
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The role of biocatalysis in the asymmetric synthesis of alkaloids – an update
Emmanuel Cigan†
 ,
Bettina Eggbauer†
,
Joerg H. Schrittwieser
* and
Wolfgang Kroutil
,
Bettina Eggbauer†
,
Joerg H. Schrittwieser
* and
Wolfgang Kroutil
Institute of Chemistry, University of Graz, NAWI Graz, BioTechMed Graz, BioHealth, Heinrichstrasse 28/II, 8010, Graz, Austria. E-mail: joerg.schrittwieser@uni-graz.at
First published on 20th August 2021
Abstract
Alkaloids are a group of natural products with interesting pharmacological properties and a long history of medicinal application. Their complex molecular structures have fascinated chemists for decades, and their total synthesis still poses a considerable challenge. In a previous review, we have illustrated how biocatalysis can make valuable contributions to the asymmetric synthesis of alkaloids. The chemo-enzymatic strategies discussed therein have been further explored and improved in recent years, and advances in amine biocatalysis have vastly expanded the opportunities for incorporating enzymes into synthetic routes towards these important natural products. The present review summarises modern developments in chemo-enzymatic alkaloid synthesis since 2013, in which the biocatalytic transformations continue to take an increasingly ‘central’ role.
 Emmanuel Cigan | Emmanuel Cigan studied chemistry at the University of Graz and completed his MSc in 2018. For his master's thesis he worked in Prof. Wolfgang Kroutil's group on the evaluation of the substrate scope of the enzyme strictosidine synthase. Staying in the same group, he is currently working on his PhD dealing with the synthesis of morphinan alkaloids in a chemo-enzymatic fashion. His main research interests are alkaloid pathway enzymes for the use in organic synthesis of natural products, chemo-enzymatic cascade systems and the heterologous protein expression and characterization of enzymes for the use in biocatalytic applications. |
 Bettina Eggbauer | Bettina Eggbauer obtained her bachelor's degree in chemistry and biochemistry from the University of Graz in 2017. After completing her master's thesis dealing with imine reductases in cyanobacterial biotransformations in 2019 at the Technical University of Graz and an Erasmus internship at the University of Turku focussing on immobilization of cyanobacteria, she joined Prof. Wolfgang Kroutil's group for her PhD thesis. With the main aim of reconstituting the early steps of the ergot alkaloid biosynthesis pathway to establish an in vitro production platform, she focuses on organic synthesis but also biocatalytic approaches towards essential intermediates. |
 Joerg H. Schrittwieser | Joerg H. Schrittwieser, born 1984, studied chemistry at the University of Graz, Austria. In 2007, he joined Prof. Wolfgang Kroutil's research group, where he completed his master's studies in 2008 and his PhD in 2011. After a post-doctoral stay at TU Delft (The Netherlands) with Dr Frank Hollmann, financed through an ‘Erwin Schrödinger’ fellowship from the Austrian Science Fund, he returned to the University of Graz in 2013 as a university assistant and became a permanent staff scientist in 2018. His research interests include biocatalytic asymmetric reduction, multi-enzymatic and chemo-enzymatic cascade reactions, and the chemo-enzymatic synthesis of natural products, in particular alkaloids. |
 Wolfgang Kroutil | Wolfgang Kroutil received his undergraduate training in chemistry at the University of Technology in Graz (Austria) and conducted his doctoral research with Prof. Kurt Faber in Graz at the University of Technology as well as in Exeter (6 months U.K., Prof. S. M. Roberts). Afterwards, he worked in the Biocatalysis Research Group at Novartis CP in Basel/CH and then as a R&D manager at Krems Chemie Chemical Services (Austria). In 2000 he joined the research group of Prof. Faber at the Department of Chemistry at the University of Graz as an assistant professor. After his habilitation he became associate professor in 2004 and full professor in 2013 at the University of Graz. He was also a visiting scientist at the University of California Los Angeles (2015), the University of Stuttgart/Germany (2003), the University of Cambridge (2004), and the University of York (2009). He has published more than 260 papers on biocatalysis and contributed to more than 20 patents concerning biocatalysis. In addition to other awards, he received the Biocat award in 2012 and the Biotrans award in 2015. His research focuses on biocatalytic redox reactions, C–C bond formation, and cascades. |
1 Introduction
The alkaloids are a large and structurally diverse group of nitrogen-containing secondary metabolites that are produced by a variety of organisms and that often possess potent biological activities.1 Although no universally accepted definition of the term ‘alkaloid’ has yet emerged,2 modern conceptions tend to be broad and include not only nitrogen compounds of basic character – the property from which the name was originally derived – but also amides, nitro, and nitroso compounds, while excluding primary metabolites such as amino acids, proteins, and porphyrins.1a,d Between 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and more than 40000 known structures can be classified as alkaloids according to this broad definition, and examples of these natural products are found in almost all kingdoms of life.1a–d The largest number of representatives, including those that are most widely known, originate from higher plants.
000 and more than 40000 known structures can be classified as alkaloids according to this broad definition, and examples of these natural products are found in almost all kingdoms of life.1a–d The largest number of representatives, including those that are most widely known, originate from higher plants.
Humanity's interest in alkaloids is spurred mainly by the strong pharmacological effects that many of these compounds exhibit, and their use as medicinal and recreational drugs predates their identification by several thousand years.1c,f,g Although most modern pharmaceuticals are synthetic, alkaloids continue to play an important role in present-day healthcare, and some have also found application in other fields, for instance, as flavour compounds or as chiral reagents in synthetic chemistry.1a–c,1e–g Consequently, many alkaloids have significant commercial value (for examples, see Table 1) and are produced at volumes of several hundred to several thousand tonnes per year.
| Alkaloid | Applications | Major production methods | Annual production volume |
|---|---|---|---|
| a Part of the World Health Organization's Model List of Essential Medicines (21st edn, 2019).3 | |||
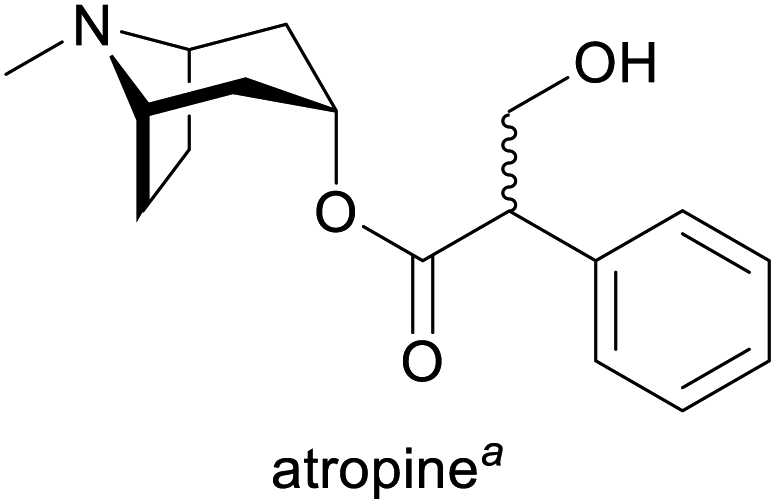 |
Treatment of organophosphate poisoning and bradycardia, as mydriatic and cycloplegic in ophthalmology, as preoperative medication (decrease of saliva production)1g,3 | Isolation from Atropa belladonna, Duboisia and Hyoscyamus species1g | 3.9 tonnes (2017)4 |
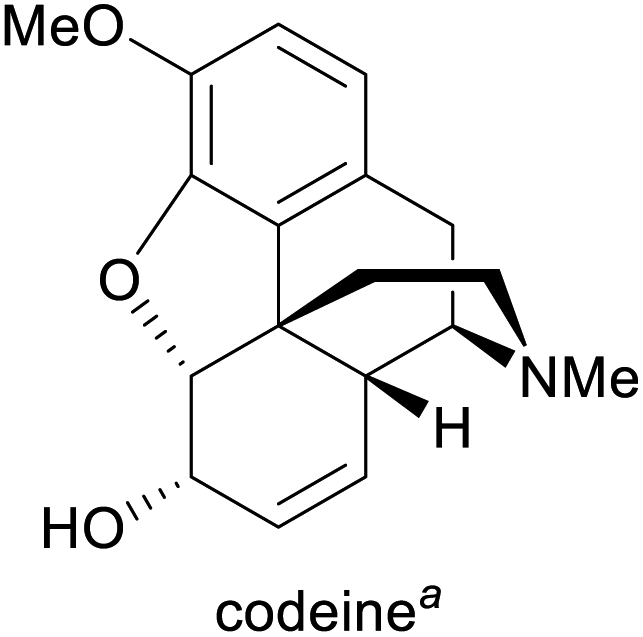 |
Pain treatment, cough suppression1g,3 | Isolation from opium poppy, semisynthesis from morphine1g | 308 tonnes (2018)5 |
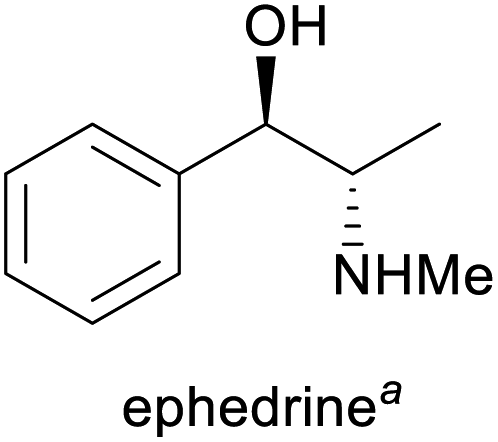 |
Prevention of hypotension during spinal anaesthesia,3 chiral auxiliary in asymmetric synthesis6 | (Chemo-enzymatic) total synthesis, isolation from Ephedra plants7 | 1500–2000 tonnes8 |
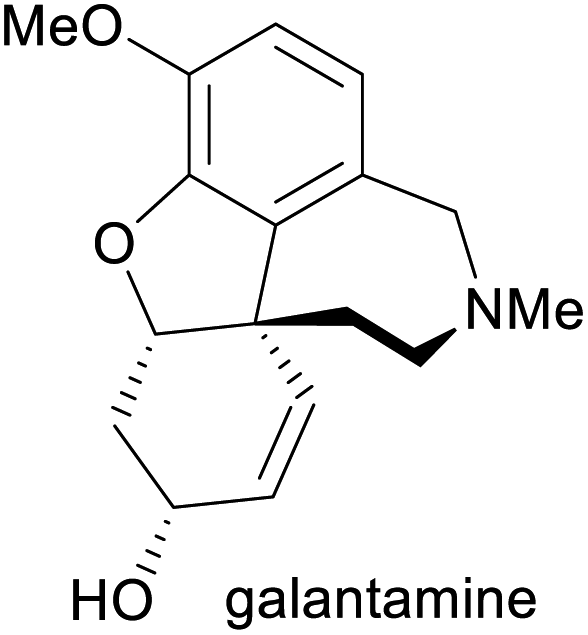 |
Treatment of Alzheimer's disease9 | Total synthesis, isolation from daffodil bulbs9,10 | 3–4 tonnes (2014)11 |
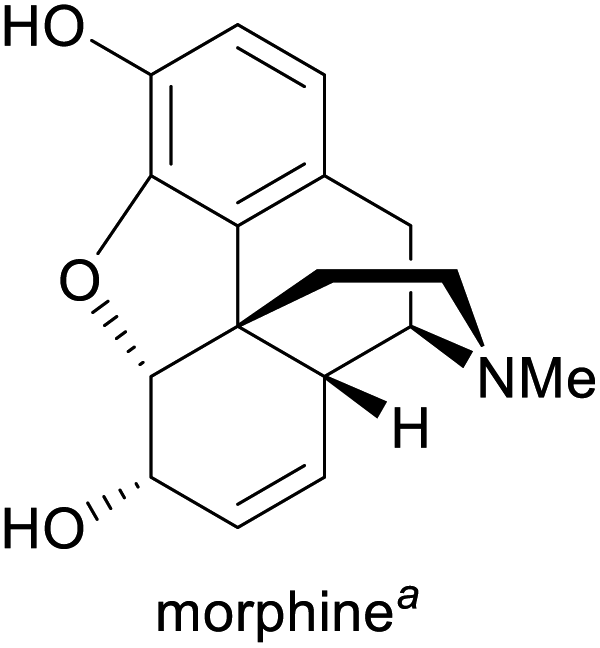 |
Pain treatment1g,3 | Isolation from opium poppy1g | 388 tonnes (2018)5 |
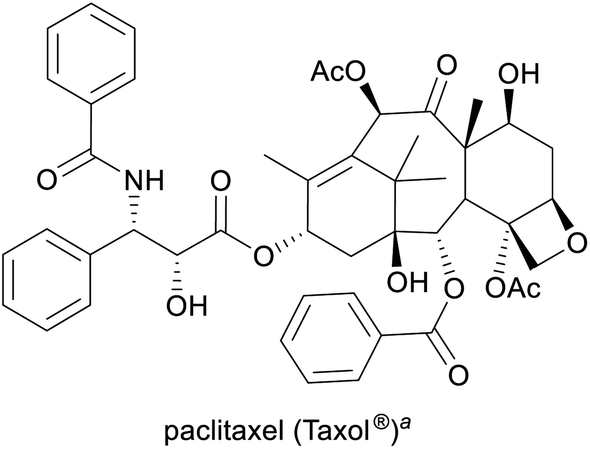 |
Cancer treatment3,12 | Semisynthesis (from 10-deacetylbaccatin-III), plant cell fermentation13 | 2.6 tonnes (2017)14 |
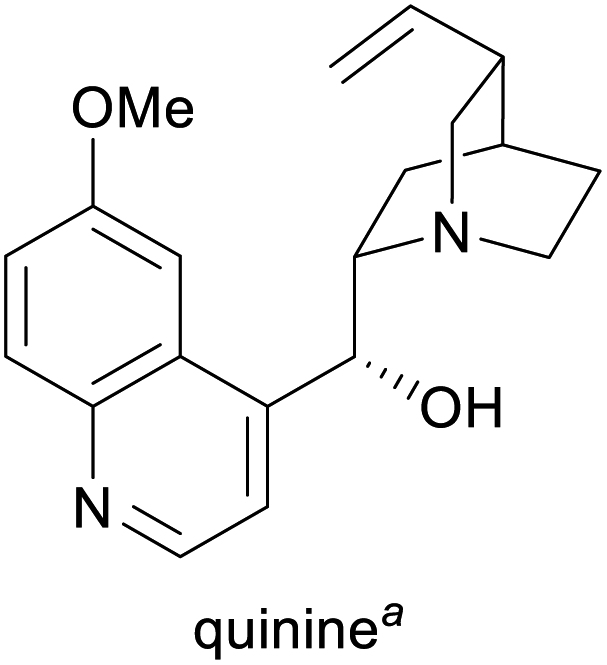 |
Treatment of malaria and babesiosis,3,15 as flavour compound,1g chiral catalyst in asymmetric synthesis16 | Isolation from cinchona bark17 | 300–500 tonnes17 |
 |
Chiral ligand in asymmetric synthesis18 | Isolation from Cytisus scoparius19 | |
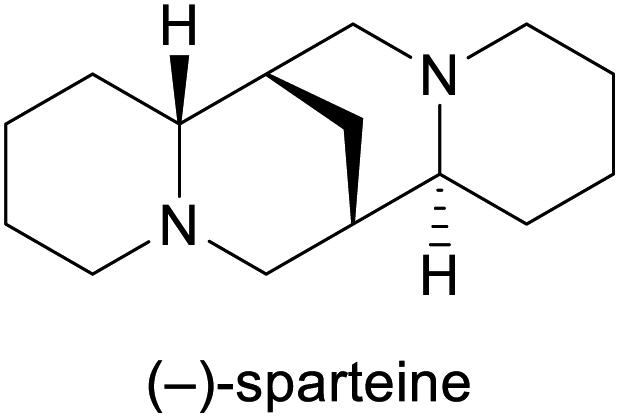
Among the employed production methods, isolation from a natural source is still the most common, but examples of alkaloids exist that are prepared on industrial scale by total synthesis, semi-synthesis or plant cell fermentation. Each of these approaches has its benefits and limitations: Isolation and semi-synthesis, for instance, predominantly rely on plants as a raw material and are hence bound to seasonal availability of crops, climate and weather conditions, and natural variations in alkaloid content. Moreover, isolation yields are often low, and the commercial exploitation of endangered species can severely interfere with conservational efforts, as has been well documented for the cases of galantamine and paclitaxel.10,13 These factors, combined with cultivation of source plants in politically unstable regions of the earth, can lead to unpredictable supply situations. One recent example that has disconcerted the chemistry community is the one of (−)-sparteine, a quinolizidine alkaloid used as chiral ligand, which became entirely unavailable for some time in the early 2010s and remains scarce for reasons that are still unclear.19 Total synthesis, on the other hand, typically starts from petrol-based raw materials, which at present are still abundantly available. However, the intricate molecular architectures of many alkaloids present considerable synthetic challenges and often require lengthy routes to assemble, resulting in low overall yields and significant generation of waste. Besides, modern synthetic methods frequently employ catalysts based on precious metals that are in high demand also by other industries and are subject to substantial price fluctuations.20 Because of these limitations, only few total syntheses of alkaloids have been successfully translated to production scale.
Methods of biotechnology could help to establish novel production routes for alkaloids and thereby alleviate existing supply limitations.21 Indeed, some biotechnological approaches have already had a major impact: Random mutagenesis of opium poppy has been used to create a variant with altered alkaloid profile, which is now cultivated on large scale for the production of thebaine, a precursor to prescription opioids.22 Plant cell fermentation23 has enabled the commercial manufacture of paclitaxel by a process whose environmental advantages over semi-synthetic routes have been recognised with the US Environmental Protection Agency's 2004 Greener Synthetic Pathways Award.13,24 Other biotechnological methods of alkaloid production have not yet been applied on industrial scale, but have delivered promising results in laboratory experiments: Metabolic pathway engineering, for instance, makes use of the biosynthetic machinery that nature has evolved for the construction of secondary metabolites, but transposes it into production hosts that are more easily cultivated and optimised than the natural source organism.25,26 Recently, this approach has been used to create transgenic yeast strains that assemble opioids,27 tetrahydroisoquinolines,28 or tropanes29 from simple sugar and amino acid building blocks, in some cases with impressive product titres [e.g., 4.6 g L−1 for (S)-reticuline].
Biocatalysis, i.e., the use of enzymes or microbial cells in preparative transformations,30 has been established as a valuable synthetic tool in the pharmaceutical industry,31 and its potential for the synthesis of complex natural products, including alkaloids, has also been recognised.32 Biocatalytic reactions typically offer high chemo-, regio-, and stereoselectivity, but also a substrate scope that is usually broad enough to allow not only the preparation of one particular product but a series of structurally related derivatives. Integrating in vitro biotransformations into the total synthesis or semi-synthesis of alkaloids can hence lead to shorter routes and a reduced need for protective groups, while avoiding limitations of productivity and flexibility that are often associated with fermentation approaches.
In a previous review, published in 2013, we have identified three main strategies for the integration of biocatalysis into alkaloid synthesis: (1) the biocatalytic preparation of chiral building blocks that are chemically transformed into the target compounds, (2) the biocatalytic kinetic resolution, desymmetrisation, or deracemisation of alkaloids that have been synthesised by chemical methods, and (3) the construction of alkaloids using biocatalytic C–N and/or C–C bond formation in the asymmetric key step.33 In the roughly eight years that have passed since then, exciting new developments have been made in all three of these areas, and the overarching trend towards a more central role of biocatalysis in chemo-enzymatic alkaloid syntheses, which we have identified in our earlier review, has continued. Moreover, the identification of imine-reducing enzymes with broad substrate scope and high stereoselectivity has enabled biocatalytic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N reduction as a fourth approach to the chemo-enzymatic asymmetric synthesis of alkaloids. In this update to our previous review, we therefore intend to show how the discovery of novel enzymes, the engineering of biocatalysts, but also the creative application of well-known biotransformations have led to many new, elegant routes towards a broad range of alkaloids, and thereby have continued to re-shape the role of biocatalysis in the asymmetric synthesis of these fascinating natural products.
N reduction as a fourth approach to the chemo-enzymatic asymmetric synthesis of alkaloids. In this update to our previous review, we therefore intend to show how the discovery of novel enzymes, the engineering of biocatalysts, but also the creative application of well-known biotransformations have led to many new, elegant routes towards a broad range of alkaloids, and thereby have continued to re-shape the role of biocatalysis in the asymmetric synthesis of these fascinating natural products.
2 Biocatalytic asymmetric synthesis of chiral building blocks
The use of enantiomerically pure building blocks for constructing complex target molecules with substrate-based stereocontrol is a classical approach to asymmetric total synthesis. The traditional source for these building blocks – the ‘chiral pool’ of naturally occurring amino acids, sugars, and terpenes – has recently enjoyed a renewed interest from organic chemists as they strive to incorporate renewable feedstocks into their synthesis routes.34 Alternatively, biocatalytic transformations can be used to prepare a wide range of chiral building blocks, which can often be tailored more closely to the desired target structure than the compounds available in nature.35 This strategy is frequently employed in alkaloid synthesis, and lipases and esterases as well as toluene dioxygenase remain the most important enzymes in this context.2.1 Lipases and esterases
The enantioselective hydrolysis and formation of ester bonds catalysed by lipases and esterases has been an early focus of biocatalysis research and has found broad application in asymmetric synthesis.36 As discussed in our previous review,33 chiral building blocks prepared using lipases and esterases have also been employed frequently in chemo-enzymatic syntheses of alkaloids. While early examples have focused on a few selected classes of building blocks, in particular cyclic alcohols and piperidine derivatives, recent contributions have made use of a much more diverse range of structures. Moreover, besides kinetic resolution and desymmetrisation, dynamic kinetic resolution has recently been explored as an additional option for the lipase-catalysed synthesis of chiral building blocks in alkaloid chemistry.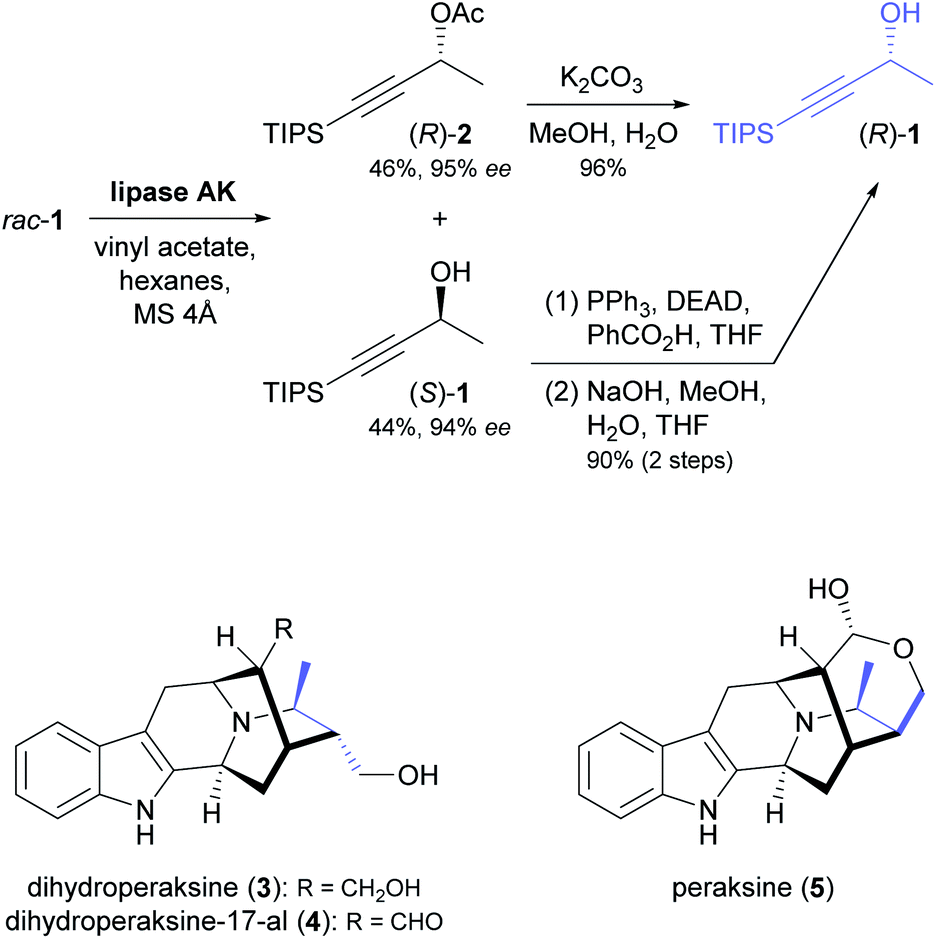 | ||
| Scheme 1 Synthesis of building block (R)-1 by lipase-catalysed kinetic resolution, and structures of indole alkaloids prepared from this building block.39 | ||
The lipase-catalysed kinetic resolution of two small, olefinic sec-alcohols, 6 and 13 (Scheme 2a), was employed in the preparation of polyhydroxylated pyrrolizidines and an indolizidine that are close structural analogues of hyacinthamines and steviamine, respectively.40 Acylation of the alcohols using Chirazyme L-2 (ref. 37) and vinyl acetate in diethyl ether at room temperature proceeded with only moderate enantioselectivity (6: E = 34, 13: E = 5), but the resulting limitations in optical purity of the reaction products could be tolerated, since their subsequent coupling with the enantiomerically pure nitrone 8 produced separable mixtures of diastereomers. Limited enantioselectivity was also observed in the kinetic resolution of alcohol 16 by lipase B from Candida antarctica (Scheme 2b, E = 29), carried out as part of a chemo-enzymatic synthesis of both enantiomers of the quinazoline alkaloid febrifugine (19) and its non-natural derivative halofuginone (20).41 The (R)-enantiomer of 16 was isolated from the lipase biotransformation in 46% yield and 88% ee and used as such in further steps. The corresponding (S)-acetate (17), obtained in 47% yield and 82% ee, was hydrolysed back to the alcohol and its optical purity upgraded to 98% ee by a second round of kinetic resolution. Alkene cross-metathesis, a diastereoselective aza-Michael addition, and α-functionalisation of methyl ketone 18 completed the syntheses of febrifugine and halofuginone.
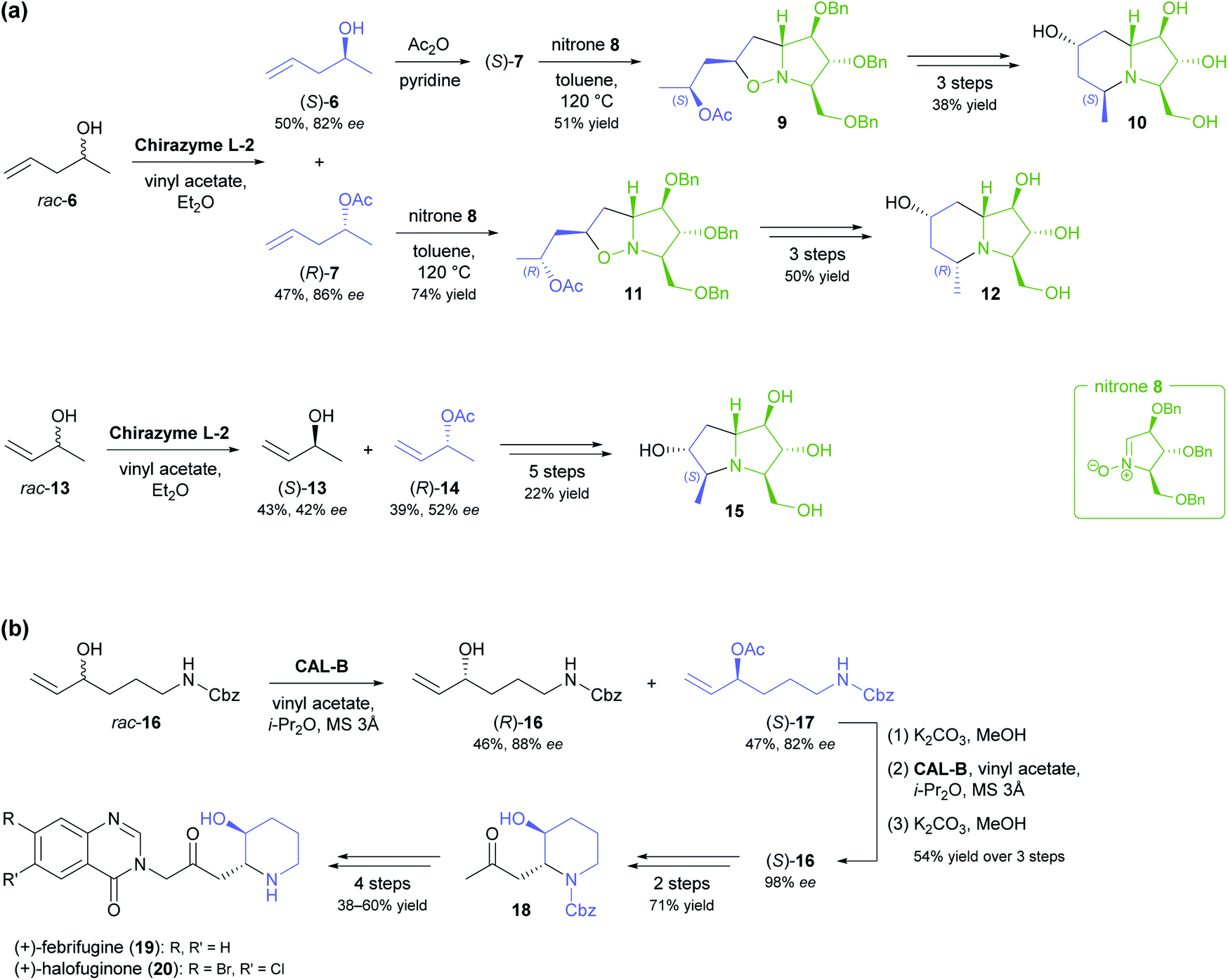 | ||
| Scheme 2 Olefinic secondary alcohols as building blocks in alkaloid synthesis: (a) Kinetic resolution of alcohols 6 and 13 and their conversion into polyhydroxylated pyrrolizidines and an indolizidine. (b) Chemo-enzymatic synthesis of (+)-febrifugine (19) and (+)-halofuginone (20) based on the kinetic resolution of alcohol 16. | ||
A recent synthesis of the hexacyclic Daphniphyllum alkaloid longeracinphyllin A (also known as daphniynnine C;42 23, Scheme 3) uses hydroxyketone (S)-21, prepared in 45% yield and 99% ee by a 74 gram-scale kinetic resolution of the racemate with immobilised CAL-B, as starting point and precursor to the central cyclohexanone ring of the target structure.43 From there, the complex alkaloid scaffold was constructed with remarkable efficiency, arriving at a tricyclic intermediate (22) in only three operations (six steps) that include a Toste-type silyl enol ether cyclisation44 and a Michael addition/aldol condensation cascade. The synthesis was completed in 14 more steps, with the remaining three ring closures being achieved by intramolecular conjugate addition, a (3 + 2) enone–allene cycloaddition, and a Horner–Wadsworth–Emmons reaction.
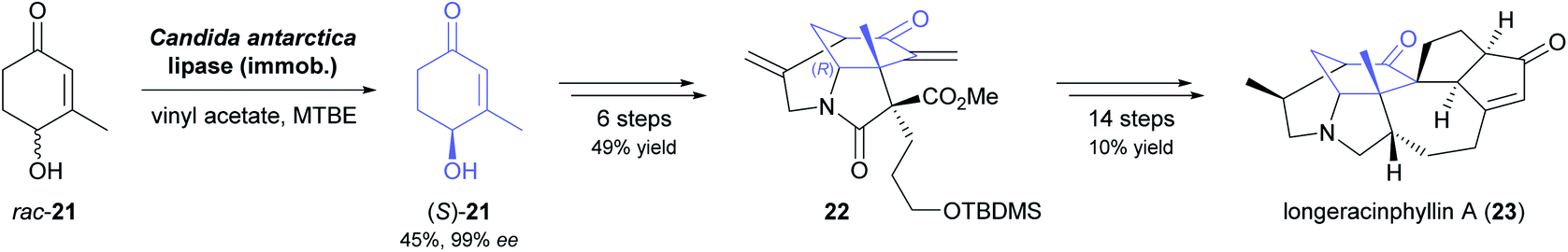 | ||
| Scheme 3 Chemo-enzymatic asymmetric synthesis of longeracinphyllin A (23). | ||
The kinetic resolution of racemic primary alcohols is more challenging than that of sec-alcohols because the stereogenic centre of the substrate is further away from the reacting hydroxyl group, which often results in lower enantioselectivity. Nevertheless, the lipase-catalysed kinetic resolution of β-chiral primary alcohols was successfully used in two recent syntheses of Nuphar alkaloids. In the first of these studies, the allenic benzoate ester 24 (Scheme 4a) was enantioselectively hydrolysed by lipase PS37 to obtain alcohol (S)-25 in 38% yield and 80% ee (E = 15).45 Transformation of the hydroxyl group into a Boc-protected hydroxylamine set the stage for a silver(I)-catalysed cyclisation that gave isoxazolidine 27 with excellent diastereoselectivity (22:1 dr). From this intermediate, (−)-deoxynupharidine (30) was prepared in nine steps, whereby the third chirality centre was formed with complete diastereocontrol in an intramolecular reductive amination reaction. In unrelated work, (−)-nupharamine (32), a piperidine Nuphar alkaloid, was synthesised from alcohol (S)-31, which was prepared by acylative kinetic resolution of the racemate, again using lipase PS (Scheme 4b).46 The enantioselectivity of the biotransformation was excellent in this case (E ≥50), resulting in 45% yield and 99% ee of (S)-31, while the corresponding (R)-acetate was not isolated. The optically pure alcohol was elaborated into the target alkaloid in nine steps with an overall yield of 12%.
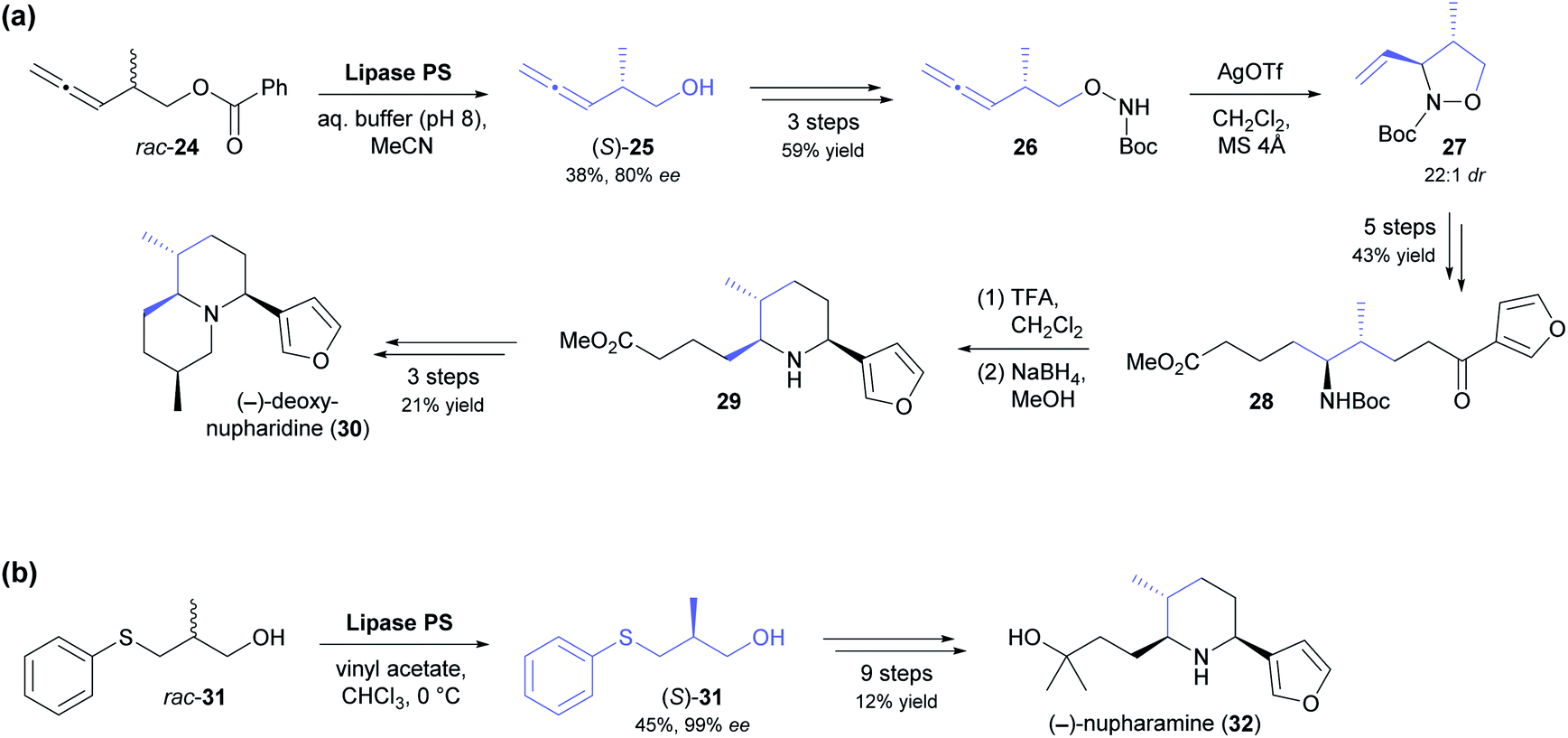 | ||
| Scheme 4 Lipase-catalysed kinetic resolution of β-chiral primary alcohols in the asymmetric synthesis of Nuphar alkaloids: (a) chemo-enzymatic synthesis of (−)-deoxynupharidine (30), (b) chemo-enzymatic synthesis of (−)-nupharamine (32). | ||
Two recent studies demonstrate that fairly complex chiral building blocks can be prepared by the kinetic resolution of primary alcohols. Attaining a sufficient enantioselectivity proved difficult also in these examples, making two consecutive rounds of kinetic resolution necessary to obtain both enantiomers in satisfactory optical purity. As part of their recent synthesis of two pumiliotoxins, Okada, Toyooka, and co-workers carried out the kinetic resolution of alcohol 33 using lipase PL37 and vinyl acetate in MTBE at room temperature (Scheme 5).47 While the unreacted alcohol (5R,6R)-33 was recovered in 36% yield and >98% ee, the corresponding acetate (2S,3S)-34 was isolated in 55% yield but only 58% ee, indicating moderate enantioselectivity (E = 16). After ester hydrolysis, enantioenriched (5S,6S)-33 was subjected to a second round of kinetic resolution under identical conditions, which raised the enantiomeric excess of (2S,3S)-34 to 98% (at 47% yield). The optically pure building blocks thus obtained were converted into both enantiomers of 8-deoxypumiliotoxin 193H (35) and 9-deoxyhomopumiliotoxin 207O (36) in seven and eight chemical steps, respectively.
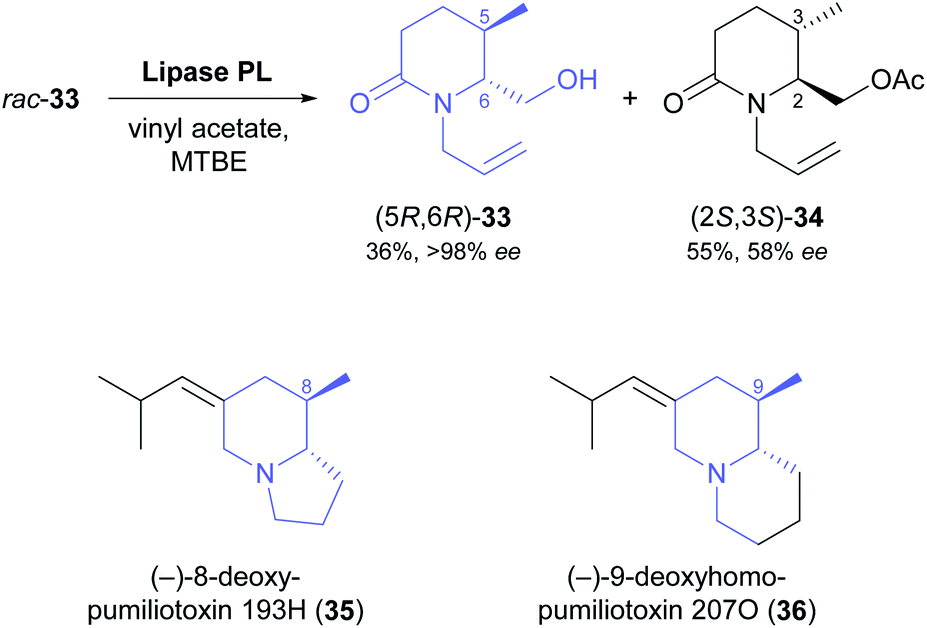 | ||
| Scheme 5 Lipase-catalysed kinetic resolution of building block 33, and structures of alkaloids prepared from this building block. | ||
Yamamoto and co-workers, in their synthesis of (−)-thallusin (39), took a slightly more elaborate approach for upgrading the optical purity of the desired enantiomer of building block 38 (Scheme 6).48 After hydrolytic kinetic resolution of acetate rac-37 under literature-known conditions (lipase PS,37 phosphate buffer/dichloromethane = 6:1, Tween 80, 30 °C)49 had provided the unreacted ester in 52% yield and 48% ee, along with the corresponding alcohol in 36% yield and >98% ee (E = 160), the authors screened five alternative lipases for a second round of kinetic resolution of enantioenriched 37. Lipase M turned out to preferentially hydrolyse the major enantiomer (i.e., it showed opposite enantioselectivity to lipase PS), giving (1S,2S,4aR,8aR)-38 in 92% ee and 29% yield. With all stereogenic centres of the target molecule thus in place, the synthesis of (−)-thallusin was completed in a 10-step sequence.
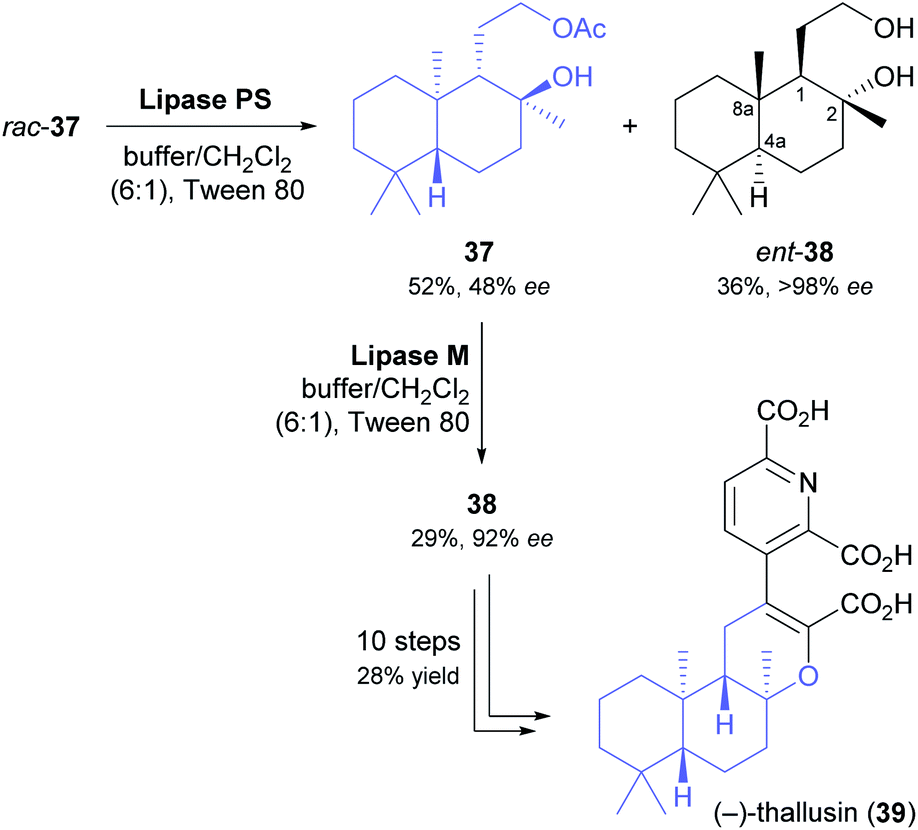 | ||
| Scheme 6 Preparation of building block 38 by double kinetic resolution, and its use in the chemo-enzymatic asymmetric synthesis of (−)-thallusin (39). | ||
:1 mixture of diastereomers (Scheme 7c).52 Treatment with DBU in dichloromethane at room temperature quantitatively converted this isomeric mixture into diene 48, and this intermediate was further elaborated into (−)-himbacine in 16 steps.
