DOI:
10.1039/D0SC04238B
(Perspective)
Chem. Sci., 2021,
12, 1964-1981
Catalysis using transition metal complexes featuring main group metal and metalloid compounds as supporting ligands
Received
3rd August 2020
, Accepted 9th September 2020
First published on 10th September 2020
Abstract
Recent development in catalytic application of transition metal complexes having an M–E bond (E = main group metal or metalloid element), which is stabilized by a multidentate ligand, is summarized. Main group metal and metalloid supporting ligands furnish unusual electronic and steric environments and molecular functions to transition metals, which are not easily available with standard organic supporting ligands such as phosphines and amines. These characteristics often realize remarkable catalytic activity, unique product selectivity, and new molecular transformations. This perspective demonstrates the promising utility of main group metal and metalloid compounds as a new class of supporting ligands for transition metal catalysts in synthetic chemistry.
 Jun Takaya | Jun Takaya received his PhD from Tokyo Institute of Technology (2004) under the supervision of Prof. Nobuharu Iwasawa. He joined Prof. John Hartwig at Yale University as a postdoctoral researcher in 2004. He was appointed as Assistant Professor at Tokyo Institute of Technology in 2005, and promoted to Associate Professor in 2014. He is also working as a PRESTO researcher of JST from 2017. He received Incentive Award in Synthetic Organic Chemistry (2013), Merck-Banyu Lectureship Award (2014), and The Young Scientist's Prize (2016). His research interests include the development and catalytic application of new transition metal complexes. |
1. Introduction
Transition metal catalysts have become widely adopted as useful tools in modern synthetic organic chemistry because of their diverse reactivity in enabling various molecular transformations. The chemistry has expansively grown along with the development of supporting ligands, which significantly affect the reactivity and stability of the metal complexes in the primary coordination sphere. Various organic compounds such as phosphines, amines, ethers, and carbenes are employed as supporting ligands for p-block organic elements in the periodic table.1 As a representative example, N-heterocyclic carbenes (NHCs) have been utilized as strong σ-donor ligands for various transition metals since the appearance of an isolable NHC in 1991,2a realizing highly efficient catalysis and sometimes enabling new molecular transformations.2b–d Development of new supporting ligands that supply unusual electronic and steric environments and molecular functions to transition metals has been an important challenge in expanding the chemistry of transition metal catalysts.
Recently, main group metal and metalloid compounds have emerged as a new class of supporting ligands for transition metals. These compounds often display characteristic coordination behaviour and electronic properties originating from their Lewis acidity and low electronegativity, which are usually not available with standard organic ligands. The redox activity and cooperative reactivity of these compounds are also of great interest. They are highly promising for exploring the new reactivity of transition metal catalysts for efficient molecular transformations. However, an M–E bond between a transition metal (M) and a main group metal or metalloid element (E) is usually unstable and not necessarily easy to form regardless of whether it is a covalent bond or a coordination bond. Therefore, development of new M–E complexes that are easily accessible and furnish sufficient stability and unique catalysis is highly desirable.
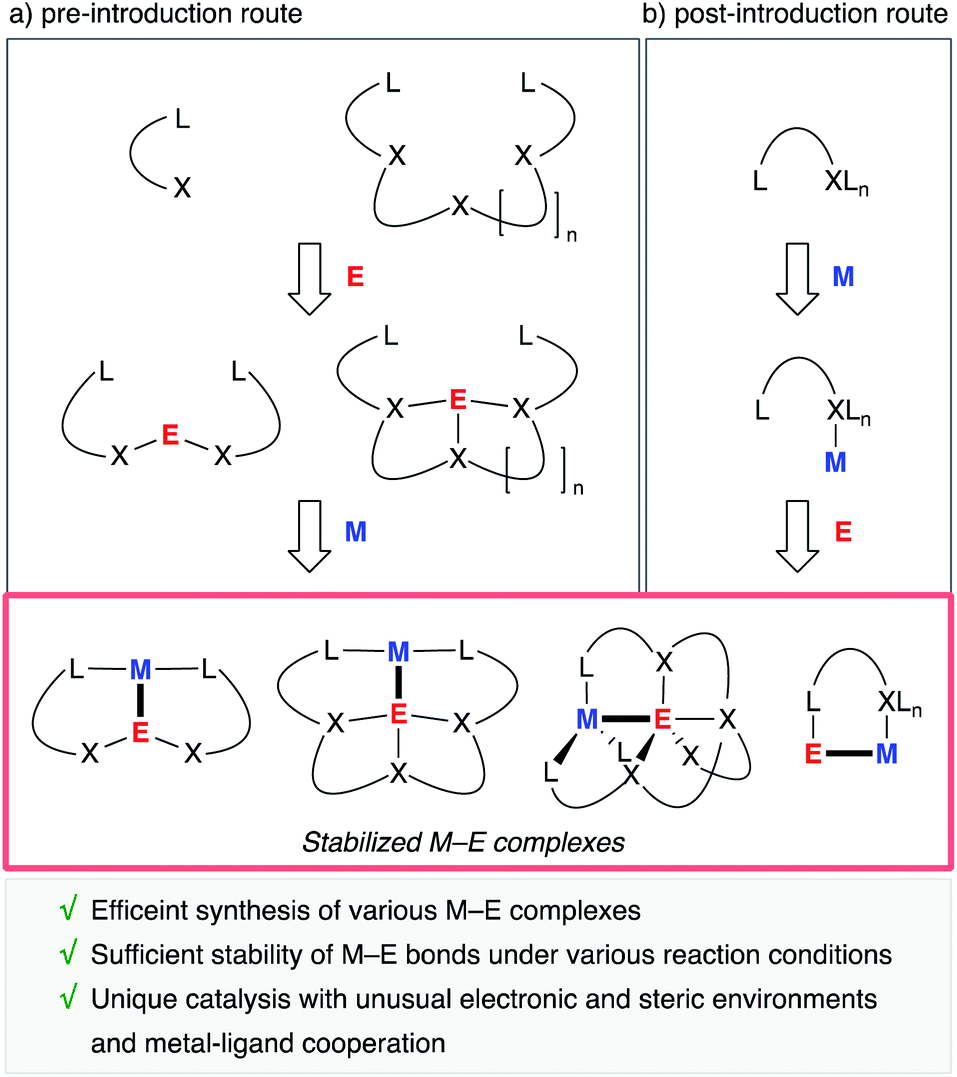
A useful strategy is to utilize rationally designed multidentate ligands as scaffolds for the M–E bonds (Fig. 1). A main group metal or metalloid element E is pre-incorporated into an organic ligand (XmLn) to form a multidentate ligand containing an E atom as one of the components of coordinating moieties. Following complexation with a transition metal an M–E complex is afforded through coordination of E to M or oxidative addition of a bond between E and its anionic ligand to M (Fig. 1, route a). As another approach, post-introduction of a main group metal or metalloid element E to a transition metal complex M bearing a rationally designed pendant moiety to coordinate to E is also possible (route b). These strategies enable efficient synthesis of a variety of M–E complexes, which are stable enough to be utilized as catalysts under various reaction conditions.
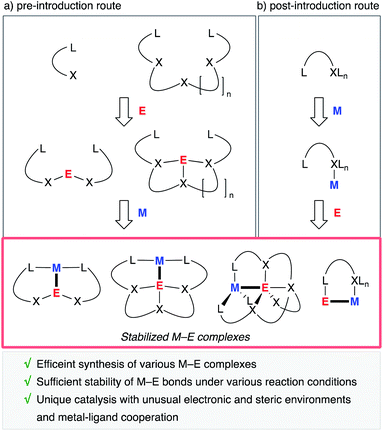 |
| | Fig. 1 Synthesis and catalytic application of stabilized M–E complexes. | |
This perspective article summarizes recent development of transition metal complexes having an M–E bond (E = main group metal or metalloid element), which is stabilized by a multidentate ligand, with an emphasis on catalysis where the main group metal or metalloid supporting ligand plays a crucial role in controlling the reactivity of the transition metal. E covers p-block metal and metalloid elements (B, Al, Ga, In, Si, Ge, Sb, and Bi) and a group 12 metal (Zn). We focus on the catalytic reactivity of M–E complexes employing E-containing tri- or tetradentate ligand structures (L2E- or L3E-type ligands), in which the direct interaction of the E–ligand with M certainly occurs in catalysts or catalytically relevant intermediates. Examples where the E–ligand acts as a Lewis acidic pendant moiety for activation of substrates in the secondary coordination sphere are not included. They are often seen with rather flexible E-containing bidentate ligands.3
2. How does E act as a supporting ligand? – general classification of the functions
2.1 Acting as a σ-acceptor ligand (Z-type ligand)
Lewis acidic metal and metalloid elements act as σ-acceptor ligands (Z-type ligand) via dative bonding with transition metals (Fig. 2A).4,5 This is often observed in group 13 element supporting ligands having a vacant p-orbital.3,6 Lewis acidic group 12 metals and some of the electronically positive heavier group 14 and 15 compounds can also be σ-acceptors for transition metals.7–9 The Z-type coordination activates the transition metal electrophilically by withdrawing electron density from the filled d-orbital, sometimes leading to change of the oxidation state of the transition metal not only in terms of formal description but also in terms of redox properties and reactivity. This is one of the characteristic properties of metal and metalloid supporting ligands distinct from standard organic ligands such as phosphines and amines, which usually coordinate to transition metals mainly through σ-donation along with partial π-back donation. Furthermore, the catalytic reactivity is modulative by exploiting the coordination and redox reactivity of the E–ligand. In particular, these characteristics are highly beneficial for electrophilic activation reactions of organic substrates such as alkynes and small molecules such as dihydrogen.
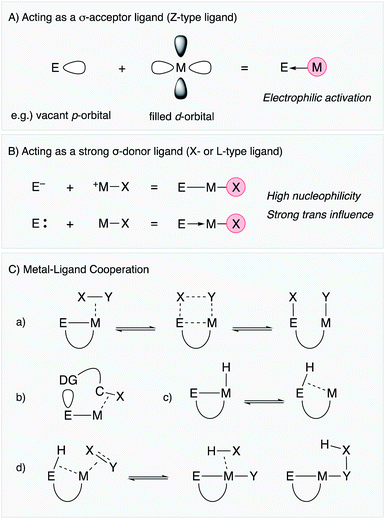 |
| | Fig. 2 General classification of the functions of M–E bonds. | |
2.2 Acting as a strong σ-donor ligand (X- or L-type ligand)
There are several examples of the X-type ligation of anionic group 13 and 14 atoms such as boron and silicon to transition metals to form covalent M–E bonds (Fig. 2B).10–14 These metal and metalloid ligands act as strong σ-donors and often exhibit strong trans influence, which destabilizes the M–X bond at the trans position.15,16 These characteristics lead to generation of highly reactive transition metal catalysts for various bond activation and nucleophilic addition reactions. Furthermore, metallylenes, which are neutral, low valent group 13 or 14 element compounds having +1 or +2 oxidation states, respectively, have also been utilized as L-type supporting ligands for transition metals.17–22 These metallylenes also act as strong σ-donors to facilitate various molecular transformations.
2.3 Metal–ligand cooperation (MLC)
In addition to the unique electronic characteristics described above, cooperative molecular activation and transformation are often enabled with M–E bonds through a kind of metal–ligand cooperation (MLC).23 As a representative example, an M–B complex having a Z-type boron–ligand cooperatively activates a substrate X–Y such as dihydrogen with a vacant p-orbital on B, promoting unusual oxidative addition of the X–Y bond across the M–B bond (Fig. 2C(a)).7 Similar MLC for activation of dihydrogen can also be considered with X-type boryl and alumanyl ligands, and they are successfully applied to various hydrogenation reactions. Moreover, the site-selective C–H bond activation of coordinated Lewis basic substrates such as pyridines is also considered as another type of MLC using the Lewis acidity of the E–ligand (Fig. 2C(b)). An M–E bond with an X-type heavier group 14 ligand, in particular a silyl ligand, undergoes facile interconversion between an η2-(Si–H)M complex and an Si–M–H complex through reversible reductive elimination/oxidative addition of the Si–H bond due to its high reactivity (Fig. 2C(c)).13,14 This is often involved in several catalytic cycles to generate coordinatively unsaturated metal species for substrate activation and transformations. Furthermore, unusual σ-bond metathesis reactions and metallation/elimination reactions that directly proceed between an η2-(Si–H)M complex and substrates without formation of a metal hydride intermediate have also been reported accompanied by formation/dissociation of the silyl ligand (Fig. 2C(d)). These processes can be regarded as a kind of σ-bond assisted metathesis (σ-CAM),24 which is distinct from usual oxidative addition/reductive elimination reactions or σ-bond metathesis reactions, demonstrating the privileged reactivity of M–E complexes.
3. Catalysis using M–E complexes in synthetic reactions
3.1 Group 12 (E = Zn)
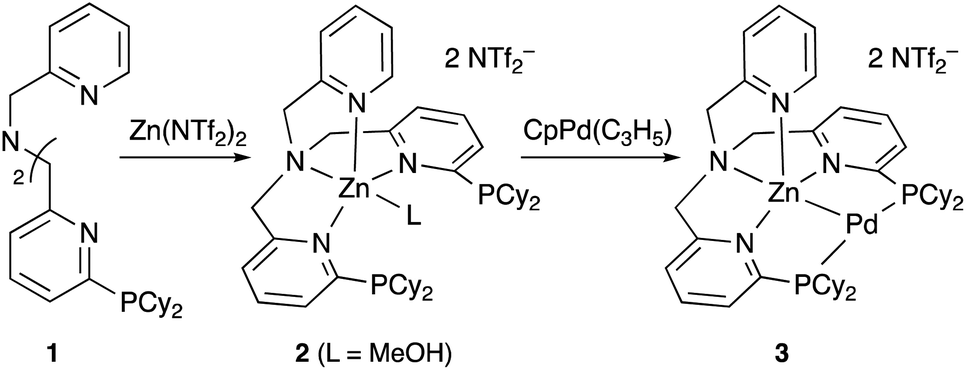
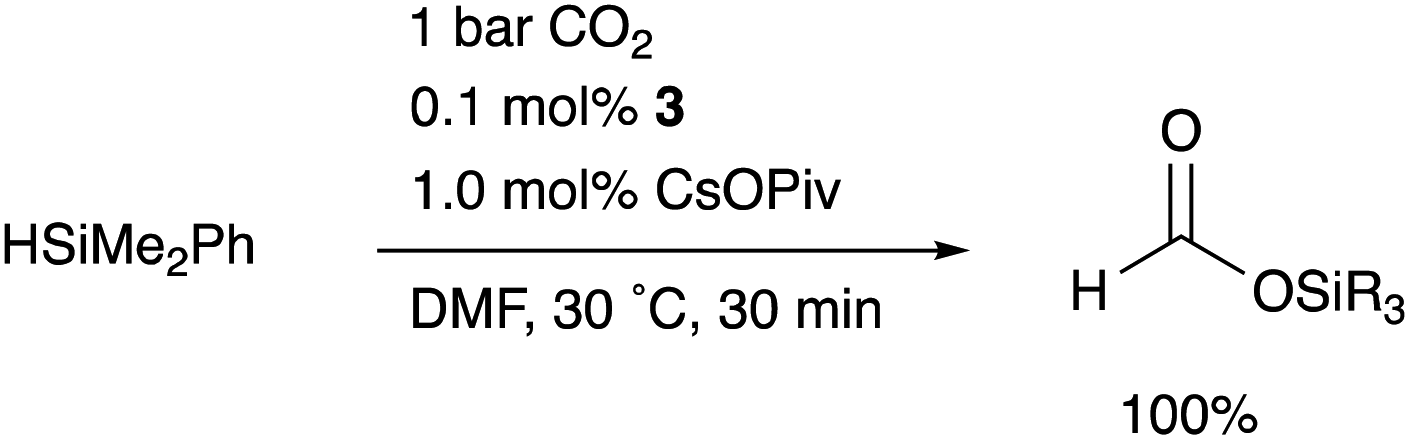
Although there are several reports on the synthesis and stoichiometric reactivity of M–E complexes with group 12 ligands (E = Zn, Cd, and Hg), their catalytic applications have scarcely been investigated to date.25 Tauchert reported the synthesis of a Pd complex 3 having Zn as a supporting ligand (a Zn-metalloligand) by utilizing a tris(pyridylmethyl)amine derivative 1 bearing two phosphine side arms as a scaffold for the Pd–Zn bond (Scheme 1).26 Pre-introduction of Zn(NTf)2 into 1 afforded the Zn-metalloligand 2, which reacted with CpPd(C3H5) to form a Pd–Zn bond. Theoretical calculations indicated that the Zn-metalloligand acts as a Z-type ligand for Pd via an acceptor s-orbital, which is a relatively weak acceptor compared with empty p-orbitals of group 13 elements such as B and Al. Corresponding Pd–Cu and Pd–Li complexes were also synthesized similarly, and the magnitude of σ-accepting ability was clarified to be in the order of Zn > Cu > Li. The Pd–Zn complex 3 exhibited high catalytic activity for hydrosilylation of CO2 to give silyl formate (Scheme 2). A corresponding Pd complex without the Zn-metalloligand and Pd–Cu and Pd–Li complexes resulted in low catalytic activity, thus demonstrating the importance of the Zn-metalloligand for efficient catalysis.
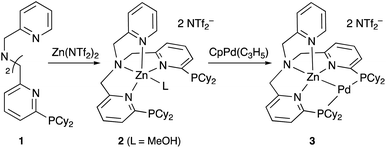 |
| | Scheme 1 Synthesis of the Pd–Zn complex 3. | |
 |
| | Scheme 2 Hydrosilylation of CO2 catalyzed by the Pd–Zn complex 3. | |
3.2 Group 13 (E = B, Al, Ga, and In)
A lot of rationally designed B-containing tri- or tetradentate ligands have been developed to date, and selected examples are listed in Fig. 3.3,5,6 These multidentate B–ligands are usually isolable and storable, enabling efficient synthesis of various M–B complexes by reacting with suitable transition metal precursors. These complexes display the unusual electronic nature and stoichiometric cooperative reactivity of the M–B bond for activation of substrates such as dihydrogen. Regarding catalytic applications, several reports briefly mentioned their catalytic activity for hydrogenation etc. although the role of the B–ligands is not necessarily clarified and advantageous.27
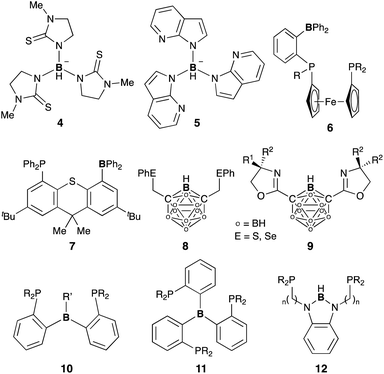 |
| | Fig. 3 Representative B-containing tri- or tetradentate ligands for M–B complexes. | |
3.2.1 Z-type borane ligand.
Important examples demonstrating the privileged reactivity of the M–B bond in catalysis have been reported using an o-phosphinophenyl linkage as a scaffold for the M–B bond. Bourissou developed bis- and tris(o-phosphinophenyl)boron 10 and 11 as boron-containing multidentate ligands and realized efficient syntheses and structural analyses of various transition metal complexes with them.28 The boron atom in 10 and 11 usually acts as a σ-accepting Z-type ligand with its vacant p-orbital, which is fixed proximally to the metal center due to the rigid tri- or tetradentate structure. This Z-type ligation leads to efficient stabilization of an electron-rich metal center and generation of a highly electrophilic metal center. Cooperative activation of substrates via MLC of the M–B bond is also possible.
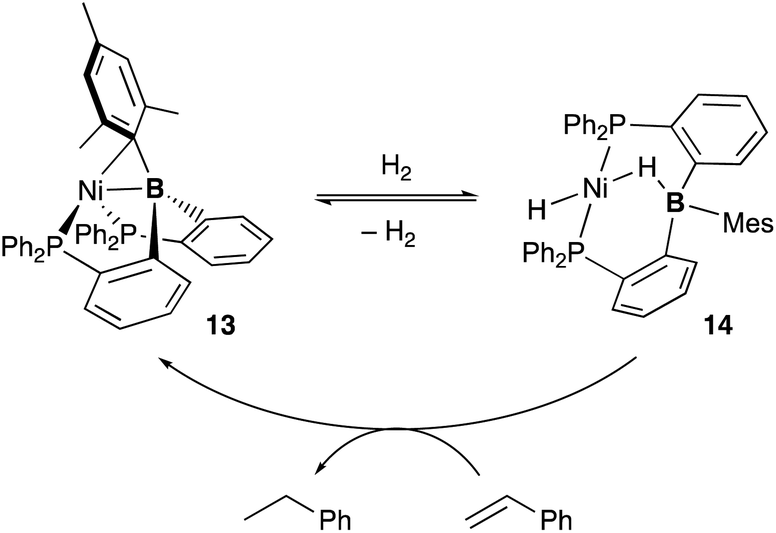
Several catalytic reactions where the PBP–ligand 10 functions as a Z-type ligand and exhibits MLC have been reported. Peters developed a Ni complex 13 with the PBP–ligand, in which the Ni coordinates to the B-Mes moiety in an η2-B,C fashion (Scheme 3).29 The high catalytic activity of 13 for hydrogenation of alkenes was demonstrated under 1 atm of H2. As a part of a plausible reaction mechanism, reversible oxidative addition of H2 across the Ni–B bond of 13 was evidenced by formation and structural characterization of a borohydride–hydride Ni complex 14.30 The Ni–B bond functioned as a kind of a frustrated Lewis pair for the heterolytic activation of H2, where the Lewis acidic boron atom accepted H− and the Lewis basic Ni accepted H+. The details of the reaction mechanism and the electronic process for the activation of the H–H bond across the Ni–B bond are investigated theoretically, clarifying that the B atom assists in the H–H σ-bond activation via charge transfer from H2 to the Ni–B bond and stabilizes the Ni–H–B bridging structure.31 A similar cooperative activation of the Si–H bond of H2SiPh2 was also reported with the Ni–B complex 13, realizing an efficient catalytic hydrosilylation reaction of aldehydes to obtain silyl ethers.32
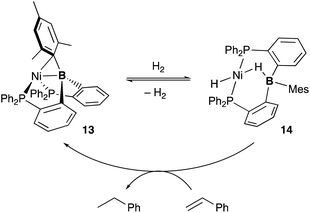 |
| | Scheme 3 Catalytic hydrogenation of alkenes via H–H σ-bond activation across the Ni–B bond of the PBP–Ni complex 13. | |
Peters also demonstrated the high utility of the P3B–ligand 11, tris(o-phosphinophenyl)boron, to stabilize electron rich, low valent Fe complexes for the catalytic conversion of dinitrogen to ammonia (Scheme 4). An anionic N2-coordinated Fe complex 15 was successfully prepared and structurally characterized by utilizing the P3B–ligand.33 It was demonstrated that 15 is catalytically active for ammonia generation from N2 in the presence of KC8 and HBAr4 (Ar = 3,5-(CF3)2C6H3) at low temperature. 7.0 equiv. of NH3 was obtained per Fe atom of 15, and 44% of the added protons were delivered to N2.34 This is the first example of a molecular Fe catalyst for NH3 generation from N2. This value was further updated to ca. 88 equiv. by carrying out the reaction with increased loading of the acid and the reductant under photoirradiation conditions.35 Several Fe–NxHy complexes were isolated and structurally characterized, and their stoichiometric reactivities were also obtained.36 It was confirmed that a cationic NH3-coordinated P3B–Fe complex 16 was formed from 15 by treatment with HBArF4·2Et2O and proton sponge, and 15 was regenerated by reduction of 16 with KC8 under N2 followed by further reduction with Na/Hg. It is proposed that the flexible Fe–B interaction, which varies the Fe–B distance, is important to stabilize various Fe–NxHy intermediates for efficient catalysis. The catalytic reactivity of the P3B–Fe complex for hydrogenation of alkenes is also reported via heterolytic H2 cleavage of the Fe–B bond.37
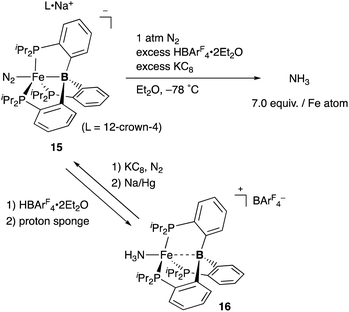 |
| | Scheme 4 Generation of ammonia from N2 catalyzed by the P3B–Fe complex 15. | |
Recently, Kameo and Bourissou reported another type of Pd–B cooperation involving anionic Pd species in a hydrodechlorination reaction of aryl chlorides.38 The reaction of a Pd complex 17 having a Z-type PBP–ligand with KH and [2.2.2]-cryptand afforded an anionic Pd complex 18, which possessed a bridging hydride ligand between Pd and B (Scheme 5). The anionic Pd complex 18 smoothly reacted with chlorobenzene to give benzene and the neutral Pd–B complex 17 whereas the same oxidative addition of PhCl to 17 did not proceed at all. This was extended to a catalytic hydrodechlorination reaction of aryl chlorides with 17 or 18 using HCOOK as a reductant in the presence of [2.2.2]-cryptand (Scheme 6). Control experiments using Pd(PPh3)4 with or without BPh3 as an external Lewis acid resulted in low yield, suggesting a crucial cooperative effect of Pd and B on the PBP–ligand. Mechanistic studies clarified that an anionic pathway is operative; the anionic Pd0 hydride complex 18 is formed first and reacts with aryl halides to give an aryl PdII complex 19. Intramolecular transfer of the hydride ligand from B to Pd followed by reductive elimination of a C–H bond affords the product with regeneration of the neutral Pd0 complex 17.
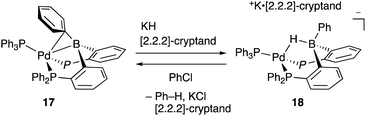 |
| | Scheme 5 Formation and reaction of the anionic PBP–Pd complex 18. | |
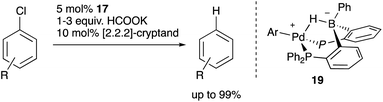 |
| | Scheme 6 Catalytic hydrodechlorination of aryl chlorides. | |
Inagaki developed a cationic Au complex 20 having a PBP–ligand, which was derived from a corresponding neutral PBP–AuCl complex originally reported by Bourissou (Scheme 7). 20 exhibited high catalytic activity for cyclization reactions of 1,n-enynes to give carbocycles in high yields.39 The same reaction using PPh3-coordinated cationic Au complex 21 or 22 without the B–ligand resulted in low yields of products or required long reaction time for completion of reactions. These results clearly demonstrate a positive effect of the Z-type boron ligand in promoting the electrophilic activation of alkynes by accepting electrons from the Au center. Related electrophilic activation reactions of alkynes catalyzed by Au–B complexes were also reported.40
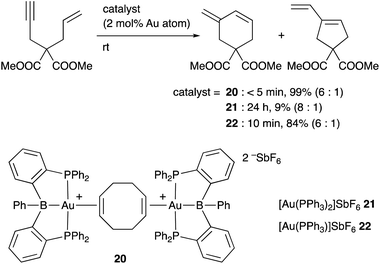 |
| | Scheme 7 Catalytic electrophilic activation of alkynes accelerated by the PBP–ligand. | |
3.2.2 X-type boryl ligand.
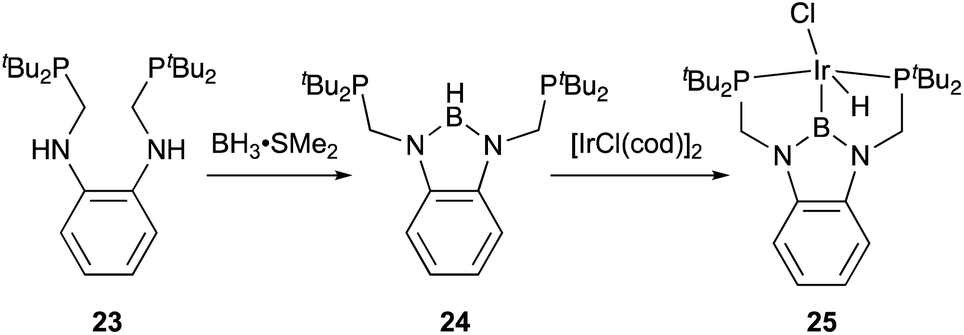
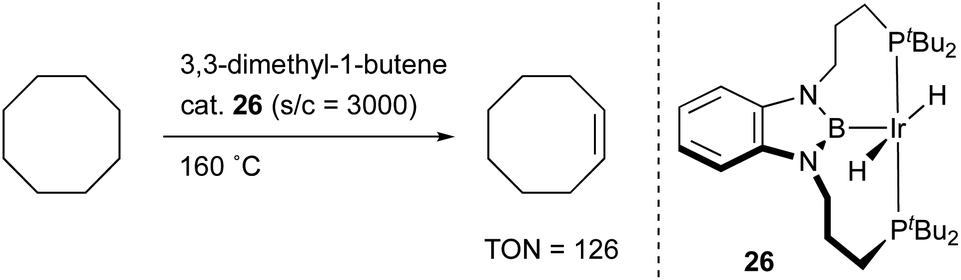
X-type boryl ligands are strong σ-donors and exhibit high trans influence.15,16 These characteristics are highly promising for realizing efficient transition metal catalysis for various molecular transformations. For the synthesis of M–B complexes having an X-type boryl ligand, the tridentate PBP–ligand 12 was developed by Yamashita and Nozaki, and the synthesis and stoichiometric reactivity of its Ir complex were demonstrated.41 A 1,2-bis(phosphinomethylamino)benzene derivative 23 reacted with BH3 to afford bis(phosphino)-hydridoborane 24 as the PBP–ligand, which underwent oxidative addition of the B–H bond to IrI smoothly to give the Ir–B complex 25 (Scheme 8). This method is widely applicable to the synthesis of various M–B complexes as demonstrated by the authors and other research groups later.12 Yamashita reported several catalytic applications of PBP–M complexes in dehydrogenation of alkanes or dimethylamine-borane, hydrosilylation of alkenes, and hydrogenation of aldehydes.42 In particular, the PBP–Ir complex 26 having long-tethered P,B-linkages exhibited good catalytic activity for dehydrogenation of cyclooctane to cyclooctene (TON = 126) although the activity is lower than those with widely employed PCP–Ir complexes (Scheme 9).43 López-Serrano and Rodriguez reported a hydrosilylation reaction of CO2 catalyzed by a PBP–Ni complex 27 to give a bis(silyl)acetal selectively with the highest TOF (55.8 h−1) ever reported for the reaction (Fig. 4).44 Zhang and Chen demonstrated the superior catalytic activity of a PBP–Pd complex 28 for a Suzuki coupling reaction to that of a corresponding PCP–Pd complex.45 The strong trans influence and σ-donor ability of the boryl ligand might positively affect these reactions although the details of the reaction mechanism and the role of the boryl ligand were not clarified.
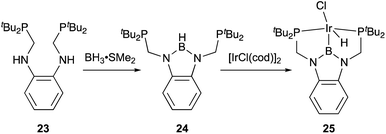 |
| | Scheme 8 Synthesis of transition metal complexes having an X-type boryl ligand. | |
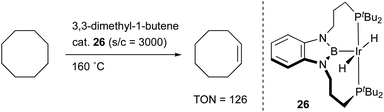 |
| | Scheme 9 Catalytic dehydrogenation of alkenes using the PBP–Ir complex 26. | |
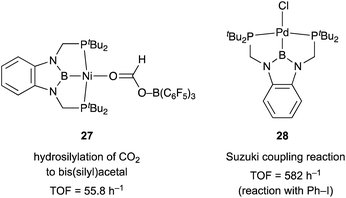 |
| | Fig. 4 Catalytic activity of PBP–Ni and PBP–Pd complexes 27 and 28. | |
Peters utilized the PBP–ligand 24 to synthesize a PBP–Co complex 29 having an X-type boryl ligand. The Co–B complex 29 was reversibly converted to a dihydridoborate–cobalt dihydride complex 30 in the presence of 1 atm H2 (Scheme 10).46 This is indicative of the cooperative activation of H2 across the Co–B bond via MLC. The PBP–Co complex 29 showed high catalytic activity for hydrogenation of octene and styrene, and the TOF reached 1000 per h (Scheme 11). This is a rare example of homogeneous Co-catalysis for alkene hydrogenation with high catalytic activity. The author also synthesized a bimetallic (Co–B)2 complex with PCy2 side arms that exhibited better catalytic activity for hydrogenation of internal alkenes than 29.47 Synthesis and reactions of corresponding Ni–B and (Ni–B)2 complexes were also demonstrated.
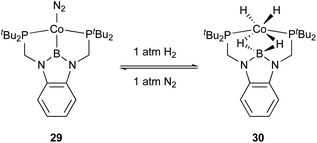 |
| | Scheme 10 Activation of H2 across the Co–B bond. | |
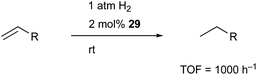 |
| | Scheme 11 Hydrogenation of alkenes catalyzed by the PBP–Co complex 29. | |
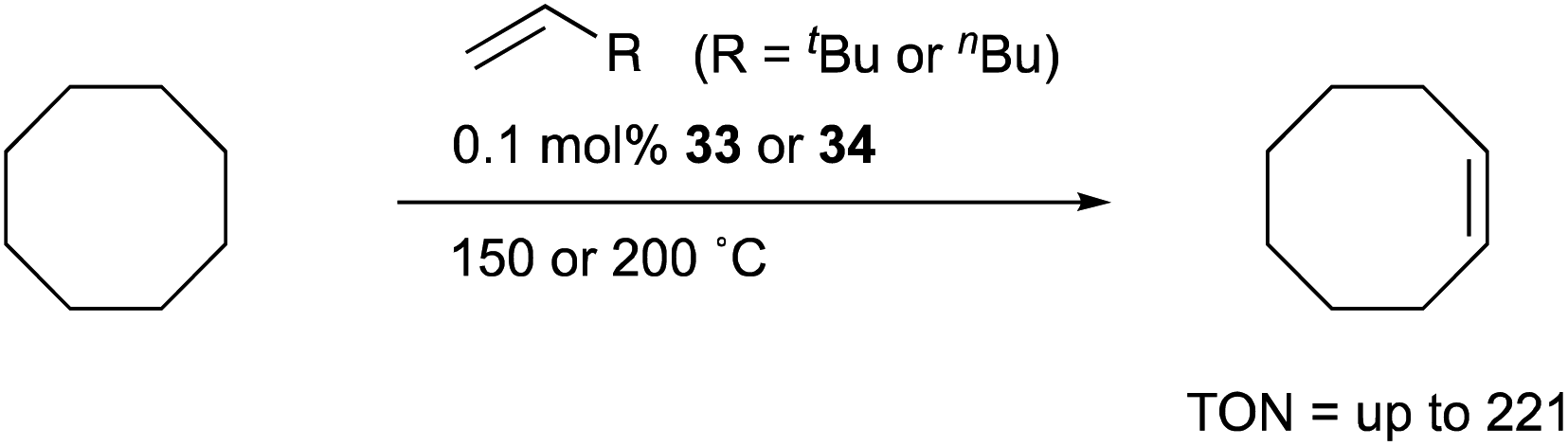
Transition metal complexes having an X-type boryl ligand stabilized by o-phosphinophenyl linkers have also been developed,48 and their stoichiometric reactivity and catalysis were reported by Ozerov. Treatment of a PBP–ligand 31 with [IrCl(coe)2]2 afforded a PBP–Ir complex 32via insertion of IrI into a B–Ph bond (Scheme 12).4932 was converted to Ir hydride complexes 33 and 34 having bridging hydride ligands between an Ir and a B atom through reactions with H2 or NaEt3BH. These complexes exhibited good catalytic activity for dehydrogenation of cyclooctane to cyclooctene with the maximum TON of 221, which is a better value than that for the PBP–Ir complex 26 (Scheme 13).
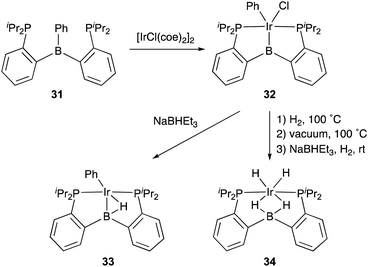 |
| | Scheme 12 Synthesis and reaction of the Ir–B complex 32 stabilized by o-phosphinophenyl linkers. | |
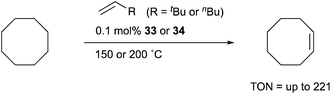 |
| | Scheme 13 Catalytic dehydrogenation using the PBP–Ir complexes 33 and 34. | |
3.2.3 L-type borylene ligand.
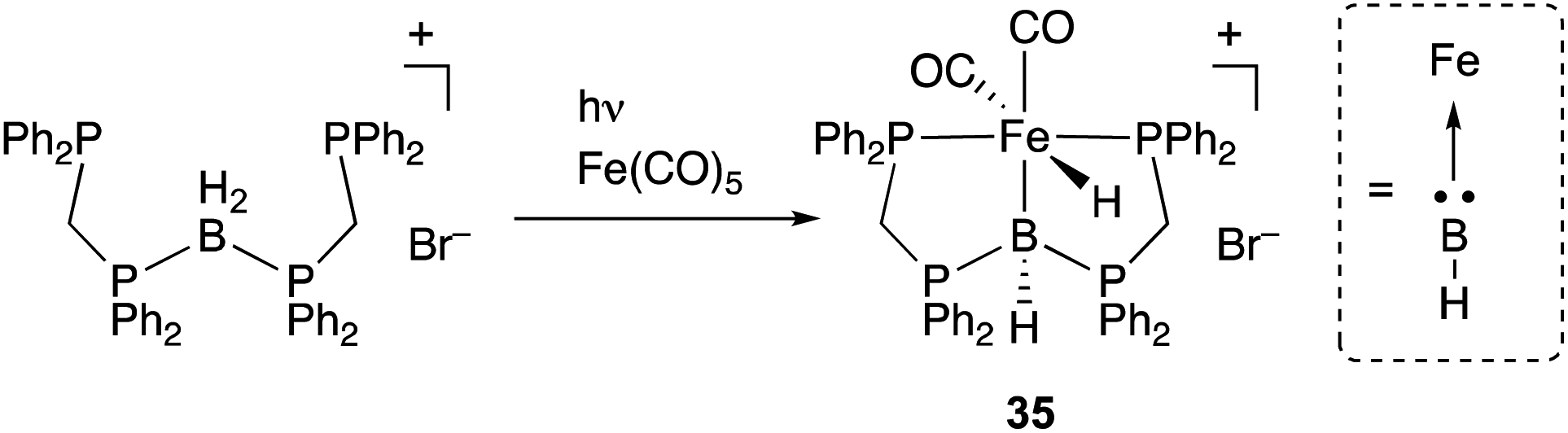
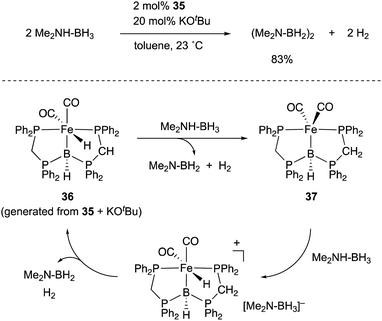
A borylene is a group 13 analogue of a carbene and coordinates to transition metals as an L-type, strong σ-donor ligand. There are numerous reports on the synthesis of borylene–metal complexes; however, most of them act as highly reactive borylating reagents due to the instability and high reactivity of a M–borylene moiety.18,19 Therefore, suitable design of the scaffold for a M–borylene bond is necessary to stabilize and utilize a borylene as a supporting ligand in catalysis. Langer reported the synthesis of a PBP–Fe complex 35 by reaction of a boronium salt with Fe(CO)5, which proceeded via dissociation of CO ligands and oxidative addition of the B–H bond to Fe under photoirradiation conditions (Scheme 14).50,51 Structural analysis showed that the central boron atom of the Fe–B complex 35 can be best regarded as a borylene stabilized by bis(phosphino)methane linkers. The PBP–Fe complex 35 catalyzed a dehydrogenation reaction of dimethylamine-borane efficiently (Scheme 15).52 Experimental and theoretical investigations indicated that the reaction mechanism involves two important steps; concerted hydrogen liberation at the anionic methine and Fe–H moieties of 36 with Me2NH–BH3 to generate a neutral PBP–Fe complex 37, and protonation at the Fe center of 37 with Me2NH–BH3 to regenerate 36. It is proposed that, in particular, the latter step is enhanced due to the strong electron donating ability of the borylene ligand.
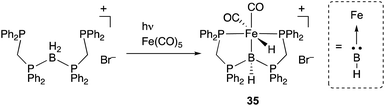 |
| | Scheme 14 Synthesis of the PBP–Fe complex 35 bearing a borylene as a supporting ligand. | |
 |
| | Scheme 15 Dehydrogenation of amine-borane catalyzed by the PBP–Fe complex 35. | |
3.2.4 Al, Ga, and In as supporting ligands.
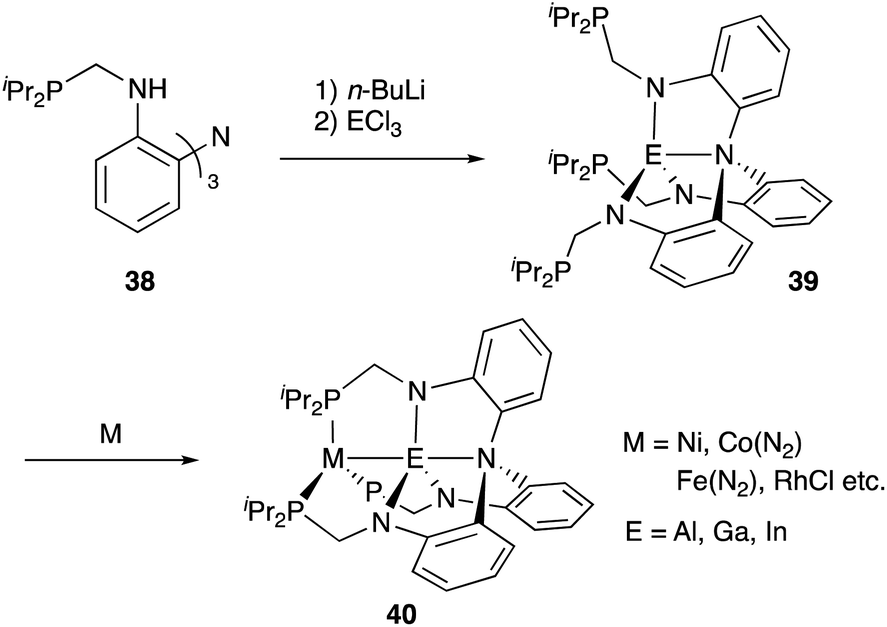
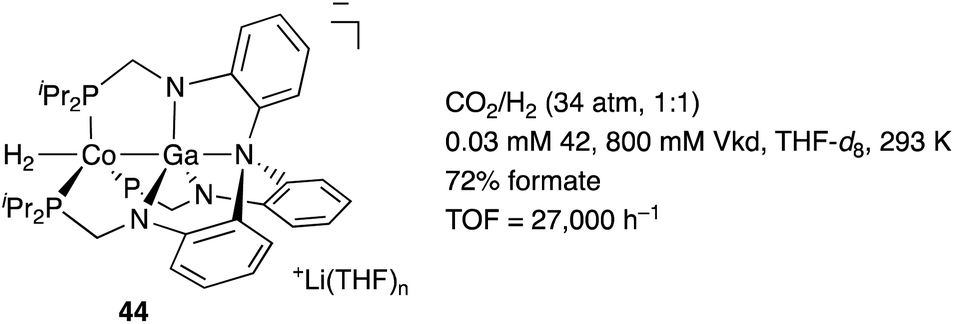
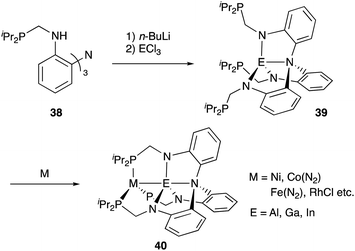
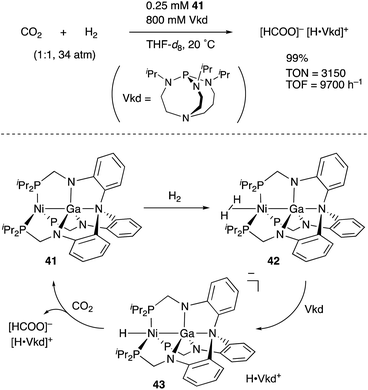
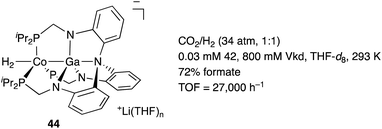
In contrast to that of M–B complexes, the catalytic application of the corresponding heavier congener M–E, where E = Al, Ga, In, and Tl, has been rather limited. This is partly due to the difficulty of incorporation and handling of highly Lewis acidic, reactive group 13 metals as supporting ligands for transition metals. In particular, the systematic synthesis of a series of M–E complexes with group 13 metals and evaluation of their catalytic reactivity have remained a formidable challenge.53 Lu solved such problems by utilizing a 3-fold N,P-multidentate ligand 38 as an efficient scaffold for the M–E bond (E = Al, Ga, and In) (Scheme 16).54 The successive introduction of E and M into the scaffold 38 enables facile syntheses and catalytic application of a variety of M–E complexes 40, in which the group 13 metal E acts as a σ-accepting Z-type ligand. They demonstrated that fine tuning of Ni for catalytic hydrogenation reactions of alkenes and carbon dioxide is possible by varying the group 13 metalloligands E, leading to development of a highly active Ni–Ga catalyst 41 for these hydrogenation reactions (Scheme 17).55,56 Gallium was a better metalloligand than Al and In. In particular, a high TOF (9700 h−1) was achieved for the hydrogenation of CO2 to formate under 34 atm of H2/CO2 in the presence of a proazaphosphatrane base. Detailed mechanistic studies clarified that the Z-type Ga–ligand plays a crucial role in activating H2 electrophilically on Ni to be deprotonated by the base in an H2-coordinated Ni–Ga complex 42. The generated anionic [H–Ni–Ga]− complex 43 is a highly hydridic metal hydride species, which is efficiently stabilized through a strong Ni–>Ga dative interaction. Furthermore, a related dihydrogen-coordinated anionic Ga–Co complex 44 was also synthesized and structurally characterized as another rare example of d10 η2-(H2)M complexes (Fig. 5).57 It was demonstrated that 44 catalyzed hydrogenation of CO2 efficiently via a CoI−/CoI redox cycle, achieving a high TOF (27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1).
000 h−1).
 |
| | Scheme 16 Efficient synthesis of M–E complexes with group 13 metalloligands 39 (E = Al, Ga, and In). | |
 |
| | Scheme 17 Catalytic hydrogenation of CO2 using the Ni–Ga complex 41. | |
 |
| | Fig. 5 Synthesis and catalytic activity of the H2-coordinated anionic Ga–Co complex 44. | |
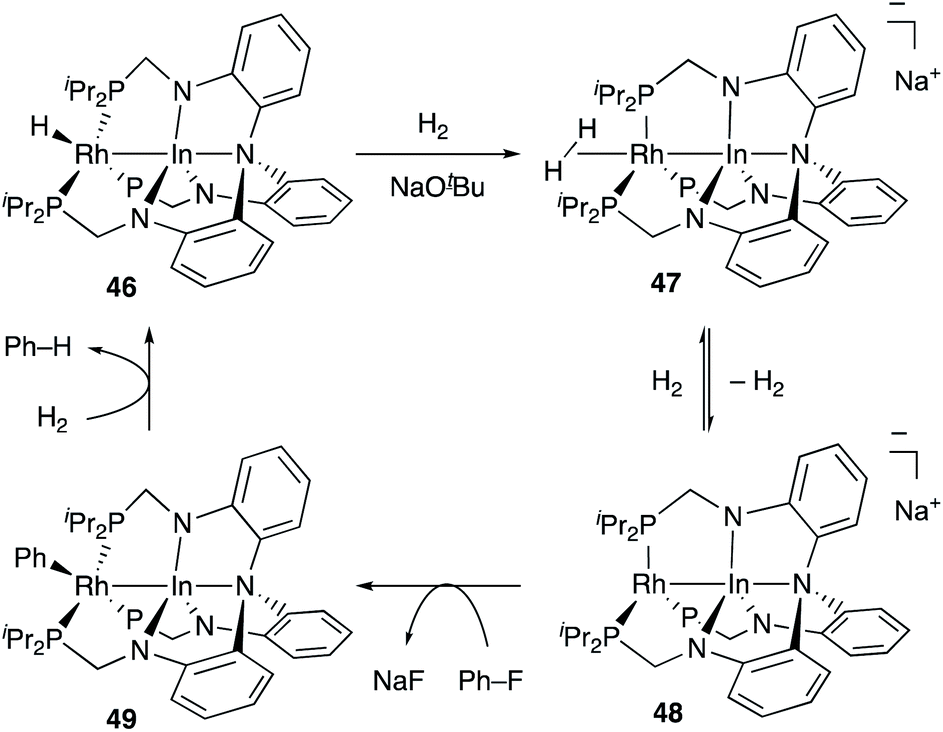
Recently, Lu reported catalytic hydrogenolysis of the sp2C–F bonds of aryl fluorides enabled using a Rh–In complex 45 (Scheme 18).58 The reaction proceeded with a stoichiometric amount of NaOtBu under 1 atm H2 to give defluorinated arenes in good yields. Mechanistic studies supported that the Rh–In complex 45 is converted to a monohydride Rh–In complex 46 under the reaction conditions, which reacts with NaOtBu in the presence of H2 to generate an H2-coordinated anionic Rh–In complex 47 (Scheme 19). The C–F bond cleavage occurs with an unsaturated rhodate complex 48 to give a phenylrhodium complex 49, and the following reaction with H2 regenerates the monohydride complex 46. These results demonstrate the high utility of the direct Rh–In interaction for the activation of strong C–F σ-bonds.
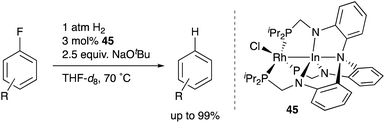 |
| | Scheme 18 Hydrogenolysis of aryl fluorides catalyzed by the Rh–In complex 45. | |
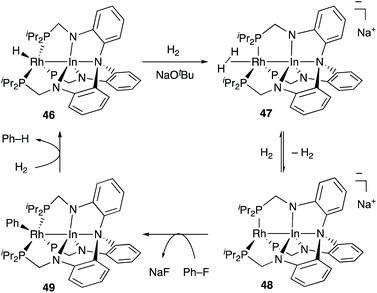 |
| | Scheme 19 Proposed reaction mechanism via the anionic Rh–In complex 48. | |
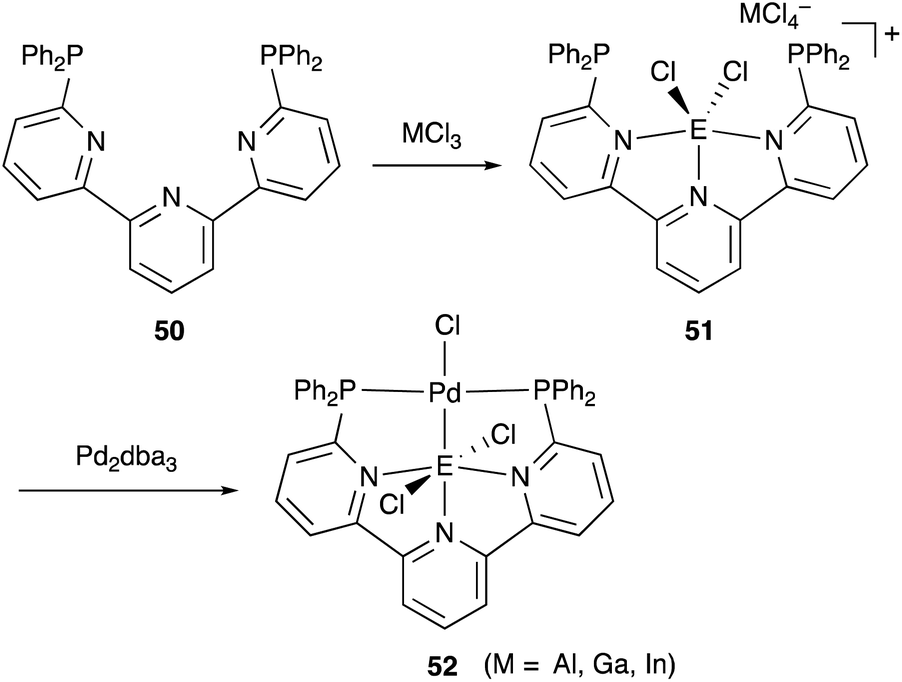
We have developed an efficient method for the synthesis of a variety of M–E complexes (E = Al, Ga, and In) by utilizing a 6,6′′-bis(phosphino)terpyridine derivative 50 as a scaffold for the M–E bond.59 The terpyridine-based rigid and planar N3P2-structure efficiently stabilizes the M–E bond by keeping sufficient coordination space and reactivity at the M center. The cationic [terpyridine–ECl2]+ complexes 51 act as highly electrophilic Z-type ligands, and complexation with Pd0 affords a series of Pd–E complexes 52 (Scheme 20).59a They seem to be PdII complexes having anionic Cl and PEP-pincer type ligands generated via redox between trivalent group 13 metals EIII and Pd0. Structural analyses showed that the Pd–Cl bond in the Pd–Al complex 52-Al is significantly elongated, suggesting the strong trans influence of the Al-metalloligand. The Pd–Al complex 52-Al exhibited remarkable catalytic activity for hydrosilylation of carbon dioxide to give silyl formate (Scheme 21). The reaction smoothly proceeded at room temperature under atmospheric pressure of CO2, and the TOF reached 19300 h−1, which is the highest value ever reported for this reaction. It is proposed that the trans-destabilizing Al-metalloligand generates highly anionic, nucleophilic Pd–H species for hydrometallation of CO2.
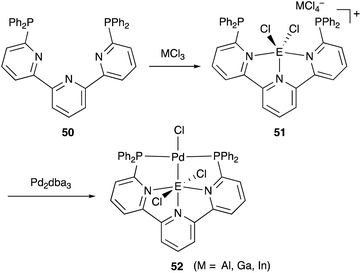 |
| | Scheme 20 6,6′′-Bis(phosphino)terpyridine 50 as an efficient scaffold for the Pd–E bond. | |
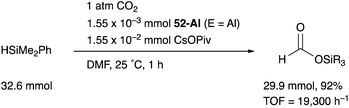 |
| | Scheme 21 Hydrosilylation of CO2 catalyzed by the Pd–Al complex 52-Al. | |
Nakao developed a Rh complex 56 bearing an X-type alumanyl ligand stabilized by a bis(2-(phosphinomethylamino)-phenyl)amine derivative 53.60 Introduction of AlCl3 and RhI afforded a Rh complex having a Z-type Al–ligand 54, which was further reduced using KC8 in the presence of norbornadiene to give the Rh–Al complex 56 (Scheme 22).60a The C–H activation of pyridine with 56 in the presence of HSiEt3 occurred selectively at the 2-position to give a C–H cleavage product 57. This result demonstrates the sufficient Lewis acidity and high σ-donating ability of the Al-metalloligand for the efficient activation of a proximal C–H bond through such a Lewis acid–Lewis base interaction. According to these observations, a C2-selective mono-alkylation reaction of pyridine with alkenes catalyzed by the Rh–Al complex 57 was developed (Scheme 23).
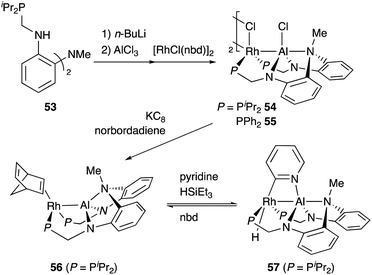 |
| | Scheme 22 Synthesis and reaction of the PAlP–Rh complex 56 having an X-type alumanyl ligand. | |
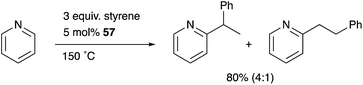 |
| | Scheme 23
57-catalyzed monoalkylation of pyridine. | |
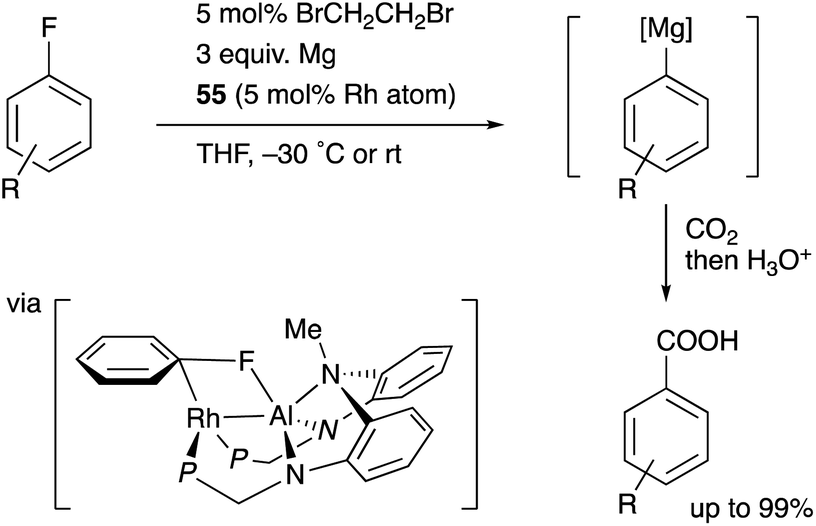
Nakao recently reported a catalytic magnesiation reaction of aryl fluorides with Mg powder using the Rh–Al complexes 54 and 55 (Scheme 24).61 The Rh–Al complex 55 having –PPh2 side arms catalytically converted various aryl fluorides to arylmagnesium compounds efficiently in the presence of Mg powder and 1,2-dibromoethane, which were trapped with various electrophiles such as CO2 to give carboxylic acids. This is a quite rare example of facile preparation of arylmagnesium reagents from aryl fluorides using readily available Mg powder, demonstrating the high synthetic utility of the Rh–Al catalyst. The catalyst 54 was confirmed to be reduced with Mg to generate the low-valent Rh–Al complex 56 in the presence of norbornene. Based on these experimental studies and theoretical calculations, it was proposed that the oxidative addition of the inert sp2C–F bond occurred across the Rh–Al bond of a low valent Rh–Al complex.
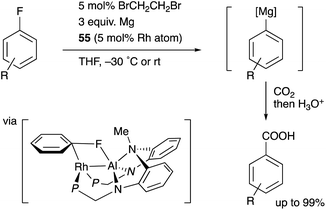 |
| | Scheme 24 PAlP–Rh complex-catalyzed magnesiation of aryl fluorides. | |
Yamashita reported the synthesis and catalytic activity of an Ir complex 58 with an X-type PAlP–ligand using the same scaffold employed for the PBP–Ir complexes 26 (Scheme 25).62 The tricoordinate Al–ligand is demonstrated to act as a Lewis acid to capture a Lewis base such as DMAP. Catalytic application of the Ir–Al complex 58 to a dehydrogenation reaction of cyclooctane to cyclooctene was investigated although the activity was not satisfactory.
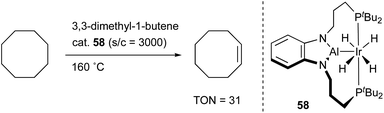 |
| | Scheme 25 PAlP–Ir 58-catalyzed dehydrogenation of alkanes. | |
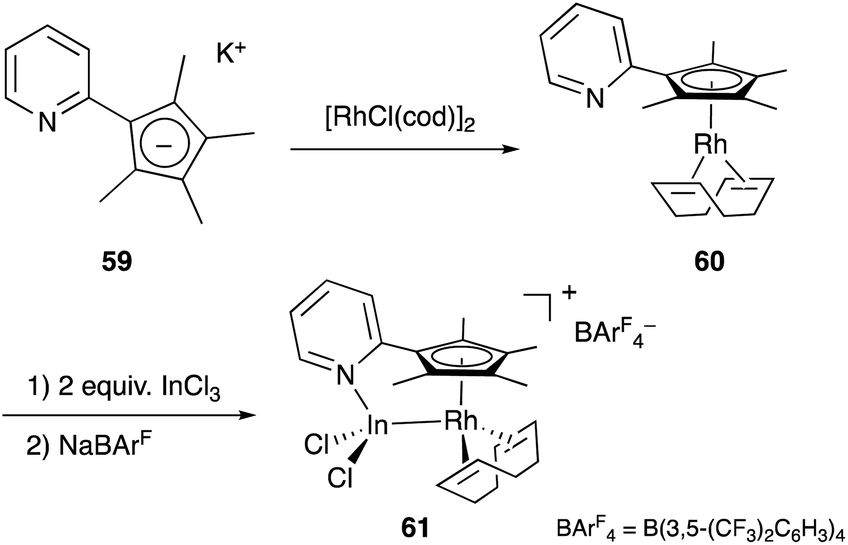
Recently, a different approach to M–E complexes with heavier group 13 elements has been established by our group. A Cp–rhodium complex 61 having a cationic InCl2+ metalloligand was synthesized through the reaction of 2 equivalents of InCl3 and a RhI complex 60, which was prepared from a pyridine-tethered Cp derivative 59 (Scheme 26).63 This method enables post-introduction of a group 13 metalloligand to a Cp–metal complex to form an M–E bond, which is distinct from the pre-introduction approaches described before. Therefore, screening of metalloligands becomes possible for the development of catalytic reactions. The cationic In-metalloligand acts as a strong σ-acceptor for Rh, and theoretical calculations and CV measurements revealed that the electronic and redox properties of the Rh in 61 become similar to those of trivalent Cp*RhIII rather than those of the original monovalent RhI complex 60 through the formation of the Rh–In bond. This was also demonstrated in catalysis. The Rh–In complex 61 catalyzed sp2C–H amidation of benzo[h]quinoline with TsN3 to afford an N-aryltosylamide derivative in good yield (Scheme 27). Control experiments employing Cp*Rh(cod) or 60 with or without InCl3 as catalysts supported the unique reactivity of the Rh–In bond to turn on the C–H activation catalysis. The reaction was also applicable to various arenes having pyridine or pyrimidine as a directing group. These results demonstrate the promising utility of this approach in exploring new transition metal catalysis with M–E bonds in synthetic chemistry.
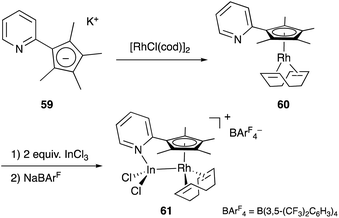 |
| | Scheme 26 Synthesis of the Rh–In complex 61via post-introduction of a group 13 metalloligand. | |
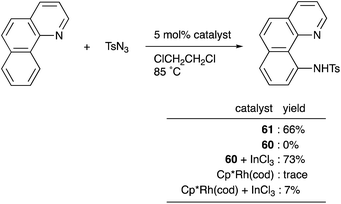 |
| | Scheme 27 sp2C–H activation enabled using the In-metalloligand. | |
3.3 Group 14 (E = Si and Ge)
Transition metal complexes having a silyl ligand as an anionic X-type ligand have been extensively studied since they are important intermediates in various catalytic silylation reactions such as hydrosilylation and C–H silylation.64 The silyl ligand is expected to act as a strong σ-donor and a trans influencing ligand.15,16 MLC on the M–Si bond can also be considered.24
Various Si-containing multidentate ligands and their transition metal complexes have been developed including tri- and tetradentate phosphinosilyl ligands tethered using alkyl or o-phenylene linkers, an NSiN-type bis(8-quinolyl)silyl ligand, poylsilyl ligands and so on.14 Although catalytic applications were demonstrated in hydroformylation, hydrogenation, hydrosilylation, hydroboration, C–H silylation reactions etc., the efficiency and selectivity of the catalysis were not necessarily advantageous compared with those of standard transition metal catalysts without silyl ligands.65 Selected examples of unusual molecular transformations and catalytic activity enabled using silyl ligands are described.
One of the most versatile multidentate Si–ligands used for transition metal catalysts is a bis(phosphinophenyl)silyl derivative (phenylene-bridged PSiP–ligand). Turculet reported the synthesis of various transition metal complexes having a PSiP–ligand in 2007 for the first time. Several catalytic applications were also examined by Turculet and other research groups, and high catalytic activity and unusual molecular transformations taking advantage of the silyl ligand are achieved in some cases. For example, a PSiP–Pt complex 62 catalyzed the reduction of CO2 to methane with HSiMe2Ph under mild reaction conditions (1 atm, 65 °C), and the TON reached 2156 (Scheme 28).66 The pincer-type PSiP–ligand was also applicable to first row transition metals such as Fe, and the catalytic activity of a PSiP–Fe hydride complex 63 for hydrogenation of alkenes with H2 (10 atm) was demonstrated as a relatively rare example of Fe-catalysts for the reaction.67
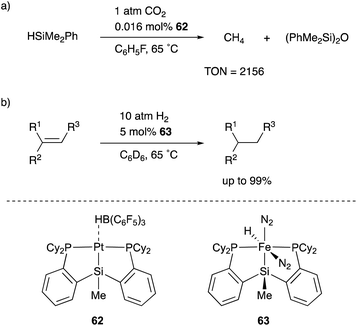 |
| | Scheme 28 Hydrosilylation and hydrogenation reactions catalyzed by PSiP–M complexes. | |
In 2008, we developed a PSiP–Pd complex 64-catalyzed hydrocarboxylation reaction of allenes and 1,3-dienes under atmospheric pressure of CO2 using AlEt3 as a stoichiometric reductant (Scheme 29).68 This is a new CO2-fixation reaction using simple unsaturated hydrocarbons as substrates to give synthetically useful β,γ-unsaturated carboxylic acids selectively in high yields. Hydrometallation of a C–C double bond generates a highly nucleophilic σ-allylpalladium complex 65, which smoothly reacts with a less reactive molecule, CO2, even at room temperature because of the strong σ-donating ability and trans influence of the silyl ligand.69 Furthermore, we also realized atom-economical hydrocarboxylation reactions of allenes and alkenes employing a formate salt as a reductant as well as a CO2 source enabled using newly developed Pd complexes 66 and 67 bearing a PGeP-pincer type ligand (Scheme 30).70 This reaction proceeds under mild conditions and provides an alternative strategy for utilizing formate salts as a C1 source. These reactions did not proceed with standard Pd complexes and PCP– and PNP–Pd complexes, thus clearly demonstrating the privileged reactivity of the Pd–Si and Pd–Ge bonds in catalysis.
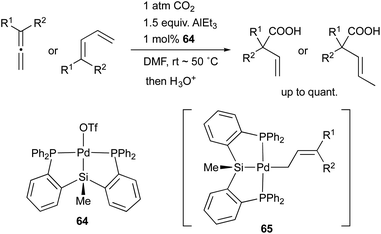 |
| | Scheme 29 Hydrocarboxylation of simple unsaturated hydrocarbons catalyzed by the PSiP–Pd complex 64. | |
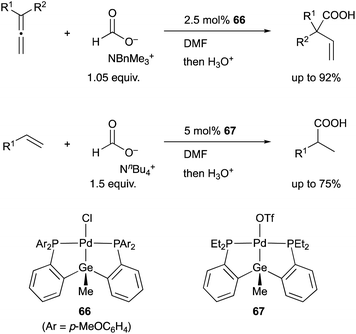 |
| | Scheme 30 PGeP–Pd complex 66 or 67-catalyzed hydrocarboxylation using a formate salt as both a reductant and CO2 source. | |
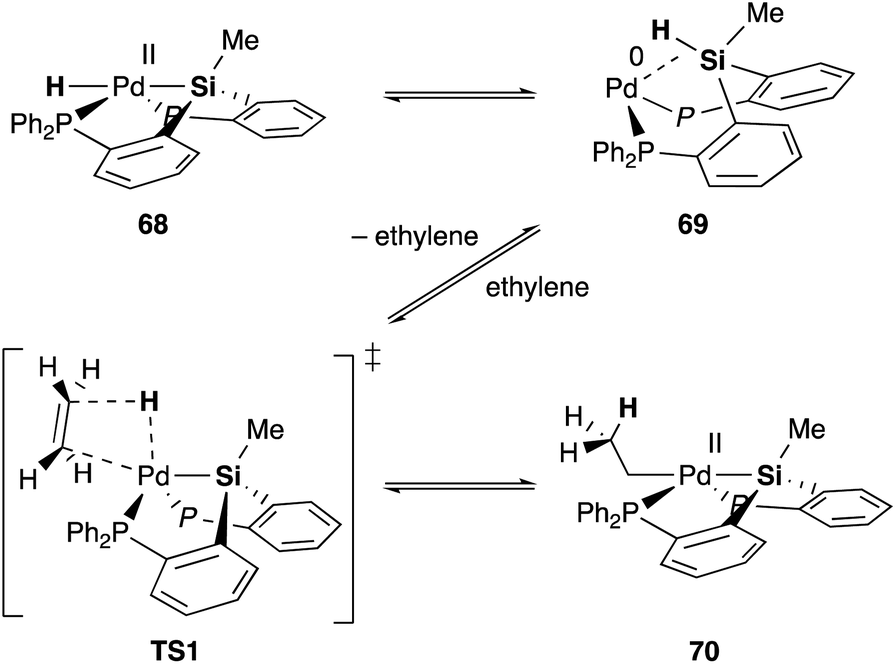
Silyl and germyl ligands play important roles not only in promoting the nucleophilic addition step, but also in enabling a new hydrometallation mechanism via MLC. Mechanistic studies using ethylene as a model substrate clarified that an η2-(E–H)Pd0 complex 69, which is reversibly generated via reductive elimination of an E–H bond from a PdII hydride complex 68, undergoes associative hydrometallation via a 5-coordinated TS1 to afford an ethyl–PdII complex 70 directly without dissociation of one of the –PR2 side arms (Scheme 31).71 During the reaction, the PEP-pincer ligand acts as an efficient scaffold for delivering the hydrogen atom as a hydride ligand between E and Pd accompanied by the formation and cleavage of the Pd–E bond. This is an unusual mechanism for hydrometallation and β-hydrogen elimination via MLC of the Pd–Si bond, demonstrating the promising utility of the Si–ligand in transition metal catalysis. It is also clarified that the germyl ligand stabilizes the ethyl–PdII state more efficiently than the silyl ligand and is suitable for reactions with formate salts under heating conditions.72
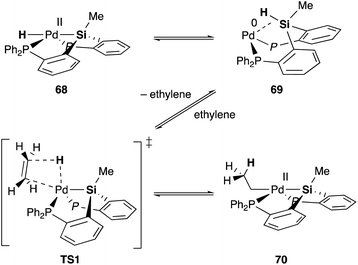 |
| | Scheme 31 Associative hydrometallation/β-hydrogen elimination via MLC of the Pd–Si bond. | |
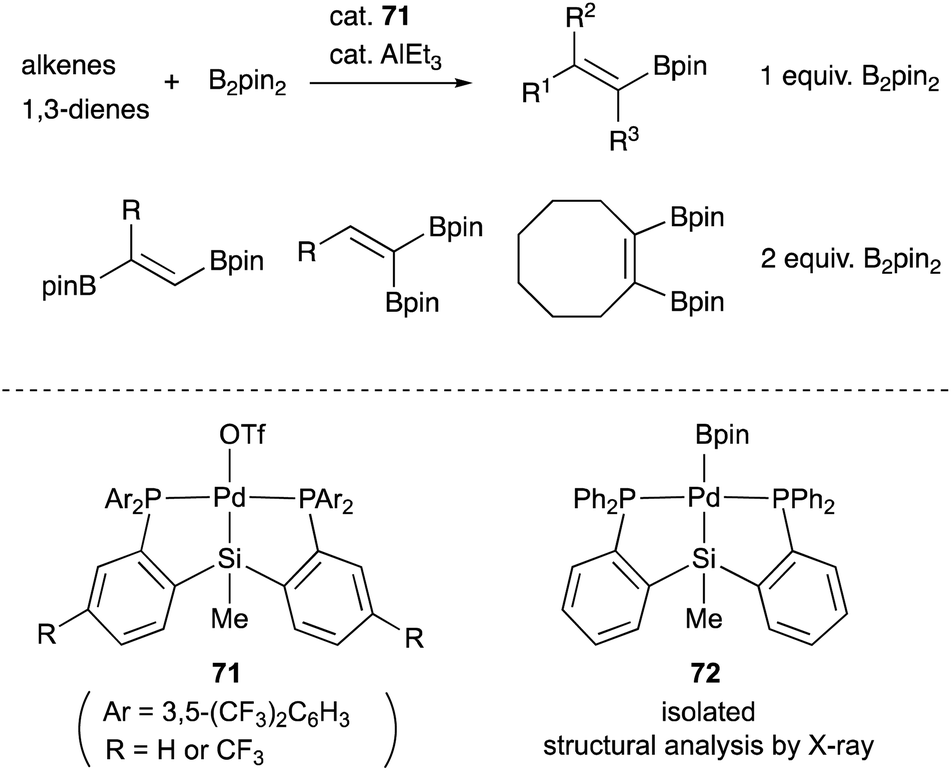
We have also developed a selective dehydrogenative borylation reaction of alkenes with B2pin2 catalyzed by PSiP–Pd complex 71 having electron deficient Ar substituents on phosphorus side arms (Scheme 32). The reaction with 2 equivalents of B2pin2 enables an efficient synthesis of a variety of double borylation products involving 1,1-, trans-1,2-, and cyclic-1,2-diborylalkenes starting from alkenes and B2pin2, which are synthetically highly useful but not accessible with standard transition metal-catalyzed borylation reactions.73 It is clarified that the reaction proceeds via a borylpalladium complex 72 having a PSiP-pincer type ligand, which possesses an elongated and highly reactive Pd–B bond due to the strong trans influence of the silyl ligand as evidenced by X-ray analysis and stoichiometric reactivity studies. Detailed mechanistic studies revealed that a reversible σ-bond metathesis reaction is operative between an η2-(Si–H)Pd0 complex 73 and B2pin2 to form a borylpalladium complex 74 through MLC similar to the case of the hydrocarboxylation reaction (Scheme 33).74
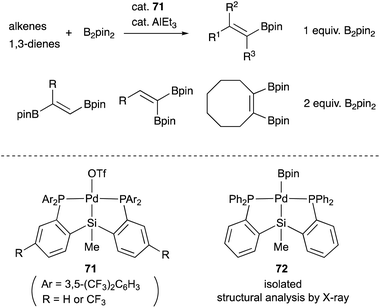 |
| | Scheme 32 Selective single or double dehydrogenative borylation of alkenes catalyzed by the PSiP–Pd complex 71. | |
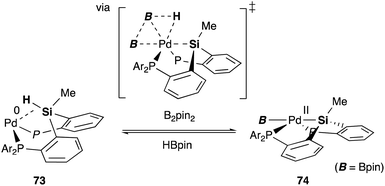 |
| | Scheme 33 Reversible σ-bond metathesis of the η2-(Si–H)Pd0 complex 73 with B2pin2. | |
We have designed and synthesized a platinum complex 75 having a PSiN-pincer type ligand, in which one of the o-phosphinophenyl linkages is replaced by an o-(aminomethyl)phenyl group expecting the lability of the Pt–N bond to generate a coordinatively unsaturated 14e Pt center (Scheme 34).75 The PSiN–Pt complex catalyzed sp2C–H borylation of various electron deficient arenes such as fluoro- or chloroarenes efficiently, affording halogenated arylboronic esters in good yields. This is a quite rare example of Pt-catalyzed sp2C–H borylation.76 Interestingly, the borylation occurs at a C–H bond adjacent to the halogen substituent preferentially. This regioselectivity is in sharp contrast to that of Ir-catalyzed borylation, in which borylation tends to occur at less sterically hindered C–H bonds as shown in the table. Although the reaction mechanism and the origin of the regioselectivity are not clear yet, these results demonstrate the unique catalytic activity of Pt–Si complexes in synthetic chemistry.
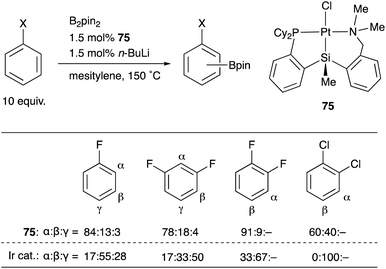 |
| | Scheme 34 Regioselective C–H borylation of fluoro- and chloroarenes catalyzed by the PSiN–Pt complex 75. | |
Tobita developed a bis(silyl)-chelate ligand, xantsil 76, based on a xanthene backbone, which possesses a wide bite-angle and exhibits strong σ-donation ability and trans influence. The xantsil–Ru complex 77 catalyzed an unusual hydrosilylation reaction of arylacetylenes with silane, in which ortho-C–H silylation of the aryl moiety occurs accompanied by trans-hydrogenation of the C–C triple bond to give trans-(o-silylaryl)alkenes (Scheme 35).77 It is proposed that one of the silyl ligands fluxionally dissociates from Ru after oxidative addition of a C–H bond and reforms after the formation of the product during the reaction. Slight modification of the substituents on the phosphorus ligand switched the reaction pathway to tandem hydrosilylation/C–H silylation of alkynes to give bis(silylated) stilbene derivatives.78
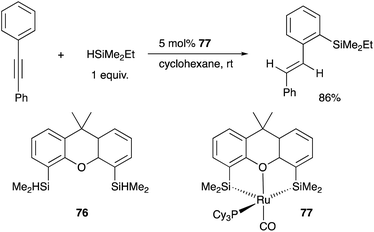 |
| | Scheme 35 Tandem hydrogenation/C–H silylation catalyzed by the xantsil–Ru complex 77. | |
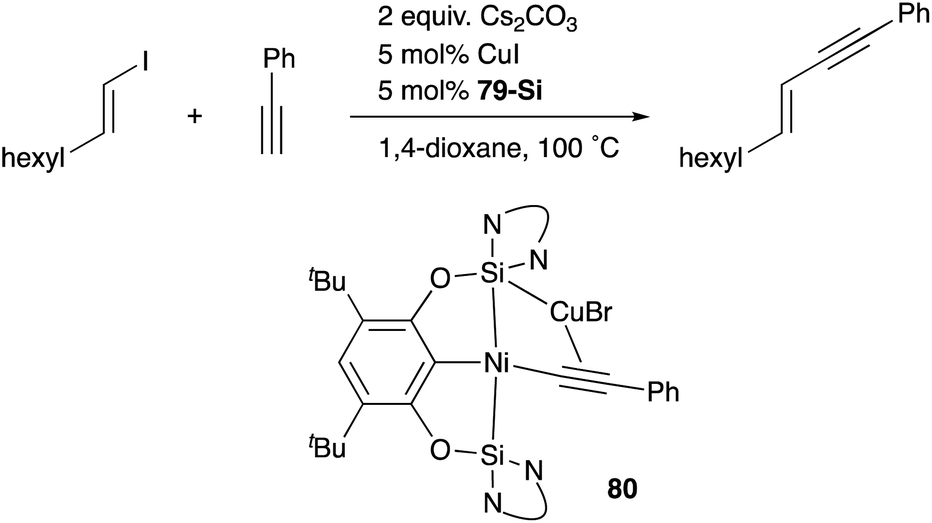
A silylene and a germylene, which are heavier analogues of a carbene, have also been utilized as a supporting ligand for transition metal catalysts. Driess and Hartwig developed ECE-type multidentate ligands possessing two base-stabilized silylene or germylene moieties as side arms (E = Si or Ge), and catalytic application of their Ir and Ni complexes was reported (Fig. 6).79 The SiCSi–Ir complex 78-Si exhibited higher efficiency for a catalytic C–H borylation reaction of benzene than the corresponding Ir complex having a PCP-pincer type ligand possibly due to the stronger σ-donating ability of the SiII ligand than the PIII ligand (Scheme 36).79a Moreover, the SiCSi–Ni complex 79-Si catalyzed the Sonogashira coupling reaction of an alkenyl iodide and an alkyne (Scheme 37).79b Mechanistic studies clarified that the reaction proceeded through the sequence of transmetallation, oxidative addition, and reductive elimination. As an important intermediate, an alkynylnickel complex 80 was isolated and structurally characterized, in which CuBr coordinates to one of the silylene ligands and an alkyne moiety. Although the catalysis in these reactions is not tremendous yet, these results demonstrate the promising utility of group 14 metallylenes as a unique donor ligand in transition metal catalysis.
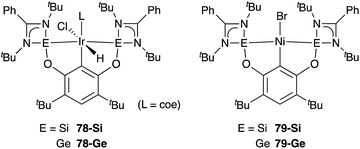 |
| | Fig. 6 Silylenes and germylenes as supporting ligands for transition metal catalysts. | |
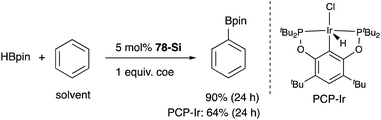 |
| | Scheme 36 sp2C–H borylation catalyzed by the bis(silylene)–Ir complex 78-Si. | |
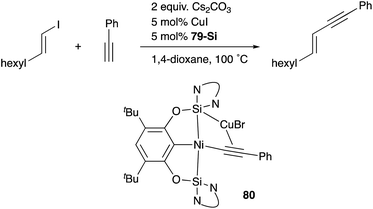 |
| | Scheme 37 The SiCSi–Ni complex 79-Si-catalyzed Sonogashira coupling reaction. | |
3.4 Group 15 (E = Sb and Bi)
Heavier group 15 elements such as Sb and Bi possess distinct electronic characteristics and reactivities from those of lighter pnictogens such N and P. The σ-donating ability of trivalent Sb and Bi compounds ER3 (E = Sb and Bi) is weak due to the stabilized 5s and 6s electrons by the relativistic contraction. Scrambling and transfer of the substituent R on E are often observed due to the relatively weak and reactive E–C bond. Therefore, the number of transition metal complexes having L-type, trivalent Sb- and Bi–ligands and their catalytic applications have been rather limited compared with those of phosphine ligands.80 On the other hand, trivalent Sb and Bi compounds often behave as Lewis acids, enabling Z-type coordination to Lewis basic transition metals.81 Moreover, redox between EIII and EV and coordination/dissociation of ligands on E enable modular tuning of electronic and steric properties of these compounds. These characteristics are highly promising for developing multi-functional transition metal catalysts, where the reactivity of the transition metal is tunable with chemical stimuli on E.
Gabbaï reported the synthesis and catalytic application of various Au and Pt complexes having an Sb–ligand.8,9,82 The o-phosphinophenyl linkage is often utilized as a support for the M–Sb bond. The Sb atom acts as a non-innocent Z-type ligand, where its σ-accepting ability is tunable by changing the oxidation state and substituents on Sb for various electrophilic activation reactions of C–C triple bonds. For example, the cationic PSbP–Au complex 81 having a trivalent SbIII–ligand scarcely catalyzed hydroamination of alkynes to give an imine product in very low yield (Scheme 38).83 On the other hand, the catalytic activity dramatically increased by oxidizing the SbIII–ligand to a pentavalent SbV–ligand in 82, giving the product in 92% yield within 40 min. It is conceivable that the electronically positive, pentavalent SbV–ligand makes the Au atom more electrophilic than the trivalent SbIII–ligand to promote the electrophilic activation of alkynes efficiently.
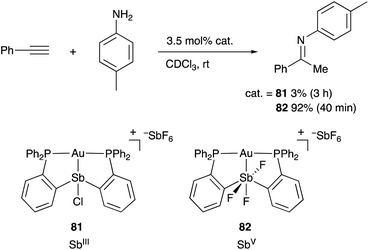 |
| | Scheme 38 Electrophilic activation of alkynes catalyzed by the Au–Sb complex 82 having the redox non-innocent PSbP–ligand. | |
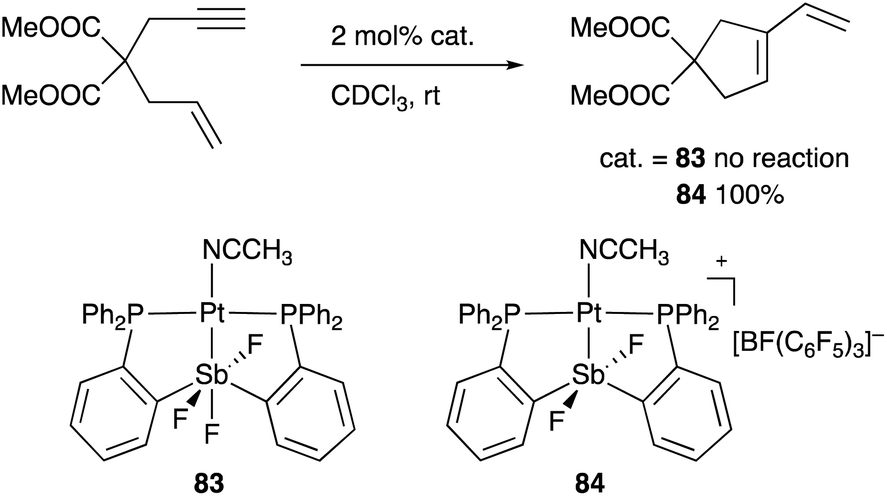
A neutral PSbP–Pt complex 83 bearing a pentavalent SbV–ligand was activated electrophilically by abstracting one of the fluorine ligands on SbV (Scheme 39).84 The activated form, a cationic PSbP–Pt complex 84, efficiently catalyzed an electrophilic cyclization reaction of 1,6-enynes to form carbocycles in good yields. The cationic Sb moiety serves as a strong α-acceptor, increasing the π-acidity of the Pt complex for efficient electrophilic activation of alkynes. Similar reactivity control on Sb was also demonstrated in the case of a PSbP–Au complex 85. Introduction of a phosphine oxide side arm on Sb enables the generation of a dicationic SbV–ligand, which makes the Au atom highly electrophilic. The dicationic PSbP–Au complex 85 catalyzed hydroamination and polymerization of styrene derivatives efficiently (Scheme 40).85 Furthermore, self-activation of the catalytic activity of a PSbP–Pt complex 86 for electrophilic activation reactions was also reported (Scheme 41).86 It is proposed that abstraction of a chloride ligand on Pt using the electrophilic Sb–ligand generates a cationic PSbP–Pt complex 87 as a reactive intermediate, which was supported by isolation and structural analysis of a related dicationic PSbP–Pt complex with L = CyCN. These examples showed that efficient control of transition metal catalysis is possible by utilizing characteristic Lewis acidity and redox and coordination behaviors of the Sb ligand, demonstrating the practical utility of the non-innocent Sb–ligand in synthetic chemistry.
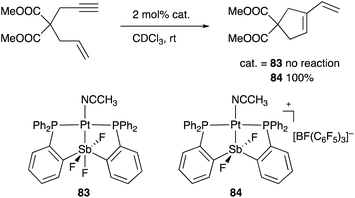 |
| | Scheme 39 The PSbP–Pt complex 84 activated by abstraction of a fluoride ligand on Sb. | |
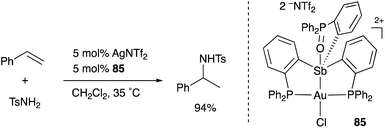 |
| | Scheme 40 Catalytic electrophilic activation of alkenes enabled using the dicationic Sb–ligand in the PSbP–Au complex 85. | |
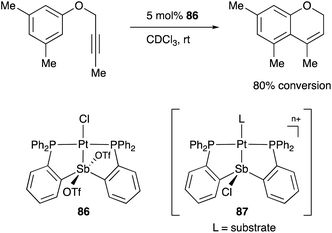 |
| | Scheme 41 Self-activation of Pt-catalysis with the Sb–ligand. | |
Regarding Bi-containing multidentate ligands, a related analogue, bis(o-phosphinophenyl)chlorobismuth(III), has been developed, and synthesis and structural analysis of Au, Pt, Pd, Cu, and Ag complexes having the PBiP–ligand were reported by Gabbaï and Limberg.87,88 It was revealed that the central BiIII atom acts as a σ-acceptor for Au, Pt, and Pd rather than as a σ-donor while the Bi–>M interaction is dominant for Cu and Ag. Limberg also developed another Bi-containing multidentate ligand based on xanthene.89 However, the catalytic reactivity of these complexes for synthetic reactions has not been investigated.
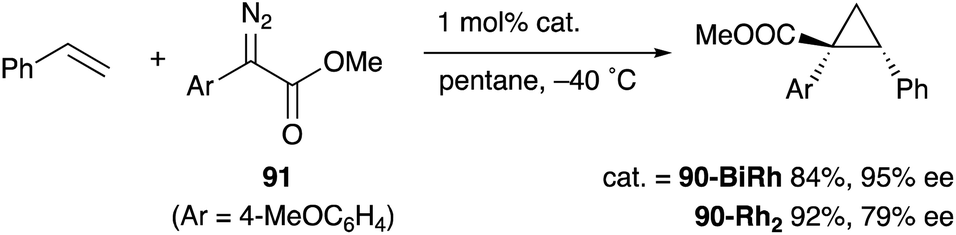
As a related example of M–Bi complexes, heterobimetallic paddlewheel Bi–Rh carboxylate complexes BiRh(X)4 have been developed by Dikarev,90 where one of the 4d Rh atoms of the well-known paddlewheel Rh2(X)4 complex was replaced by a 6p Bi (Fig. 7). The parent Rh2(X)4 complexes are widely utilized as efficient catalysts for carbene transfer reactions of diazo compounds. The catalytic activity of the BiRh(X)4 complexes has been evaluated in the carbene transfer reactions to elucidate the effect of the Bi–ligand on the reactivity. Davies reported the catalytic activity of various paddlewheel Bi–Rh complexes in cyclopropanation and C–H activation reactions and commented that decomposition of a diazo compound became slower with the Bi–Rh complex 88-BiRh than 88-Rh2.91 Recently, Fürstner developed a Bi–Rh complex 89-BiRh and realized catalytic cyclopropanation of highly electron-deficient polychlorinated alkenes such as trichloroethylene, which were not employable substrates in the previously reported Rh2-catalyzed reactions (Scheme 42).92 Experimental analyses and theoretical calculations on the structure and electronic properties of the Bi–Rh bimetallic complexes showed that the carbene moiety becomes electrophilic because back donation of electrons from Rh to the carbene is reduced in the Bi–Rh complex. Furthermore, the chiral version of the Bi–Rh complex 90-BiRh having a tert-leucine derivative as bridging carboxylate ligands was also developed and applied to an asymmetric cyclopropanation reaction of styrenes (Scheme 43).93 The Bi–Rh complex 90-BiRh achieved better enantioselectivity in the reactions of a diazo compound 91 than that with a standard Rh2 catalyst 90-Rh2 due to the more confined, effective chiral space produced by the Bi–Rh core.
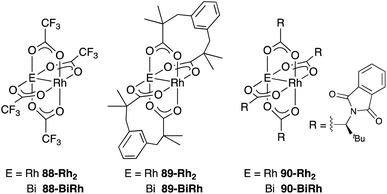 |
| | Fig. 7 Paddlewheel Bi–Rh complexes. | |
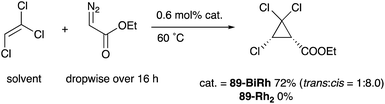 |
| | Scheme 42 Cyclopropanation of electron deficient alkenes catalyzed by the Bi–Rh complex 89-BiRh. | |
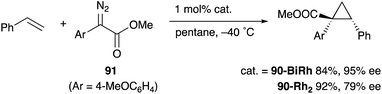 |
| | Scheme 43 The Bi–Rh complex 90-BiRh-catalyzed asymmetric cyclopropanation of styrenes. | |
4. Conclusions and perspectives
Recent progress on catalytic application of transition metal complexes having an M–E bond (E = main group metal or metalloid element) is summarized. Several important scaffolds for M–E bonds have been established to enable efficient synthesis of a variety of M–E complexes and their utilization as catalysts in synthetic reactions. Main group metal and metalloid supporting ligands E furnish unusual electronic and steric environments and molecular functions to the transition metals, in particular through Z-type ligation and MLC of the M–E bonds, which are not easily available with standard organic supporting ligands such as phosphines and amines. These characteristics lead to remarkable catalytic activity and product selectivity in some cases. The reactivity control with the chemical stimuli on E–ligands is also intriguing to develop a multi-functional transition metal catalyst that changes its reactivity depending on the environment. This progress demonstrates the importance and synthetic utility of main group metal and metalloid supporting ligands in organometallic and synthetic chemistry.
Besides the examples described in this article, there are numerous reports on the synthesis and stoichiometric reactivity of M–E complexes, which are potentially applicable to catalytic reactions. Investigation on the catalytic activity of other M–E complexes including group 16 elements or other heavier elements as E–ligands is also of great interest. An important future challenge is to realize new and synthetically valuable molecular transformations, which are not easily achieved with the standard transition metal catalysts, exploiting the characteristic reactivity of M–E complexes from the viewpoint of synthetic chemistry. In particular, strategic utilization of MLC for activation of unreactive chemical bonds and molecules based on the rational design of the ligand structure will be important. Further exploration will ensure successful utilization of M–E complexes as a new class of transition metal catalysts.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The author thanks JSPS KAKENHI grant numbers 17H03019, 20H02732, 18H04646, 20H04806, and JST, PRESTO grant number JY290145, Japan for financial support.
Notes and references
-
M. Stradiotto, Ligand Design in Metal Chemistry: Reactivity and Catalysis, Wiley, 2016 Search PubMed.
-
(a) A. J. Arduengo, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef CAS;
(b) W. A. Herrmann and C. Köcher, Angew. Chem., Int. Ed., 1997, 36, 2162–2187 CrossRef CAS;
(c) F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122–3172 CrossRef CAS PubMed;
(d)
S. P. Nolan, N-Heterocyclic Carbenes, Wiley, 2014 Search PubMed.
- G. Bouhadir and D. Bourissou, Chem. Soc. Rev., 2015, 45, 1065–1079 RSC.
- G. Parkin, Organometallics, 2006, 25, 4744–4747 CrossRef CAS.
- A. Amgoune and D. Bourissou, Chem. Commun., 2011, 47, 859–871 RSC.
- H. Kameo and H. Nakazawa, Chem.–Asian J., 2013, 8, 1720–1734 CrossRef CAS PubMed.
- H. Kameo and H. Nakazawa, Chem. Rec., 2017, 17, 268–286 CrossRef CAS PubMed.
- S. J. Jones and F. P. Gabbaï, Acc. Chem. Res., 2016, 49, 857–867 CrossRef PubMed.
- D. You and F. P. Gabbaï, Trends Chem., 2019, 1, 485–496 CrossRef CAS.
- S. Aldridge and D. L. Coombs, Coord. Chem. Rev., 2004, 248, 535–559 CrossRef CAS.
- H. Braunschweig, R. D. Dewhurst and A. Schneider, Chem. Rev., 2010, 110, 3924–3957 CrossRef CAS PubMed.
- M. Yamashita, Bull. Chem. Soc. Jpn., 2016, 89, 269–281 CrossRef CAS.
- M. S. Balakrishna, P. Chandrasekaran and P. P. George, Coord. Chem. Rev., 2003, 241, 87–117 CrossRef CAS.
- M. Simon and F. Breher, Dalton Trans., 2017, 46, 7976–7997 RSC.
- T. G. Appleton, H. C. Clark and L. E. Manzer, Coord. Chem. Rev., 1973, 10, 335–422 CrossRef CAS.
- J. Zhu, Z. Lin and T. B. Marder, Inorg. Chem., 2005, 44, 9384–9390 CrossRef CAS PubMed.
- M. Asay, C. Jones and M. Driess, Chem. Rev., 2011, 111, 354–396 CrossRef CAS PubMed.
- H. Braunschweig, R. D. Dewhurst and V. H. Gessner, Chem. Soc. Rev., 2013, 42, 3197–3208 RSC.
- H. Braunschweig and R. Shang, Inorg. Chem., 2015, 54, 3099–3106 CrossRef CAS PubMed.
- J. T. Goettel and H. Braunschweig, Coord. Chem. Rev., 2019, 380, 184–200 CrossRef CAS.
- P. W. Roesky, Dalton Trans., 2009, 1887–1893 RSC.
- D. Vidovic and S. Aldridge, Chem. Sci., 2011, 2, 601–608 RSC.
- J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef CAS PubMed.
- R. N. Perutz and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2007, 46, 2578–2592 CrossRef CAS PubMed.
- For selected examples, see;
(a) M. Bennett, M. Contel, D. C. R. Hockless and L. L. Welling, Chem. Commun., 1998, 2401–2402 RSC;
(b) A. C. Kuate, R. A. Lalancette, T. Bannenberg and F. Jäkle, Angew. Chem., Int. Ed., 2018, 57, 6552–6557 CrossRef PubMed and references cited therein.
- P. Steinhoff, M. Paul, J. P. Schroers and M. E. Tauchert, Dalton Trans., 2018, 48, 1017–1022 RSC.
-
(a) N. Tsoureas, G. R. Owen, A. Hamilton and A. G. Orpen, Dalton Trans., 2008, 6039–6044 RSC;
(b) N. Tsoureas, Y.-Y. Kuo, M. F. Haddow and G. R. Owen, Chem. Commun., 2010, 47, 484–486 RSC;
(c) M. E. El-Zaria, H. Arii and H. Nakamura, Inorg. Chem., 2011, 50, 4149–4161 CrossRef CAS PubMed;
(d) H. Kameo and H. Nakazawa, Organometallics, 2012, 31, 7476–7484 CrossRef CAS;
(e) T. Schindler, M. Lux, M. Peters, L. T. Scharf, H. Osseili, L. Maron and M. E. Tauchert, Organometallics, 2015, 34, 1978–1984 CrossRef CAS.
-
(a) S. Bontemps, G. Bouhadir, K. Miqueu and D. Bourissou, J. Am. Chem. Soc., 2006, 128, 12056–12057 CrossRef CAS PubMed;
(b) S. Bontemps, H. Gornitzka, G. Bouhadir, K. Miqueu and D. Bourissou, Angew. Chem., Int. Ed., 2006, 45, 1611–1614 CrossRef CAS PubMed;
(c) S. Bontemps, G. Bouhadir, W. Gu, M. Mercy, C. Chen, B. M. Foxman, L. Maron, O. V. Ozerov and D. Bourissou, Angew. Chem., Int. Ed., 2008, 47, 1481–1484 CrossRef CAS PubMed.
- H. W. Harman and J. C. Peters, J. Am. Chem. Soc., 2012, 134, 5080–5082 CrossRef PubMed.
- W. H. Harman, T.-P. Lin and J. C. Peters, Angew. Chem., Int. Ed., 2014, 53, 1081–1086 CrossRef CAS PubMed.
-
(a) G. Zeng and S. Sakaki, Inorg. Chem., 2013, 52, 2844–2853 CrossRef CAS PubMed;
(b) Y. Li, C. Hou, J. Jiang, Z. Zhang, C. Zhao, A. J. Page and Z. Ke, ACS Catal., 2016, 6, 1655–1662 CrossRef CAS.
- S. N. MacMillan, H. W. Harman and J. C. Peters, Chem. Sci., 2013, 5, 590–597 RSC.
- M.-E. Moret and J. C. Peters, Angew. Chem., Int. Ed., 2011, 50, 2063–2067 CrossRef CAS PubMed.
- J. S. Anderson, J. Rittle and J. C. Peters, Nature, 2013, 501, 84 CrossRef CAS PubMed.
- T. M. Buscagan, P. H. Oyala and J. C. Peters, Angew. Chem., Int. Ed., 2017, 56, 6921–6926 CrossRef CAS PubMed.
-
(a) M. Moret and J. C. Peters, J. Am. Chem. Soc., 2011, 133, 18118–18121 CrossRef CAS PubMed;
(b) J. S. Anderson, M.-E. Moret and J. C. Peters, J. Am. Chem. Soc., 2013, 135, 534–537 CrossRef CAS PubMed.
- H. Fong, M.-E. Moret, Y. Lee and J. C. Peters, Organometallics, 2013, 32, 3053–3062 CrossRef CAS PubMed.
- H. Kameo, J. Yamamoto, A. Asada, H. Nakazawa, H. Matsuzaka and D. Bourissou, Angew. Chem., Int. Ed., 2019, 58, 18783–18787 CrossRef CAS PubMed.
- F. Inagaki, C. Matsumoto, Y. Okada, N. Maruyama and C. Mukai, Angew. Chem., Int. Ed., 2015, 54, 818–822 CrossRef CAS PubMed.
-
(a) F. Inagaki, K. Nakazawa, K. Maeda, T. Koseki and C. Mukai, Organometallics, 2017, 36, 3005–3008 CrossRef CAS;
(b) C. Matsumoto, M. Yamada, X. Dong, C. Mukai and F. Inagaki, Chem. Lett., 2018, 47, 1321–1323 CrossRef CAS;
(c) F. Inagaki, K. Maeda, K. Nakazawa and C. Mukai, Eur. J. Org. Chem., 2018, 2972–2976 CrossRef CAS.
- Y. Segawa, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2009, 131, 9201–9203 CrossRef CAS PubMed.
-
(a) H. Ogawa and M. Yamashita, Dalton Trans., 2012, 42, 625–629 RSC;
(b) T. Miyada, E. Kwan and M. Yamashita, Organometallics, 2014, 33, 6760–6770 CrossRef CAS;
(c) H. Ogawa and M. Yamashita, Chem. Lett., 2014, 43, 664–666 CrossRef CAS;
(d) K. Tanoue and M. Yamashita, Organometallics, 2015, 34, 4011–4017 CrossRef CAS;
(e) E. Kwan, H. Ogawa and M. Yamashita, ChemCatChem, 2017, 9, 2457–2462 CrossRef CAS.
- E. Kwan, Y. Kawai, S. Kamakura and M. Yamashita, Dalton Trans., 2016, 45, 15931–15941 RSC.
- P. Ríos, N. Curado, J. López-Serrano and A. Rodríguez, Chem. Commun., 2015, 52, 2114–2117 RSC.
- Y. Ding, Q.-Q. Ma, J. Kang, J. Zhang, S. Li and X. Chen, Dalton Trans., 2019, 48, 17633–17643 RSC.
- T.-P. Lin and J. C. Peters, J. Am. Chem. Soc., 2013, 135, 15310–15313 CrossRef CAS PubMed.
- T.-P. Lin and J. C. Peters, J. Am. Chem. Soc., 2014, 136, 13672–13683 CrossRef CAS.
-
(a) W.-C. Shih, W. Gu, M. C. MacInnis, S. D. Timpa, N. Bhuvanesh, J. Zhou and O. V. Ozerov, J. Am. Chem. Soc., 2016, 138, 2086–2089 CrossRef CAS PubMed;
(b) W.-C. Shih, W. Gu, M. C. MacInnis, D. E. Herbert and O. V. Ozerov, Organometallics, 2017, 36, 1718–1726 CrossRef CAS;
(c) W.-C. Shih and O. V. Ozerov, J. Am. Chem. Soc., 2017, 139, 17297–17300 CrossRef CAS PubMed.
- W.-C. Shih and O. V. Ozerov, Organometallics, 2016, 36, 228–233 CrossRef.
-
(a) L. Vondung, N. Frank, M. Fritz, L. Alig and R. Langer, Angew. Chem., Int. Ed., 2016, 55, 14450–14454 CrossRef CAS;
(b) L. Vondung, L. Alig, M. Ballmann and R. Langer, Chem.–Eur. J., 2018, 24, 12346–12353 CrossRef CAS PubMed.
-
(a) M. Grätz, A. Bäcker, L. Vondung, L. Maser, A. Reincke and R. Langer, Chem. Commun., 2017, 53, 7230–7233 RSC;
(b) L. Maser, C. Schneider, L. Alig and R. Langer, Inorganics, 2019, 7, 61 CrossRef CAS.
- A. Bäcker, Y. Li, M. Fritz, M. Grätz, Z. Ke and R. Langer, ACS Catal., 2019, 9, 7300–7309 CrossRef.
- Catalytic hydroboration and hydrosilylation reactions utilizing [Fe(CH3CN)6][Fe(CO)4(InCl3)2] were reported, where an Fe–In bond is proposed to remain intact during the catalysis without a multidentate support. See:
(a) M. Ito, M. Itazaki and H. Nakazawa, ChemCatChem, 2016, 8, 3323–3325 CrossRef CAS;
(b) M. Ito, M. Itazaki and H. Nakazawa, Inorg. Chem., 2017, 56, 13709–13714 CrossRef CAS PubMed.
-
(a) P. Rudd, S. Liu, L. Gagliardi, V. G. Young and C. C. Lu, J. Am. Chem. Soc., 2011, 133, 20724–20727 CrossRef CAS PubMed;
(b) P. A. Rudd, N. Planas, E. Bill, L. Gagliardi and C. C. Lu, Eur. J. Inorg. Chem., 2013, 3898–3906 CrossRef CAS;
(c) J. T. Moore, N. E. Smith and C. C. Lu, Dalton Trans., 2017, 46, 5689–5701 RSC;
(d) M. V. Vollmer, J. Xie and C. C. Lu, J. Am. Chem. Soc., 2017, 139, 6570–6573 CrossRef CAS PubMed;
(e) M. V. Vollmer, J. Xie, R. C. Cammarota, V. G. Young, E. Bill, L. Gagliardi and C. C. Lu, Angew. Chem., Int. Ed., 2018, 57, 7815–7819 CrossRef CAS PubMed;
(f) M. V. Vollmer, R. C. Cammarota and C. C. Lu, Eur. J. Inorg. Chem., 2019, 2140–2145 CrossRef CAS;
(g) R. C. Cammarota, L. J. Clouston and C. C. Lu, Coord. Chem. Rev., 2017, 334, 100–111 CrossRef CAS.
-
(a) R. C. Cammarota and C. C. Lu, J. Am. Chem. Soc., 2015, 137, 12486–12489 CrossRef CAS;
(b) R. C. Cammarota, M. V. Vollmer, J. Xie, J. Ye, J. C. Linehan, S. A. Burgess, A. M. Appel, L. Gagliardi and C. C. Lu, J. Am. Chem. Soc., 2017, 139, 14244–14250 CrossRef CAS PubMed;
(c) J. Ye, R. C. Cammarota, J. Xie, M. V. Vollmer, D. G. Truhlar, C. J. Cramer, C. C. Lu and L. Gagliardi, ACS Catal., 2018, 8, 4955–4968 CrossRef CAS.
- Lu reported the preparation of a metal–organic framework consisting of well-defined Rh–Ga catalytic sites and its catalysis for a semihydrogenation reaction of alkynes to give trans-alkenes. See:
(a) S. P. Desai, J. Ye, J. Zheng, M. S. Ferrandon, T. E. Webber, A. E. Platero-Prats, J. Duan, P. Garcia-Holley, D. M. Camaioni, K. W. Chapman, M. Delferro, O. K. Farha, J. L. Fulton, L. Gagliardi, J. A. Lercher, R. L. Penn, A. Stein and C. C. Lu, J. Am. Chem. Soc., 2018, 140, 15309–15318 CrossRef CAS PubMed;
(b) S. P. Desai, J. Ye, T. Islamoglu, O. K. Farha and C. C. Lu, Organometallics, 2019, 38, 3466–3473 CrossRef CAS.
- M. V. Vollmer, J. Ye, J. C. Linehan, B. J. Graziano, A. Preston, E. S. Wiedner and C. C. Lu, ACS Catal., 2020, 10, 2459–2470 CrossRef CAS.
- J. T. Moore and C. C. Lu, J. Am. Chem. Soc., 2020, 142, 11641–11646 CrossRef CAS PubMed.
-
(a) J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2017, 139, 6074–6077 CrossRef CAS PubMed;
(b) N. Saito, J. Takaya and N. Iwasawa, Angew. Chem., Int. Ed., 2019, 58, 9998–10002 CrossRef CAS;
(c) J. Takaya, M. Hoshino, K. Ueki, N. Saito and N. Iwasawa, Dalton Trans., 2019, 48, 14604–14610 RSC.
-
(a) N. Hara, T. Saito, K. Semba, N. Kuriakose, H. Zheng, S. Sakaki and Y. Nakao, J. Am. Chem. Soc., 2018, 140, 7070–7073 CrossRef CAS PubMed;
(b) N. Kuriakose, J.-J. Zheng, T. Saito, N. Hara, Y. Nakao and S. Sakaki, Inorg. Chem., 2019, 58, 4894–4906 CrossRef CAS PubMed;
(c) T. Saito, N. Hara and Y. Nakao, Chem. Lett., 2017, 46, 1247–1249 CrossRef CAS.
- I. Fujii, K. Semba, Q.-Z. Li, S. Sakaki and Y. Nakao, J. Am. Chem. Soc., 2020, 142, 11647–11652 CrossRef CAS PubMed.
- S. Morisako, S. Watanabe, S. Ikemoto, S. Muratsugu, M. Tada and M. Yamashita, Angew. Chem., Int. Ed., 2019, 58, 15031–15035 CrossRef CAS PubMed.
- R. Yamada, N. Iwasawa and J. Takaya, Angew. Chem., Int. Ed., 2019, 58, 17251–17254 CrossRef CAS PubMed.
-
(a) J. Y. Corey and J. Braddock-Wilking, Chem. Rev., 1999, 99, 175–292 CrossRef CAS;
(b) J. Y. Corey, Chem. Rev., 2011, 111, 863–1071 CrossRef CAS;
(c) J. Y. Corey, Chem. Rev., 2016, 116, 11291–11435 CrossRef CAS PubMed.
- For selected examples; see:
(a) D. E. Hendriksen, A. A. Oswald, G. B. Ansell, S. Leta and R. V. Kastrup, Organometallics, 1989, 8, 1153–1157 CrossRef CAS;
(b) P. Sangtrirutnugul and T. D. Tilley, Organometallics, 2007, 26, 5557–5568 CrossRef CAS;
(c) M. C. MacInnis, D. F. MacLean, R. J. Lundgren, R. McDonald and L. Turculet, Organometallics, 2007, 26, 6522–6525 CrossRef CAS;
(d) Y.-H. Li, Y. Zhang and X.-H. Ding, Inorg. Chem. Commun., 2011, 14, 1306–1310 CrossRef CAS;
(e) H.-W. Suh, L. M. Guard and N. Hazari, Chem. Sci., 2014, 5, 3859–3872 RSC;
(f) H.-W. Suh, L. M. Guard and N. Hazari, Polyhedron, 2014, 84, 37–43 CrossRef CAS;
(g) T. Komuro, T. Arai, K. Kikuchi and H. Tobita, Organometallics, 2015, 34, 1211–1217 CrossRef CAS;
(h) Z. Xiong, X. Li, S. Zhang, Y. Shi and H. Sun, Organometallics, 2016, 35, 357–363 CrossRef CAS;
(i) B. A. Connor, J. Rittle, D. VanderVelde and J. C. Peters, Organometallics, 2016, 35, 686–690 CrossRef CAS;
(j) L. J. Murphy, H. Hollenhorst, R. McDonald, M. Ferguson, M. D. Lumsden and L. Turculet, Organometallics, 2017, 36, 3709–03720 CrossRef CAS;
(k) L. J. Murphy, A. J. Ruddy, R. McDonald, M. J. Ferguson and L. Turculet, Eur. J. Inorg. Chem., 2018, 4481–4493 CrossRef CAS;
(l) P. Zhang, X. Li, X. Qi, H. Sun, O. Fuhr and D. Fenske, RSC Adv., 2018, 8, 14092–14099 RSC;
(m) T. Komuro, T. Osawa, R. Suzuki, D. Mochizuki, H. Higashi and H. Tobita, Chem. Commun., 2019, 55, 957–960 RSC.
- S. J. Mitton and L. Turculet, Chem.–Eur. J., 2012, 18, 25258–25263 CrossRef PubMed.
- L. J. Murphy, M. J. Ferguson, R. McDonald, M. D. Lumsden and L. Turculet, Organometallics, 2018, 37, 4814–4826 CrossRef CAS.
-
(a) J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2008, 130, 15254–15255 CrossRef CAS PubMed;
(b) J. Takaya, K. Sasano and N. Iwasawa, Org. Lett., 2011, 13, 1698–1701 CrossRef CAS PubMed.
- J. Takaya and N. Iwasawa, Organometallics, 2009, 28, 6636–6638 CrossRef CAS.
-
(a) C. Zhu, J. Takaya and N. Iwasawa, Org. Lett., 2015, 17, 1814–1817 CrossRef CAS PubMed;
(b) J. Takaya, K. Miyama, C. Zhu and N. Iwasawa, Chem. Commun., 2017, 53, 3982–3985 RSC.
-
(a) J. Takaya and N. Iwasawa, Chem.–Eur. J., 2014, 20, 11812–11819 CrossRef CAS PubMed;
(b) J. Takaya and N. Iwasawa, Dalton Trans., 2011, 40, 8814–8821 RSC.
- J. Takaya and N. Iwasawa, Eur. J. Inorg. Chem., 2018, 5012–5018 CrossRef CAS.
-
(a) J. Takaya, N. Kirai and N. Iwasawa, J. Am. Chem. Soc., 2011, 133, 12980–12983 CrossRef CAS PubMed;
(b) N. Kirai, S. Iguchi, T. Ito, J. Takaya and N. Iwasawa, Bull. Chem. Soc. Jpn., 2013, 86, 784–799 CrossRef CAS.
-
(a) N. Kirai, J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2013, 135, 2493–2496 CrossRef CAS PubMed;
(b) J. Takaya, N. Kirai and N. Iwasawa, Organometallics, 2014, 33, 1499–1502 CrossRef CAS.
- J. Takaya, S. Ito, H. Nomoto, N. Saito, N. Kirai and N. Iwasawa, Chem. Commun., 2015, 51, 17662–17665 RSC.
- A related Pt-catalyzed sp2C–H borylation at sterically congested positions was reported at almost the same time. See: T. Furukawa, M. Tobisu and N. Chatani, J. Am. Chem. Soc., 2015, 137, 12211–12214 CrossRef CAS PubMed.
-
(a) H. Tobita, N. Yamahira, K. Ohta, T. Komuro and M. Okazaki, Pure Appl. Chem., 2008, 80, 1155–1160 CAS;
(b) T. Komuro, T. Kitano, N. Yamahira, K. Ohta, S. Okawara, N. Mager, M. Okazaki and H. Tobita, Organometallics, 2016, 35, 1209–1217 CrossRef CAS.
- T. Kitano, T. Komuro, R. Ono and H. Tobita, Organometallics, 2017, 36, 2710–2713 CrossRef CAS.
-
(a) A. Brück, D. Gallego, W. Wang, E. Irran, M. Driess and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 11478–11482 CrossRef PubMed;
(b) D. Gallego, A. Brück, E. Irran, F. Meier, M. Kaupp, M. Driess and J. F. Hartwig, J. Am. Chem. Soc., 2013, 135, 15617–15626 CrossRef CAS PubMed.
-
(a) W. Levason and G. Reid, Coord. Chem. Rev., 2006, 250, 2565–2594 CrossRef CAS;
(b) H. Braunschweig, P. Cogswell and K. Schwab, Coord. Chem. Rev., 2011, 255, 101–117 CrossRef CAS;
(c) S. Roggan and C. Limberg, Inorg. Chim. Acta, 2006, 359, 4698–4722 CrossRef CAS.
- S. L. Benjamin and G. Reid, Coord. Chem. Rev., 2015, 297–298, 168–180 CrossRef CAS.
-
(a) T. Lin, C. R. Wade, L. M. Pérez and F. P. Gabbaï, Angew. Chem., Int. Ed., 2010, 49, 6357–6360 CrossRef CAS PubMed;
(b) C. R. Wade, T.-P. Lin, R. C. Nelson, E. A. Mader, J. T. Miller and F. P. Gabbaï, J. Am. Chem. Soc., 2011, 133, 8948–8955 CrossRef CAS PubMed;
(c) T.-P. Lin, R. C. Nelson, T. Wu, J. T. Miller and F. P. Gabbaï, Chem. Sci., 2011, 3, 1128–1136 RSC;
(d) C. R. Wade, I. Ke and F. P. Gabbaï, Angew. Chem. Int. Ed., 2012, 51, 478–481 CrossRef CAS PubMed;
(e) I.-S. Ke and F. P. Gabbaï, Inorg. Chem., 2013, 52, 7145–7151 CrossRef CAS PubMed;
(f) I. Ke, J. S. Jones and F. P. Gabbaï, Angew. Chem. Int. Ed., 2014, 53, 2633–2637 CrossRef CAS;
(g) J. S. Jones, C. R. Wade and F. P. Gabbaï, Angew. Chem. Int. Ed., 2014, 53, 8876–8879 CrossRef CAS;
(h) H. Yang and F. P. Gabbaï, J. Am. Chem. Soc., 2014, 136, 10866–10869 CrossRef CAS PubMed;
(i) J. S. Jones, C. R. Wade and F. P. Gabbaï, Organometallics, 2015, 34, 2647–2654 CrossRef CAS;
(j) S. Sen, I.-S. Ke and F. P. Gabbaï, Inorg. Chem., 2016, 55, 9162–9172 CrossRef CAS PubMed;
(k) S. J. Jones, C. R. Wade, M. Yang and F. P. Gabbaï, Dalton Trans, 2017, 46, 5598–5604 RSC;
(l) S. Sahu and F. P. Gabbaï, J. Am. Chem. Soc., 2017, 139, 5035–5038 CrossRef CAS PubMed.
-
(a) H. Yang and F. P. Gabbaï, J. Am. Chem. Soc., 2015, 137, 13425–13432 CrossRef CAS;
(b) S. Sen, I.-S. Ke and F. P. Gabbaï, Organometallics, 2017, 36, 4224–4230 CrossRef CAS.
- D. You, H. Yang, S. Sen and F. P. Gabbaï, J. Am. Chem. Soc., 2018, 140, 9644–9651 CrossRef CAS.
- Y. Lo and F. P. Gabbaï, Angew. Chem., Int. Ed., 2019, 58, 10194–10197 CrossRef CAS PubMed.
-
(a) D. You and F. P. Gabbaï, J. Am. Chem. Soc., 2017, 139, 6843–6846 CrossRef CAS PubMed;
(b)
D. You, J. E. Smith, S. Sen and F. P. Gabbaï, Organometallics, DOI:10.1021/acs.organomet.0c00193.
-
(a) T.-P. Lin, I.-S. Ke and F. P. Gabbaï, Angew. Chem., Int. Ed., 2012, 51, 4985–4988 CrossRef CAS PubMed;
(b) I.-S. Ke and F. P. Gabbaï, Aust. J. Chem., 2013, 66, 1281 CrossRef CAS.
-
(a) C. Tschersich, C. Limberg, S. Roggan, C. Herwig, N. Ernsting, S. Kovalenko and S. Mebs, Angew. Chem., Int. Ed., 2012, 51, 4989–4992 CrossRef CAS PubMed;
(b) C. Tschersich, B. Braun, C. Herwig and C. Limberg, J. Organomet. Chem., 2015, 784, 62–68 CrossRef CAS.
-
(a) K. Materne, B. Braun-Cula, C. Herwig, N. Frank and C. Limberg, Chem.–Eur. J., 2017, 23, 11797–11801 CrossRef CAS PubMed;
(b) K. Materne, S. Hoof, N. Frank, C. Herwig and C. Limberg, Organometallics, 2017, 36, 4891–4895 CrossRef CAS.
-
(a) E. V. Dikarev, T. G. Gray and B. Li, Angew. Chem., Int. Ed., 2005, 44, 1721–1724 CrossRef CAS PubMed;
(b) E. V. Dikarev, B. Li and H. Zhang, J. Am. Chem. Soc., 2006, 128, 2814–2815 CrossRef CAS PubMed.
- J. Hansen, B. Li, E. Dikarev, J. Autschbach and H. M. Davies, J. Org. Chem., 2009, 74, 6564–6571 CrossRef CAS PubMed.
- L. R. Collins, M. van Gastel, F. Neese and A. Fürstner, J. Am. Chem. Soc., 2018, 140, 13042–13055 CrossRef CAS PubMed.
- L. R. Collins, S. Auris, R. Goddard and A. Fürstner, Angew. Chem., Int. Ed., 2019, 58, 3557–3561 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab