 with FEDER funds, as well as Consejo Nacional de Ciencia y Tecnología (CONACYT, Mexico) through project 2015-01-807. D. H. also acknowledges her current contract (TRANSALUDAGRO) financed by Cabildo de Tenerife, Program TF INNOVA 2016-21 (with MEDI & FDCAN Funds). C. J. S. is currently recipient of a “Agustín de Betancourt”-Tenerife 2030 postdoctoral grant (Universidad de La Laguna-Cabildo de Tenerife, cofinanced by MEDI & FDCAN Funds). I. R-E. thanks CONACYT for Catedra contract 942.We also acknowledge support of the publication fee by CSIC Open Access Publication Support Initiative-through its Unit of Information Resources for Research (URICI).
Finally, although most of the Figures and Schemes were created for this review, some use 3D-structures obtained from the original sources, and have been acknowledged in the Scheme footer. We thank the authors and editorials for making them available for reproduction, particularly Fig. 2, which presents photos taken directly from the source (permission by Wiley-VCH). Other permissions by ACS, Wiley-VCH, Royal Society of Chemistry, Springer-Nature, Thieme, AAAS (for Science), Elsevier, USA National Academy of Sciences (for PNAS) etc., as commented in the footer references, are gratefully acknowledged.
Notes and references
- (a) D. G. Rawale, K. Thakur, S. R. Adusumalli and V. Rai, Chemical methods for selective labeling of proteins, Eur. J. Org. Chem., 2019, 6749 CrossRef CAS; (b) K. L. George and W. S. Horne, Guided protein backbone alteration, Acc. Chem. Res., 2018, 51, 1220 CrossRef CAS PubMed; (c) A. Dumas, L. Lercher, C. D. Spicer and B. G. Davis, Designing logical codon reassignment–Expanding the chemistry in biology, Chem. Sci., 2015, 6, 50 RSC; (d) A. G. Jamieson, N. Boutard, D. Sabatino and W. D. Lubell, Peptide scanning for studying structure-activity relationships in drug discovery, Chem. Biol. Drug Des., 2013, 81, 148 CrossRef CAS; (e) F. Albericio and H. G. Kruger, Therapeutic peptides, Future Med. Chem., 2012, 4, 1527 CrossRef CAS PubMed.
- J. N. de Gruyter, L. R. Malins and P. S. Baran, Residue-specific peptide modification: a chemist's guide, Biochemistry, 2017, 56, 3863 CrossRef CAS.
- (a) J. A. Shadish and C. A. DeForest, Site-selective protein modification: from functionalized proteins to functional biomaterials, Matter, 2020, 2, 50 CrossRef; (b) E. A. Hoyt, P. M. S. D. Cal, B. L. Oliveira and G. J. L. Bernardes, Contemporary approaches to site-selective protein modification, Nat. Rev. Chem., 2019, 3, 147 CrossRef CAS; (c) P. G. Isenegger and B. G. Davis, Concepts of catalysis in site-selective protein modifications, J. Am. Chem. Soc., 2019, 141, 8005 CrossRef CAS; (d) J. Ohata, S. C. Martin and Z. T. Ball, Metal-mediated functionalization of natural peptides and proteins: panning for bioconjugation gold, Angew. Chem., Int. Ed., 2019, 58, 6176 CrossRef CAS; (e) C. B. Rosen and M. B. Francis, Targeting the N-terminus for site-selective protein modification, Nat. Chem. Biol., 2017, 13, 697 CrossRef CAS; (f) N. Krall, F. P. da Cruz, O. Boutureira and G. J. L. Bernardes, Site-selective protein-modification chemistry for basic biology and drug development, Nat. Chem., 2016, 8, 103 CrossRef CAS; (g) O. Konievab and A. Wagner, Developments and recent advancements in the field of endogenous amino acid selective bond forming reactions for bioconjugation, Chem. Soc. Rev., 2015, 44, 5495 RSC; (h) O. Boutureira and G. J. L. Bernardes, Advances in chemical protein modification, Chem. Rev., 2015, 115, 2174 CrossRef CAS; (i) A. M. ElSohly and M. B. Francis, Development of oxidative coupling strategies for site-selective protein modification, Acc. Chem. Res., 2015, 48, 1971 CrossRef CAS PubMed; (j) G. Pasut and F. M. Veronese, State of the art in PEGylation: The great versatility achieved after forty years of research, J. Controlled Release, 2012, 161, 461 CrossRef CAS; (k) E. Basle, N. Joubert and M. Pucheault, Protein chemical modification on endogenous amino acids, Chem. Biol., 2010, 17, 213 CrossRef CAS; (l) J. Deska and U. Kazmaier, Peptide backbone modifications, Curr. Org. Chem., 2008, 12, 355 CrossRef CAS.
- (a) J. M. Chalker, G. J. L. Bernardes and B. G. Davis, A “Tag-and-Modify” approach to site-selective protein modification, Acc. Chem. Res., 2011, 44, 730 CrossRef CAS; (b) See also: C. D. Spicer and B. G. Davis, Selective chemical protein modification, Nat. Commun., 2014, 5, 4740 CrossRef CAS.
- (a) W. Tang and M. L. Becker, “Click” reactions: a versatile toolbox for the synthesis of peptide-conjugates, Chem. Soc. Rev., 2014, 43, 7013 RSC; (b) H. Li, R. Aneja and I. Chaiken, Click chemistry in peptide-based drug design, Molecules, 2013, 18, 9797 CrossRef CAS; (c) A. A. H. Ahmad Fuaad, F. Azmi, M. Skwarczynski and I. Toth, Peptide conjugation via CuAAC ‘click’ chemistry, Molecules, 2013, 18, 13148 CrossRef CAS.
- (a) M. S. Messina and H. D. Maynard, Modification of proteins using olefin metathesis, Mater. Chem. Front., 2020, 4, 1040 RSC; (b) Y. A. Lin, J. M. Chalker and B. G. Davis, Olefin metathesis for site-selective protein modification, ChemBioChem, 2009, 10, 959 CrossRef CAS; (c) A. Brik, Metathesis in peptides and peptidomimetics, Adv. Synth. Catal., 2008, 350, 1661 CrossRef CAS.
- S. B. Gunnoo and A. Madder, Chemical protein modification through cysteine, ChemBioChem, 2016, 17, 529 CrossRef CAS.
- D. Rabuka, Chemoenzymatic methods for site-specific protein modification, Curr. Opin. Chem. Biol., 2010, 14, 790 CrossRef CAS.
- (a) L. Zhang, R. Zhu, A. Feng, C. Zhao, L. Chen, G. Feng and L. Liu, Redox deracemization of β,γ-alkynyl α-amino esters, Chem. Sci., 2020, 11, 4444 RSC; (b) Z. Xie, X. Liu and L. Liu, Copper-catalyzed aerobic enantioselective cross-dehydrogenative coupling of N-aryl glycine esters with terminal alkynes, Org. Lett., 2016, 18, 2982 CrossRef CAS; (c) Z. Xie, J. Jia, X. Liu and L. Liu, Copper(II) triflate-catalyzed aerobic oxidative C-H functionalization of glycine derivatives with olefins and organoboranes, Adv. Synth. Catal., 2016, 358, 919 CrossRef CAS; (d) J.-P. Ebran, C. M. Jensen, S. A. Johannesen, J. Karaffa, K. B. Lindsay, R. Taaning and T. Skrydstrup, Creating carbon−carbon bonds with samarium diiodide for the synthesis of modified amino acids and peptides, Org. Biomol. Chem., 2006, 4, 3553 RSC.
- T. A. King, J. M. Kandemir, S. J. Walsh and D. R. Spring, Photocatalytic methods for amino acid modification, Chem. Soc. Rev., 2021, 50, 39 RSC.
- D. Hernández, C. Carro and A. Boto, “Doubly Customizable” Unit for the generation of structural diversity: From pure enantiomeric amines to peptide derivatives, J. Org. Chem., 2021, 86, 2796 CrossRef.
- T. J. Osberger, D. C. Rogness, J. T. Kohrt, A. F. Stepan and M. C. White, Oxidative diversification of amino acids and peptides by small-molecule iron catalysis, Nature, 2016, 537, 214 CrossRef CAS PubMed.
- J. W. Bogart and A. A. Bowers, Dehydroamino acids: chemical multi-tools for late-stage diversification, Org. Biomol. Chem., 2019, 17, 3653 RSC.
- (a) S. Kotha, V. B. Bandarugattu and N. G. Krishna, Diversity-oriented approach to unusual amino acid derivatives and heterocycles via methyl 2-acetamidoacrylate and its congeners, Tetrahedron, 2014, 70, 5361 CrossRef CAS; (b) C. Bonauer, T. Walenzyk and B. König, α,β-Dehydroamino Acids, Synthesis, 2006, 1 CAS.
- (a) D. Ajò, G. Granozzi, E. Tondello and A. Del Prà, Conformational flexibility of peptides containing α,β-unsaturated amino acid residues. Conformational analysis of N-acetyl-N′-methylamides of dehydroalanine and N-methyldehydroalanine, Biopolymers, 1980, 19, 469 CrossRef; (b) See also: H. L. Tran, K. W. Lexa, O. Julien, T. S. Young, C. T. Walsh, M. P. Jacobson and J. A. Wells, Structure–activity relationship and molecular mechanics reveal the importance of ring entropy in the biosynthesis and activity of a natural product, J. Am. Chem. Soc., 2017, 139, 2541 CrossRef CAS PubMed; (c) A. A. Bowers, M. G. Acker, A. Koglin and C. T. Walsh, Manipulation of Thiocillin variants by prepeptide gene replacement: structure, conformation, and activity of heterocycle substitution mutants, J. Am. Chem. Soc., 2010, 132, 7519 CrossRef CAS.
- (a) J. T. Mhlongo, E. Brasil, B. G. de la Torre and F. Albericio, Naturally Occurring Oxazole-Containing Peptides, Mar. Drugs, 2020, 18, 203 CrossRef CAS PubMed; (b) R. Dahiya, S. Dahiya, N. K. Fuloria, S. Kumar, R. Mourya, S. V. Chennupati, S. Jankie, H. Gautam, S. Singh, S. K. Karan, S. Maharaj, S. Fuloria, J. Shrivastava, A. Agarwal, S. Singh, A. Kishor, G. Jadon and A. Sharma, Natural bioactive thiazole-based peptides from marine resources: structural and pharmacological aspects, Mar. Drugs, 2020, 18, 329 CrossRef CAS; (c) S. Tilvi and K. S. Singh, Synthesis of oxazole, oxazoline and isoxazoline derived marine natural products: A review, Curr. Org. Chem., 2016, 20, 898 CrossRef CAS; (d) D. Davyt and G. Serra, Thiazole and oxazole alkaloids: isolation and synthesis, Mar. Drugs, 2010, 8, 2755 CrossRef CAS; (e) R. S. Roy, A. M. Gehring, J. C. Milne, P. J. Belshaw and C. T. Walsh, Thiazole and oxazole peptides: biosynthesis and molecular machinery, Nat. Prod. Rep., 1999, 16, 249 RSC.
- (a) F. Hamdan, F. Tahoori and S. Balalaie, Synthesis of novel cyclopeptides containing heterocyclic skeletons, RSC Adv., 2018, 8, 33893 RSC; (b) For a synthetic example, see: M. R. Davis, E. K. Singh, H. Wahyudis, L. D. Alexander, J. B. Kunicki, L. A. Nazarova, K. A. Fairweather, A. M. Giltrap, K. A. Jolliffe and S. R. McAlpine, Synthesis of sansalvamide A peptidomimetics: triazole, oxazole, thiazole, and pseudoproline containing compounds, Tetrahedron, 2012, 68, 1029 CrossRef CAS.
- (a) J. Chatterjee, F. Rechenmacher and H. Kessler, N-Methylation of peptides and proteins: An important element for modulating biological functions, Angew. Chem., Int. Ed., 2013, 52, 254 CrossRef CAS; (b) M. Quibell, T. Johnson and W. G. Turnell, Conformational studies on beta-amyloid protein carboxy-terminal region (residues 34-42): strategic use of amide backbone protection as a structural probe, Biomed. Pept., Proteins Nucleic Acids, 1994–1995, 1, 3 Search PubMed.
- (a) J. Blaakmeer, T. Tijsse-Klasen and G. Tesser, Enhancement of solubility by temporary dimethoxybenzyl-substitution of peptide bonds. Towards the synthesis of defined oligomers of alanine and of lysyl-glutamylglycine, Int. J. Pept. Protein Res., 1991, 37, 556 CrossRef CAS; (b) P. White, J. W. Keyte, K. Bailey and G. Bloomberg, Expediting the Fmoc solid phase synthesis of long peptides through the application of dimethyloxazolidine dipeptides, J. Pept. Sci., 2004, 10, 18 CrossRef CAS PubMed; (c) F. El Oualid, R. Merkx, R. Ekkebus, D. S. Hameed, J. J. Smit, A. de Jong, H. Hilkmann, T. K. Sixma and H. Ovaa, Chemical synthesis of ubiquitin, ubiquitin-based probes, and diubiquitin, Angew. Chem., Int. Ed., 2010, 49, 10149 CrossRef CAS.
- C. J. Saavedra, C. Carro, D. Hernández and A. Boto, Conversion of “customizable units” into N-alkyl amino acids and generation of N-alkyl peptides, J. Org. Chem., 2019, 84, 8392 CrossRef CAS PubMed.
- (a) Z. T. Ball, Protein substrates for reaction discovery: site-selective modification with boronic acid reagents, Acc. Chem. Res., 2019, 52, 566 CrossRef CAS PubMed; (b) J. Ohata, M. B. Minus, M. E. Abernathy and Z. T. Ball, Histidine-directed arylation/alkenylation of backbone N–H bonds mediated by copper(II), J. Am. Chem. Soc., 2016, 138, 7472 CrossRef CAS PubMed.
- V. Cardona, I. Eberle, S. Barthelemy, J. Beythien, B. Doerner, P. Schneeberger, J. Keyte and P. D. White, Application of DMB-dipeptides in the Fmoc SPPS of difficult and aspartimide-prone sequences, Int. J. Pept. Res. Ther., 2008, 14, 285 CrossRef CAS and references cited therein.
- For some representative examples, see: (a) C. Li, J. Wang, L. M. Barton, S. Yu, M. Tian, D. S. Peters, M. Kumar, A. W. Yu, K. A. Johnson, A. K. Chatterjee, M. Yan and P. S. Baran, Decarboxylative borylation, Science, 2017, 356, 7355 CrossRef PubMed; (b) T. Qin, L. R. Malins, J. T. Edwards, R. R. Merchant, A. J. Novak, J. Z. Zhong, R. B. Mills, M. Yan, C. Yuan, M. D. Eastgate and P. S. Baran, Nickel-catalyzed Barton decarboxylation and Giese reactions: A practical take on classic transforms, Angew. Chem., Int. Ed., 2017, 56, 260 CrossRef CAS PubMed; (c) W. M. Cheng, R. Shang and Y. Fu, Photoredox/Brønsted acid co-catalysis enabling decarboxylative coupling of amino acid and peptide redox-active esters with N-heteroarenes, ACS Catal., 2017, 7, 907 CrossRef CAS; (d) D. Hernández, A. Boto, D. Guzmán and E. Alvarez, Metal-free, direct conversion of α-amino acids into α-keto γ-amino esters for the synthesis of α,γ-peptides, Org. Biomol. Chem., 2017, 15, 7736 RSC; (e) Y. Jin, M. Jiang, H. Wang and H. Fu, Installing amino acids and peptides on N-heterocycles under visible-light assistance, Sci. Rep., 2016, 6, 20068 CrossRef PubMed; (f) J. Alsina and F. Albericio, Solid-phase synthesis of C-terminal modified peptides, Biopolymers, 2003, 71, 454 CrossRef CAS PubMed.
- For a review on N-terminal protein modification, see: (a) C. B. Rosen and M. B. Francis, Targeting the N-terminus for siteselective protein modification, Nat. Chem. Biol., 2017, 13, 697 CrossRef CAS PubMed; (b) A. O. Y. Chan, C. M. Ho, H. C. Chong, Y. C. Leung, J. S. Huang, M. K. Wong and C. M. Che, Modification of N-Terminal α-Amino Groups of Peptides and Proteins Using Ketenes, J. Am. Chem. Soc., 2012, 134, 2589 CrossRef CAS PubMed; (c) J. C. Slootweg, H. B. Albada, D. Siegmund and N. Metzler-Nolte, Efficient reagent-saving method for the N-terminal labeling of bioactive peptides with organometallic carboxylic acids by solid-phase synthesis, Organometallics, 2016, 35, 3192 CrossRef CAS.
- (a) J. Ohata and Z. T. Ball, Ascorbate as a pro-oxidant: mild N-terminal modification with vinylboronic acids, Chem. Commun., 2017, 53, 1622 RSC; (b) L. S. Witus, C. Netirojjanakul, K. S. Palla, E. M. Muehl, C. H. Weng, A. T. Iavarone and M. B. Francis, Site-specific protein transamination using N-methylpyridinium-4-carboxaldehyde, J. Am. Chem. Soc., 2013, 135, 17223 CrossRef CAS PubMed; (c) X. Ning, R. P. Temming, J. Dommerholt, J. Guo, D. B. Ania, M. F. Debets, M. A. Wolfert, G. J. Boons and F. L. van Delft, Protein modification by strain-promoted alkyne–nitrone cycloaddition, Angew. Chem., Int. Ed., 2010, 49, 3065 CrossRef CAS; (d) R. A. Scheck, M. T. Dedeo, A. T. Iavarone and M. B. Francis, Optimization of a biomimetic transamination reaction, J. Am. Chem. Soc., 2008, 130, 11762 CrossRef CAS PubMed.
- (a) A. C. Obermeyer, J. B. Jarman and M. B. Francis, N-Terminal Modification of Proteins with o-Aminophenols, J. Am. Chem. Soc., 2014, 136, 9572 CrossRef CAS; (b) H. Ren, F. Xiao, K. Zhan, Y. P. Kim, H. Xie, Z. Xia and J. Rao, A biocompatible condensation reaction for the labeling of terminal cysteine residues on protein, Angew. Chem., Int. Ed., 2009, 48, 9658 CrossRef CAS PubMed; (c) Y. Yuan and G. Liang, A biocompatible, highly efficient click reaction and its applications, Org. Biomol. Chem., 2014, 12, 865 RSC; (d) A. Bandyopadhyay, S. Cambray and J. Gao, Fast and selective labeling of N-terminal cysteines at neutral pH via thiazolidino boronate formation, Chem. Sci., 2016, 7, 4589 RSC; (e) L. R. Malins, J. N. deGruyter, K. J. Robbins, P. M. Scola, M. D. Eastgate, M. R. Ghadiri and P. S. Baran, Peptide macrocyclization inspired by non-ribosomal imine natural product, J. Am. Chem. Soc., 2017, 139, 5233 CrossRef CAS PubMed; (f) X. Li, L. Zhang, S. E. Hall and J. P. Tam, A new ligation method for N-terminal tryptophan-containing peptides using the Pictet–Spengler reaction, Tetrahedron Lett., 2000, 41, 4069 CrossRef CAS.
- L. Herrera Belén, C. de Oliveira Rangel-Yagui, J. F. Beltrán Lissabet, B. Effer, M. Lee-Estevez, A. Pessoa, R. L. Castillo and J. G. Farías, From synthesis to characterization of site-selective PEGylated proteins, Front. Pharmacol., 2019, 10, 1450 CrossRef PubMed.
- (a) J. M. Chalker, Chemoselective and bioorthogonal ligation reactions, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2017 Search PubMed; (b) See also: J. P. Tam, J. Xu and K. D. Eom, Methods and strategies of peptide ligation, Biopolymers, 2001, 60, 194 CrossRef CAS PubMed; (c) S. Kulkarni, J. Sayers, B. Premdjee and R. J. Payne, Rapid and efficient protein synthesis through expansion of the native chemical ligation concept, Nat. Rev. Chem., 2018, 2, 0122 CrossRef CAS; (d) T. Nuijens, A. Toplak, M. Schmidt, A. Ricci and W. Cabri, Natural occurring and engineered enzymes for peptide ligation and cyclization, Front. Chem., 2019, 7, 829 CrossRef CAS PubMed; (e) H. Liu and X. Li, Development and application of serine/threonine ligation for synthetic protein chemistry, Org. Biomol. Chem., 2014, 12, 3768 RSC.
- (a) D. I. Kim, S. C. Jensen, K. A. Noble, B. Kc, K. H. Roux, K. Motamedchaboki and K. J. Roux, An improved smaller biotin ligase for BioID proximity labeling, Mol. Biol. Cell, 2016, 27, 1188 CrossRef CAS PubMed; (b) B. Devadas, T. Lu, A. Katoh, N. S. Kishore, A. C. Wade, P. P. Mehta, D. A. Rudnick, M. L. Bryant, S. P. Adams and Q. Li, Substrate specificity of Saccharomyces cerevisiae myristoyl-CoA: protein N-myristoyltransferase. Analysis of fatty acid analogs containing carbonyl groups, nitrogen heteroatoms, and nitrogen heterocycles in an in vitro enzyme assay and subsequent identification of inhibitors of human immunodeficiency virus I replication, J. Biol. Chem., 1992, 267, 7224 CrossRef CAS PubMed; (c) W. P. Heal, M. H. Wright, E. Thinon and E. W. Tate, Multifunctional protein labeling via enzymatic N-terminal tagging and elaboration by click chemistry, Nat. Protoc., 2011, 7, 105 CrossRef PubMed; (d) C. Kulkarni, T. L. Kinzer-Ursem and D. A. Tirrell, Selective functionalization of the protein N- terminus with N-myristoyl transferase for bioconjugation in cell lysate, ChemBioChem, 2013, 14, 1958 CrossRef CAS PubMed.
- D. Seebach, A. K. Bech and A. Studer, in Modern Synthetic Methods, vol. 7, ed. B. Ernst and C. Leumann, VCH, Weinheim, 1995 Search PubMed.
- H. G. Bossler, P. Waldmeier and D. Seebach, Preparation and C-Alkylation of Peptides with Aminomalonate Building Blocks, Angew. Chem., Int. Ed. Engl., 1994, 33, 439 CrossRef.
- (a) H. Dialer, W. Steglich and W. Beck, 2-Cyclopentadienylglycine, a new α-amino acid and its use for η5-complex formation, Tetrahedron, 2001, 57, 4855 CrossRef CAS; (b) S. Schumann, K. Zeitler, M. Jäger, K. Polborn and W. Steglich, Stereocontrolled backbone connection of peptides by C=C double bonds, Tetrahedron, 2000, 56, 4187 CrossRef CAS.
- (a) P. Blakskjær, A. Gavrila, L. Andersen and T. Skrydstrup, An improved protocol for the SmI2-promoted C-alkylation of peptides: degradation and functionalization of serine residues in linear and cyclic peptides, Tetrahedron Lett., 2004, 45, 9091 CrossRef; (b) See also: M. Ricci, P. Blakskjær and T. Skrydstrup, Selective side chain introduction onto small peptides mediated by samarium diiodide: a potential route to peptide libraries, J. Am. Chem. Soc., 2000, 122, 12413 CrossRef CAS; (c) M. Ricci, L. Madariaga and T. Skrydstrup, Application of reductive samariation to the synthesis of small unnatural peptides, Angew. Chem., Int. Ed., 2000, 39, 242 CrossRef CAS.
- (a) J. Deska and U. Kazmaier, Highly stereoselective peptide modifications through Pd-catalyzed allylic alkylations of chelated peptide enolates, Chem. – Eur. J., 2007, 13, 6204 CrossRef CAS PubMed; (b) S. Datta and U. Kazmaier, Stereoselective palladium-catalyzed allylic alkylations of peptideamide enolates, Org. Biomol. Chem., 2011, 9, 872 RSC; (c) S. Datta, A. Bayer and U. Kazmaier, Highly stereoselective modifications of peptides via Pd-catalyzed allylic alkylation of internal peptide amide enolates, Org. Biomol. Chem., 2012, 10, 8268 RSC; (d) See also: M. Kohr and U. Kazmaier, Halogenated allyl alcohol derivatives - versatile electrophiles for palladium-catalyzed allylic alkylations, Eur. J. Org. Chem., 2019, 2843 CrossRef CAS; (e) For recent work on related complexes, see: A. Horn and U. Kazmaier, Stereoselective modification of N-(α-hydroxyacyl)-glycinesters via palladium-catalyzed allylic alkylation, Org. Lett., 2019, 21, 4595 CrossRef CAS PubMed.
- (a) L. Zhao, O. Basle and C. J. Li, Site-specific C-functionalization of free-(NH) peptides and glycine derivatives via direct C-H bond functionalization, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4106 CrossRef CAS PubMed; (b) L. Zhao, X. Liao and C. J. Li, Access to α-Functionalized Glycine Derivatives with Arylboronic Acid via Imino Amides, Synlett, 2009, 2953 CAS; (c) For reviews on the mechanism, see: T. Brandhofer and O. García Mancheño, Site-Selective C–H Bond Activation/Functionalization of α-Amino Acids and Peptide-Like Derivatives, Eur. J. Org. Chem., 2018, 6050 CrossRef CAS; (d) From C-H to C-C Bonds: Cross-Dehydrogenative-Coupling, ed. C.-J. Li, Green Chemistry Series, The Royal Society of Chemistry, 2015, 10.1039/978178262008, ISBN: 978-1-84973-797-5.
- C. Wang, M. Guo, R. Qi, Q. Shang, Q. Liu, S. Wang, L. Zhao, R. Wang and Z. Xu, Visible-light-driven, copper-catalyzed decarboxylative C(sp3)-H alkylation of glycine and peptides, Angew. Chem., Int. Ed., 2018, 57, 15841 CrossRef CAS PubMed.
- C. Wang, R. Qi, H. Xue, Y. Shen, M. Chang, Y. Chen, R. Wang and Z. Xu, Visible-light-promoted C(sp3)-H alkylation by intermolecular charge transfer: preparation of unnatural α-amino acids and late-stage modification of peptides, Angew. Chem., Int. Ed., 2020, 59, 7461 CrossRef CAS PubMed and references cited therein.
- H. Xue, M. Guo, C. Wang, Y. Shen, R. Qi, Y. Wu, Z. Xu and M. Chang, Photo-induced preparation of unnatural α-amino acids: synthesis and characterization of novel Leu5-enkephalin analogues, Org. Chem. Front., 2020, 7, 2426 RSC.
- F. B. Dyer, C. M. Park, R. Joseph and P. Garner, Aziridine-mediated ligation and site-specific modification of unprotected peptides, J. Am. Chem. Soc., 2011, 133, 20033 CrossRef CAS PubMed.
- (a) D. P. Galonić, N. D. Ide, W. A. van der Donk and D. Y. Gin, Aziridine-2-carboxylic acid-containing peptides: application to solution- and solid-phase convergent site-selective peptide modification, J. Am. Chem. Soc., 2005, 127, 7359 CrossRef PubMed; (b) D. P. Galonić, W. van der Donk and D. Y. Gin, Site-selective conjugation of thiols with aziridine-2-carboxylic acid-containing peptides, J. Am. Chem. Soc., 2004, 126, 12712 CrossRef PubMed.
- S. B. Cohen and R. L. Halcomb, Application of serine- and threonine-derived cyclic sulfamidates for the preparation of S-linked glycosyl amino acids in solution- and solid-phase peptide synthesis, J. Am. Chem. Soc., 2002, 124, 2534 CrossRef CAS PubMed.
- (a) N. Boutard, A. G. Jamieson, H. Ong and W. D. Lubell, Structure-activity analysis of the growth hormone secretagogue GHRP-6 by alpha- and beta-amino gamma-lactam positional scanning, Chem. Biol. Drug Des., 2010, 75, 40 CrossRef CAS PubMed; (b) For related work, see: N. Boutard, S. Turcotte, K. Beauregard, C. Quiniou, S. Chemtob and W. D. Lubell, Examination of the active secondary structure of the peptide 101.10, an allosteric modulator of the interleukin-1 receptor, by positional scanning using β-amino γ-lactams, J. Pept. Sci., 2011, 17, 288 CrossRef CAS PubMed; (c) A. G. Jamieson, N. Boutard, K. Beauregard, M. S. Bodas, H. Ong, C. Quiniou, S. Chemtob and W. D. Lubell, Positional scanning for peptide secondary structure by systematic solid-phase synthesis of amino lactam peptides, J. Am. Chem. Soc., 2009, 131, 7917 CrossRef CAS PubMed.
- (a) N. Mazo, I. García-González, C. D. Navo, F. Corzana, G. Jiménez-Osés, A. Avenoza, J. H. Busto and J. M. Peregrina, Synthesis of mixed α/β 2,2-peptides by site-selective ring-opening of cyclic quaternary sulfamidates, Org. Lett., 2015, 17, 5804 CrossRef CAS PubMed; (b) N. Mazo, C. D. Navo, J. M. Peregrina, J. H. Busto and G. Jiménez-Osés, Selective modification of sulfamidate-containing peptides, Org. Biomol. Chem., 2020, 18, 6265 RSC.
- (a) K. Wright, M. Sarciaux, A. de Castries, M. Wakselman, J. P. Mazaleyrat, A. Toffoletti, C. Corvaja, M. Crisma, C. Peggion, F. Formaggio and C. Toniolo, Synthesis of enantiomerically pure cis- and trans-4-amino-1-oxyl-2,2,6,6-tetramethylpiperidine-3-carboxylic acid: a spin-labelled, cyclic, chiral β-amino acid, and 3D-structural analysis of a doubly spin-labelled β-hexapeptide, Eur. J. Org. Chem., 2007, 3133 CrossRef CAS; (b) Z. X. Jiang and Y. B. Yu, The synthesis of a geminally perfluoro-tert-butylated β-amino acid and its protected forms as a potential pharmacokinetic modulator and reporter for peptide-based pharmaceuticals, J. Org. Chem., 2007, 72, 1464 CrossRef CAS PubMed; (c) F. Fülöp, T. A. Martinek and G. K. Tóth, Application of alicyclic β-amino acids in peptide chemistry, Chem. Soc. Rev., 2006, 35, 323 RSC; (d) P. I. Arvidsson, N. S. Ryder, H. M. Weiss, D. F. Hook, J. Escalante and D. Seebach, Exploring the antibacterial and hemolytic activity of shorter- and longer-chain β-, α,β-, and γ-peptides, and of β-peptides from β,2-3-aza- and β3-2-methylidene-amino acids bearing proteinogenic side chains–A survey, Chem. Biodivers., 2005, 2, 401 CrossRef CAS PubMed; (e) R. P. Cheng, S. H. Gellman and W. F. DeGrado, β-Peptides: from structure to function, Chem. Rev., 2001, 101, 3219 CrossRef CAS PubMed.
- (a) R. Rai, P. G. Vasudev, K. Ananda, S. Raghothama, N. Shamala, I. L. Karle and P. Balaram, Hybrid peptides: expanding the β turn in peptide hairpins by the insertion of β-, γ-, and δ-residues, Chem. – Eur. J., 2007, 13, 5917 CrossRef CAS PubMed; (b) S. Abele, P. Seiler and D. Seebach, synthesis, crystal structures, and modelling of β-oligopeptides consisting of 1-(aminomethyl)cyclopropanecarboxylic acid: ribbon-type arrangement of eight-membered H-bonded rings, Helv. Chim. Acta, 1999, 82, 1559 CrossRef CAS; (c) S. Abele and D. Seebach, Preparation of achiral and of enantiopure geminally disubstituted β-amino acids for β-peptide synthesis, Eur. J. Org. Chem., 2000, 1 CrossRef CAS; (d) For the biological importance of turns, see: J. D. A. Tyndall, B. Pfeiffer, G. Abbenante and D. P. Fairlie, Over one hundred peptide-activated G-protein-coupled receptors recognize ligands with turn structure, Chem. Rev., 2005, 105, 793 CrossRef CAS PubMed; (e) J. A. Kritzer, O. M. Stephens, D. A. Guarracino, S. K. Reznik and A. Schepartz, β-Peptides as inhibitors of protein–protein interactions, Bioorg. Med. Chem., 2005, 13, 11 CrossRef CAS PubMed.
- C. J. Saavedra, A. Boto and R. Hernández, Synthesis of α,γ-peptide hybrids by selective conversion of glutamic acid units, Org. Lett., 2012, 14, 3542 CrossRef CAS PubMed.
- M. Bogensttäte and W. Steglich, Synthesis of optically pure α-hydroxyglycine peptides, Tetrahedron, 1997, 53, 7267 CrossRef.
- (a) I. Romero-Estudillo, C. J. Saavedra, A. Boto and E. Alvarez, Site-selective modification of peptides: From “customizable units” to novel α-aryl and α-alkyl glycine derivatives, and components of branched peptides, Biopolymers, 2015, 104, 650 CrossRef CAS PubMed; (b) For a precedent, see: A. Boto, J. A. Gallardo, D. Hernández and R. Hernández, Synthesis of unnatural amino acids from serine derivatives by β-fragmentation of primary alkoxyl radicals, J. Org. Chem., 2007, 72, 7260 CrossRef CAS PubMed.
- (a) C. J. Saavedra, D. Hernández and A. Boto, Site-selective peptide modification by conversion of “customizable” units into β-substituted dehydroamino acids, Chem. – Eur. J., 2018, 24, 599 CrossRef CAS PubMed; (b) C. J. Saavedra, R. Hernández and A. Boto, “Customizable” units in di- and tripeptides: selective conversion into substituted dehydroamino acids, Org. Lett., 2012, 14, 3788 CrossRef CAS PubMed.
- (a) I. Romero-Estudillo and A. Boto, Creating diversity by site-selective peptide modification: a customizable unit affords amino acids with high optical purity, Org. Lett., 2013, 15, 5778 CrossRef CAS PubMed; (b) I. Romero-Estudillo and A. Boto, Domino process achieves site-selective peptide modification with high optical purity. Applications to chain diversification and peptide ligation, J. Org. Chem., 2015, 80, 9379 CrossRef CAS PubMed.
- C. J. Saavedra, F. Cuevas, I. Romero-Estudillo and A. Boto, Synthesis of diketopiperazine scaffolds with tailored N- and α-chains by selective modification of customizable units, Adv. Synth. Catal., 2020, 362, 3158 CrossRef CAS.
- F. Cuevas, C. J. Saavedra, I. Romero-Estudillo, A. Boto, M. Ordóñez and I. Vergara, Structural diversity using Hyp “customizable units”: proof of-concept, synthesis of sansalvamide-related antitumoral peptides, Eur. J. Org. Chem., 2021, 933 CrossRef CAS.
- M. Beals, L. Gross and S. Harrell, Amino Acid Frequency, in http://www.tiem.utk.edu/~gross/bioed/webmodules/aminoacid.htm.
- (a) G. J. L. Bernardes, J. M. Chalker, J. C. Errey and B. G. Davis, Facile conversion of cysteine and alkyl cysteines to dehydroalanine on protein surfaces: versatile and switchable access to functionalized proteins, J. Am. Chem. Soc., 2008, 130, 5052 CrossRef CAS PubMed; (b) See also: O. Boutureira, G. J. L. Bernardes, F. D'Hooge and B. G. Davis, Direct radiolabelling of proteins at cysteine using [18F]-fluorosugars, Chem. Commun., 2011, 47, 10010 RSC; (c) E. J. Grayson, G. J. L. Bernardes, J. M. Chalker, O. Boutureira, J. R. Koeppe and B. G. Davis, A coordinated synthesis and conjugation strategy for the preparation of homogeneous glycoconjugate vaccine candidates, Angew. Chem., Int. Ed., 2011, 50, 4127 CrossRef CAS PubMed; (d) J. M. Chalker, Y. A. Lin, O. Boutureira and B. G. Davis, Enabling olefin metathesis on proteins: chemical methods for installation of S-allyl cysteine, Chem. Commun., 2009, 3714 RSC; (e) M. Fernández-González, O. Boutureira, G. J. L. Bernardes, J. M. Chalker, M. A. Young, J. C. Errey and B. G. Davis, Site-selective chemoenzymatic construction of synthetic glycoproteins using endoglycosidases, Chem. Sci., 2010, 1, 709 RSC.
- (a) L. R. Malins, N. J. Mitchell and R. J. Payne, Peptide ligation chemistry at selenol amino acids, J. Pept. Sci., 2014, 20, 64 CrossRef CAS PubMed; (b) J. M. Chalker, S. B. Gunnoo, O. Boutureira, S. C. Gerstberger, M. Fernandez-Gonzalez, G. J. L. Bernardes, L. Griffin, H. Hailu, C. J. Schofield and B. G. Davis, Methods for converting cysteine to dehydroalanine on peptides and proteins, Chem. Sci., 2011, 2, 1666 RSC; (c) N. M. Okeley, Y. Zhu and W. A. van der Donk, Facile Chemoselective Synthesis of Dehydroalanine-Containing Peptides, Org. Lett., 2000, 2, 3603 CrossRef CAS PubMed.
- J. M. Chalker, L. Lercher, N. R. Rose, C. J. Schofield and B. G. Davis, Conversion of Cysteine into dehydroalanine enables access to synthetic histones bearing diverse post-translational modifications, Angew. Chem., Int. Ed., 2012, 51, 1835 CrossRef CAS PubMed.
- (a) F. C. Rowan, M. Richards, R. A. Bibby, A. Thompson, R. Bayliss and J. Blagg, Insights into Aurora-A kinase activation using unnatural amino acids incorporated by chemical modification, ACS Chem. Biol., 2013, 8, 2184 CrossRef CAS PubMed; (b) R. I. Nathani, P. Moody, V. Chudasama, M. E. B. Smith, R. J. Fitzmaurice and S. Caddick, A novel approach to the site-selective dual labelling of a protein via chemoselective cysteine modification, Chem. Sci., 2013, 4, 3455 RSC.
- (a) J. Guo, J. Wang, J. Seok Lee and P. G. Schultz, Site-specific incorporation of methyl- and acetyl-lysine analogues into recombinant proteins, Angew. Chem., Int. Ed., 2008, 47, 6399 CrossRef CAS PubMed; (b) See also: Y. Takaoka, A. Ojida and I. Hamachi, Protein organic chemistry and applications for labeling and engineering in live-cell systems, Angew. Chem., Int. Ed., 2013, 52, 4088 CrossRef CAS PubMed.
- Y. Zhu and W. A. van der Donk, Convergent synthesis of peptide conjugates using dehydroalanines for chemoselective ligations, Org. Lett., 2001, 3, 1189 CrossRef CAS PubMed.
- G. B. Vamisetti, G. Satish, P. Sulkshane, G. Mann, M. H. Glickman and A. Brik, On-demand detachment of succinimides on cysteine to facilitate (semi)synthesis of challenging proteins, J. Am. Chem. Soc., 2020, 142, 19558 CrossRef CAS PubMed.
- C. L. Windle, K. J. Simmons, J. R. Ault, C. H. Trinh, A. Nelson, A. R. Pearson and A. Berry, Extending enzyme molecular recognition with an expanded amino acid alphabet, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 2610 CrossRef CAS PubMed.
- J. Dadová, K.-J. Wu, P. G. Isenegger, J. C. Errey, G. J. L. Bernardes, J. M. Chalker, L. Raich, C. Rovira and B. G. Davis, Precise probing of residue roles by post-translational β,γ-C,N aza-Michael mutagenesis in enzyme active sites, ACS Cent. Sci., 2017, 3, 1168 CrossRef PubMed.
- P. M. T. Ferreira, H. L. S. Maia and L. S. Monteiro, High yielding synthesis of heterocyclic β-substituted alanine derivatives, Tetrahedron Lett., 1999, 40, 4099 CrossRef CAS.
- P. M. T. Ferreira, H. L. S. Maia, L. S. Monteiro and J. Sacramento, Michael addition of thiols, carbon nucleophiles and amines to dehydroamino acid and dehydropeptide derivatives, J. Chem. Soc., Perkin Trans. 1, 2001, 3167 RSC.
- A. de la Hoz, A. Díaz-Ortiz, M. Victoria Gómez, J. A. Mayoral, A. Moreno, A. M. Sánchez-Migallón and E. Vázquez, Preparation of α- and β-substituted alanine derivatives by α-amidoalkylation or Michael addition reactions under heterogeneous catalysis assisted by microwave irradiation, Tetrahedron, 2001, 57, 5421 CrossRef CAS.
- E. Angelini, C. Balsamini, F. Bartoccini, S. Lucarini and G. Piersanti, Switchable reactivity of acylated α,β-dehydroamino ester in the Friedel−Crafts alkylation of indoles by changing the Lewis acid, J. Org. Chem., 2008, 73, 5654 CrossRef CAS PubMed.
- M. Righi, F. Bartoccini, S. Lucarini and G. Piersanti, Organocatalytic synthesis of α-quaternary amino acid derivatives via aza-Friedel–Crafts alkylation of indoles with simple α-amidoacrylates, Tetrahedron, 2011, 67, 7923 CrossRef CAS.
- Y. A. Lin, O. Boutureira, L. Lercher, B. Bhushan, R. S. Paton and B. G. Davis, Rapid cross-metathesis for reversible protein modifications via chemical access to Se-allyl-selenocysteine in proteins, J. Am. Chem. Soc., 2013, 135, 12156 CrossRef CAS PubMed.
- J. Yang, W. Sun, Z. He, C. Yu, G. Bao, Y. Li, Y. Liu, L. Hong and R. Wang, Access to α,γ-diamino diacid derivatives via organocatalytic asymmetric 1,4-addition of azlactones and dehydroalanines, Org. Lett., 2018, 20, 7080 CrossRef CAS PubMed.
- C. Aydillo, I. Compañón, A. Avenoza, J. H. Busto, F. Corzana, J. M. Peregrina and M. M. Zurbano, S-Michael additions to chiral dehydroalanines as an entry to glycosylated cysteines and a sulfa-Tn antigen mimic, J. Am. Chem. Soc., 2014, 136, 789 CrossRef CAS PubMed.
- B. N. Naidu, M. E. Sorenson, T. P. Connolly and Y. Ueda, Michael addition of amines and thiols to dehydroalanine amides: a remarkable rate acceleration in water, J. Org. Chem., 2003, 68, 10098 CrossRef CAS PubMed.
- (a) M. P. C. Mulder, K. Witting, I. Berlin, J. N. Pruneda, K.-P. Wu, J.-G. Chang, R. Merkx, J. Bialas, M. Groettrup, A. C. O. Vertegaal, B. A. Schulman, D. Komander, J. Neefjes, F. El Oualid and H. Ovaa, A cascading activity-based probe sequentially targets E1-E2-E3 ubiquitin enzymes, Nat. Chem. Biol., 2016, 12, 523 CrossRef CAS PubMed; (b) N. Haj-Yahya, H. P. Hemantha, R. Meledin, S. Bondalapati, M. Seenaiah and A. Brik, Dehydroalanine-based diubiquitin activity probes, Org. Lett., 2013, 16, 540 CrossRef PubMed; (c) Z. U. Wang, Y.-S. Wang, P.-J. Pai, W. K. Russell, D. H. Russell and W. R. Liu, A facile method to synthesize histones with posttranslational modification mimics, Biochemistry, 2014, 51, 5232 CrossRef PubMed.
- C. Dupuy, C. Petrier, L. A. Sarandeses and J. L. Luche, Ultrasound in organic syntheses. XIX. Further studies on the conjugate additions to electron deficient olefins in aqueous media, Synth. Commun., 1991, 21, 643 CrossRef CAS.
- M. D. Simon, F. Chu, L. R. Racki, C. C. de la Cruz, A. L. Burlingame, B. Panning, G. J. Narlikar and K. M. Shokat, The site-specific installation of methyl-lysine analogs into recombinant histones, Cell, 2007, 128, 1003 CrossRef CAS PubMed.
- J. Wang, H. Lundberg, S. Asai, P. Martín-Acosta, J. S. Chen, S. Brown, W. Farrell, R. G. Dushin, C. J. O'Donnell, A. S. Ratnayake, P. Richardson, Z. Liu, T. Qin, D. G. Blackmond and P. S. Baran, Kinetically guided radical-based synthesis of C(sp3)−C(sp3) linkages on DNA, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E6404 CrossRef CAS PubMed.
- A. Yang, S. Ha, J. Ahn, R. Kim, S. Kim, Y. Lee, J. Kim, D. Soll, H. Y. Lee and H. S. Park, A chemical biology route to site-specific authentic protein modifications, Science, 2016, 354, 623C CrossRef PubMed.
- (a) T. H. Wright, B. J. Bower, J. M. Chalker, G. J. L. Bernardes, R. Wiewiora, W. L. Ng, R. Raj, S. Faulkner, M. R. J. Vallee, A. Phanumartwiwath, O. D. Coleman, M. L. Thezenas, M. Khan, S. R. G. Galan, L. Lercher, M. W. Schombs, S. Gerstberger, M. E. Palm-Espling, A. J. Baldwin, B. M. Kessler, T. D. W. Claridge, S. Mohammed and B. G. Davis, Posttranslational mutagenesis: A chemical strategy for exploring protein side-chain diversity, Science, 2016, 354, 1465 CrossRef PubMed; (b) For related work with these proteins, see also: M. R. J. Vallée, M. W. Schombs, Z. J. Balaban, J. Colyer and B. G. Davis, Ready display of antigenic peptides in a protein ‘mimogen’, Chem. Commun., 2016, 52, 3014 RSC; (c) B. Josephson, C. Fehl, P. G. Isenegger, S. Nadal, T. H. Wright, A. W. J. Poh, B. J. Bower, A. M. Giltrap, L. Chen, C. Batchelor-McAuley, G. Roper, O. Arisa, J. B. I. Sap, A. Kawamura, A. J. Baldwin, S. Mohammed, R. G. Compton, V. Gouverneur and B. G. Davis, Light-driven post-translational installation of reactive protein side chains, Nature, 2020, 585, 530 CrossRef CAS PubMed.
- For a precedent in amino acids, see: R. A. Aycock, D. Vogt and N. Jui, A practical and scalable system for heteroaryl amino acid synthesis, Chem. Sci., 2017, 8, 7998 RSC.
- R. A. Aycock, C. J. Pratt and N. T. Jui, Aminoalkyl Radicals as powerful intermediates for the synthesis of unnatural amino acids and peptides, ACS Catal., 2018, 8, 9115 CrossRef CAS.
- T. Rossolini, J. A. Leitch, R. Grainger and D. J. Dixon, Photocatalytic three-component umpolung synthesis of 1,3-diamines, Org. Lett., 2018, 20, 6794 CrossRef CAS PubMed.
- A. D. de Bruijn and G. Roelfes, Chemical modification of dehydrated amino acids in natural antimicrobial peptides by photoredox catalysis, Chem. – Eur. J., 2018, 24, 11314 CrossRef CAS PubMed.
- (a) For precedents in amino acids, see: J. Sim, M. W. Campbell and G. A. Molander, Synthesis of α-fluoro-α-amino acid derivatives via photoredox-catalyzed carbofluorination, ACS Catal., 2019, 9, 1558 CrossRef CAS PubMed; (b) T. Constantin, M. Zanini, A. Regni, N. S. Sheikh, F. Juliá and D. Leonori, Aminoalkyl radicals as halogen-atom transfer agents for activation of alkyl and aryl halides, Science, 2020, 367, 1021 CrossRef CAS PubMed.
- (a) M. R. Aronoff, B. Gold and R. T. Raines, 1,3-Dipolar cycloadditions of diazo compounds in the presence of azides, Org. Lett., 2016, 18, 1538 CrossRef CAS PubMed; (b) R. H. de Vries, J. H. Viel, R. Oudshoorn, O. P. Kuipers and G. Roelfes, Selective modification of ribosomally synthesized and post-translationally modified peptides (RiPPs) through Diels-Alder cycloadditions on dehydroalanine residues, Chem. – Eur. J., 2019, 25, 12698 CrossRef CAS PubMed; (c) G. Bao, P. Wang, G. Li, C. Yu, Y. Li, Y. Liu, Z. He, T. Zhao, J. Rao, J. Xie, L. Hong, W. Sun and R. Wang, 1,3-Dipolar cycloaddition between dehydroalanines and C,N-cyclic azomethine imines: application to late-stage peptide modification, Angew. Chem., Int. Ed., 2021, 60, 5331 CrossRef CAS PubMed.
- W. J. Wever, J. W. Bogart, J. A. Baccile, A. N. Chan, F. C. Schroeder and A. A. Bowers, Chemoenzymatic synthesis of thiazolyl peptide natural products featuring an enzyme-catalyzed formal [4+2] cycloaddition, J. Am. Chem. Soc., 2015, 137, 3494 CrossRef CAS PubMed.
- (a) B. M. Kenda, A. C. Matagne, P. E. Talaga, P. M. Pasau, E. Differding, B. I. Lallemand, A. M. Frycia, F. G. Moureau, H. V. Klitgaard, M. R. Gillard, B. Fuks and P. Michel, Discovery of 4-substituted pyrrolidone butanamides as new agents with significant antiepileptic activity, J. Med. Chem., 2004, 47, 530 CrossRef CAS PubMed; (b) R. Kuwano and Y. Ito, Asymmetric hydrogenation of 1,4,5,6-tetrahydropyrazine-2-(N-tert-butyl)carboxamide catalyzed by trans-chelating chiral diphosphine−rhodium complexes, J. Org. Chem., 1999, 64, 1232 CrossRef CAS; (c) K. B. Hansen, Y. Hsiao, F. Xu, N. Rivera, A. Clausen, M. Kubryk, S. Krska, T. Rosner, B. Simmons, J. Balsells, N. Ikemoto, Y. Sun, F. Spindler, C. Malan, E. J. J. Grabowski and J. D. Armstrong III, Highly efficient asymmetric synthesis of sitagliptin, J. Am. Chem. Soc., 2009, 131, 8798 CrossRef CAS PubMed; (d) A. Ohashi and T. Imamoto, Highly enantioselective hydrogenation of α-dehydroamino acids by rhodium complexes with new unsymmetric P−chirogenic bisphosphine ligands, Org. Lett., 2001, 3, 373 CrossRef CAS PubMed; (e) P. Etayo and A. Vidal-Ferran, Rhodium-catalysed asymmetric hydrogenation as a valuable synthetic tool for the preparation of chiral drugs, Chem. Soc. Rev., 2013, 42, 728 RSC.
- D. N. Le, E. Hansen, H. A. Khan, B. Kim, O. Wiest and V. M. Dong, Hydrogenation catalyst generates cyclic peptide stereocentres in sequence, Nat. Chem., 2018, 10, 968 CrossRef CAS PubMed.
- C. J. Chapman, J. D. Hargrave, G. Bish and C. G. Frost, Peptide modification through site-selective residue interconversion: application of the rhodium-catalysed 1,4-addition of aryl siloxanes and boronates, Tetrahedron, 2008, 64, 9528 CrossRef CAS.
- C. J. Chapman, A. Matsuno, C. G. Frost and M. C. Willis, Site-selective modification of peptides using rhodium and palladium catalysis: complementary electrophilic and nucleophilic arylation, Chem. Commun., 2007, 3903 RSC.
- H. M. Key and S. J. Miller, Site- and stereoselective chemical editing of thiostrepton by Rh-catalyzed conjugate arylation: new analogues and collateral enantioselective synthesis of amino acids, J. Am. Chem. Soc., 2017, 139, 15460 CrossRef CAS PubMed.
- A. D. de Bruijn and G. Roelfes, Catalytic modification of dehydroalanine in peptides and proteins by palladium-mediated cross-coupling, Chem. – Eur. J., 2018, 24, 12728 CrossRef CAS PubMed.
- (a) L. M. Repka, J. R. Chekan, S. K. Nair and W. A. van der Donk, Mechanistic understanding of lanthipeptide biosynthetic enzymes, Chem. Rev., 2017, 117, 5457 CrossRef CAS PubMed; (b) X. Zhang, W. Ni and W. A. van der Donk, On the regioselectivity of thioether formation by lacticin 481 synthetase, Org. Lett., 2007, 9, 3343 CrossRef CAS PubMed; (c) H. Wang and W. A. van der Donk, Biosynthesis of the class III lantipeptide catenulipeptin, ACS Chem. Biol., 2012, 7, 1529 CrossRef CAS PubMed; (d) G. H. Völler, J. M. Krawczyk, A. Pesic, B. Krawczyk, J. Nachtigall and R. D. Süssmuth, Involvement and unusual substrate specificity of a prolyl oligopeptidase in class III Lanthipeptide maturation, ChemBioChem, 2012, 13, 1174 CrossRef PubMed.
- (a) B. Li, J. P. J. Yu, J. S. Brunzelle, G. N. Moll, W. A. van der Donk and S. K. Nair, Structure and mechanism of the lantibiotic cyclase involved in nisin biochemistry, Science, 2006, 311, 1464 CrossRef CAS PubMed; (b) Y. Zhu, M. D. Gieselman, H. Zhou, O. Averin and W. A. van der Donk, Biomimetic studies on the mechanism of stereoselective lanthionine formation, Org. Biomol. Chem., 2003, 1, 330 RSC.
- B. Li, D. Sher, L. Kelly, Y. Shi, K. Huang, P. J. Knerr, I. Joewono, D. Rusch, S. W. Chisholm and W. A. van der Donk, Catalytic promiscuity in the biosynthesis of cyclic peptide secondary metabolites in planktonic marine cyanobacteria, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 10430 CrossRef CAS PubMed.
- L. An, D. P. Cogan, C. D. Navo, G. Jiménez-Osés, S. K. Nair and W. C. van der Donk, Substrate-assisted enzymatic formation of lysinoalanine in duramycin, Nat. Chem. Biol., 2018, 14, 928 CrossRef CAS PubMed.
- (a) P. D. Cotter, P. M. O'Connor, L. A. Draper, E. M. Lawton, L. H. Deegan, C. Hill and R. P. Ross, Posttranslational conversion of L-serines to D-alanines is vital for optimal production and activity of the lantibiotic lacticin 3147, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 18584 CrossRef CAS PubMed; (b) C. T. Lohans, J. L. Li and J. C. Vederas, Structure and biosynthesis of carnolysin, a homologue of enterococcal cytolysin with D-amino acids, J. Am. Chem. Soc., 2014, 136, 13150 CrossRef CAS PubMed; (c) C. T. Lohans and J. C. Vederas, Structural Characterization of Thioether-Bridged Bacteriocins, J. Antibiot., 2013, 67, 23 CrossRef PubMed; (d) L. Huo and W. A. van der Donk, Discovery and characterization of bicereucin, an unusual D-amino acid-containing mixed two-component lantibiotic, J. Am. Chem. Soc., 2016, 138, 5254 CrossRef CAS PubMed.
- F. Hemmerling and F. Hahn, Biosynthesis of oxygen and nitrogen-containing heterocycles in polyketides, Beilstein J. Org. Chem., 2016, 12, 1512 CrossRef CAS PubMed.
- Heterocycles in Natural Product Synthesis, ed. K. C. Majumdar and S. K. Chattopadhyay, Wiley-VCH, Weinheim, 2011, ISBN: 978-3-527-32706-5 Search PubMed.
- J. Deeley, A. Bertram and G. Pattenden, Novel polyoxazole-based cyclopeptides from Streptomyces sp. Total synthesis of the cyclopeptide ym-216391 and synthetic studies towards telomestatin, Org. Biomol. Chem., 2008, 6, 1994 RSC.
- B. McKeever and G. Pattenden, Total synthesis of the prenylated cyclopeptide trunkamide A, a cytotoxic metabolite from Lissoclinum sp., Tetrahedron Lett., 2001, 42, 2573 CrossRef CAS.
- (a) N. Mahanta, D. M. Szantai-Kis, E. J. Petersson and D. A. Mitchell, Biosynthesis and chemical applications of thioamides, ACS Chem. Biol., 2019, 14, 142 CrossRef CAS PubMed; (b) For articles on their importance for peptide structure, see: R. W. Newberry, B. VanVeller and R. T. Raines, Thioamides in the collagen triple helix, Chem. Commun., 2015, 51, 9624 RSC.
- I. Romero-Estudillo, V. R. Batchu and A. Boto, One-Pot conversion of amino acids into 2,5-disubstituted oxazoles: no metals needed, Adv. Synth. Catal., 2014, 356, 3742 CrossRef CAS.
- J. Petersen, K. E. Christensen, M. T. Nielsen, K. T. Mortensen, V. V. Komnatnyy, P. K. Nielsen and K. Qvortrup, Oxidative modification of tryptophan-containing peptides, ACS Comb. Sci., 2018, 20, 344 CrossRef CAS PubMed.
- (a) S. Barik, The uniqueness of tryptophan in biology: properties, metabolism, interactions and localization in proteins, Int. J. Mol. Sci., 2020, 21, 8776 CrossRef CAS PubMed; (b) L. Palego, L. Betti, A. Rossi and G. Giannaccini, Tryptophan biochemistry: structural, nutricional, metabolic and medical aspects in humans, J. Amino Acids, 2016, 8952520 Search PubMed.
- (a) D. Skropeta, K. A. Jolliffe and P. Turner, Pseudoprolines as removable turn inducers: tools for the cyclization of small peptides, J. Org. Chem., 2004, 69, 8804 CrossRef CAS PubMed; (b) T. Wöhr, F. Wahl, A. Nefzi, B. Rohwedder, T. Sato, X. Sun and M. Mutter, Pseudo-prolines as a solubilizing, structure-disrupting protection technique in peptide synthesis, J. Am. Chem. Soc., 1996, 118, 9218 CrossRef; (c) D. A. Senko, N. D. Timofeev, I. E. Kasheverov and I. A. Ivanov, Scope and limitations of pseudoprolines as individual amino acids, Amino Acids, 2021, 53, 665 CrossRef CAS PubMed.
- (a) L. Song, G. M. Ojeda-Carralero, D. Parmar, D. A. González-Martínez, L. Van Meervelt, J. van der Eycken, J. Goeman, D. G. Rivera and E. van der Eycken, Chemoselective peptide backbone diversification and bioorthogonal ligation by ruthenium-catalyzed C-H activation/annulation, Adv. Synth. Catal., 2021, 363, 3297 CrossRef CAS; (b) For a review: M. Oliva, G. A. Coppola, E. V. Van der Eycken and U. K. Sharma, Photochemical and electrochemical strategies towards benzylic C-H functionalization: A recent update, Adv. Synth. Catal., 2021, 363, 1810 CrossRef CAS.
- A. I. Fernández-Llamazares, J. Spengler and F. Albericio, Backbone N-modified peptides: how to meet the challenge of secondary amine acylation, Biopolymers, 2015, 104, 435 CrossRef PubMed.
- L. Aurelio and A. B. Hughes, Synthesis of N-alkyl amino acids in proteins, in Organic Chemistry: Origins and synthesis of amino acids, ed. A. B. Hughes, Wiley-VCH, Weinheim, 2010, Vol. 1 Search PubMed.
- (a) H. Rodríguez, M. Suárez and F. Albericio, A Convenient microwave-enhanced solid-phase synthesis of short chain N-methyl-rich peptides, J. Pept. Sci., 2010, 16, 136 CrossRef PubMed; (b) J. Wang, M. Kwiatkowski, M. Y. Pavlov, M. Ehrenberg and A. C. Forster, Peptide formation by N-methyl amino acids in translation is hastened by higher pH and tRNAPRO, ACS Chem. Biol., 2014, 9, 1303 CrossRef CAS PubMed.
- (a) C. Labrière, N. Kondori, J. S. Caous, M. Boomgaren, K. Sandholm, K. N. Ekdahl, J. H. Hansen and J. Svenson, Development and evaluation of cationic amphiphilic antimicrobial 2,5-diketopiperazines, J. Pept. Sci., 2018, e3090 CrossRef PubMed; (b) J. S. M. Svendsen, T. M. Grant, D. Rennison, M. A. Brimble and J. Svenson, Very short and stable lactoferricin-derived antimicrobial peptides: design principles and potential uses, Acc. Chem. Res., 2019, 52, 749 CrossRef CAS PubMed; (c) M. Tullberg, M. Grøtli and K. Luthman, Synthesis of functionalized, unsymmetrical 1,3,4,6-tetrasubstituted 2,5-diketopiperazines, J. Org. Chem., 2007, 72, 195 CrossRef CAS PubMed.
- Y.-C. Huang, C.-J. Guan, X.-L. Tan, C.-C. Chen, Q.-X. Guo and Y.-M. Li, Accelerated Fmoc solid-phase synthesis of peptides with aggregation-disrupting backbones, Org. Biomol. Chem., 2015, 13, 1500 RSC.
- (a) F. Weygand, W. Steglich, J. Bjarnason, R. Akhtar and N. M. Khan, Leicht abspaltbare schutzgruppen für säureamidfunktionen 1. Mitteilung, Tetrahedron Lett., 1966, 7, 3483 CrossRef; (b) M. Narita, T. Fukunaga, A. Wakabayashi, K. Ishikawa and H. Nakano, Syntheses and properties of tertiary peptide bond-containing polypeptides, Int. J. Pept. Protein Res., 1984, 23, 306 CrossRef CAS.
- (a) T. Wöhr and M. Mutter, Pseudo-prolines in peptide synthesis: direct insertion of serine and threonine derived oxazolidines in dipeptides, Tetrahedron Lett., 1995, 36, 3847 CrossRef; (b) J. Offer, T. Johnson and M. Quibell, Application of reversible amide-bond protection to suppress peptide segment epimerization, Tetrahedron Lett., 1997, 38, 9047 CrossRef CAS; (c) A.-B. M. Abdel-Aal, G. Papageorgiou, M. Quibell and J. Offer, Automated synthesis of backbone protected peptides, Chem. Commun., 2014, 50, 8316 RSC; (d) L. P. Miranda, W. D. F. Meutermans, M. L. Smythe and P. F. Alewood, An activated O-N acyl transfer auxiliary: efficient amide-backbone substitution of hindered “difficult” peptides, J. Org. Chem., 2000, 65, 5460 CrossRef CAS PubMed; (e) T. Johnson, M. Quibell, D. Owen and R. C. Sheppard, A reversible protecting group for the amide bond in peptides. Use in the synthesis of “difficult sequences”, J. Chem. Soc., Chem. Commun., 1993, 369 RSC.
- E. C. B. Johnson, J. D. Lanning and S. C. Meredith, Peptide backbone modification in the bend region of amyloid-β inhibits fibrillogenesis but not oligomer formation, J. Pept. Sci., 2016, 22, 368 CrossRef CAS PubMed.
- (a) A.-B. M. Abdel-Aal, G. Papageorgiou, R. Raz, M. Quibell, F. Burlina and J. Offer, A backbone amide protecting group for overcoming difficult sequences and suppressing aspartimide formation, J. Pept. Sci., 2016, 22, 360 CrossRef CAS PubMed; (b) S. Yamagami, Y. Okada, Y. Kitano and K. Chiba, Peptide Head-to-Tail cyclization: a “molecular claw” approach, Eur. J. Org. Chem., 2021, 3133 CrossRef CAS.
- C. Zuo, S. Tang, Y.-Y. Si, Z. A. Wang, C.-L. Tian and J.-S. Zheng, Efficient synthesis of longer Aβ Peptides via removable backbone modification, Org. Biomol. Chem., 2016, 14, 5012 RSC.
- S. Laps, G. Satish and A. Brik, Harnessing the power of transition metals in solid-phase peptide synthesis and key steps in the (semi)synthesis of proteins, Chem. Soc. Rev., 2021, 50, 2367 RSC.
- (a) M. Jbara, S. Laps, S. K. Maity and A. Brik, Palladium-assisted cleavage of peptides and proteins containing a backbone with thiazolidine linkage, Chem. – Eur. J., 2016, 22, 14851 CrossRef PubMed; (b) G. Mann, G. Satish, R. Meledin, G. B. Vamisetti and A. Brik, Palladium-mediated cleavage of proteins with thiazolidine-modified backbone in live cells, Angew. Chem., 2019, 58, 13540 CrossRef CAS PubMed.
- M. Jbara, E. Eid and A. Brik, Gold(I)-mediated decaging or cleavage of propargylated peptide bond in aqueous conditions for protein synthesis and manipulation, J. Am. Chem. Soc., 2020, 142, 8203 CrossRef CAS PubMed.
- (a) D. M. T. Chan, K. L. Monaco, R. P. Wang and M. P. Winters, New N- and O-Arylation with phenylboronic acids and cupric acetate, Tetrahedron Lett., 1998, 39, 2933 CrossRef CAS; (b) J. X. Qiao and P. Y. S. Lam, Copper-promoted carbon-heteroatom bond cross-coupling with boronic acids and derivatives, Synthesis, 2011, 829 CrossRef CAS; (c) D. M. T. Chan and P. Y. S. Lam, Boronic acids: preparation and application in organic synthesis and medicine, Wiley-VCH, 2006, p. 205 Search PubMed; (d) P. Y. S. Lam, Chan-Lam coupling reaction: copper-promoted C-element bond oxidative coupling reaction with boronic acids, in Synthetic Methods in Drug Discovery, 2016, vol. 1, p. 242 Search PubMed.
- A. E. Mangubat-Medina, S. C. Martin, K. Hanaya and Z. T. Ball, A vinylogous photocleavage strategy allows direct photocaging of backbone amide structure, J. Am. Chem. Soc., 2018, 140, 8401 CrossRef CAS PubMed.
- K. P. Neupane, A. R. Aldous and J. A. Kritzer, Metal-binding and redox properties of substituted linear and cyclic ATCUN motifs, J. Inorg. Biochem., 2014, 139, 65 CrossRef CAS PubMed.
- K. P. Neupane, A. R. Aldous and J. A. Kritzer, Macrocyclization of the ATCUN motif controls metal binding and catalysis, Inorg. Chem., 2013, 52, 2729 CrossRef CAS PubMed.
- T. Abura, S. Ogo, Y. Watanabe and S. Fukuzumi, Isolation and crystal structure of a water-soluble iridium hydride: a robust and highly active catalyst for acid-catalyzed transfer hydrogenations of carbonyl compounds in acidic media, J. Am. Chem. Soc., 2003, 125, 4149 CrossRef CAS PubMed.
- A. E. Mangubat-Medina, H. O. Trial, R. D. Vargas, M. T. Setegne, T. Bader, M. D. Distefano and Z. T. Ball, Red-shifted backbone N-H photocaging agents, Org. Biomol. Chem., 2020, 18, 5110 RSC.
- N. M. Milović and N. M. Kostić, Interplay of terminal amino group and coordinating side chains in directing regioselective cleavage of natural peptides and proteins with palladium(II) complexes, Inorg. Chem., 2002, 41, 7053 CrossRef PubMed.
- (a) D. Uraguchi, Y. Asai and T. Ooi, Site-directed asymmetric quaternization of a peptide backbone at a C-terminal azlactone, Angew. Chem., Int. Ed., 2009, 48, 733 CrossRef CAS PubMed; (b) See also: D. Uraguchi, Y. Ueki, A. Sugiyama and T. Ooi, Highly stereoselective Michael addition of azlactones to electron-deficient triple bonds under P-spiro chiral iminophosphorane catalysis: importance of protonation pathway, Chem. Sci., 2013, 4, 1308 RSC.
- X. Wang and J. A. Porco Jr., Modification of C-terminal peptides to form peptide enamides: synthesis of chondriamides A and C, J. Org. Chem., 2001, 66, 8215 CrossRef CAS PubMed.
- (a) S. H. Choi, I. A. Guzei, L. C. Spencer and S. H. Gellman, Crystallographic characterization of helical secondary structures in 2:1 and 1:2 α/β-peptides, J. Am. Chem. Soc., 2009, 131, 2917 CrossRef CAS PubMed; (b) S. H. Choi, I. A. Guzei and S. H. Gellman, Crystallographic characterization of the α/β-peptide 14/15-helix, J. Am. Chem. Soc., 2007, 129, 13780 CrossRef CAS PubMed; (c) J. L. Price, W. S. Horne and S. H. Gellman, Discrete heterogeneous quaternary structure formed by α/β-peptide foldamers and α-peptides, J. Am. Chem. Soc., 2007, 129, 6376 CrossRef CAS PubMed; (d) W. S. Horne, J. L. Price, J. L. Keck and S. H. Gellman, helix bundle quaternary structure from α/β-peptide foldamers, J. Am. Chem. Soc., 2007, 129, 4178 CrossRef CAS PubMed.
- M. I. Aguilar, A. W. Purcell, R. Devi, R. Lew, J. Rossjohn, A. I. Smith and P. Perlmutter, β-Amino acid-containing hybrid peptides-new opportunities in peptidomimetics, Org. Biomol. Chem., 2007, 5, 2884 RSC.
- C. Saavedra, R. Hernández, A. Boto and E. Álvarez, Catalytic, One-pot synthesis of β-amino acids from α-amino acids. Preparation of α,β-peptide derivatives, J. Org. Chem., 2009, 74, 4655 CrossRef CAS PubMed.
- C. J. Saavedra, A. Boto, R. Hernández, J. I. Miranda and J. M. Aizpurua, Conformation and chiral effects in α,β,α-tripeptides, J. Org. Chem., 2012, 77, 5907 CrossRef CAS PubMed.
- S. Bloom, C. Liu, D. K. Kölmel, J. X. Qiao, Y. Zhang, M. A. Poss, W. R. Ewing and D. W. C. MacMillan, Decarboxylative alkylation for site-selective bioconjugation of native proteins via oxidation potentials, Nat. Chem., 2018, 10, 205 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Visible light photoredox catalysis with transition metal complexes: applications in organic synthesis, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed.
- Y. Yu, L.-K. Zhang, A. V. Buevich, G. Li, H. Tang, P. Vachal, S. L. Colletti and Z.-C. Shi, Chemoselective peptide modification via photocatalytic tryptophan β-position conjugation, J. Am. Chem. Soc., 2018, 140, 6797 CrossRef CAS PubMed.
- G. Marino, U. Eckhard and C. M. Overall, Protein termini and their modifications revealed by positional proteomics, ACS Chem. Biol., 2015, 10, 1754 CrossRef CAS PubMed.
- (a) M. Garreau, F. Le Vaillant and J. Waser, C-Terminal bioconjugation of peptides through photoredox catalyzed decarboxylative alkynylation, Angew. Chem., 2019, 58, 8182 CrossRef CAS PubMed; (b) E. Le Du, M. Garreau and J. Waser, Small peptide diversification through photoredox-catalyzed oxidative C-terminal modification, Chem. Sci., 2021, 12, 2467 RSC.
- D. C. Marcote, R. Street-Jeakings, E. Dauncey, J. J. Douglas, A. Ruffoni and D. Leonori, Photoinduced Decarboxylative Azidation of Cyclic Amino Acids, Org. Biomol. Chem., 2019, 17, 1839 RSC.
- V. Haridas, S. Sadanandan and N. U. Dheepthi, Sortase-based bio-organic strategies for macromolecular synthesis, ChemBioChem, 2014, 15, 1857 CrossRef CAS PubMed.
- S. Lin and C. He, A Mini-review on the enzyme-mediated manipulation of proteins/peptides, Chin. Chem. Lett., 2018, 29, 1017 CrossRef CAS.
- (a) M. W. Popp, J. M. Antos and H. L. Ploegh, Site-specific protein labelling via sortase-mediated transpeptidation, Curr. Protoc. Protein Sci., 2009, 56, 15.3.1 Search PubMed; (b) X. Dai, A. Böker and U. Glebe, Broadening the scope of sortagging, RSC Adv., 2019, 9, 4700 RSC.
- (a) X. Hemu, X. Zhang, X. Bi, C.-F. Liu and J. P. Tam, Butelase-1 mediated ligation of peptides and proteins, Methods Mol. Biol., 2019, 2012, 83 CrossRef CAS PubMed; (b) G. K. T. Nguyen, S. Wang, Y. Qiu, X. Hemu, Y. Lian and J. P. Tam, Butelase 1 is an Asx-specific ligase enabling peptide macrocyclization and synthesis, Nat. Chem. Biol., 2014, 10, 732 CrossRef CAS PubMed; (c) G. K. T. Nguyen, A. Kam, S. Loo, A. E. Jansson, L. X. Pan and J. P. Tam, Butelase 1: a versatile ligase for peptide and protein macrocyclization, J. Am. Chem. Soc., 2015, 137, 15398 CrossRef CAS PubMed.
- G. K. T. Nguyen, Y. Cao, W. Wang, C. F. Liu and J. P. Tam, Site-specific N-terminal labelling of peptides and proteins using butelase 1 and thiodepsipeptide, Angew. Chem., Int. Ed., 2015, 54, 15694 CrossRef CAS PubMed.
- U. Tomita, S. Yamaguchi, Y. Maeda, K. Chujo, K. Minamihata and T. Nagamune, Protein cell-surface display through in situ enzymatic modification of proteins with a poly(ethylene glycol)-lypid, Biotechnol. Bioeng., 2013, 110, 2785 CrossRef CAS PubMed.
- H. Shi, Q. Shi, J. T. Oswald, Y. Gao, L. Li and Y. Li, Site-specific PEGylation of human growth hormone by mutated sortase A, Chem. Res. Chin. Univ., 2018, 34, 428 CrossRef CAS.
- T. J. Harmand, D. Bousbaine, A. Chan, X. Zhang, D. R. Liu, J. P. Tam and H. L. Ploegh, One-pot dual labeling of IgG1 and preparation of C-to-C fusion proteins through a combination of sortase A and butelase 1, Bioconjugate Chem., 2018, 29, 3245 CrossRef CAS PubMed.
- (a) N. Asiimwe, M. F. Al Mazid, D. P. Murale, Y. K. Kim and J.-S. Lee, Recent advances in protein modifications techniques for the targeting N-terminal cysteine, Pept. Sci., 2021, e24235 Search PubMed; (b) G. Hermanson, Bioconjugate Techniques Ed. 2nd, Academic Press, San Diego, CA, 2008, pp. 1–1323 Search PubMed.
- (a) Bioconjugation: protein coupling techniques for the biomedical sciences, ed. M. Aslam and A. Dent, Macmillan Reference Ltd, London, 1998, pp. 1–634 Search PubMed; (b) See also: K. Maruoka, E. Tamaya and T. Ooi, Stereoselective terminal functionalization of small peptides for catalytic asymmetric synthesis of unnatural peptides, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5824 CrossRef CAS PubMed.
- D. Chen, M. M. Disotuar, X. Xiong, Y. Wang and D. H-C. Chou, Selective N-terminal functionalization of native peptides and proteins, Chem. Sci., 2017, 8, 2717 RSC.
- H. Wu, G. Mousseau, S. Mediouni, S. T. Valente and T. Kodadek, Cell-permeable peptides containing cycloalanine residues, Angew. Chem., Int. Ed., 2016, 55, 12637 CrossRef CAS PubMed.
- J. M. McFarland and M. B. Francis, Reductive alkylation of proteins using iridium catalyzed transfer hydrogenation, J. Am. Chem. Soc., 2005, 127, 13490 CrossRef CAS PubMed.
- J. I. MacDonald, H. K. Munch, T. Moore and M. B. Francis, One-step site-specific modification of native protein with 2-pyridinecarboxayaldehydes, Nat. Chem. Biol., 2015, 11, 326 CrossRef CAS PubMed.
- G. J. L. Bernardes, M. Steiner, I. Hartmann, D. Neri and G. Casi, Site-specific chemical modification of antibody fragments using traceless cleavable linkers, Nat. Protoc., 2013, 8, 2079 CrossRef CAS PubMed.
- A. C. Obermeyer, J. B. Jarman, C. Netirojjanakul, K. El Muslemany and M. B. Francis, Mild bioconjugation through the oxidative coupling of ortho-aminophenols and anilines with ferricyanide, Angew. Chem., Int. Ed., 2014, 53, 1057 CrossRef CAS PubMed.
- Y. E. Sim, O. Nwajiobi, S. Mahesh, R. D. Cohen, M. Y. Reibarkh and M. Raj, Secondary amine selective Petasis (SASP) bioconjugation, Chem. Sci., 2020, 11, 53 RSC.
- L. Purushottam, S. R. Adusumalli, U. Singh, V. B. Unnikrishnan, D. G. Rawale, M. Gujrati, R. K. Mishra and V. Rai, Single-site glycine-specific labelling of proteins, Nat. Commun., 2019, 10, 2539 CrossRef PubMed.
- E. Reid, Relative importance of free α- and ε-amino groups for the biological activity of the growth hormone, Nature, 1951, 1, 955 CrossRef PubMed.
- (a) X. Chen, K. Muthoosamy, A. Pfisterer, B. Neumann and T. Weil, Site-selective lysine modification of native proteins and peptides via kinetically controlled labelling, Bioconjugate Chem., 2012, 23, 500 CrossRef CAS PubMed; (b) R. Singudas, S. R. Adusumalli, P. N. Joshi and V. Rai, A phthalimidation protocol that follows protein defined parameters, Chem. Commun., 2015, 51, 473 RSC; (c) M. Raj, H. Wu, S. L. Blosser, M. A. Vittoria and P. S. Arora, Aldehyde capture ligation for synthesis of native peptide bonds, J. Am. Chem. Soc., 2015, 137, 6932 CrossRef CAS PubMed.
- J. Xiao, A. Burn and T. J. Tolbert, Increasing solubility of proteins and peptides by site-specific modification with betaine, Bioconjugate Chem., 2008, 19, 1113 CrossRef CAS PubMed.
- (a) D. E. Metzler and E. E. Snell, Some transamination reactions involving vitamin B61, J. Am. Chem. Soc., 1952, 74, 979 CrossRef CAS; (b) C. Cennamo, B. Carafoli and E. P. Bonetti, Non-enzymatic transamination between peptides and pyridoxal. Isolation of the 2,4-dinitrophenylhyrazones of some ketopeptides, J. Am. Chem. Soc., 1956, 78, 3523 CrossRef CAS.
- (a) H. Dixon, Transamination of peptides, Biochem. J., 1964, 92, 661 CrossRef CAS PubMed; (b) H. Dixon and V. Moret, Removal of the N-terminal residue of a protein after transamination, Biochem. J., 1965, 94, 463 CrossRef CAS PubMed.
- (a) L. S. Witus and M. Francis, Site-specific protein bioconjugation via a pyridoxal 5′-phosphate-mediated N-terminal transamination reaction, Curr. Protoc. Chem. Biol., 2010, 2, 125 CrossRef PubMed; (b) J. M. Gilmore, R. A. Scheck, A. P. Esser-Kahn, N. S. Joshi and M. B. Francis, N-Terminal protein modification through a biomimetic transamination reaction, Angew. Chem., Int. Ed., 2006, 45, 5307 CrossRef CAS PubMed.
- M. Zhang, X. Zhang, J. Li, Q. Guo and Q. Xiao, A new pyridoxal derivative for transamination of N-Terminus of Proteins, Chin. J. Chem., 2011, 29, 1715 CrossRef CAS.
- A. P. Crochet, M. M. Kabir, M. B. Francis and C. D. Paavola, Site-selective dual modification of periplasmic binding proteins for sensing appliations, Biosens. Bioelectron., 2010, 26, 55 CrossRef CAS PubMed.
- K. S. Palla, L. S. Witus, K. J. Mackenzie, C. Netirojjanakul and M. B. Francis, Optimization and expansion of a site-selective N-methylpyridinium-4-carboxaldehyde-mediated transamination for bacterially expressed proteins, J. Am. Chem. Soc., 2015, 137, 1123 CrossRef CAS PubMed.
- (a) R. Fields and H. B. Dixon, A Spectrophotometric method for the microdetermination of periodate, Biochem. J., 1968, 106, 883 CrossRef PubMed; (b) K. F. Geoghegan and J. G. Stroh, Site-directed conjugation of nonpeptide groups to peptides and proteins via periodate oxidation of a 2-amino alcohol. Application to modification at N-terminal serine, Bioconjugate Chem., 1992, 3, 138 CrossRef CAS PubMed.
- O. El Mahdi and O. Melnyk, α-Oxo aldehyde or glyoxylyl group chemistry in peptide bioconjugation, Bioconjugate Chem., 2013, 24, 735 CrossRef CAS PubMed.
- S. Schoffelen, M. B. Van Eldijk, B. Rooijakkers, R. Raijmakers, A. J. R. Heck and J. C. M. Van Hest, Metal-free and pH-controlled introduction of azides in proteins, Chem. Sci., 2011, 2, 701 RSC.
- E. Hernando, J. Villalva, A. M. Martínez, I. Alonso, N. Rodríguez, R. Gómez Arrayás and J. C. Carretero, Palladium-catalyzed carbonylative cyclization of amines via γ-C(sp3)-H activation: late-stage diversification of amino acids and peptides, ACS Catal., 2016, 6, 6868 CrossRef CAS.
- B.-B. Zhan, M.-X. Jiang and B.-F. Shi, Late-stage functionalization of peptides via a palladium-catalyzed C(sp3)-H activation strategy, Chem. Commun., 2020, 56, 13950 RSC.
- A. M. Weeks and J. A. Wells, Subtiligase-catalyzed peptide ligation, Chem. Rev., 2020, 120, 3127 CrossRef CAS PubMed.
- (a) B. Wang, T. Dai, W. Sun, Y. Wei, J. Ren, L. Zhang, M. Zhang and F. Zhou, Protein N-Myristoylation: functions and mechanisms in control of innate immunity, Cell. Mol. Immunol., 2021, 18, 878 CrossRef CAS PubMed; (b) D. I. Udenwobele, R.-C. Su, S. V. Good, T. B. Ball, S. V. Shrivastav and A. Shrivastav, Myristoylation: an important protein modification in the immune response, Front. Immunol., 2017, 8, 751 CrossRef PubMed.
- K. M. Luginbuhl, D. Mozhdehi, M. Dzuricky, P. Yousefpour, F. C. Huang, N. R. Mayne, K. L. Buehne and A. Chilkoti, Recombinant synthesis of hybrid lipid-peptide polymer fusions that self-assemble and encapsulate hydrophobic drugs, Angew. Chem., Int. Ed., 2017, 56, 13979 CrossRef CAS PubMed.
- D. Mozhdehi, K. M. Luginbuhl, J. R. Simon, M. Dzuricky, R. Berger, H. S. Varol, F. C. Huang, K. L. Buehne, N. R. Mayne, I. Weitzhandler, M. Bonn, S. H. Parekh and A. Chilkoti, Genetically encoded lipid-polypeptide hybrid biomaterials that exhibit temperature-triggered hierarchical self-assembly, Nat. Chem., 2018, 10, 496 CrossRef CAS PubMed.
- I. T. Song, M. Lee, H. Lee, J. Han, J.-H. Jang, M. S. Lee, G. Y. Koh and H. Lee, PEGylation and HAylation via catechol: α-amine-specific reaction at N-terminus of peptides and proteins, Acta Biomater., 2016, 43, 50 CrossRef CAS PubMed.
- S. Vutti, S. Schoffelen, J. Bolinsson, N. Buch-Manson, N. Bovet, J. Nygard, K. L. Martínez and M. Meldal, Click chemistry mediated functionalization of vertical nanowires for biological applications, Chem. – Eur. J., 2016, 22, 496 CrossRef CAS PubMed.
- (a) The following 3D protein structures are public domain: Rnase by Vossman (CC BY-SA 2.5), https://commons.wikimedia.org/w/index.php?curid=683307 and; (b) interleukin1 by Boghog: https://commons.wikimedia.org/w/index.php?curid=21247671.
| This journal is © the Partner Organisations 2021 |