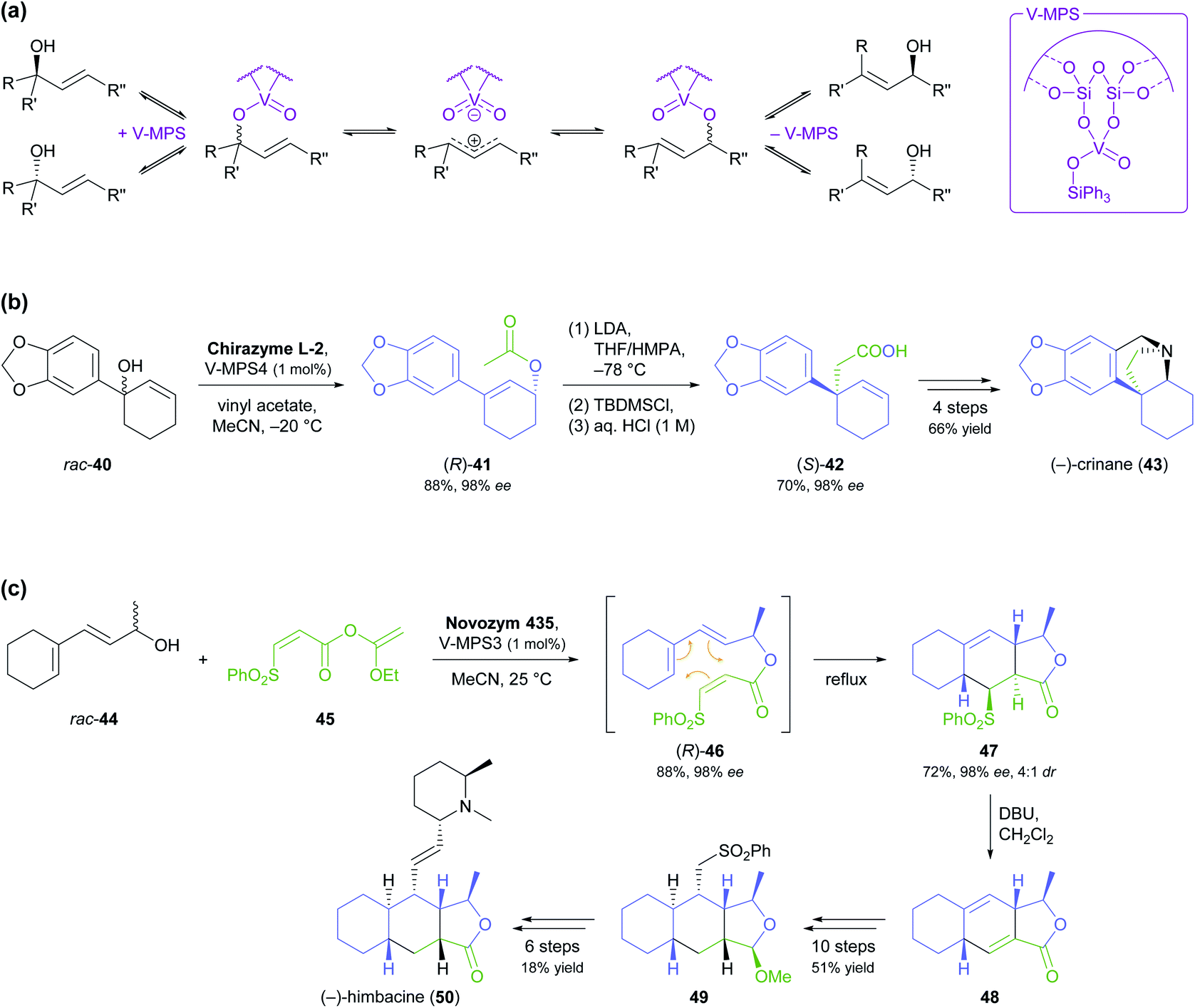 | ||
| Scheme 7 Dynamic kinetic resolution (DKR) of allylic alcohols in the asymmetric synthesis of alkaloids: (a) general racemisation mechanism of oxovanadium catalyst immobilised on mesoporous silica (V-MPS), (b) DKR of 40 followed by Ireland–Claisen rearrangement in the synthesis of (−)-crinane (43), (c) DKR of 44 followed by Diels–Alder cycloaddition in the synthesis of (−)-himbacine (50). | ||
:1 excess over the cis-isomer, most likely via epimerisation by imine–enamine tautomerism. Conversion of 57 into (−)-yohimbane (58) was achieved by the same sequence of steps used for 54.
 | ||
| Scheme 8 Preparation of building block 52 by hydrolase-catalysed desymmetrisation and its use in the synthesis of yohimbe alkaloids: (a) synthesis of (−)-alloyohimbane (54) from (1S,6R)-52, (b) synthesis of (−)-alloyohimbane (54) and (−)-yohimbane (58) from (1R,6S)-52. | ||
Desymmetrisation of the tricyclic meso-compound 60 (Scheme 9) is the asymmetric key step in Fukuyama's recent synthesis of the neurotoxic puffer fish alkaloid (−)-tetrodotoxin.56 The diol 60, prepared by a Diels–Alder reaction of para-benzoquinone (59) with 5-TMS-cyclopentadiene followed by Luche reduction, was stereoselectively acetylated by lipase PS-IM37 under previously reported conditions57 to give monoacetate 61 in 89% yield and >99% ee. From there, the target compound was assembled in a 29-step sequence that exploits facial selectivity in the tricyclic intermediate 61 for controlling three stereogenic centres of the central cyclohexane ring, while the remaining three were installed by a [3,3]-sigmatropic rearrangement of an allylic cyanate and an intramolecular 1,3-dipolar cycloaddition of a nitrile oxide.
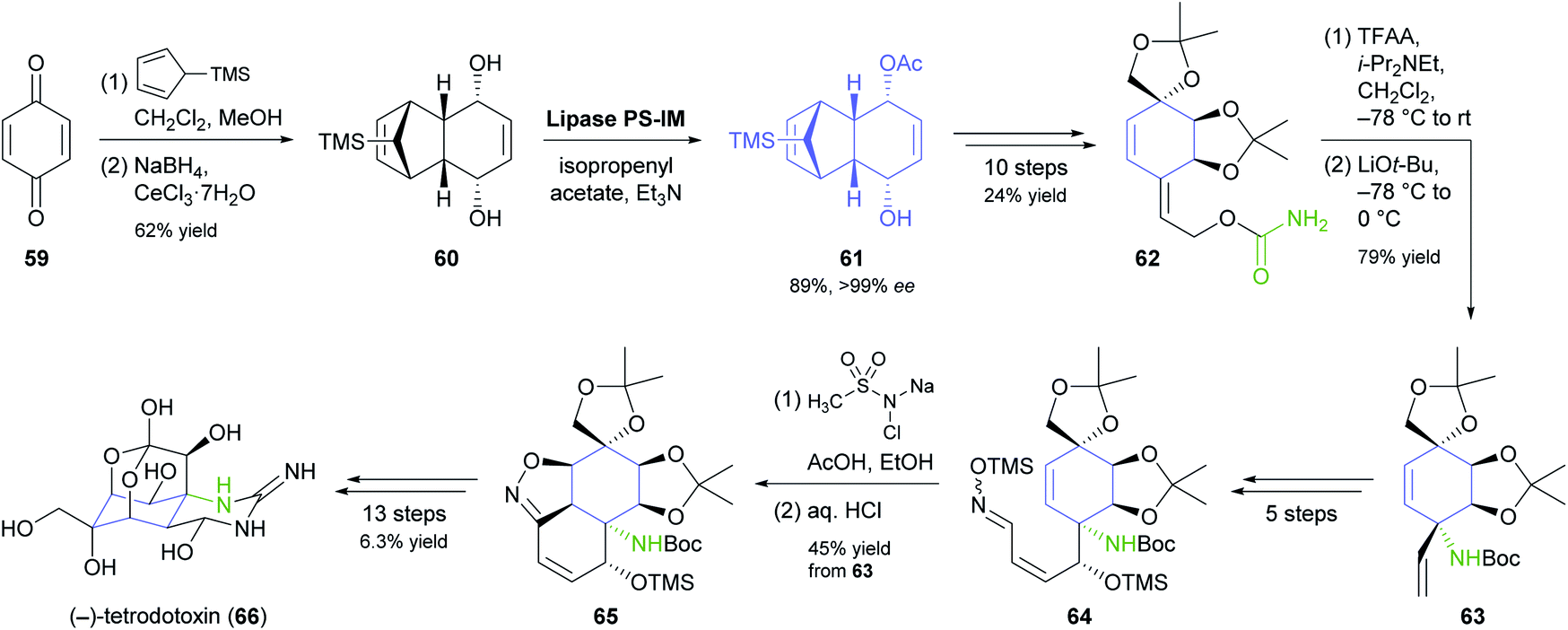 | ||
| Scheme 9 Fukuyama's chemo-enzymatic total synthesis of (−)-tetrodotoxin (66). | ||
In the 1990s, disubstituted piperidine derivatives were used extensively as substrates for lipase-catalysed desymmetrisation and as building blocks for alkaloid synthesis.58 One of the reactions studied during this time is the enantiotoposelective acetylation of meso-diol 67 by Candida antarctica lipase, which affords the monoacetate (2S,6R)-68 in 80% yield and 95% ee (Scheme 10).59 In a more recent study, this building block was employed in the synthesis of (+)-methyl dihydropalustramate (71), a degradation product of the alkaloid (+)-palustrine.60 Key transformations of the 8-step synthetic sequence are a Wittig reaction, a Seyferth–Gilbert homologation, and the ruthenium(II)-catalysed anti-Markovnikov hydration of a terminal alkyne. The acetylative desymmetrisation of a structurally related diol, 69 (Scheme 10), employing a commercial lipase from Pseudomonas fluorescens, was used to access the tricyclic enaminone (−)-72, a known intermediate61 in the synthesis of the poison frog alkaloid (−)-gephyrotoxin.62 The biotransformation furnished the (2R,5S)-enantiomer of monoacetate 70 in 48% yield and 88% ee, indicating that compared to 67 the increased distance of the reactive atom from the meso “core” of the molecule did not only lead to lower selectivity but also to a switch in the recognised enantiotopic group. From (2R,5S)-70, the target enaminone was obtained in a straightforward sequence comprised of Swern oxidation of the free alcohol, a proline-catalysed, reductive Knoevenagel condensation, and two deprotection steps.
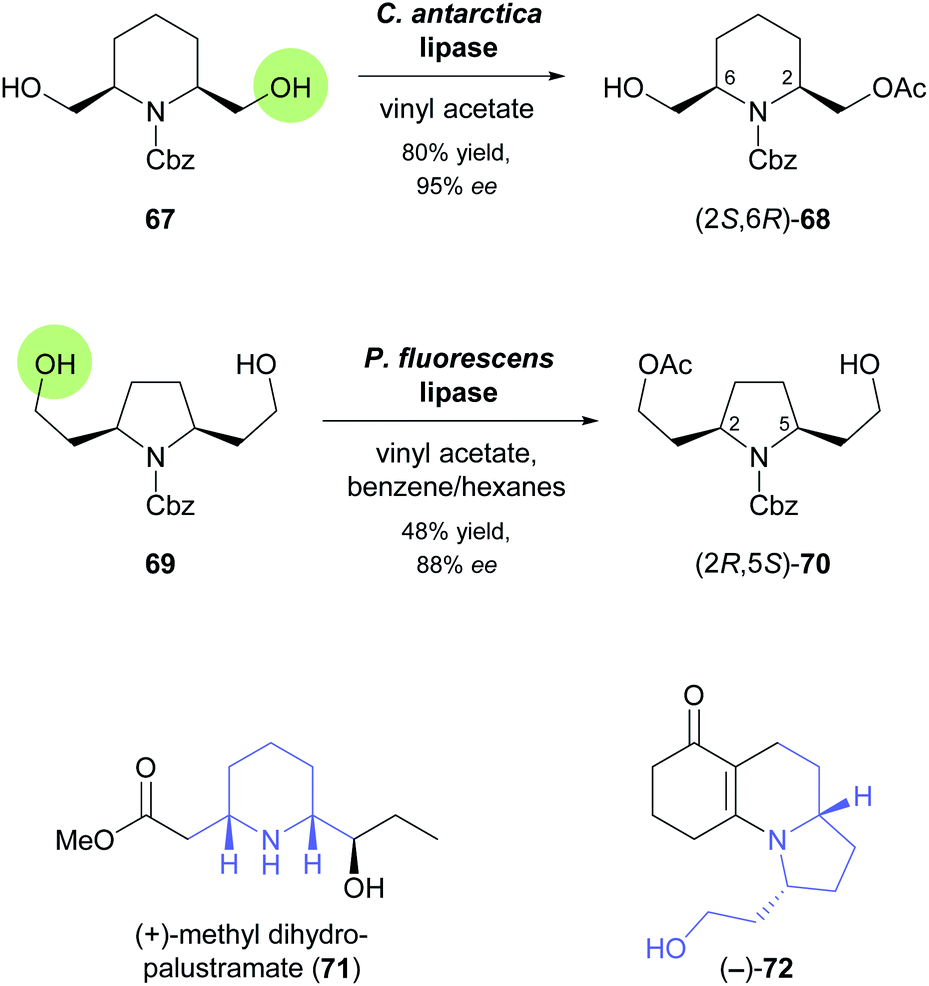 | ||
| Scheme 10 Biocatalytic preparation of piperidine building block 68 and pyrrolidine building block 70, and structures of target molecules prepared from these building blocks. | ||
Open-chain meso-1,3-diols are challenging substrates for desymmetrisation because racemisation of the resulting monoacetates by acyl group migration can occur, leading to reduced enantiomeric purity.63 In their chemo-enzymatic synthesis of the marine sponge alkaloid (−)-petrosin (79, Scheme 11), Hidetoshi Tokuyama and co-workers managed to prevent this side reaction by immediate silyl-protection of the free hydroxyl group.64 Hence, they reacted diol 73 with vinyl acetate under lipase PS37 catalysis, and – after filtration and evaporation of excess acyl donor – treated the crude monoacetate with TBDMS chloride to give compound (S)-74 in 84% yield (two steps) and 99% ee. This building block was elaborated into the piperidine derivative 75, which in turn served as precursor to vinylic iodide 76 and borane 77. Suzuki–Miyaura coupling of the two latter compounds gave olefin 78, from which the bisquinolizidine alkaloid 79 was prepared in 12 additional steps.
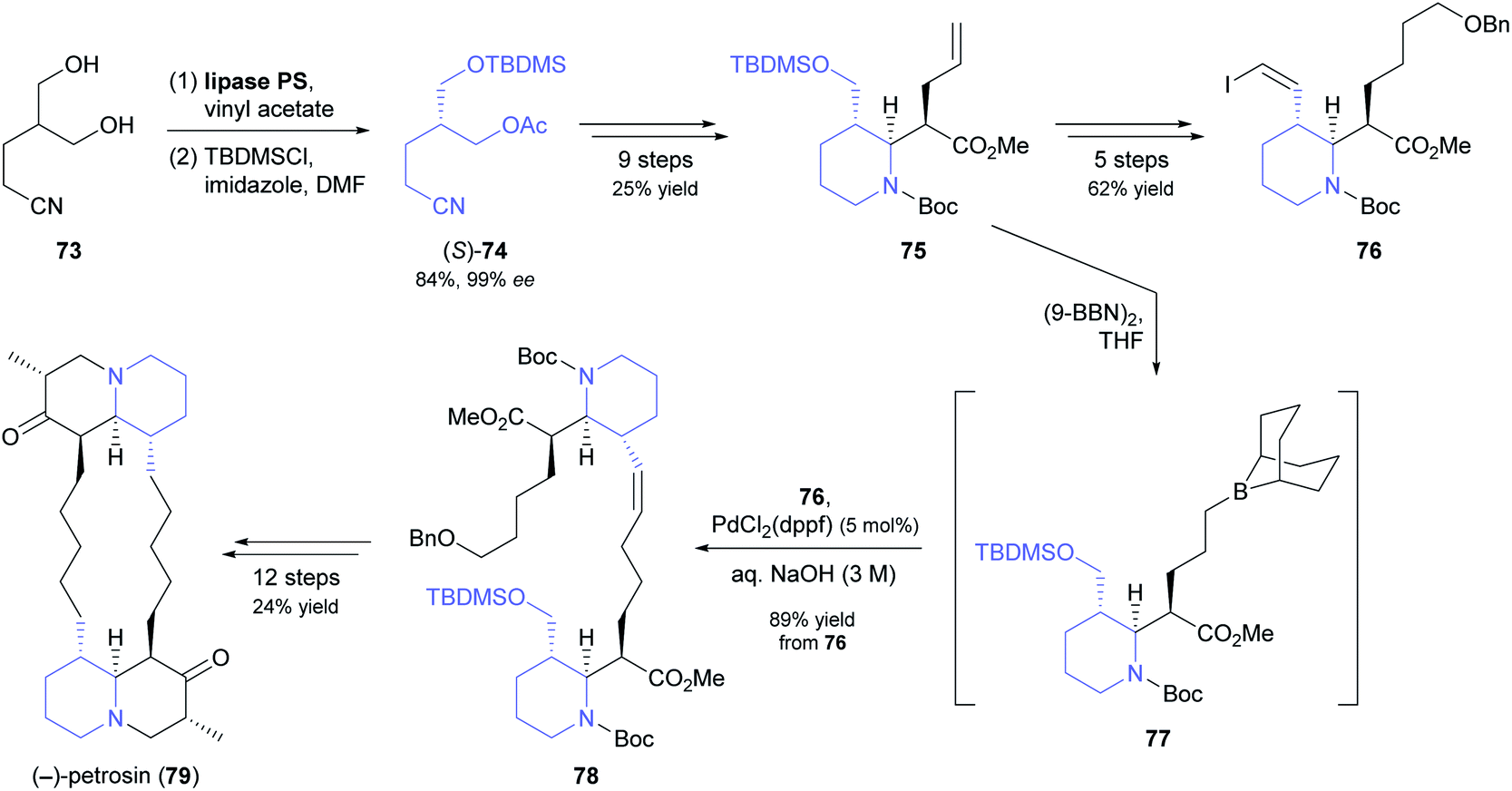 | ||
| Scheme 11 Lipase-catalysed desymmetrisation of 1,3-diol 73 in the total synthesis of the bisquinolizidine alkaloid (−)-petrosin (79). | ||
The lipase-catalysed desymmetrisation of cyclic anhydrides is much less common than the desymmetrisation of diols or diesters, perhaps due to the intrinsically high reactivity of anhydrides that can give rise to undesired non-enzymatic background reactivity.65 Nevertheless, the alcoholysis of 3-methylglutaric anhydride (80, Scheme 12) by lipase PS37 is known to provide monoester 81 in excellent yield (95–98%) and high optical purity (92–93%).66 Matthew Shair and co-workers, of Harvard University, recently used this biotransformation in constructing the quinolizidine subunit of the proposed structure of (−)-himeradine A (84), a Lycopodium alkaloid.66b The pentacyclic core of the target molecule, on the other hand, was assembled from ‘chiral pool’ building blocks (S)-epichlorohydrin (82) and (R)-pulegone (83) in a multi-step sequence that the authors adapted for the synthesis of other Lycopodium alkaloids sharing the same scaffold.
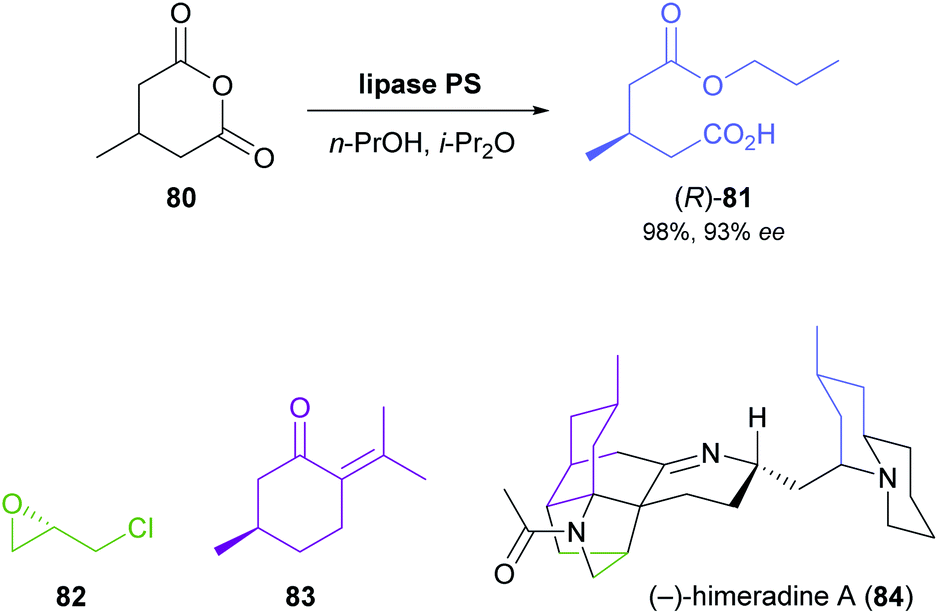 | ||
| Scheme 12 Lipase-catalysed desymmetrisation of cyclic anhydride 80, and the structures of (S)-epichlorohydrin (82), (R)-pulegone (83), and (−)-himeradine A (84). | ||
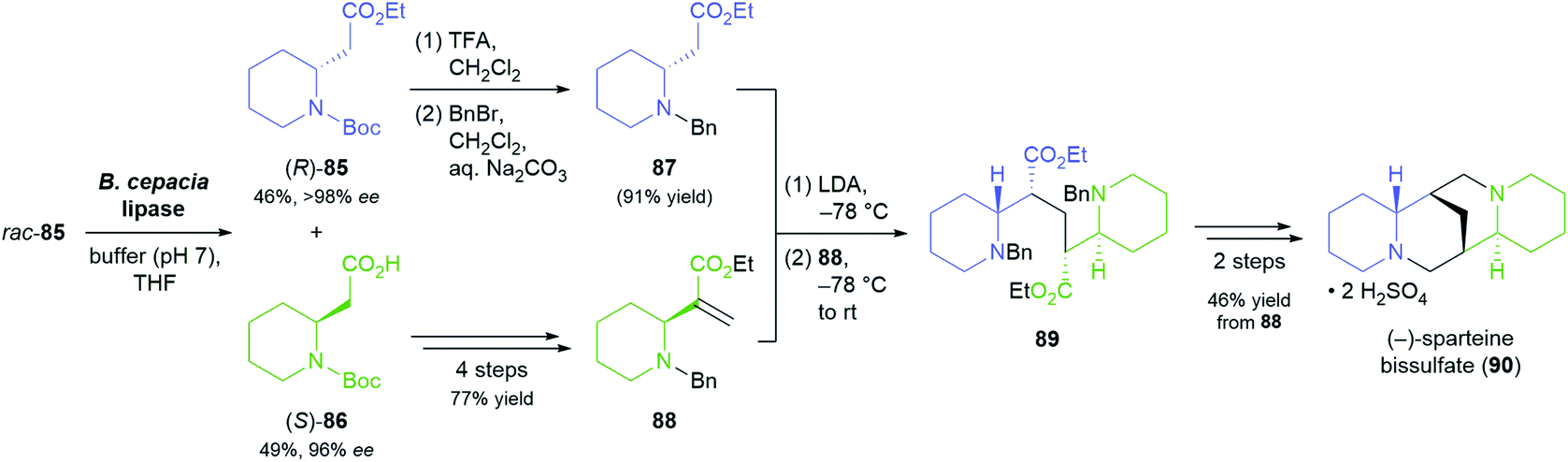 | ||
| Scheme 13 Lipase-catalysed kinetic resolution of building block 85 in the asymmetric synthesis of (−)-sparteine bissulfate (90). | ||
Another rather unusual type of hydrolase reaction, the kinetic resolution of a lactone, was used in a recent synthesis of (R)-harmonine (94, Scheme 14), the toxic principle of the Asian lady beetle.70 The nine-membered chiral lactone 91 was hydrolysed by horse liver esterase with excellent selectivity for the (R)-enantiomer (E >200), affording the (R)-hydroxy acid 92 in 39% yield along with 43% of the unreacted (S)-lactone, both in optically pure form (ee >99%). The latter compound was reduced to the hydroxy aldehyde and directly subjected to a Wittig reaction to give a hydroxy ester (93) that already contained the complete carbon skeleton of the target compound. Reduction to the diol and conversion of both hydroxyl groups to amines via the bis-azide completed the synthesis of (R)-harmonine.
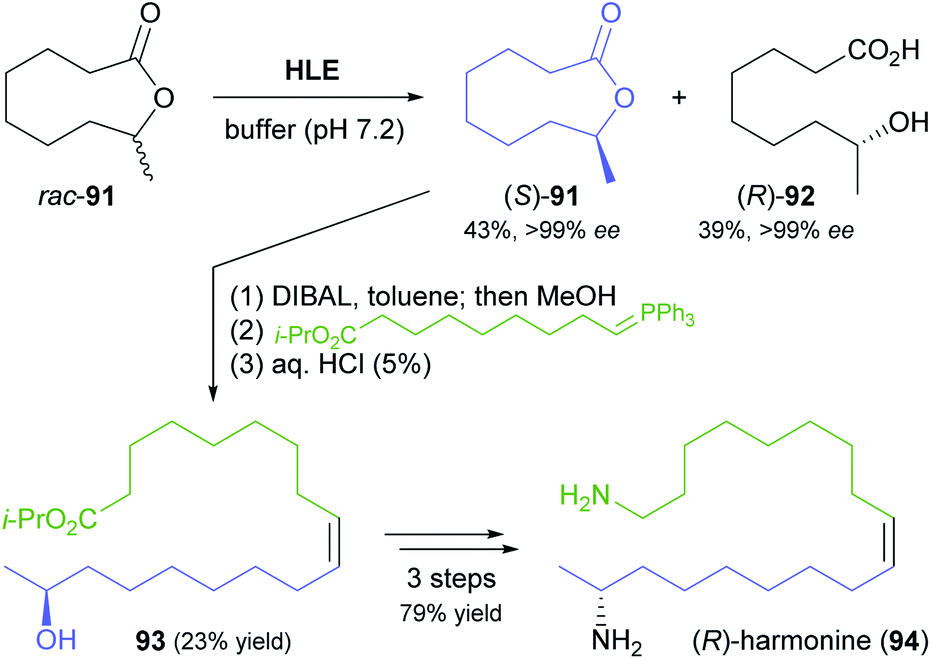 | ||
| Scheme 14 Hydrolytic kinetic resolution of lactone 91 by horse liver esterase (HLE) used in the chemo-enzymatic synthesis of (R)-harmonine (94). | ||
2.2 Toluene dioxygenase
Toluene dioxygenase (TDO) from Pseudomonas putida, an iron–sulfur protein first reported and characterised in the late 1960s,71 oxidises toluene and other mono- and disubstituted arenes to the corresponding cis-dihydrodiols, usually with excellent stereoselectivity (Scheme 15a). The versatile reactivity of these compounds has enabled their exploitation in the asymmetric synthesis of a broad range of complex target molecules, and consequently TDO has become the most widely used dioxygenase in organic synthesis.72 Due to the success of arene cis-dihydrodiols as chiral building blocks, several of these compounds are now commercially available (for examples, see Scheme 15b), making them accessible also to researchers that lack the necessary equipment or expertise for carrying out TDO biotransformations.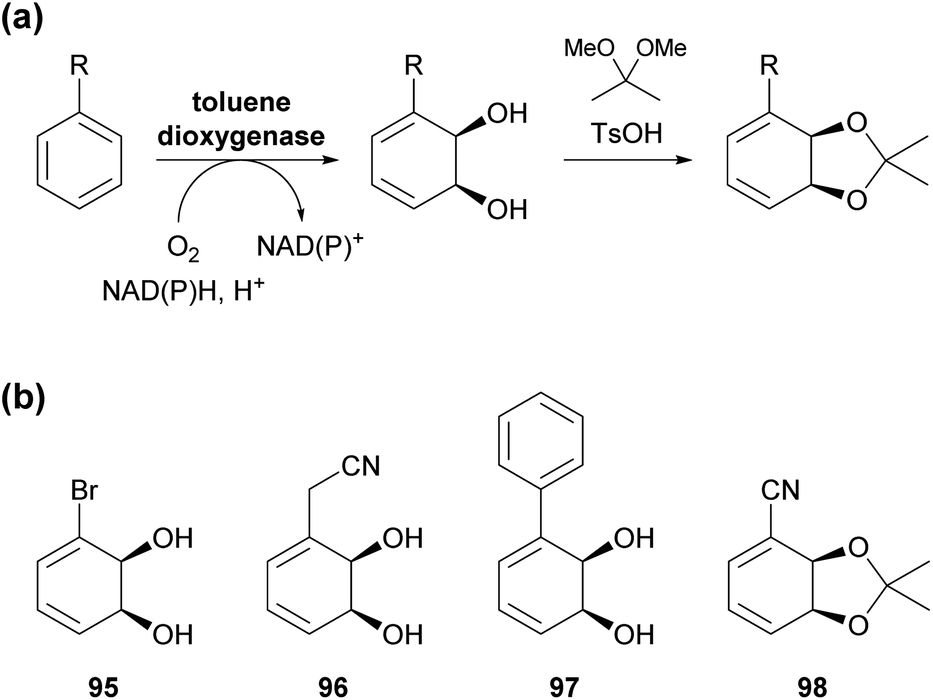 | ||
| Scheme 15 Arene cis-dihydroxylation by toluene dioxygenase (TDO): (a) general reaction scheme for the dihydroxylation of a monosubstituted arene by TDO and for the conversion of the resulting diol into an acetonide. (b) Examples of commercially available arene cis-dihydrodiols or the corresponding acetonides (as of January 2021). | ||
In the context of alkaloid synthesis, TDO-derived building blocks have been most intensively used by the groups of Tomas Hudlicky, who has developed several TDO-based chemo-enzymatic approaches to morphinan and isocarbostyril alkaloids over the course of the last 30 years, and of Martin Banwell, who has focused on Amaryllidaceae alkaloids of the montanine and lycorenine classes (Fig. 1).73 Both groups have continued these efforts in recent years, with a shift of focus to the preparation of non-natural alkaloid derivatives following synthetic strategies developed in earlier work. This trend is exemplified by the preparation of 7-aza-nornarciclasine (114) and 10-aza-narciclasine (119), two isocarbostyril analogues featuring a nitrogen atom in the A-ring (Scheme 16a).74 Both syntheses commenced with the transformation of TDO-derived cis-diol 95 into the conduramine derivative 109 via an acyl-nitroso-diene [4 + 2] cycloaddition (nitroso-Diels–Alder reaction) followed by reduction with aluminium amalgam and protective-group manipulations. Compound 109, which contains the four contiguous stereogenic centres of the target alkaloids in the correct configuration, was then coupled with the lithium carboxylates 110 and 115 to give the amides 111 and 116, respectively. Closure of ring B by a Heck reaction proceeded in moderate yield in both cases (31% for 113 and 58% for 118), and deprotection furnished the target compounds. Biological evaluation showed the 7-aza-derivative 114 to be inactive against human cancer cell lines,74a while 10-aza-narciclasine (119) did show significant cytotoxic activity.74b A related synthetic approach was used to prepare 2-epi-narciclasine (126, Scheme 16b), a trace constituent of snowdrop (Galanthus) bulb extracts.75 In this work, TDO-catalysed oxidation of meta-dibromobenzene afforded the diol 120, which was protected as the usual acetonide and transformed into ketone 122 via a nitroso-Diels–Alder reaction, Suzuki coupling, and Mo(CO)6 reduction. Luche reduction then established the desired (R)-configuration at C7 (123; C2 in isocarbostyril numbering). After protection of the three vicinal hydroxyl groups as acetates, a modified Bischler–Napieralski cyclisation was used to construct ring B, and deprotection gave the target alkaloid. The allylic alcohol 123 that is a central intermediate in this sequence also proved useful in a formal total synthesis of (+)-pancratistatin (101) that uses 1,3-chirality transfer by Myers' reductive transposition as a key step (Scheme 16b).76 While this transposition could be accomplished in satisfactory yield (68–79%), trans-dihydroxylation of the resulting olefin 127 by literature-known methods was unsuccessful. The authors, therefore, chose to establish the required (1R,2S)-stereochemistry (isocarbostyril numbering) through cis-dihydroxylation followed by preparation and ring-opening of a cyclic sulfate. Closure of ring B was again accomplished by a modified Bischler–Napieralski cyclisation, and although both of the latter two reactions produced regioisomeric mixtures, the desired isomers were favoured in both cases.
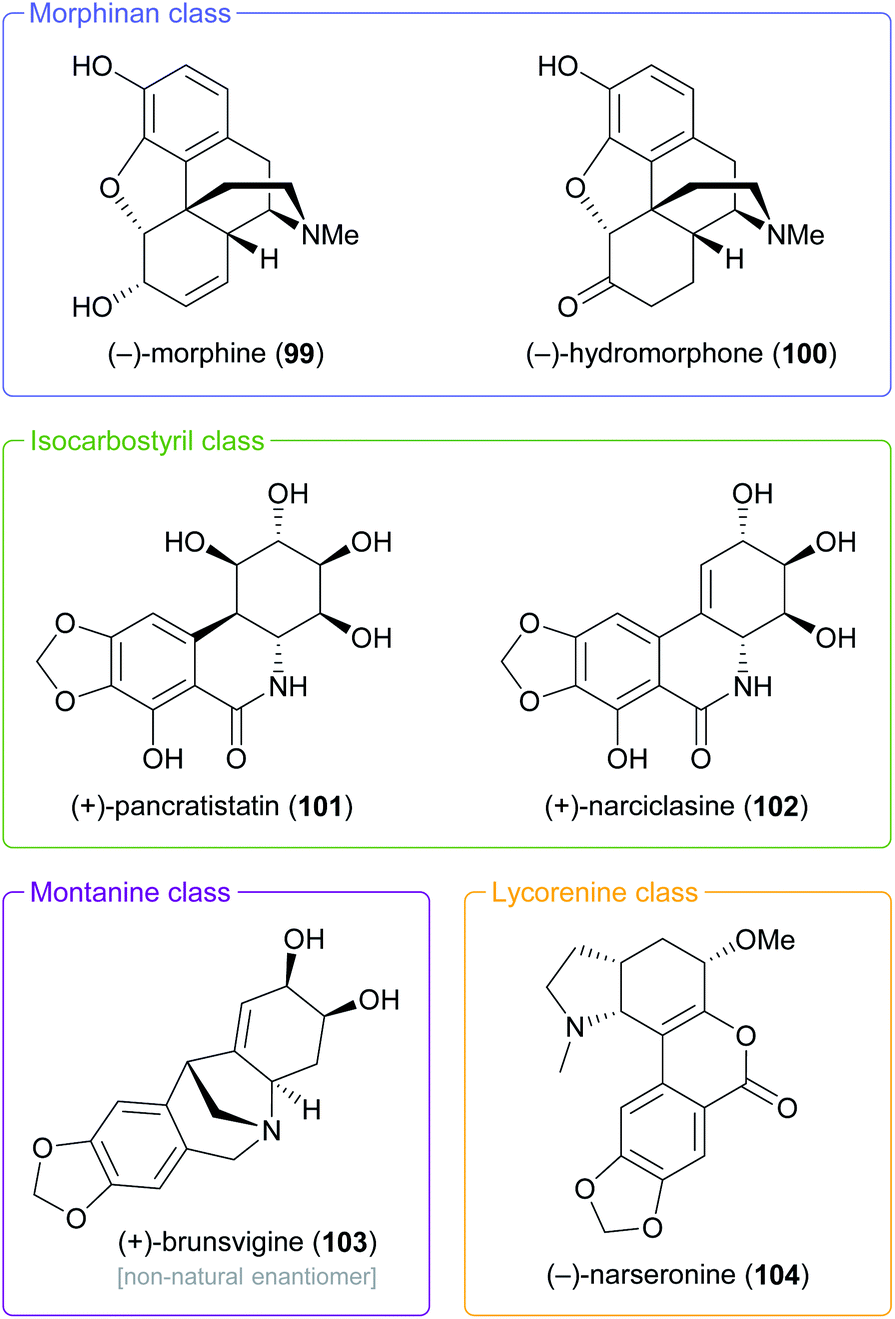 | ||
| Fig. 1 Examples of alkaloid classes targeted by TDO-based chemo-enzymatic syntheses. | ||
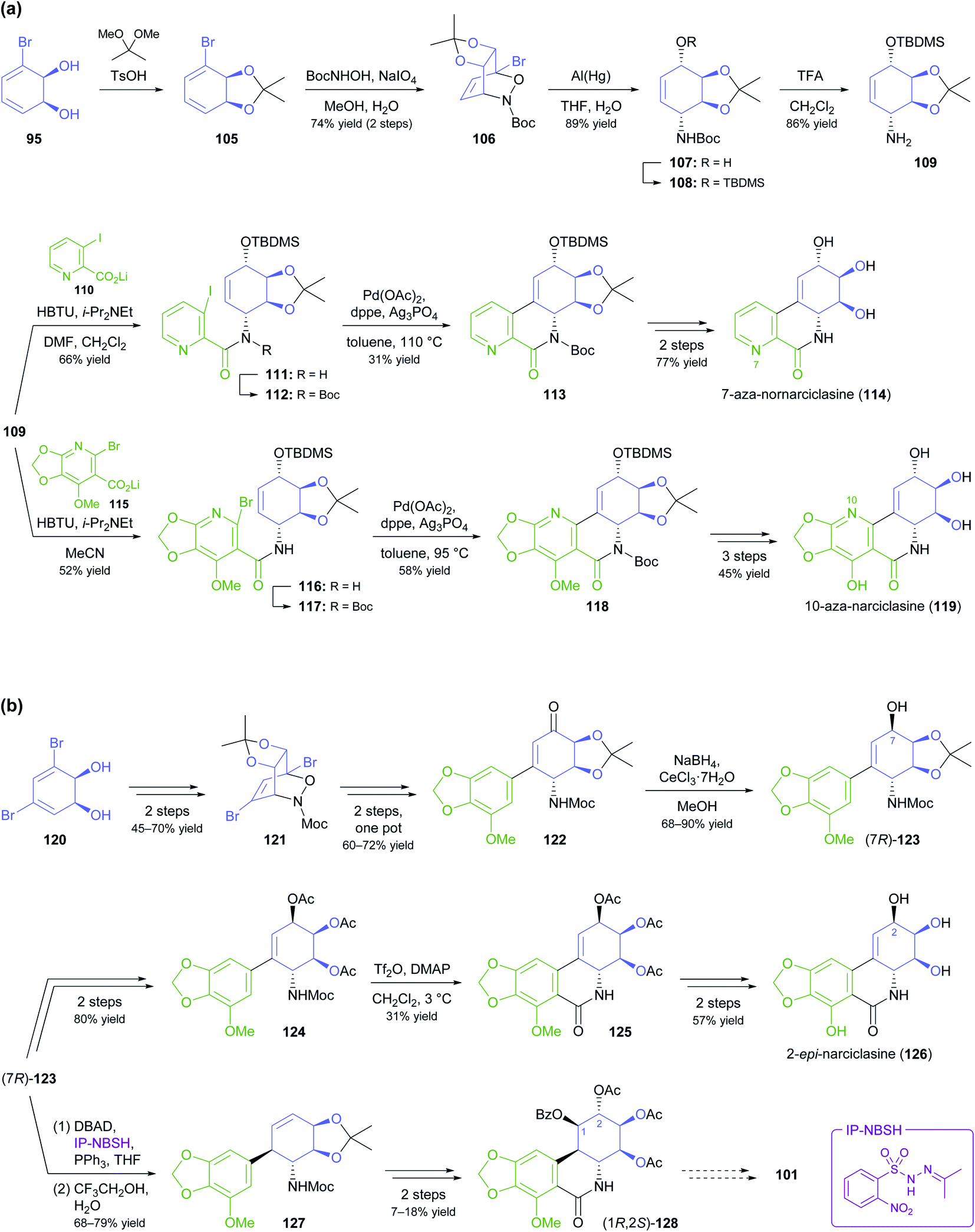 | ||
| Scheme 16 Recent application of TDO-derived chiral building blocks in the synthesis of isocarbostyril alkaloids: (a) transformation of building block 95 into 7-aza-nornarciclasine (114) and 10-aza-narciclasine (119), (b) transformation of building block 120 into 2-epi-narciclasine (126) and a late synthetic intermediate 128 of (+)-pancratistatin (101). | ||
As part of their ongoing efforts to develop practical syntheses of morphinan alkaloids,77 Hudlicky and co-workers have recently presented a novel chemo-enzymatic approach to both enantiomers of hydromorphone (100) starting from the TDO-derived building block 129 (Scheme 17a).78 The cornerstone of their synthetic strategy is the construction of ring B of the morphinan skeleton by a Diels–Alder reaction of intermediate 135, produced in situ by oxidative dearomatisation of the styrene derivative 134. This is followed by closure of ring D through radical cyclisation of tosylamide 137. The feasibility of this sequence was first demonstrated for the non-natural enantiomer, ent-100, using lead(IV) acetate as oxidant in the dearomatisation step and affording the target alkaloid in 2% overall yield over 12 steps from 129.78a A follow-up study explored alternative oxidation methods, including the use of electrochemistry and of hypervalent iodine reagents, and found that Pb(OAc)4 can be replaced with the less environmentally problematic (diacetoxyiodo)benzene (DAIB), albeit at the cost of a reduced yield (20–30% for the cycloaddition product, compared to 50%).78b In the same publication, the authors formally extended their synthetic strategy to the natural enantiomer, as they could demonstrate the conversion of diol 130 into ent-133 via a five-step sequence (Scheme 17b).
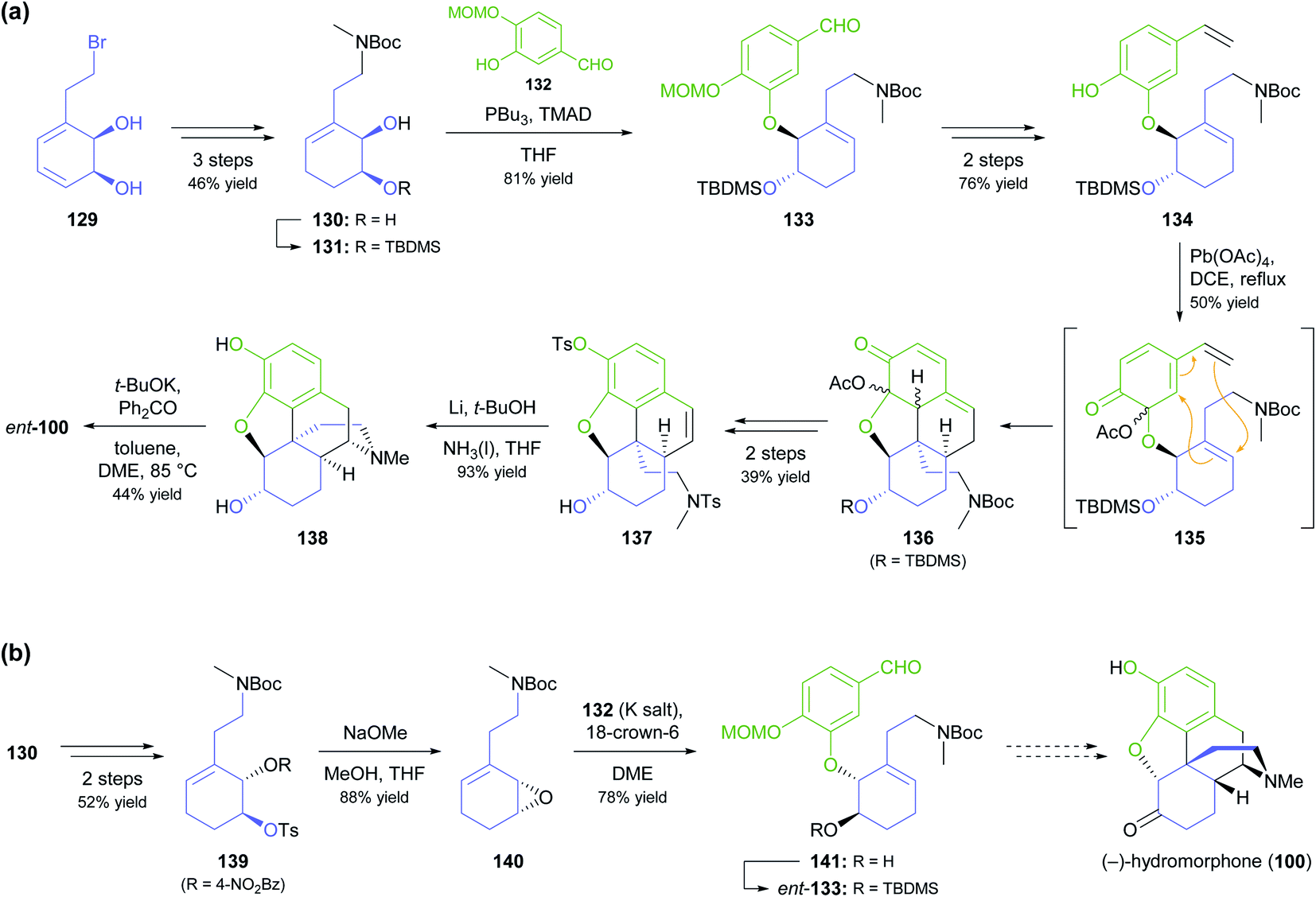 | ||
| Scheme 17 Synthesis of both enantiomers of hydromorphone (100) from TDO-derived building block 129: (a) transformation of 129 into the non-natural enantiomer, (+)-hydromorphone (ent-100), via Mitsunobu coupling, Diels–Alder cycloaddition, and N-centred radical cyclisation as key steps, (b) formal synthesis of the natural enantiomer, (−)-hydromorphone (100), using formation and ring-opening of epoxide 140 for stereoinversion. | ||
A more common, alternative strategy for morphinan synthesis is the construction of ring E by a sequence of Mitsunobu coupling and Heck cyclisation (cf. Schemes 6 and 18d of our previous review, ref. 33), followed by the successive closure of the remaining two rings. In recent years, the Hudlicky group has extensively explored this approach in synthetic studies that are based on building block 142 (Scheme 18a), a compound obtained in product titres of up to 5 g L−1 by the TDO-catalysed oxidation of phenethyl acetate.79 Partial reduction and silyl protection converted 142 into 144, which underwent Mitsunobu coupling with phenol 145 and subsequent Heck cyclisation to give the key intermediate 146, from which various routes to the complete morphinan scaffold were developed.80 In one study,80 conversion of 146 into the nitrone 148 triggered a (3 + 2)-cycloaddition that was intended to close ring B and establish the C9 amine stereocentre in a single operation. Unfortunately, the reaction produced isoxazolidine 149 with an incorrect C9,C14-stereoconfiguration, and an alternative cyclisation of a nitrile oxide followed by reduction gave the same result. Consequently, the authors decided to cleave the C9–N bond by a Hofmann elimination to enable installation of the correct stereochemistry at a later point in the synthesis. Within five more steps, they arrived at compound 150, which can be transformed into the non-natural enantiomers of codeine and codeinone by literature-known methods. Later studies established several options for converting intermediate 146 and related compounds into the non-natural (+)-forms of the semi-synthetic opioid analgesics oxycodone and 10-keto-oxycodone.81 Moreover, a structurally similar tricyclic intermediate, 151, served as precursor to the non-natural antipodes of the snowdrop alkaloids narwedine (153) and galantamine (154; Scheme 18b).82 The authors investigated two alternative Mitsunobu–Heck sequences, which both afforded 151 in good yield (61% over three steps, 68% over two steps) from diol 143. Elimination of the hydroxyl group, followed by iodoacetoxylation (Prévost reaction), reductive deiodination, and closure of the seven-membered D ring gave a 2:1 mixture of galantamine epimers (152), which was oxidised to (+)-narwedine (153) using pyridinium chlorochromate. Reduction of 153 with L-selectride at low temperature stereoselectively afforded (+)-galantamine (154).
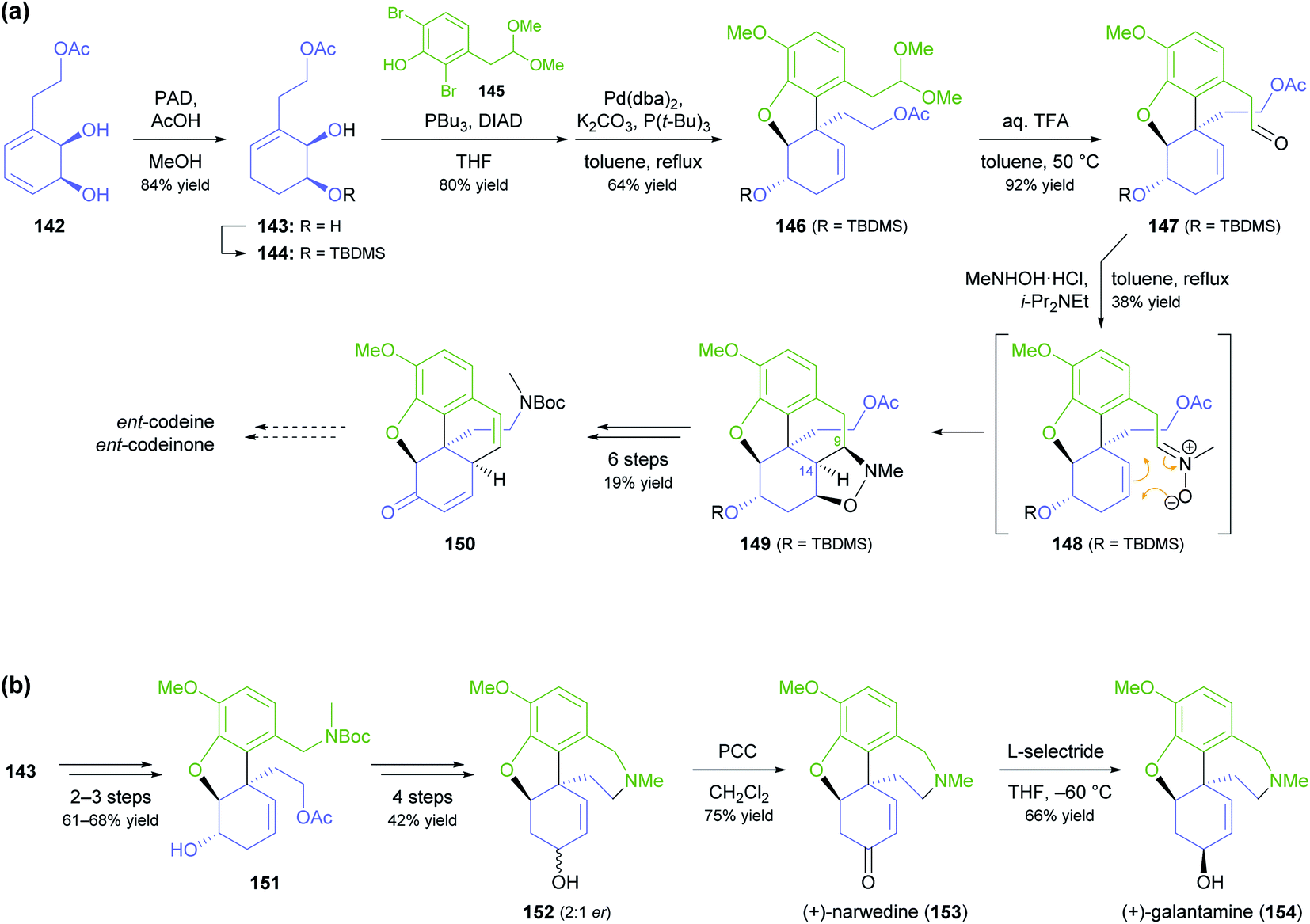 | ||
| Scheme 18 Examples for the use of TDO-derived building block 142 in alkaloid synthesis: (a) formal synthesis of ent-codeine and ent-codeinone via Mitsunobu coupling, Heck cyclisation, and nitrone–alkene (3 + 2) cycloaddition as key steps, (b) total synthesis of (+)-narwedine (153) and (+)-galantamine (154; non-natural enantiomers in both cases). | ||
In an interesting example of stereodivergent synthesis, Banwell and co-workers prepared (+)-narseronine (ent-104) from the commercially available diol 95,83 after having previously used the same building block to obtain the (−)-enantiomer of this Amaryllidaceae alkaloid.84 The sequence followed to arrive at ent-104, however, resembles more closely the one developed in another synthetic endeavour of Banwell's group, the preparation of (+)-clividine:85 Diol 95 was converted into the vinylic bromide 155 via a five-step sequence before being coupled with boronate 156 in a Suzuki reaction (Scheme 19). Protective-group manipulations and hydrogenation of the nitrile moiety led to primary amine 158, which underwent radical cyclisation to give the C5a–C12b-unsaturated compound 159. This outcome is in contrast to the clividine synthesis, where a similar reaction resulted in formation of the saturated product. The synthesis of (+)-narseronine was completed by inversion of the C5-stereocentre through an oxidation–reduction sequence and N,O-bismethylation. Other complex target molecules recently prepared by the Banwell group from building block 95 include non-natural analogues of the alkaloids galantamine and vindoline.86
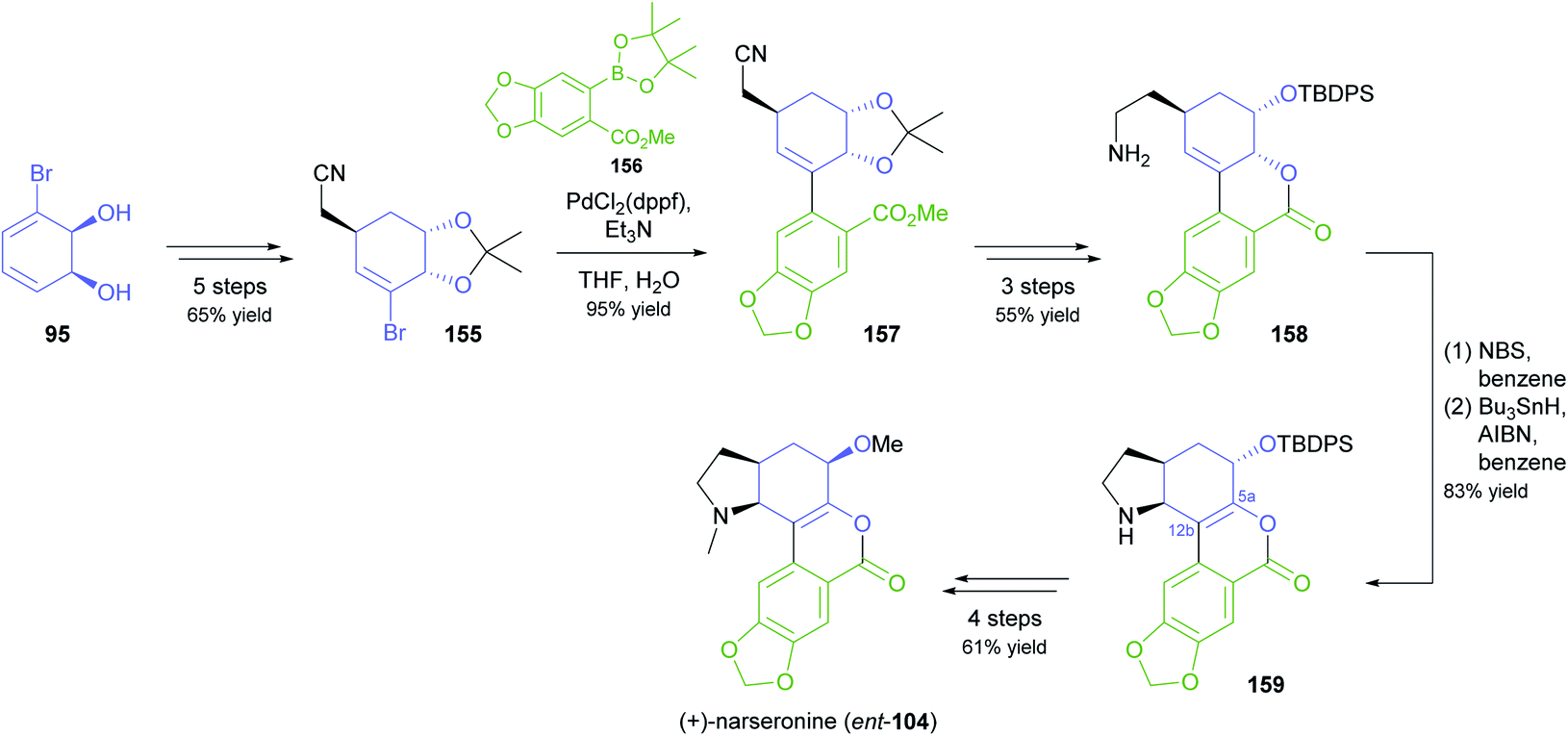 | ||
| Scheme 19 Synthesis of the non-natural enantiomer of narseronine (ent-104) from TDO-derived diol 95. | ||
2.3 Baker's yeast
Despite the advances in molecular biology that have made recombinant enzymes broadly accessible,87 wild-type microorganisms remain relevant as biocatalysts. This is particularly true for baker's yeast, as it is cheap, easily available, and simple to use. In two recent studies, baker's yeast was employed for generating chiral building blocks that constituted the starting points for multi-step syntheses of highly complex alkaloids (Scheme 20).88 In both cases, the yeast reduced an α-substituted β-dicarbonyl compound, thereby controlling two vicinal chirality centres, one of which is quaternary. Hanessian and co-workers, in their synthesis of isodaphlongamine H (167), made use of the literature-known89 reductive kinetic resolution of β-ketoester 160 to prepare hydroxyester (1S,2S)-161 (50% yield, single isomer), which they elaborated into the enone 162 in a 12-step sequence (Scheme 20a). From 162, they constructed the hexacyclic framework of the target compound in 9 more steps, which include a copper(I)-mediated conjugate addition of vinyllithium reagent 163 and an intramolecular aldol addition.88c The baker's yeast catalysed reductive desymmetrisation of diketone 168, inspired by similar reactions reported in literature,90 was used by Sharpe and Johnson in their total synthesis of paspaline (173), an indole diterpenoid from ergot fungi (Scheme 20b).88a,b Reduction product 169, obtained in 66% yield, 10:1 dr, and >98% ee, served as basis for constructing the elongated, hexacyclic target compound with substrate-based stereocontrol of five additional chirality centres. Key transformations of the 27-step reaction sequence include an epoxidation–etherification cascade, an Ireland–Claisen rearrangement, a palladium(II)-catalysed, diastereoselective C–H acetoxylation of intermediate 171, and a Gassmann indole synthesis.
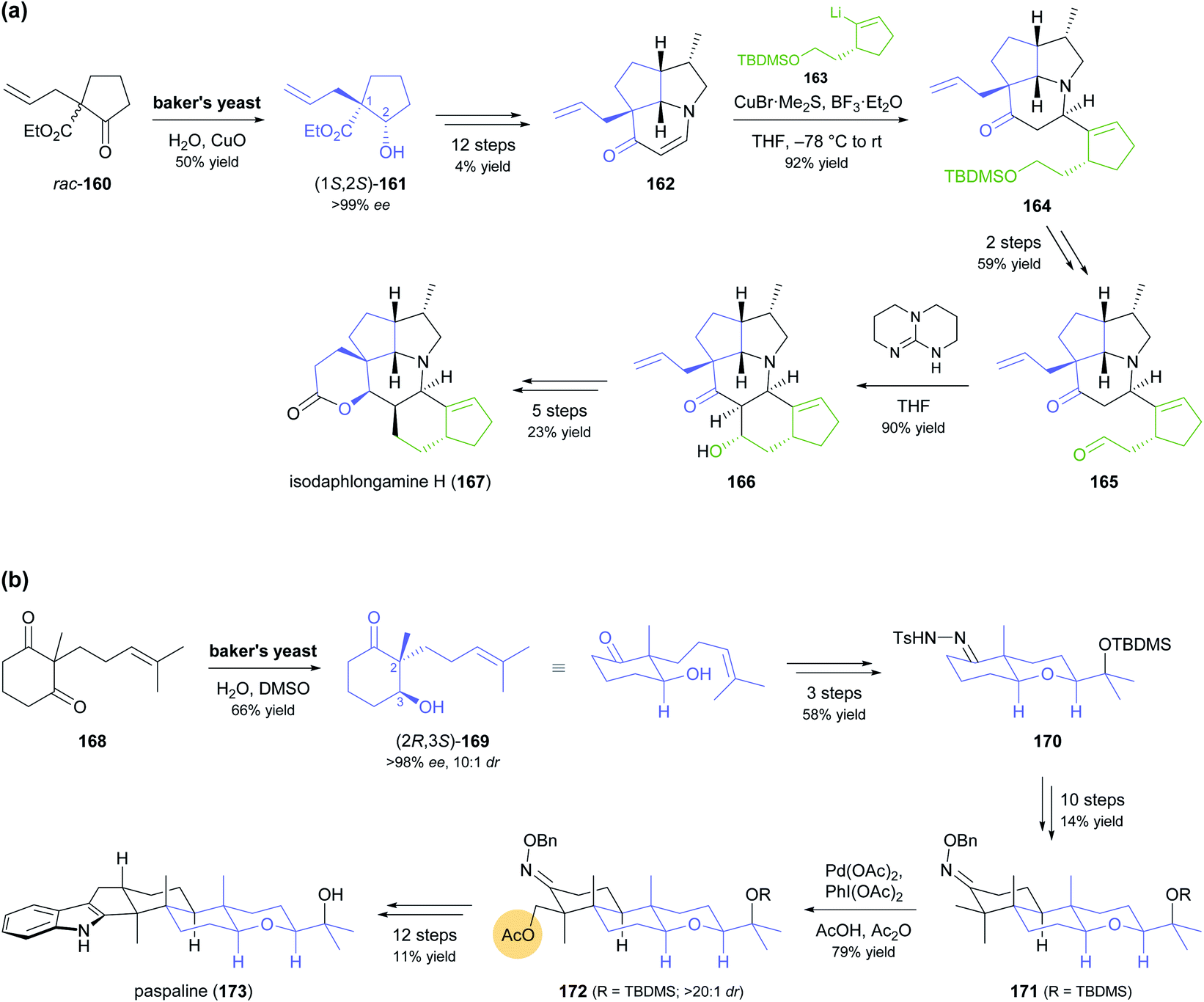 | ||
| Scheme 20 Recent application of baker's yeast in the total synthesis of alkaloids: (a) reductive kinetic resolution of β-ketoester rac-160 in the synthesis of isodaphlongamine H (167), (b) synthesis of paspaline (173) featuring reductive desymmetrisation of β-diketone 168 by baker's yeast and local desymmetrisation of 171 by Pd2+-catalysed diastereoselective C–H acetoxylation. | ||
3 Biocatalytic kinetic resolution, dynamic kinetic resolution, and deracemisation of alkaloids
The examples in the previous section have shown how a chiral building block prepared by an enzymatic transformation early in a synthetic sequence can be used to construct complex alkaloids with multiple chirality centres through substrate-based stereocontrol. For simpler target structures, in particular those that contain only a single stereogenic centre, it can often be more efficient to incorporate the asymmetric key step at a later point in the route. Biocatalysis has hence been used for the kinetic resolution, dynamic kinetic resolution, and deracemisation of alkaloids or advanced synthetic intermediates, with the majority of examples relying on lipases and amine oxidases.3.1 Lipase-catalysed kinetic resolution and dynamic kinetic resolution
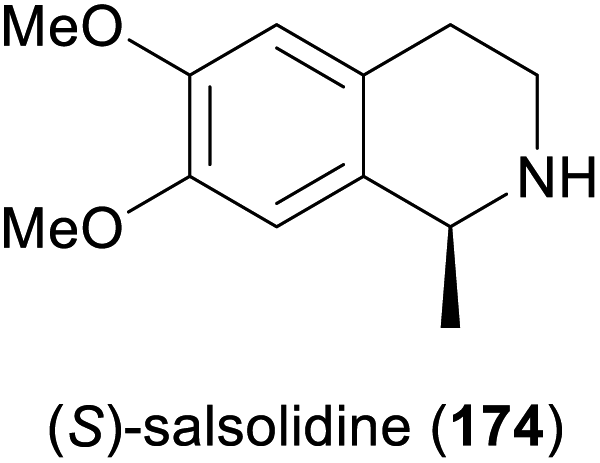
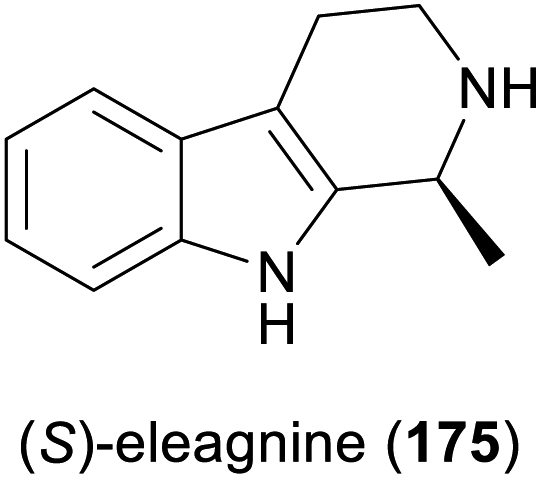
Some simple alkaloids containing a chiral secondary amine moiety are amenable to kinetic resolution by lipase-catalysed N-acylation. Excellent results in this context were achieved for salsolidine (174) and eleagnine (175; Fig. 2) using lipase B from Candida antarctica (CAL-B) as biocatalyst and allyl phenyl carbonate as acyl donor.91 A strong dependence of enantioselectivity on reaction temperature and solvent was observed, but in methyl tert-butyl ether (MTBE) at 50 °C the kinetic resolution of rac-174 proceeded fast (50% conv. in 2.5 h) and with high selectivity (E >200), affording the acylated (R)-enantiomer in 97% ee along with unreacted (S)-174 in 99% ee. Interestingly, under identical conditions the acylation of rac-175 was prohibitively slow (47% conv. after one week), but substoichiometric addition of triethylamine (ca. 29 mol%) led to an increased reaction rate (50% conv. in 48 h) while maintaining an excellent enantioselectivity (E >200). In a follow-up study, the kinetic resolution of tetrahydro-β-carbolines with different C1 substituents (ethyl, n-propyl, isopropyl) was investigated, and excellent enantioselectivities were found in all cases.92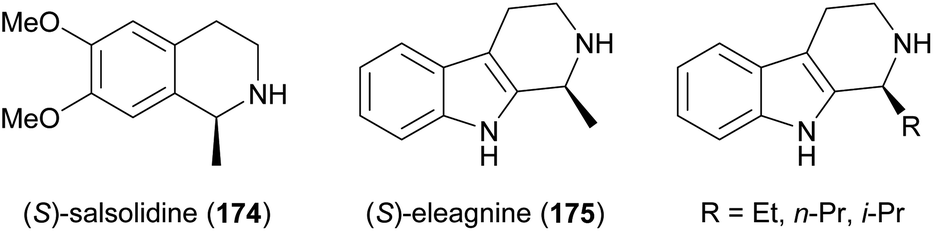 | ||
| Fig. 2 Structures of alkaloids obtained by kinetic resolution using lipase B from Candida antarctica (CAL-B). | ||
Recent progress in the mild racemisation of chiral secondary amines has also rendered the dynamic kinetic resolution (DKR) of simple alkaloids possible. In a recent example, the literature-known93 DKR of rac-174 using lipase from Candida rugosa in combination with an iridium(III)-based racemisation catalyst was used to obtain the carbamate (R)-176 in 68% yield and 99% ee (Scheme 21).94 Hydrolysis of 176 gave (R)-salsolidine, which was further transformed into (+)-bernumidine (177), an alkaloid of Berberis nummularia.
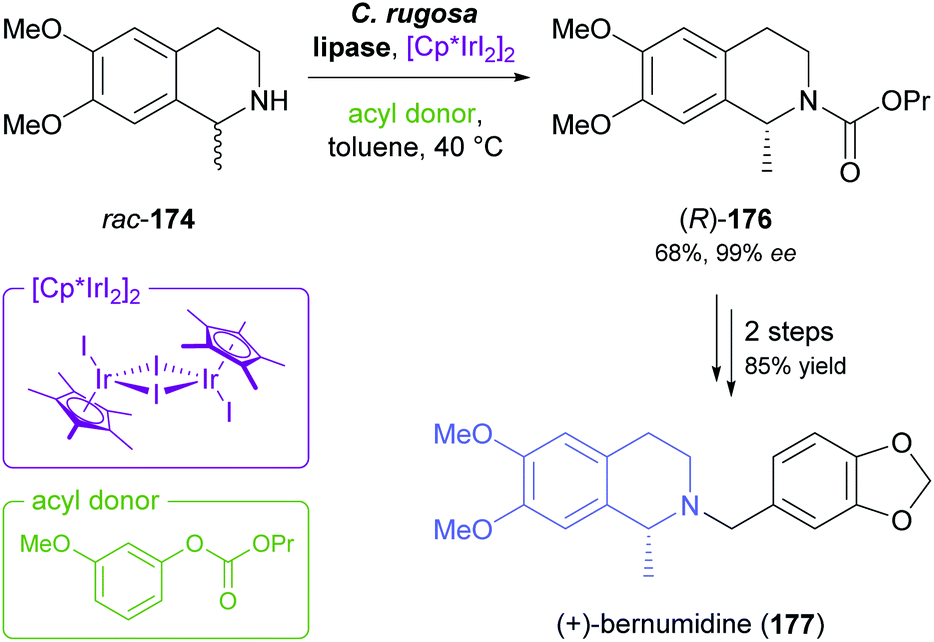 | ||
| Scheme 21 Chemo-enzymatic dynamic kinetic resolution of salsolidine (rac-174) and conversion of the resulting carbamate (R)-176 into (+)-bernumidine (177). | ||
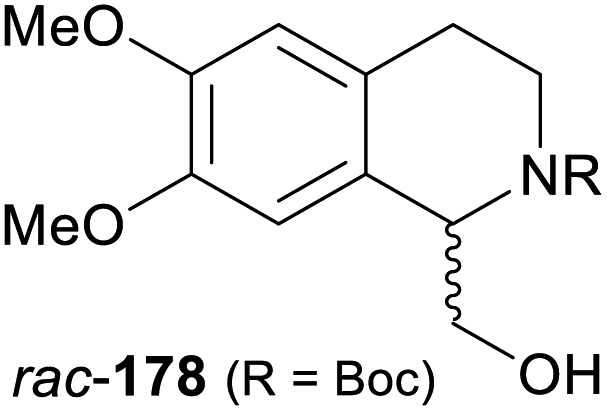
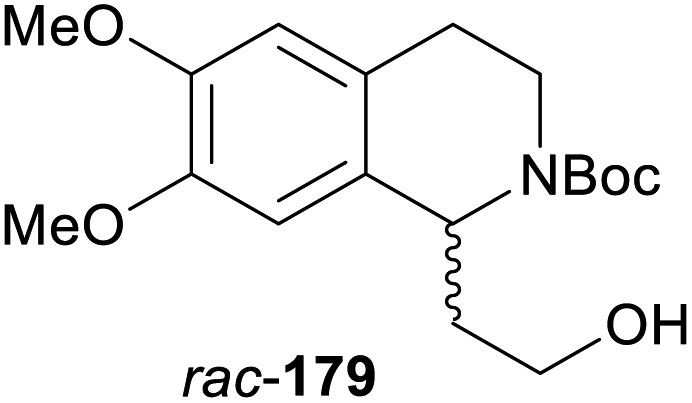
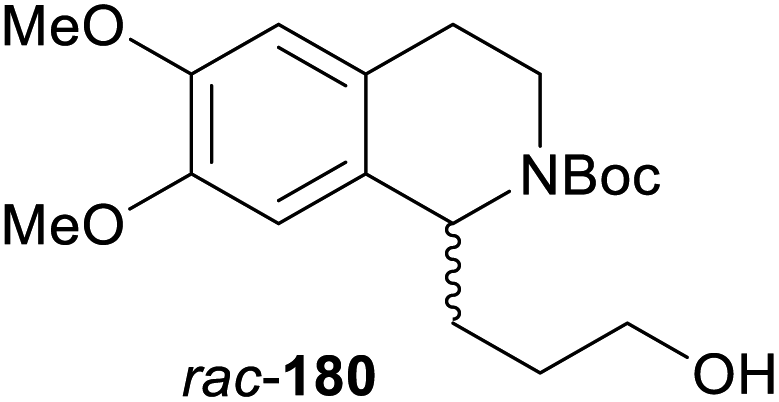
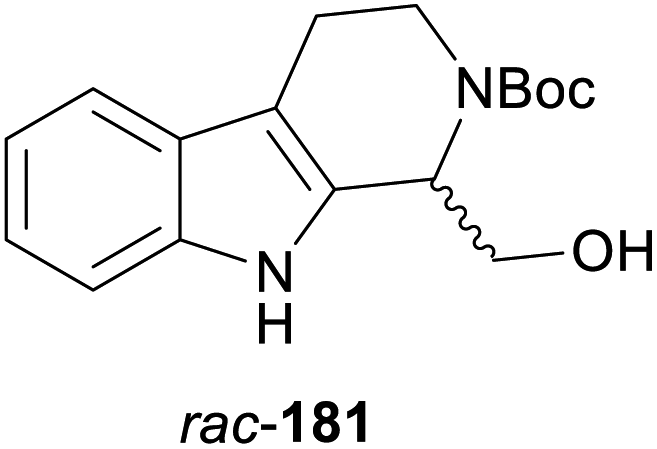
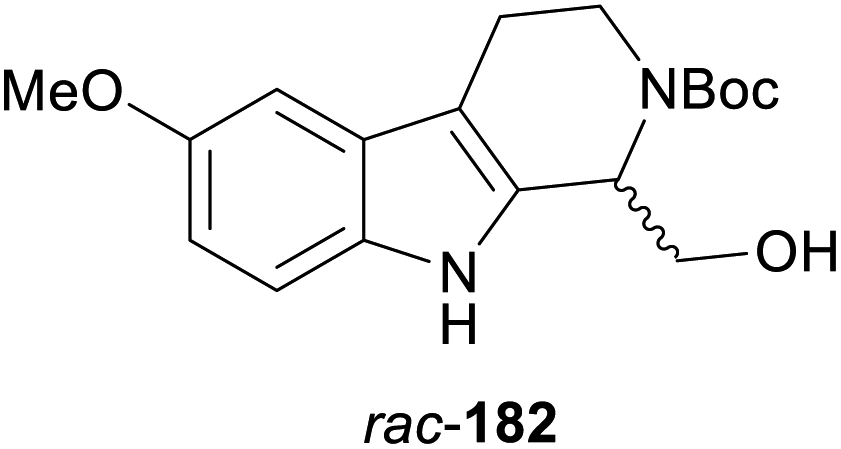
In cases where the (dynamic) kinetic resolution of alkaloids by lipase-catalysed N-acylation is not possible, it may be achieved through the acylation of side-chain OH groups. Fülöp and co-workers have previously followed this strategy in their synthesis of crispine A, which used the kinetic resolution of the primary alcohol 180 as a key step (see Section 3.1 of our earlier review, ref. 33).95 More recently, the same authors have extended this method to tetrahydroisoquinolines with shorter C1 substituents – the natural product calycotomine (178, R = H) and its non-natural homologue 179.96 Kinetic resolution reactions were carried out using CAL-B as biocatalyst, vinyl acetate as acyl donor, and toluene as solvent in both batch and flow systems. Under flow conditions, (R)-178 (R = Boc) and its acylated (S)-enantiomer could be obtained in 99% ee at 50% conversion (E >200), while the kinetic resolution of 179 required the addition of triethylamine and sodium sulfate under batch conditions to reach a practically useful enantioselectivity (E = 88; Table 2). Three hydroxymethyl-substituted tetrahydro-β-carbolines (181–183) were subjected to CAL-B-catalysed kinetic resolution under similar reaction conditions, with excellent results in all cases (Table 2).97
| Substrate | Conditions | Products | ee [%] | Ea | Ref. |
|---|---|---|---|---|---|
| a Enantioselectivity, calculated from ee of substrate and product. | |||||
 |
Toluene, 12 mM 178, 2 eq. vinyl acetate | (R)-178 | 99 | >200 | 96a |
| (S)-acetate | 99 | ||||
| Flow (80 bar, 0.1 mL min−1), 60 °C | |||||
 |
Toluene, 12 mM 179, 4 eq. vinyl acetate | (R)-179 | 30 | 88 | 96b |
| Batch, Et3N, Na2SO4, 3 °C | (S)-acetate | 97 | |||
 |
Toluene, 12 mM 180, 2 eq. vinyl acetate | (R)-180 | rac | 1 | 96a |
| Flow (80 bar, 0.1 mL min−1), 60 °C | (S)-acetate | rac | |||
 |
Toluene, 12.5 mM 181, 2 eq. Ac2O | (R)-181 | 98 | >200 | 97 |
| Batch, 60 °C | (S)-acetate | 98 | |||
 |
Toluene, 12.5 mM 182, 8 eq. Ac2O | (R)-182 | 98 | >200 | 97 |
| Batch, 60 °C | (S)-acetate | 98 | |||
 |
Toluene, 12.5 mM 183, 6 eq. Ac2O | (R)-183 | 96 | >200 | 97 |
| Batch, 60 °C | (S)-acetate | 98 | |||
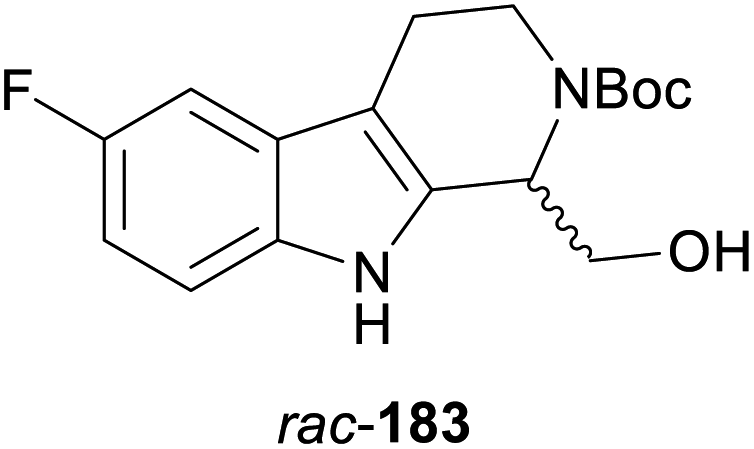
A particularly impressive example of a lipase-catalysed kinetic resolution of a primary alcohol constitutes the asymmetric key step in a recent synthesis of five Aspidosperma alkaloids:98 Lactam 184 (Scheme 22), containing a quaternary stereogenic centre in γ-position of the reactive hydroxyl group, was kinetically resolved using lipase PS37 and vinyl acetate. Although the enantioselectivity of this biotransformation was moderate (E = 22), the desired enantiomer (R)-(−)-184 could be obtained in 36% yield and >98% ee at 64% conversion. This advanced chiral intermediate was transformed into the alkaloids (+)-limaspermidine (186) and (+)-fendleridine (187) via electrophilic activation of the lactam moiety for cyclisation, followed by reduction and intramolecular hemiaminal formation, respectively. Acetylation of the secondary nitrogen atom led to (+)-acetylaspidoalbidine (188), while a palladium(II)-catalysed, amide-directed ortho-oxidation provided access to the C17-hydroxylated natural products (+)-haplocidine (189) and (+)-haplocine (190).
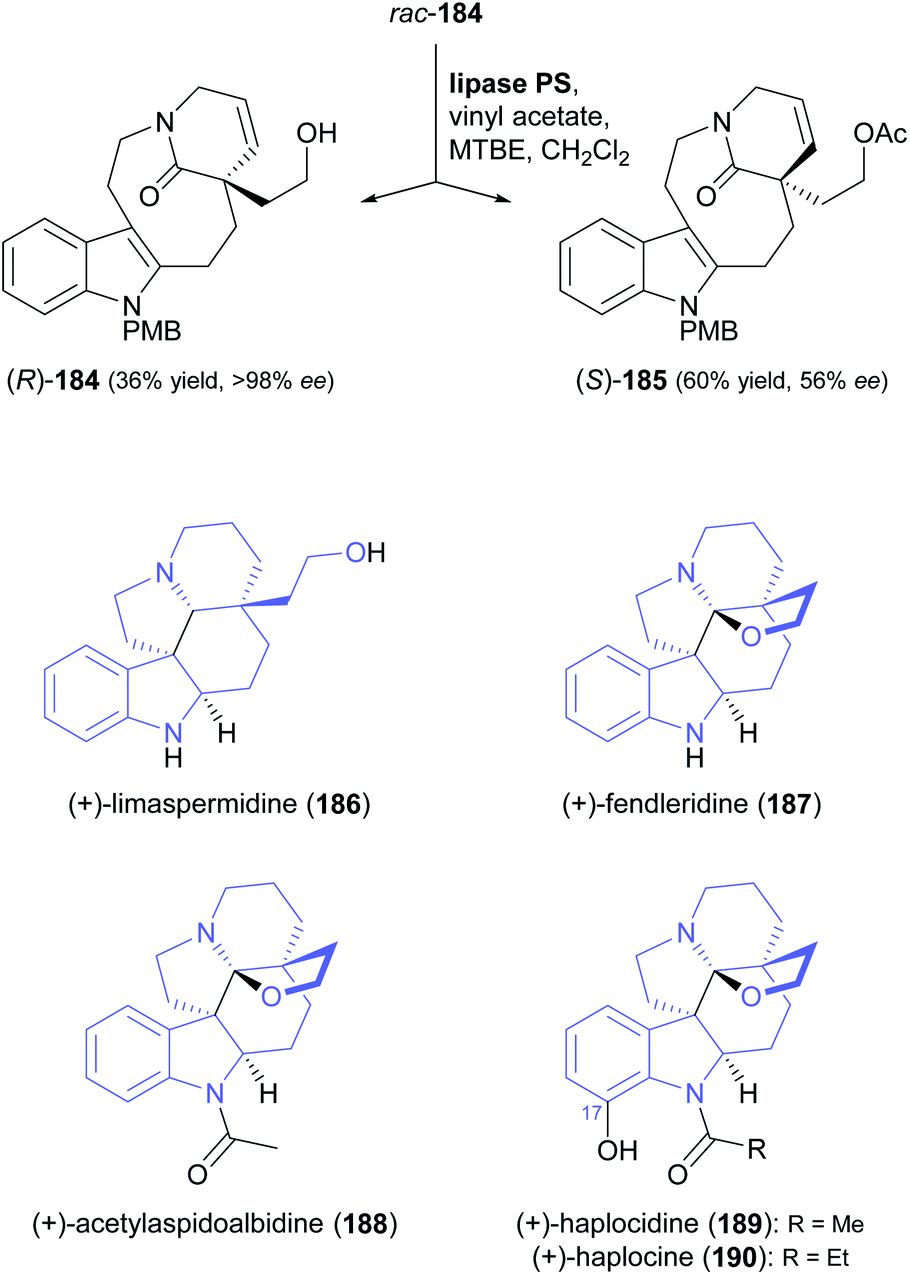 | ||
| Scheme 22 Kinetic resolution of lactam 184, and structures of alkaloids prepared from its (R)-enantiomer. | ||
The lipase-catalysed acylation of secondary alcohols was used in two recent studies for the kinetic resolution of alkaloids and for elucidating the absolute configuration of natural products derived from them. The methyl ester of dichotomine A (191, Scheme 23a), a stitchwort alkaloid, was enantioselectively acetylated by lipase QLM37 to afford the (R)-acetate in 50% yield and 96% ee along with the (S)-alcohol in 45% yield and 96% ee (E = 194).99 Saponification of the methyl ester in (S)-191 gave (−)-dichotomine A (193), while aminolysis and a copper(I)-catalysed C–N-coupling – among other steps – were used to obtain (+)-dichotomide II (194) from (R)-192. The spectral characteristics and optical rotation of both target compounds matched those reported for material isolated from the natural source,100 confirming that the two natural products 193 and 194 have opposite absolute configuration. The kinetic resolution of maleimycin (195, Scheme 23b), an antibiotic metabolite of Streptomyces showdoensis, was accomplished using lipase PS37 and vinyl acetate in THF, giving (+)-195 in 47% yield and the corresponding (−)-acetate 196 in 38% yield, both in optically pure form (ee >98%, E >200). Hydrolysis of (−)-196 by lipase PS was used to access (−)-195. Both out of a synthetic interest and to elucidate the absolute configuration of (+)- and (−)-195, the authors then converted the two enantiomers into nitrosporeusines A and B (197 and 198), two alkaloids first isolated from an Arctic actinomycete.101 Interestingly, the non-natural enantiomer of maleimycin, (−)-(S)-195, was the one that led to the naturally occurring isomers of the nitrosporeusines.
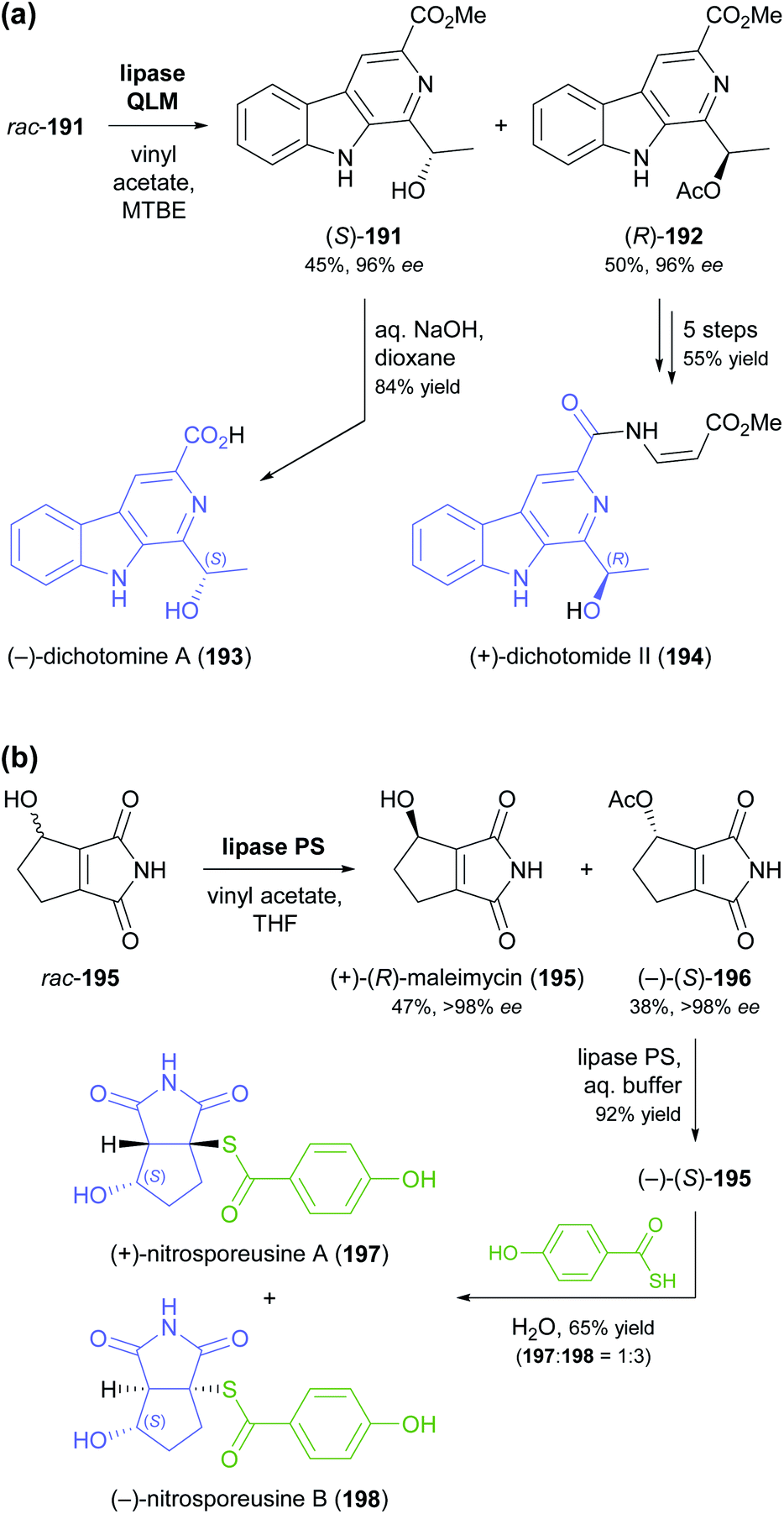 | ||
| Scheme 23 Lipase-catalysed kinetic resolution of sec-alcohol moieties in alkaloids: (a) kinetic resolution of rac-191 and its conversion into (−)-dichotomine A (193) and (+)-dichotomide II (194), (b) kinetic resolution of maleimycin (195) and its conversion into nitrosporeusines A and B (197, 198). | ||
3.2 Amine oxidases in chemo-enzymatic deracemisation systems
Monoamine oxidases (MAOs) constitute a large class of flavoenzymes that oxidise amines to the corresponding imine or iminium species. Combining this transformation with a non-stereoselective reducing agent that converts the imine back to the racemic amine establishes a ‘cyclic deracemisation’, in which the less reactive amine enantiomer accumulates (Scheme 24).102 As discussed in our previous review,33 this deracemisation concept has been applied to a broad range of amines, including several alkaloids. In particular, Nicholas Turner's group at the University of Manchester has used directed evolution to develop several highly enantioselective variants of MAO from Aspergillus niger (MAO-N), which were shown to be applicable to the asymmetric synthesis of tetrahydroisoquinoline and tetrahydro-β-carboline alkaloids.103 Variants from this protein engineering campaign were recently tested with a broader set of substrates of these structural classes, with interesting stereochemical outcome.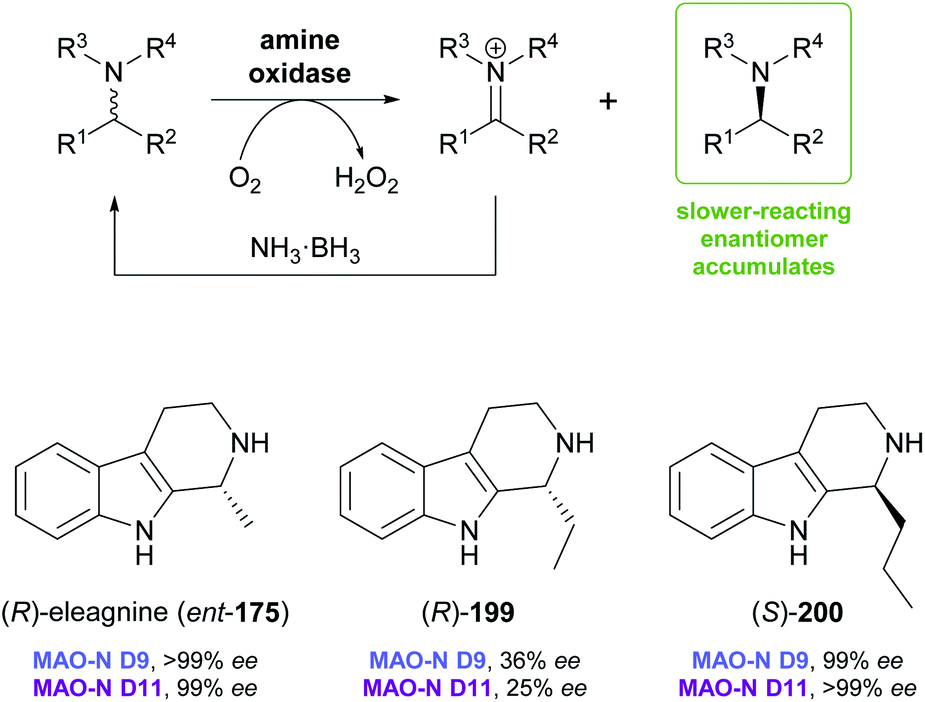 | ||
| Scheme 24 General reaction scheme for the ‘cyclic deracemisation’ of amines, and examples of tetrahydro-β-carbolines that have been deracemised using engineered variants of MAO-N. | ||
Screening MAO-N variants D9 and D11 against eleagnine (175) and 10 non-natural tetrahydro-β-carbolines revealed a substrate-dependent switch in enantioselectivity for both variants (Scheme 24).104 While deracemisation of eleagnine and its ethyl analogue (199) afforded the (R)-enantiomers (in ≥99% and ≤36% ee, respectively), derivatives with longer-chain, branched, or cyclic C1-substituents led to the (S)-configured products. This observation was rationalised based on docking simulations, which indicated two different binding modes of the substrate enantiomers in the active site. (S)-Tetrahydro-β-carbolines with sterically demanding groups at C1 failed to dock in a productive conformation, explaining the switch in selectivity with increasing substituent size. The synthetic value of the method was demonstrated by preparative-scale (0.4 mmol) deracemisation of the propyl (200), butyl, isobutyl and phenyl derivatives, leading to the (S)-enantiomers in 90–99% ee and >85% yield.
In another study, MAO-N variants D5, D9 and D11 were screened against 15 benzylisoquinolines with diverse substitution patterns, and variant D11 showed activity on eight of these compounds (Scheme 25).105 Cyclic deracemisation led to the (S)-enantiomers in all cases, consistent with the earlier example of 1-phenyl-1,2,3,4-tetrahydroisoquinoline,103 but the enantioselectivity (E-value) of the MAO-N oxidation varied between 5 and >200 depending on the substrate structure. Docking simulations were again invoked to rationalise the experimental observations, revealing substituents on the substrates' isoquinoline ring to govern the enantioselectivity through steric interactions with active-site residues. After optimisation of biocatalyst production and reaction conditions, three deracemisations were run on a preparative scale (0.5 mmol), affording the natural product (S)-reticuline (202) and two non-natural analogues in 77–85% isolated yield and excellent optical purity (>97% ee).
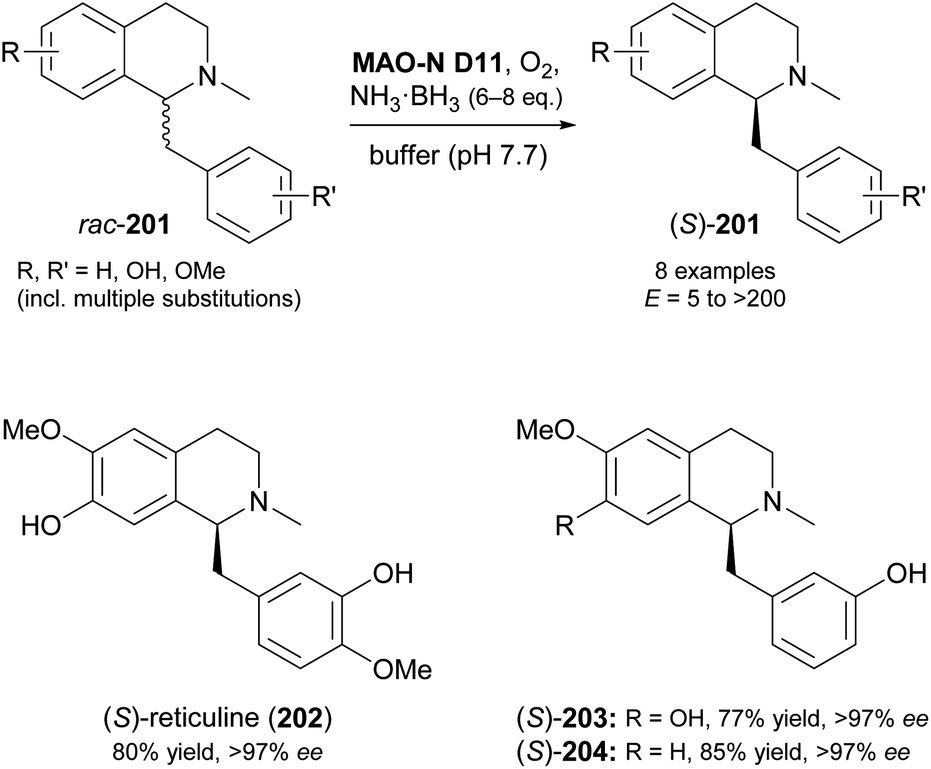 | ||
| Scheme 25 Deracemisation of benzylisoquinoline alkaloids 201 employing MAO-N variant D11 in combination with ammonia–borane as reducing agent. | ||
While these recent findings demonstrate a certain stereochemical flexibility of engineered MAO-N variants, most deracemisation reactions using this enzyme afford (R)-configured products. A general access to (S)-amines via ‘cyclic deracemisation’ requires alternative amine oxidases with an enantioselectivity complementary to MAO-N. Turner and co-workers have identified 6-hydroxy-D-nicotine oxidase (6-HDNO) from Arthrobacter nicotinovorans as a suitable candidate in this regard and have engineered the enzyme for increased substrate tolerance.106,107 Wild-type 6-HDNO enabled the stereoinversion of (R)-nicotine to its naturally occurring (S)-enantiomer (205, Fig. 3), but was only active on eight of 34 tested amines. Analysis of a previously published model108 of enzyme–substrate interaction suggested residues for mutagenesis by a CAST (combinatorial active-site saturation test)109 approach, and a solid-phase colorimetric assay developed for MAO-N110 proved suitable for the fast screening of 6-HDNO variants. The mutation of only two active-site residues, E350L and E352D, resulted in a considerable expansion of the substrate scope (19 of 34 substrates accepted), which contrasts with the case of MAO-N, where up to 11 mutations were required to achieve a similar effect. Preparative deracemisation reactions afforded the nightshade alkaloids (S)-nicotine (205) and (S)-anabasine (206), as well as the N-methyl derivative of the latter (207), in high yields (55–93%) and excellent optical purities (97% to >99% ee). Moreover, the formation of (S)-salsolidine (174) and (S)-crispine A (208; non-natural enantiomer) from the racemates was demonstrated in analytical-scale reactions.
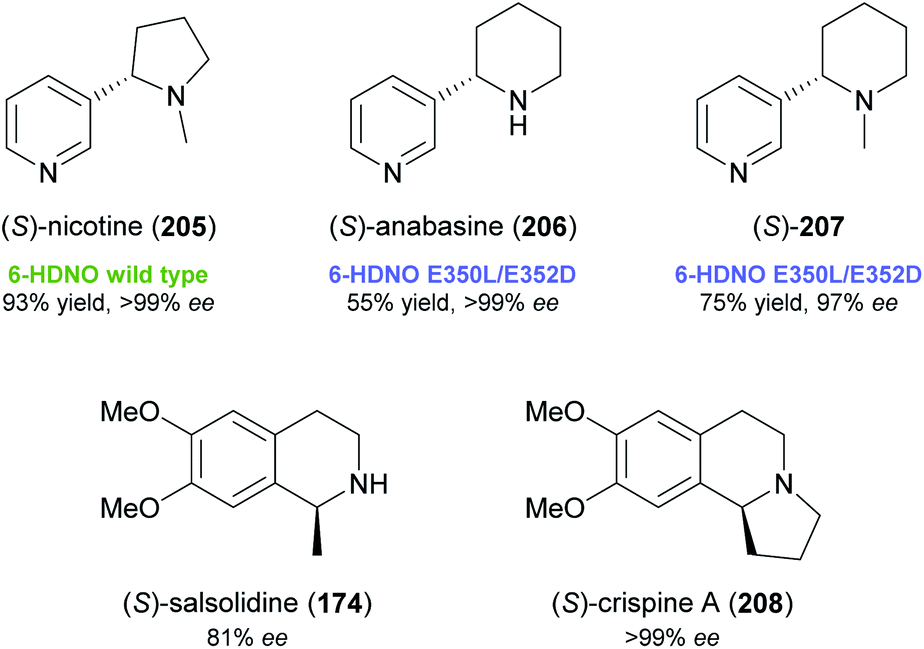 | ||
| Fig. 3 Alkaloids deracemised using 6-hydroxy-D-nicotine oxidase (6-HDNO). | ||
In a follow-up study, 6-HDNO was combined with an imine reductase (cf. Section 4) instead of ammonia-borane as reducing agent.111 The resulting two-enzyme system allowed the deracemisation of various 2-substituted pyrrolidines and piperidines, but alkaloids were not among the successful examples.
3.3 Oxidative kinetic resolution of benzylisoquinolines by berberine bridge enzyme
Berberine bridge enzyme (BBE, EC 1.21.3.3) is a flavoprotein oxidase that occurs in various alkaloid-producing plants, mainly from the families of Papaveraceae (poppy) and Fumariaceae (fumitory).112 Identified as a branch-point enzyme in the biosynthesis of benzylisoquinoline alkaloids, it catalyses the oxidative cyclisation of (S)-reticuline (209, R = OMe; Scheme 26) to (S)-scoulerine (210, R = OMe), forming the so-called ‘berberine bridge’. This transformation consumes O2 as stoichiometric oxidant and is enabled by an FAD cofactor that is bicovalently attached to the protein via a histidine and a cysteine residue.113 As discussed in our previous review,33 the efficient heterologous expression of BBE in Pichia pastoris has paved the way for detailed investigations of its structure and function, as well as for its exploitation as a biocatalyst. Racemic, non-natural benzylisoquinolines were transformed with high enantioselectivity, leading to kinetic resolution into (S)-berbines (tetrahydroprotoberberines) and the remaining (R)-configured substrates.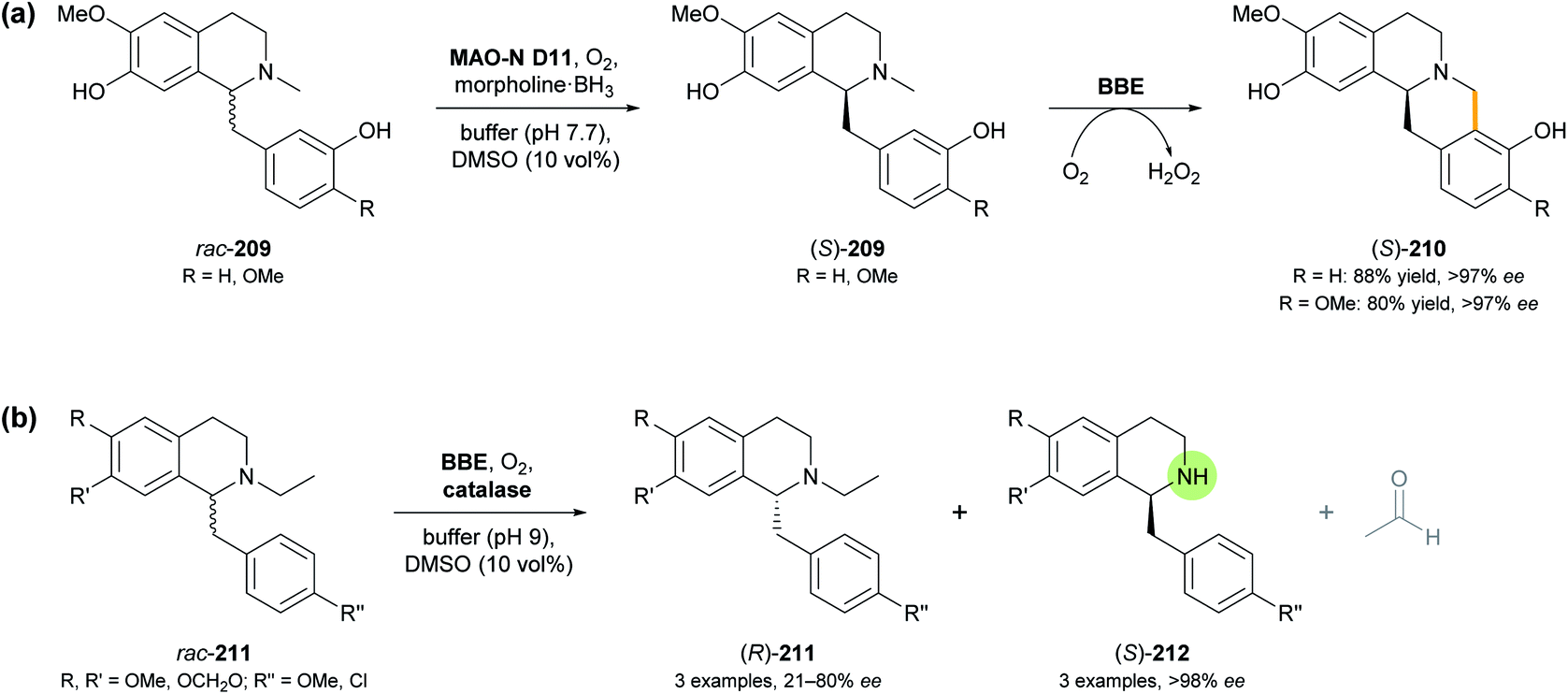 | ||
| Scheme 26 Recent applications of BBE in alkaloid synthesis: (a) transformation of rac-benzylisoquinolines 209 into (S)-berbines 210 by combining BBE with a monoamine oxidase variant (MAO-N D11) and a borane reducing agent, and (b) enantioselective dealkylation of N-ethyl-benzylisoquinolines 211 by BBE. | ||
As a continuation of this work, and to overcome the intrinsic 50% yield limit of kinetic resolution, the BBE-catalysed oxidative C–C coupling was combined with ‘cyclic deracemisation’ of the benzylisoquinoline substrate using a monoamine oxidase variant (MAO-N D11) and a borane reducing agent (cf. Section 3.2).114 Critical for the success of this concept, which makes (S)-berbines 210 accessible from rac-benzylisoquinolines 209 (Scheme 26a), was the finding that MAO-N D11 oxidises several known BBE substrates with high (R)-selectivity, while being inactive towards the BBE reaction products. Morpholine-borane was chosen as reductant for the ‘cyclic deracemisation’ step, as it was found to be more compatible with the two oxidases than the much more commonly used ammonia-borane. The MAO- and BBE-catalysed transformations could be performed in step-wise or concurrent fashion with similar success, and the latter option was chosen for preparative-scale reactions (0.5 mmol of rac-209). Thus, the natural product (S)-scoulerine (210, R = OMe) and a close structural analogue were obtained in 80% and 88% isolated yield, respectively, and in optically pure form (>97% ee).
An attempt to extend the scope of BBE-catalysed C–C coupling from N-methyl to N-ethyl-benzylisoquinolines surprisingly revealed an enantioselective N-dealkylation activity of BBE. Instead of undergoing cyclisation to berbines, several N-ethyl-benzylisoquinolines (211) with varied substitution at the two aromatic rings were oxidatively cleaved to give the secondary amines (212) and acetaldehyde (Scheme 26b).115 Like the natural BBE activity, this dealkylation was highly selective for the (S)-enantiomer of the substrates (E >100), leaving the (R)-enantiomer untouched, but due to the relatively low reaction rate, maximum conversion (50%) was not attained in this kinetic resolution process. Consequently, the products (S)-212 were formed in >98% ee, while the optical purity of the remaining substrates (R)-211 varied between 21% and 80% ee, depending on the extent of conversion. Two preparative transformations on 80–170 mg scale afforded the dealkylation products in 16–24% isolated yield and >98% ee. Interestingly, one N-methyl-benzylisoquinoline that lacks the structural prerequisites for C–C-coupling by BBE (N-methyl analogue of 211 with R, R′, R′′ = OMe) also underwent dealkylation with high conversion (47%) and enantioselectivity (E >100), while two other tested N-methyl derivatives were not transformed at all.
4 Biocatalytic reduction of imines and iminium ions
Since the publication of our last review on the role of biocatalysis in the asymmetric synthesis of alkaloids in 2013,33 a large, novel group of enzymes that show applicability for this purpose has emerged: Imine reductases (IREDs), as their name suggests, catalyse the reduction of a wide range of cyclic imines to the corresponding amines, consuming a reduced nicotinamide cofactor (usually NADPH) in the process (Scheme 27).102c,116 In addition, these enzymes are capable of catalysing the reductive amination of carbonyl compounds,117 a reactivity that has only very recently been harnessed for alkaloid synthesis.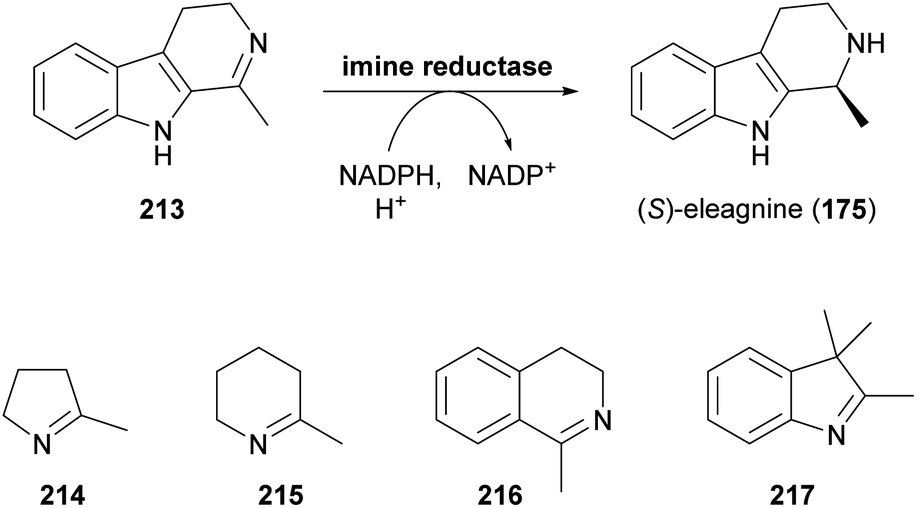 | ||
| Scheme 27 Formation of the alkaloid (S)-eleagnine (175) from the corresponding imine 213 by an imine reductase (IRED), and examples of non-alkaloid substrates of IREDs. | ||
While the enzymatic reduction of CN double bonds is common in primary metabolism, this reaction has long remained elusive in a biocatalytic context.116d Early examples have relied on whole microbial cells or cell-free extracts as catalysts, without identification of the responsible enzymes. This is also true for two studies in which the yeast Saccharomyces bayanus and a homogenisate of red Californian earthworms (Eisenia foetida) were used to obtain the (S)- and (R)-enantiomer, respectively, of the alkaloid eleagnine (175, cf. Scheme 27).118 Several non-natural tetrahydro-β-carbolines were also synthesised in these studies in 45–83% yield and 50% to >99% enantiomeric excess.
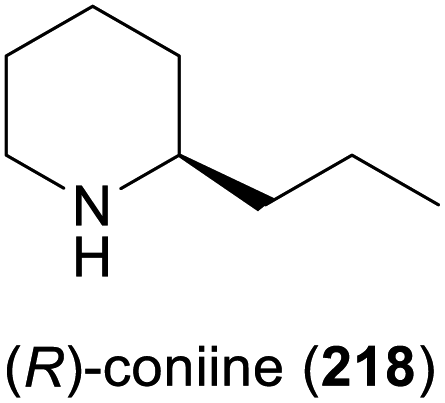
In 2010, Koichi Mitsukura and co-workers discovered imine reductase activity in two Streptomyces species, termed GF3587 and GF3546, by screening more than 680 microbial strains using 2-methyl-1-pyrroline (214, Scheme 27) as substrate.119 The responsible enzymes, which display complementary stereoselectivity, were identified soon afterwards120 and became the first characterised members of the IRED enzyme family, which now comprises more than 1400 known sequences.121 The discovery of this new enzyme class has sparked an immense interest from both academic and industrial research groups, and consequently, over the course of the last decade, many IREDs have been thoroughly studied regarding their substrate scope and stereoselectivity. The investigated substrate panels typically include small prochiral imines like the examples 214–217 shown in Scheme 27, but several IREDs have also proven useful for the asymmetric synthesis of simple alkaloids (Table 3). For instance, the hemlock neurotoxin (R)-coniine (218)122 was obtained in 90% isolated yield and >98% ee by gram-scale reduction of the corresponding imine, γ-coniceine, using the original (R)-selective IRED from Streptomyces sp. GF3587, heterologously expressed in E. coli.123 In situ regeneration of the required NADPH cofactor was achieved via the metabolism of the expression host at the expense of D-glucose. (R)-Coniine is also accessible in high conversion and enantiomeric excess using the IREDs from Nocardiopsis halophila (Nh-IRED) and Streptomyces kanamyceticus (Q1EQE0, IRED-B),124 but interestingly no IRED that would afford the (S)-enantiomer (ent-218) with good stereoselectivity has been identified yet.
| Product | IREDa | Conditions | Convb. [%] | ee [%] | Ref. |
|---|---|---|---|---|---|
| a The IRED abbreviations are adopted from the original research articles and refer to the enzymes from the following source organisms: (R)-IRED GF3587, Streptomyces sp. GF3587; (S)-IRED GF3546, Streptomyces sp. GF3546; Ao-IRED, Amycolatopsis orientalis; Bc-IRED, Bacillus cereus BAG3X2; Nh-IRED, Nocardiopsis halophila; Sa-IRED, Streptomyces aurantiacus JA 4570; Sn-IRED, Stackebrandtia nassauensis; IRED-B, Streptomyces kanamyceticus; IRED-D, Mesorhizobium sp. L2C089B000; IRED-G, Streptomyces rimosus ATCC 10970; IRED-L, Nocardia brasiliensis ATCC 700358; IRED-M, Saccharothrix espanaensis ATCC 51144.b Conversion according to GC or HPLC analysis. The values in parentheses are isolated yields of preparative-scale transformations. | |||||
 |
Nh-IRED | 5 mM imine, 0.25 mg mL−1 IRED (purified enzyme), buffer not reported, 30 °C | 90 | 82 | 124a |
| IRED-B | 50 mM imine, 2 mg mL−1 IRED (crude cell-free preparation), buffer (pH 7.5), 30 °C | 98 | >99 | 124b | |
| (R)-IRED GF3587 | 25 mM imine, whole cells expressing IRED (OD600 = 30), buffer (pH 7.0), 30 °C | >98 (90 isol.) | >98 | 123 | |
| (S)-coniine (ent-218) | Bc-IRED | 5 mM imine, 0.25 mg mL−1 IRED (purified enzyme), buffer not reported, 30 °C | 57 | 35 | 124a |
| IRED-G | 50 mM imine, 2 mg mL−1 IRED (crude cell-free preparation), buffer (pH 6.0), 30 °C | 75 | 70 | 124b | |
 |
(S)-IRED GF3546 | 5 mM imine, whole cells expressing IRED (OD600 = 30), buffer (pH 7.0), 30 °C | 92 | >98 | 125a |
| Ao-IRED (wt) | 5 mM imine, 0.4 mg mL−1 IRED (purified enzyme), buffer (pH 7.5), 30 °C | 50 | 79 | 125c | |
| IRED-M | 10 mM imine, 2 mg mL−1 IRED (crude cell-free preparation), buffer (pH 7.5), 30 °C | >99 | >99 | 125e | |
| Sn-IRED | 100 mM imine, 10 mg mL−1 IRED (crude cell-free preparation), buffer (pH 7.0), 30 °C | >99 (76 isol.) | >99 | 125d | |
| (R)-salsolidine (ent-174) | Ao-IRED (Y179A) | 5 mM imine, 0.4 mg mL−1 IRED (purified enzyme), buffer (pH 7.5), 30 °C | 15 | >99 | 125c |
 |
(S)-IRED GF3546 | 5 mM imine, whole cells expressing IRED (OD600 = 30), buffer (pH 7.0), 30 °C | >98 | >98 | 125a |
| IRED-M | 10 mM imine, 2 mg mL−1 IRED (crude cell-free preparation), buffer (pH 7.5), 30 °C | 92 | >99 | 125e | |
| Sa-IRED | 20 mM, 0.08 U mL−1 IRED (crude cell-free preparation), buffer (pH 7.0) | 91 | 99 | 125b | |
| (R)-eleagnine (ent-175) | Ao-IRED (wt) | 5 mM imine, 0.4 mg mL−1 IRED (purified enzyme), buffer (pH 7.5), 30 °C | 5 | >99 | 125c |
| IRED-D | 10 mM imine, 2 mg mL−1 IRED (crude cell-free preparation), buffer (pH 7.5), 30 °C | 27 | 78 | 125e | |
 |
(S)-IRED GF3546 | 5 mM imine, whole cells expressing IRED (OD600 = 30), buffer (pH 7.0), 30 °C | 50 | >98 | 125a |
| Ao-IRED (wt) | 5 mM imine, 0.4 mg mL−1 IRED (purified enzyme), buffer (pH 7.5), 30 °C | 15 | 79 | 125c | |
| IRED-L | 10 mM imine, 2 mg mL−1 IRED (crude cell-free preparation), buffer (pH 7.5), 30 °C | 88 | >99 | 125e | |
| (R)-leptaflorine (ent-219) | Ao-IRED (N241A) | 5 mM imine, 0.4 mg mL−1 IRED (purified enzyme), buffer (pH 7.5), 30 °C | 96 | 60 | 125c |
Among the amines most commonly prepared by IRED catalysis are the isoquinoline alkaloid (S)-salsolidine (174) and the tetrahydro-β-carbolines (S)-eleagnine (175) and (S)-leptaflorine (219; Table 3). Several enzymes that are suitable for the synthesis of these natural products have been identified in screenings,125 and 174 has been prepared on a 150 mg scale (76% yield, >99% ee) using an IRED from Stackebrandtia nassauensis (Sn-IRED).125d Access to the corresponding (R)-enantiomers is less straightforward, which reflects the general trend that (S)-selective IREDs seem to be more tolerant towards sterically demanding substrates than their (R)-selective counterparts.125e Of particular note in this context is an IRED from Amycolatopsis orientalis (Ao-IRED), whose stereoselectivity is unusually sensitive to small variations in substrate structure.125c The wild-type enzyme affords (S)-salsolidine (174) and (S)-leptaflorine (219) in moderate optical purity (79% ee in both cases), while eleagnine – which differs from 219 only by the absence of a methoxy substituent – is formed with low conversion (5%) but perfect (R)-selectivity (>99% ee). Even more surprisingly, imine 216 (Scheme 27) was reduced to the corresponding (S)-amine in 81–85% ee by fresh biocatalyst preparations, but storage of Ao-IRED at 4 °C for 24 h completely reversed its selectivity, leading to formation of the (R)-product in 98% ee. Neither extensive investigation of additives and reaction conditions nor the elucidation of the enzyme's crystal structure provided any explanations for this peculiar behaviour, but the crystal structure served as a basis for site-directed mutagenesis, which led to interesting (R)-selective enzyme variants. Ao-IRED Y179A produced optically pure (R)-salsolidine (ent-174), albeit at only 15% conversion, and variant N241A gave (R)-leptaflorine (ent-219) with near-complete conversion (96%) and 60% ee.
More complex natural products than those shown in Table 3 have rarely been targeted with IRED-based routes. One notable example is a recent study by Xudong Qu and co-workers, who engineered an IRED from Streptomyces aurantiacus (IR45) for acceptance of sterically demanding substrates and used it in combination with D-glucose dehydrogenase and an N-methyltransferase to access phenyl- and benzylisoquinoline alkaloids (Scheme 28).126 Protein engineering of the imine reductase, identified in a previous study by the same group,127 was guided by docking simulations and led to two double variants (F190L–W191F and F190M–W191F) that reduced several phenyl- and benzyl-substituted imines 220 with fair activity (kcat = 0.003–0.248 s−1) and excellent stereoselectivity (>99% ee). For preparative biotransformations, the appropriate IRED variant was either co-expressed with D-glucose dehydrogenase (GDH; for cofactor regeneration) and the third required enzyme, coclaurine N-methyltransferase (CNMT) from Coptis japonica, in a single E. coli host, or IRED and GDH were added to an E. coli culture expressing CNMT alone. In both systems, S-adenosyl methionine (SAM), the cofactor required by CNMT, was supplied and regenerated in situ by the expression host. Thus, (S)-cryptostylines I–III (223–225) and (S)-laudanosine (226) were obtained from the corresponding imines (50 mg L−1) in 93–98% isolated yield and in optically pure form (>99% ee).
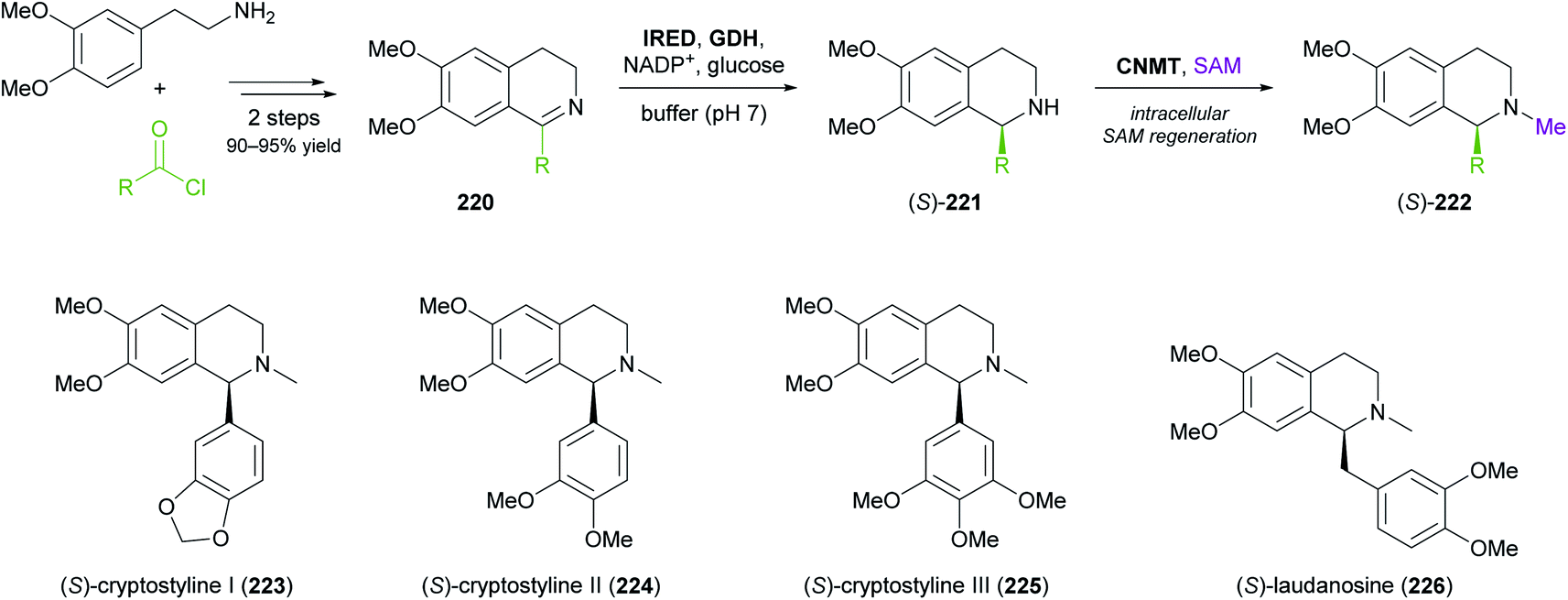 | ||
| Scheme 28 Three-enzyme cascade for the production of phenylisoquinoline and benzylisoquinoline alkaloids employing an IRED, D-glucose dehydrogenase (GDH) and coclaurine N-methyltransferase (CNMT). | ||
A very recent publication by Cosgrove, Flitsch, and co-workers marks the first time that an intermolecular reductive amination catalysed by an IRED was used in the context of alkaloid synthesis. As part of a larger study that explored multi-enzyme flow systems for the preparation of secondary amines, the authors prepared 4′-O-methylnorbelladine (230), a central biosynthetic precursor of Amaryllidaceae alkaloids,128 from tyramine (229) and isovanillyl alcohol (227) in an oxidation–amination cascade (Scheme 29).129 The oxidation of 227 to isovanillin (228) was performed by a galactose oxidase variant in a previously described130 multipoint injection reactor (MPIR). As soon as this continuous-flow biotransformation had reached a steady state, the MPIR effluent was mixed with a solution of 229, NADP+, and D-glucose, and pumped through a packed-bed reactor containing immobilised IRED and GDH. The two-step flow system produced a stable output of >90% 230 for four hours at 36 min total residence time, corresponding to a space-time yield (STY) of 2.26 g L−1 h−1.
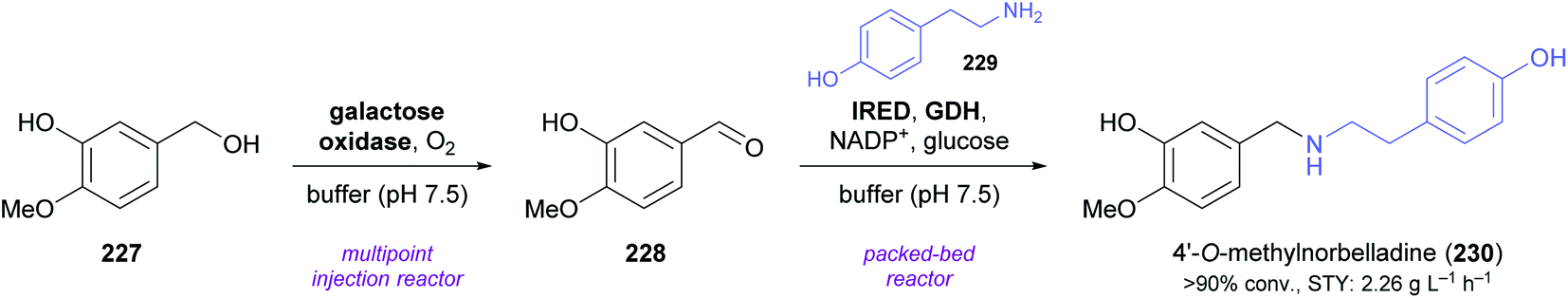 | ||
| Scheme 29 Synthesis of 4′-O-methylnorbelladine (230) in a three-enzyme continuous-flow system. | ||
Other recent examples of multi-enzyme reaction systems that involve imine reductases and bear some relevance to alkaloid synthesis are the combination of ene-reductases and IREDs for the preparation of cyclic amines with branched alkyl substituents, including non-natural coniine derivatives,131 and the combination of carboxylic acid reductases, transaminases and IREDs for the synthesis of diverse piperidines and pyrrolidines, including the natural product dihydropinidine (cf. Scheme 33a, Section 5.2).132
5 Biocatalytic asymmetric key C–C and C–N bond formation
The generation of molecular complexity from simple precursors is a key challenge in the synthesis of all natural products. In the context of alkaloid chemistry, carbon–carbon and carbon–nitrogen bond formation are of particular importance for constructing the scaffold of the target molecule. Biocatalytic methods that achieve these bond formations in stereoselective fashion hence have immense potential for enabling concise and often entirely novel synthesis routes to alkaloids. In our earlier review we have noted a shift of research focus towards such methods, and this trend has continued in recent years, as aldolases, transaminases, and Pictet–Spenglerases were employed in the preparation of a broad variety of alkaloids and non-natural alkaloid derivatives.5.1 Aldolases
Aldolases are probably the most thoroughly studied class of C–C bond forming enzymes, and they have been applied in the preparation of a vast range of saccharides and other polyhydroxylated target molecules.133 In the field of alkaloid synthesis, enzymatic aldol additions to Cbz-amino-substituted aldehydes have been widely used in combination with catalytic hydrogenation to access iminocyclitols (‘aza-sugars’),134 as discussed in our previous review.33 Many of the approaches developed in this context gave rise to recent follow-up studies, which often focus on the investigation of novel aldolases or aldolase variants, and on the preparation of non-natural derivatives of known aza-sugars. For instance, a thermostable L-rhamnulose-1-phosphate aldolase (RhuA) from Thermotoga maritima was used to prepare a variety of non-natural piperidine, pyrrolidine, and pyrrolizidine iminocyclitols.135 The high solvent tolerance of this enzyme enabled an efficient aldol reaction between the poorly water-soluble aldehyde 231 and dihydroxyacetone phosphate (DHAP) in the presence of 35% (v/v) of DMF (Scheme 30a). The resulting aldol product 232 was dephosphorylated and subjected to catalytic hydrogenation to give compounds 234 and 235, which are diastereomers of the natural aza-sugar D-fagomine, in 32% combined yield and a 53:47 ratio. This stereochemical outcome indicates that the aldol addition proceeded with high (R)-selectivity at C3 of 232, but with low selectivity at the neighbouring C4 stereocentre. Similarly, the coupling of DHAP with racemic aldehyde 236 catalysed by variant F131A of L-fuculose-1-phosphate aldolase (FucA) from E. coli yielded, after dephosphorylation, two diastereomers of aldol 237 in a 1:1 ratio (Scheme 30b).136 Catalytic hydrogenation of this diastereomeric mixture afforded the non-natural benzopyrrolizidines 238 and 239, which proved separable by chromatography and were isolated in 12% and 11% overall yield, respectively.
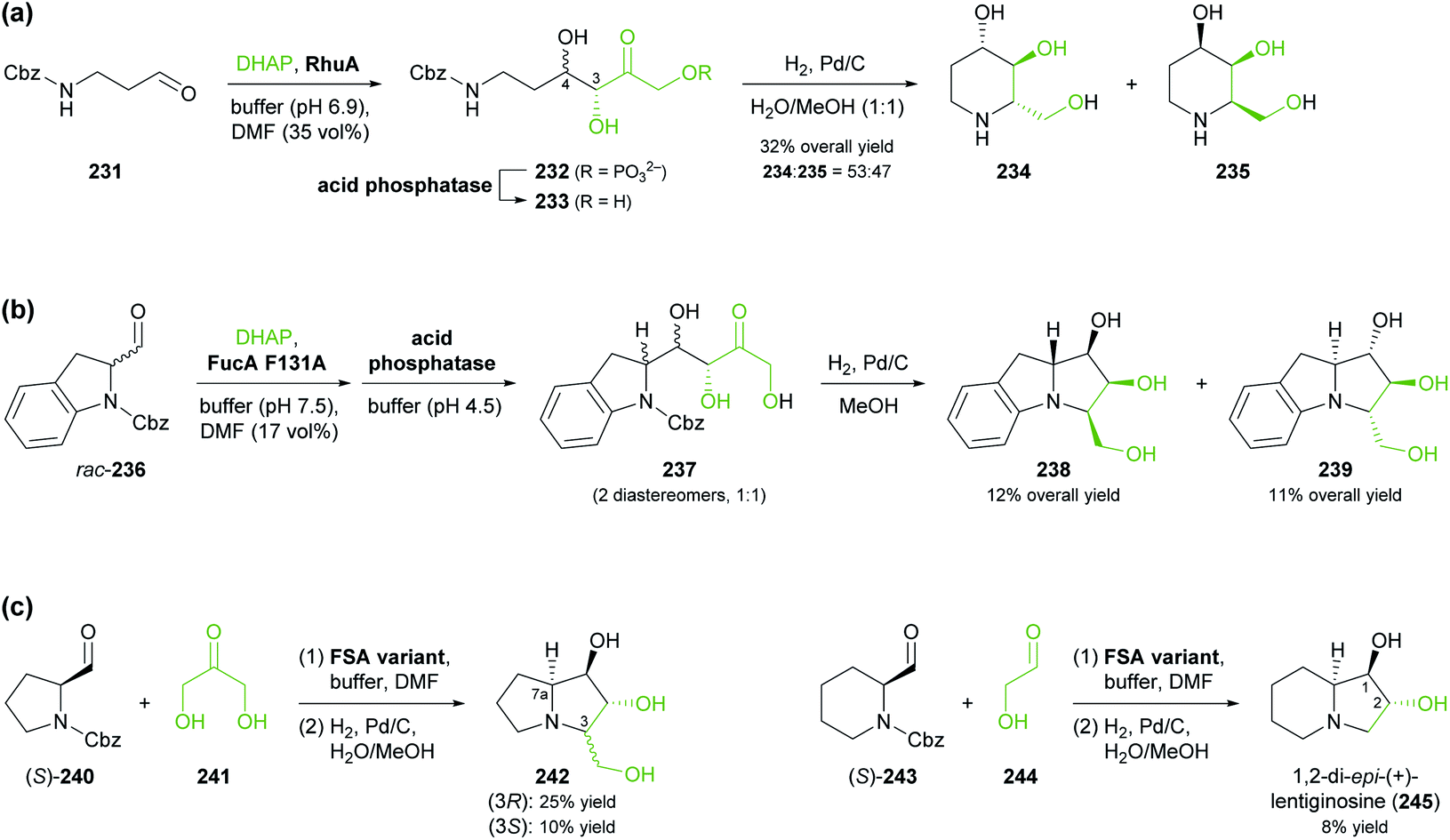 | ||
| Scheme 30 Recent applications of aldolases in the synthesis of non-natural iminocyclitols: (a) preparation of fagomine stereoisomers 234 and 235 using L-rhamnulose-1-phosphate aldolase (RhuA) from Thermotoga maritima, (b) synthesis of benzopyrrolizidines using an L-fuculose-1-phosphate aldolase (FucA) variant, and (c) preparation of (+)-hyacinthacine A2 stereoisomers 242 and 1,2-di-epi-(+)-lentiginosine (245) using variants of D-fructose-6-phosphate aldolase (FSA). | ||
In contrast to DHAP-dependent aldolases like RhuA and FucA, D-fructose-6-phosphate aldolase (FSA) from E. coli is known to accept non-phosphorylated carbonyl compounds as donor substrates.133c This offers many advantages in chemo-enzymatic syntheses, including a lower cost of the donor, no requirement for phosphatase addition, and a generally broader donor scope, which expands the range of accessible products. Pere Clapés and co-workers recently carried out extensive protein engineering of FSA with the goal to improve its acceptance of α-substituted and α-cyclic aldehydes.137 The structure-guided engineering campaign started from previously reported single and double variants of FSA and explored the effect of various active-site mutations on aldol reactions using dihydroxyacetone (241), hydroxyacetone, and glycolaldehyde (244) as donor substrates. For the latter two donors, saturation mutagenesis of position R134 proved particularly effective and led to a panel of seven 4-point and 5-point variants capable of generating no less than 47 structurally diverse iminocyclitols. Of these, the ones most closely related to natural products are 7a-epi-(+)-hyacinthacine A2 [(3R)-242] and 7a,3-di-epi-(+)-hyanthacine A2 [(3S)-242], obtained via the aldol coupling of 241 and aldehyde (S)-240, and 1,2-di-epi-(+)-lentiginosine (245), prepared from 244 and aldehyde (S)-243 (Scheme 30c).
While all the examples discussed so far rely on acceptor substrates obtained by chemical synthesis, two recent studies have established a stereodivergent access to iminocyclitols via two sequential aldolase reactions, one of which yields the acceptor substrate for the other. In the first case, a screening of glycine-utilising aldolases revealed L-serine hydroxymethyltransferase (L-SHMT) from Streptococcus thermophilus to give the threonine derivative (2S,3R)-248 in good yield (69%) and high stereoselectivity (98:2 dr) after Cbz protection, while D-threonine aldolase (D-ThrA) from Achromobacter xylosoxidans led to the (2R,3R)-diastereomer with comparable efficiency (62% yield, 95:5 dr; Scheme 31).138 Acidic hydrolysis of the dimethyl acetal in (2R,3R)-248 liberated aldehyde (2R,3R)-249, which underwent aldol coupling with DHAP, catalysed by FucA variant F131A, to yield compound 250 with full stereocontrol. The usual sequence of dephosphorylation and catalytic hydrogenation then afforded the polyhydroxylated pipecolic acid derivative 252 in good yield (29% from 249). Using epimeric (2S,3R)-248, aldolases other than FucA, and alternative donor substrates, a panel of 10 further pipecolic acid derivatives was also prepared. Moreover, both epimers of 248 could be transformed into the corresponding dihydroxyaldehydes 253 and further into (homo)iminocyclitols. For example, the aldol addition of (2R,3R)-253 and glycolaldehyde catalysed by FSA variant A129G gave – after hydrogenation – the iminocyclitol D-1-deoxyaltronojirimycin (255) in high yield (43% from 248) on multi-milligram scale.
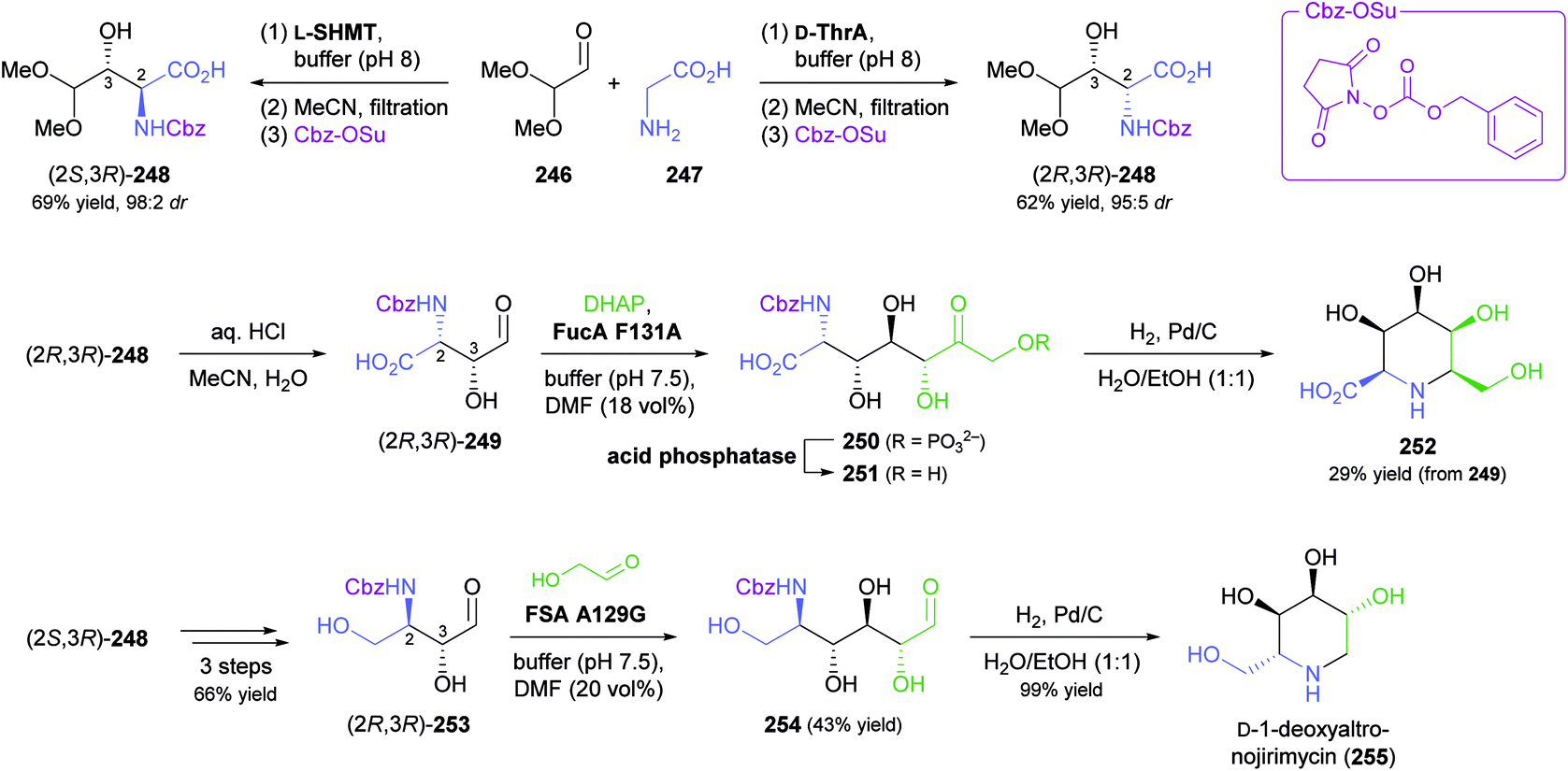 | ||
| Scheme 31 Chemo-enzymatic preparation of (2S,3R)-248 and its (2R,3R)-diastereomer using L-serine hydroxymethyltransferase (L-SHMT) and D-threonine aldolase (D-ThrA), respectively, and their further conversion into a pipecolic acid derivative (252) and the iminocyclitol D-1-deoxyaltronojirimycin (255) via a second aldol reaction. | ||
In the second study, a related ‘double aldol’ approach gave access to four stereoisomers of the polyhydroxylated pyrrolizidine alkaloid casuarine.139 The aldol addition of dihydroxyacetone (241) to aldehyde 256, catalysed either by FSA variant A129S–A165G or by RhuA, and followed by hydrogenation, Cbz protection, and oxidation, yielded aldehyde 257 and its enantiomer, as described in a previous study (Scheme 32).140 The two aldehydes 257 and ent-257 were coupled with dihydroxyacetone phosphate by FucA variant F131A with (R)-selectivity at the 2′ carbon atom, irrespective of the aldehyde stereochemistry. The neighbouring C1′ stereocentre, on the other hand, was under substrate-based stereocontrol, leading to the exclusive formation of aldol adducts 258 and 261 after dephosphorylation. The final catalytic hydrogenation step was not stereoselective, but afforded epimeric mixtures that were separable by ion-exchange chromatography. Thus, ent-3-epi-casuarine (259), ent-casuarine (260), 2-epi-casuarine (262) and 2,3-di-epi-casuarine (263) were obtained in yields of 5.3–9.0% (from 257/ent-257).
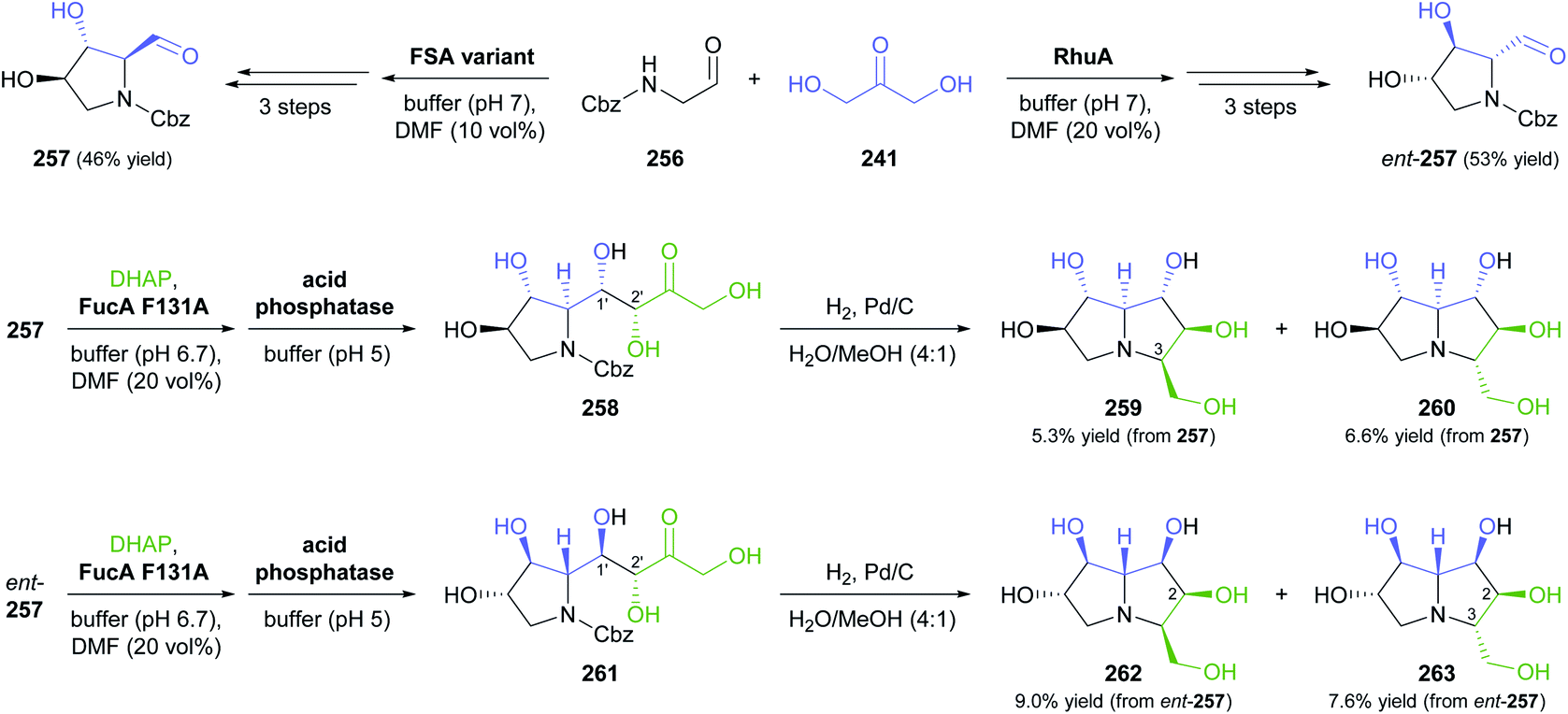 | ||
| Scheme 32 Chemo-enzymatic synthesis of ent-3-epi-casuarine (259), ent-casuarine (260), 2-epi-casuarine (262) and 2,3-di-epi-casuarine (263) via stereodivergent aldol reactions using D-fructose-6-phosphate aldolase (FSA), L-rhamnulose-1-phosphate aldolase (RhuA), and L-fuculose-1-phosphate aldolase (FucA). | ||
5.2 Transaminases
Transaminases are pyridoxal 5′-phosphate (PLP)-dependent enzymes that catalyse the reversible transfer of an amino group from an amine donor to a carbonyl acceptor. Over the course of the last decade, ω-transaminases (also referred to as amine transaminases, ATAs) have become the biocatalysts of choice for the synthesis of chiral primary amines from the corresponding prochiral ketones, a reaction with countless applications in academia and industry.141 Thanks to their broad substrate scope and their high stereoselectivity, ω-transaminases are also promising biocatalysts for the synthesis of alkaloids, and their use in this context has been significantly expanded since the publication of our previous review.33 There, we had discussed that the regioselective mono-amination of 1,5-diketones (e.g., 265, Scheme 33) by transaminases leads to chiral, cyclic imines that can be reduced with excellent cis-selectivity by catalytic hydrogenation or with moderate trans-selectivity by LiAlH4 in the presence of Et3Al, affording 2,6-disubstituted piperidine derivatives.142 More recently, imine reductases (IREDs, cf. Section 4) were identified as an alternative to chemical reducing agents for the second step in this sequence, rendering a one-pot biotransformation of 1,5-diketones into disubstituted piperidines possible. This concept was first explored by Nicholas Turner and co-workers as part of a larger study aimed at the formation of diverse N-heterocycles from dicarbonyl precursors.132 Among several 2,6-disubstituted piperidines prepared in this work, the spruce tree alkaloid (−)-dihydropinidine (267) was obtained with 23% conversion, >99% ee and 13% de, via mono-amination of diketone 265 using a commercial, (R)-selective transaminase and subsequent reduction of imine (R)-266 using (R)-IRED GF3587 (Scheme 33a; cf. Section 4).143 Employing a commercial (S)-transaminase and the same IRED, the opposite enantiomer, ent-267, was formed with quantitative conversion and excellent stereoselectivity (>98% ee, 95% de). In both cases an established pyruvate removal system (LDH, GDH, glucose) was used to displace the transamination equilibrium, and the two enzymatic steps were performed sequentially in the same pot, adding the IRED after completion of the transaminase reaction. An even more integrated, multi-enzymatic approach to the same target molecule was later developed by our group (Scheme 33b).124b In this work, the transamination of 265 was performed with isopropylamine as the amine donor, eliminating the need for auxiliary enzymes, and reduction of imine (R)-266 by an IRED from Nocardiopsis alba was carried out concurrently in the same reaction vessel. The synthesis of the substrate required for the biotransformation, diketone 265, was also modified: The previously used Grignard reaction of lactone 264 was replaced with a sequence of Lewis acid-catalysed Michael addition of compounds 268 and 269, ester hydrolysis by pig liver esterase (PLE), and acid-promoted decarboxylation. None of these steps required chromatographic purification of intermediates, making isolation of the target alkaloid by a single hydrochloride precipitation possible. Thus, 267·HCl was obtained in 57% overall yield (from 269) and stereoisomerically pure form (>99% ee and de) on a 5 gram scale. Similarly, 265 was transformed into ent-267·HCl (71% yield, >99% ee, >99% de) by an (S)-transaminase and an IRED from Mesorhizobium sp. on a 1 g scale.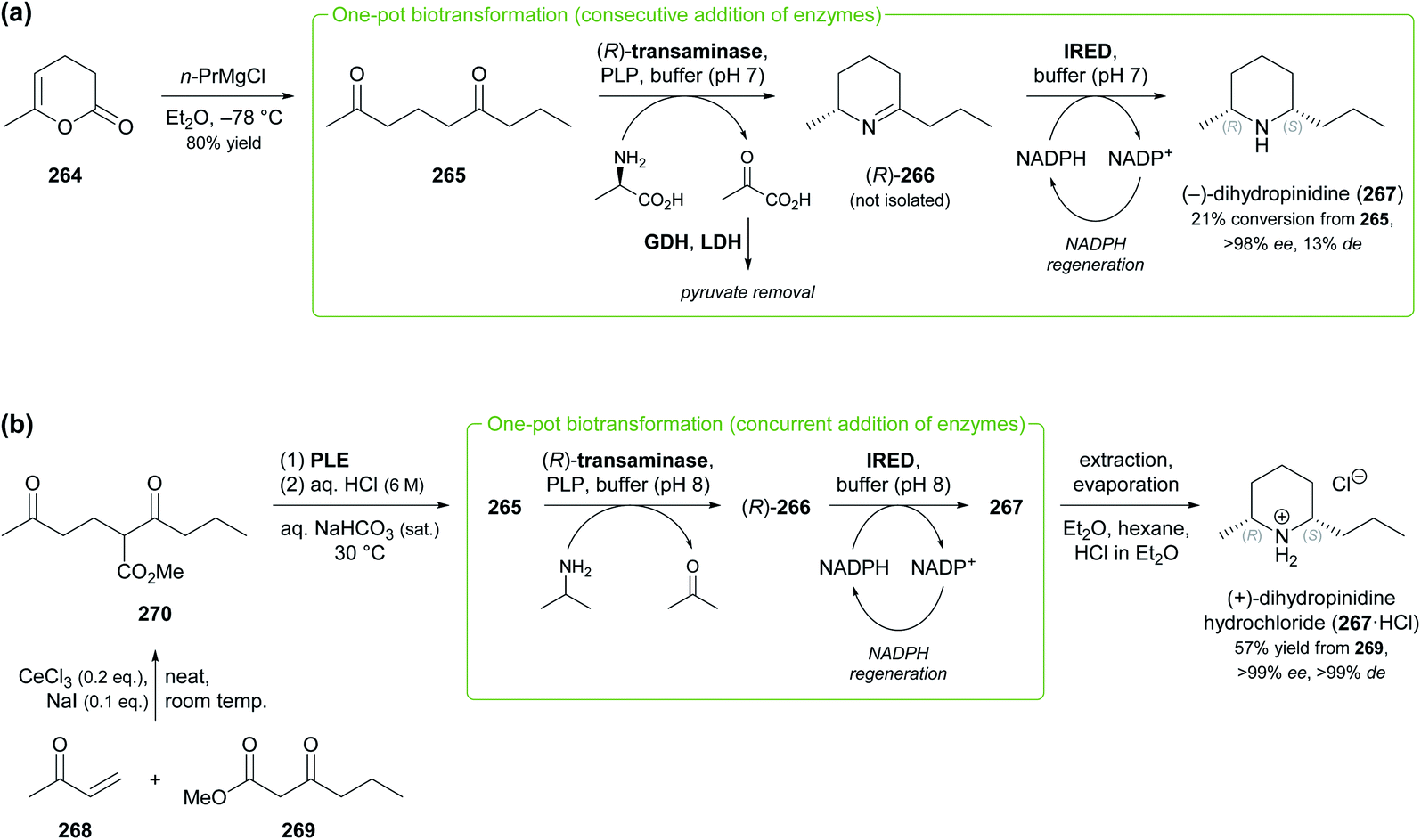 | ||
| Scheme 33 Chemo-enzymatic synthesis of dihydropinidine (267): (a) combination of transaminase and IRED biocatalysis in a one-pot system with consecutive addition of enzymes, (b) combination of transaminase and IRED biocatalysis in a concurrent one-pot system. Note that 267 and its hydrochloride (267·HCl) have opposite signs of optical rotation. | ||
The original concept of combining a regioselective, enzymatic transamination with a diastereoselective, chemical reduction was recently extended to a triketone, thereby establishing access to both enantiomers of the ant venom alkaloid xenovenine (274, Scheme 34).144 Triketone 271, obtained in two steps from 2-heptylfuran, was aminated at the least sterically hindered of the three carbonyl groups by various transaminases, giving imine (R)- or (S)-272 with optical purities ranging from 54% to >99% ee. The use of n-heptane (30% v/v) and DMSO (5% v/v) as co-solvents was required to achieve high conversions of the poorly water-soluble substrate 271. Although imine 272 could be reduced to the target alkaloid by NaCNBH3, this reaction gave low yield, as large amounts of pyrrole 273 were formed as side product. The authors, therefore, decided to transform 272 into 273 quantitatively by treatment with acetic acid in methanol, followed by catalytic hydrogenation with camphorsulfonic acid (CSA) as additive. Despite attempts at optimisation, the hydrogenation could not be rendered completely stereoselective, but the obtained diastereomeric mixtures proved separable by chromatography. Performing the chemo-enzymatic sequence on 100 mg scale gave (+)-xenovenine (274) in 30% overall yield (from 271) and its enantiomer in 48% overall yield, both as single stereoisomers.
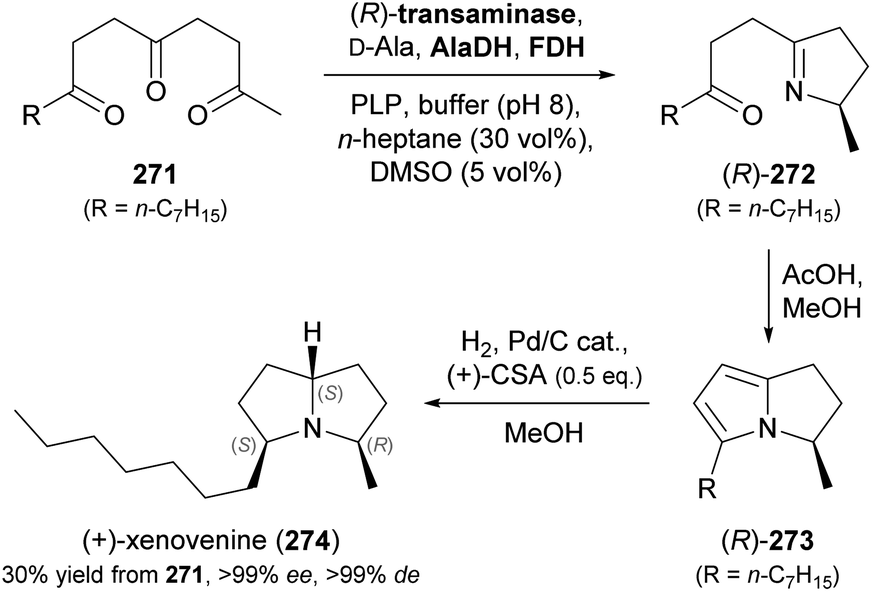 | ||
| Scheme 34 Chemo-enzymatic synthesis of (+)-xenovenine (274) via regioselective mono-amination of triketone 271, acid-catalysed pyrrole formation, and acid-promoted catalytic hydrogenation. | ||
In another elegant combination of enzymatic transamination with follow-up chemistry, the spruce alkaloid (−)-pinidinone (277) was obtained in essentially a single step from the diketone 275 (Scheme 35a).145 A commercial, (R)-selective transaminase converted 275 into the amine (R)-276, which underwent a spontaneous, intramolecular aza-Michael reaction to give 277 as a mixture of C2 epimers. This mixture equilibrated to the more stable cis-configuration by simple stirring in methanol for 24 h, affording (−)-pinidinone in 86% yield and optically pure form (>99% ee and de) on a half-gram scale. Several non-natural derivatives – including the opposite enantiomer of 277 – were obtained in analogous reactions. Notably, the transamination only required ≤2 equivalents of the amine donor, isopropylamine, as the Michael reaction withdraws amine 276 from the equilibrium. Since the authors did not expect the amination of 275 to be fully regioselective for the C8 carbonyl group, they independently synthesised the allylic amine (R)-278 and incubated it with the transaminase in the absence of amine donor. Complete conversion of 278 into 277 was observed, which demonstrates that the amine group can be transposed within the molecule. A recent follow-up study explored the extension of the transamination/aza-Michael sequence to yne-diones 279, giving β-enaminones (Z)-280 as products (Scheme 35b).146 Several examples of the general structure 280 were obtained in good yields (up to 95%) and optical purities ranging from 83% to >99% ee, with access to both enantiomeric forms through the use of stereocomplementary transaminases. The products were shown to be amenable to various annulation reactions, which afforded bicyclic compounds such as 281 and 282.
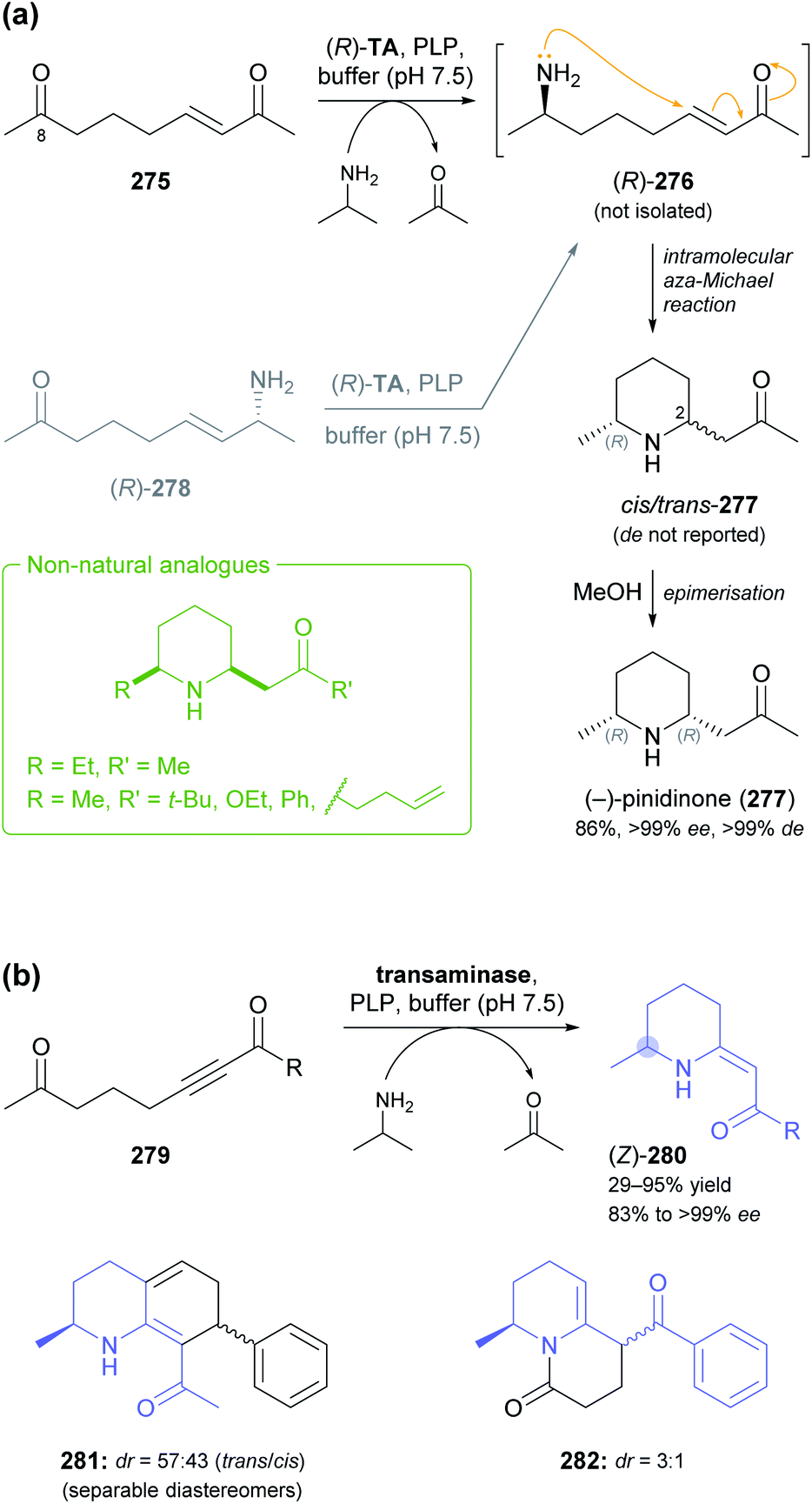 | ||
| Scheme 35 Reductive amination of ene-diones and yne-diones by transaminases (TAs) triggers an intramolecular aza-Michael reaction: (a) asymmetric synthesis of (−)-pinidinone (277) and several non-natural analogues, (b) preparation of (Z)-β-enaminones 280 and examples of annulation products that can be obtained from these building blocks. | ||
Stereocomplementary transaminases have also been used to access all four stereoisomers of the Ephedra constituents147 norephedrine and norpseudoephedrine via different two-enzyme sequences. The first strategy, developed in 2013 by Dörte Rother and co-workers, relies on the thiamine diphosphate (ThDP)-dependent carboligase acetohydroxyacid synthase I (AHAS-I) from E. coli for formation of the hydroxyketone (R)-285 from benzaldehyde (283) and pyruvic acid (284, Scheme 36a) as the first step.148 Subsequently, the stereocomplementary transaminases from Chromobacterium violaceum (Cv-TA) and Aspergillus terreus (At-TA) convert (R)-285 into (−)-norephedrine [(1R,2S)-286] or (−)-norpseudoephedrine [(1R,2R)-286], using L- or D-alanine as amine donor. The two steps can be combined in one pot, whereby a consecutive addition of the enzymes is advantageous, as it avoids the undesired transamination of benzaldehyde. Thus, (1R,2S)-286 was formed in 78% conversion and (1R,2R)-286 in 96% conversion with excellent stereoselectivity in both cases (>99% ee, >98% de). Since the transaminase generates pyruvic acid as co-product, which is one of the two substrates for AHAS-I, the carboligation can be re-initiated by adding benzaldehyde after completion of the transamination step. This establishes what the authors call a “recycling cascade”, but the preparative value of this option was found to be limited. In a follow-up study, the same authors investigated a stereocomplementary carboligase and alternative reaction setups with the aim of extending the scope of the method to the remaining two isomers of 286 (Scheme 36b).149 The E469G variant of a pyruvate decarboxylase from Acetobacter pasteurianus (Ap-PDC-E469G) generated (S)-285 with near-complete conversion (95%) but only moderate ee (70%). Consequently, since the used transaminases accept both enantiomers of 285 as substrate, the final products (1S,2R)-286 and (1S,2S)-286 were obtained with quantitative conversion and >99% ee but only 70% diastereomeric excess. To overcome this limitation, the authors turned towards an entirely different approach that relies on alcohol dehydrogenases in addition to transaminases for the formation of the four 286 stereoisomers from diketone 288 (Scheme 36c).149 In this setup, the transamination of 288 using Cv-TA or At-TA as catalyst and 1-phenylethylamine as amine donor first leads to the (S)- and (R)-enantiomer, respectively, of amino ketone 289. Thereafter, the stereocomplementary alcohol dehydrogenases from Lactobacillus brevis (Lb-ADH) and Ralstonia sp. (R-ADH) were employed in combination with formate dehydrogenase (FDH) for cofactor regeneration to reduce 289 to the target compound 286. Inactivation of the transaminase by pH shift before ADH addition was necessary to avoid an undesired side reaction, the formation of 1-phenylpropane-1,2-diol. In this way, (1S,2S)-286 was generated in >98% ee and >99% de at 80% conversion, and an alternative entry to (1R,2S)-286 was also established (82% conv., >99% ee, 97% de). Generation of the (2R)-isomers by this approach was less successful: (1R,2R)-286 was formed in quantitative conversion but only 55% de by At-TA and R-ADH, while the combination of At-TA and Lb-ADH did not afford the expected (1S,2R)-product at all, producing (1S,2S)-286 instead (40% conv., >99% ee, 60% de). The authors propose an isomerisation of intermediate 289 as a possible explanation for this unexpected result.
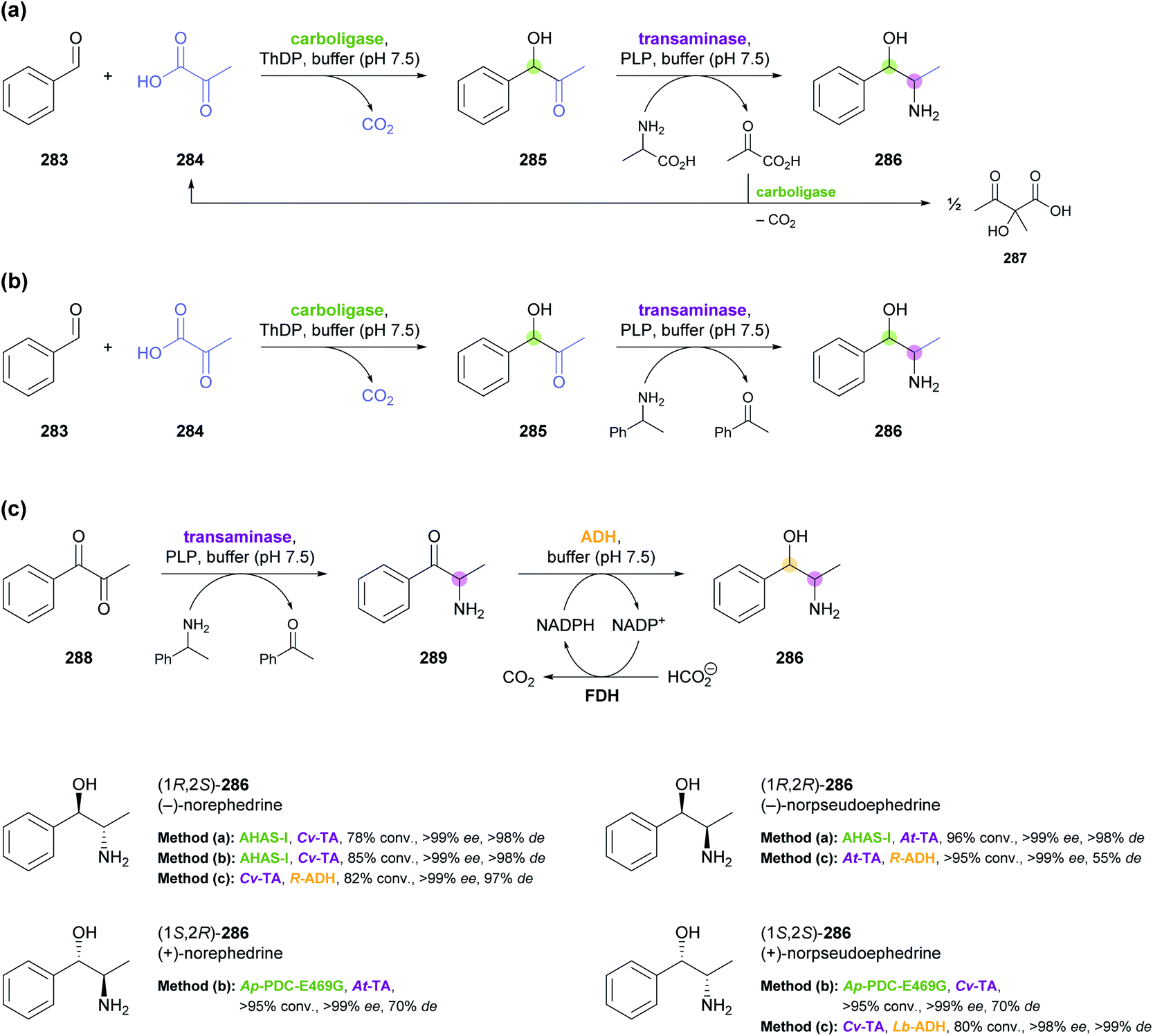 | ||
| Scheme 36 Biocatalytic two-step strategies for the synthesis of all four stereoisomers of nor(pseudo)ephedrine (286): (a) combination of carboligation and transamination, using L- or D-alanine as amine donor in a “recycling cascade”, (b) combination of carboligation and transamination, using (R)- or (S)-1-phenylethylamine as amine donor, and (c) combination of transamination and ketone reduction. Coloured circles represent stereogenic centres controlled by the enzyme shown in the same colour. For explanation of the enzyme abbreviations, see the main text. | ||
Unfortunately, no preparative-scale biotransformations were conducted in either of these studies, but the synthesis of (−)-norephedrine [(1R,2S)-286] by the original carboligase–transaminase approach on a half-gram scale was independently reported by Yijun Chen and co-workers.150 They employed an (R)-selective pyruvate decarboxylase from Saccharomyces cerevisiae and an (S)-selective transaminase from Vibrio fluvialis as biocatalysts and obtained stereoisomerically pure (−)-norephedrine in 57% yield after column chromatography. In another independent report, AHAS-I and Cv-TA were used in the form of two individual E. coli whole-cell preparations to generate (1R,2S)-286 with 62% conversion, excellent stereoselectivity (>99% ee and de), and good reusability of the biocatalyst.151
An even more elaborate multi-enzymatic approach to all four stereoisomers of 286 was recently developed by Francesco Mutti and co-workers.152 Their synthetic sequence starts from β-methylstyrene (290), which was oxidised by a flavin-dependent styrene monooxygenase (SMO), used in combination with formate dehydrogenase (FDH) for NADH regeneration, to give the corresponding epoxide 291 (Scheme 37). This biotransformation proceeded with high (S)-stereoselectivity at the benzylic carbon atom (C1),153 while the configuration at C2 depended on the double bond geometry of 290: the (E)-isomer led to (1S,2S)-291, while the (Z)-isomer was converted into (1S,2R)-291. The epoxide was not isolated but directly transformed into the corresponding diol 292 by epoxide hydrolases from Sphingomonas sp. HXN-200 (Sp-EH) or Solanum tuberosum (potato; St-EH), thereby achieving further stereodivergence. The former enzyme catalysed water attack at C2 of the epoxide, resulting in retention of the C1 stereocentre and consequently to the formation of (1S,2R)-292 or (1S,2S)-292, depending on the C2 stereochemistry of 291. In contrast, epoxide hydrolysis by St-EH proceeded with attack at the benzylic carbon atom (C1), which led to stereochemical inversion at this position and afforded the (1R,2S)- or (1R,2R)-isomer of diol 292. The two biotransformations were carried out concurrently in one pot, using equal volumes of aqueous buffer and n-heptane as reaction medium. Thus, all four isomers of diol 292 could be accessed in good isolated yields (78–85%) and high optical purity (91% to >99% ee, >99% de) by proper choice of the starting material [(E)- or (Z)-290] and the epoxide hydrolase.152a In a separate reaction step, the different stereoisomers of 292 were converted into nor(pseudo)ephedrines 286 by a redox-neutral biocatalytic network154 comprised of an alcohol dehydrogenase (ADH), a transaminase, and L-alanine dehydrogenase (AlaDH).152b Since the stereoinformation at C2 of the diol is lost in this process, each isomer of 286 can in principle be accessed from two diastereomers of 292, but not all of the resulting cascade biotransformations proved equally effective. The best results were obtained when oxidation of (1R,2R)-292 by ADH from Leifsonia sp. or oxidation of (1S,2R)-292 by ADH from Bacillus subtilis was combined with transamination using enzymes from Aspergillus terreus (R-selective), Bacillus megaterium (S-selective) or Chromobacterium violaceum (S-selective). All four stereoisomers of 286 could be generated with high conversions (76–95%) and excellent optical purities (>99% ee, 92% to >99% de), whereby the authors ascribe the small limitations in de observed for the (1S)-products to an imperfect ee of diol (1S,2R)-292. For two representative cases, the entire sequence from β-methylstyrene (290) to 286 was carried out on a preparative scale (>100 mg), affording (+)-norpseudoephedrine (>99% ee, 94% de) and (−)-norephedrine (>99% ee and de) in isolated yields of 71% and 53%, respectively.
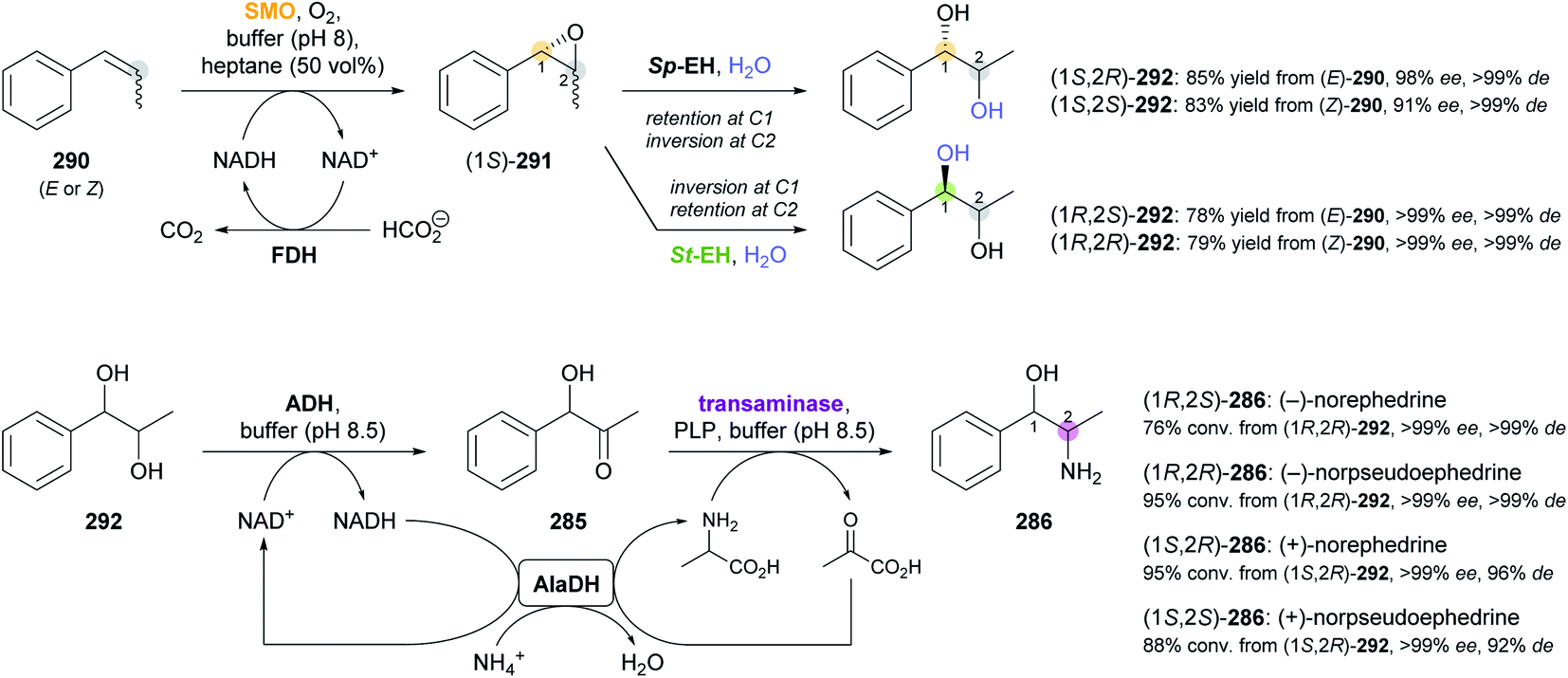 | ||
| Scheme 37 Asymmetric synthesis of all four stereoisomers of nor(pseudo)ephedrine (286) from β-methylstyrene (290) using a total of six enzymes in two biocatalytic one-pot reactions. Coloured circles represent stereogenic centres controlled by the enzyme shown in the same colour. For explanation of the enzyme abbreviations, see the main text. | ||
While the main strength of transaminases is the stereoselective generation of α-chiral primary amines from prochiral ketones, as used in all examples discussed so far, the reverse reaction, i.e., the deamination of amines, has also found application in alkaloid synthesis. A putrescine transaminase from E. coli was used to form the imines 1-pyrroline (294a) and 1-piperideine (294b) from the corresponding linear diamines 293 and pyruvic acid as amine group acceptor (Scheme 38a).155 The hydrolysis of β-ketoesters 295 by lipase B from Candida antarctica was carried out concurrently in the same reaction vessel and generated β-ketoacids 296, which combined with the imines 294 in a non-enzymatic, decarboxylative Mannich reaction that afforded racemic aminoketones 297 as final products. Biotransformations on 1.5 mmol scale gave the alkaloids ruspolinone (298), norsedaminone (299), hygrine (300), and norhygrine (301) in yields of 50–75%. Moreover, the formation of pelletierine (302) was confirmed analytically, but the compound could not be isolated in pure form. In a related approach, imine 294a was formed by putrescine transaminase and coupled with two molecules of aldehyde 303 to give the antibacterial and antifungal Ficus constituent ficuseptine (304) in 50% isolated yield (Scheme 38b).156
 | ||
| Scheme 38 Use of putrescine transaminase in alkaloid synthesis: (a) biocatalytic formation of cyclic imines 294 from linear diamines 293 and decarboxylative Mannich reaction with in situ generated β-ketoacids 296 affords racemic pyrrolidine and piperidine alkaloids 297. (b) 1-Pyrroline (294a) formed by transaminase catalysis reacts with two molecules of aldehyde 303 to give the 2,3-dihydroindolizinium alkaloid ficuseptine (304). | ||
Additional examples of the use of transaminases in alkaloid synthesis, encompassing both amination and deamination reactions, come in the form of multi-enzyme sequences centred on Pictet–Spenglerases. These approaches are discussed in the next section.
5.3 Pictet–Spenglerases
In our previous review from 2013, we described the Pictet–Spenglerases as an emerging class of biocatalysts with vast application potential in the asymmetric synthesis of alkaloids.33 The progress achieved since then, however, has even exceeded our expectations. Large efforts were made to further investigate the substrate scope of the two established Pictet–Spenglerases, norcoclaurine synthase (NCS) and strictosidine synthase (STR), and to engineer variants with improved activities, which was often made possible by advancements in the mechanistic understanding of these enzymes.157 In particular, the non-natural substrate scope of NCS was expanded greatly, and novel synthetic approaches to natural alkaloids using NCS and STR were developed.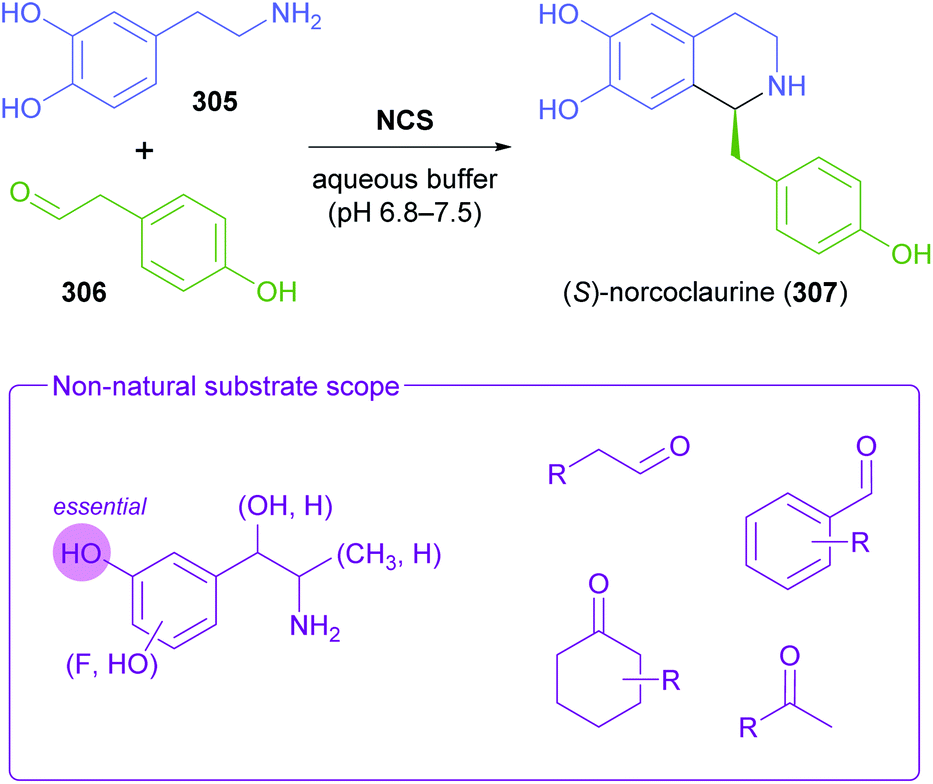 | ||
| Scheme 39 Natural reaction and non-natural substrate scope of norcoclaurine synthase (NCS). | ||
Early investigations into the substrate scope of NCS, carried out using the enzymes from Thalictrum flavum (Tf-NCS) and Coptis japonica (Cj-NCS), have found the meta-hydroxyphenethylamine core of dopamine to be a strict structural requirement, in line with the currently accepted mechanism of NCS catalysis.159 This leaves opportunities for varied substitution in other positions of the aromatic ring and in the aliphatic side chain, some of which were explored in recent studies. Dörte Rother and co-workers used the carboligation–transamination sequence developed in earlier work by their group (cf. Scheme 36c, Section 5.2) to generate the amino alcohol metaraminol (310) with excellent stereocontrol (>99% ee, 97% de; Scheme 40a).160 This intermediate was then subjected to Pictet–Spengler reactions catalysed either by the A79I variant of NCS from Thalictrum flavum or by phosphate buffer. In the first case, using phenylacetaldehyde (311) as carbonyl substrate, the reaction delivered the benzylisoquinoline (1S,3S,4R)-312 with 88% overall conversion and >97% stereoisomeric purity thanks to the reliable (1S)-selectivity of norcoclaurine synthase. The phosphate-catalysed reaction, on the other hand, requires substrate-based stereocontrol, which was found to be limited (30–89% de) with 311 and several aromatic aldehydes. 2-Bromobenzaldehyde (313), however, underwent the Pictet–Spengler cyclisation to (1S,3S,4R)-314 with 77% overall conversion and complete diastereoselectivity. Notably, the (1S)-configuration of this compound reflects a switch in Cahn–Ingold–Prelog priorities, while the stereoselectivity of the reaction is actually opposite to that of the NCS biotransformation.
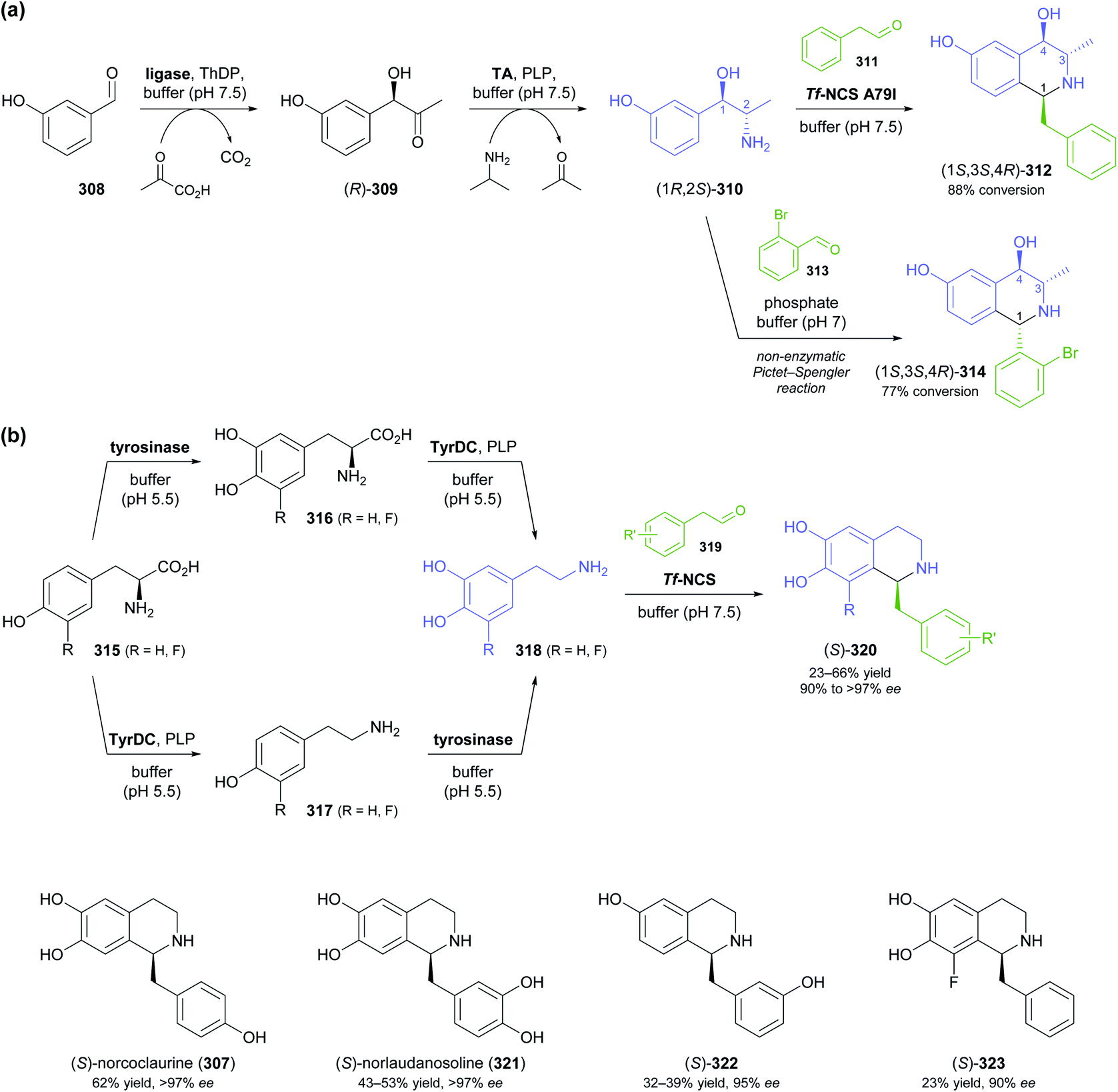 | ||
| Scheme 40 In situ formation of amine substrates for norcoclaurine synthase: (a) combination of biocatalytic carboligation and transamination leads to metaraminol (310), which can undergo enzymatic or phosphate-catalysed Pictet–Spengler reactions. (b) Tyrosinase and tyrosine decarboxylase (TyrDC) can be used to prepare dopamine and its derivatives from the corresponding amino acids. | ||
Ward, Hailes and co-workers investigated the formation of dopamine and close structural analogues 318 from the corresponding tyrosine derivatives 315 by the combined action of tyrosinases and tyrosine decarboxylases (TyrDCs) from various microbial sources (Scheme 40b).161 The best-performing enzymes showed a relatively broad substrate tolerance, and biotransformations that use both enzymes concurrently are therefore likely to convert 315 into 318 via two competing pathways with 316 and 317 as the respective intermediates. Coupling of these hydroxylation and decarboxylation reactions with Tf-NCS-catalysed Pictet–Spengler cyclisation established multi-enzyme cascades, whereby the authors followed what they call a “mix and match” strategy: The carbonyl substrates for NCS were either added to the biotransformation or were generated in situ from amines 318 by a transaminase (cf. Scheme 41c below). Moreover, tyrosinase and TyrDC were used either in combination or individually, hence giving rise to a variety of (S)-benzylisoquinoline products. These include the natural alkaloids (S)-norcoclaurine (307) and (S)-norlaudanosoline (321), obtained as single enantiomers (>97% ee) in isolated yields of 62% (307) and 43–53% (321, depending on the batch size). Several non-natural derivatives (e.g., 322 and 323) were also prepared, with yields ranging from 23% to 66% and ee values from 75% to >97%.
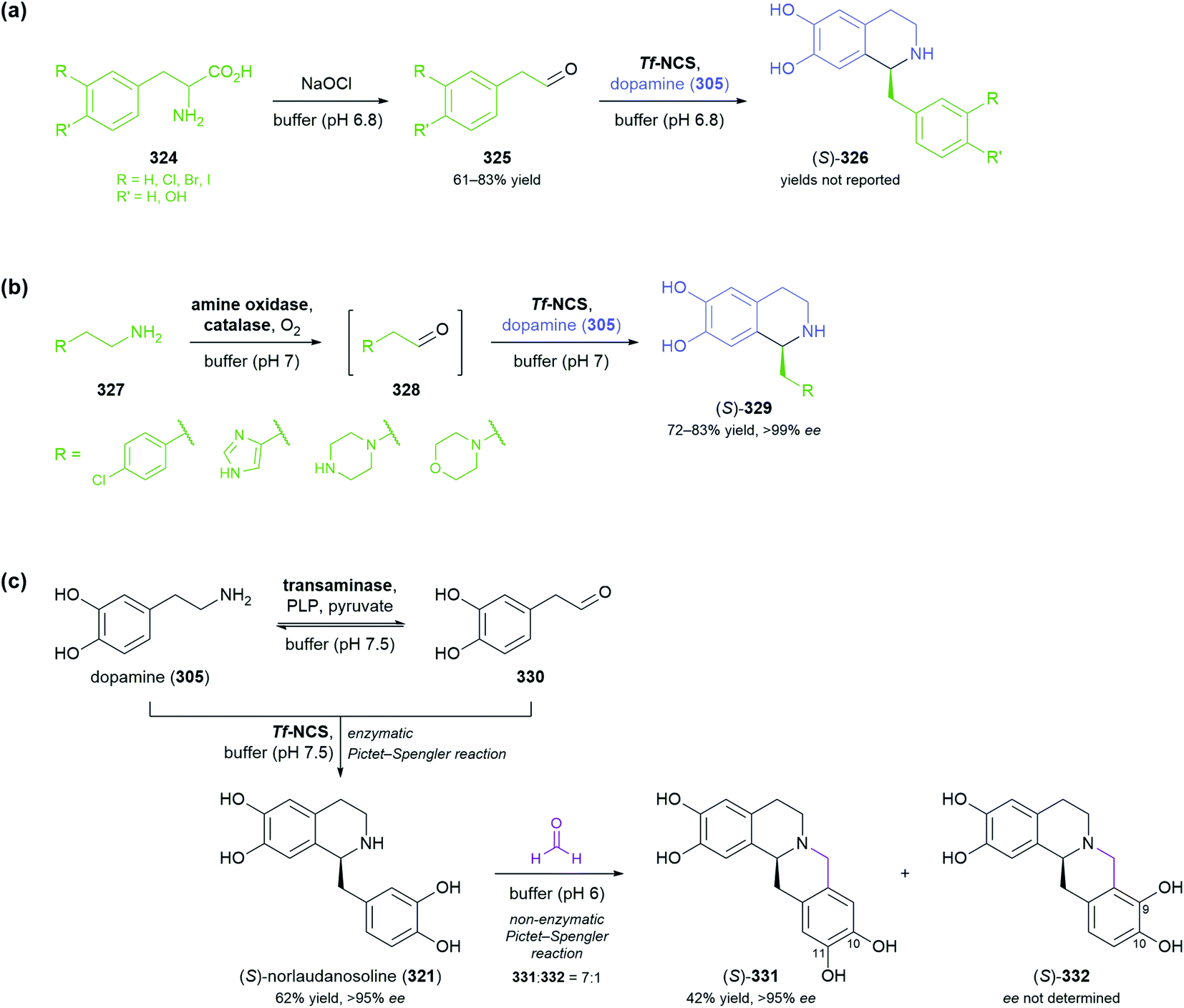 | ||
| Scheme 41 In situ formation of aldehyde substrates for norcoclaurine synthase: (a) oxidative decarboxylation of amino acids 324 with sodium hypochlorite, (b) enzymatic oxidation of primary amines 327 by an amine oxidase, and (c) enzymatic transamination of dopamine (305) in a “triangular cascade” leading to (S)-norlaudanosoline (321) and tetrahydroprotoberberines (S)-331 and (S)-332. | ||
Compared to the amine substrate scope, the carbonyl scope of norcoclaurine synthase has been established as relatively broad already in early investigations, comprising a range of arylaliphatic and aliphatic aldehydes. Since many of these are unstable and difficult to synthesise, several studies have explored their in situ formation under the conditions of the NCS biotransformation. For instance, a previously reported method162 that generates arylacetaldehyde derivatives 325 from the corresponding amino acids 324 using sodium hypochlorite was extended to a broader range of substrates (Scheme 41a).163 The aldehydes 325 thus generated were either isolated in 61–83% yield or directly transformed further into (S)-benzylisoquinolines 326 using NCS from Thalictrum flavum. Unfortunately, only relative reaction rates but no conversion or stereoselectivity data were reported for the chemo-enzymatic sequence. Alternatively, primary amines can serve as precursors to aldehyde substrates of NCS. The quantitative conversion of four amines 327 into the corresponding aldehydes 328 using a diamine oxidase from Lathyrus cicera (red pea) was combined with a Pictet–Spengler reaction catalysed by Tf-NCS, affording non-natural (S)-benzylisoquinolines 329 in 72–83% yield and >99% ee (Scheme 41b).164 To prevent the undesired oxidation of dopamine, the second NCS substrate, this one-pot sequence was carried out in step-wise fashion and phenylhydrazine was used to inactivate the amine oxidase once its reaction was complete. In a related approach, a transaminase from Chromobacterium violaceum was used to deaminate dopamine (305) to aldehyde 330 (Scheme 41c).165 Only 0.5 equivalents of the amine acceptor, pyruvate, were employed so that the reaction remained incomplete, allowing the coupling of 330 and remaining 305 to (S)-norlaudanosoline (321) by Tf-NCS in what the authors have termed a ‘triangular cascade’. A preparative reaction on 0.5 mmol scale gave 321 in 62% isolated yield and excellent optical purity (>95% ee). The cascade was easily extended by adding formaldehyde after completion of the biotransformation, which triggered a non-enzymatic Pictet–Spengler reaction that led to the tetrahydroprotoberberines 331 and 332 as final products. Notably, this cyclisation proceeded with a 7:1 selectivity for the 10,11-disubstituted 331 over its 9,10-disubstituted regioisomer 332, making the method complementary to enzymatic cyclisation by berberine bridge enzyme (cf. Section 3.3). Implementing the three-step chemo-enzymatic process on 0.5 mmol scale resulted in the formation of 331 in 42% overall yield (from 47% conversion) and >95% ee.
A non-enzymatic cyclisation was also exploited in the chemo-enzymatic synthesis of the globeflower alkaloid (S)-trolline (335) from dopamine (305) and aldehyde 333 (Scheme 42).166 The aliphatic aldehyde was readily accepted as substrate by Tf-NCS, which produced the expected tetrahydroisoquinoline 334 along with minor amounts of the target compound 335 in a combined 86% analytical yield. Subsequent basification of the reaction mixture by addition of aqueous Na2CO3 solution liberated the nucleophilic nitrogen atom in 334 by deprotonation and thus promoted its quantitative cyclisation to 335. (S)-Trolline was obtained by this simple one-pot procedure in 74% isolated yield and 95% ee. Through variation of the amine and carbonyl substrates, several non-natural derivatives of 335, including compounds 336–339, were prepared in analogous fashion. Notably, tetrahydroisoquinoline 339 could not be cyclised, reflecting the general difficulties associated with the formation of 7-membered rings.
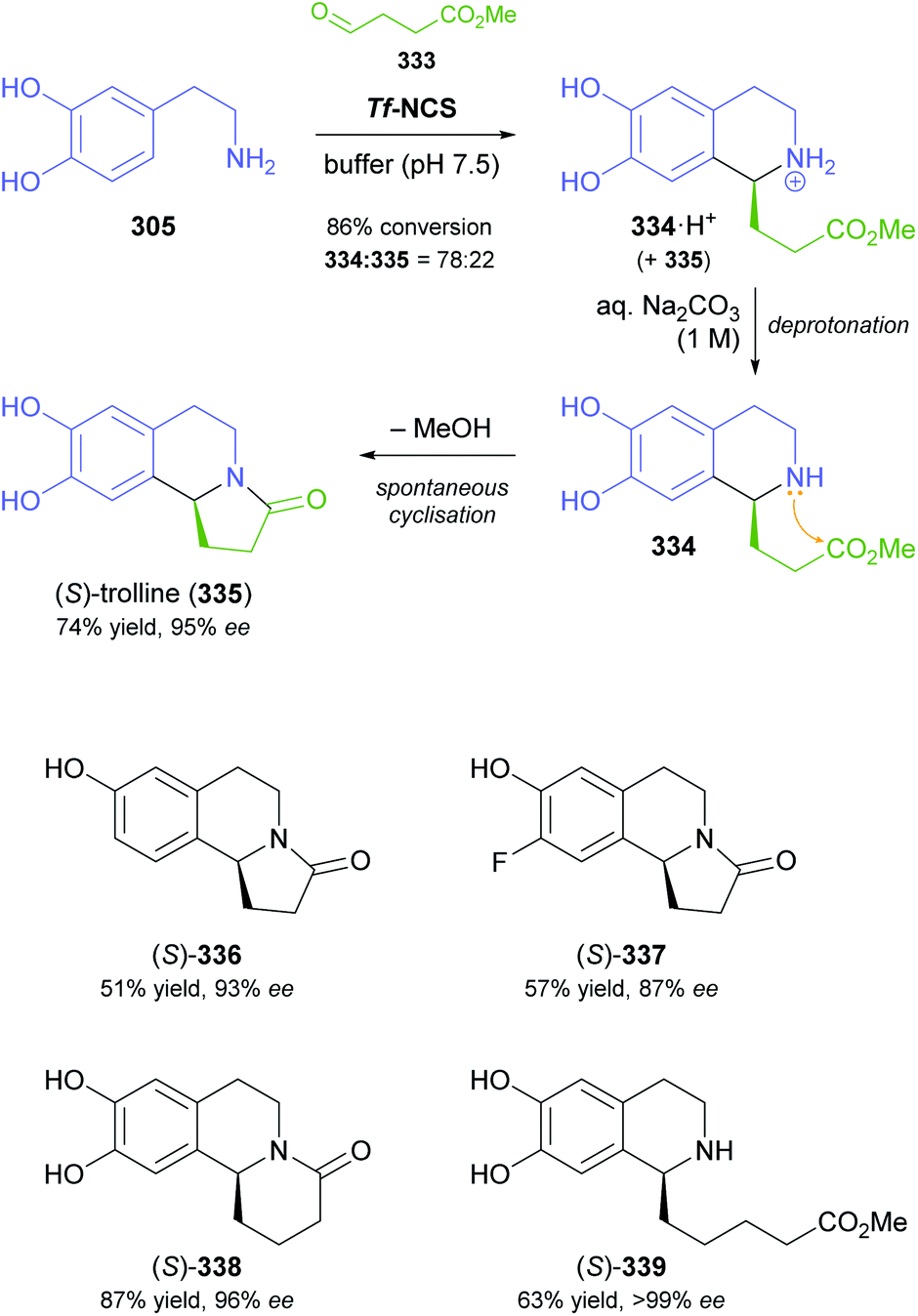 | ||
| Scheme 42 Synthesis of (S)-trolline (335) by enzymatic Pictet–Spengler reaction and base-promoted cyclisation, and examples of non-natural analogues prepared in the same way. | ||
A significant recent development regarding the substrate scope of norcoclaurine synthases is the finding that α-branched and α-cyclic aldehydes are accepted by wild-type NCSs and that this activity can be readily enhanced by protein engineering. First hints towards a potential acceptance of α-branched aldehydes were provided by studies using NCS from Coptis japonica, where small activities with aldehydes such as 2-methylpropanal (isobutanal) and 2-phenylpropanal were detected, but no reaction products were isolated.167 Much more detailed investigations were recently performed by Helen Hailes and co-workers, who found that NCS from Thalictrum flavum converts various α-methyl-substituted aliphatic and arylaliphatic aldehydes with good efficiency and stereoselectivity.168 For instance, 2-methylpropanal and dopamine were converted into the tetrahydroisoquinoline (S)-340 with near-complete conversion (97%) and >95% ee (Fig. 4). When chiral, racemic aldehydes were employed, the enzyme controlled not only the C1-configuration of the tetrahydroisoquinoline, but also the neighbouring side-chain stereocentre, typically affording (1S,1′R)-products in diastereomeric ratios of 82:18 and higher. Experiments performed with the individual enantiomers of 2-methylbutanal confirmed that this diastereoselectivity results from kinetic resolution of the racemates. Notably, the acceptance of α-branched aldehydes was not limited to reactions with the natural amine substrate, dopamine, as demonstrated by the formation of products such as 344 and 345. For some of the investigated reactions, the conversions and diastereoselectivities observed with wild-type Tf-NCS were even surpassed by previously reported enzyme variants, and the co-crystallisation of Tf-NCS with a reaction intermediate mimic, (R)-346, provided insights into the structural basis of these experimental findings. Moreover, a preparative biotransformation towards product (S)-341 showed good scalability (88% isolated yield on 0.5 g scale) and only a slight erosion of diastereoselectivity (87:13 dr) compared to the analytical-scale reaction (94:6 dr).
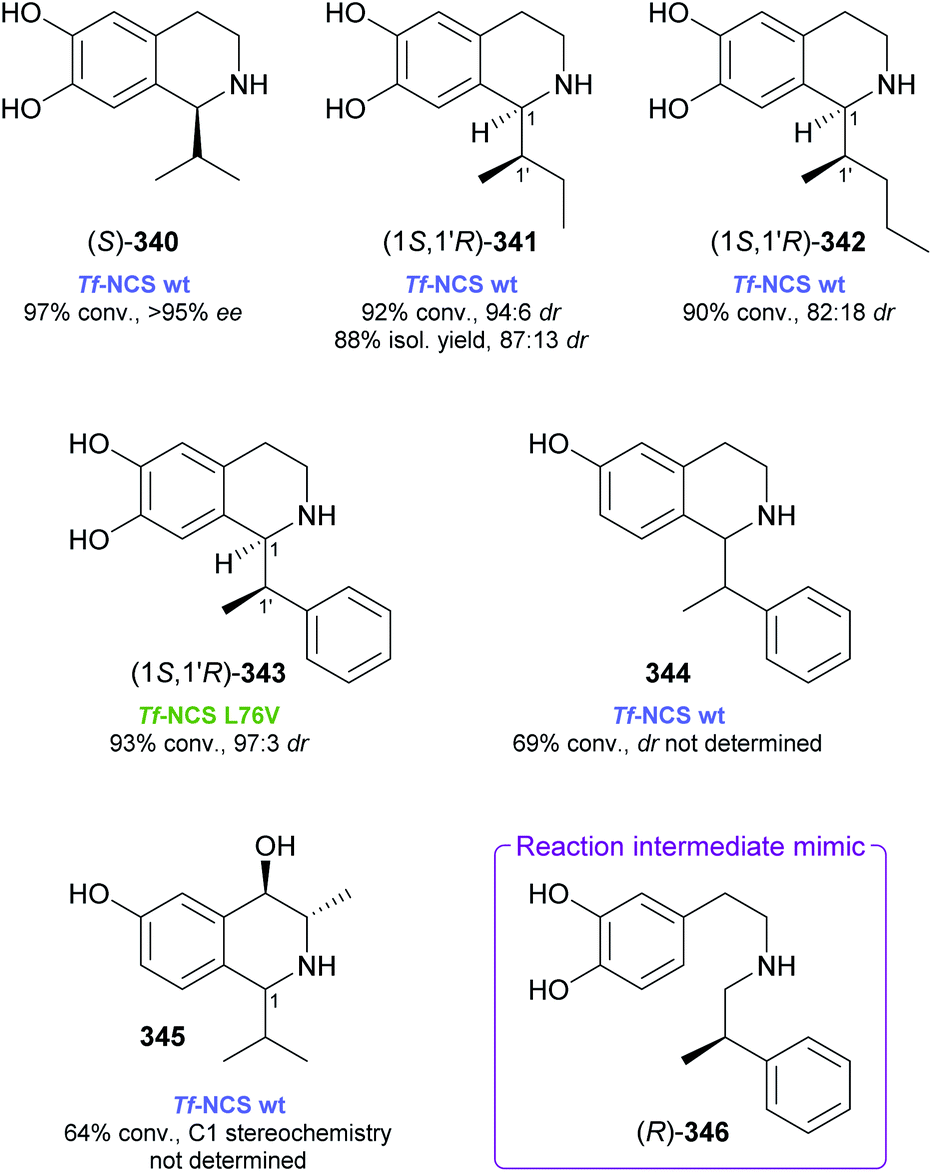 | ||
| Fig. 4 Examples of tetrahydroisoquinolines obtained by NCS reactions of α-branched aldehydes, and structure of a reaction intermediate mimic used for mechanistic studies. | ||
In a follow-up study published one year later, the authors could show that wild-type Tf-NCS and its variants also catalyse Pictet–Spengler reactions of cyclohexanecarbaldehyde, benzaldehyde, and many monosubstituted benzaldehyde derivatives (Scheme 43).169 In particular, the variant M97V proved to be very suitable for this class of substrates, generating a panel of (S)-1-aryl-tetrahydroisoquinolines 348 with moderate to high conversions (45% to >99%) and high to excellent stereoselectivities (79% to >97% ee). For the halogenated benzaldehydes, carrying out the biotransformation at pH 6 instead of the usual pH 7.5 led to improved ee values, which is partly due to a lower non-enzymatic background reactivity under the slightly acidic conditions. However, the change to a lower pH also improved analytical yields, indicating an increase in enzyme activity. Unsurprisingly, the substrates that led to the highest non-enzymatic background and consequently to the lowest product ee were the o-F and o-Cl derivative, in which the carbonyl group experiences significant activation by the electron-withdrawing effect of the halogen atom. To further increase the range of accessible products, the authors also investigated the methylation of two tetrahydroisoquinolines 348 by SAM-dependent O-methyltransferases from Rattus norvegicus and Myxococcus xanthus. Both enzymes were found to be active in these reactions, and the latter also proved completely regioselective for the C6 hydroxyl group.
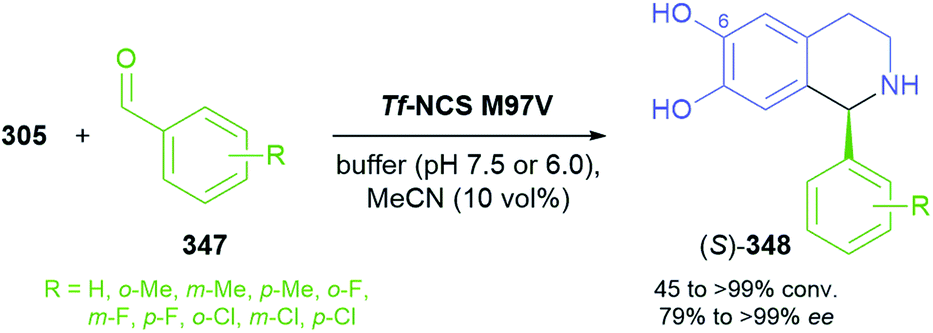 | ||
| Scheme 43 Synthesis of 1-aryl-tetrahydroisoquinolines 348 using variant M97V of NCS from Thalictrum flavum. | ||
Perhaps even more surprising than the acceptance of α-branched aldehydes by norcoclaurine synthase is the finding that ketones are viable substrates for this enzyme class. This discovery was first made in 2017, when Ward, Hailes, and co-workers investigated the Pictet–Spengler reaction between dopamine and 18 structurally diverse ketones, using Tf-NCS and some of its variants as biocatalysts.170 Out of the diverse substrate panel, phenylacetone and its para-hydroxy and para-methoxy derivatives, cyclohexanone, and four monosubstituted cyclohexanone congeners were transformed by the enzymes, leading to 1,1-disubstituted and spirocyclic tetrahydroisoquinolines (Fig. 5). The products were formed with up to 99% conversion and could be isolated from preparative biotransformations (50 μmol scale) as their hydrochloride salts in fair to excellent yields. For instance, (S)-349 was obtained in 87% yield and 95% ee, and a reaction between dopamine and (R)-3-methylcyclohexanone gave (1R,3R)-351 in 27% yield and stereoisomerically pure form. Very recently, a broad and systematic investigation of the ketone substrate scope of Tf-NCS revealed many more interesting products to be accessible.171 Aliphatic ketones and 4-phenylbutan-2-one were found to be accepted, although the conversions to tetrahydroisoquinolines such as 352 and 353 were moderate (37%, 61%) and the stereoselectivities were poor (14%, 15%). Reactions with cyclic ketones gave generally good conversions (52–73%) and were also successful with non-natural amine substrates. The bicyclic 2-tetralone and its derivatives were transformed with surprising efficiency, as exemplified by the formation of (S)-354 with 79% conversion and 86% ee. The most impressive example, however, is probably the conversion of dopamine and cyclohexane-1,4-dione into the unusual dimeric spiro-tetrahydroisoquinoline 355, which was isolated in 1.4% yield and fully characterised by NMR, proving it to be stereoisomerically pure.
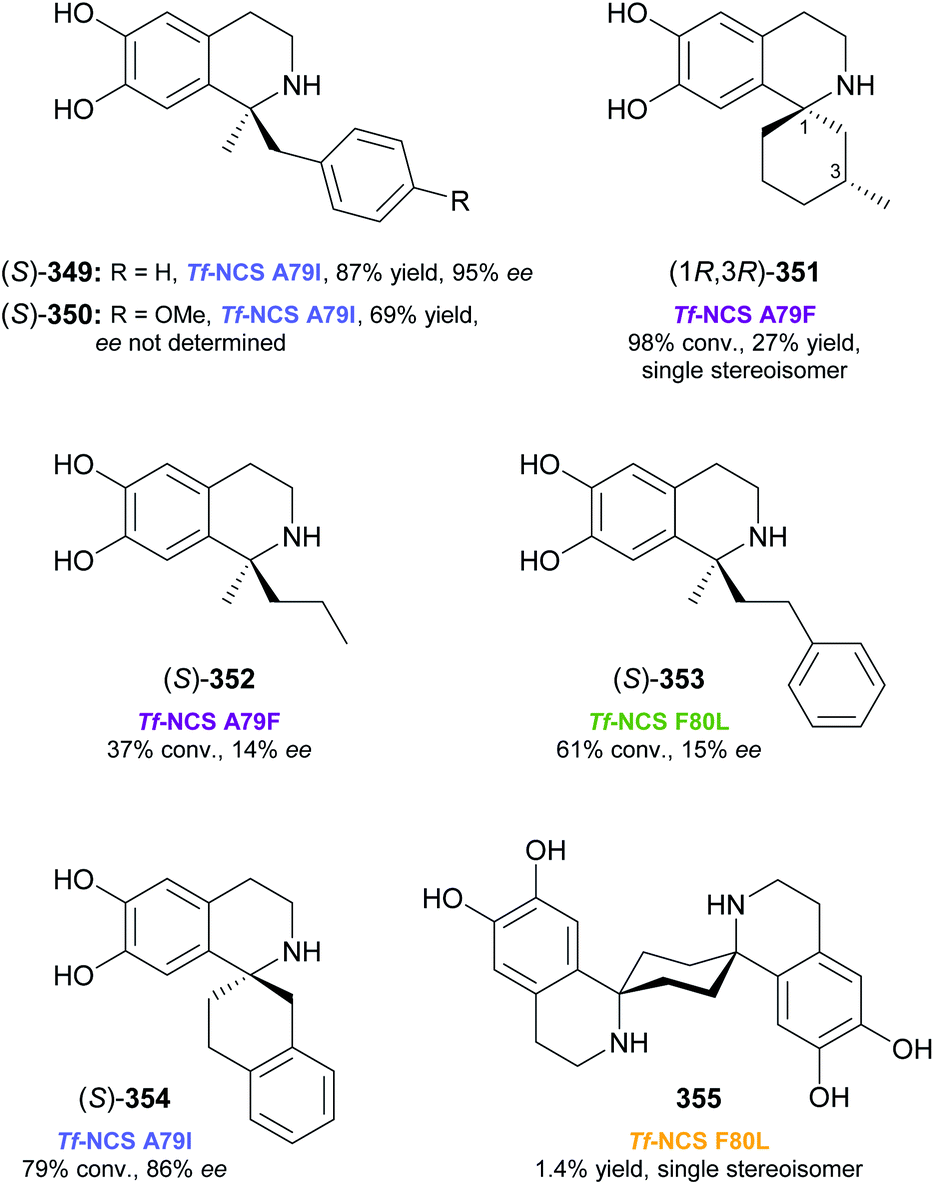 | ||
| Fig. 5 Examples of 1,1-disubstituted and spirocyclic tetrahydroisoquinolines obtained by NCS reactions of open-chain or cyclic ketones. | ||
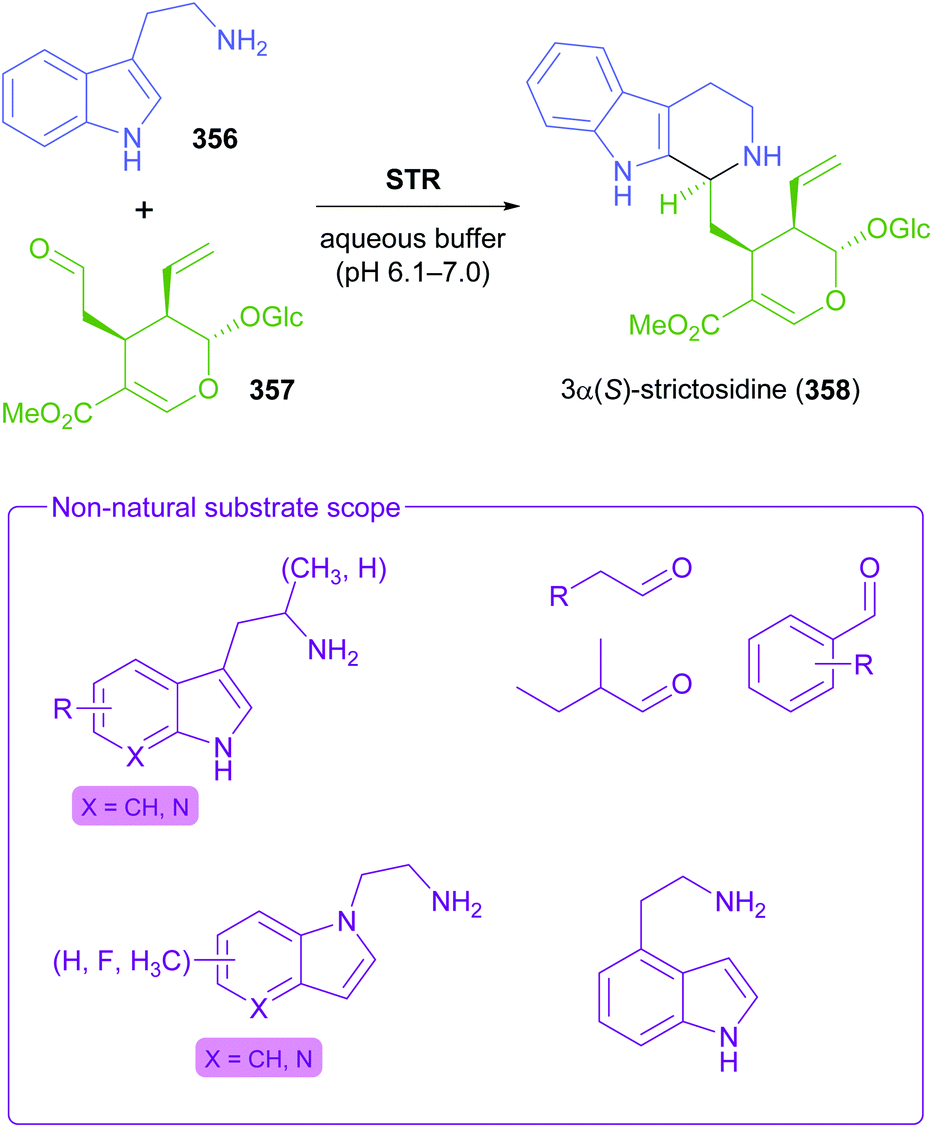 | ||
| Scheme 44 Natural reaction and non-natural substrate scope of strictosidine synthase (STR). | ||
In contrast to norcoclaurine synthase, early investigations into the substrate scope of STRs focused on variations of the amine partner, while largely relying on secologanin (357) and some of its close structural analogues as the aldehyde substrate. The same is true for a more recent study, in which (R)- or (S)-configured α-methyltryptamines 360, obtained from ketones 359 by transaminase reactions, were coupled with 357 by STR from Ophiorrhiza pumila (Op-STR) to give C3-methylated strictosidine derivatives 361 (Scheme 45).172 The amines 360 were prepared in 20–57% isolated yield and high optical purity (97% to >98% ee) for an initial activity screening with five STRs and detailed investigations of diastereoselectivity with Op-STR, the best-performing enzyme. The latter experiments revealed that Op-STR converts both amine enantiomers at comparable rates (E = 5–7 for 360, R = H) and that it controls the configuration at C1 of the tetrahydro-β-carboline product 361 to give the (1S)-diastereomer (>98% de) irrespective of the amine's absolute configuration. For preparative biotransformations, the transaminase and STR reactions were combined in a one-pot process, whereby a step-wise approach (addition of STR and 357 after completion of the transamination step) was slightly superior in terms of overall conversion compared to the simultaneous addition of all required enzymes and reagents. Three of the investigated indolylacetones (359, R = H, 5-OH, 7-Me) could thus be transformed into either diastereomer of (1S)-361 with high efficiency (56% to >99% overall conversion), and multi-milligram amounts of three products were isolated from pooled small-scale reactions in 70–80% yield and >98% de.
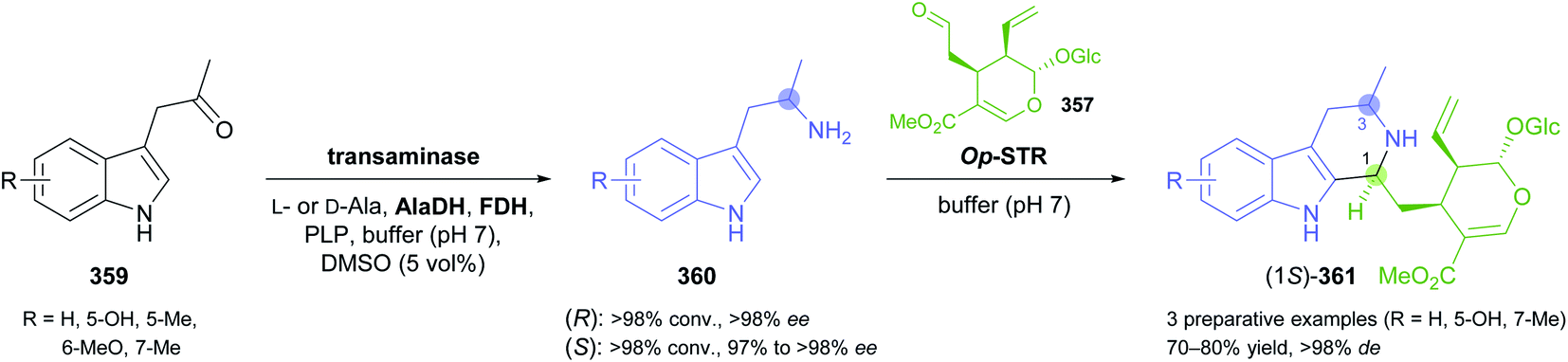 | ||
| Scheme 45 Biocatalytic one-pot synthesis of C3-methylated strictosidine derivatives 361 by the combination of transaminases and STR from Ophiorrhiza pumila. The stereogenic centres controlled by the transaminase and by STR are highlighted with blue and green circles, respectively. | ||
Two recent studies by Hongbin Zou and co-workers have focussed on the transformation of STR products into derivatives with potential medicinal application, while in one case also expanding the amine substrate scope of strictosidine synthase to a previously not investigated scaffold. In the first of these reports, the authors used the physiological reaction of STR from Rauvolfia serpentina to generate 3α(S)-strictosidine (358), which upon heating in aqueous Na2CO3 solution underwent cyclisation to (3S)-362, a known compound also referred to as ‘strictosamide’ (Scheme 46a).173 Its (3R)-epimer (‘vincosamide’), on the other hand, is directly accessible from tryptamine (356) and secologanin (357) through a non-enzymatic, acid-promoted Pictet–Spengler reaction. The lactams (3S)- and (3R)-362 were elaborated into a panel of 21 N-substituted tetrahydroangustines 363 via a five-step sequence that involved an enzymatic deglycosylation, and the products were investigated as topoisomerase I inhibitors. In related work, the authors discovered that Rs-STR catalyses the coupling of secologanin (357) and 1H-indole-4-ethanamine (364) to azepinoindole (4S)-365 (Scheme 46b).174 Next to the natural tetrahydro-β-carbolines and piperazinoindoles generated in earlier studies,175 this represents the third distinct scaffold accessible through STR catalysis. The enzyme product (4S)-365 was not isolated but directly transformed into the more stable lactam (4S)-366, which was obtained in 24% isolated yield over the two steps and in >98% de. Catalytic hydrogenation, enzymatic deglycosylation, and condensation with benzene-1,2-diamine then furnished compound (4S)-367, which was shown to exhibit antimalarial activity. Similar to the case of ‘vincosamide’ discussed above, the (4R)-epimers (85% de) were accessible through a non-enzymatic Pictet–Spengler reaction, which in this work was performed using phosphate catalysis.
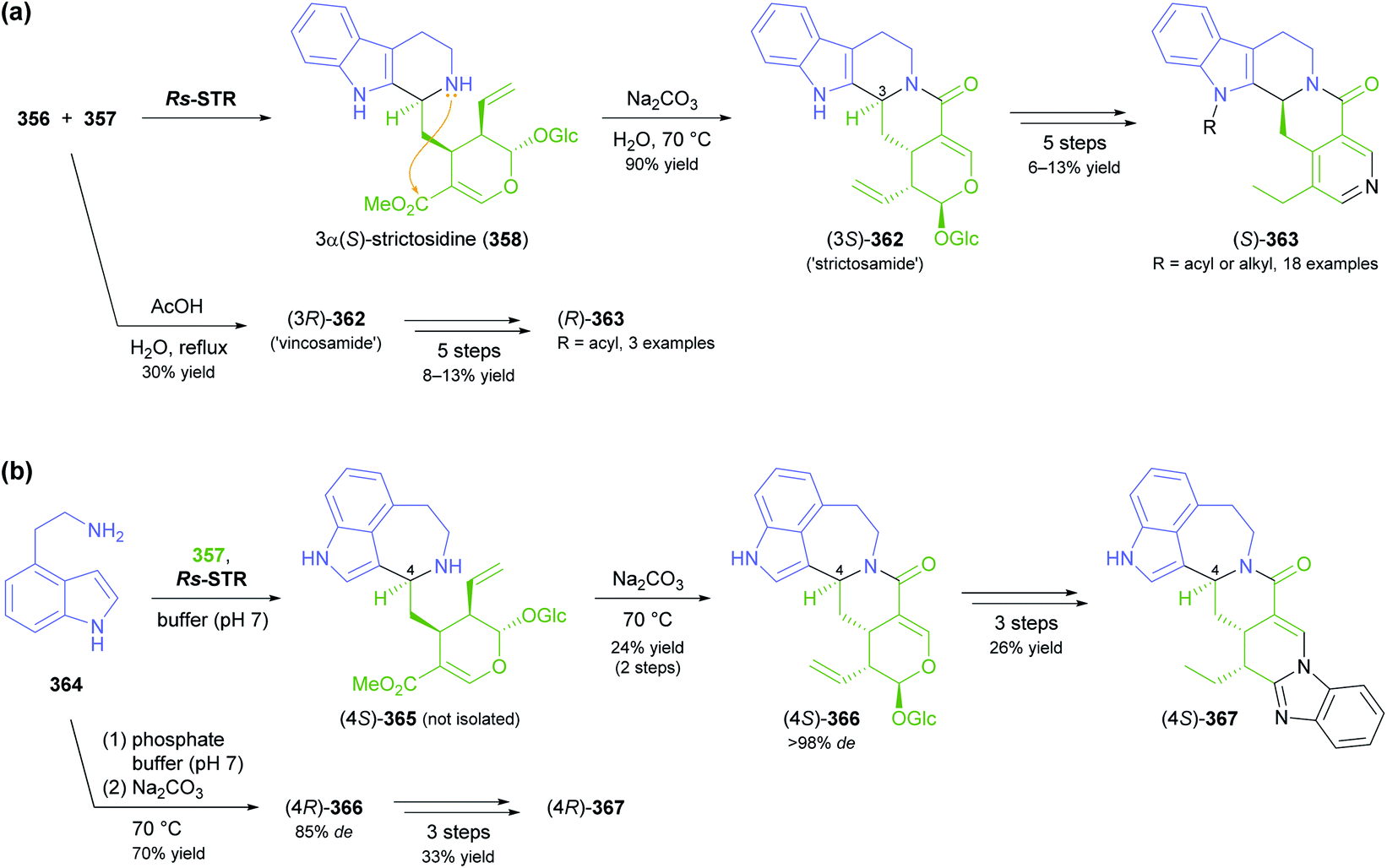 | ||
| Scheme 46 Applications of strictosidine synthase in medicinal chemistry: (a) chemo-enzymatic synthesis of N-substituted 3,14,18,19-tetrahydroangustines (363) as topoisomerase 1 inhibitors, (b) condensation of secologanin (357) with non-natural amine substrate 364 to give azepinoindole derivative (4S)-366, and its conversion into the antimalarial 367. | ||
Substantial variations of the aldehyde substrate of strictosidine synthase were first reported in 2018, when it was shown that the STRs from Catharanthus roseus, Ophiorrhiza pumila, and Rauvolfia serpentina (as well as the V208A variant of the latter) readily accept small aliphatic aldehydes.176 Surprisingly, the resulting tetrahydro-β-carbolines 175 and 368–370 (Scheme 47a) were formed with (R)-stereoconfiguration, in several cases even with >98% ee.177 Rs-STR and its V208A variant gave the best conversions and stereoselectivities across the tested range of substrates, and the wild type was selected for a preparative application: the condensation of tryptamine (356) and aldehyde 333 catalysed by Rs-STR afforded – after spontaneous cyclisation of the immediate reaction product 371 – the lactam (R)-372 in 67% isolated yield and >98% ee (Scheme 47b; cf. Scheme 42, Section 5.3.1). Subsequent reduction of 372 by lithium aluminium hydride provided the alkaloid (R)-harmicine (373) and hence completed an asymmetric synthesis of this compound in only two steps (62% yield overall) from commercially available starting materials.
 | ||
| Scheme 47 (R)-Selective Pictet–Spengler reactions of strictosidine synthases (STRs): (a) 1-alkyl-tetrahydro-β-carbolines obtained from tryptamine (356) and small, aliphatic aldehydes, (b) two-step chemo-enzymatic synthesis of (R)-harmicine (373). | ||
A further expansion of the carbonyl substrate scope of strictosidine synthase to benzaldehyde derivatives was recently achieved through protein engineering.178 After an initial screening of three wild-type STRs with benzaldehyde found no activity, and 2-methylbutanal gave barely detectable conversions (0.2–1.0%), the authors followed a ‘substrate walking’ approach:179,180 First, variants with increased activity towards 2-methylbutanal were identified and only the best of these were then tested with benzaldehyde as substrate. Moreover, the protein engineering was initially performed on STR from Ophiorrhiza pumila and beneficial mutations were later transferred to the Rauvolfia serpentina enzyme. The best variant, Rs-STR V176L–V208A, was active on nine benzaldehyde derivatives 374 and furnished the corresponding 1-aryltetrahydro-β-carbolines (R)-375 with 8–68% conversion and 90–99% ee (Scheme 48). In particular, ortho- and meta-substitution was well tolerated, while aldehydes with a para-substituent larger than fluorine were not converted. Four biotransformations were carried out on preparative scale (5 mmol of tryptamine) and afforded products (R)-375 in moderate yields (4–31%) but high optical purities (96–98%).
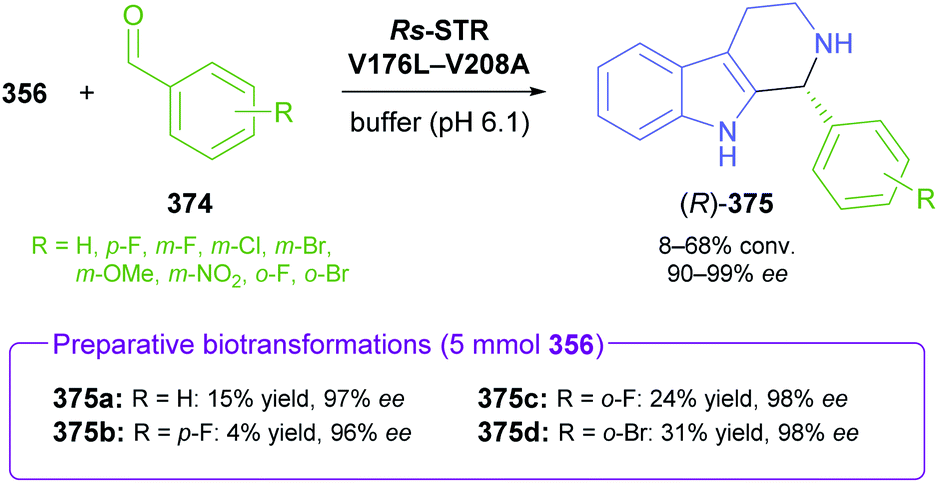 | ||
| Scheme 48 Synthesis of 1-aryl-tetrahydro-β-carbolines 375 using variant V176L–V208A of STR from Rauvolfia serpentina. | ||
5.4 Other enzymes
The biocatalysts discussed in the previous subsections – aldolases, transaminases, and Pictet–Spenglerases – have enabled the preparation of a wide range of alkaloids. More specialised enzymatic key bond formations can also be used in alkaloid synthesis with good success, as demonstrated by three recent studies. Interestingly, all three fall in the broad category of C–H functionalisation. In the first of these reports, the crape myrtle constituents (+)-dihydrolyfoline (380; non-natural enantiomer) and (−)-5-epi-dihydrolyfoline (381) were obtained via horseradish peroxidase-catalysed oxidative cyclisation of the bis-phenols 378 and 379, respectively (Scheme 49).181 Although a single coupling product was formed in both cases, indicating high regio- and diastereoselectivity, the reaction towards 381 proceeded with significantly higher yield (50–66%) than that leading to 380 (15–21%). The coupling precursors 378 and 379 were prepared in a 4–5 step sequence that involved a lipase-catalysed Mannich reaction between O-benzylated isovanillin (376) and (S)-pelletierine (302, itself an alkaloid) as another enzymatic transformation. | ||
| Scheme 49 Synthesis of (+)-dihydrolyfoline (380) and (−)-5-epi-dihydrolyfoline (381) via lipase-catalysed Mannich reaction and horseradish peroxidase (HRP)-catalysed oxidative phenol coupling. | ||
In recent years, the scope of biocatalytic carbon–carbon and carbon–heteroatom bond formation has been expanded impressively through non-natural reactions of engineered heme proteins that exploit the diverse reactivity of iron(II) carbene or nitrene intermediates.182 In particular, by replacing the conserved axial cysteine ligand in cytochromes P450 with serine, Frances Arnold's research group has created a novel class of heme-dependent enzymes, termed cytochromes P411, which are capable of catalysing cyclopropanation, aziridination, and C–H amination reactions.183 In a recent study, Arnold and co-workers have demonstrated that cytochromes P411 also catalyse C–C bond formation via sp3 C–H activation.184 While this work mainly focused on benzylic and allylic substrates, alkylation of amines in α-position of the nitrogen atom was also achieved, and the preparative value of this transformation was demonstrated by using it as the key step in the asymmetric synthesis of the alkaloid (+)-(R)-cuspareine (386; non-natural enantiomer; Scheme 50a). The main P411 variant investigated in this study, P411-CHF, catalysed the coupling of N-methyl-1,2,3,4-tetrahydroquinoline (382) and ethyl diazoacetate (383) with good regioselectivity (9:1 for C2 over the N-methyl group) and moderate preference for the (R)-enantiomer (46% ee). An intermediate variant from the directed evolution campaign (P411-gen5) displayed better regio- (>50:1) and stereoselectivity (82% ee) with inverted stereopreference, affording (S)-384 in 85% yield on half-gram scale. Reduction of the ester to a primary alcohol, conversion of the latter into a bromide, and Suzuki coupling completed the synthesis of (+)-cuspareine (386).
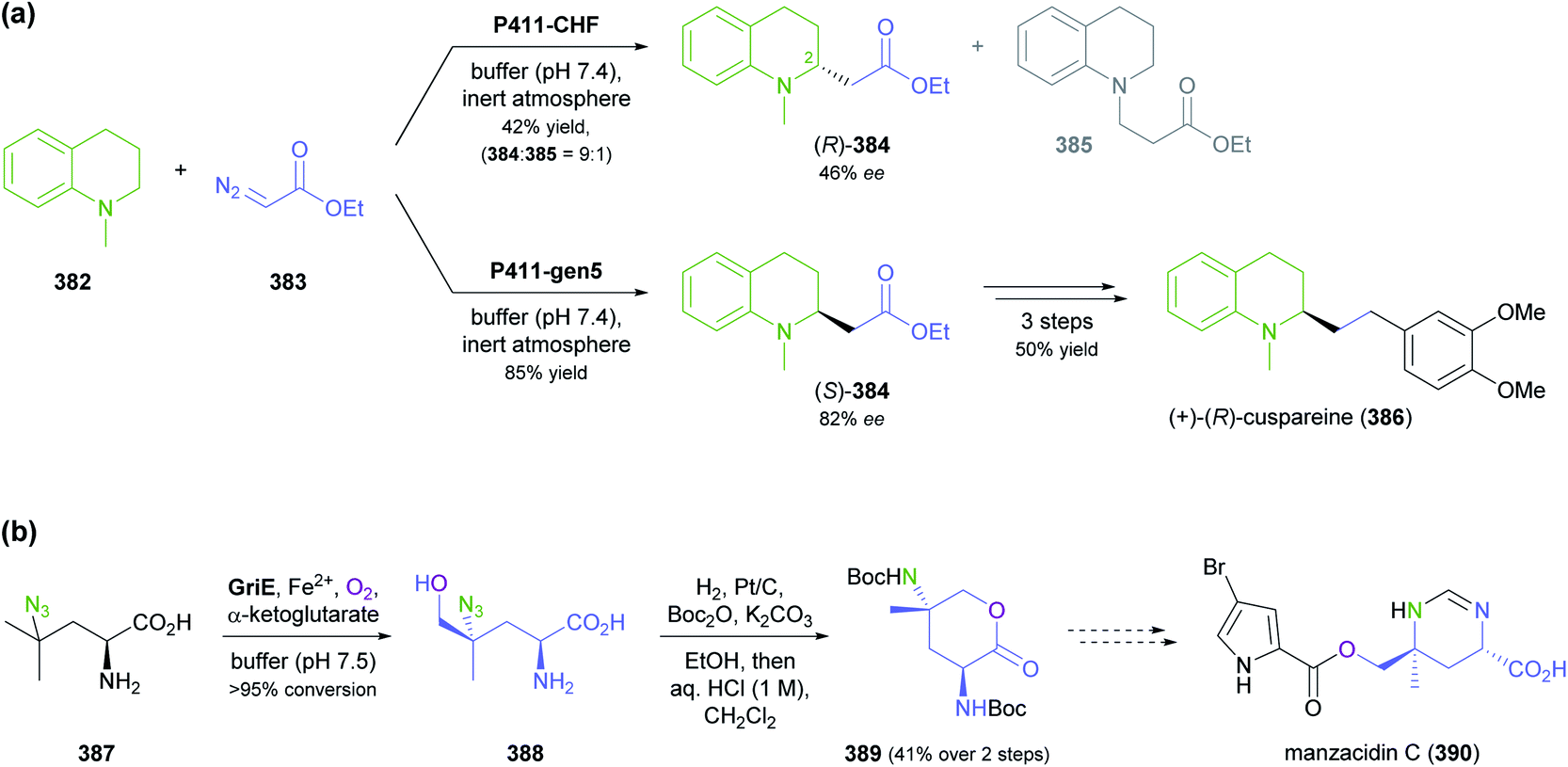 | ||
| Scheme 50 Recent applications of biocatalytic sp3 C–H functionalisation in alkaloid synthesis: (a) α-alkylation of N-methyl-1,2,3,4-tetrahydroquinoline (382) using cytochrome P411 variants, (b) terminal hydroxylation of amino acid 387 catalysed by an iron(II)/α-ketoglutarate-dependent oxidase, GriE. | ||
Biocatalytic C–H oxidation has occasionally been used in alkaloid synthesis, usually for late-stage functionalisation.185,186 Zwick and Renata, however, have recently used C–H hydroxylation catalysed by an iron(II)/α-ketoglutarate-dependent dioxygenase, GriE, as the asymmetric key step in the formal synthesis of manzacidin C (390), a sea sponge alkaloid (Scheme 50b).187 Compound 387, obtained by a photocatalytic C–H azidation of L-leucine, was locally desymmetrised by the biocatalytic terminal hydroxylation, giving alcohol 388 along with minor amounts of the bis-hydroxylation product. Directly subjecting this crude mixture to hydrogenation and Boc protection led to lactone 389 in 41% yield, a compound that can be converted into manzadicin C (390) via two literature-known188 steps. Hence, a remarkably concise formal synthesis of 390 was enabled by two consecutive C–H functionalisations of an unprotected amino acid.
6 Emerging biocatalysts from alkaloid biosynthetic pathways
A quick survey of the reactions discussed in the previous sections shows that the majority of chemo-enzymatic alkaloid syntheses (approx. 60%) make use of generalist enzymes with broad availability, such as lipases, transaminases, amine oxidases, or imine reductases. However, the secondary metabolic pathways through which alkaloids are assembled in nature hold in store a dazzling array of more specialised enzymes that could be applied in synthesis.189 Indeed, the successful examples of berberine bridge enzyme, norcoclaurine synthase, and strictosidine synthase demonstrate that once the protein expression issues often associated with plant enzymes are solved, these can become valuable biocatalysts with broad application potential.Recent advances in genomics, proteomics, and metabolomics have accelerated the elucidation of biosynthetic pathways and the identification of enzymes on a sequence level.190 This development, combined with the ever-decreasing costs of DNA synthesis,191 makes a wealth of prospective biocatalysts accessible for experimental characterisation and synthetic application, which we believe will further expand the scope of biocatalysis in alkaloid synthesis.
One area with considerable potential for diversification are enzymatic Pictet–Spengler reactions: As discussed in Section 5.3.1, norcoclaurine synthase (NCS) from Thalictrum flavum has proven outstandingly flexible in terms of its carbonyl substrate acceptance. NCS sequences have been identified in many other organisms, and some have recently been heterologously expressed and studied in their natural reaction,192 but their non-natural substrate scope remains to be explored. Moreover, Pictet–Spenglerases other than NCS and STR are known, but most of them have not yet been evaluated in a biocatalytic context.157a,193
Oxidative cyclisations are common in alkaloid biosynthetic pathways, where they usually proceed with levels of regio- and stereoselectivity that are unmatched by chemical catalysts. Some of the responsible enzymes are close structural homologues of BBE,194 a flavin-dependent oxidase that has proven its preparative applicability (cf. Section 3.3), while another large group are heme-dependent oxidoreductases. The recently elucidated transformations of N-methyl-4-(dimethylallyl)tryptophan (391, R = Me) to chanoclavine I (393) and of 4-(dimethylallyl)tryptophan (391, R = H) to aurantioclavine (394) both require one enzyme from each of these groups and involve not only a cyclisation but also a decarboxylation (Scheme 51a).195 Oxidative phenol coupling reactions catalysed by cytochromes P450 constitute key steps in the biosynthesis of morphinans (formation of salutaridine, 395)196 and Amaryllidaceae alkaloids (e.g., noroxomaritidine, 396),197 among others. The enzymes responsible for these transformations are membrane-bound and their natural redox partners are not yet known, which complicates their functional heterologous expression and their use in vitro. However, if these hurdles could be overcome and these enzymatic cyclisations could be translated to preparatively useful conditions, they would unlock new synthetic approaches to the respective families of natural products.
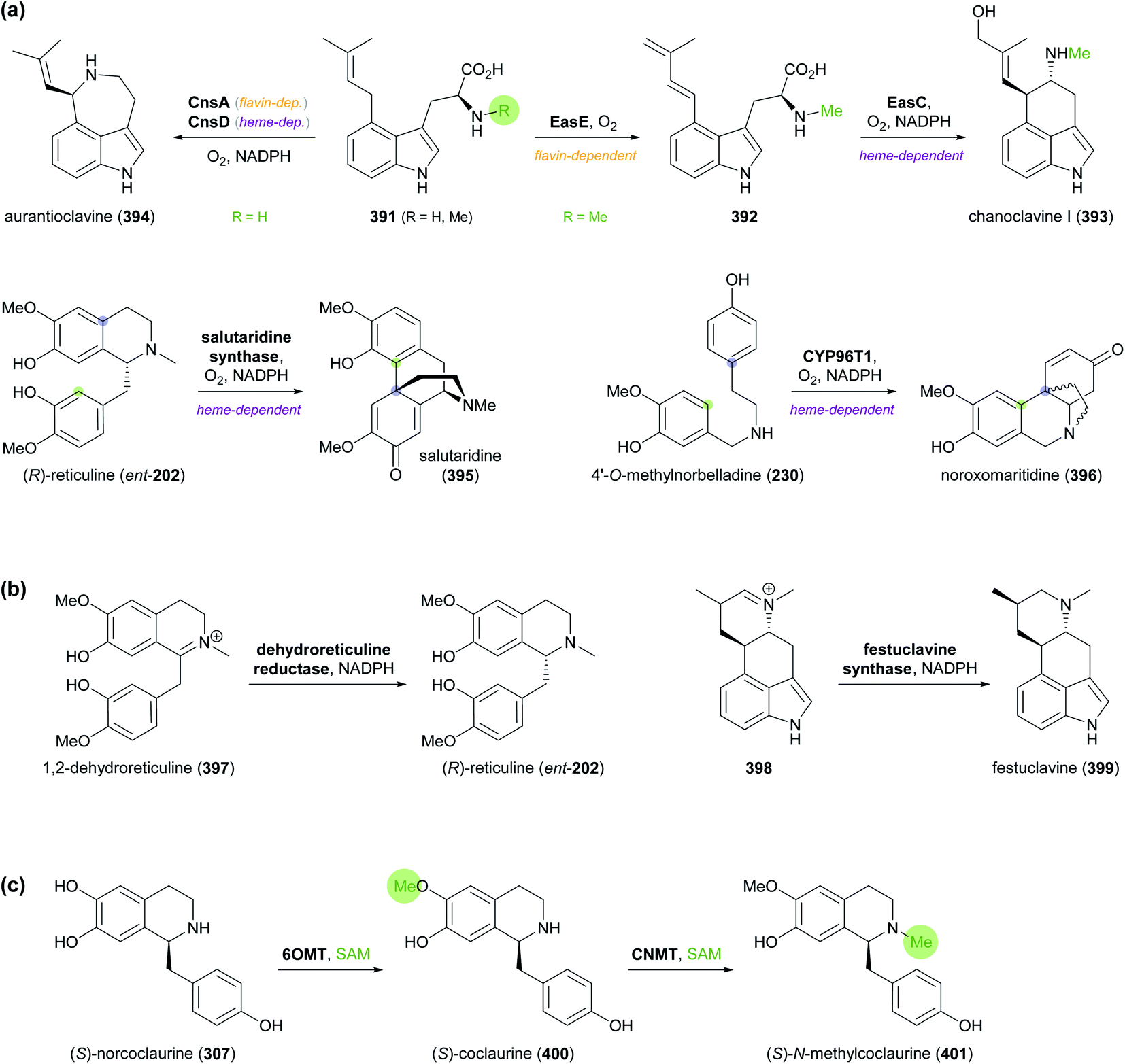 | ||
| Scheme 51 Examples of enzymatic transformations from alkaloid biosynthetic pathways that have potential for biocatalytic application: (a) oxidative cyclisations, (b) reduction of iminium ions, and (c) regioselective methylations. | ||
Imine reduction has become an established part of the biocatalytic toolbox of reactions over the last ten years, but virtually all work in this area relies on enzymes from a single protein fold class. Alkaloid biosynthesis in plants and fungi involves CN reduction by enzymes that are structurally unrelated to the common IREDs.198 Since these reductases have evolved to transform relatively large and complex substrates (e.g., 397, 398, Scheme 51b), their use in preparative biotransformations could expand the scope of biocatalytic imine reduction.
Finally, regioselective methylations catalysed by S-adenosyl methionine (SAM)-dependent methyltransferases are commonplace in secondary metabolism (for two examples from benzylisoquinoline alkaloid biosynthesis, see Scheme 51c),199 and some of the involved enzymes have already found first applications in biocatalysis (cf. Scheme 28).126,200 Since recent studies have introduced practical options for an in situ regeneration of the SAM cofactor and some analogues with modified alkyl groups,201 the importance of biocatalytic alkylations can be expected to grow,202 and methyltransferases from alkaloid biosynthetic pathways might play a significant role in this context.
7 Conclusions and outlook
Over the last eight years, the role of biocatalysis in the asymmetric synthesis of alkaloids has continued to evolve. Chiral building block approaches relying on lipases, esterases, and toluene dioxygenase still account for the largest number and the greatest structural diversity of alkaloids prepared through chemo-enzymatic routes, but biocatalytic key C–C and C–N bond formations have steadily grown in importance and are now almost equally common (Fig. 6). Kinetic resolution, dynamic kinetic resolution, and deracemisation of alkaloids continue to be explored, but are largely restricted to target molecules with only a single centre of chirality, which severely limits their scope. The same is true for biocatalytic imine reduction, which has recently emerged as a novel option for alkaloid synthesis, but has not yet had a significant impact in a preparative context. | ||
| Fig. 6 Chemo-enzymatic total syntheses of alkaloids published per year. The colours indicate the different chemo-enzymatic approaches as discussed in this review: (a) biocatalytic preparation of chiral building blocks (blue), (b) biocatalytic kinetic resolution, dynamic kinetic resolution, and deracemisation of alkaloids (green), (c) biocatalytic reduction of imines or iminium ions (purple), and (d) chemo-enzymatic syntheses that use enzymes for key asymmetric C–C and/or C–N bond formation (orange). | ||
The observed shift towards key bond formations can mainly be attributed to the vastly increased use of two enzyme classes in alkaloid synthesis: transaminases and norcoclaurine synthases (Fig. 7). Neither of the two groups has found application in the chemo-enzymatic synthesis of alkaloids before 2010, but both have quickly risen in importance after that point. The broad substrate scope of both enzyme families is a significant factor in this development, which becomes particularly evident when comparing norcoclaurine synthase with strictosidine synthase: Both enzymes catalyse Pictet–Spengler reactions that directly lead to alkaloid scaffolds, but the former accepts a much more diverse range of carbonyl substrates and has hence found more widespread use than the latter. Irrespective of this difference, both Pictet–Spenglerases can be considered prime examples of plant enzymes that have become successful biocatalysts, and we anticipate that many more enzymes of alkaloid biosynthesis – in particular those forming carbon–carbon bonds – will find their way into preparative applications in the future.
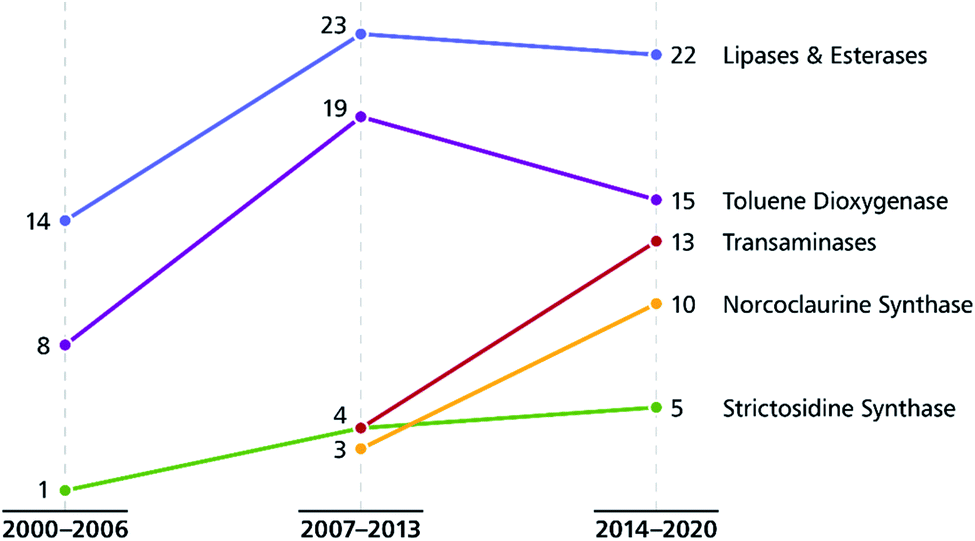 | ||
| Fig. 7 Use of selected enzyme classes in chemo-enzymatic alkaloid synthesis over the last 21 years, expressed as the number of published reports grouped into 7 year periods. | ||
The increasingly ‘central’ use of biocatalytic transformations in chemo-enzymatic routes towards alkaloids has given rise to many syntheses that are considerably shorter than purely chemical alternatives and that require little or no use of protective groups. These characteristics bode well for applications on larger scale, and the example of (−)-ephedrine, produced industrially via biocatalytic carboligation and transition metal-catalysed reductive amination since almost a century,203 shows that chemo-enzymatic alkaloid syntheses can indeed be economically viable. Compared to the traditional isolation of natural products, a significant benefit of synthetic methods is their independence of plant material, while their main advantages over fermentation are higher space-time yields and a broader product portfolio.
Perhaps the last step required for an increased transfer of the chemo-enzymatic approaches discussed herein to production scale is a more intense communication and collaboration between synthetic chemists and researchers in biocatalysis, biotechnology and enzymology. Recognising the often complementary strengths of chemo- and biocatalytic methods and strategically exploiting them in synthetic planning should enable concise and efficient routes towards many target compounds, including alkaloids of practical relevance.204 We hope that this review, by showcasing a broad spectrum of chemo-enzymatic strategies for alkaloid synthesis ranging from chemistry-dominated to primarily biocatalytic, makes a small contribution to achieving this goal.
Abbreviations
| 9-BBN | 9-Borabicyclo[3.3.1]nonane |
| Ac | Acetyl |
| AIBN | Azobisisobutyronitrile |
| ADH | Alcohol dehydrogenase |
| Ala | Alanine |
| AlaDH | L-Alanine dehydrogenase |
| BBE | Berberine bridge enzyme |
| Bn | Benzyl |
| Boc | tert-Butoxycarbonyl |
| Bz | Benzoyl |
| CAL-B | Lipase B from Candida antarctica |
| Cbz | Carboxybenzyl (benzyloxycarbonyl) |
| CNMT | Coclaurine N-methyltransferase |
| CSA | Camphorsulfonic acid |
| DAIB | (Diacetoxyiodo)benzene |
| DBAD | Di-tert-butyl azodicarboxylate |
| DBU | 1,8-Diazabicyclo[5.4.0]undec-7-ene |
| DCE | 1,2-Dichloroethane |
| DEAD | Diethyl azodicarboxylate |
| DHAP | Dihydroxyacetone phosphate |
| DIAD | Diisopropyl azodicarboxylate |
| DIBAL | Diisobutylaluminium hydride |
| DKR | Dynamic kinetic resolution |
| DMAP | 4-Dimethylaminopyridine |
| DME | 1,2-Dimethoxyethane |
| DMF | N,N-Dimethylformamide |
| DMSO | Dimethyl sulfoxide |
| dba | Dibenzylideneacetone |
| dppe | 1,2-Bis(diphenylphosphino)ethane |
| dppf | 1,1′-Bis(diphenylphosphino)ferrocene |
| de | Diastereomeric excess |
| dr | Diastereomeric ratio |
| E | Enantioselectivity |
| E. coli | Escherichia coli |
| ee | Enantiomeric excess |
| FDH | Formate dehydrogenase |
| FSA | D-Fructose-6-phosphate aldolase |
| FucA | L-Fuculose-1-phosphate aldolase |
| GDH | D-Glucose dehydrogenase |
| Glc | D-Glucopyranosyl |
| HBTU | 2-(1H-Benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate |
| HLE | Horse liver esterase |
| HRP | Horseradish peroxidase |
| IP-NBSH | N′-Isopropylidene-2-nitrobenzenesulfonohydrazide |
| IRED | Imine reductase |
| kcat | Turnover number (Michaelis–Menten kinetics) |
| LDA | Lithium diisopropylamide |
| LDH | Lactate dehydrogenase |
| Moc | Methoxycarbonyl |
| MOM | Methoxymethyl |
| MS | Molecular sieves |
| MTBE | Methyl tert-butyl ether |
| NADH | β-Nicotinamide adenine dinucleotide |
| NADPH | β-Nicotinamide adenine dinucleotide phosphate |
| NBS | N-Bromosuccinimide |
| NCS | Norcoclaurine synthase |
| NMR | Nuclear magnetic resonance |
| PAD | Potassium azodicarboxylate |
| PCC | Pyridinium chlorochromate |
| PDC | Pyruvate decarboxylase |
| PLE | Pig liver esterase |
| PLP | Pyridoxal 5′-phosphate |
| PMB | para-Methoxybenzyl |
| RhuA | L-Rhamnulose-1-phosphate aldolase |
| rt | Room temperature |
| SAM | S-Adenosyl methionine |
| STR | Strictosidine synthase |
| STY | Space-time yield |
| TA | Transaminase |
| TBD | 1,5,7-Triazabicyclo[4.4.0]dec-5-ene |
| TBDMS | tert-Butyldimethylsilyl |
| TBDPS | tert-Butyldiphenylsilyl |
| TDO | Toluene dioxygenase |
| TFA | Trifluoroacetic acid |
| TFAA | Trifluoroacetic anhydride |
| ThDP | Thiamine diphosphate |
| THF | Tetrahydrofuran |
| TIPS | Triisopropylsilyl |
| TMAD | Tetramethylazodicarboxamide |
| TMS | Trimethylsilyl |
| Ts | Tosyl (p-toluenesulfonyl) |
| TyrDC | L-Tyrosine decarboxylase |
| wt | Wild type |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
B. E. and W. K. acknowledge the Austrian Science Fund (FWF) for funding within the project CATALOX (DOC 46-B21). The University of Graz and the Field of Excellence BioHealth are acknowledged for financial support.Notes and references
- (a) W. A. Kukula-Koch and J. Widelski, in Pharmacognosy, ed. S. Badal and R. Delgoda, Academic Press, Boston, 2017, pp. 163–198 Search PubMed; (b) S. Funayama and G. A. Cordell, Alkaloids: A Treasury of Poisons and Medicines, Academic Press, London, 2015 Search PubMed; (c) T. Aniszewski, Alkaloids: Chemistry, Biology, Ecology, and Applications, Elsevier, Boston, 2nd edn, 2015 Search PubMed; (d) Dictionary of Alkaloids with CD-ROM, ed. J. Buckingham, K. H. Baggaley, A. D. Roberts and L. F. Szábo, CRC Press, Boca Raton, FL, 2nd edn, 2010 Search PubMed; (e) M. Mahajan, V. Kumar and S. K. Yadav, in Alkaloids: Properties, Applications, and Pharmacological Effects, ed. N. M. Cassiano, Nova Science Publishers, Hauppauge, NY, 2010, pp. 1–36 Search PubMed; (f) M. Hesse, Alkaloids: Nature's Curse Or Blessing?, Wiley-VCH, Weinheim, 2002 Search PubMed; (g) Alkaloids: Biochemistry, Ecology, and Medicinal Applications, ed. M. F. Roberts and M. Wink, Plenum Press, New York, 1998 Search PubMed.
- For terminological discussions, see ref. 1b and 1c..
- World Health Organization, World Health Organization Model List of Essential Medicines, 21st List, World Health Organization, Geneva, 2019, https://apps.who.int/iris/handle/10665/325771 Search PubMed.
- Atropine Market 2021, https://www.marketwatch.com/press-release/atropine-market-2021-business-status-by-top-key-companies-market-size-growth-and-geographical-forecast-till-2026-with-top-countries-data-2021-03-03, accessed on 21 03 2021 Search PubMed.
- Narcotic Drugs, Estimated World Requirements for 2020, Statistics for 2018, United Nations International Narcotics Control Board, 2019, https://www.incb.org/documents/Narcotic-Drugs/Technical-Publications/2019/Narcotic_Drugs_Technical_Publication_2019_web.pdf Search PubMed.
- S. R. Hitchcock and D. D. Dore, in Comprehensive Chirality, ed. E. M. Carreira and H. Yamamoto, Elsevier, Amsterdam, 2012, pp. 248–283 Search PubMed.
- (a) A. Parsaeimehr and E. Sargsyan, in Natural Products: Phytochemistry, Botany and Metabolism of Alkaloids, Phenolics and Terpenes, ed. K. G. Ramawat and J.-M. Mérillon, Springer, Berlin, Heidelberg, 2013, pp. 909–922 Search PubMed; (b) P. L. Rogers, H. S. Shin and B. Wang, in Biotreatment, Downstream Processing and Modelling, Springer, Berlin, Heidelberg, 1997, pp. 33–59 Search PubMed.
- Technology Profile, Clean Green Ephedrine Manufacturing Process, https://www.vu.edu.au/sites/default/files/research/pdfs/Technology%20Profile%20Ephedrine%20(2).pdf, accessed on 24 05 2021 Search PubMed.
- B. Janssen and B. Schäfer, ChemTexts, 2017, 3, 7 CrossRef.
- J. Marco-Contelles, M. do Carmo Carreiras, C. Rodríguez, M. Villarroya and A. G. García, Chem. Rev., 2006, 106, 116–133 CrossRef CAS PubMed.
- Yellow Gold, Innovative systems for sustainable daffodil-derived galanthamine production in the uplands, https://gtr.ukri.org/projects?ref=BB%2FM027120%2F1, accessed on 24 05 2021 Search PubMed.
- M. Markman and T. M. Mekhail, Expert Opin. Pharmacother., 2002, 3, 755–766 CrossRef PubMed.
- P. G. Mountford, in Green Chemistry in the Pharmaceutical Industry, ed. P. J. Dunn, A. S. Wells and M. T. Williams, Wiley-VCH, Weinheim, 2010, pp. 145–160 Search PubMed.
- Global Paclitaxel Sales Market Report 2018, https://www.marketresearch.com/QYResearch-Group-v3531/Global-Paclitaxel-Sales-11375346/, accessed on 21 03 2021 Search PubMed.
- (a) J. Achan, A. O. Talisuna, A. Erhart, A. Yeka, J. K. Tibenderana, F. N. Baliraine, P. J. Rosenthal and U. D'Alessandro, Malar. J., 2011, 10, 144 CrossRef CAS PubMed; (b) L. M. Weiss, Expert Opin. Pharmacother., 2002, 3, 1109–1115 CrossRef CAS PubMed.
- Cinchona Alkaloids in Synthesis and Catalysis: Ligands, Immobilization and Organocatalysis, ed. C. E. Song, Wiley-VCH, Weinheim, 2009 Search PubMed.
- D. Weiß, China-Alkaloide, https://roempp.thieme.de/lexicon/RD-03-01281, accessed on 26 04 2021 Search PubMed.
- (a) S. R. Hitchcock, J. A. Wolfe and D. D. Dore, in Comprehensive Chirality, ed. E. M. Carreira and H. Yamamoto, Elsevier, Amsterdam, 2012, pp. 284–296 Search PubMed; (b) A. Gómez-SanJuan, N. Sotomayor and E. Lete, Beilstein J. Org. Chem., 2013, 9, 313–322 CrossRef PubMed.
- S. K. Ritter, Chem. Eng. News, 2017, 95, 18–20 Search PubMed.
- (a) A. J. Hunt, T. J. Farmer and J. H. Clark, in Element Recovery and Sustainability, ed. A. J. Hunt, The Royal Society of Chemistry, Cambridge, 2013, pp. 1–28 RSC; (b) P. Fröhlich, T. Lorenz, G. Martin, B. Brett and M. Bertau, Angew. Chem., Int. Ed., 2017, 56, 2544–2580 CrossRef PubMed.
- (a) S. Srivastava and A. K. Srivastava, in Natural Products: Phytochemistry, Botany and Metabolism of Alkaloids, Phenolics and Terpenes, ed. K. G. Ramawat and J.-M. Mérillon, Springer, Berlin, Heidelberg, 2013, pp. 213–250 Search PubMed; (b) S. Schläger and B. Dräger, Curr. Opin. Biotechnol., 2016, 37, 155–164 CrossRef PubMed.
- (a) A. G. Millgate, B. J. Pogson, I. W. Wilson, T. M. Kutchan, M. H. Zenk, W. L. Gerlach, A. J. Fist and P. J. Larkin, Nature, 2004, 431, 413–414 CrossRef CAS PubMed; (b) P. Whoriskey and S. Georges, How Johnson & Johnson companies used a ‘super poppy’ to make narcotics for America's most abused opioid pills, The Washington Post, 26 03 2020, https://www.washingtonpost.com/graphics/2020/business/opioid-crisis-johnson-and-johnson-tasmania-poppy/, accessed on 28 04 2021 Search PubMed.
- (a) S. Ahmad, M. Garg, E. T. Tamboli, M. Z. Abdin and S. H. Ansari, Pharmacogn. Rev., 2013, 7, 27–33 CrossRef PubMed; (b) H. Chandran, M. Meena, T. Barupal and K. Sharma, Biotechnol. Rep., 2020, 26, e00450 CrossRef PubMed.
- (a) V. Bringi, P. G. Kadkade, C. L. Prince and B. L. Roach, US Pat., US2011/0086397 A1, Phyton Holdings LLC, 2011; (b) Presidential Green Chemistry Challenge, 2004 Greener Synthetic Pathways Award, https://www.epa.gov/greenchemistry/presidential-green-chemistry-challenge-2004-greener-synthetic-pathways-award, accessed on 28 04 2021 Search PubMed.
- For a general discussion of metabolic engineering, see: (a) The Metabolic Pathway Engineering Handbook, Two Volume Set, ed. C. Smolke, CRC Press, Boca Raton, 2010 Search PubMed; (b) W. Niu, J. Guo and S. Van Dien, in Metabolic Engineering for Bioprocess Commercialization, ed. S. Van Dien, Springer, Cham, 2016, pp. 31–52 Search PubMed; (c) G. Stephanopoulos, ACS Synth. Biol., 2012, 1, 514–525 CrossRef CAS PubMed.
- For recent reviews, see: (a) B. Hoff, J. Plassmeier, M. Blankschien, A.-C. Letzel, L. Kourtz, H. Schröder, W. Koch and O. Zelder, Angew. Chem., Int. Ed., 2021, 60, 2258–2278 CrossRef CAS PubMed; (b) R. Chen, S. Yang, L. Zhang and Y. J. Zhou, iScience, 2020, 23, 100879 CrossRef CAS PubMed; (c) N. Xu, Y. Liu, H. Jiang, J. Liu and Y. Ma, Curr. Opin. Biotechnol., 2020, 66, 27–35 CrossRef CAS PubMed; (d) A. Cravens, J. Payne and C. D. Smolke, Nat. Commun., 2019, 10, 2142 CrossRef PubMed; (e) C. Dziggel, H. Schäfer and M. Wink, Biotechnol. J., 2017, 12, 1600145 CrossRef PubMed; (f) A. Ehrenworth, Nat. Chem. Biol., 2017, 13, 243 CrossRef PubMed.
- S. Galanie, K. Thodey, I. J. Trenchard, M. Filsinger Interrante and C. D. Smolke, Science, 2015, 349, 1095–1100 CrossRef CAS PubMed.
- M. E. Pyne, K. Kevvai, P. S. Grewal, L. Narcross, B. Choi, L. Bourgeois, J. E. Dueber and V. J. J. Martin, Nat. Commun., 2020, 11, 3337 CrossRef CAS PubMed.
- P. Srinivasan and C. D. Smolke, Nature, 2020, 585, 614–619 CAS.
- C. K. Winkler, J. H. Schrittwieser and W. Kroutil, ACS Cent. Sci., 2021, 7, 55–71 CrossRef CAS PubMed.
- (a) S. Wu, R. Snajdrova, J. C. Moore, K. Baldenius and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2021, 60, 88–119 CrossRef CAS PubMed; (b) A. Fryszkowska and P. N. Devine, Curr. Opin. Chem. Biol., 2020, 55, 151–160 CrossRef CAS PubMed; (c) J. P. Adams, M. J. B. Brown, A. Diaz-Rodriguez, R. C. Lloyd and G.-D. Roiban, Adv. Synth. Catal., 2019, 361, 2421–2432 CAS; (d) P. N. Devine, R. M. Howard, R. Kumar, M. P. Thompson, M. D. Truppo and N. J. Turner, Nat. Rev. Chem., 2018, 2, 409–421 CrossRef; (e) R. N. Patel, Bioorg. Med. Chem., 2018, 26, 1252–1274 CrossRef CAS PubMed.
- (a) S. Chakrabarty, E. O. Romero, J. B. Pyser, J. A. Yazarians and A. R. H. Narayan, Acc. Chem. Res., 2021, 54, 1374–1384 CrossRef CAS PubMed; (b) B. S. Moore and T. A. M. Gulder, Nat. Prod. Rep., 2020, 37, 1292–1293 RSC; (c) S. Galanie, D. Entwistle and J. Lalonde, Nat. Prod. Rep., 2020, 37, 1122–1143 RSC; (d) R. Tanifuji, A. Minami, H. Oguri and H. Oikawa, Nat. Prod. Rep., 2020, 37, 1098–1121 RSC; (e) J. Li, A. Amatuni and H. Renata, Curr. Opin. Chem. Biol., 2020, 55, 111–118 CrossRef CAS PubMed; (f) T. Sugai, S. Higashibayashi and K. Hanaya, Tetrahedron, 2018, 74, 3469–3487 CrossRef CAS; (g) S. Friedrich and F. Hahn, Tetrahedron, 2015, 71, 1473–1508 CrossRef CAS.
- J. H. Schrittwieser and V. Resch, RSC Adv., 2013, 3, 17602–17632 RSC.
- (a) J. Kühlborn, J. Groß and T. Opatz, Nat. Prod. Rep., 2020, 37, 380–424 RSC; (b) W.-L. Ng, A. W. H. Wong and T. K. M. Shing, in Efficiency in Natural Product Total Synthesis, Wiley, Hoboken, NJ, 2018, pp. 345–393 Search PubMed.
- C. N. Stout and H. Renata, Acc. Chem. Res., 2021, 54, 1143–1156 CrossRef CAS PubMed.
- U. T. Bornscheuer and R. J. Kazlauskas, Hydrolases in Organic Synthesis, Wiley-VCH, Weinheim, 2nd edn, 2005 Search PubMed.
- The following commercial lipase preparations are mentioned in this review (source organism and formulation details in parentheses): from Amano Enzyme Inc.: lipase AK (Pseudomonas fluorescens), lipase M (Mucor javanicus), lipase PS (Burkholderia cepacia, previously classified as Pseudomonas cepacia), lipase PS-IM (Burkholderia cepacia, immobilised on diatomaceous earth); from Meito Sangyo Co., Ltd.: lipase PL (Alcaligenes sp.), lipase QLM (Alcaligenes sp.); from Novozymes A/S: Novozym 435 (lipase B from Candida antarctica, immobilised on acrylic resin); from Roche Applied Science: Chirazyme L-2 (lipase B from Candida antarctica)..
- R. V. Edwankar, C. R. Edwankar, J. R. Deschamps and J. M. Cook, J. Org. Chem., 2014, 79, 10030–10048 CrossRef CAS PubMed.
- In the schemes of this section, atoms directly derived from the biocatalytically prepared building block are highlighted in blue, and enantiotopic groups recognised by the used enzyme are highlighted by a green circle..
- D. Martella, F. Cardona, C. Parmeggiani, F. Franco, J. A. Tamayo, I. Robina, E. Moreno-Clavijo, A. J. Moreno-Vargas and A. Goti, Eur. J. Org. Chem., 2013, 2013, 4047–4056 CrossRef CAS.
- S. Smullen and P. Evans, Tetrahedron, 2017, 73, 5493–5499 CrossRef CAS.
- J. Li, W. Zhang, F. Zhang, Y. Chen and A. Li, J. Am. Chem. Soc., 2018, 140, 2384 CrossRef CAS PubMed.
- J. Li, W. Zhang, F. Zhang, Y. Chen and A. Li, J. Am. Chem. Soc., 2017, 139, 14893–14896 CrossRef CAS PubMed.
- S. T. Staben, J. J. Kennedy-Smith, D. Huang, B. K. Corkey, R. L. LaLonde and F. D. Toste, Angew. Chem., Int. Ed., 2006, 45, 5991–5994 CrossRef CAS PubMed.
- R. W. Bates, C. J. Lim, S. J. Collier and J. Sukumaran, Asian J. Org. Chem., 2015, 4, 652–658 CrossRef CAS.
- S. Raghavan and S. Rajendar, Org. Biomol. Chem., 2016, 14, 131–137 RSC.
- T. Okada, T. Yamamoto, D. Kato, M. Kawasaki, R. A. Saporito and N. Toyooka, Tetrahedron Lett., 2018, 59, 3797–3800 CrossRef CAS.
- H. Yamamoto, Y. Takagi, T. Oshiro, T. Mitsuyama, I. Sasaki, N. Yamasaki, A. Yamada, H. Kenmoku, Y. Matsuo, Y. Kasai and H. Imagawa, J. Org. Chem., 2014, 79, 8850–8855 CrossRef CAS PubMed.
- H. Tanimoto and T. Oritani, Tetrahedron: Asymmetry, 1996, 7, 1695–1704 CrossRef CAS.
- M. Egi, K. Sugiyama, M. Saneto, R. Hanada, K. Kato and S. Akai, Angew. Chem., Int. Ed., 2013, 52, 3654–3658 CrossRef CAS PubMed.
- S. Kawanishi, K. Sugiyama, Y. Oki, T. Ikawa and S. Akai, Green Chem., 2017, 19, 411–417 RSC.
- K. Sugiyama, S. Kawanishi, Y. Oki, M. Kamiya, R. Hanada, M. Egi and S. Akai, Bioorg. Med. Chem., 2018, 26, 1378–1386 CrossRef CAS PubMed.
- A. Patti and C. Sanfilippo, Symmetry, 2020, 12, 1454 CrossRef CAS.
- R. Riva, L. Banfi, B. Danieli, G. Guanti, G. Lesma and G. Palmisano, J. Chem. Soc., Chem. Commun., 1987, 299–300 RSC.
- A. K. Ghosh and A. Sarkar, Eur. J. Org. Chem., 2016, 2016, 6001–6009 CrossRef CAS PubMed.
- T. Maehara, K. Motoyama, T. Toma, S. Yokoshima and T. Fukuyama, Angew. Chem., Int. Ed., 2017, 56, 1549–1552 CrossRef CAS PubMed.
- D. A. Kummer, D. Li, A. Dion and A. G. Myers, Chem. Sci., 2011, 2, 1710–1718 RSC.
- For an overview, see Section 2.1.2 of our previous review, ref. 33..
- R. Chênevert, G. M. Ziarani, M. P. Morin and M. Dasser, Tetrahedron: Asymmetry, 1999, 10, 3117–3122 CrossRef.
- C. Mayer, A. Romek and T. Bach, Synlett, 2015, 26, 1505–1509 CrossRef CAS.
- (a) R. Fujimoto, Y. Kishi and J. F. Blount, J. Am. Chem. Soc., 1980, 102, 7154–7156 CrossRef CAS; (b) R. Fujimoto and Y. Kishi, Tetrahedron Lett., 1981, 22, 4197–4198 CrossRef CAS.
- S. Pichette, D. K. Winter, J. Lessard and C. Spino, J. Org. Chem., 2013, 78, 12532–12544 CrossRef CAS PubMed.
- S. Akai, T. Naka, T. Fujita, Y. Takebe, T. Tsujino and Y. Kita, J. Org. Chem., 2002, 67, 411–419 CrossRef CAS PubMed.
- H. Toya, T. Satoh, K. Okano, K. Takasu, M. Ihara, A. Takahashi, H. Tanaka and H. Tokuyama, Tetrahedron, 2014, 70, 8129–8141 CrossRef CAS.
- W. Liu, Y. Hu, Y. Zhang, Y. Ma and H. Huang, Biotechnol. Bioprocess Eng., 2014, 19, 449–455 CrossRef CAS.
- (a) Y. Yamamoto, K. Yamamoto, T. Nishioka and J. i. Oda, Agric. Biol. Chem., 1988, 52, 3087–3092 CAS; (b) A. S. Lee, B. B. Liau and M. D. Shair, J. Am. Chem. Soc., 2014, 136, 13442–13452 CrossRef CAS PubMed; (c) H. Ito, T. Inoue and K. Iguchi, Org. Lett., 2008, 10, 3873–3876 CrossRef CAS PubMed.
- J. D. Firth, S. J. Canipa, L. Ferris and P. O'Brien, Angew. Chem., Int. Ed., 2018, 57, 223–226 CrossRef CAS PubMed.
- C. Pousset, R. Callens, M. Haddad and M. Larchevêque, Tetrahedron: Asymmetry, 2004, 15, 3407–3412 CrossRef CAS.
- P. O'Brien, Chem. Commun., 2008, 655–667 RSC.
- N. C. Nagel, A. Masic, U. Schurigt and W. Boland, Org. Biomol. Chem., 2015, 13, 5139–5146 RSC.
- (a) D. T. Gibson, J. R. Koch and R. E. Kallio, Biochemistry, 1968, 7, 2653–2662 CrossRef CAS PubMed; (b) D. T. Gibson, J. R. Koch, C. L. Schuld and R. E. Kallio, Biochemistry, 1968, 7, 3795–3802 CrossRef CAS PubMed; (c) D. T. Gibson, M. Hensley, H. Yoshioka and T. J. Mabry, Biochemistry, 1970, 9, 1626–1630 CrossRef CAS PubMed; (d) V. Subramanian, T.-N. Liu, W. K. Yeh and D. T. Gibson, Biochem. Biophys. Res. Commun., 1979, 91, 1131–1139 CrossRef CAS PubMed.
- (a) U. Rinner, in Comprehensive Chirality, ed. E. M. Carreira and H. Yamamoto, Elsevier, Amsterdam, 2012, pp. 240–267 Search PubMed; (b) T. Hudlicky and J. W. Reed, Synlett, 2009, 2009, 685–703 CrossRef; (c) R. A. Johnson, in Organic Reactions, ed. L. E. Overman, 2004, vol. 63, pp. 117–264 Search PubMed.
- For an overview, see section 2.2 of our previous review (ref. 33), as well as two recent reviews on TDO, and references cited therein: (a) E. S. Taher, M. G. Banwell, J. N. Buckler, Q. Yan and P. Lan, Chem. Rec., 2018, 18, 239–264 CrossRef CAS PubMed; (b) T. Hudlicky, ACS Omega, 2018, 3, 17326–17340 CrossRef CAS PubMed.
- (a) S. Vshyvenko, M. R. Reisenauer, S. Rogelj and T. Hudlicky, Bioorg. Med. Chem. Lett., 2014, 24, 4236–4238 CrossRef CAS PubMed; (b) S. Vshyvenko, Z. W'Giorgis, A. Weber, N. Neverova, B. Hedberg and T. Hudlicky, Adv. Synth. Catal., 2015, 357, 83–87 CrossRef CAS.
- S. Borra, R. Lapinskaite, C. Kempthorne, D. Liscombe, J. McNulty and T. Hudlicky, J. Nat. Prod., 2018, 81, 1451–1459 CrossRef CAS PubMed.
- R. Lapinskaite, M. Ghavre, C. L. Rintelmann, K. Bedard, H. E. Dela Paz and T. Hudlicky, Synlett, 2017, 28, 2896–2900 CrossRef CAS.
- J. W. Reed and T. Hudlicky, Acc. Chem. Res., 2015, 48, 674–687 CrossRef CAS PubMed.
- (a) V. Varghese and T. Hudlicky, Angew. Chem., Int. Ed., 2014, 53, 4355–4358 CrossRef CAS PubMed; (b) L. Rycek, J. J. Hayward, M. A. Latif, J. Tanko, R. Simionescu and T. Hudlicky, J. Org. Chem., 2016, 81, 10930–10941 CrossRef CAS PubMed.
- (a) T. Hudlicky, M. A. A. Endoma and G. Butora, J. Chem. Soc., Perkin Trans. 1, 1996, 2187–2192 RSC; (b) M. A. Endoma, V. P. Bui, J. Hansen and T. Hudlicky, Org. Process Res. Dev., 2002, 6, 525–532 CrossRef CAS.
- M. A. A. Endoma-Arias, J. R. Hudlicky, R. Simionescu and T. Hudlicky, Adv. Synth. Catal., 2014, 356, 333–339 CrossRef CAS.
- (a) M. A. A. Endoma-Arias, M. Makarova, H. E. Dela Paz and T. Hudlicky, Synthesis, 2019, 51, 225–232 CrossRef CAS; (b) M. Makarova, M. A. A. Endoma-Arias, H. E. Dela Paz, R. Simionescu and T. Hudlicky, J. Am. Chem. Soc., 2019, 141, 10883–10904 CrossRef CAS PubMed; (c) M. A. A. Endoma-Arias, M. Makarova, H. E. Dela Paz and T. Hudlicky, ChemistrySelect, 2020, 5, 8241–8245 CrossRef CAS; (d) M. A. Endoma-Arias, H. Dela Paz and T. Hudlicky, Molecules, 2019, 24, 3477 CrossRef PubMed.
- M. A. A. Endoma-Arias and T. Hudlicky, Chem. – Eur. J., 2016, 22, 14540–14543 CrossRef PubMed.
- S. Yang, M. G. Banwell, A. C. Willis and J. S. Ward, Aust. J. Chem., 2015, 68, 241–247 CrossRef CAS.
- B. D. Schwartz, M. G. Banwell and I. A. Cade, Tetrahedron Lett., 2011, 52, 4526–4528 CrossRef CAS.
- (a) L. V. White, B. D. Schwartz, M. G. Banwell and A. C. Willis, J. Org. Chem., 2011, 76, 6250–6257 CrossRef CAS PubMed; (b) Compare Scheme 19 of our previous review, ref. 33..
- (a) J. N. Buckler, E. S. Taher, N. J. Fraser, A. C. Willis, P. D. Carr, C. J. Jackson and M. G. Banwell, J. Org. Chem., 2017, 82, 7869–7886 CrossRef CAS PubMed; (b) L. V. White and M. G. Banwell, J. Org. Chem., 2016, 81, 1617–1626 CrossRef CAS PubMed.
- C. M. Heckmann and F. Paradisi, ChemCatChem, 2020, 12, 6082–6102 CrossRef CAS PubMed.
- (a) R. J. Sharpe and J. S. Johnson, J. Am. Chem. Soc., 2015, 137, 4968–4971 CrossRef CAS PubMed; (b) R. J. Sharpe and J. S. Johnson, J. Org. Chem., 2015, 80, 9740–9766 CrossRef CAS PubMed; (c) A. K. Chattopadhyay, V. L. Ly, S. Jakkepally, G. Berger and S. Hanessian, Angew. Chem., Int. Ed., 2016, 55, 2577–2581 CrossRef CAS PubMed.
- C. A. M. Fraga and E. J. Barreiro, Chirality, 1996, 8, 305–310 CrossRef CAS PubMed.
- (a) H. Watanabe, M. Iwamoto and M. Nakada, J. Org. Chem., 2005, 70, 4652–4658 CrossRef CAS PubMed; (b) T. Katoh, S. Mizumoto, M. Fudesaka, Y. Nakashima, T. Kajimoto and M. Node, Synlett, 2006, 2006, 2176–2182 CrossRef; (c) T. Katoh, S. Mizumoto, M. Fudesaka, M. Takeo, T. Kajimoto and M. Node, Tetrahedron: Asymmetry, 2006, 17, 1655–1662 CrossRef CAS.
- B. Kovács, R. Megyesi, E. Forró and F. Fülöp, Tetrahedron: Asymmetry, 2017, 28, 1829–1833 CrossRef.
- B. Kovács, E. Forró and F. Fülöp, Tetrahedron, 2018, 74, 6873–6877 CrossRef.
- M. Stirling, J. Blacker and M. I. Page, Tetrahedron Lett., 2007, 48, 1247–1250 CrossRef CAS.
- B. K. Corrêa, T. R. C. Silva and C. Raminelli, Tetrahedron Lett., 2018, 59, 3583–3585 CrossRef.
- E. Forró, L. Schönstein and F. Fülöp, Tetrahedron: Asymmetry, 2011, 22, 1255–1260 CrossRef.
- (a) L. Schönstein, E. Forró and F. Fülöp, Tetrahedron: Asymmetry, 2013, 24, 202–206 CrossRef; (b) L. Schönstein, E. Forró and F. Fülöp, Tetrahedron: Asymmetry, 2013, 24, 1059–1062 CrossRef.
- R. Megyesi, E. Forró and F. Fülöp, ChemistryOpen, 2016, 5, 254–260 CrossRef CAS PubMed.
- K. L. White and M. Movassaghi, J. Am. Chem. Soc., 2016, 138, 11383–11389 CrossRef CAS PubMed.
- S. Tagawa, T. Choshi, A. Okamoto, T. Nishiyama, S. Watanabe, N. Hatae, M. Ishikura and S. Hibino, Eur. J. Org. Chem., 2013, 2013, 1805–1810 CrossRef CAS.
- B. Sun, T. Morikawa, H. Matsuda, S. Tewtrakul, L. J. Wu, S. Harima and M. Yoshikawa, J. Nat. Prod., 2004, 67, 1464–1469 CrossRef CAS PubMed.
- (a) S. C. Philkhana, G. R. Jachak, V. B. Gunjal, N. M. Dhage, A. H. Bansode and D. S. Reddy, Tetrahedron Lett., 2015, 56, 1252–1254 CrossRef CAS; (b) S. C. Philkhana, A. K. Verma, G. R. Jachak, B. Hazra, A. Basu and D. S. Reddy, Eur. J. Med. Chem., 2017, 135, 89–109 CrossRef CAS PubMed.
- (a) J. Duan, B. Li, Y. Qin, Y. Dong, J. Ren and G. Li, Bioresources and Bioprocessing, 2019, 6, 37 CrossRef; (b) V. F. Batista, J. L. Galman, D. C. G. A. Pinto, A. M. S. Silva and N. J. Turner, ACS Catal., 2018, 8, 11889–11907 CrossRef CAS; (c) M. D. Patil, G. Grogan, A. Bommarius and H. Yun, ACS Catal., 2018, 8, 10985–11015 CrossRef CAS.
- D. Ghislieri, A. P. Green, M. Pontini, S. C. Willies, I. Rowles, A. Frank, G. Grogan and N. J. Turner, J. Am. Chem. Soc., 2013, 135, 10863–10869 CrossRef CAS PubMed.
- D. Ghislieri, D. Houghton, A. P. Green, S. C. Willies and N. J. Turner, ACS Catal., 2013, 3, 2869–2872 CrossRef CAS.
- J. H. Schrittwieser, B. Groenendaal, S. C. Willies, D. Ghislieri, I. Rowles, V. Resch, J. H. Sattler, E.-M. Fischereder, B. Grischek, W.-D. Lienhart, N. J. Turner and W. Kroutil, Catal. Sci. Technol., 2014, 4, 3657–3664 RSC.
- R. S. Heath, M. Pontini, B. Bechi and N. J. Turner, ChemCatChem, 2014, 6, 996–1002 CrossRef CAS.
- For a different approach to developing an amine oxidase that is stereocomplementary to MAO-N, see: K. Yasukawa, S. Nakano and Y. Asano, Angew. Chem., Int. Ed., 2014, 53, 4428–4431 CrossRef CAS PubMed.
- J. W. A. Koetter and G. E. Schulz, J. Mol. Biol., 2005, 352, 418–428 CrossRef CAS PubMed.
- M. T. Reetz, L.-W. Wang and M. Bocola, Angew. Chem., Int. Ed., 2006, 45, 1236–1241 CrossRef CAS PubMed.
- (a) I. Rowles, K. J. Malone, L. L. Etchells, S. C. Willies and N. J. Turner, ChemCatChem, 2012, 4, 1259–1261 CrossRef CAS; (b) R. Carr, M. Alexeeva, M. J. Dawson, V. Gotor-Fernández, C. E. Humphrey and N. J. Turner, ChemBioChem, 2005, 6, 637–639 CrossRef CAS PubMed.
- R. S. Heath, M. Pontini, S. Hussain and N. J. Turner, ChemCatChem, 2016, 8, 117–120 CrossRef CAS.
- S. Wallner, C. Dully, B. Daniel and P. Macheroux, in Handbook of Flavoproteins, ed. R. Hille, S. M. Miller and B. Palfey, De Gruyter, Berlin, 2013, vol. 1, pp. 1–30 Search PubMed.
- (a) A. Winkler, F. Hartner, T. M. Kutchan, A. Glieder and P. Macheroux, J. Biol. Chem., 2006, 281, 21276–21285 CrossRef CAS PubMed; (b) A. Winkler, A. Lyskowski, S. Riedl, M. Puhl, T. M. Kutchan, P. Macheroux and K. Gruber, Nat. Chem. Biol., 2008, 4, 739–741 CrossRef CAS PubMed.
- J. H. Schrittwieser, B. Groenendaal, V. Resch, D. Ghislieri, S. Wallner, E. M. Fischereder, E. Fuchs, B. Grischek, J. H. Sattler, P. Macheroux, N. J. Turner and W. Kroutil, Angew. Chem., Int. Ed., 2014, 53, 3731–3734 CrossRef CAS PubMed.
- S. Gandomkar, E. M. Fischereder, J. H. Schrittwieser, S. Wallner, Z. Habibi, P. Macheroux and W. Kroutil, Angew. Chem., Int. Ed., 2015, 54, 15051–15054 CrossRef CAS PubMed.
- For recent reviews on IREDs, see: (a) M. Lenz, N. Borlinghaus, L. Weinmann and B. M. Nestl, World J. Microbiol. Biotechnol., 2017, 33, 199 CrossRef PubMed; (b) J. Mangas-Sanchez, S. P. France, S. L. Montgomery, G. A. Aleku, H. Man, M. Sharma, J. I. Ramsden, G. Grogan and N. J. Turner, Curr. Opin. Chem. Biol., 2017, 37, 19–25 CrossRef CAS PubMed; (c) G. Grogan and N. J. Turner, Chem. – Eur. J., 2016, 22, 1900–1907 CrossRef CAS PubMed; (d) J. H. Schrittwieser, S. Velikogne and W. Kroutil, Adv. Synth. Catal., 2015, 357, 1655–1685 CrossRef CAS.
- For recent reviews on biocatalytic reductive amination, see: (a) L. Ducrot, M. Bennett, G. Grogan and C. Vergne-Vaxelaire, Adv. Synth. Catal., 2021, 363, 328–351 CrossRef CAS; (b) M. Sharma, J. Mangas-Sanchez, N. J. Turner and G. Grogan, Adv. Synth. Catal., 2017, 359, 2011–2025 CrossRef CAS PubMed.
- (a) M. Espinoza-Moraga, T. Petta, M. Vasquez-Vasquez, V. F. Laurie, L. A. B. Moraes and L. S. Santos, Tetrahedron: Asymmetry, 2010, 21, 1988–1992 CrossRef CAS; (b) Y. Mirabal-Gallardo, M. d. P. C. Soriano and L. S. Santos, Tetrahedron: Asymmetry, 2013, 24, 440–443 CrossRef CAS.
- K. Mitsukura, M. Suzuki, K. Tada, T. Yoshida and T. Nagasawa, Org. Biomol. Chem., 2010, 8, 4533–4535 RSC.
- (a) K. Mitsukura, T. Kuramoto, T. Yoshida, N. Kimoto, H. Yamamoto and T. Nagasawa, Appl. Microbiol. Biotechnol., 2013, 97, 8079–8086 CrossRef CAS PubMed; (b) K. Mitsukura, M. Suzuki, S. Shinoda, T. Kuramoto, T. Yoshida and T. Nagasawa, Biosci., Biotechnol., Biochem., 2011, 75, 1778–1782 CrossRef CAS PubMed.
- An online database of IRED sequences is maintained by researchers from the University of Stuttgart: https://ired.biocatnet.de/.
- Coniine isolated from poison hemlock is a mixture of the two enantiomers, of which the (R)-form has been shown to be the more potent neurotoxin. See: S. T. Lee, B. T. Green, K. D. Welch, J. A. Pfister and K. E. Panter, Chem. Res. Toxicol., 2008, 21, 2061–2064 Search PubMed.
- S. Hussain, F. Leipold, H. Man, E. Wells, S. P. France, K. R. Mulholland, G. Grogan and N. J. Turner, ChemCatChem, 2015, 7, 579–583 Search PubMed.
- (a) H. Man, E. Wells, S. Hussain, F. Leipold, S. Hart, J. P. Turkenburg, N. J. Turner and G. Grogan, ChemBioChem, 2015, 16, 1052–1059 CrossRef CAS PubMed; (b) N. Alvarenga, S. E. Payer, P. Petermeier, C. Kohlfuerst, A. L. Meleiro Porto, J. H. Schrittwieser and W. Kroutil, ACS Catal., 2020, 10, 1607–1620 CrossRef CAS.
- (a) F. Leipold, S. Hussain, D. Ghislieri and N. J. Turner, ChemCatChem, 2013, 5, 3505–3508 CrossRef CAS; (b) T. Huber, L. Schneider, A. Präg, S. Gerhardt, O. Einsle and M. Müller, ChemCatChem, 2014, 6, 2248–2252 CrossRef CAS; (c) G. A. Aleku, H. Man, S. P. France, F. Leipold, S. Hussain, L. Toca-Gonzalez, R. Marchington, S. Hart, J. P. Turkenburg, G. Grogan and N. J. Turner, ACS Catal., 2016, 6, 3880–3889 CrossRef CAS; (d) H. Li, P. Tian, J.-H. Xu and G.-W. Zheng, Org. Lett., 2017, 19, 3151–3154 CrossRef CAS PubMed; (e) S. Velikogne, V. Resch, C. Dertnig, J. H. Schrittwieser and W. Kroutil, ChemCatChem, 2018, 10, 3236–3246 CrossRef CAS PubMed.
- L. Yang, J. Zhu, C. Sun, Z. Deng and X. Qu, Chem. Sci., 2020, 11, 364–371 RSC.
- J. Zhu, H. Tan, L. Yang, Z. Dai, L. Zhu, H. Ma, Z. Deng, Z. Tian and X. Qu, ACS Catal., 2017, 7, 7003–7007 CrossRef CAS.
- I. Desgagné-Penix, Phytochem. Rev., 2021, 20, 409–431 CrossRef.
- A. P. Mattey, G. J. Ford, J. Citoler, C. Baldwin, J. R. Marshall, R. B. Palmer, M. Thompson, N. J. Turner, S. C. Cosgrove and S. L. Flitsch, Angew. Chem., Int. Ed., 2021, 60(34), 18660–18665 CrossRef PubMed.
- M. R. Chapman, S. C. Cosgrove, N. J. Turner, N. Kapur and A. J. Blacker, Angew. Chem., Int. Ed., 2018, 57, 10535–10539 CrossRef CAS PubMed.
- T. W. Thorpe, S. P. France, S. Hussain, J. R. Marshall, W. Zawodny, J. Mangas-Sanchez, S. L. Montgomery, R. M. Howard, D. S. B. Daniels, R. Kumar, F. Parmeggiani and N. J. Turner, J. Am. Chem. Soc., 2019, 141, 19208–19213 CrossRef CAS PubMed.
- S. P. France, S. Hussain, A. M. Hill, L. J. Hepworth, R. M. Howard, K. R. Mulholland, S. L. Flitsch and N. J. Turner, ACS Catal., 2016, 6, 3753–3759 CrossRef CAS.
- (a) N. G. Schmidt, E. Eger and W. Kroutil, ACS Catal., 2016, 6, 4286–4311 CrossRef CAS PubMed; (b) C. L. Windle, M. Müller, A. Nelson and A. Berry, Curr. Opin. Chem. Biol., 2014, 19, 25–33 CrossRef CAS PubMed; (c) M. Brovetto, D. Gamenara, P. S. Mendez and G. A. Seoane, Chem. Rev., 2011, 111, 4346–4403 CrossRef CAS PubMed.
- These molecules are inhibitors of glycosidases and glycosyltransferases and have found various therapeutic applications. For reviews, see: (a) I. Conforti and A. Marra, Org. Biomol. Chem., 2021, 19, 5439–5475 RSC; (b) R. J. Nash, A. Kato, C.-Y. Yu and G. W. Fleet, Future Med. Chem., 2011, 3, 1513–1521 CrossRef CAS PubMed; (c) B. G. Winchester, Tetrahedron: Asymmetry, 2009, 20, 645–651 CrossRef CAS.
- I. Oroz-Guinea, K. Hernández, F. Camps Bres, C. Guérard-Hélaine, M. Lemaire, P. Clapés and E. García-Junceda, Adv. Synth. Catal., 2015, 357, 1951–1960 CrossRef CAS.
- P. Laborda, F. J. Sayago, C. Cativiela, T. Parella, J. Joglar and P. Clapes, Org. Lett., 2014, 16, 1422–1425 CrossRef CAS PubMed.
- A. Soler, M. L. Gutiérrez, J. Bujons, T. Parella, C. Minguillon, J. Joglar and P. Clapés, Adv. Synth. Catal., 2015, 357, 1787–1807 CrossRef CAS.
- A. Soler, X. Garrabou, K. Hernández, M. L. Gutiérrez, E. Busto, J. Bujons, T. Parella, J. Joglar and P. Clapés, Adv. Synth. Catal., 2014, 356, 3007–3024 CrossRef CAS.
- A. L. Concia, L. Gomez, T. Parella, J. Joglar and P. Clapes, J. Org. Chem., 2014, 79, 5386–5389 CrossRef CAS PubMed.
- A. L. Concia, L. Gomez, J. Bujons, T. Parella, C. Vilaplana, P. J. Cardona, J. Joglar and P. Clapes, Org. Biomol. Chem., 2013, 11, 2005–2021 RSC.
- For recent reviews, see: (a) S. A. Kelly, S. Mix, T. S. Moody and B. F. Gilmore, Appl. Microbiol. Biotechnol., 2020, 104, 4781–4794 CrossRef CAS PubMed; (b) A. Gomm and E. O'Reilly, Curr. Opin. Chem. Biol., 2018, 43, 106–112 CrossRef CAS PubMed; (c) S. A. Kelly, S. Pohle, S. Wharry, S. Mix, C. C. R. Allen, T. S. Moody and B. F. Gilmore, Chem. Rev., 2018, 118, 349–367 CrossRef CAS PubMed; (d) M. D. Patil, G. Grogan, A. Bommarius and H. Yun, Catalysts, 2018, 8, 254 CrossRef; (e) E. E. Ferrandi and D. Monti, World J. Microbiol. Biotechnol., 2017, 34, 13 CrossRef PubMed; (f) F. Guo and P. Berglund, Green Chem., 2017, 19, 333–360 RSC; (g) I. Slabu, J. L. Galman, R. C. Lloyd and N. J. Turner, ACS Catal., 2017, 7, 8263–8284 CrossRef CAS; (h) M. Fuchs, J. E. Farnberger and W. Kroutil, Eur. J. Org. Chem., 2015, 2015, 6965–6982 CrossRef CAS PubMed.
- (a) R. C. Simon, B. Grischek, F. Zepeck, A. Steinreiber, F. Belaj and W. Kroutil, Angew. Chem., Int. Ed., 2012, 51, 6713–6716 CrossRef CAS PubMed; (b) R. C. Simon, F. Zepeck and W. Kroutil, Chem. – Eur. J., 2013, 19, 2859–2865 CrossRef CAS PubMed; (c) R. C. Simon, C. S. Fuchs, H. Lechner, F. Zepeck and W. Kroutil, Eur. J. Org. Chem., 2013, 2013, 3397–3402 CrossRef CAS PubMed.
- The reduction step in this sequence is interesting from a stereochemical perspective, as a typically (R)-selective IRED is used to generate an (S)-configured stereogenic centre, albeit in only 13% de. In general, the authors observed that all 2,6- and 2,4-disubstituted piperidines investigated in their study were formed with a preference for the cis-isomer regardless of the IRED used, indicating substrate-based stereocontrol..
- S. E. Payer, J. H. Schrittwieser, B. Grischek, R. C. Simon and W. Kroutil, Adv. Synth. Catal., 2016, 358, 444–451 CrossRef CAS.
- J. Ryan, M. Siauciulis, A. Gomm, B. Macia, E. O'Reilly and V. Caprio, J. Am. Chem. Soc., 2016, 138, 15798–15800 CrossRef CAS PubMed.
- F. Taday, J. Ryan, S. P. Argent, V. Caprio, B. Macia and E. O'Reilly, Chem. – Eur. J., 2020, 26, 3729–3732 CrossRef CAS PubMed.
- R. Krizevski, E. Bar, O. Shalit, Y. Sitrit, S. Ben-Shabat and E. Lewinsohn, Phytochemistry, 2010, 71, 895–903 CrossRef CAS PubMed.
- T. Sehl, H. C. Hailes, J. M. Ward, R. Wardenga, E. von Lieres, H. Offermann, R. Westphal, M. Pohl and D. Rother, Angew. Chem., Int. Ed., 2013, 52, 6772–6775 CrossRef CAS PubMed.
- T. Sehl, H. C. Hailes, J. M. Ward, U. Menyes, M. Pohl and D. Rother, Green Chem., 2014, 16, 3341–3348 RSC.
- X. Wu, M. Fei, Y. Chen, Z. Wang and Y. Chen, Appl. Microbiol. Biotechnol., 2014, 98, 7399–7408 CrossRef CAS PubMed.
- Y.-C. Lee, Y.-Y. Chen, J.-S. Lin, Y.-W. Chen, C.-C. Li, K.-X. Liang, H.-H. Chan, W.-D. Lin and C.-H. Kao, Process Biochem., 2018, 73, 74–81 CrossRef CAS.
- (a) M. L. Corrado, T. Knaus and F. G. Mutti, Green Chem., 2019, 21, 6246–6251 RSC; (b) M. L. Corrado, T. Knaus and F. G. Mutti, ChemBioChem, 2021, 22, 2345–2350 CrossRef CAS PubMed.
- This numbering, which is also used in the original publications, is based on 1-phenylpropylene oxide as the name of compound 291. If the preferred IUPAC name, 2-methyl-3-phenyloxirane, is used, the benzylic carbon atom is assigned number 3..
- J. H. Sattler, M. Fuchs, K. Tauber, F. G. Mutti, K. Faber, J. Pfeffer, T. Haas and W. Kroutil, Angew. Chem., Int. Ed., 2012, 51, 9156–9159 CrossRef CAS PubMed.
- J. L. Galman, I. Slabu, F. Parmeggiani and N. J. Turner, Chem. Commun., 2018, 54, 11316–11319 RSC.
- I. Slabu, J. L. Galman, N. J. Weise, R. C. Lloyd and N. J. Turner, ChemCatChem, 2016, 8, 1038–1042 CrossRef CAS.
- (a) R. Roddan, J. M. Ward, N. H. Keep and H. C. Hailes, Curr. Opin. Chem. Biol., 2020, 55, 69–76 CrossRef CAS PubMed; (b) M. D. Patil, G. Grogan and H. Yun, ChemCatChem, 2018, 10, 4783–4804 CrossRef.
- J. Stöckigt, A. P. Antonchick, F. Wu and H. Waldmann, Angew. Chem., Int. Ed., 2011, 50, 8538–8564 CrossRef PubMed.
- (a) B. R. Lichman, M. C. Gershater, E. D. Lamming, T. Pesnot, A. Sula, N. H. Keep, H. C. Hailes and J. M. Ward, FEBS J., 2015, 282, 1137–1151 CrossRef CAS PubMed; (b) B. R. Lichman, A. Sula, T. Pesnot, H. C. Hailes, J. M. Ward and N. H. Keep, Biochemistry, 2017, 56, 5274–5277 CrossRef CAS PubMed; (c) X. Sheng and F. Himo, J. Am. Chem. Soc., 2019, 141, 11230–11238 CrossRef CAS PubMed.
- V. Erdmann, B. R. Lichman, J. Zhao, R. C. Simon, W. Kroutil, J. M. Ward, H. C. Hailes and D. Rother, Angew. Chem., Int. Ed., 2017, 56, 12503–12507 CrossRef CAS PubMed.
- Y. Wang, N. Tappertzhofen, D. Méndez-Sánchez, M. Bawn, B. Lyu, J. M. Ward and H. C. Hailes, Angew. Chem., Int. Ed., 2019, 58, 10120–10125 CrossRef CAS PubMed.
- A. Bonamore, I. Rovardi, F. Gasparrini, P. Baiocco, M. Barba, C. Molinaro, B. Botta, A. Boffi and A. Macone, Green Chem., 2010, 12, 1623–1627 RSC.
- J. J. Maresh, S. O. Crowe, A. A. Ralko, M. D. Aparece, C. M. Murphy, M. Krzeszowiec and M. W. Mullowney, Tetrahedron Lett., 2014, 55, 5047–5051 CrossRef CAS.
- A. Bonamore, L. Calisti, A. Calcaterra, O. H. Ismail, M. Gargano, I. D'Acquarica, B. Botta, A. Boffi and A. Macone, ChemistrySelect, 2016, 1, 1525–1528 CrossRef CAS.
- B. R. Lichman, E. D. Lamming, T. Pesnot, J. M. Smith, H. C. Hailes and J. M. Ward, Green Chem., 2015, 17, 852–855 RSC.
- J. Zhao, B. R. Lichman, J. M. Ward and H. C. Hailes, Chem. Commun., 2018, 54, 1323–1326 RSC.
- (a) T. Pesnot, M. C. Gershater, J. M. Ward and H. C. Hailes, Adv. Synth. Catal., 2012, 354, 2997–3008 CrossRef CAS; (b) M. Nishihachijo, Y. Hirai, S. Kawano, A. Nishiyama, H. Minami, T. Katayama, Y. Yasohara, F. Sato and H. Kumagai, Biosci., Biotechnol., Biochem., 2014, 78, 701–707 CrossRef CAS PubMed.
- R. Roddan, G. Gygli, A. Sula, D. Méndez-Sánchez, J. Pleiss, J. M. Ward, N. H. Keep and H. C. Hailes, ACS Catal., 2019, 9, 9640–9649 CrossRef CAS.
- R. Roddan, A. Sula, D. Méndez-Sánchez, F. Subrizi, B. R. Lichman, J. Broomfield, M. Richter, J. N. Andexer, J. M. Ward, N. H. Keep and H. C. Hailes, Commun. Chem., 2020, 3, 170 CrossRef CAS.
- B. R. Lichman, J. Zhao, H. C. Hailes and J. M. Ward, Nat. Commun., 2017, 8, 14883 CrossRef CAS PubMed.
- J. Zhao, D. Méndez-Sánchez, R. Roddan, J. M. Ward and H. C. Hailes, ACS Catal., 2021, 11, 131–138 CrossRef CAS.
- E.-M. Fischereder, D. Pressnitz and W. Kroutil, ACS Catal., 2016, 6, 23–30 CrossRef CAS.
- Y. Cai, H. Zhu, Z. Alperstein, W. Yu, A. Cherkasov and H. Zou, ACS Chem. Biol., 2017, 12, 3086–3092 CrossRef CAS PubMed.
- Y. Cai, N. Shao, H. Xie, Y. Futamura, S. Panjikar, H. Liu, H. Zhu, H. Osada and H. Zou, ACS Catal., 2019, 9, 7443–7448 CrossRef CAS.
- F. Wu, H. Zhu, L. Sun, C. Rajendran, M. Wang, X. Ren, S. Panjikar, A. Cherkasov, H. Zou and J. Stöckigt, J. Am. Chem. Soc., 2012, 134, 1498–1500 CrossRef CAS PubMed.
- D. Pressnitz, E.-M. Fischereder, J. Pletz, C. Kofler, L. Hammerer, K. Hiebler, H. Lechner, N. Richter, E. Eger and W. Kroutil, Angew. Chem., Int. Ed., 2018, 57, 10683–10687 CrossRef CAS PubMed.
- This switch in stereopreference compared to the natural reaction was later rationalised based on X-ray crystal structures, mutational studies, and molecular dynamics simulations, which suggest an inverted mode of substrate binding as the underlying cause. Quantum chemical calculations, on the other hand, provided no support for this interpretation and instead point towards steric interactions in the last transition state as the determinants of stereoselectivity. See: (a) E. Eger, A. Simon, M. Sharma, S. Yang, W. B. Breukelaar, G. Grogan, K. N. Houk and W. Kroutil, J. Am. Chem. Soc., 2020, 142, 792–800 CrossRef CAS PubMed; (b) X. Sheng and F. Himo, ACS Catal., 2020, 10, 13630–13640 CrossRef CAS.
- E. Eger, J. H. Schrittwieser, D. Wetzl, H. Iding, B. Kuhn and W. Kroutil, Chem. – Eur. J., 2020, 26, 16281–16285 CrossRef CAS PubMed.
- For the origin of this protein engineering strategy, see: (a) P. Dalby, World Pat., WO2004/024918 A1, UCL Biomedica, 2004; (b) Z. Chen and H. Zhao, J. Mol. Biol., 2005, 348, 1273–1282 CrossRef CAS PubMed.
- For selected recent examples, see: (a) C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman and G. J. Hughes, Science, 2010, 329, 305–309 CrossRef CAS PubMed; (b) H. Li and J. C. Liao, ACS Synth. Biol., 2014, 3, 13–20 CrossRef CAS PubMed; (c) J. T. Payne, C. B. Poor and J. C. Lewis, Angew. Chem., Int. Ed., 2015, 54, 4226–4230 CrossRef CAS PubMed; (d) P. E. Affaticati, S.-B. Dai, P. Payongsri, H. C. Hailes, K. Tittmann and P. A. Dalby, Sci. Rep., 2016, 6, 35716 CrossRef CAS PubMed.
- R. Mei, D. Xu, H. Hu, D. Song, H. Zhang, D. Ma, X. Xie and X. She, Org. Lett., 2015, 17, 2230–2233 CrossRef CAS PubMed.
- (a) F. H. Arnold, Angew. Chem., Int. Ed., 2018, 57, 4143–4148 CrossRef CAS PubMed; (b) Y. Yang and F. H. Arnold, Acc. Chem. Res., 2021, 54, 1209–1225 CrossRef CAS PubMed.
- (a) P. S. Coelho, Z. J. Wang, M. E. Ener, S. A. Baril, A. Kannan, F. H. Arnold and E. M. Brustad, Nat. Chem. Biol., 2013, 9, 485–487 CrossRef CAS PubMed; (b) C. C. Farwell, R. K. Zhang, J. A. McIntosh, T. K. Hyster and F. H. Arnold, ACS Cent. Sci., 2015, 1, 89–93 CrossRef CAS PubMed; (c) C. K. Prier, R. K. Zhang, A. R. Buller, S. Brinkmann-Chen and F. H. Arnold, Nat. Chem., 2017, 9, 629–634 CrossRef CAS PubMed.
- R. K. Zhang, K. Chen, X. Huang, L. Wohlschlager, H. Renata and F. H. Arnold, Nature, 2019, 565, 67–72 CrossRef CAS PubMed.
- For recent examples, see: (a) Y.-D. Cao, Y.-C. He, H. Li, G.-Y. Kai, J.-H. Xu and H.-L. Yu, J. Biotechnol., 2015, 211, 123–129 CrossRef CAS PubMed; (b) A. B. Cardillo, M. Perassolo, M. Sartuqui, J. Rodríguez Talou and A. M. Giulietti, Biochem. Eng. J., 2017, 125, 180–189 CrossRef CAS; (c) S. A. Loskot, D. K. Romney, F. H. Arnold and B. M. Stoltz, J. Am. Chem. Soc., 2017, 139, 10196–10199 CrossRef CAS PubMed.
- For a general review of enzymatic late-stage functionalisation, see: E. Romero, B. S. Jones, B. N. Hogg, A. Rué Casamajo, M. A. Hayes, S. L. Flitsch, N. J. Turner and C. Schnepel, Angew. Chem., Int. Ed., 2021, 60, 16824–16855 CrossRef CAS PubMed.
- (a) C. R. Zwick and H. Renata, J. Am. Chem. Soc., 2018, 140, 1165–1169 CrossRef CAS PubMed; (b) C. R. Zwick and H. Renata, J. Org. Chem., 2018, 83, 7407–7415 CrossRef CAS PubMed.
- K. Namba, T. Shinada, T. Teramoto and Y. Ohfune, J. Am. Chem. Soc., 2000, 122, 10708–10709 CrossRef CAS.
- For a recent review focused on plant enzymes, see: H. Kries and S. E. O'Connor, Curr. Opin. Chem. Biol., 2016, 31, 22–30 CrossRef CAS PubMed.
- For recent reviews discussing the role of omics in the elucidation of secondary metabolism, see: (a) A. Rai, K. Saito and M. Yamazaki, Plant J., 2017, 90, 764–787 CrossRef CAS PubMed; (b) S. Zeilinger, C. García-Estrada and J.-F. Martín, in Biosynthesis and Molecular Genetics of Fungal Secondary Metabolites, ed. S. Zeilinger, J.-F. Martín and C. García-Estrada, Springer, New York, 2015, vol. 2, pp. 1–12 CrossRef; (c) E. Palazzotto and T. Weber, Curr. Opin. Microbiol., 2018, 45, 109–116 CrossRef CAS PubMed.
- R. Carlson, Nat. Biotechnol., 2009, 27, 1091–1094 CrossRef CAS PubMed.
- H. Lechner, P. Soriano, R. Poschner, H. C. Hailes, J. M. Ward and W. Kroutil, Biotechnol. J., 2018, 13, 1700542 CrossRef PubMed.
- For a notable exception, see: (a) R. Tanifuji, K. Koketsu, M. Takakura, R. Asano, A. Minami, H. Oikawa and H. Oguri, J. Am. Chem. Soc., 2018, 140, 10705–10709 CrossRef CAS PubMed; (b) R. Tanifuji, K. Tsukakoshi, K. Ikebukuro, H. Oikawa and H. Oguri, Bioorg. Med. Chem. Lett., 2019, 29, 1807–1811 CrossRef CAS PubMed.
- B. Daniel, B. Konrad, M. Toplak, M. Lahham, J. Messenlehner, A. Winkler and P. Macheroux, Arch. Biochem. Biophys., 2017, 632, 88–103 CrossRef CAS PubMed.
- (a) Y. Yao, C. An, D. Evans, W. Liu, W. Wang, G. Wei, N. Ding, K. N. Houk and S. S. Gao, J. Am. Chem. Soc., 2019, 141, 17517–17521 CrossRef CAS PubMed; (b) K.-L. Chen, C.-Y. Lai, M.-T. Pham, R.-J. Chein, Y. Tang and H.-C. Lin, Org. Lett., 2020, 22, 3302–3306 CrossRef CAS PubMed.
- A. Gesell, M. Rolf, J. Ziegler, M. L. Díaz Chávez, F.-C. Huang and T. M. Kutchan, J. Biol. Chem., 2009, 284, 24432–24442 CrossRef CAS PubMed.
- M. B. Kilgore, M. M. Augustin, G. D. May, J. A. Crow and T. M. Kutchan, Front. Plant Sci., 2016, 7, 225 Search PubMed.
- (a) S. C. Farrow, J. M. Hagel, G. A. W. Beaudoin, D. C. Burns and P. J. Facchini, Nat. Chem. Biol., 2015, 11, 728–732 CrossRef CAS PubMed; (b) T. Winzer, M. Kern, A. J. King, T. R. Larson, R. I. Teodor, S. L. Donninger, Y. Li, A. A. Dowle, J. Cartwright, R. Bates, D. Ashford, J. Thomas, C. Walker, T. A. Bowser and I. A. Graham, Science, 2015, 349, 309–312 CrossRef CAS PubMed; (c) M. Matuschek, C. Wallwey, B. Wollinsky, X. Xie and S.-M. Li, RSC Adv., 2012, 2, 3662–3669 RSC.
- For reviews, see: (a) J. S. Morris and P. J. Facchini, Front. Plant Sci., 2019, 10, 1058 CrossRef PubMed; (b) M. R. Bennett, S. A. Shepherd, V. A. Cronin and J. Micklefield, Curr. Opin. Chem. Biol., 2017, 37, 97–106 CrossRef CAS PubMed; (c) A.-W. Struck, M. L. Thompson, L. S. Wong and J. Micklefield, ChemBioChem, 2012, 13, 2642–2655 CrossRef CAS PubMed; (d) D. K. Liscombe, G. V. Louie and J. P. Noel, Nat. Prod. Rep., 2012, 29, 1238–1250 RSC.
- (a) F. Subrizi, Y. Wang, B. Thair, D. Méndez-Sánchez, R. Roddan, M. Cárdenas-Fernández, J. Siegrist, M. Richter, J. N. Andexer, J. M. Ward and H. C. Hailes, Angew. Chem., Int. Ed., 2021, 60, 18673–18679 CrossRef PubMed; (b) M. R. Bennett, M. L. Thompson, S. A. Shepherd, M. S. Dunstan, A. J. Herbert, D. R. M. Smith, V. A. Cronin, B. R. K. Menon, C. Levy and J. Micklefield, Angew. Chem., Int. Ed., 2018, 57, 10600–10604 CrossRef CAS PubMed; (c) E. Matsumura, A. Nakagawa, Y. Tomabechi, T. Koyanagi, H. Kumagai, K. Yamamoto, T. Katayama, F. Sato and H. Minami, Biosci., Biotechnol., Biochem., 2017, 81, 396–402 CrossRef CAS PubMed.
- (a) C. Liao and F. P. Seebeck, Nat. Catal., 2019, 2, 696–701 CrossRef CAS; (b) S. Mordhorst, J. Siegrist, M. Müller, M. Richter and J. N. Andexer, Angew. Chem., Int. Ed., 2017, 56, 4037–4041 CrossRef CAS PubMed; (c) Q. Tang, C. W. Grathwol, A. S. Aslan-Üzel, S. Wu, A. Link, I. V. Pavlidis, C. P. S. Badenhorst and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2021, 60, 1524–1527 CrossRef PubMed.
- I. J. W. McKean, P. A. Hoskisson and G. A. Burley, ChemBioChem, 2020, 21, 2890–2897 CrossRef CAS PubMed.
- (a) G. Hildebrandt and W. Klavehn, German Pat., DE548459C, Knoll A.-G. Chemische Fabriken, 1930; (b) C. Neuberg and J. Hirsch, Biochem. Z., 1921, 115, 282–310 CAS.
- For a related argument, see: M. Hönig, P. Sondermann, N. J. Turner and E. M. Carreira, Angew. Chem., Int. Ed., 2017, 56, 8942–8973 CrossRef PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |