
Immunoaffinity monoliths for multiplexed extraction of preterm birth biomarkers from human blood serum in 3D printed microfluidic devices†
Haifa M.
Almughamsi
a,
Makella K.
Howell
a,
Samuel R.
Parry
a,
Joule E.
Esene
a,
Jacob B.
Nielsen
a,
Gregory P.
Nordin
 b and
Adam T.
Woolley
*a
b and
Adam T.
Woolley
*a
aDepartment of Chemistry and Biochemistry, Brigham Young University, Provo, UT 84602, USA. E-mail: atw@byu.edu; Tel: +1-801-422-1701
bDepartment of Electrical and Computer Engineering, Brigham Young University, Provo, UT 84602, USA
First published on 20th January 2022
Abstract
In an effort to develop biomarker-based diagnostics for preterm birth (PTB) risk, we created 3D printed microfluidic devices with multiplexed immunoaffinity monoliths to selectively extract multiple PTB biomarkers. The equilibrium dissociation constant for each monoclonal antibody toward its target PTB biomarker was determined. We confirmed the covalent attachment of three different individual antibodies to affinity monoliths using fluorescence imaging. Three different PTB biomarkers were successfully extracted from human blood serum using their respective single-antibody columns. Selective binding of each antibody toward its target biomarker was observed. Finally, we extracted and eluted three PTB biomarkers from depleted human blood serum in multiplexed immunoaffinity columns in 3D printed microfluidic devices. This is the first demonstration of multiplexed immunoaffinity extraction of PTB biomarkers in 3D printed microfluidic devices.
Introduction
Multiplexed assays are important for drug screening,1 cytotoxicity,2 and biomarker detection.3,4 Biomarkers can serve as indicators for the presence of disease, but are usually present in low concentrations in blood serum.5 Particularly, because blood serum contains up to 50 g L−1 of albumin and immunoglobulins,6 these matrix components can interfere with biomarker analysis, making this a challenging endeavor.7Risk for a preterm birth (PTB), defined as birth prior to 37 weeks of gestation, can be correlated with a panel of nine biomarkers found in maternal blood serum.8 These biomarkers include four proteins and five peptides, and allow for prediction of PTB risk with 87% selectivity and 81% specificity. However, detecting these biomarkers from human blood serum is challenging due to their very low concentrations relative to other proteins in serum. To achieve successful early diagnosis of PTB risk, there is a need for effective and rapid sample preparation methods to address these issues.
An immunoaffinity column selectively retains analytes using antigen–antibody interaction. This strong and selective interaction is desirable for purifying target biomarkers from complex specimens. Porous polymer monoliths are useful in sample preparation9–11 and show promise in immunoaffinity extraction of biomarkers from blood serum using one or more antibodies attached to a column.12,13 The biophysical characteristics of antigen–antibody binding play a significant role in immunoaffinity extraction. Understanding this interaction provides information about the stability of binding, which is useful for development of multiplexed immunoaffinity extraction. Two complementary methods, biolayer interferometry (BLI)14 and surface plasmon resonance (SPR),15 are both used to determine affinity binding strength between antigen–antibody complexes.
Microfluidics is a promising platform for multiplex sample preparation. The small amounts of sample and reagents needed can reduce costs and analysis time.16,17 3D printing of microfluidic devices can overcome challenges associated with traditional fabrication techniques, and can enable novel designs that are otherwise inaccessible.12,18–21 These 3D printed microfluidic devices can be used to facilitate sample preparation and biomarker analysis. For example, Bickham et al.19 used solid phase extraction monoliths to concentrate and label a panel of nine PTB biomarkers. Although this is a good method for enriching and labeling samples, PTB biomarkers must first be purified from blood serum, which cannot be performed on this type of monolith. Parker et al.12 purified ferritin from human blood serum as a step toward a PTB risk diagnostic, but only one PTB biomarker was captured on a single-antibody column. Multiple, single-antibody capture elements have also been used to determine fertility-related biomarkers in a miniaturized device.22 Extending these approaches to immobilize multiple antibodies and extract multiple PTB biomarkers on a single immunoaffinity column would be advantageous for rapid measurement.23
Here, we overcome these earlier limitations with immunoaffinity columns to purify multiple PTB biomarkers simultaneously from human blood serum in 3D printed microfluidic devices. Immunoaffinity columns were modified with a mixture of monoclonal antibodies that were initially evaluated using dot blots, BLI, and SPR. Selectivity of each biomarker toward its respective antibody was first studied, and each biomarker was individually extracted from a single-antibody column. We then selectively enriched three PTB biomarkers from blood serum on a single multiplexed immunoaffinity monolith in a 3D printed microfluidic device. These three biomarkers are representative of the entire panel of proteins and peptides, but provide a simpler system for initial study. This study is the first use of a well-characterized, high-specificity immunoaffinity column that simultaneously extracts several PTB biomarkers in a 3D printed microfluidic device. Immunoaffinity extraction, which will be subsequently combined with other on-chip analysis processes, is a key part of future 3D printed microfluidic devices for rapid PTB risk assessment.
Materials and methods
Chemicals
Glycidyl methacrylate (GMA), ethylene glycol dimethacrylate (EDMA), 1-dodecanol, 2,2-dimethoxy-2-phenylacetophenone (DMPA), poly(ethylene glycol)diacrylate (PEGDA, MW 250), Tris hydrochloride, 3-(trimethoxysilyl)propyl methacrylate, dimethyl sulfoxide (DMSO), phenylbis(2,4,6 trimethylbenzoyl)phosphine oxide (Irgacure 819), Amicon ultra 0.5 mL centrifugal filters (10 and 50 kDa cutoff), boric acid, sodium bicarbonate, and sodium carbonate were obtained from Thermo Fisher (St Louis, MO). Alexa Fluor 532 (carboxylic acid, succinimidyl ester) and Tris base were purchased from Fisher Scientific (Fair Lawn, NJ). Solutions were made using deionized water (18.3 MΩ) filtered by a Barnstead EASY-pure UV/UF system (Dubuque, IA). Isopropyl alcohol (IPA) was from Macron (Center Valley, PA). Cyclohexanol was obtained from Spectrum (New Brunswick, NJ). 2-nitrophenyl phenyl sulfide (NPS) came from TCI (Portland, OR). Amicon ultra-4 centrifugal filters (15 mL, 10 and 30 kDa cutoff), and 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) were purchased from EMD Millipore (Billerica, MA). Glass slides for 3D printing were purchased from VWR (Radnor, PA).Biological components
Corticotropin releasing factor (CRF) came from GenScript (Piscataway, NJ), and tumor necrosis factor-α receptor type 1 (TNF) was purchased from ProSpec (East Brunswick, NJ). Thrombin and antithrombin were obtained from Haematologic Technology (Essex Junction, VT), and heparin was obtained from Alfa-Aesar (Haverhill, MA). Antibodies to thrombin-antithrombin complex (anti-TAT, produced in mice) were purchased from Abcam (ab191378, Cambridge, MA). Anti-CRF (2B11, produced in mice), anti-TNF (H398, produced in mice), EZ-Link Sulfo-NHS-LC-Biotin, and high select top 14 abundant protein depletion midi spin columns were obtained from Thermo Fisher. Dry milk was purchased from Walmart (Bentonville, AR). Nitrocellulose paper was obtained from Bio-Rad (Hercules, CA), and IRDye 800CW labeled secondary goat anti-mouse IgG came from LICOR (Lincoln, NE). Female human blood serum (off-the-clot, sterile filtered) was purchased from Zen-Bio (Research Triangle Park, NC). Streptavidin biosensors were purchased from ForteBio (Fremont, CA) and bovine serum albumin (BSA) was purchased from EMD Millipore.3D printed microfluidic devices
The microfluidic devices (Fig. 1A) were adapted from Bickham et al.19 and designed to have five separated channels, each with a 50 μm × 45 μm cross section. Each channel had a reservoir on one side of the device and a port on the other side for PTFE tubing (0.22 in ID × 0.042 in OD; Cole Parmer, Vernon Hills, IL) to connect to vacuum to flow analyte through the channel. A 600 μm wide monolith polymerization window (MPW), a region with fewer absorber-containing layers in the 3D print, was designed in each device to allow monolith polymerization.12 97% PEGDA, 2% NPS UV absorber, and 1% Irgacure 819 photoinitiator composed the resin used to fabricate the devices in a custom 3D printer.24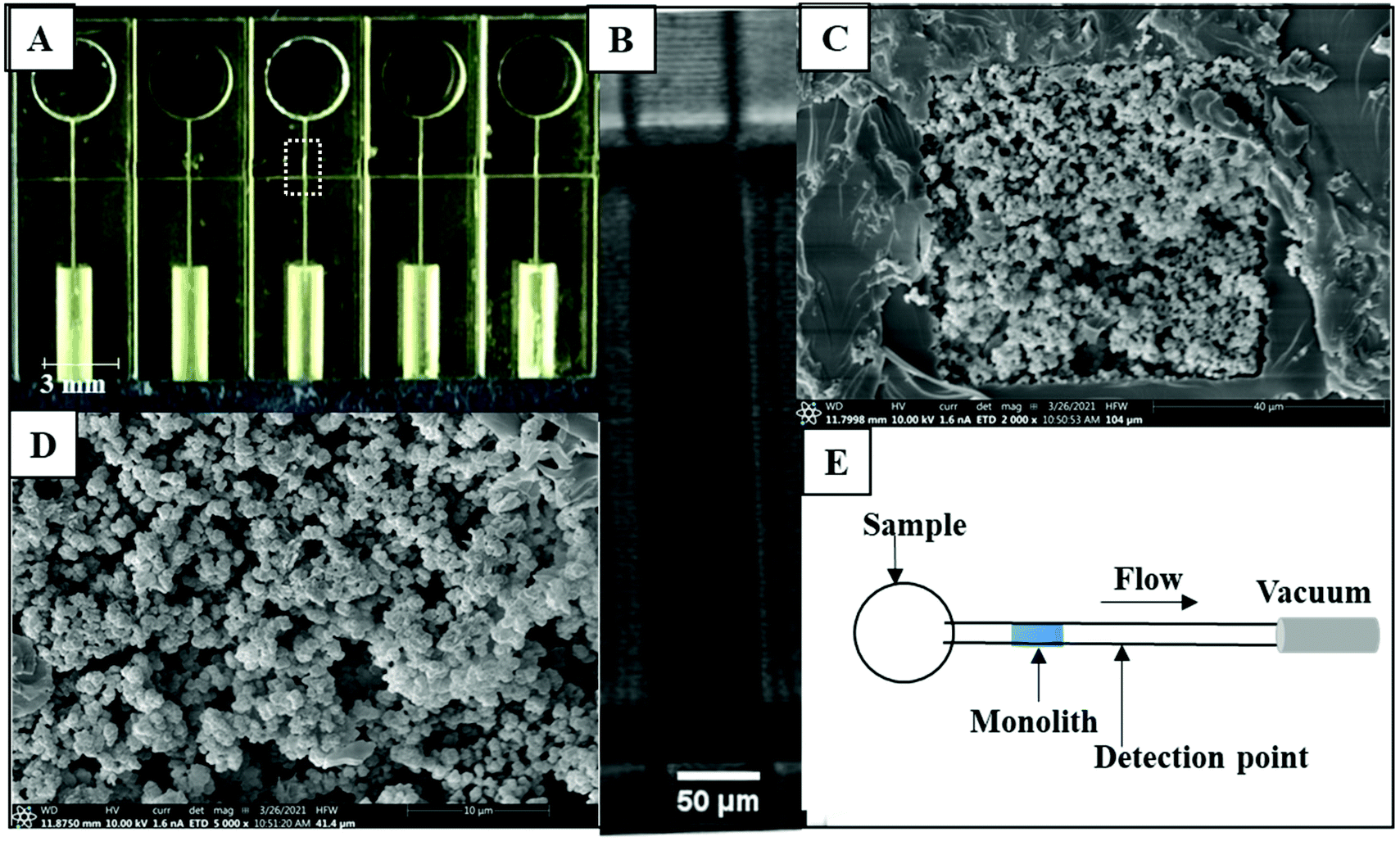 | ||
| Fig. 1 3D printed devices with monoliths for affinity extraction. (A) Photograph of 3D printed device. (B) Photograph of monolith inside channel, corresponding to the dotted white box region in (A). (C and D) SEM images of monoliths prepared in a 3D printed microfluidic device; (C) channel view and (D) zoom view. (E) Device schematic for PTB biomarker extraction. The labeled biomarker flows through the channel via vacuum; as labeled analytes pass the detection point, the signal is recorded. | ||
PTB biomarker preparation
The PTB biomarkers were fluorescently labeled; CRF and TNF were dissolved in 10 mM bicarbonate buffer (BCB, pH 10), and TAT was prepared as in Nielsen et al.25 Alexa Fluor 532-succinimdyl ester was dissolved in DMSO, added to each biomarker and incubated at room temperature overnight. CRF (100 μM), TNF (40 μM), and TAT (26 μM), were labeled at a dye![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :biomarker molar ratio of 3:2, 20:1, and 20:1, respectively. Next, TNF and TAT were filtered four times at 14000 rpm (22000g) for 15 min using a 10 kDa or 50 kDa cutoff filter, respectively, to remove excess dye. Samples were diluted to the desired concentration in 20 mM HEPES buffer pH 7 (biomarkers in buffer). To prepare depleted blood serum, we followed the Thermo Fisher protocol for their columns (Cat No. A36371). Briefly, 100 μL of serum was added into a pre-filled midi spin column that contained 1000 μL of a 50% slurry in 10 mM PBS, 0.02% sodium azide, pH 7.4. Then, the column was gently inverted to form a homogenous mixture, which was incubated in the column for 10 min with gentle agitation. We placed the column in a 15 mL Falcon tube and centrifuged the column at 1000 g for 2 min. Afterward, this sample, now 10-fold diluted, was collected and stored at −20 °C until further use. Labeled biomarker samples were spiked into the depleted serum to yield 100 nM CRF, 30 nM TNF, and 60 nM TAT (spiked depleted serum). Labeled biomarkers were similarly spiked into five-fold diluted serum that had not been depleted of abundant proteins (spiked diluted serum).
:biomarker molar ratio of 3:2, 20:1, and 20:1, respectively. Next, TNF and TAT were filtered four times at 14000 rpm (22000g) for 15 min using a 10 kDa or 50 kDa cutoff filter, respectively, to remove excess dye. Samples were diluted to the desired concentration in 20 mM HEPES buffer pH 7 (biomarkers in buffer). To prepare depleted blood serum, we followed the Thermo Fisher protocol for their columns (Cat No. A36371). Briefly, 100 μL of serum was added into a pre-filled midi spin column that contained 1000 μL of a 50% slurry in 10 mM PBS, 0.02% sodium azide, pH 7.4. Then, the column was gently inverted to form a homogenous mixture, which was incubated in the column for 10 min with gentle agitation. We placed the column in a 15 mL Falcon tube and centrifuged the column at 1000 g for 2 min. Afterward, this sample, now 10-fold diluted, was collected and stored at −20 °C until further use. Labeled biomarker samples were spiked into the depleted serum to yield 100 nM CRF, 30 nM TNF, and 60 nM TAT (spiked depleted serum). Labeled biomarkers were similarly spiked into five-fold diluted serum that had not been depleted of abundant proteins (spiked diluted serum).
Kinetic characterization
A single determination of Kd values was done for CRF, TNF, and TAT with their respective monoclonal antibodies. Assays were conducted using SPR (Biacore, Creative Biolabs) for CRF and TNF, and BLI (OCTET RED96, ForteBio) for TAT in PBS pH 7 at 30 °C with 1000 rpm shaking. TAT was biotinylated as described in the Thermo Fisher protocol for product no. A39257. Then, the BLI assay was run on a standard microwell plate when the streptavidin biosensors were loaded with the biotinylated TAT at 5–10 μg mL−1 at 300 s. Five sensors were used, three for antibody binding, and two for the reference control. The loaded sensors were equilibrated first with PBS pH 7 to generate the baseline. Then, the association step was performed for 30 s for different concentrations ranging from 62 to 250 nM. The dissociation step was performed in the assay buffer for 800 s in PBS pH 7. The data for association and dissociation were analyzed by Octet Data Analysis 8.2 software and fit to a 1:1 binding model to obtain kinetic parameters. SPR data were obtained on samples shipped to Creative Biolabs.
Monolith formation
Modified monoliths were prepared as reported by Parker et al.12 using 24% GMA as the monomer, 11% EDMA as cross linker, 10% cyclohexanol and 55% 1-dodecanol as porogens, and 1% DMPA as the photoinitiator. The monolith mixture was sonicated for 10 min and loaded into the microfluidic channel via capillary action. Then, the entire device was placed under a UV light (SunRay 600, Uvitron, West Springfield, MA) for 10 min allowing the monolith to polymerize at the desired channel location in the MPW. After polymerization, PTFE tubing was inserted into the device port and hot glued in place. The unpolymerized mixture was removed from the channel by flushing with IPA for 30 min using vacuum. Scanning electron microscopy (SEM) images of the monolith were taken by removing the 3D-printed channel from the glass slide using a razor blade. Then, the monoliths were cut through the MPW and affixed on stubs using carbon tape. Next, 80:20 Au:Pd was sputter coated on the surface of the monolith using a Q150 T ES Sputterer (Quorum Technologies, Lewes, East Sussex, UK). Finally, the SEM images were taken using an Apreo C Low-Vacuum SEM instrument (Thermo) in high vacuum mode at 10 kV.
Antibody immobilization
Compatibility of the PTB biomarker antibodies towards their targets and off-target analytes was tested using dot blots. Solutions of CRF, TNF, and TAT (2 μL, 1 mg ml−1) were dotted on nitrocellulose paper and left to dry for 30 min. Then, a blocking buffer of either 5% milk or 5% BSA in 10× Tris buffer saline (TBS) was applied for 1 h to prevent nonspecific binding. Next, primary antibodies (1 μg mL−1 in TBS plus 0.05% Tween 20; TBST) were added and incubated for 1 h for binding to the biomarker. TBST was used to rinse away unbound antibodies for 15 min. Finally, the labeled secondary antibody (1 μg mL−1 in TBS) was incubated for 1 h, and the paper was washed again with TBST for 15 min. The dot blots were scanned via a LI-COR ODYSSEY imaging system. The cross reactivity between each antibody and off-target biomarker was also tested using the process above.The attachment of each antibody to the GMA monolith was verified using fluorescence images. To measure attachment to monoliths, anti-CRF, anti-TNF, and anti-TAT were labeled at a dye:antibody molar ratio of 10:1 and were filtered four times at 14000 rpm (22000g) for 15 min using a 10 kDa cutoff filter to remove excess dye. We directed a 532 nm laser through a 4× objective and captured images with a Hamamatsu ORCA-Fusion CMOS camera (Bridgewater, NJ) using 10, 100, or 300 ms exposure times. First, a blank image of the monolith was taken before attaching the antibody. Then, the monoclonal anti-CRF, anti-TNF, or anti-TAT labeled with Alexa Fluor 532 was immobilized onto the monolith overnight. Next, the monolith was rinsed with 20 mM borate buffer pH 8 for 30 min and another monolith image was captured. Background-subtracted fluorescence of the monolith with the labeled antibody and the control channel was determined using Image J (imagej.nih.gov). To measure the background-subtracted fluorescence for extracted PTB biomarkers, the same steps were used with monolith images taken at each stage of the experimental process: loading, rinsing, and elution. Fluorescence values were normalized to the signal after loading, and the average signal was determined from three replicates.
For the attachment of multiple antibodies to the monolith, a mixture consisting of 4 μL each of anti-CRF, anti-TNF, and anti-TAT (1 mg mL−1) was added to the device and allowed to flow through the channel via capillary action. 20 mM borate buffer pH 8 was next added to the reservoirs, and the entire device was covered with parafilm and placed in a humid chamber overnight to prevent the channel from drying. This reaction time allowed amine groups in the antibody to bind covalently to the epoxy groups in the immunoaffinity monolith. Following overnight incubation, 0.1 M Tris buffer (pH 8.5) was flowed through the channel and then incubated for 1 h in a humid chamber to block any remaining epoxy groups. Finally, the channel was rinsed with 20 mM HEPES buffer pH 7 for 5 min. For control experiments, the monolith was blocked using Tris buffer and no antibodies were incubated.
Immunoaffinity extraction
The experimental set up was as follows: a 532 nm laser was used to induce fluorescence. The fluorescence signal was recorded with a photomultiplier tube and digitized using LabVIEW software. Experiments were then carried out using the following steps. First, channels were filled with 20 mM HEPES buffer pH 7 and vacuum was applied for 1 min through the device as seen in Fig. 1E. Then, the vacuum was paused, the reservoir was emptied, the fluorescently labeled analyte was loaded into the reservoir, and analyte was drawn through the channel by applying vacuum for ∼40 s. The labeled analyte was incubated in the monolith for 10 min, after which the reservoir was washed three times with 20 mM HEPES buffer pH 7, and the monolith had 20 mM HEPES buffer drawn through until the signal went back to baseline. Finally, the reservoir was filled with 50 mM BCB and vacuum was applied for 1 min to elute the sample.Results and discussion
Antibody characterization
Dot blots were used to confirm the compatibility between CRF, TNF, and TAT and their respective antibodies, as well as their cross reactivity toward off-target antibodies. Fig. S1 in the ESI† shows three replicate dot blots in each panel. Fig. S1A† displays a positive control for ferritin and anti-ferritin binding. Fig. S1B–D† similarly shows binding between CRF and anti-CRF, TNF and anti-TNF, and TAT and anti-TAT. Fig. S1E† shows that binding also occurs between thrombin and anti-TAT. Very little binding was observed between antithrombin and anti-TAT as seen in Fig. S1F.† We hypothesize that rather than affinity interaction, thrombin nonspecifically sticks to primary antibodies, and in Fig. S1G,† we confirmed that thrombin also binds non-specifically to anti-ferritin. These dot blots demonstrate that the anti-CRF, anti-TNF, and anti-TAT antibodies selected are appropriate for CRF, TNF, and TAT, but that thrombin by itself may stick nonspecifically to antibodies.We further studied the cross reactivity between CRF, TNF, and TAT, and off-target antibodies, as shown in the dot blots in Fig. S1H–M.† No fluorescent dots were seen in any of these experiments, which confirmed a lack of affinity binding between CRF, TNF, and TAT, and off-target antibodies. These data further support the use of these anti-CRF, anti-TNF, and anti-TAT antibodies for multiplex immunoaffinity experiments.
SPR and BLI of PTB biomarkers with their target monoclonal antibodies
We obtained Kd data for CRF, TNF, and TAT with their monoclonal antibodies using either SPR or BLI. Fig. 2 shows the fitting curves for association and dissociation of CRF, TNF, and TAT with their corresponding antibodies. The Kd values for these three PTB biomarkers and their monoclonal antibodies were calculated as seen in Table 1. The Kd values in Table 1 show that of these three, TAT has the strongest binding affinity toward its antibody, and CRF has the lowest binding affinity toward anti-CRF. This means in a multiplexed affinity column, CRF will dissociate more readily from its target compared to TNF and TAT. The lower affinity for CRF and anti-CRF likely is due to the smaller size of CRF, relative to TNF and TAT.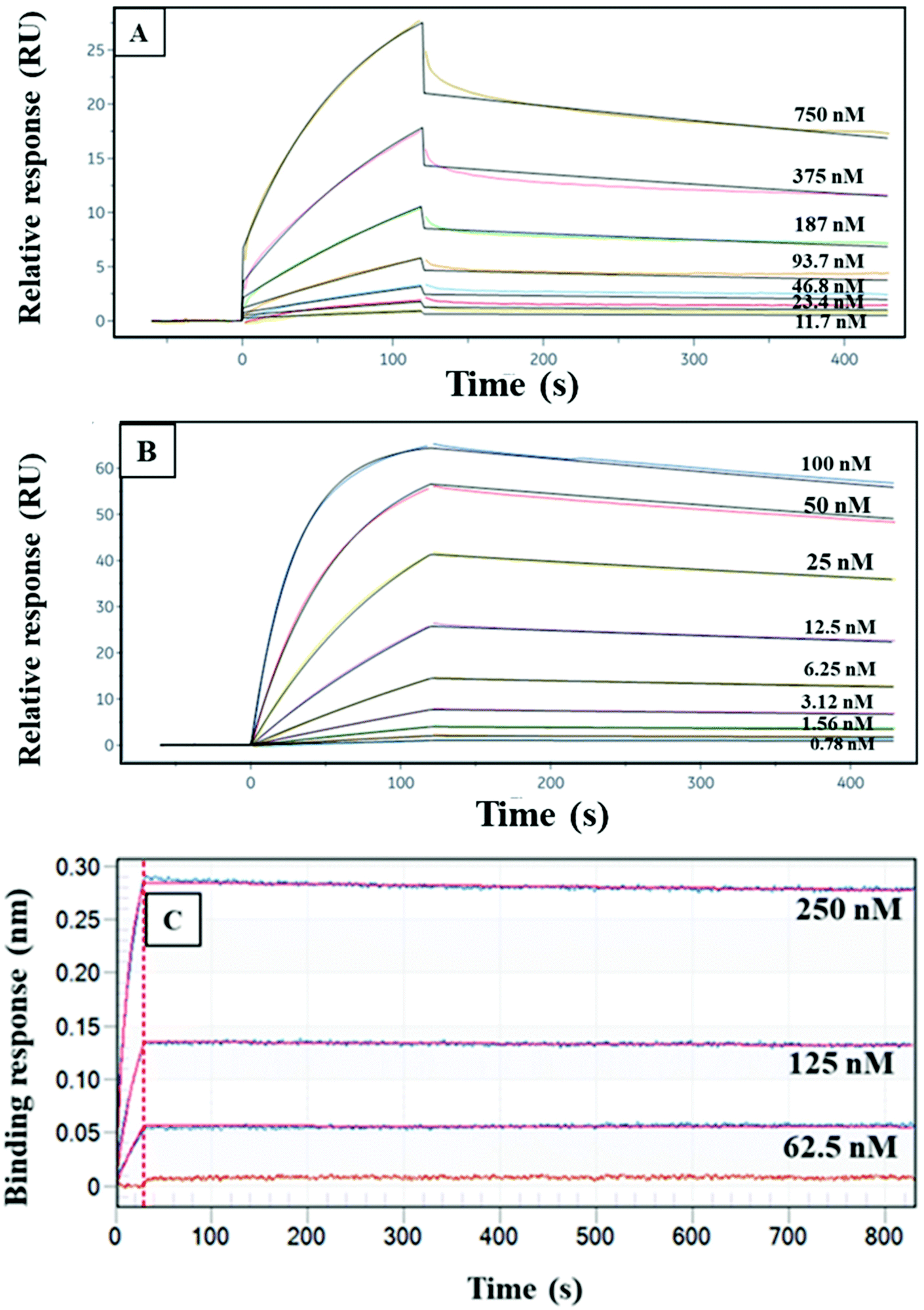 | ||
| Fig. 2 Association and dissociation data and fitting for CRF, TNF, and TAT binding to their respective monoclonal antibodies. CRF and TNF data were collected from SPR. TAT data were obtained with BLI. Different concentrations of (A) CRF, (B) TNF, or (C) anti-TAT. | ||
| PTB biomarkers | K d (nM) |
|---|---|
| CRF and anti-CRF | 41 |
| TNF and anti-TNF | 1.4 |
| TAT and anti-TAT | 0.11 |
Monolith characterization and modification
We used the same monolith formulation that Parker et al.12 developed to retain the PTB biomarker, ferritin, using a single-antibody column. GMA monoliths were successfully formed inside channels as seen in Fig. 1B. The morphology of a representative monolith was determined using SEM as shown in Fig. 1C and D. The SEM data in Fig. 1C show that the monolith is fully attached to the microfluidic channel wall. The monolith pores are randomly distributed as seen in Fig. 1D, and there is sufficient surface area for antibody immobilization. We analyzed the pore and nodule sizes for these monoliths, which were 0.5 μm ± 0.1 μm (n = 30) and 0.7 μm ± 0.1 μm (n = 30), respectively.We used fluorescence imaging to verify the attachment of antibodies to the affinity monoliths, as seen in Fig. S2 in the ESI.† The direct reaction of antibody amines with the GMA epoxy groups on the monoliths results in non-oriented antibody attachment; although this may yield some antibodies arranged with an inaccessible active site, we have not found this to be a major limitation in prior studies.12,13,26–28 In the control monoliths in Fig. S2A, C, and E† the fluorescence was much lower compared to the fluorescence observed when labeled anti-CRF, anti-TNF, or anti-TAT was attached to the column in Fig. S2B, D, and F.† Fig. S2G–I† shows the background-subtracted fluorescence on the monolith before and after antibody attachment; fluorescence signal significantly increased for labeled anti-CRF, anti-TNF, and anti-TAT columns compared to the control columns. This clear increase in fluorescence confirmed the attachment of antibodies to PTB biomarkers on monoliths.
Immunoaffinity extraction of PTB biomarkers
Briefly, immunoaffinity extraction first entails flowing buffer through the channel to equilibrate it, followed by loading of biomarkers until column breakthrough is observed. A buffer rinse removes unbound material from the monolith, after which flow of higher pH eluent disrupts antigen–antibody interaction and removes all retained biomarkers in a band that is detected upon passing through the focused laser spot ∼1 mm beyond the monolith. The immunoaffinity extraction columns are designed for the first step in an integrated on-chip analysis workflow (where the entire eluted band is transferred to a solid-phase extraction and labeling monolith), rather than for chromatographic resolution of all components. We measured the fluorescence during elution after extraction of CRF, TNF, and TAT on their respective, single-antibody-modified columns and control columns, from both buffer and depleted human blood serum, as seen in Fig. 3. During the elution step a peak appeared as an increase in fluorescence signal at 5–10 s after flow started for each biomarker loaded from buffer (Fig. 3A–C). In contrast, little or no increase in the fluorescence signal occurred for the control monoliths lacking attached antibodies, supporting that the elution peaks seen for the antibody columns are not due to fluorophores in the void fraction. The elution peaks appeared at similar times for the three biomarkers, but they exhibited different shapes. In the depleted blood serum experiments (Fig. 3D–F), the elution peaks were seen at 5 s, 15 s, and 20 s for CRF, TNF, and TAT, respectively. These peaks were generally wider and had higher signal compared to the peaks observed for analyte loaded from buffer. The fast retention times (<1 min) are an intentional result of design for an integrated analysis workflow with a short (600 μm) monolith column and ∼1 mm distance to the detection location. Some variability in retention time and fluorescence intensity was observed between the biomarkers in buffer and in spiked depleted serum. Retention time and fluorescence signal variability are likely due to the manual nature of loading and elution in these devices. The higher fluorescence and greater tailing for spiked depleted serum may also be attributed to interactions of biomarkers with proteins that still remain in depleted serum leading to stronger retention. Our data demonstrate the ability to retain CRF, TNF, and TAT individually on single-antibody columns after loading from either buffer or blood serum.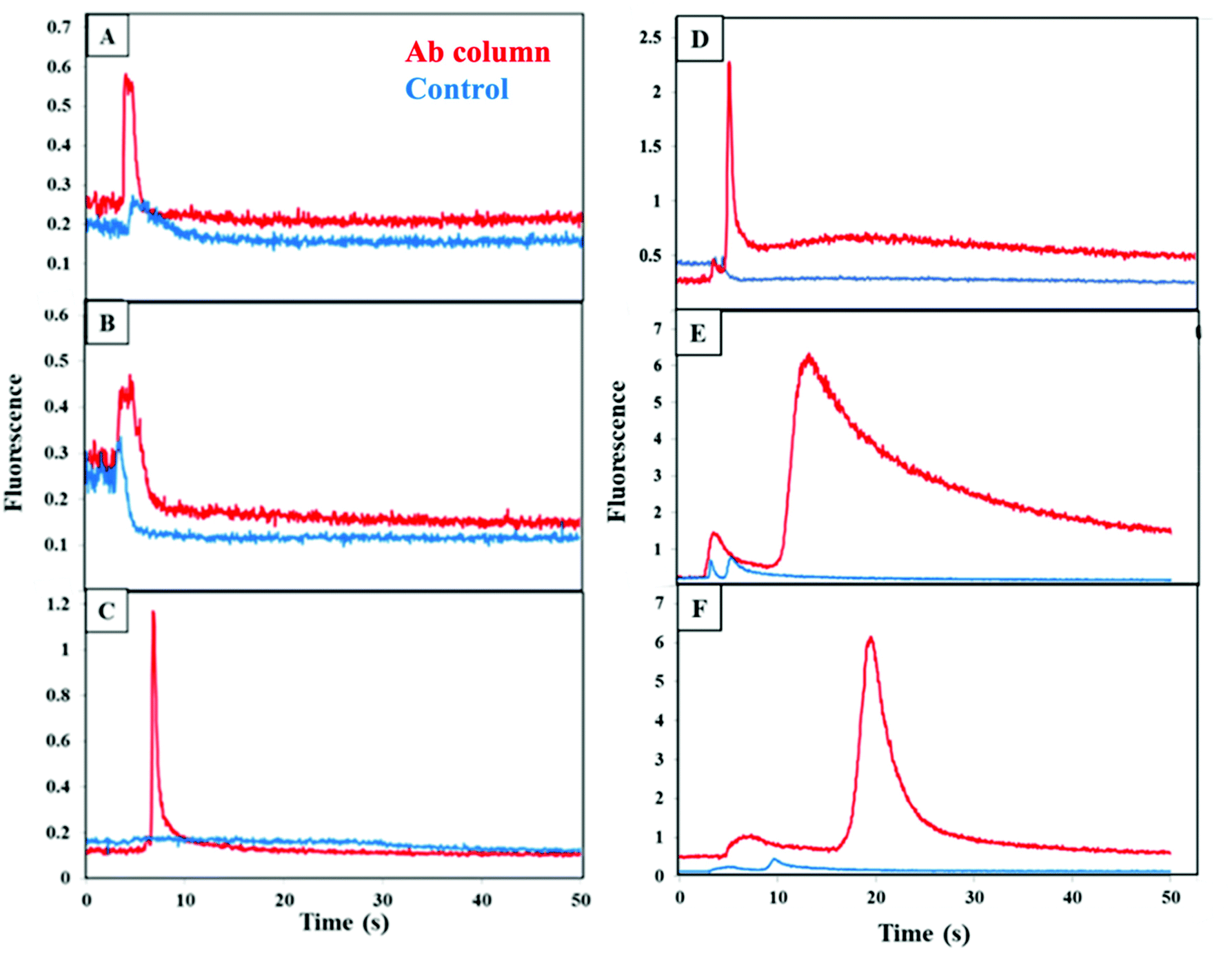 | ||
| Fig. 3 Fluorescence during elution after extraction of labeled PTB biomarkers from their respective single-antibody-modified columns (red) or a control monolith lacking attached antibody (blue) for (A and D) 100 nM CRF; (B and E) 30 nM TNF; and (C and F) 60 nM TAT for biomarkers either in (A–C) buffer or (D-F) spiked depleted serum. Similar elution data were obtained for (A–C) 4 or (D–F) 3 replicate experiments on different columns. | ||
Cross reactivity of each of the PTB biomarkers with off-target antibodies was tested as shown in Fig. 4 for fluorescence during elution from a column with the two off-target antibodies for each biomarker, which was loaded from buffer. A small increase in the fluorescence was seen for both the antibody and blocked column, which possibly resulted from limited, nonspecific adsorption of each biomarker on the affinity column. This data clearly confirms that there is little cross reactivity occurring between each biomarker and the other two off-target antibodies.
 | ||
| Fig. 4 Fluorescence during elution after extraction of biomarkers in buffer on an off-target multiplexed affinity monolith (red) or a control monolith lacking attached antibodies (blue). Labeled (A) 100 nM CRF on anti-TNF and anti-TAT; (B) 30 nM TNF on anti-CRF and anti-TAT; (C) 60 nM TAT on anti-CRF and anti-TNF. Similar elution data were obtained for 3 replicate experiments on different columns. | ||
We then tested extraction of individual PTB biomarkers spiked into human blood serum on multiplexed antibody columns. As can be seen in Fig. 5 small, narrow peaks were detected between 5–10 s for the blocked monoliths, which likely resulted from limited adsorption of the biomarker to the blocked column. However, a large peak in the fluorescence was observed at 5–10 s when each biomarker was eluted after extraction on a multiplexed antibody column. These results show the ability to individually extract CRF, TNF, and TAT on multiplexed antibody columns.
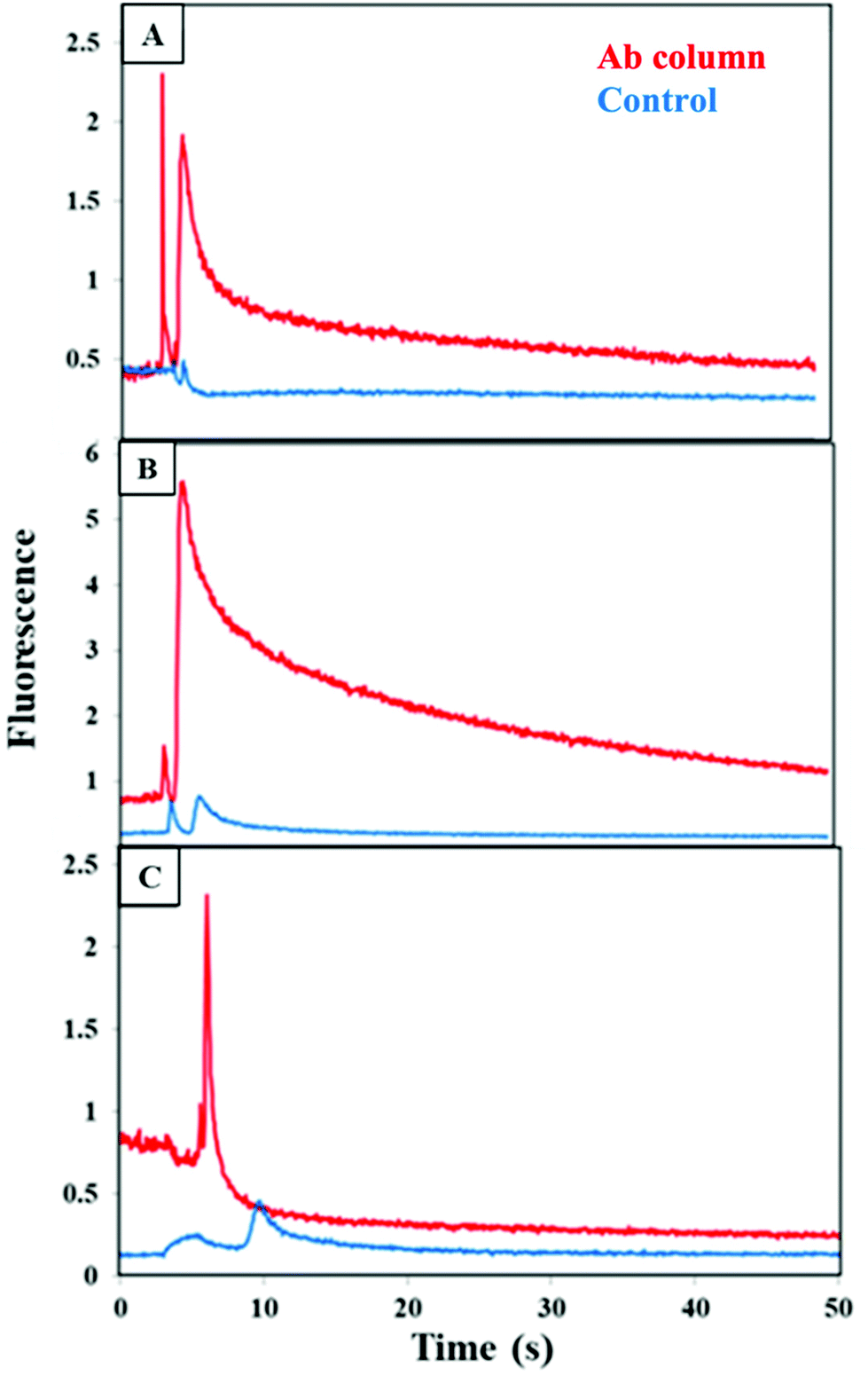 | ||
| Fig. 5 Fluorescence during elution of a single PTB biomarker loaded from spiked depleted serum and eluted from a multiplexed anti-CRF, anti-TNF, and anti-TAT monolith (red) or a control monolith lacking attached antibodies (blue). (A) 100 nM CRF, (B) 30 nM TNF, and (C) 60 nM TAT. Similar elution data were obtained for 3 replicate experiments on different columns. | ||
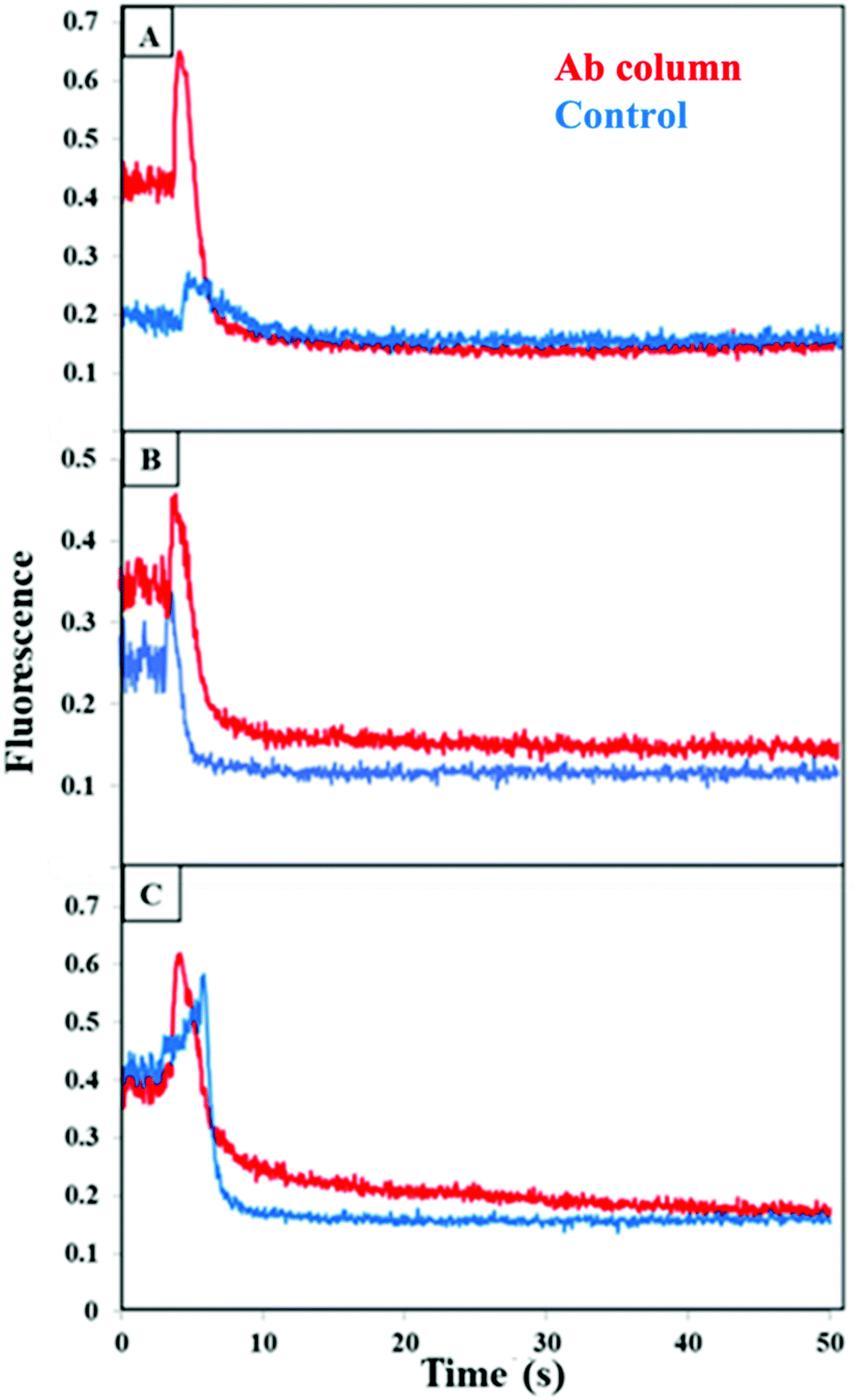
When we tested the individual protein biomarkers TNF and TAT, spiked into serum diluted 5-fold and loaded on a blocked column, only a small elution peak was observed as seen in the ESI Fig. S3A and B.† This indicates that, similar to the result in Fig. 5B and C, neither TNF nor TAT interacted significantly with the blocked column, even in the serum matrix that had not been depleted of abundant proteins. In contrast, a much larger elution peak was observed when both TNF and TAT were spiked into diluted blood serum and loaded on a blocked column as seen in the ESI Fig. S3C.† This result infers that some sort of combined interaction occurs between TNF, TAT and components in blood serum, even on a blocked column. We hypothesized that one or more of the most abundant proteins in serum might facilitate this nonspecific interaction, perhaps by binding to both TNF and TAT and interacting with the blocked column. Thus, we used five-fold diluted serum that had been depleted of abundant proteins in further multiplexing studies.
We studied the retention and elution from a combined anti-CRF, anti-TNF, and anti-TAT monolith by running a mixture of CRF, TNF, and TAT on either a multiplexed antibody column or a blocked column. Fig. 6A shows an increase in the fluorescence signal during elution of all three biomarkers loaded from buffer on a combined anti-CRF, anti-TNF, and anti-TAT column as compared to a blocked column. The elution peak appeared at 7 s and was taller and wider than the peak from the blocked column. These data indicate successful retention and elution of these three PTB biomarkers in buffer using a multiplexed antibody column.
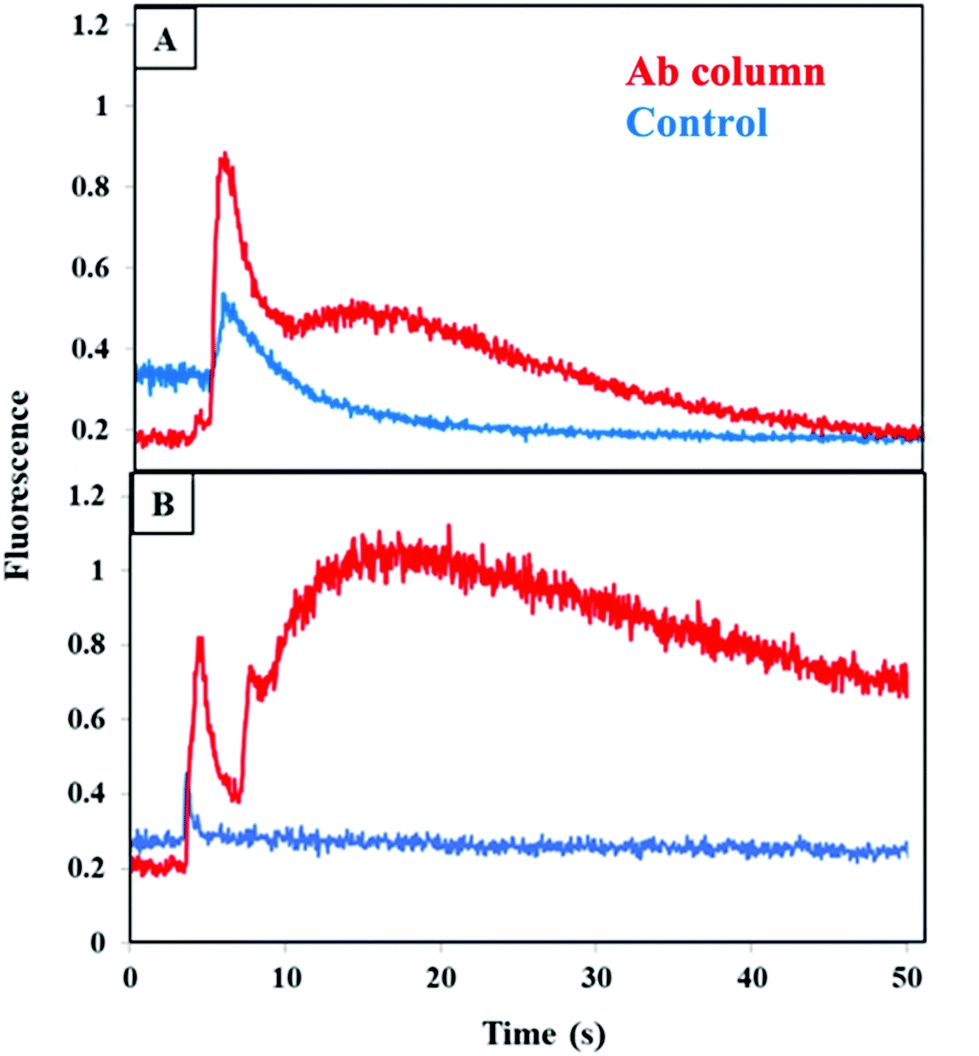 | ||
| Fig. 6 Fluorescence during elution after extraction on a combined anti-CRF, anti-TNF, and anti-TAT monolith (red) or a control monolith lacking attached antibodies (blue) for a mixture of labeled 100 nM CRF, 30 nM TNF, and 60 nM TAT for (A) biomarkers in buffer and (B) spiked depleted serum. Similar elution data were obtained for 4 replicate experiments on different columns. | ||
A similar experiment was performed wherein the biomarkers were spiked into depleted blood serum, loaded on a monolith, and then eluted as seen in Fig. 6B. An elution peak was observed initially at 5 s with a second, broader peak starting at 10 s. In contrast, a much smaller peak appeared at 5 s and the signal quickly went back to the baseline in the control experiment. This contrast with the large peak seen in Fig. S3C† supports the idea that the abundant serum proteins were responsible for the nonspecific binding observed in multiplexing experiments in serum when those proteins had not been removed. These results confirm successful retention and elution of these PTB biomarkers from a multiplexed monolith compared to a column lacking antibodies in a 3D printed device. These results also show promise for future extension to working with all nine PTB biomarkers on a single multiplexed antibody column.
Further confirmation of the selective retention and elution of PTB biomarkers on a multiplexed immunoaffinity monolith is found in Fig. 7. We took fluorescence images of a monolith after loading, rinsing, and elution of these three PTB biomarkers in depleted human blood serum on multiplexed immunoaffinity columns. The background-subtracted fluorescence seen in Fig. 7 demonstrates that after the biomarker mixture was first loaded into a column the fluorescence signal was at its highest. The control columns showed a drop in signal after rinsing to remove any nonspecifically bound analyte, indicating that the biomarkers were not strongly retained on blocked columns. However, the fluorescence signal was considerably higher after the rinsing step for antibody columns, showing specific retention of the biomarkers on multiplexed affinity monoliths compared to control columns. The signal on the monolith dropped more than twofold after elution from the antibody columns, indicating removal of retained PTB biomarkers. Elution from the control columns led to a small decrease in the already near background signal. These data show limited biomarker retention for the control columns with considerably more retention and elution of the biomarkers on the antibody columns. This further supports the use of these multiplexed immunoaffinity columns for PTB biomarker sample preparation.
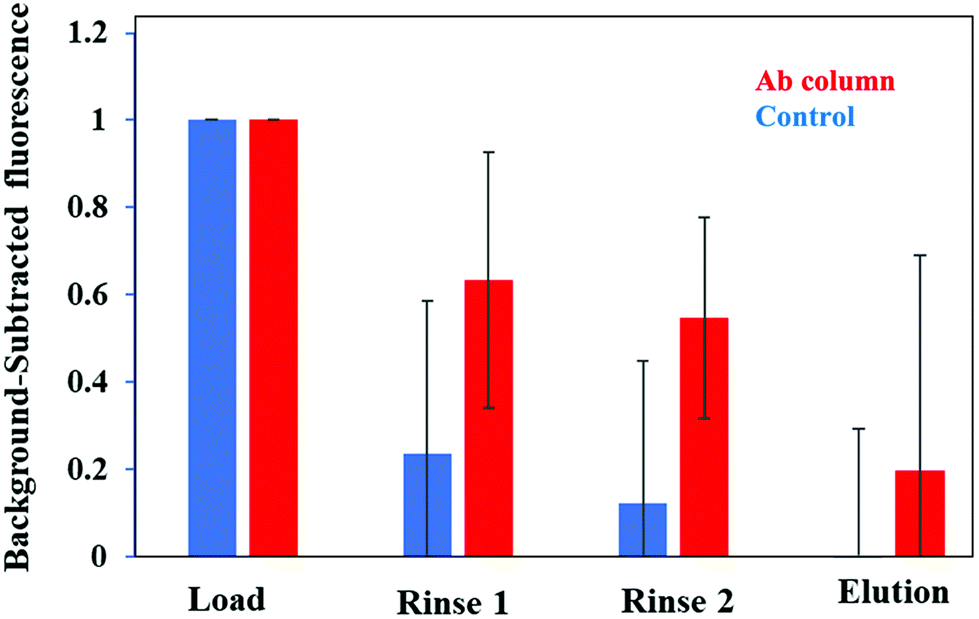 | ||
| Fig. 7 Normalized fluorescence signal on a monolith during the extraction of a mixture of 300 nM CRF, 90 nM TNF, and 180 nM TAT for spiked depleted serum. (Blue) control monolith without attached antibodies; (red) monolith with attached antibodies. Error bars show the standard deviation for three replicates. The p value for the change in signal between rinse 2 and elution is 0.28 for the control monolith and 0.14 for the antibody-modified monolith. | ||
We were able to extract CRF spiked into human blood serum with an antibody having moderate affinity (see Table 1). However, extraction of native CRF from human blood serum may require an antibody with higher affinity binding compared to the current one, as serum CRF levels are lower than the Kd for this antibody. In contrast, TAT has high affinity toward anti-TAT, and with a PTB risk level of ∼5 μM,8 we should readily be able to extract TAT from blood serum at clinical levels.
We found the monoliths to be stable in our 3D printed devices for at least one week; future experiments could focus on optimization of device storage conditions for even longer times. Although our long-term objective is to utilize these 3D prints as single-use, point-of-care devices, we have also shown that individual 3D prints can be reused for multiple analyses.29 Future integration of pumps and valves30,31 in our 3D printed devices would allow automation of loading and elution to improve retention time and fluorescence signal reproducibility.
Conclusion
To improve sample preparation for analysis of biomarkers for maternal and fetal wellness, we created 3D printed microfluidic devices with multiplexed immunoaffinity monoliths. We characterized antibodies to target three PTB biomarkers. We also verified attachment of these antibodies to immunoaffinity columns using fluorescent imaging. Furthermore, we demonstrated successful retention and elution of individual PTB biomarkers on single-antibody columns from both buffer and depleted human blood serum. Minimal cross reactivity was observed between each biomarker and the two off-target antibodies. Each of the three individual biomarkers were successfully extracted from multiplexed antibody columns. Finally, we demonstrated selective retention and elution of three PTB biomarkers on a multiplexed immunoaffinity column. In the future, we plan to multiplex additional antibodies on these columns, which should allow extraction of the whole panel of nine PTB biomarkers using a single immunoaffinity column. Additionally, these immunoaffinity monoliths form a significant part of a potential integrated chip that could be used in point-of-care early identification of the risk of preterm birth.Conflicts of interest
A. T. W. and G. P. N. both own shares in Acrea 3D, a company that is commercializing 3D printing of microfluidics.Acknowledgements
We thank NIH for funding of this work (R01 EB027096). H. M. A. acknowledges a scholarship from Taif University in the Kingdom of Saudi Arabia. We thank the BYU College of Physical and Mathematical Sciences for Undergraduate Research Awards to M. K. H. and S. R. P.References
- M. Platchek, Q. Lu, H. Tran and W. Xie, SLAS Discovery, 2020, 25, 1197–1213 CrossRef CAS PubMed.
- A. Mori, T. Saito, M. Takahashi, M. Shibata, G. Tsuji, S. Hatachi, S. Takahashi and S. Kumagai, PLoS One, 2020, 15, e0243729 CrossRef CAS PubMed.
- H. Furuya, L. Tabula, R. Lee, P. Kralovec, M. Ramsden, R. Wong and C. J. Rosser, Pract. Lab. Med., 2020, 22, e00189 CrossRef PubMed.
- K. R. Mitchell, J. E. Esene and A. T. Woolley, Anal. Bioanal. Chem., 2022, 414, 167–180 CrossRef CAS PubMed.
- R. T. Souza, E. J. McKenzie, B. Jones, J. V. de Seymour, M. M. Thomas, E. Zarate, T. L. Han, L. McCowan, K. Sulek, S. Villas-Boas, L. C. Kenny, J. G. Cecatti and P. N. Baker, Sci. Rep., 2019, 23(9), 13701 CrossRef PubMed.
- P. G. Hill, Ann. Clin. Biochem., 1985, 22, 565–578 CrossRef CAS PubMed.
- V. Polaskova, A. Kapur, A. Khan, M. P. Molloy and M. S. Baker, Electrophoresis, 2010, 31, 471–482 CrossRef CAS PubMed.
- M. S. Esplin, K. Merrell, R. Goldenberg, Y. Lai, J. D. Iams, B. Mercer, C. Y. Spong, M. Miodovnik, H. N. Simhan, P. van Dorsten and M. Dombrowski, Am. J. Obstet. Gynecol., 2011, 204, 391.e1–391.e8 CrossRef PubMed.
- R. Knob, V. Sahore, M. Sonker and A. T. Woolley, Biomicrofluidics, 2016, 10, 032901 CrossRef PubMed.
- F. Svec and Y. Lv, Anal. Chem., 2015, 87, 250–273 CrossRef CAS PubMed.
- U. Andelkovic, S. Tufegdzic and P. Milica, Electrophoresis, 2017, 38, 2851–2869 CrossRef PubMed.
- E. K. Parker, A. V. Nielsen, M. J. Beauchamp, H. M. Almughamsi, J. B. Nielsen, M. Sonker, H. Gong, G. P. Nordin and A. T. Woolley, Anal. Bioanal. Chem., 2019, 411, 5405–5413 CrossRef CAS PubMed.
- M. Sonker, E. K. Parker, A. V. Nielsen, V. Sahore and A. T. Woolley, Analyst, 2017, 143, 224–231 RSC.
- Y. Abdiche, D. Malashock, A. Pinkerton and J. Pons, Anal. Biochem., 2008, 377, 209–217 CrossRef CAS PubMed.
- P. Estep, F. Reid, C. Nauman, Y. Liu, T. Sun, J. Sun and Y. Xu, mAbs, 2013, 5, 270–278 CrossRef PubMed.
- J. B. Nielsen, R. L. Hanson, H. M. Almughamsi, C. Pang, T. R. Fish and A. T. Woolley, Anal. Chem., 2020, 92, 150–168 CrossRef CAS PubMed.
- C. M. Pandey, S. Augustine, S. Kumar, S. Kumar, S. Nara, S. Srivastava and B. D. Malhotra, Biotechnol. J., 2018, 13, 1700047 CrossRef PubMed.
- A. V. Nielsen, M. J. Beauchamp, G. P. Nordin and A. T. Woolley, Annu. Rev. Anal. Chem., 2020, 13, 45–65 CrossRef PubMed.
- A. V. Bickham, C. Pang, B. Q. George, D. J. Topham, J. B. Nielsen, G. P. Nordin and A. T. Woolley, Anal. Chem., 2020, 92, 12322–12329 CrossRef CAS PubMed.
- H. Gong, M. Beauchamp, S. Perry, A. T. Woolley and G. P. Nordin, RSC Adv., 2015, 5, 106621–106632 RSC.
- B. M. C. Costa, A. G. Coelho, M. J. Beauchamp, J. B. Nielsen, G. P. Nordin, A. T. Woolley and J. A. F. da Silva, Anal. Bioanal. Chem., 2022, 414, 545–550 CrossRef CAS PubMed.
- S. Kalme, S. Kandaswamy, A. Chandrasekharmath, R. Katiyar, G. P. Rajamanickam, S. Kumara and D. Dendukur, Anal. Methods, 2019, 11, 1639–1650 RSC.
- C. Dincer, R. Bruch, A. Kling, P. S. Dittrich and G. A. Urban, Trends Biotechnol., 2017, 35, 728–742 CrossRef CAS PubMed.
- H. Gong, B. P. Bickham, A. T. Woolley and G. P. Nordin, Lab Chip, 2017, 17, 2899–2909 RSC.
- J. B. Nielsen, A. V. Nielsen, R. H. Carson, H. L. Lin, R. L. Hanson, M. Sonker, D. N. Mortensen, J. C. Price and A. T. Woolley, Electrophoresis, 2019, 40, 2853–2859 CrossRef CAS PubMed.
- W. Yang, X. Sun, T. Pan and A. T. Woolley, Electrophoresis, 2008, 29, 3429–3435 CrossRef CAS PubMed.
- W. Yang, X. Sun, H.-Y. Wang and A. T. Woolley, Anal. Chem., 2009, 81, 8230–8235 CrossRef CAS PubMed.
- W. Yang, M. Yu, X. Sun and A. T. Woolley, Lab Chip, 2010, 10, 2527–2533 RSC.
- M. J. Beauchamp, A. V. Nielsen, H. Gong, G. P. Nordin and A. T. Woolley, Anal. Chem., 2019, 91, 7418–7425 CrossRef CAS PubMed.
- H. Gong, A. T. Woolley and G. P. Nordin, Biomicrofluidics, 2019, 13, 014106 CrossRef PubMed.
- J. L. Sanchez Noriega, N. A. Chartrand, J. C. Valdoz, C. G. Cribbs, D. A. Jacobs, D. Poulson, M. S. Viglione, A. T. Woolley, P. M. Van Ry, K. A. Christensen and G. P. Nordin, Nat. Commun., 2021, 12, 5509 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1an01365c |
| This journal is © The Royal Society of Chemistry 2022 |