
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCoordination-based self-assembled capsules (SACs) for protein, CRISPR–Cas9, DNA and RNA delivery
Lukman O.
Alimi
,
Mram Z.
Alyami
,
Santanu
Chand
,
Walaa
Baslyman
and
Niveen M.
Khashab
 *
*
Smart Hybrid Materials Laboratory (SHMs), Advanced Membranes and Porous Materials Center, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia. E-mail: niveen.khashab@kaust.edu.sa
First published on 1st February 2021
Abstract
Biologics, such as functional proteins and nucleic acids, have recently dominated the drug market and comprise seven out of the top 10 best-selling drugs. Biologics are usually polar, heat sensitive, membrane impermeable and subject to enzymatic degradation and thus require systemic routes of administration and delivery. Coordination-based delivery vehicles, which include nanosized extended metal–organic frameworks (nMOFs) and discrete coordination cages, have gained a lot of attention because of their remarkable biocompatibility, in vivo stability, on-demand biodegradability, high encapsulation efficiency, easy surface modification and moderate synthetic conditions. Consequently, these systems have been extensively utilized as carriers of biomacromolecules for biomedical applications. This review summarizes the recent applications of nMOFs and coordination cages for protein, CRISPR–Cas9, DNA and RNA delivery. We also highlight the progress and challenges of coordination-based platforms as a promising approach towards clinical biomacromolecule delivery and discuss integral future research directions and applications.
1. Introduction
Traditionally, the common treatment of human diseases has been dominated by small molecule drugs. Over the past few decades, advances in biotechnology have enabled the engineering and preparation of specific biological molecules (primarily proteins) in microorganisms and other living cells using recombinant DNA technology. From these early discoveries and subsequent innovations, the realm of biologics has expanded to include a wide range of products. In addition to therapeutic proteins like peptides and antibodies, biologics also include nucleic acid-based therapies (e.g. RNAi, gene therapy, and gene editing). Such therapies have the potential to treat genetic disorders such as severe combined immunodeficiency,1 cystic fibrosis2 and Parkinson's disease3 in addition to drastically improving cancer treatment.4,5 However, the utilization of these highly effective biomolecules is dramatically hindered by their physicochemical properties such as fragile and tertiary structure, large size and inadequate cell internalization.6 Such limitations mark the direct delivery of biomolecules into cell compartments as an unsatisfactory technique. Hence, there is a huge need for safe and efficient delivery techniques for biologics to eventually bridge the gap between innovative nano-therapeutics and translational clinical trials. A fundamental engineering challenge for developing an effective delivery system is the development of a safe “nanocarrier” that has the ability to transmit intact therapeutic materials into a wide variety of cells, tissues and whole organs, with fully preserved bioactivity.7 Basically, nanocarriers are categorized into two types: viral and synthetic (non-viral). Although highly efficient and heavily used in vaccination, researchers have relentlessly tried to avoid using viruses due to their inherent immunogenicity, complicated synthetic routes and pre-modification of proteins.8 Synthetic approaches are judiciously considered as a promising alternative with no or negligible immunogenicity, high biocompatibility, ease of production, programmed targetability and the possibility to be administered multiple times.9–11Inspired by nature, a variety of synthetic self-assembled structures have been discovered for medical and industrial purposes.12–14 Coordination-based intercalation and delivery platforms are crystalline materials with voids created via the self-assembly and coordination interactions of molecular building blocks, which can accommodate biologically relevant guest molecules. We refer to this class of materials in this review as coordination-based supramolecular assembled capsules (SACs). SACs include extended frameworks such as nanosized metal–organic frameworks and discrete molecular entities such as coordination cages. Metal–organic frameworks (MOFs), as a class of hybrid materials, have attracted considerable attention recently due to their intriguing structural motifs and various potential applications.15 Although the main focus of MOF research is gas storage and selective gas adsorption, there are other potential applications of MOFs where a well-controlled small or nano size is required such as imaging,16 biosensing,17 and drug delivery.18 MOFs with bulk crystal size of several micrometers to millimeters are not usually suitable for practical applications in the biomedical field.19 Easy transportation through capillaries or the lymphatic system as well as effective cellular uptake requires customized small-sized (nano-scale) crystals. Consequently, studies on nano-sized metal–organic frameworks (nMOFs) in recent years have led to considerable innovation in the design and applicability of these systems.20 On another front, progress on coordination cages for biomolecule encapsulation has been rather limited. Coordination cages are discrete molecular constructs that are usually obtained by combining soluble metal and ligand building blocks that spontaneously generate metal–ligand bonds to form a single thermodynamically favored product.21 Coordination cages with different compositions, discrete size and shape and a variety of applications have been prepared using the coordination driven self-assembly approach.22,23 They demonstrated a high potential to purposefully combine the properties of metal ions with those of organic ligands for practical use in therapy and/or diagnostics.24 In this review, we focus on nMOFs (≤200 nm) and coordination cages that show great prospects for biomedical translation in terms of biocompatibility, biodegradability, encapsulation efficiency and targeted delivery of RNA, DNA and proteins (Fig. 1). We will highlight the size of the cargo compared to the nanocarrier as well as the type of cell where optimal results were achieved (Table 1). This review conjoins the various challenges facing the development of SACs and expectantly stimulates new ideas within the bio-supramolecular chemistry field to achieve actual clinical translation.
 | ||
| Fig. 1 Delivery of biologics using coordination-based supramolecular assembled capsules (SACs). | ||
| SACs | Size | Biomacromolecules (cargo) | Size | Biomedicine studies | Ref. |
|---|---|---|---|---|---|
| UiO-66 | 40–270 nm | Cytochrome c (Cyt c) | 12 kDa | In vitro: 4T1 cells | 49 |
| ZIF-90/GFP | 146 ± 3.8 | GFP | 27 kDa | HeLa cells | 50 |
| ZIF-90/BSA | 472 ± 33 | BSA | 66 kDa | ||
| ZIF-90/RNase A–NBC | 429 ± 27 | RNase A–NBC | 13 kDa | ||
| ZIF-90/SOD | 321 ± 64 | SOD | 16 kDa | ||
| ZIF-90/Cas9 | 583 ± 18 | Cas9–EGFP | 188 kDa | ||
| NU-1000 | 150 nm | Insulin | 13 Å × 13 Å | In vitro: SKOV-3 cells, B16-F10 cells | 51 |
| NU-1000 | 10 μm | ||||
| PCN-222 | ∼200 nm in length | ||||
| OVA@ZIF-8 | 200 nm | Ovalbumin | 45 kDa | In vitro: RAW264.7 cells | 52 |
| OVA@ZIF-8–CpG | 262.5 nm | In vivo study | |||
| CC-ZIFs | 100 nm | Cas9 + sgRNA = CRISPR | Cas9 with a size of 160 kDa and 150 nucleotides per sgRNA | In vitro: CHO cells | 57 |
| C3-ZIFs | 125 ± 3.32 nm | CRISPR/Cas9 | 160 kDa and 150 nucleotides per sgRNA | In vitro: HeLa, MCF-7, HDFn and a aTC cells + in vivo study | 60 |
| plGFP@ZIF-8 | Heterogeneous population (200–750 nm) | plGFP (plasmid DNA) | 6.5 kilo base-pairs | In vitro: PC-3 cells | 73 |
| pEGFP-C1@ZIF-8 | 279.1 ± 31.5 nm | pEGFP-C1 (plasmid DNA) | 4700 base-pairs | In vitro: MCF-7 cells | 77 |
| pEGFP-C1@ZIF-8–PEI | 275.7 ± 27.8 nm | ||||
| Ce6-DNAzyme@ZIF-8 | 167 nm | Ce6-DNAzyme | — | In vitro: MCF-7 cells + in vivo study | 78 |
| Ni-IRMOF-74-II to -V | 100–300 nm | Single-stranded DNA (ssDNA) | 11, 22, 33, and 53 nucleotides | In vitro: CD4+ T cells, B cells RAW264.7, THP-1 and MCF-7 cells | 79 |
| DNA/UiO-66 | 207.4 ± 0.4 | DNA | — | In vitro: HeLa and RAW264.7 cells | 80 |
| nNU-1000 | 150 nm | siRNA | 21–23 nt length (25 kDa) | In vitro: HEK293 cells | 81 |
| UiO | 98 ± 11 nm | siRNA + cisplatin prodrug | — | In vitro: SKOV-3 cells | 82 |
| UiO-Cis | 103 ± 17 nm | ||||
| siRNA/UiO-Cis | 128 ± 3 nm | ||||
| MIL-101 | 150 nm | siRNA | — | In vitro: MCF-7/T + in vivo study | 83 |
| Ru@MIL-101 | 160 nm | ||||
| Se@MIL-101 | 180 nm | ||||
| Pd12L24 cage | 7.3 nm | Ubiquitin | 8.6 kDa | N/A | 106 |
| Pd2L4 cage | —, exo-functionalization | Ac-NLEFK-Am | — | N/A | 114 |
| Di-iron(II) supramolecular cylinder | —, RNA–metal complexes binding through crystallization | RNA 3WJ | — | N/A | 117 |
| ZIF-8 | 0.5–1 μm | Catalase | 250 kDa | N/A | 132 |
| ZIF-90 | 0.5–2 μm | ||||
| MAF-7 | 0.6–2 μm | ||||
| ZIF-8/proteins (MP) | 76.2 ± 32.7 | BSA, cytochrome c, gelonin | 66 kDa, 12 kDa, 30 kDa | In vitro: MDA-MB-231, 293T, 3T3, CAD, MCF7, and SY5Y cells + in vivo study | 133 |
| MP-EVM (EMP) | 94.6 ± 32.5 |
2. nMOFs for biologics delivery
nMOFs have emerged as a potentially significant platform for biomacromolecule encapsulation and delivery due to their combined merits including tuneable chemical and physical properties and enhanced biocompatibility. Furthermore, the recent growth of nMOFs critically influenced their utility in biomedical applications such as efficient cell internalization and organelle interaction. Consequently, many studies have been conducted to investigate the validity of nMOFs in bioapplications.16–19,25–27 Furthermore, nMOFs are considered as smart materials as their structures respond to internal and external stimuli making them excellent candidates for controlled release delivery systems.28Considering the high demand for the prospective biomedical applications of nMOFs, it is important to evaluate their biocompatibility, stability, targetability and pharmacokinetics. Particle size plays a critical role in the intended biological application as particles with size larger than 200 nm are usually removed by macrophages29 while particles with a very small size (>100 nm) show successful transport in vivo without accumulation in specific organs.30 nMOFs should be toxicologically compatible; therefore, it is essential that the molecular building blocks are non-toxic. To date, in vitro toxicological experiments are being conducted to assess the cytotoxicity of different nMOF-based nanocarriers determined by cell viability on various cell lines. However, a valid question that is always raised when it comes to biocompatibility is whether we should go directly to in vivo testing since in most cases, synthetic delivery vehicles fail to go through pharmaceutical translation even after showing excellent in vitro profiles.31
Compared to their bulk counter materials, nMOFs' profile is no longer exclusively determined by their inner surface but also by their external surface properties through their high external surface area to volume ratio.4,5 In addition to the high loading capacities ensured by the large inner surface area, the precise and selective chemical modification of the internal pores is key to the achievement of proper encapsulation, driving the diffusional transport, delivery kinetics and stability of the designated cargo molecules.6 Loading through physical adsorption by immersing the prepared MOF nanocarriers into cargo-containing solutions typically applies to molecules that are smaller than the pore size of nMOFs. When the size of the cargo molecules is greater, physical adsorption on the surface of the MOF nanocarrier is obtained. To resolve this limitation regarding size-dependent loading of cargo molecules, one-pot synthesis of zeolitic imidazolate frameworks (ZIFs) has been established.32 With this facile in situ synthesis, larger sized guest molecules that cannot diffuse into nMOFs can be encapsulated for efficient target delivery without premature release.
2.1 nMOFs for protein and CRISPR–Cas9 delivery
The past decades have witnessed rapid development in protein delivery for several diagnostic and therapeutic purposes.5,33–37 Proteins are large biomacromolecules that display a set of dynamic and complex functions in critical biological processes such as biochemical catalysis, molecular transportation, formation of cellular structures, and DNA replication.33 In the intracellular microenvironment, the presence of insufficient or functionally altered proteins is the reason behind many human diseases, including cancer, neurological disorders, etc.36 Unlike small molecule delivery, delivery of large molecules such as proteins and peptides is more challenging due to their size, component effects and surface charge. Physical adsorption by immersing the prepared nMOFs with large pore size into a guest protein-containing solution can significantly improve the protein loading efficiency.38 In addition, storage of the protein inside the nMOF structure can protect the protein from enzymatic degradation during transportation.38 Most importantly, the intracellular uptake of proteins can be controlled further by surface modification of nMOFs, such as controlling the size and adjusting the surface charge.38a,39 For example, insulin, the most important protein drug for the treatment of type I diabetes, cannot be directly applied by oral delivery because of the extremely poor bioavailability and low diffusion rate through the mucus layer. In the stomach acid environment, free insulin can be denatured by strong acid and digested by pepsin. However, when using nMOF as a carrier for oral delivery of insulin, the ultra-stable MOF in the stomach acid environment can maintain the integrity of insulin while simultaneously excluding pepsin from getting access to the insulin, therefore limiting its proteolysis.38aGuest loading and/or encapsulation is the very first step and entails most consideration. Since sufficient biomolecule loading is the prerequisite for biomolecule delivery, much effort has been made on designing nMOFs with high biomolecule loading.14,18 The most common strategies are designing large pore frameworks by using longer linkers or etching the pores of an already prepared framework. Deng et al. made a series of IRMOF-74 MOFs with large aperture sizes for protein encapsulation by substituting the original linker of MOF-74, dioxidoterephthalate (DOT), with longer carboxylic acid-terminated palindromic oligophenylene derivatives.40 The pore aperture ranged from 1.4 nm to 9.8 nm. They successfully incorporated myoglobin (MB) and green fluorescent protein (GFP) into the MOF pores by simply immersing these MOFs in protein solutions. MB or GFP diffuses through the aperture, and is retained inside the framework due to hydrophilic interactions (MB) or hydrophobic interactions (GFP). It is remarkable that the crystallinity of the IRMOF materials is fully maintained throughout the inclusion process; the diffraction lines in the PXRD patterns of the included samples are coincident with those of the starting materials.
Along the same line, Feng et al. performed parallel work to synthesize PCN-333(Al) MOFs through linker extension and obtained mesoporous cages inside nanosized MOFs.41 These ultra-large mesoporous cages are suitable for the encapsulation of three enzymes, including HRP, Cyt c and microperoxidase-11 (MP-11). Successful enzyme loading was observed, for which they proposed that there might be only one HRP and Cyt c protein molecule in each cage, but multiple MP-11 enzymes in single cages due to the size mismatch. PCN-333 exhibits the largest cage (5.5 nm) and one of the highest void volumes (3.84 cm3 g−1) among all reported nanosized MOFs. PCN-333(Al, Fe) shows high stability in aqueous solutions with pH 3 to 9, making it an extraordinary candidate for enzyme encapsulation. It is worth mentioning that the extension of organic linkers, which mostly serve as the faces or edges of pores in MOFs, is the main approach to achieve large pores in MOFs.42 However, the pore shape or symmetry plays a very important role in determining the overall pore size.
Ma and coworkers carried out extensive systematic studies on protein diffusion inside MOF structures.43–45 The pre-synthesized Tb-mesoMOFs are immersed in protein buffer solutions. Proteins are taken up by MOFs after a given amount of time of incubation. Microperoxidase-11 (MP-11; having dimensions of ∼3.3 × 1.7 × 1.1 nm)43 has for the first time been successfully immobilized into a mesoporous MOF residing in nanoscopic cages.45 It consists of an iron-heme group linked to an α-helical undecapeptide chain via two thioether bonds of cysteine residues and a coordinated histidine residue at an axial position of the Fe(III)-heme center. It is able to oxidize a wide range of organic molecules using hydrogen peroxide.46 The mesoporous MOF selected for MP-11 immobilization is Tb-TATB (hereafter denoted as Tb-mesoMOF), which contains nanoscopic cages of diameter 3.9 and 4.7 nm.47,48 It exhibits characteristic type-IV N2 sorption isotherms with pore sizes dominantly distributed around 3.0 and 4.1 nm in addition to a small portion with a micropore size of around 0.9 nm. MP-11 resides in the nanocages due to its small size, and has catalytic activity toward alcohol oxidation to the corresponding ketones. Interestingly, no MP-11 leaching is observed once immobilized in the Tb-mesoMOF. This is attributed to the strong π–π interactions between triazine/benzene rings on the linker and the heme structure on MP-11.47
Similarly, cytochrome c (Cyt c) protein has been reported to enter Tb-mesoMOF via conformational changes, which is supported by fluorescence studies.44 Cyt c is a relatively small, structurally robust heme protein with molecular dimensions of ∼2.6 nm × 3.2 nm × 3.3 nm that serves as a component of the electron transport chain in the mitochondria and is involved in apoptosis. The protein consists of a single polypeptide chain of 104 amino acid residues containing a covalently attached heme group. Cyt c undergoes conformational changes during the immobilization process and adopts a new conformation,44 (Fig. 2) which resembles the translocation of some proteins via nanopores during metabolism. Considering the stability of its framework in buffer solutions, Tb-mesoMOF provides an excellent platform for investigating the interaction between Cyt c and MOF nanopores. In view of the dimensions of the Cyt c molecule as well as the hydrophobic properties of the Tb-mesoMOF framework, importing Cyt c into the MOF interior would require the protein to undergo a change in conformation initiated first by surface contacts between the protein and the MOF crystal. Migration of the protein through the relatively small nanopores to enter the 3.9 and 4.7 nm diameter cages would require a partial unfolding of the protein's tertiary structure.44
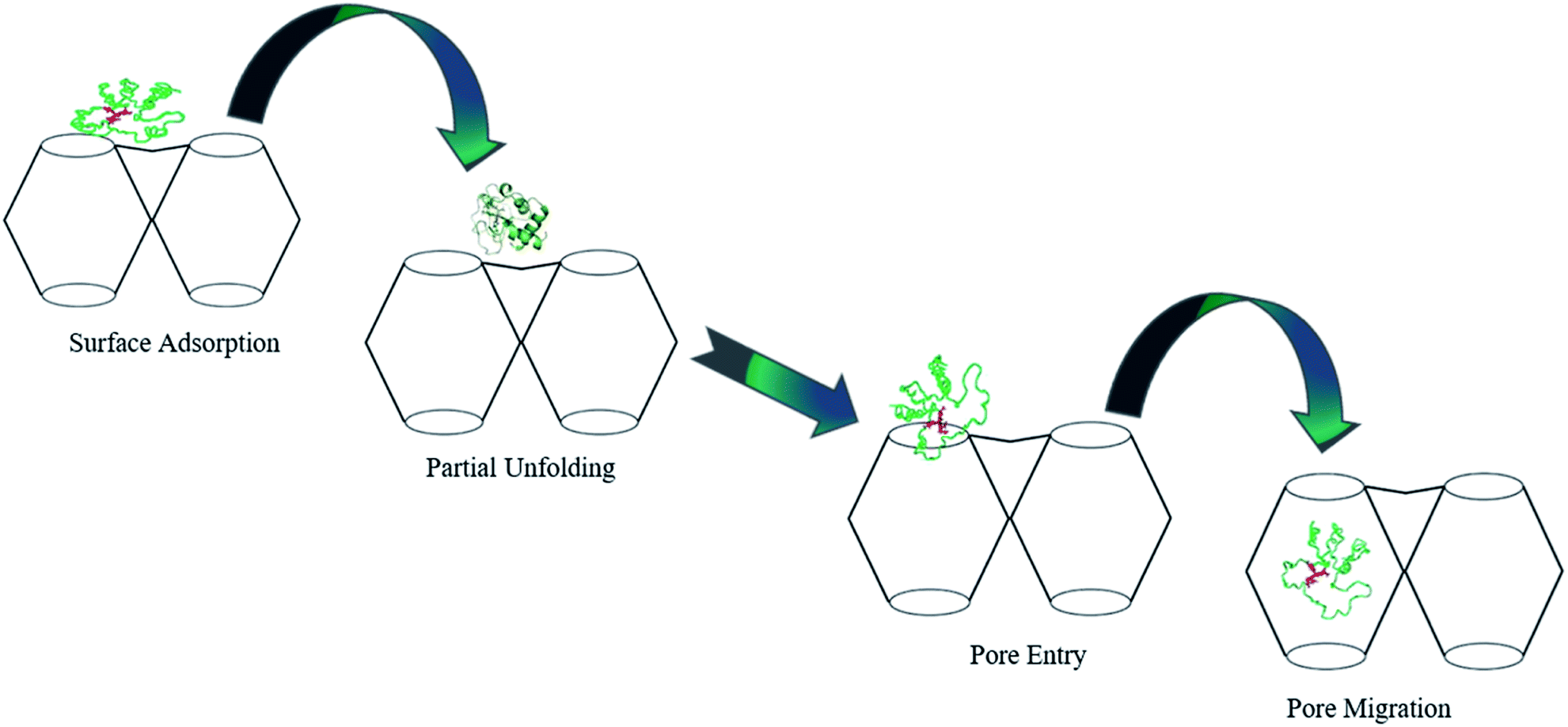 | ||
| Fig. 2 Proposed mechanism for the translocation of Cyt c into Tb-mesoMOF. Adapted with permission from ref. 44. Copyright (2012) American Chemical Society. | ||
Recently, Wang et al. encapsulated cytochrome c (Cyt c) into a series of hierarchically porous UiO-66(Zr) that differ in their nano-sizes.49 Size-related cellular uptake was observed with the nanoparticle size of 90 nm showing the highest uptake in comparison with others in the range of 40–270 nm. The endosomal escape was also monitored in a time-controlled manner due to the unique dual pore systems (micro and meso) of the UiO-66(Zr).
Another study utilized the possible competition between MOF components and specific intracellular compositions, which was reported by Yang et al. using ZIF-90 as the protein delivery vehicle. This approach relies on the presence of the intracellular ATP (adenosine triphosphate) component that has different concentrations in healthy and diseased cells and an affinity to coordinate with 2-imidazole carboxaldehyde (ICA). Consequently, ATP serves as an intracellular stimulus that disassembles the MOF framework by competing with Zn2+ to coordinate with the ICA linker. The result of the intracellular delivery of the green fluorescent protein (GFP) encapsulated inside ZIF-90 shows a high encapsulation efficiency of 90% when exposed to HeLa cells while no notable cellular uptake was observed with the free GFP (Fig. 3).50
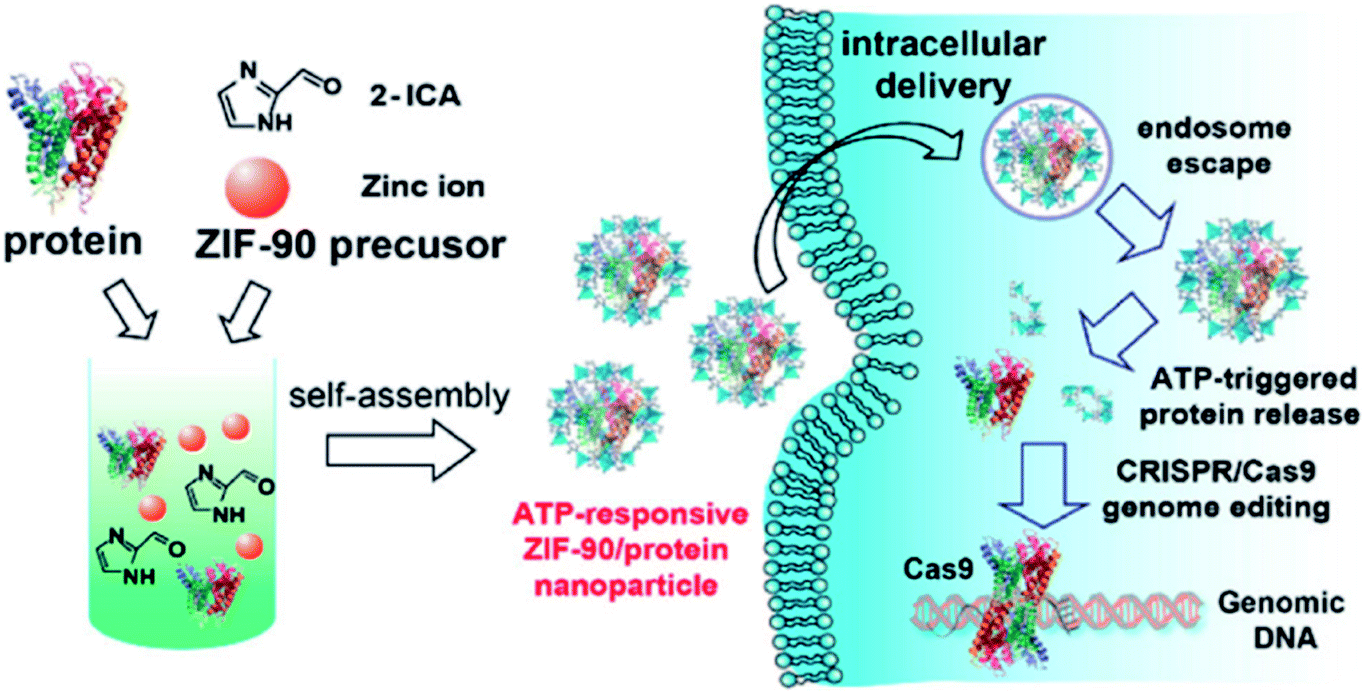 | ||
| Fig. 3 Schematic illustration of the self-assembly of ZIF-90/protein nanoparticles and ATP-triggered protein release from ZIF-90 nanoparticles inside cells. Adapted with permission from ref. 50. Copyright (2019) American Chemical Society. | ||
DNA-functionalized MOF nanocarriers were introduced as a promising strategy for improving colloidal-stability with facile cellular uptake. Wang et al. encapsulated insulin into two water-stable MOF nanoparticles, NU-1000 and PCN-222, and then functionalized their surfaces with terminal phosphate-modified DNA to yield insulin@DNA-MOF NPs.51 The nanoparticle sizes of the two MOFs are 150 nm for NU-1000 and 200 nm for PCN-222. The results demonstrated that high loading and enzymatic activity of insulin were achieved with ten-fold enhancement of the cellular uptake.
Zhang et al. reported another delivery system of protein embedded MOF vaccine, in which a model antigen was encapsulated into ZIF-8, with an average particle size of 200 nm, followed by adsorption of an immune adjuvant, cytosine–phosphate–guanine oligodeoxynucleotides (CpG ODNs), into the encapsulated MOF NPs by strong electrostatic interaction. The role of the immune adjuvant (CpG ODNs) was showcased to improve the biocompatibility and immunogenicity of the nanocarrier. The in vitro and in vivo results highlight the excellent biocompatibility and controlled release to deliver the antigen protein into the targeted cell, inducing a strong humoral and cellular immune response.52,53
Protein loading into nMOF nanoparticles can also take place via pore encapsulation, de novo approach54 or biomimetic mineralization.55 The term biomimetic mineralization was first introduced by Doonan, Falcaro and coworkers as a novel strategy to encapsulate biomacromolecules inside MOFs under physiological conditions. Biomimetic mineralization allows encapsulation of various proteins, with high loading (90%) and no requirements for pre-modification to either proteins or synthetic procedures. Interestingly, under the physiological environment, proteins, DNA and enzymes influence the formation of the nMOFs by concentrating the molecular building blocks, which ultimately leads to the nucleation of the porous crystals around the biomacromolecules. Significantly, after biomineralization, the subsequent biocomposite is quite stable even after exposure to harsh conditions such as high temperature and enzymatic degradation.55
With the improvement of the preparation of nMOF, the size, biocompatibility, zeta potential, and biodegradability of some protein-encapsulated nMOFs could contest the prerequisites of the biomedical field. In 2018, Chen et al. synthesized BSA@ZIF-8 NPs via biomineralization with a specific size of 92 ± 7.9 nm. The BSA loading capacity was ∼52.2 μg and the encapsulation efficiency was 93%, in 1 mg BSA@ZIF-8 NPs.56 The BSA delivery system was also extended to other proteins and even multiple proteins in one single ZIF-8 NP for co-delivery with high loading capacities where only the size changed. This might be due to the different pre-nucleated clusters surrounding the proteins.
Most recently, nMOFs have emerged as a unique platform to deliver clusters combining proteins and genetic materials for cytosolic delivery and genome editing.57 Clustered regularly interspaced short palindromic repeat (CRISPR)-associated proteins (Cas9) have recently triggered a revolution in biological research. CRISPR/Cas9 can precisely target a DNA sequences via sgRNA within the protein complex. A key property of Cas9 protein is its ability to cleave DNA strands. The break is then repaired by non-homologous end-joining (NHEJ) or homology-directed repair (HR).58,59 However, for this technique to be effective, the Cas9 protein and sgRNA need to be perfectly protected from endosomal degradation.
Our group reported the first example of CRISPR/Cas9 complex delivery (size of Cas9 ≈ 160 KDa and two sgRNAs of 150 nucleotides) by the co-encapsulation of Cas9 protein and guide RNA into the ZIF-8 (CC-ZIF) framework in situ with an impressive loading efficiency of 17% (Fig. 4a).57 The stability study of CC-ZIFs shows that less than 3% of CRISPR/Cas9 was released under physiological conditions (pH 7) and the optimum conditions for the maximum release (99%) were at pH 5 after 3 h. The transmission electron microscopy (TEM) image showed that the CC-ZIFs had an average size of ≈100 nm, which is the ideal size for intercellular delivery and the in vivo study. The CC-ZIFs displayed superior efficiency for knocking down gene expression (GFP) that reached 37% over a period of four days in Chinese hamster ovary (CHO) cells due to the enhanced endosomal rupture as a result of the degraded ZIF-8.57 Furthermore, we improved the cell specific delivery of CRISPR/Cas9 by the essential homotypic binding phenomenon of tumor cells.60 CC-ZIFs were coated with MCF-7 cell membrane through coextrusion to produce C3-ZIFMCF with an average size of ≈120 nm (Fig. 4b). C3-ZIFMCF displayed great efficiency for specific and targeted delivery. The cellular uptake studies of C3-ZIFMCF show the highest uptake in MCF-7 cells compared to other cell types due to the inherent homotypic binding phenomenon (Fig. 5c). EGFP expression was knocked down by three fold with C3-ZIFMCF, while C3-ZIFHELA knocked down the expression in MCF-7 cells by one fold.60
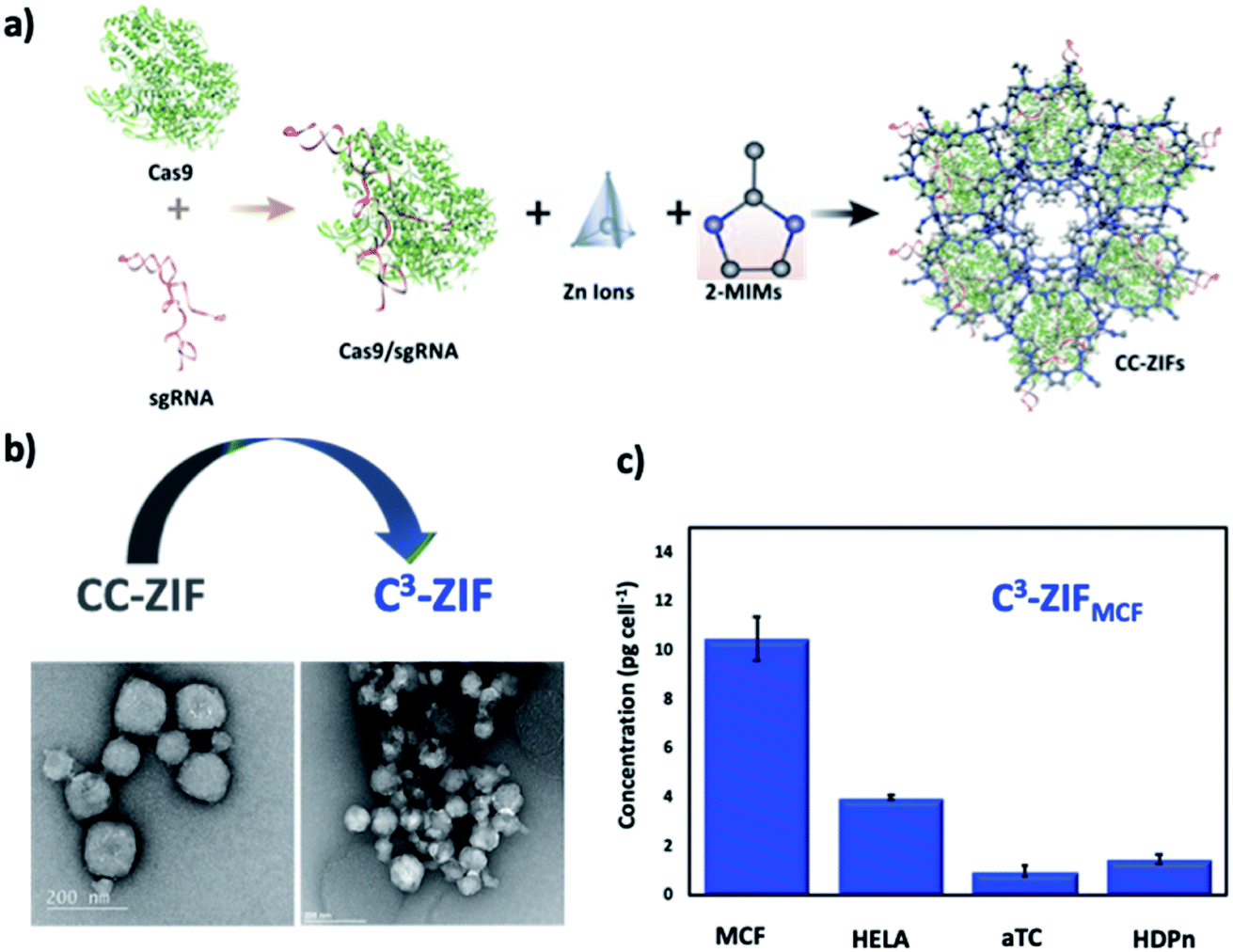 | ||
| Fig. 4 Nanosized extended frameworks for CRISPR/Cas9 delivery. (a) Illustration of CC-ZIF synthesis and cellular uptake. Adapted with permission from ref. 57. Copyright (2018) American Chemical Society. (b) TEM images of CC-ZIF and C3-ZIF. (c) ICP-MS of the Zn concentration of different cell lines incubated by C3-ZIFMCF. Adapted with permission from ref. 60. Copyright (2020) American Chemical Society. | ||
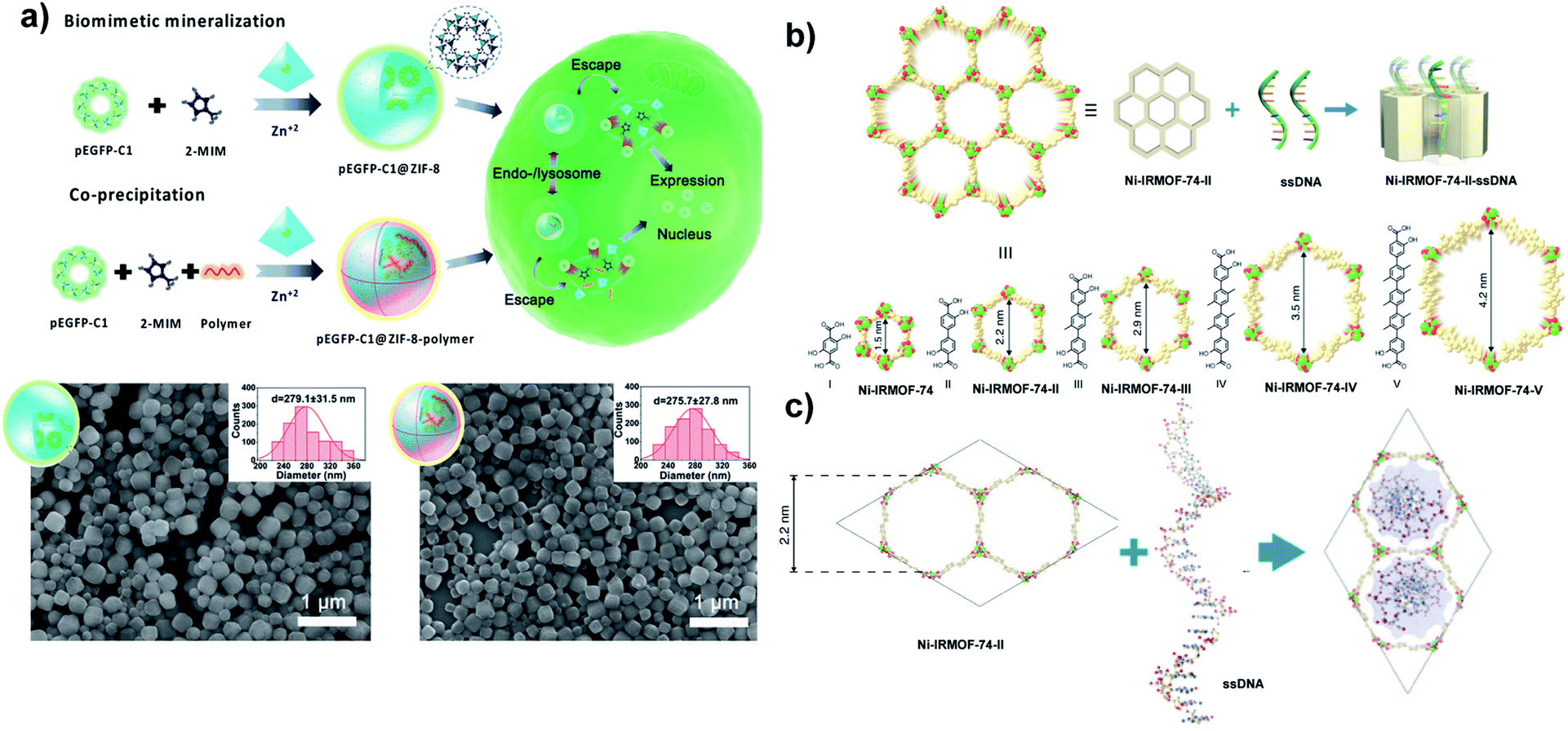 | ||
| Fig. 5 Nanosized extended frameworks for DNA delivery. (a) Schematic representation for the synthesis of pEGFP-C1@ZIF-8 via biomimetic mineralization and pEGFP-C1@ZIF-8–polymer using a coprecipitation method and their cellular delivery, expression process and SEM images. Adapted with permission from ref. 77. Copyright (2019) Wiley-VCH. (b) Illustration of ssDNA inclusion in the Ni-IRMOF-74 series. Ni, C, and O atoms were labelled with green, gold, and red color, respectively. (c) Visualization of ssDNA inclusion in the pore of Ni-IRMOF-74-II. Adapted with permission from ref. 80. Copyright (2018) Springer Nature. | ||
2.2 nMOFs for RNA and DNA delivery
In general, gene therapy refers collectively to methods aimed at prevention or treatment of diseases associated with defective gene expression including autoimmune disease,61 hemophilia62 and cancer,63–68 replacing damaged genes or expressing inhibition of undesired genes through the insertion of integrating or non-integrating exogenous DNA or RNA. Gene delivery for therapeutic application currently involves two strategies that affect the genetics of the targeted cells: (1) DNA delivery with the aim of providing a functional copy of a defective gene. (2) The delivery of therapeutic ribonucleic acids which include microRNA (miRNA), short hairpin RNA (shRNA) and small interfering RNA (siRNA). The RNA species are processed via the Dicer complex, and loaded into the RNA-induced silencing complex (RISC), which then binds to messenger RNA (mRNA) molecules to either degrade them or modulate their expression.69–71 Indeed, an efficient delivery system is required for successful gene therapy, which allows the transfer and expression of the therapeutic gene in the target organ or tissue. nMOFs have attracted increasing attention for RNA and DNA delivery due to the possibility of tuning their porosity by appropriate molecular design compared to other synthetic gene delivery vehicles.72 Also, nMOFs have successfully overcome the obstacles of genetic material delivery since they maintain their chemical and physical integrity and facilitate their cell penetration compared to naked genetic materials.Much effort has been focused recently on large DNA delivery using nMOFs with an average size of 200–300 nm (based on DNA size). Shukla and coworkers have demonstrated successful delivery of an entire plasmid with a size of ≈6.5 kilo base-pairs via encapsulation utilizing nano-sized ZIF-8.73 ZIFs can be easily synthesized at room temperature in water through the coordination of zinc ions and imidazole rings as chemical stimuli responsive nanocarriers which can rapidly degrade in an acidic environment or chelating agents.74 plGFP@ZIF-8 with a loading efficiency of 82% and heterogeneous population was synthesized by mixing plasmid DNA (500 ng) dissolved in DNase free water with aqueous solutions of 2 mIM (10 μl); then zinc acetate dihydrate (10 μl) was added at room temperature for 10 min. To investigate whether plGFP is encapsulated within ZIF-8 or remains in the supernatant, both plGFP@ZIF-8 and the supernatant were treated with propidium iodide (PI) for DNA staining with an emission peak at 612 nm. The PI emission peak was absent in both PI treated supernatant and plGFP@ZIF-8. In comparison, plGFP@ZIF-8 treated with EDTA shows an emission peak at 612 nm after chelating Zn2+ with EDTA, confirming successful encapsulation. As a confirmation that plasmid DNA (plGFP) remains functionally intact and is not negatively affected by the biomimetic mineralization process, human prostate cancer epithelial cells (PC-3) incubated with plGFP@ZIF-8 show green fluorescence after 96 h, which indicates successful gene expression.73
Most recently, Shukla and coworkers in a separate study reported for the first time the use of ZIF-C as a suitable gene therapy delivery system for prostate cancer (PC).75 Previously, the authors reported the discovery of ZIF-C which was described as ZIF-8 with aggregated plate-like morphology.76 They demonstrated that when targeting the ribosomal protein SA (RPSA) gene, which is overexpressed in prostate cancer, there was a significant increase in the toxicity due to the disruption of the gene. Transcriptional efficiency following transfection with a ZIF-C based delivery system was determined by calculating the % RPSA knockdown (% KD). When siRPSA@ZIF-8 was used, 22% KD was observed but this increased to 40% KD upon using EsiRPSA@ZIF-C.
Tang and coworkers reported the delivery of pDNA with ≈4700 bp by coprecipitation with different molecular weights of polyethyleneimine (PEI) as a capping agent. The influence of PEI on pDNA delivery has been evaluated. pEGFP-C1@ZIF-8–PEI nanoparticles (PEI MW ≈ 25 kD) exhibit better stability of pEGFP-C1 against DNase degradation (≈82.3%) compared to pEGFP-C1@ZIF-8. The excellent loading capacity (≈3.4 wt%) refers to the strong electrostatic interaction between pDNA and PEI (MW ≈ 25 kD). Scanning electron microscopy (SEM) images indicate that pEGFP-C1@ZIF-8 and pEGFP-C1@ZIF-8–PEI display a uniform size of around 280 nm (Fig. 5a). In contrast, SEM images of plGFP@ZIF-8 in a previous report show a heterogeneous population referring to cargo size (≈6.5 kbp vs. ≈4700 bp). However, the size could be controlled by tuning the reaction time and the concentration of precursors.77 The surface functionalization of pEGFP-C1@ZIF-8 by PEI led to higher cellular uptake than for pEGFP-C1@ZIF-8, due to the negatively charged cell membrane interaction with positively charge PEI which facilitates the internalization of the nanoparticles.
Wang et al. reported ZIF-8 as a nanocarrier for effective photodynamic and gene therapy by encapsulating DNAzyme functionalized chlorin e6 (DNAzyme@ZIF-8). ZIF-8 enhanced the biocatalytic activity of the DNAzyme via supplying the requisite Zn2+ ions. DNAzyme@ZIF-8 (with an average size of ≈167 nm and loading capacity of 10 wt%) was incubated with MCF-7 cells to evaluate EGR-1 mRNA expression. qRT-PCR and western blot studies indicate that the gene silencing has been successfully dominated by DNAzyme. Interestingly, the apoptotic ratio of Ce6-DNAzyme@ZIF-8 was higher compared to that of the gene therapy and PDT alone (44.9%, 19.85%, and 33.6%, respectively).78 Efficient delivery of DNAzyme and different sizes of plasmid DNA have been reported in previously mentioned studies through biomimetic mineralization where the DNA has been added in situ during the nMOF synthesis which facilitates large size DNA accommodation. Kong and Fan reported DNA delivery via coordination with unsaturated zirconium centers and DNA backbone phosphate without specific modification. Immunostimulatory nMOFs (isMOFs) have been prepared by coordinating cytosine–phosphate–guanosine on the surface of UiO-66-NH2 constructed from amino-terephthalic acid and ZrCl4 followed by biomineralization with calcium phosphate to facilitate CpG release in a cellular environment. isMOFs with size of ≈207.4 nm show high loading capacity, and each single MOF particle was able to load 2500 CpG strands. isMOFs exhibit a higher secretion level of IL-6 compared to CpG-MOF.79
A new strategy was developed by Deng and Zhou for DNA delivery based on the inclusion of single-stranded DNA (ssDNA) with different lengths (11, 22, 33, and 53 nucleotides) into a series of nMOFs (Ni-IRMOF-74-II to -V) with hexagonal topology and different suitable pore sizes that allow single-stranded DNA delivery via van der Waals interactions (Fig. 5b and c). ssDNA was released via strong interaction with cDNA to form dsDNA without disassociation from Ni-IRMOF-74. A dynamic light scattering (DLS) study and SEM images show that Ni-IRMOF-74-II and Ni-IRMOF-74-III have a size of ≈146.4 nm and ≈89.91 nm, respectively. The small size of Ni-IRMOF-74-III could make it a good candidate for future in vivo study. Both Ni-IRMOF-74-II and Ni-IRMOF-74-III displayed weak interactions with ssDNA that shows great transfection efficiency for primary mouse immune cells (CD4+ T cells), 60% and 92%, respectively.80
Furthermore, nMOFs with an average size of 100 nm to 200 nm were efficiently employed for small interfering RNA (siRNA) delivery, which is typically 20–27 base pairs in length, via nMOF surface functionalization or siRNA loading within the appropriate nMOF size. Fairen-Jimenez and coworkers reported the successful delivery of unmodified siRNA with 21–23 nt to the cytoplasm employing mesoporous channels of zirconium-based MOFs (nNU-1000).81 siRNA was loaded in nNU-1000 with an average size of ≈150 nm after activation to ensure the removal of residual solvent. nNU-1000 successfully protects siRNAs from RNase degradation, as relevant siRNA bands were still observed on a polyacrylamide gel after an enzymatic attack. siRNA@nNU-1000 treated HEK293-mC cells show an inconsistent change in mCherry expression due to the degradation in the endosome. Then, cofactors such as proton-sponges (PS) and membrane opening peptides were used to overcome endosomal entrapment. Moreover, in vitro studies proved that nNU-1000@siRNA incubated with PS and amphipathic KALA cell-penetrating peptide decreased mCherry expression to ca. 78% and 73%.81
Lin and colleagues reported the co-delivery of siRNAs and cisplatin utilizing nMOFs, to enhance the therapeutic efficacy through the use of silencing multiple drug resistance (MDR) gene and overcoming resistant ovarian cancer cells to cisplatin treatment.82 UiO nMOFs were prepared by mixing DMF solutions of ZrCl4 and amino-TPDC, followed by adding 750 μl of acetic acid. The mixture was kept in an 80 °C oven for 5 days. Then, cisplatin was loaded into UiO nMOFs via diffusion forming UiO-Cis followed by coordination of MDR gene-silencing siRNA with vacant Zr ions on the surface of UiO-Cis. siRNA/UiO-Cis with a size of ≈128 nm enhanced the cellular uptake of siRNA by 11-fold compared to naked siRNA. siRNAs/UiO-Cis induced cell apoptosis in different cell lines by combining chemotherapeutics and the synergistic effects of down-regulating the expressions of MDR-relevant genes.82
Liu and coworkers utilized MIL-101 for co-delivery of chemotherapy agents and siRNA. Se@MIL-101 and Ru@MIL-101 with a remarkable antitumor activity were prepared by reducing SeO32− and RuCl3 into the cavities of MIL-101 at a low temperature followed by adding 100 nM siRNA forming Ru@MIL-101–siRNA and Se@MIL-101–siRNA at room temperature through the coordination between phosphate on the backbone of siRNA and Fe(III) sites of MIL-101. Se/Ru@MIL-101–siRNA had an average size of ≈180 nm and ≈160 nm and loading efficiency of 6.92 wt% for SeNPs and 8.13 wt% for RuNPs. Both Se@MIL-101 and Ru@MIL-101 exhibit efficient endosomal/lysosomal escape via the interaction between Fe ions resulting from the MIL-101 decomposition in the lysosomal membrane.83
It is important to note at this stage that controlling the particle size could be difficult when biomacromolecule-MOFs were prepared by the one-pot synthesis strategy. Most recently, Falcaro and coworkers addressed this issue by encapsulating different proteins (bovine serum albumin, BSA) and a clinical therapeutic (α1-antitrypsin, AAT) in ZIF-8 using a continuous flow synthetic method. In this study, BSA was used as a model protein to develop a continuous flow procedure and to understand the growth kinetics for ZIF-8 based composites. The nucleation and growth of BSA@ZIF-8 and the influence of the residence time using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ethanol/water ratio was examined by synchrotron time-resolved SAXS and atomic force microscopy (AFM) and SEM, respectively. By varying the residence time from 0.33 to 120 s they were able to synthesize BSA@ZIF-8 crystals with particle sizes ranging from 40 to 100 nm. Interestingly, the particle size can be precisely tuned by controlling the residence time prior to injection of ethanol into the mixed ZIF precursor solution. This phenomenon was attributed to an abrupt amorphous-to-crystalline transition. The application of the flow system to test for the encapsulation of AAT, a biotherapeutic with anti-inflammatory and immunomodulatory properties, was also demonstrated. AAT@ZIF-8 samples with different particle sizes of 90, 110 and 180 nm were prepared and after dissolution of the ZIF matrix in 1 mM HCl and exposure of the released AAT to a trypsin solution and incubation at 48 °C for 30 min, the protease inhibitor fully retained its bioactivity. From the study it was concluded that continuous flow synthesis of ZIF-8-based composites afforded excellent control over the particle size that is suitable for intravenous drug delivery administration (particle size ≤ 200 nm).84
:1 ethanol/water ratio was examined by synchrotron time-resolved SAXS and atomic force microscopy (AFM) and SEM, respectively. By varying the residence time from 0.33 to 120 s they were able to synthesize BSA@ZIF-8 crystals with particle sizes ranging from 40 to 100 nm. Interestingly, the particle size can be precisely tuned by controlling the residence time prior to injection of ethanol into the mixed ZIF precursor solution. This phenomenon was attributed to an abrupt amorphous-to-crystalline transition. The application of the flow system to test for the encapsulation of AAT, a biotherapeutic with anti-inflammatory and immunomodulatory properties, was also demonstrated. AAT@ZIF-8 samples with different particle sizes of 90, 110 and 180 nm were prepared and after dissolution of the ZIF matrix in 1 mM HCl and exposure of the released AAT to a trypsin solution and incubation at 48 °C for 30 min, the protease inhibitor fully retained its bioactivity. From the study it was concluded that continuous flow synthesis of ZIF-8-based composites afforded excellent control over the particle size that is suitable for intravenous drug delivery administration (particle size ≤ 200 nm).84
In a separate study, Ren and coworkers reported the synthesis of enzyme–MOF composites with high substrate accessibility and enzyme activity using a microfluidic gradient mixing strategy. They demonstrated that by continuously changing the concentrations of MOF precursors in the gradient mixing of the microfluidic flow through a double-Y-shaped microfluidic channel, structural defects were induced into the enzyme–MOF composites. These structural defects were attributed to the loss of Zn atoms as R-space EXAFS data showed a smaller amount of Zn compared to the Zn amount in the bulk solution synthesis method leading to the formation of mesopores in enzyme–MOF composites. This promoted the substrate accessibility and enzyme activity where the enzymatic activity reached 98% which was higher than the enzymatic activity when the bulk solution synthesis method was used. This value was said to be the highest record of enzymatic activity of enzyme–MOF composites. In addition, enzyme–MOF composites prepared by microfluidic flow synthesis were reported to have a wide pore distribution from 1 to 6 nm as a result of change in the flow rate. They concluded that the unique gradient mixing nature of the microfluidic laminar flow synthesis strategy allows controllable, continuous, and fast synthesis of enzyme–MOF composites with improved activity, which is expected to become a new general approach for inducing defects into enzyme–MOF composites and other biocomposites.85
3. Coordination cages for biologics delivery
Molecules that form supramolecular capsules are defined by two fundamental properties: the mode of self-assembly and the emergent host–guest interactions. Self-assembly is based on capsule components with functional groups that are capable of reversible, noncovalent interactions. Hydrogen bonds and metal–ligand interactions are the most useful noncovalent forces in constructing capsules. These interactions enjoy facile reversibility and reliable directionality, but hydrogen bonds show greater plasticity and faster equilibration, while metal–ligand bonds typically offer greater strength and more rigidity. High-symmetry designs are used to multiply these individually weak and reversible interactions into coherent structures with lifetimes that range from microseconds to hours.The subsequent encapsulation of guest molecules is dependent on the complementarity of the guest's size, shape, and chemical surface with the cavity of the host.86 One of the major challenges for designing/constructing these capsules is being able to isolate guest molecules from the bulk solvent. The medium must not disrupt the interactions that hold the components of the capsule together. Capsules constructed through metal–ligand interactions are typically disrupted by strongly ligating solvents, while they may remain stable in water. In contrast, solvent competition for hydrogen bonds prevents capsules constructed using these forces from being stable in aqueous media. The properties of the solvent to fill space must also be taken into consideration when dealing with encapsulation complexes. While the encapsulation of the solvent itself is sometimes desirable, the use of a large solvent that is physically excluded from the cavity can be an important tactic when encapsulating other guest molecules.87
A popular strategy for designing large multicomponent cage assemblies is the metal-directed self-assembly approach, advocated chiefly by Fujita and coworkers88–90 and Stang and coworkers.91–93 Coordination cages, with various compositions, shapes, and sizes, have been constructed to establish the main principles in the design and synthesis of the metallo-supramolecular unit with predefined shapes by exploiting the bond directionality and coordination properties of the metal ions.21
Metallo-supramolecular units with well-defined internal voids or inner cavities have attracted a great deal of attention because of their appealing structures and potential applications in biocatalysis, biotechnology, drug encapsulation and delivery systems.94–96 In general, normal assembly of highly directional ligands and appropriate metal ions can produce elaborate metal-coordinated cavitands and cages with various sizes, shapes, and cavities.95 Moreover, the hybrid nature of these molecular entities could make them great candidates as imaging and chemotherapeutic agents. As a result, the growing interest in exploring the biomedical applications of these types of 3D metallo-supramolecules has progressed to building coordination cages that are either capable of encapsulating bioactive molecules or can be structurally decorated with bio-functional fragments.97
There are critical properties of coordination cages that make them particularly attractive for biological applications.98,99 First, the internal cavity of the coordination cages shows host/guest capabilities, which makes them excellent candidates for sensing applications100 as well as drug-delivery scaffolds.101 Second, the nanoscale dimensions of most ensembles allow for rapid cellular uptake.102 Third, the size tunable, modular nature of coordination-driven self-assembly enables the construction of carefully designed architectures that can interact spatially and electrostatically with specific biological targets. Finally, the specific metal ion used is versatile since coordination geometries are always predictable for a given element in a controllable oxidation state. The demonstrated high potential to purposefully combine the properties of metal ions with those of organic ligands has recently attracted the attention of bioinorganic chemists which has added more facets to the range of practical uses of metallo-supramolecular capsules and their biological application for therapy and/or diagnostics.24 In the following section we will discuss some of these coordination cages and focus on recent studies and future aspects of their protein, DNA and RNA encapsulation and delivery.
3.1 Coordination cages for protein delivery
Encapsulation of proteins in synthetic hosts may enable the control of protein functions. With the expectation of enhanced stability and enzymatic activities, some proteins have been accommodated in synthetic host materials such as solid supports,103 polymer matrices,104 and reverse micelles.105 In these structurally nonuniform hosts, however, the protein functions are dispersed. Furthermore, functional control and elaboration of the encapsulated proteins are difficult because they could no longer be analyzed by conventional spectroscopic or crystallographic methods. However, until now, protein delivery by coordination cages is not very well known. This could be attributed to some unattractive properties connected with metal complexes such as instability in an aqueous environment and toxicity problems of both the ligand and metal-ion components as well as the nature of the host–guest interactions.24In 2012, Fujita and co-workers made a significant achievement by encapsulating an entire protein within a self-assembled coordination capsule.106 In their study they have reported that a glycine-to-cysteine mutation of the carboxyl terminus of ubiquitin does not affect the protein's tertiary structure but does allow attachment of a dipyridyl maleimide linker. So, the encapsulation proceeded with the addition of 30 equivalents of a dipyridyl ligand and 17 equivalents of Pd(NO3)2. The cavity volume of the resulting capsule is 63500 Å3, which is large enough to accommodate one ubiquitin molecule. The protein encapsulation was also confirmed by diffusion ordered NMR spectroscopy (DOSY), analytical ultracentrifugation (AUC), gel electrophoretic analysis (SDS-PAGE) and X-ray crystallography coupled with maximum entropy measurement (MEM) analysis. They showcased the encapsulation of proteins in a structurally well-defined host, where the protein functions are not dispersed but distinctly controlled and can be observed using spectroscopic and crystallographic methods.106 This chemical approach has a great advantage over biological strategies with natural cages such as viruses which present difficulties in preparation in addition to the need for structural modification.107,108
Raymond and coworkers have also conveyed an example of both edge-directed (M4L6) and face-directed (M4L4) gallium-based tetrahedral coordination cages.109 The interactions between both types of coordination cage host structures and a range of guest molecules have been explored,109–112 and the edge directed M4L6 cages were found to exhibit a greater scope of host–guest chemistry compared to the face-directed M4L4.109,110 It was postulated that the broader range of host–guest chemistry of the M4L6-type architecture could be attributed to the increased flexibility of the edge-directed scaffold. This flexibility allows slight conformational changes of the host cage favoring stronger association with the guest molecule, similar to that observed for highly specific protein–ligand interactions in biological systems. However, this concept has been scarcely explored so far. For example, one study has shown a noncovalent peptide coating on self-assembled M12L24 coordination spheres.113 In contrast, the encapsulation of a protein within a Pd12L14 cage (L = bidentate ligand) has been achieved by appropriate endo-functionalization of the ligands.111 Ligands were first tethered to the protein followed by the addition of metal ions and other ligands; coordination nanocages then self-assembled around the protein.
To implement supramolecular coordination cages as drug delivery systems, self-assembled Pd2L4 cages bio-conjugated to a model linear peptide were recently reported as the first example.114 The methodology of the bioconjugation of coordination cages was based on the formation of amide bonds between the amine (or carboxylic acid) serving as an exo-functionalized cage/ligand and the carboxylic acid (or amine) groups of the model peptide side chains. Accordingly, the bioconjugation was accomplished using two different techniques: (approach I) direct hitching of the coordination cage to the peptide or (approach II) initial anchoring of the ligand to the peptide, followed by coordination cage self-assembly (Fig. 6a).114 To date, the best results were realized with approach II, where firstly, the coupling of the peptide to the ligands organizing the cages was completed, followed by in situ re-formation of the Pd2L4 cages via self-assembly. The acquired results demonstrate the opportunity for the effective bioconjugation of coordination cages to peptides, which might be extended to targeting moieties such as affirmers, and feasibly also to antibodies.
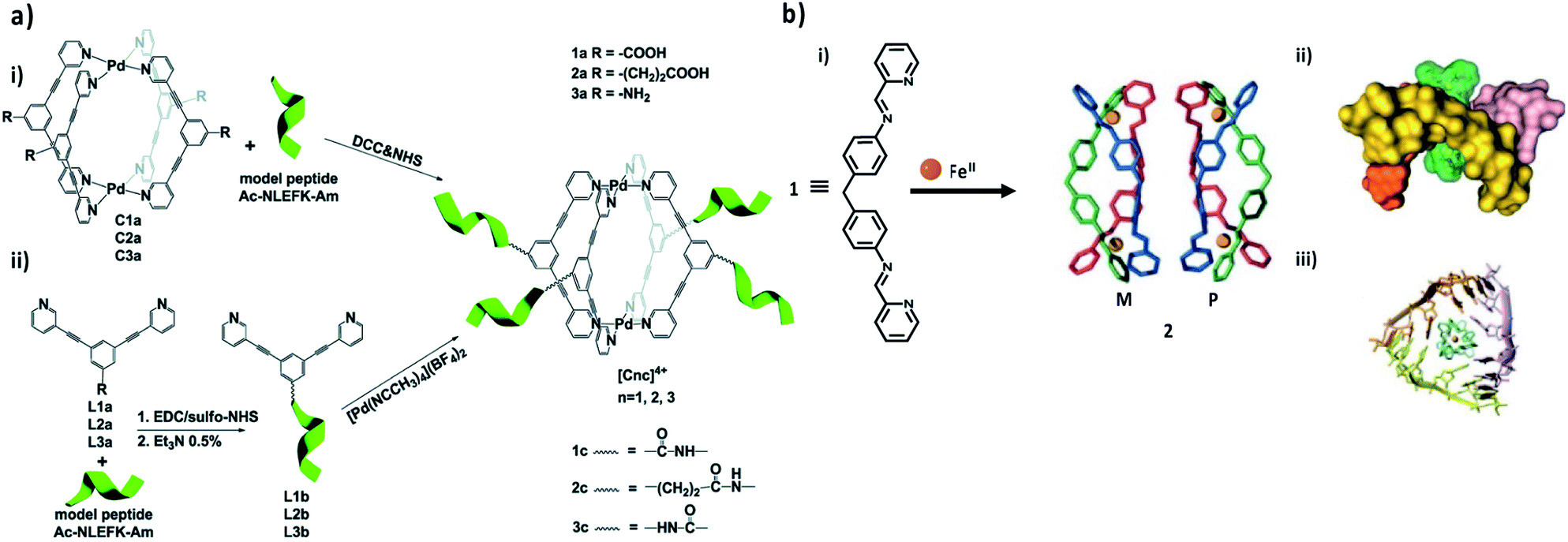 | ||
| Fig. 6 Metal–organic/biomacromolecule complex. (a) Scheme of the two different bioconjugation approaches: (i) direct tethering of the coordination cage to the peptide (approach I); or (ii) initial anchoring of the ligand to the peptide, followed by coordination cage self-assembly in situ (approach II). Adapted with permission from ref. 106. Copyright (2017) The Royal Society of Chemistry. (b) Synthesis of a pair of enantiomeric helicates for RNA-binding (i), as well as top view (ii) and side view (iii) of the X-ray crystal structure of the M enantiomer bound to an afgRNA three-way junction. Adapted with permission from ref. 117. Copyright (2013) Wiley-VCH. | ||
3.2 Coordination cages for RNA and DNA delivery
RNA and DNA delivery using coordination cages has not been very well established so far. However, establishing ways to control specific gene expression by synthesizing agents which can bind selectively to specific genes and turn their expression either on or off has attracted remarkable attention. Coordination cages could be new synthetic agents that could bind strongly to DNA and cause intramolecular DNA coiling.115 Also, the interaction between coordination cages and RNA could play a key role in regulating the activity of the corresponding gene.116 Freisinger and collaborators have reported for the first time a study that involves the complexation of a triple-stranded helicate within the central cavity of an RNA three-way junction (3WJ). A high-resolution X-ray crystal structure of the helicate within the 3WJ (Fig. 6b) reveals π-stacking interactions between the bases of RNA and the central phenylene rings of the helicate. Gel electrophoresis, circular dichroism (CD) and UV-Vis absorption experiments all confirmed the formation of the complex between the helicate and the 3WJ. Interestingly, RNA binding and stabilization assays showed that the RNA had no preference for either helical enantiomer, yet the M enantiomer was the only one to co-crystallize with the RNA. Further studies and development of RNA-binding helicates such as these may produce effective anticancer therapeutics.117In another study, Mishra et al. reported the preparation of two new self-assembled heterobimetallacycles (HBMCs). In their study they explored the unique structures and properties of metallo-supramolecular complexes to construct two HBMCs by using a bis-pyridine amide ligand and two metal (Pd and Pt) acceptors. Because DNA is the primary pharmacological target of many metal complex antitumor compounds, the interaction between DNA and metal complexes is of paramount importance in understanding their mechanism of action; therefore, the DNA-binding mode of these HBMCs was examined by UV-Vis spectroscopy. The investigation of the binding properties of these HBMCs revealed that they efficiently interact with DNA, as established by photophysical and gel electrophoresis studies. These results are similar to those reported previously for metal–macrocycles and suggest that the complexes used in this study bind to DNA by intercalation.118
On the other hand, employing macrocycle cavities to host biomacromolecules through host–guest interaction besides the cage cavity could be another approach for bioconjugation of coordination cages. Calixarenes, as a special family of macrocyclic host compounds, are excellent ligand candidates,119 which have been extensively utilized in supramolecular chemistry.120–123 Moreover, calixarenes have been widely studied in biological and medical applications due to their biocompatibility and low immunogenicity.124 The progress in calixarene coordination chemistry has resulted in an increasing number of reports relating to the use of calix[n]arenes as supporting ligands in metal-containing components especially for catalysis.125 The conformational flexibility, presence of cavities, and the ability to simultaneously coordinate numerous metal centers are proving to be extremely good attributes. Most of the work carried out has been conducted on the easily prepared, and relatively cheap, calix[4]arene system, which upon metalation tends to retain the cone conformation.21
Pasquale et al. have described giant,126 highly symmetrical polyhedral capsules that self-assemble from calixarene carboxylate 3 and the uranyl cation UO22+ in the presence of pyridine base in DMF. In their study both calix[4]arene and calix[5]arene were used during the investigation. Calix[4]arene and calix[5]arene produced an octahedral complex and an icosahedral complex 4 (Fig. 7a), respectively. Both ligands show a wide-open cone conformation at the vertices of the structure and bridge the metal centres which reside on the faces.127 The structures are anionic, with each metal contributing only one negative charge. Few counter cations are required for neutrality, which leaves a large space for guest encapsulation with a radius of 1.4 Å for the larger, icosahedral capsule. Based on the size of the cavity, the capsule could be a potential candidate for encapsulation of some biomacromolecules even though this was not further studied.
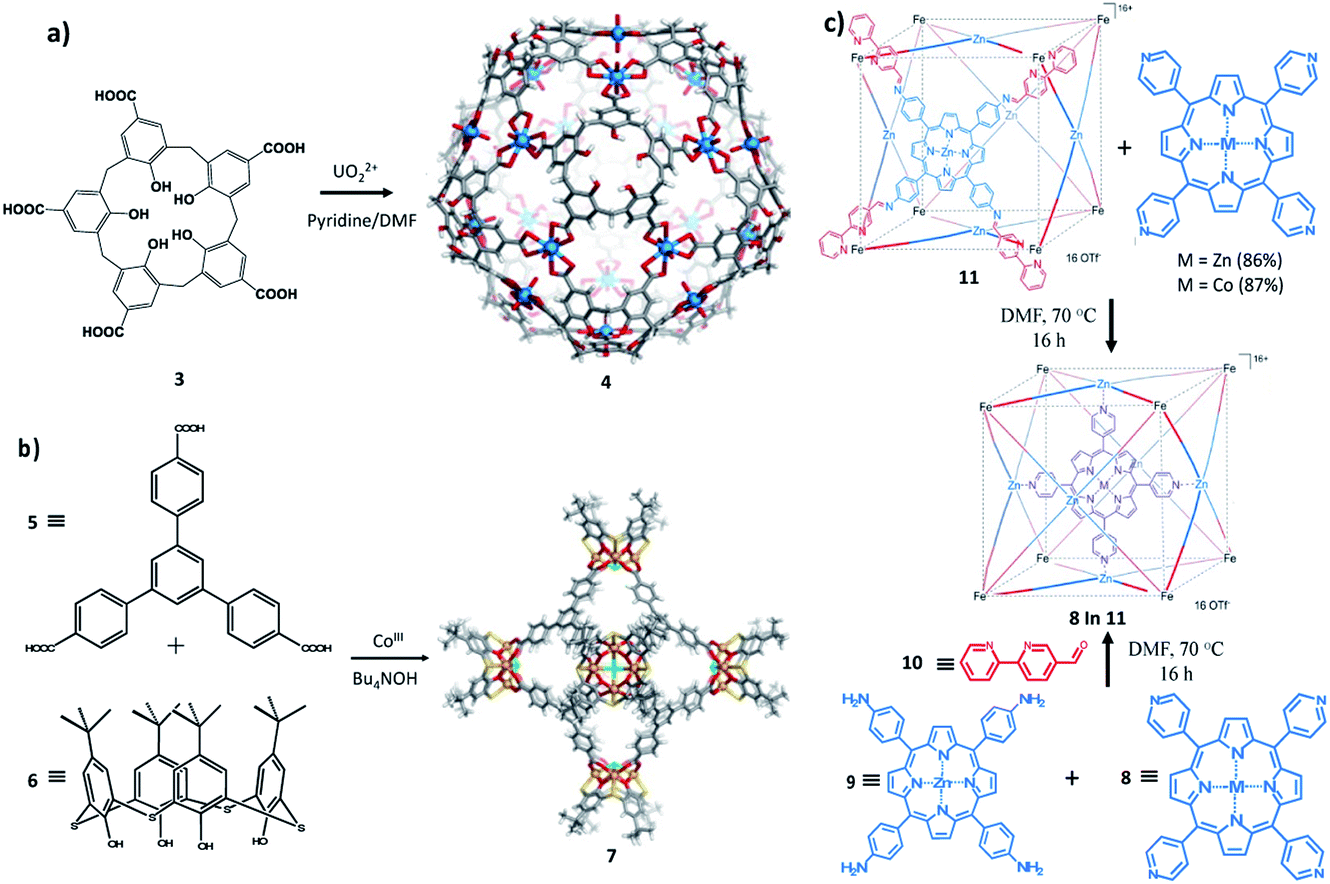 | ||
| Fig. 7 Molecular entities. (a) Synthesis and X-ray crystal structure of 4: icosahedral coordination capsules from calix[5]arene 3 and uranyl ions. (b) Synthesis and X-ray crystal structure of 7: a calix[4]arene-based coordination capsule. Adapted with permission from ref. 129. Copyright (2014) Elsevier. (c) Subcomponent self-assembly of capsule 12 through two synthetic routes. Adapted with permission from ref. 130. Copyright (2013) Wiley-VCH. | ||
Zhang and colleagues128 have also described a series of calixarene-based coordination capsules including a p-tert-butylthiacalix[4]arene, a tripodal ternary acid (either 1,3,5-benzenetricarboxylic acid) and cobalt(III) chloride. The characteristic feature of these capsules is the presence of Co4/calixarene nodes, as the secondary building units (SBUs), at their octahedral vertices. The X-ray crystal structure (7) of the large capsule is shown in Fig. 7b. The diameter of its internal cavity is 1.7 nm. Its external diameter, considering the projecting calixarenes, is 4.4 nm. These cavities have sizes that are comparable with those of some nMOFs reported for DNA encapsulation/delivery and as such could be a better candidate for this biomedical application.129
Apart from calixarenes, other cavitand like porphyrins have been employed in the formulation of self-assembled coordination capsules. Bruin and collaborators have described the synthesis of a large porphyrin based M6L8 capsule. The capsule was prepared by mixing 5-bipyridine-aldehyde with tetra(4-aminophenyl)porphyrinatozinc(II) (10) in the presence of Fe(OTf)2 as illustrated in Fig. 7c. The cube (12) obtained was characterized by NMR and Cryo-UHR-ESI-ToF mass spectrometry experiments. The MM-minimized model of the cube indicates that the average Zn–Zn distance between two opposite faces is 19.5 Å and reflects a large cavity volume relative to other face-defined cubes. In solution, the cube was found to be capable of selectively encapsulating transition-metal complexes, such as tetra(4-pyridyl)metalloporphyrins (MII(TPyP), MII = Co, Zn). The catalytic activity of the encapsulated CoII(TPyP) complex was enhanced relative to the free CoII(TPyP).130
Biomacromolecules are intercalated and delivered by nMOF especially through biomimetic mineralization in which the size will be adjusted to accommodate the biomacromolecules. In contrast, coordination cage intercalation occurs through the ligand interaction with the guest followed by metal coordination in which the size could accommodate small molecules such as ubiquitin. Most biomacromolecules cannot be encapsulated because of their incommensurably larger size compared with common molecular entities. However, enhancing the encapsulation of biomacromolecules utilizing coordination cages could be possible by adjusting the ligand length.131
4. Conclusion and future outlook
The field of coordination-based self-assembled capsules (SACs) has seen a tremendous growth and attracted great attention over the past decade. We have classified SACs into two main categories: nano-sized metal–organic frameworks (nMOFs) and molecularly discrete coordination cages. Compared to cages, nMOFs showed better results in overcoming the limitations of conventional biologics delivery, mainly poor cellular uptake and rapid degradation. nMOFs can successfully protect biomacromolecules with different sizes and charges against enzymatic degradation via diffusion, biomimetic mineralization or co-precipitation methods. However, the uniform size of the nMOFs and their colloidal stability have been greatly overlooked so far. As for cytotoxicity, the nature of the metal and the building blocks used in the preparation of these coordination-based platforms play a crucial role and can hinder transformation into clinical trials. Minimum toxicity and high selectivity for the targeted cells or tissues are the ultimate goals for any delivery system. Employing calcium and magnesium as the coordinating metal center is a great step towards improving biocompatibility. Moreover, systems composed of biologically derived motifs such as amino acids and carbohydrates could be a highly promising alternative. In addition to the proper choice of metal center and organic linkers, coating the delivery platforms with biocompatible polymers and surface functionalization with specific groups or antibodies could improve the selectivity and targetability of the system. Furthermore, it is essential to focus on improving the current promising structures with more detailed in vivo studies which would help take nMOFs one step closer to clinical trials and potentially FDA approval and pharmaceutical production (Table 1). Comparably, the biological application of coordination cages is still at an early stage. We have highlighted some of the few examples in the recent literature where coordination cages have been employed for the encapsulation of small molecules as guests with only one example on the encapsulation of a small protein. This lack of substantial research into biological applications especially with regards to biomacromolecule encapsulation and delivery may emanate from their small size relative to the cargo molecules as well as their strong host–guest interactions. A different perspective for coordination cages would be to be involved directly as a therapeutic or imaging agent rather than an encapsulating or intercalating agent. Moreover, the multifunctional host and guest approach, in which both players possess distinct qualities and specificities, is certainly a promising strategy for the development of these molecular entities with synergistic effects. Coordination-based platforms show great promise for biomedical translation; however, much work remains to be done to fine-tune these systems in terms of improving biocompatibility, colloidal stability, pharmacokinetics and biodegradation. Consequently, this research area is practically in the initiation stage and would benefit from new and disruptive research ideas in addition to in-depth in vivo studies on the interface of these smart hybrid materials with living organisms and biological tissues.Conflicts of interest
There are no conflicts to declare.Notes and references
- M. Cavazzana-Calvo, S. Hacein-Bey, G. De, S. Basile, F. Gross, E. Yvon, P. Nusbaum, F. Selz, C. Hue, S. Certain, J. L. Casanova, P. Bousso, F. Le Deist and A. Fischer, Science, 2000, 288, 669 CrossRef CAS
.
-
A. C. Boyd, Gene and Stem Cell Therapy, Karger, Basel, New York, 2006 Search PubMed
- M. G. Kaplitt, A. Feigin, C. Tang, H. L. Fitzsimons, P. Mattis, P. A. Lawlor, R. J. Bland, D. Young, K. Strybing, D. Eidelberg and M. J. During, Lancet, 2007, 369, 2097 CrossRef CAS
- Z. R. Yang, H. F. Wang, J. Zhao, Y. Y. Peng, J. Wang, B.-A. Guinn and L. Q. Huang, Cancer Gene Ther., 2007, 14, 599 CrossRef CAS
-
(a) X. Qin, C. Yu, J. Wei, L. Li, C. Zhang, Q. Wu, J. Liu, S. Q. Yao and W. Huang, Adv. Mater., 2019, 31, 1902791 CrossRef CAS
- Z. Gu, A. Biswas, M. Zhaoab and Y. Tang, Chem. Soc. Rev., 2011, 40, 3638 RSC
- R. C. Rao and D. N. Zacks, Dev. Ophthalmol., 2014, 53, 167 Search PubMed
- A. Fu, R. Tang, J. Hardie, M. E. Farkas and V. M. Rotello, Bioconjugate Chem., 2014, 25, 1602 CrossRef CAS
- M. S. Al-Dosari and X. Gao, AAPS J., 2009, 11, 671 CrossRef CAS
- D. J. Gloves and H. J. Lipps, Nat. Rev. Genet., 2005, 6, 299 CrossRef
- I. Roy, S. Mitra, A. Maitra and S. Mozumdar, Int. J. Pharm., 2003, 250, 25 CrossRef CAS
- H. Amouri, C. Desmarets and J. Moussa, Chem. Rev., 2012, 112, 2015 CrossRef CAS
- A. Pöthig and A. Casini, Theranostics, 2019, 9, 3150 CrossRef
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Ferey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232 CrossRef CAS
- M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319 CrossRef CAS
- W. J. Rieter, K. M. L. Taylor, H. An, W. Lin and W. Lin, J. Am. Chem. Soc., 2006, 128, 9024 CrossRef CAS
- W. Lin, W. J. Rieter and K. L. M. Taylor, Angew. Chem., Int. Ed., 2009, 48, 650 CrossRef CAS
- P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. F. Eubank, D. Heurtaux, P. Clayette, C. Kreuz, J.-S. Chang, Y. K. Hwang, V. Marsaud, P.-N. Bories, L. Cynober, S. Gil, G. Ferey, P. Couvreur and R. Gref, Nat. Mater., 2010, 9, 172 CrossRef CAS
- A. M. Spokoyny, D. Kim, A. Sumrein and C. A. Mirkin, Chem. Soc. Rev., 2009, 38, 1218 RSC
-
(a) S. Rojas, A. Arenas-Vivo and P. Horcajada, Coord. Chem. Rev., 2019, 388, 202–226 CrossRef CAS
- T. R. Cook, Y. R. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734 CrossRef CAS
- A. Schmidt, A. Casini and F. E. Kühn, Coord. Chem. Rev., 2014, 275, 19 CrossRef CAS
- T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001 CrossRef CAS
- A. Casini, B. Woods and M. Wenzel, Inorg. Chem., 2017, 56, 14715 CrossRef CAS
- G. Lan, K. Ni and W. Lin, Coord. Chem. Rev., 2019, 379, 65 CrossRef CAS
- I. A. Lázaro and R. S. Forgan, Coord. Chem. Rev., 2019, 380, 230 CrossRef
- W. Cai, C.-C. Chu, G. Liu and Y.-X. J. Wáng, Small, 2015, 11, 4806 CrossRef CAS
- A. J. McConnell, C. S. Wood, P. P. Neelakandan and J. R. Nitschke, Chem. Rev., 2015, 115, 7729 CrossRef CAS
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941 CrossRef CAS
- N. Hoshyar, S. Gray, H. Han and G. Bao, Nanomedicine, 2016, 11, 673 CrossRef CAS
- C. Tamames-Tabar, D. Cunha, E. Imbuluzqueta, F. Ragon, C. Serre, M. J. Blanco-Prieto and P. Horcajada, J. Mater. Chem. B, 2014, 2, 262 RSC
-
(a) F. Lyu, Y. Zhang, R. N. Zare, J. Ge and Z. Liu, Nano Lett., 2014, 14, 5761 CrossRef CAS
- T. Sun, Y. S. Zhang, B. Pang, D. C. Hyun, M. Yang and Y. Xia, Angew. Chem., Int. Ed., 2014, 53, 12320 CAS
- X. Hu, B. Lim, S. R. Torati, J. Ding, V. Novosad, M. Y. Im, V. Reddy, K. Kim, E. Jung, A. I. Shawl, E. Kim and C. Kim, Small, 2018, 14, 1800504 CrossRef
-
(a) B. Leader, Q. J. Baca and D. E. Golan, Nat. Rev. Drug Discovery, 2008, 7, 21 CrossRef CAS
- H. An, M. Li, J. Gao, Z. Zhang, S. Ma and Y. Chen, Coord. Chem. Rev., 2019, 384, 90 CrossRef CAS
- X. Lian, Y. Fang, E. Joseph, Q. Wang, J. Li, S. Banerjee, C. Lollar, X. Wang and H. C. Zhou, Chem. Soc. Rev., 2017, 46, 3386 RSC
-
(a) Y. Chen, P. Li, J. A. Modica, R. J. Drout and O. K. Farha, J. Am. Chem. Soc., 2018, 140, 5678 CrossRef CAS
- N. K. Maddigan, A. Tarzia, D. M. Huang, C. J. Sumby, S. G. Bell, P. Falcaro and C. J. Doonan, Chem. Sci., 2018, 9, 4217 RSC
- H. Deng, S. Grunder, K. E. Cordova, C. Valente, H. Furukawa, M. Hmadeh, F. Gándara, A. C. Whalley, Z. Liu, S. Asahina, H. Kazumori, M. O'Keeffe, O. Terasaki, J. F. Stoddart and O. M. Yaghi, Science, 2012, 336, 1018 CrossRef CAS
- D. Feng, T. F. Liu, J. Su, M. Bosch, Z. Wei, W. Wan, D. Yuan, Y. P. Chen, X. Wang, K. Wang, X. Lian, Z. Y. Gu, J. Park, X. Zou and H. C. Zhou, Nat. Commun., 2015, 6, 5979 CrossRef
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230 CrossRef
- V. Lykourinou, Y. Chen, X.-S. Wang, L. Meng, T. Hoang, L.-J. Ming, R. L. Musselman and S. Ma, J. Am. Chem. Soc., 2011, 133, 10382 CrossRef CAS
- Y. Chen, V. Lykourinou, C. Vetromile, T. Hoang, L.-J. Ming, R. W. Larsen and S. Ma, J. Am. Chem. Soc., 2012, 134, 13188 CrossRef CAS
- Y. Chen, S. Han, X. Li, Z. Zhang and S. Ma, Inorg. Chem., 2014, 53, 10006 CrossRef CAS
- C. J. Veeger, Inorg. Biochem., 2002, 91, 35 CrossRef CAS
- Y. K. Park, S. B. Choi, H. Kim, K. Kim, B.-H. Won, K. Choi, J.-S. Choi, W.-S. Ahn, N. Won, S. Kim, D. H. Jung, S.-H. Choi, G.-H. Kim, S.-S. Cha, Y. H. Jhon, J. K. Yang and J. Kim, Angew. Chem., Int. Ed., 2007, 46, 8230 CrossRef CAS
-
(a) R. J. Drout, L. Robison and O. K. Farha, Coord. Chem. Rev., 2019, 381, 151 CrossRef CAS
- Z. Wang, S. Hu, J. Yang, A. Liang, Y. Li, Q. Zhuang and J. Gu, Adv. Funct. Mater., 2018, 28, 1707356 CrossRef
- X. Yang, Q. Tang, Y. Jiang, M. Zhang, M. Wang and L. Mao, J. Am. Chem. Soc., 2019, 141, 3782 CrossRef CAS
- S. Wang, Y. Chen, S. Wang, P. Li, C. A. Mirkin and O. K. Farha, J. Am. Chem. Soc., 2019, 141, 2215 CrossRef CAS
- Y. Zhang, F. Wang, E. Ju, Z. Liu, Z. Chen, J. Ren and X. Qu, Adv. Funct. Mater., 2016, 26, 6454 CrossRef CAS
- M. Giménez-Marqués, T. Hidalgo, C. Serre and P. Horcajada, Coord. Chem. Rev., 2016, 307, 342 CrossRef
- F. K. Shieh, S. C. Wang, C. I. Yen, C. C. Wu, S. Dutta, L. Y. Chou, J. V. Morabito, P. Hu, M. H. Hsu, K. C. Wu and C. K. Tsung, J. Am. Chem. Soc., 2015, 137, 4276 CrossRef CAS
- K. Liang, R. Ricco, C. M. Doherty, M. J. Styles, S. Bell, N. Kirby, S. Mudie, D. Haylock, A. J. Hill, C. J. Doonan and P. Falcaro, Nat. Commun., 2015, 6, 7240 CrossRef CAS
- T.-T. Chen, J.-T. Yi, Y.-Y. Zhao and X. Chu, J. Am. Chem. Soc., 2018, 140, 9912 CrossRef CAS
- S. K. Alsaiari, S. Patil, M. Alyami, K. O. Alamoudi, F. A. Aleisa, J. S. Merzaban, M. Li and N. M. Khashab, J. Am. Chem. Soc., 2018, 140, 143 CrossRef CAS
- P. D. Hsu, E. S. Lander and F. Zhang, Cell, 2014, 157, 1262 CrossRef CAS
- J. A. Doudna and E. Charpentier, Science, 2014, 346, 1258096 CrossRef
- M. Z. Alyami, S. K. Alsaiari, Y. Li, S. S. Qutub, F. A. Aleisa, R. Sougrat, J. S. Merzaban and N. M. Khashab, J. Am. Chem. Soc., 2020, 142, 1715 CrossRef CAS
- G. Courties, J. Presumey, I. Duroux-Richard, C. Jorgensen and F. Apparailly, Expert Opin. Biol. Ther., 2009, 9, 535 CrossRef CAS
- S. S. Dhadwar, J. Kiernan, J. Wen and G. Hortelano, J. Thromb. Haemostasis, 2010, 8, 2743 CrossRef CAS
- G. Devi, Cancer Gene Ther., 2006, 13, 819 CrossRef CAS
- M. Abdelrahim, S. Safe, C. Baker and A. Abudayyeh, J. RNAi Gene Silencing, 2006, 2, 136 CAS
- A. L. Gartel and E. S. Kandel, Biomol. Eng., 2006, 23, 17 CrossRef CAS
- L. A. Martinez, I. Naguibneva, H. Lehrmann, A. Vervisch, T. Tchénio, G. Lozano and A. Harel-Bellan, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14849 CrossRef CAS
- S. Pai, Y. Lin, B. Macaes, A. Meneshian, C.-F. Hung and T. C. Wu, Gene Ther., 2006, 13, 464 CrossRef CAS
- F. Takeshita and T. Ochiya, Cancer Sci., 2006, 97, 689 CrossRef CAS
- Y. Tomari and P. D. Zamore, Genes Dev., 2005, 19, 517 CrossRef CAS
- A. J. Hamilton and D. C. Baulcombe, Science, 1999, 286, 950 CrossRef CAS
- S. M. Elbashir, J. Harborth, W. Lendeckel, A. Yalcin, K. Weber and T. Tuschl, Nature, 2001, 411, 494 CrossRef CAS
-
(a) S. Keskin and S. Kızılel, Ind. Eng. Chem. Res., 2011, 50, 1799 CrossRef CAS
- A. Poddar, J. J. Conesa, K. Liang, S. Dhakal, P. Reineck, G. Bryant, E. Pereiro, R. Ricco, H. Amenitsch, C. Doonan, X. Mulet, C. Doherty, P. Falcaro and R. Shukla, Small, 2019, 15, 1902268 CrossRef
- W. Cai, J. Wang, C. Chu, W. Chen, C. Wu and G. Liu, Adv. Sci., 2019, 6, 1801526 CrossRef
- A. Poddar, S. Pyreddy, F. Carraro, S. Dhakal, A. Rassell, M. R. Field, T. S. Reddy, P. Falcaro, C. M. Doherty and R. Shukla, Chem. Commun., 2020, 56, 15406 RSC
- F. Carraro, M. d. J. Velásquez-Hernández, E. Astria, W. Liang, L. Twight, C. Parise, M. Ge, Z. Huang, R. Ricco, X. Zou, L. Villanova, C. O. Kappe, C. Doonan and P. Falcaro, Chem. Sci., 2020, 11, 3397 RSC
- Y. Li, K. Zhang, P. Liu, M. Chen, Y. Zhong, Q. Ye, M. Q. Wei, H. Zhao and Z. Tang, Adv. Mater., 2019, 31, 1901570 CrossRef
- H. Wang, Y. Chen, H. Wang, X. Liu, X. Zhou and F. Wang, Angew. Chem., Int. Ed., 2019, 58, 7380 CrossRef CAS
- Z. Wang, Y. Fu, Z. Kang, X. Liu, N. Chen, Q. Wang, Y. Tu, L. Wang, S. Song, D. Ling, H. Song, X. Kong and C. Fan, J. Am. Chem. Soc., 2017, 139, 15784 CrossRef CAS
- S. Peng, B. Bie, Y. Sun, M. Liu, H. Cong, W. Zhou, Y. Xia, H. Tang, H. Deng and X. Zhou, Nat. Commun., 2018, 9, 1293 CrossRef
- M. H. Teplensky, M. Fantham, C. Poudel, C. Hockings, M. Lu, A. Guna, M. Aragones-Anglada, P. Z. Moghadam, P. Li, O. K. Farha, S. B. de Quirós Fernández, F. M. Richards, D. I. Jodrell, G. K. Schierle, C. F. Kaminski and D. Fairen-Jimenez, Chem, 2019, 5, 2926 CAS
- C. He, K. Lu, D. Liu and W. Lin, J. Am. Chem. Soc., 2014, 136, 5181 CrossRef CAS
- Q. Chen, M. Xu, W. Zheng, T. Xu, H. Deng and J. Liu, ACS Appl. Mater. Interfaces, 2017, 9, 6712 CrossRef CAS
- F. Carraro, J. D. Williams, M. Linares-Moreau, C. Parise, W. Liang, H. Amenitsch, C. Doonan, C. O. Kappe and P. Falcaro, Angew. Chem., Int. Ed., 2020, 59, 8123–8127 CrossRef CAS
- C. Hu, Y. Bai, M. Hou, Y. Wang, L. Wang, X. Cao, C.-W. Chan, H. Sun, W. Li, J. Ge and K. Ren, Sci. Adv., 2020, 6, eaax5785 CrossRef CAS
- D. A. Evans and J. S. Evans, J. Org. Chem., 1998, 63, 8027 CrossRef CAS
- K. T. Chapman and W. C. Still, J. Am. Chem. Soc., 1989, 111, 3075 CrossRef CAS
- M. Fujita, Chem. Soc. Rev., 1998, 27, 417 RSC
- N. Takeda, K. Umemoto, K. Yamaguchi and M. Fujita, Nature, 1999, 398, 794 CrossRef CAS
- M. Fujita, K. Umemoto, M. Yoshizawa, N. Fujita, T. Kusukawa and K. Biradha, Chem. Commun., 2001, 509 RSC
- B. Olenyuk, A. Fechtenkötter and P. J. Stang, J. Chem. Soc., Dalton Trans., 1998, 1707 RSC
- B. Olenyuk, M. D. Levin, J. A. Whiteford, J. E. Shield and P. J. Stang, J. Am. Chem. Soc., 1999, 121, 10434 CrossRef CAS
- B. Olenyuk, J. A. Whiteford, A. Fechtenkötter and P. J. Stang, Nature, 1999, 398, 796 CrossRef CAS
- J. R. Nitschke, Acc. Chem. Res., 2007, 40, 103 CrossRef CAS
-
(a) X. Hang, B. Liu, X. Zhu, S. Wang, H. Han, W. Liao, Y. Liu and C. Hu, J. Am. Chem. Soc., 2016, 138, 2969 CrossRef CAS
- P. Jin, S. J. Dalgarno and J. L. Atwood, Coord. Chem. Rev., 2010, 254, 1760 CrossRef CAS
- J. E. M. Lewis, E. L. Gavey, S. A. Cameron and J. D. Crowley, Chem. Sci., 2012, 3, 778 RSC
- T. R. Cook, V. Vajpayee, M. H. Lee, P. J. Stang and K. W. Chi, Acc. Chem. Res., 2013, 46, 2464 CrossRef CAS
- N. Ahmad, H. A. Younus, A. H. Chughtai and F. Verpoort, Chem. Soc. Rev., 2015, 44, 9 RSC
- M. Wang, V. Vajpayee, S. Shanmugaraju, Y.-R. Zheng, Z. Zhao, H. Kim, P. S. Mukherjee, K.-W. Chi and P. J. Stang, Inorg. Chem., 2011, 50, 1506 CrossRef CAS
-
B. Therrien, Drug Delivery by Water-Soluble Organometallic Cages, in Chemistry of Nanocontainers. Topics in Current Chemistry, ed. M. Albrecht and E. Hahn, Springer, Berlin, Heidelberg, 2011, vol. 319 Search PubMed
-
(a) H. Maeda, G. Y. Bharate and J. Daruwalla, Eur. J. Pharm. Biopharm., 2009, 71, 409 CrossRef CAS
- L. M. Ellerby, C. R. Nishida, F. Nishida, S. A. Yamanaka, B. Dunn, J. S. Valentine and J. I. Zink, Science, 1992, 255, 1113 CrossRef CAS
- G. Zhu, S. R. Mallery and S. P. Schwendeman, Nat. Biotechnol., 2000, 18, 52 CrossRef CAS
- G. A. Ayala, S. Kamat, E. J. Beckman and A. J. Russell, Biotechnol. Bioeng., 1992, 39, 806 CrossRef CAS
- D. Fujita, K. Suzuki, S. Sato, M. Yagi-Utsumi, Y. Yamaguchi, N. Mizuno, T. Kumasaka, M. Takata, M. Noda, S. Uchiyama, K. Kato and M. Fujita, Nat. Commun., 2012, 3, 1093 CrossRef
- M. Comellas-Aragones, H. Engelkamp, V. I. Claessen, N. A. Sommerdijk, A. E. Rowan, P. C. Christianen, J. C. Maan, B. J. Verduin, J. J. Cornelissen and R. J. Nolte, Nat. Nanotechnol., 2007, 2, 635 CrossRef CAS
- B. Wörsdörfer, K. J. Woycechowsky and D. Hilvert, Science, 2011, 331, 589 CrossRef
- C. M. Hong, D. M. Kaphan, R. G. Bergman, K. N. Raymond and F. D. Toste, J. Am. Chem. Soc., 2017, 139, 8013 CrossRef CAS
- R. M. Yeh, J. Xu, G. Seeber and K. N. Raymond, Inorg. Chem., 2005, 44, 6228 CrossRef CAS
- M. D. Pluth, D. Fiedler, J. S. Mugridge, R. G. Bergman and K. N. Raymond, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10438 CrossRef CAS
- J. S. Mugridge, A. Zahl, R. van Eldik, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2013, 135, 4299 CrossRef CAS
- M. Ikemi, T. Kikuchi, S. Matsumura, K. Shiba, S. Sato and M. Fujita, Chem. Sci., 2010, 1, 68 RSC
- J. Han, A. Schmidt, T. Zhang, H. Permentier, G. M. Groothuis, R. Bischoff, F. E. Kuhn, P. Horvatovich and A. Casini, Chem. Commun., 2017, 53, 1405 RSC
- M. J. Hannon and L. J. Childs, Supramol. Chem., 2004, 16, 7 CrossRef CAS
- A. Ramesh, C. A. Wakeman and W. C. Winkler, J. Mol. Biol., 2011, 407, 556 CrossRef CAS
- S. Phongtongpasuk, S. Paulus, J. Schnabl, R. K. O. Sigel, B. Spingler, M. J. Hannon and E. Freisinger, Angew. Chem., Int. Ed., 2013, 52, 11513 CrossRef CAS
- A. Mishra, S. Ravikumar, S. H. Hong, H. Kim, V. Vajpayee, H. Lee, B. C. Ahn, M. Wang, P. J. Stang and K.-W. Chi, Organometallics, 2011, 30, 6343 CrossRef CAS
- R. S. Patil, D. Banerjee, C. Zhang, P. K. Thallapally and J. L. Atwood, Angew. Chem., Int. Ed., 2016, 55, 4523 CrossRef CAS
- N. Morohashi, F. Narumi, N. Iki, T. Hattori and S. Miyano, Chem. Rev., 2006, 106, 5291 CrossRef CAS
- H. Kumari, C. L. Dennis, A. V. Mossine, C. A. Deakyne and J. L. Atwood, J. Am. Chem. Soc., 2013, 135, 7110 CrossRef CAS
- K. S. Iyer, M. Norret, S. J. Dalgarno, J. L. Atwood and C. L. Raston, Angew. Chem., Int. Ed., 2008, 47, 6362 CrossRef CAS
-
S. Steyer, C. Jeunesse, D. Armspach, D. Matt and J. Harrowfield, in Calixarenes 2001, ed. Z. Asfari, V. Böhmer, J. Harrowfield and J. Vicens, Kluwer Academic Publishers, Dordrecht, 2001, ch. 28 Search PubMed
- M.-H. Paclet, C. F. Rousseau, C. Yannick, F. Morel and A. W. Coleman, J. Inclusion Phenom. Macrocyclic Chem., 2006, 55, 353 CrossRef CAS
- D. M. Homden and C. Redshaw, Chem. Rev., 2008, 108, 5086 CrossRef CAS
- S. Pasquale, S. Sattin, E. C. Escudero-Adan, M. Martinez-Belmonte and J. de Mendoza, Nat. Commun., 2012, 3, 1 Search PubMed
- Q.-F. Sun, J. Iwasa, D. Ogawa, Y. Ishido, S. Sato, T. Ozeki, Y. Sei, K. Yamaguchi and M. Fujita, Science, 2010, 328, 1144 CrossRef CAS
- M. Liu, W. Liao, C. Hu, S. Du and H. Zhang, Angew. Chem., Int. Ed. Engl., 2012, 51, 1585 CrossRef CAS
- R. A. Bilbeisi, J.-C. Olsen, L. J. Charbonnière and A. Trabolsi, Inorg. Chim. Acta, 2014, 417, 79 CrossRef CAS
- M. Otte, P. F. Kuijpers, O. Troeppner, I. Ivanović-Burmazović, J. N. H. Reek and B. de Bruin, Chem.–Eur. J., 2013, 19, 10170 CrossRef CAS
- C. R. K. Glasson, G. V. Meehan, C. A. Motti, J. K. Clegg, P. Turner, P. Jensen and L. F. Lindoy, Dalton Trans., 2011, 40, 10481 RSC
- W. Liang, H. Xu, F. Carraro, N. K. Maddigan, Q. Li, S. G. Bell, D. M. Huang, A. Tarzia, M. B. Solomon, H. Amenitsch, L. Vaccari, C. J. Sumby, P. Falcaro and C. J. Doonan, J. Am. Chem. Soc., 2019, 141, 2348 CrossRef CAS
- G. Cheng, W. Li, L. Ha, X. Han, S. Hao, Y. Wan, Z. Wang, F. Dong, X. Zou, Y. Mao and S. Y. Zheng, J. Am. Chem. Soc., 2018, 140, 7282 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2021 |