
Antioxidant Response Activating nanoParticles (ARAPas) localize to atherosclerotic plaque and locally activate the Nrf2 pathway†
Sophie
Maiocchi
 abcd,
Ana
Cartaya
cde,
Sydney
Thai
f,
Adam
Akerman
f and
Edward
Bahnson
*abcde
abcd,
Ana
Cartaya
cde,
Sydney
Thai
f,
Adam
Akerman
f and
Edward
Bahnson
*abcde
aDepartment of Cell Biology and Physiology, University of North Carolina at Chapel Hill, NC 27599, USA. E-mail: edward_bahnson@med.unc.edu
bCurriculum of Toxicology and Environmental Medicine, University of North Carolina at Chapel Hill, NC 27599, USA
cCenter for Nanotechnology in Drug Delivery, University of North Carolina at Chapel Hill, NC 27599, USA
dMcAllister Heart Institute, University of North Carolina at Chapel Hill, NC 27599, USA
eDepartment of Pharmacology, University of North Carolina at Chapel Hill, NC 27599, USA
fDepartment of Surgery, University of North Carolina at Chapel Hill, NC 27599, USA
First published on 13th January 2022
Abstract
Atherosclerotic disease is the leading cause of death world-wide with few novel therapies available despite the ongoing health burden. Redox dysfunction is a well-established driver of atherosclerotic progression; however, the clinical translation of redox-based therapies is lacking. One of the challenges facing redox-based therapies is their targeted delivery to cellular domains of redox dysregulation. In the current study, we sought to develop Antioxidant Response Activating nanoParticles (ARAPas), encapsulating redox-based interventions, that exploit macrophage biology and the dysfunctional endothelium in order to selectively accumulate in atherosclerotic plaque. We employed flash nanoprecipitation (FNP) to synthesize bio-compatible polymeric nanoparticles encapsulating the hydrophobic Nrf2 activator drug, CDDO-Methyl (CDDOMe-ARAPas). Nuclear factor erythroid 2-related factor 2 (Nrf2)-activators are a promising class of redox-active drug molecules whereby activation of Nrf2 results in the expression of several antioxidant and cyto-protective enzymes that can be athero-protective. In this study, we characterize the physicochemical properties of CDDOMe-ARAPas as well as confirm their in vitro internalization by murine macrophages. Drug release of CDDOMe was determined by Nrf2-driven GFP fluorescence. Moreover, we show that these CDDOMe-ARAPas exert anti-inflammatory effects in classically activated macrophages. Finally, we show that CDDOMe-ARAPas selectively accumulate in atherosclerotic plaque of two widely-used murine models of atherosclerosis: ApoE−/− and LDLr−/− mice, and are capable of increasing gene expression of Nrf2-transcriptional targets in the atherosclerotic aortic arch. Future work will assess the therapeutic efficacy of intra-plaque Nrf2 activation with CDDOMe-ARAPas to inhibit atherosclerotic plaque progression. Overall, our present studies underline that targeting of atherosclerotic plaque is an effective means to enhance delivery of redox-based interventions.
Introduction
According to the World Health Organization, cardiovascular disease (CVD) is the most common cause of death in the entire world.1,2 A common underlying cause of clinical events is a chronic, long-term process called atherosclerosis, which involves progressive changes to arterial structure and function.3 Atherosclerosis is characterized by focal lesions formed in the sub-intimal space of large and mid-sized arteries, composed of lipids, fibrotic tissue and inflammatory cells that result in narrowing of the blood vessel lumen.3 Atherosclerotic lesion rupture and thrombosis leads to embolism that manifests as acute coronary syndrome, myocardial infarction or stroke. A significant early event occurring at vascular sites of disturbed blood flow is endothelial dysfunction, which permits the increased infiltration of activated immune cells, such as monocytes, and low-density lipoprotein (LDL) into the sub-intimal space, where LDL is susceptible to oxidative modification. Infiltrating monocytes differentiate into macrophages, which recognize modified LDL, by surface scavenger receptors. This results in the excessive accumulation of lipids in macrophages, which become foam cells, the hallmark of early fatty-streaks.4 Ongoing chronic inflammation within the intima results in the eventual formation of more complex atherosclerotic plaques, consisting of an overlying fibrous cap, composed of collagen and smooth muscle cells (SMCs), and a necrotic core, derived from dying foam cells, calcium deposits and cholesterol crystals.5,6 Overall, both vascular inflammation and dysregulated redox signalling play important roles in the development of endothelial dysfunction, the progression of atherosclerosis and its clinical manifestations.7,8 Although a considerable body of pre-clinical studies indicate that atherosclerosis is driven by oxidative processes,7,8 the clinical translation of redox-based therapies is lacking. One challenge related to therapeutically targeting redox dysregulation in atherosclerosis, is the specific delivery of redox-based interventions to cellular domains where redox dysregulation is occurring.8 Targeted nanomedicine is a promising, and under-studied approach to counter this challenge in CVD.9–13 Encapsulation of pharmacological therapeutics into nanoparticles can endow them with the capacity to exploit the enhanced permeability retention effect present in atherosclerotic plaque due to gaps in the dysfunctional endothelium,14 as well as exploit internalization by plaque-resident macrophages.15,16A promising mechanistic approach to limit oxidative stress and inflammation associated with atherosclerosis is activation of the Kelch-like ECH-associated protein 1/Nuclear factor erythroid 2-related factor 2 (Keap1/Nrf2) transcription factor pathway.9,17 Nrf2 is a master regulator of the cellular response to oxidative or electrophilic stress through the transcription of numerous cytoprotective and antioxidant genes including glutamate-cysteine ligase catalytic subunit (GCLC), NADPH Quinone dehydrogenase (NQO1), heme oxygenase 1 (HO1), and superoxide dismutase 1 (SOD1). Under homeostatic conditions, Keap1 binds Nrf2 in the cytoplasmic compartment and promotes its ubiquitination and downstream proteasomal degradation. However, upon oxidative or electrophilic stress, specific Keap1 cysteine resides are modified, resulting in nuclear translocation of newly generated Nrf2 where it binds to the Antioxidant Response Element (ARE) and/or the Electrophilic Response Element (EPRE) and drives expression of its target cytoprotective genes. Nrf2 and its target genes have local anti-atherogenic effects in the vascular wall, including endothelial cells, vascular smooth muscle cells, and macrophages.18–21 Nrf2 also limits inflammation by directly impeding transcription of pro-inflammatory cytokines such as IL-1β and Il-6.20 Moreover, compounds known as Nrf2-inducers (tBHQ,22 Ebselen,23 CDDO-Me analogue dh404,24 and oleanolic acid25) augmented endogenous antioxidant systems and limited inflammation to prevent atherosclerosis development or progression in diabetes-aggravated atherosclerosis. One potent nanomolar Nrf2 activator of interest is CDDO-Me, a synthetic triterpenoid analogue of oleanolic acid, which is currently under-going clinical trials.26 Herein we sought to generate Antioxidant Response Activating Particles (ARAPas), encapsulating the Nrf2-actvator (CDDOMe), for the selective delivery of this Nrf2 activator to atherosclerotic plaque.
Experimental
Further detailed methods of all experiments can be found in the ESI.†General materials
CDDO-methyl (Sigma Aldrich, St Louis, MO. SMB00376–100MG), Synperonic-PE-P84 pluronic tri-block co-polymer (Sigma Aldrich, St. Louis, MO. 713538-1Kg), DMSO (Fisher Scientific, D128-1), DMEM (11885-084; Gibco, Grand Island, NY). DMSO (BP231; Thermo-Fisher Scientific, Waltham, MA), F-12 nutrient mix Ham's media (11765-054; Gibco), Glucose (50-99-7, Sigma-Aldrich), DMEM High glucose (4.5 g L−1) (11995-065, Gibco), DMEM low glucose (1 g L−1) (11885092, Gibco), RPMI 1640 media (11875135, Gibco), heat-inactivated fetal bovine serum (FBS) (16140071; Gibco), penicillin–streptomycin 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 U mL−1 (15140122, Gibco), Glutamine (200 mM, 25030081; Gibco), Paraformaldehyde (158127; Sigma-Aldrich). PBS (20–134; Apex Bioresearch Products, San Diego, CA). Trypsin-EDTA (0.05%) (25300054; Gibco).
000 U mL−1 (15140122, Gibco), Glutamine (200 mM, 25030081; Gibco), Paraformaldehyde (158127; Sigma-Aldrich). PBS (20–134; Apex Bioresearch Products, San Diego, CA). Trypsin-EDTA (0.05%) (25300054; Gibco).
Cell culture
All cells were cultured in an incubator at 37 °C with 5% CO2. RAW 264.7 macrophage cells (ATCC, TIB-71) and bone marrow derived macrophages (BMDMs) were cultured in DMEM, high glucose (4.5 g L−1) supplemented with 10% FBS, 1% penicillin–streptomycin. H1299 cells containing a GFP fragment retrovirally inserted into the second intron of the NQO1 gene (130207PL1G9) were a gift from Tigist Yibeltal and the Major Lab (UNC, LCCC), Uri Alon and the Kahn Protein Dynamics group.27,28 These cells were cultured in RPMI 1640 media supplemented with 10% FBS and 1% penicillin–streptomycin. CDDO-Me was stored in stock concentrations in DMSO at −20 °C and diluted directly in media for treatment. Equal volume DMSO was included for all controls. CDDOMe-ARAPas were stored in 1XPBS and diluted into media. Equal volumes of PBS and equal mass concentrations of polymer were included as a control.Synthesis of CDDOMe-ARAPas
CDDOMe-ARAPas were synthesized via flash nanoprecipitation (FNP) with a confined impinging jet mixer (CIJ) as previously described.29 Briefly, CDDO-Me and Synperonic PE-P84 were dissolved in tetrahydrofuran (THF, Fisher Scientific, T425-1) at a final total mass concentration (TMC) of 5 mg mL−1 (2.5 mg mL−1 CDDOMe, 2.5 mg mL−1 Synperonic PE-P84). PBS was used as the aqueous solvent. The two solvent streams were mixed together in the CIJ into 4 mL of PBS. This suspension was then dialyzed overnight against PBS (Spectra-por 25 mm × 16 mm 2 mL cm−1 12–14 kDa MWCO, 15 m tubing (Fisher Scientific 08-670-3BB). To generate fluorescent nanoparticles, DiD (Fisher Scientific, D7757, final concentration 0.25 mg mL−1) was added to the organic solvent prior to injecting the solvent streams through the CIJ.Nrf2 activation assay
H1299 cells were seeded at 20000 cells per well in a 96 well black glass-bottom plate (Greiner 96 well plates, 655891, VWR). Media was replaced with media containing treatments (1% DMSO, CDDO-Me (10–200 nM), CDDOMe-ARAPas (10–200 nM) and equivalent concentrations of polymer). Cells were then either imaged continuously over a 40 h period (intervals of 4 h), or were imaged once at 24 hours following incubation with treatment. Wells were imaged using Gen 5 software (BioTek Instruments, Winooski, VT) on a Cytation 5 plate reader (BioTek Instruments) (37 °C, 5% CO2) with a GFP filter cube (BioTek Instruments, part #: 1225101) and a Texas Red filter cube (BioTek Instruments: Part # 1225102). Exposure times were fixed for each well. Cells were counted automatically by the Gen 5 software by thresholding in the Texas Red channel.
Confocal microscopy
Fluorescence images were obtained by using an inverted laser scanning confocal microscope (Zeiss LSM 780; Zeiss, Oberkochen Germany) through a 63× oil immersion objective lens (numerical aperture 1.40, catalog# 420782-9900; Zeiss, Oberkochen, Germany) with a zoom of 2, pixel size: 0.0659 × 0.0659 μm2. Nuclear DAPI was excited by a 405 nm laser diode and images obtained through a detection wavelength of 410–585 nm with a conventional PMT detector. CD11b-Alexa Fluor 555 was excited by an Argon laser (514 nm) and images obtained through a detection wavelength of 524–656 nm with a conventional PMT detector. DiD-CDDOMe-ARAPas were excited by a helium–neon laser (633 nm) and images obtained through a detection wavelength of 638–755 nm with a conventional PMT detector. Pinhole size for all channels was set to 1 airy unit. Scanning mode was set to frame. 3D stacks were obtained with dz = 1.00 μm. Three dimensional rendering and related surfaces were obtained with IMARIS software v9.7.2 (Bitplane AG, Zürich, Switzerland). Orthogonal views and representative image (slice, z6/13) images were obtained using Fiji (Fiji Is Just ImageJ; NIH).Animals and diet
All animal handling and experimental procedures were approved by the Institutional Animal Care and Use Committee at the University of North Carolina – Chapel Hill (IACUC ID: 18-303). 4–6-week-old male ApoE−/− mice (B6.129P2-Apoetm1Unc, stock number: 002052), LDLr−/− (B6.129S7-Ldlrtm1Her/J, stock number: 002207) and C57Bl/6 mice (stock number: 000664) were purchased from Jackson laboratory. C57Bl/6 mice were fed standard chow. Mice were allowed ad libitum access to food and water throughout the study. After 1 week acclimation in the Division of Comparative Medicine (DCM) facility, ApoE−/− and LDLr−/− were placed on a western high-fat diet containing 40% fat, 17% protein, 43% carbohydrate by kcal and 0.15% cholesterol by weight (RD Western Diet, catalog number: D12079Bi). apoE−/− and LDLr−/− mice were fed with the high-fat diet over the course of 8–15 weeks. Mice underwent experimental procedures and were sacrificed when they were approximately either 13–15 weeks old or 20–22 weeks old.Statistical analysis
Numerical data are represented as means ± standard deviation. Statistical analyses were performed using an unpaired Student's t-test, one-way ANOVA, or factorial ANOVA, or ANCOVA followed by Tukey's post-hoc test, as appropriate with a p-value <0.05 considered statistically significant. Multivariate analysis was performed using MANOVA with subsequent univariate analysis and post hoc testing. Statistical analyses were performed either using OriginLab, Northampton, MA or SPSS, Armonk, NY.Results and discussion
Generation of CDDOMe-ARAPas
We utilized a novel nanoparticle synthesis technique, flash nanoprecipitation (FNP), to generate polymeric ARAPas encapsulating CDDOMe, a potent nanomolar activator of the Nrf2 transcription factor, as well the lipophilic dye, DiD.29 FNP allows for the encapsulation of hydrophobic drugs into the core of a protective polymer shell. It is a scalable, continuous synthesis technique which relies upon the rapid mixing of high-velocity streams of liquid, containing the polymers and therapies, in a specially engineered device (confined impinging jet mixer (CIJ)) (Fig. 1A). The rapidity of the mixing results in robust generation of homogenous nanoparticles (similar size and therapeutic loading). We chose this technique due to its capacity for high loading efficiency, use of bio-compatible polymers, its scalability, and translational applicability. Indeed, Feng and co-workers have shown that this technique can be scaled up directly from laboratory milligram scale to a 1 kg day−1 nanoparticle production scale, without changes in the size and homogeneity of nanoparticles.30 | ||
| Fig. 1 Synthesis and characterization of CDDOMe-ARAPas. (A) Schematic overview of the Flash Nanoprecipitation (FNP) method to generate CDDOMe-ARAPas using a confined impinging jet (CIJ) mixer. (B) Hydrodynamic diameter and polydispersity index (PDI) of CDDOMe-ARAPas with or without the lipophilic fluorescent dye, DiD (25 μg mL−1 final concentration). These were measured via an intensity distribution with Dynamic Light Scattering (DLS). (C) Change in hydrodynamic diameter of CDDOMe-ARAPas following 6 days of incubation at 4 °C, pH 7.4. DLS was used to show that the size was unchanged relative to freshly prepared CDDOMe-ARAPas. (D) Hydrodynamic diameter of CDDOMe-ARAPas determined via nanosight nanotracking analysis. The data for (A)–(D) represent the mean of 3–24 separate preparations of CDDOMe-ARAPas. (E) TEM of CDDOMe-ARAPas. Particles were adsorbed onto copper 400 mesh TEM grids were negatively stained with 2% uranyl acetate and imaged via TEM at 5000× magnification (scale bar = 5 μm) and at (F) 100000× magnification (scale bar = 200 nm). (G) Particle concentration as a function of temperature, pH and time. CDDOMe-ARAPas were diluted 5-fold either into 10 mM PBS, pH 7.4 or into 5 mM citrate buffer, pH 5 and maintained at either 4 °C or 37 °C for up to 48 h. Total particle concentration was assayed at both 24 and 48 h incubation via Zetaview Nanoparticle Tracking. A 3-way ANOVA to assess the effect of pH, time and temperature was conducted, followed by post-hoc Tukey Pairwise comparisons (**P < 0.01). | ||
We generated CDDOMe-ARAPas using the tri-block copolymer synperonic-PE-84 and measured their hydrodynamic diameter by dynamic light scattering (DLS). We determined that they had an average size of 234 ± 10 nm (Fig. 1B, Table 1). DLS also revealed a polydispersity index (PDI) of 0.07 ± 0.03, demonstrating that they were highly uniform, which is indicated by a PDI < 0.2.31 To make these nanoparticles fluorescent for visualization in cells and in vivo, we included the lipophilic dye, DiD, which resulted in nanoparticles of similar size and polydispersity (Fig. 1B, Table 1). Additionally, we found that the nanoparticle size remained unchanged over a period of up to 6 days, when the nanoparticles were stored at 4 °C in pH 7.4 10 mM PBS (Fig. 1C). As an orthogonal measurement, we also performed nanosight nanotracking analysis (NTA) and found a similar average diameter for CDDOMe-ARAPas of 228 ± 24 nm (Fig. 1D, Table 1). Moreover, we confirmed a spherical shape and uniform size via transmission electron microscopy (TEM) (Fig. 1E and F). Finally, we examined particle stability of CDDOMe-ARAPas, by using total particle concentration as an index, measured with the Zetaview nanoparticle tracking instrument (Fig. 1G). CDDOMe-ARAPas were incubated for up to 48 h at either 4 or 37 °C, and at pH 5 or pH 7.4. We analyzed the effect of temperature, time, and pH upon CDDOMe-ARAPas particle concentration with a factorial ANOVA. The 3-way ANOVA model was significant (F(8, 36) = 3.5, P = 0.004). A simple main effects analysis revealed that time (F(1, 36) = 5.3, P = 0.03) and temperature (F(1, 36) = 15.4, P < 0.0001) significantly affected particle concentration, whilst pH (F(1, 36) = 3.9, P = 0.06) was not significant. Amongst these variables, temperature had the largest effect size ωp2 = 0.24, with the effect size of pH and time being ωp2 = 0.06 and ωp2 = 0.09, respectively. None of the interactions of each of the variables was significant. A post-hoc pairwise comparison with Tukey correction found a significant difference between the particle concentration at 24 h and 4 °C vs. 48 h and 37 °C with particles at either pH 5 or pH 7 (Fig. 1G). Moreover, we found that although there is a drop in concentration, this does not relate to particle aggregation as DLS measurements continue to show homogenous populations of the same size (data not shown). To determine the loading efficiency and loading capacity, we performed dialysis to remove unincorporated CDDOMe, and then measured the remaining CDDOMe concentration by HPLC-UV-VIS analysis of the nanoparticles. Loading efficiency was determined by measuring the amount of CDDOMe encapsulated in freshly synthesized nanoparticles vs. those that had been dialyzed overnight. Loading capacity was determined by measuring the mass of CDDOMe present in a solution of solubilized nanoparticles and dividing this by the total maximum mass of CDDOMe and polymer present (i.e. assuming there was no loss of polymer in the nanoparticle generation). In this manner, the loading efficiency was found to be 95.6 ± 5.3%, with a calculated loading capacity of 32 ± 5%. FNP is highly useful due to the rapid formulation as well as the high cargo loading.29 Our studies support this capacity for high loading efficiency resulting in high loading capacity. At the time of writing there appears to be only one other literature report of a polymeric formulation of CDDO-Me with PLGA, where the drug loading capacity was reported to be 2.9 ± 0.2%, after being generated by the solvent displacement method.32
| Nanoparticle | Hydrodynamic diameter (d, nm) | Polydispersity index (PDI) ± 1 S.D. | % Loading efficiency | Loading capacity (wt%) |
|---|---|---|---|---|
| a Intensity distribution, dynamic light scattering (DLS). b Nanosight Nanotracking Analysis (NTA). Values represent the means ± 1 standard deviation of 3–12 independent measurements. | ||||
| CDDOMe-ARAPas | 234 ± 10a | 0.07 ± 0.03 | 95.6 ± 5.3 | 32 ± 5 |
| 228 ± 28b | ||||
| DiD-CDDOMe-ARAPas | 236 ± 10a | 0.08 ± 0.04 | ||
Time-dependent internalization of CDDOMe-ARAPas by RAW macrophages
Macrophages comprise the bulk of atherosclerotic plaque. The CDDOMe-ARAPas were designed to be close to 200 nm as the literature shows optimal macrophage internalization of nanoparticles around this size.15,16 Previous literature has also supported that size is an important determinant for accumulation in atherosclerotic lesions, with 200nm-sized liposomes providing the best accumulation.16,33 We utilized a murine macrophage cell line, RAW 264.7 macrophages, to examine the association and internalization of CDDOMe-ARAPas with macrophages. CDDOMe-ARAPas were loaded with DiD lipophilic dye and then incubated with RAW 264.7 macrophages over a period of up to 18 hours. Firstly, their association with macrophage cells was followed by kinetic live cell fluorescence imaging (Fig. 2A and B), plotting the fluorescence intensity per cell against time. In previous literature, human macrophages internalized 200 nm human serum albumin nanoparticles over 4–6 h, and then plateaued from 10 h, with measurements extending out to 24 h.34 Similarly, in a study by Yu and colleagues35 using THP-1 macrophages and studying the internalization of 30, 40 and 100 nm polymer-coated iron oxide nanoparticles over 24 h, they found that nanoparticle internalization plateaued following 10 h, and was described via an exponential function. We also fitted our results to an exponential growth function. The regression was significant (F(3, 9) = 11.2, P = 0.004 R2 = 0.56), indicating that with time, RFU per cell due to CDDOMe-ARAPas association increases. Our results showed that the nanoparticle internalization remained in a growth phase and had not plateaued between 12 and 18 h. Differences in our study that may explain this discrepancy with published literature include that the kinetic assay presented quantifies both nanoparticle external association and internalization, which may not follow the same kinetics as simply internalization. Additionally, we did not follow internalization up to 24 h, where it may have plateaued from the 18 h time point. To confirm internalization, we also visualized the CDDOMe-ARAPas, following an 18 h incubation in RAW macrophages, using confocal microscopy and co-staining for an external cell surface marker, cd11b (Fig. 2C). Three-dimensional surface rendering of the confocal fluorescence images indicated that the DiD-CDDOMe-ARAPas were inside the murine macrophages (Fig. 2D). To quantify internalization by murine macrophages, we also performed a temperature dependent assay whereby DiD-CDDOMe-ARAPas were incubated with RAW macrophages over 18 h at either 4 or 37 °C (Fig. 2E). At 4 °C, energy-dependent internalization processes are inhibited, therefore the signal derived from RAW macrophages incubated at this temperature is CDDOMe-ARAPas that are associated with the external cell surface. We analyzed the effect of temperature upon μg DiD/μg cell protein using a t-test. This revealed a significant difference between 4 °C vs. 37 °C (F(1, 4) = 39.7, P = 0.003), with an effect size of ω2 = 0.9, indicating that the majority of signal is due to internalized CDDOMe-ARAPas rather than externally associated nanoparticle. Overall, our studies confirm that murine macrophages significantly internalize CDDOMe-ARAPas in a time-dependent manner.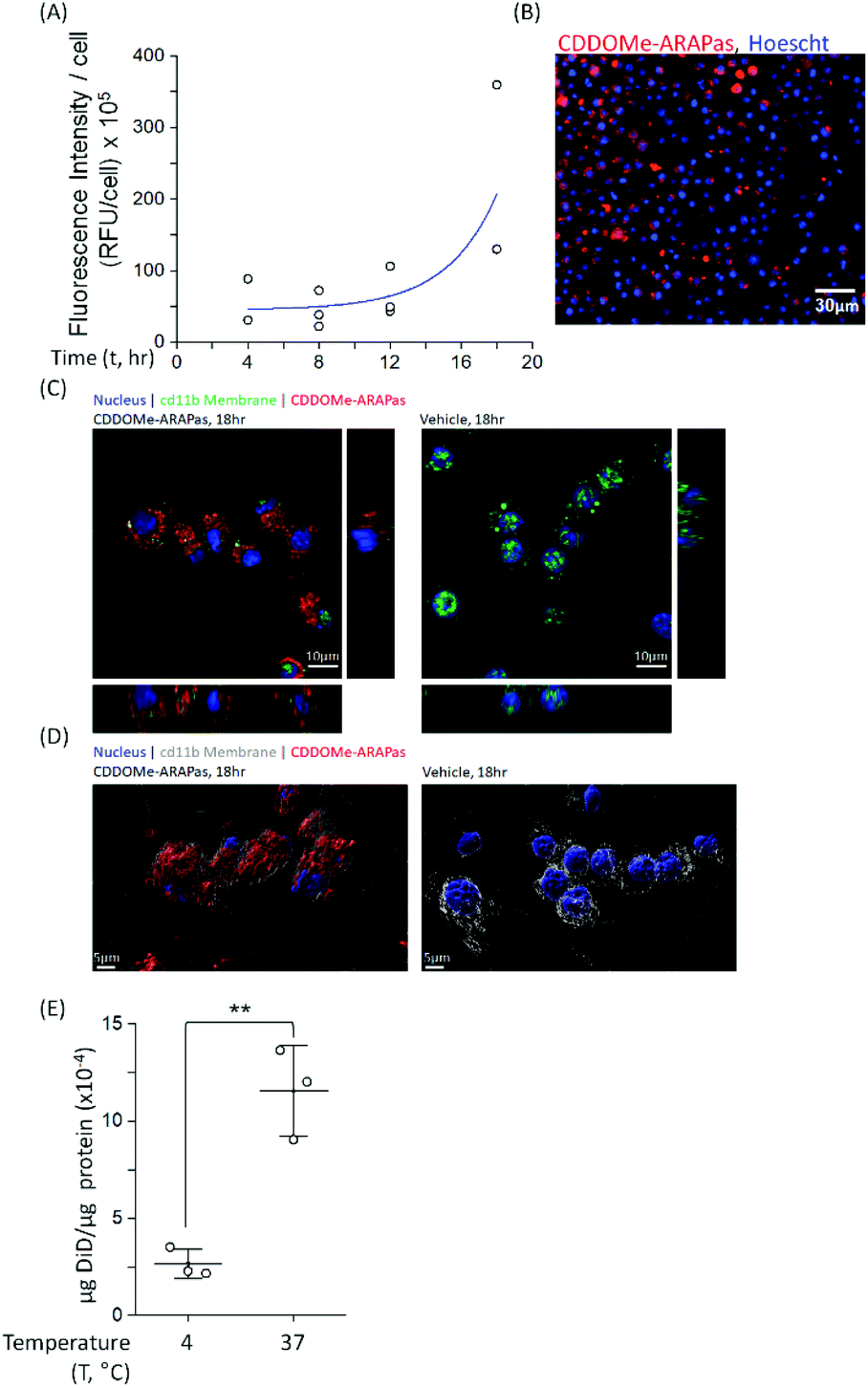 | ||
| Fig. 2 Internalization of CDDOMe-ARAPas by murine macrophages. (A) Kinetic association of CDDOMe-ARAPas with murine macrophages. RAW macrophages stained with Hoechst dye (10 μg mL−1, 30 minutes) were treated with fluorescent DiD-CDDOMe-ARAPas (0.1 mg mL−1 final, 20% PBS) over the course of 18 h (37 °C, 5% CO2). Fluorescent images of RAW macrophages were taken at 4 hours intervals and thresholded fluorescence area was quantified per cell, an average of 800 cells were counted per well. Data represents N = 3 independent experiments in 6–12-tuplicate. (B) A representative image of macrophage-associated fluorescent DiD-CDDOMe-ARAPas at 18 h. Scale bar is 30 μm. (C) Z-Projection and orthogonal slices of internalized DiD-CDDO-Me-ARAPas in cd11b stained macrophages. RAW macrophages were incubated in the presence or absence of fluorescent DiD-CDDOMe-ARAPas (0.1 mg mL−1 final, 20% PBS) for 18 h (37 °C, 5% CO2), and then fixed and counter-stained with cd11b antibody and DAPI. Scale bar is 10 μm. (D) 3D rendered surface image of cd11b membrane, and internalized DiD-CDDO-Me-ARAPas in murine macrophages. Scale bar is 5 μm. (E) Temperature-dependent internalization of DiD-CDDOMe-ARAPas at 18 h by RAW macrophages. Macrophages were incubated with DiD-CDDOMe-ARAPas as described in (C), and then washed of un-associated ARAPas and the cell lysate assayed for fluorescence intensity and protein content. Data represents means ± 1 S.D. (N = 3 independent experiments in triplicate). **, p < 0.01. | ||
Activation of Nrf2 transcription factor by CDDOMe-ARAPas
Various methods exist to determine the release kinetic profile of hydrophobic drugs from nanoparticles,36 however irrespective of the method, the underlying assumption is that the experiment satisfies sink conditions, where the volume of medium must be at least three times that required to form a saturated solution of the drug. This condition is commonly not satisfied,37,38 as for highly insoluble drugs, such as CDDOMe, this can be quite challenging due to large volumes of release medium required and subsequent difficulties analyzing low concentrations of drug. Abouelmagd and co-workers illustrated the issues with two main methods (dialysis and centrifugation) leading to different conclusions of paclitaxel release from nanoparticles.38 A problem arising from these methods is a mismatch of expected bioactivity either in vitro or in vivo. Instead, in order to confirm that drug was released by the CDDOMe-ARAPas, we chose to utilize an in vitro functional assay, which reflects Nrf2 activation.27,28 In this assay, H1299 cells containing a GFP fragment retrovirally inserted into the second intron of the NQO1 gene, a canonical Nrf2-regulated gene,39 are used to determine Nrf2 activation. Upon Nrf2 activation, GFP fluorescence is observed, which can be quantified as thresholded fluorescence intensity and normalized to cell number due to a Cherry Red nuclear fluorescence. The cells were incubated for 24 hours with increasing doses of either CDDOMe or CDDOMe-ARAPas and then GFP fluorescence intensity per cell measured, of which representative images are shown in Fig. 3A and quantified in Fig. 3B. To analyze the effect of treatment (CDDOMe-ARAPas and CDDOMe) on Nrf2 activation we conducted an ANCOVA to control for the effect of concentration. The results show that the model fit was significant (F(2, 76) = 27, P < 0.0001). Simple main effects analysis showed that dose had a statistically significant effect upon fluorescence intensity per cell ((F(1, 76) = 47.5, P < 0.0001)) with an effect size of ωp2 = 0.4, whilst treatment with either CDDOMe-ARAPas vs. CDDOMe alone also had a statistically significant effect upon fluorescence intensity per cell ((F(1, 76) = 12.5, P = 0.001)), with an effect size of ωp2 = 0.1. Thus, we show that that CDDO-Me, a well-known Nrf2 activator, activates the transcription of the canonical down-stream target of Nrf2, NQO1, in a dose-dependent manner (10–400 nM). We additionally show that CDDOMe-ARAPas activate Nrf2, albeit in a significantly different, lesser manner compared to CDDOMe, suggestive of delayed release of CDDOMe from the ARAPas. In previous literature, D'Addio and colleagues indeed reported release of lipophilic compounds from polymeric nanoparticles generated by flash nanoprecipitation,37 thus our results are consistent with the reported behavior of polymeric nanoparticles generated in a similar manner.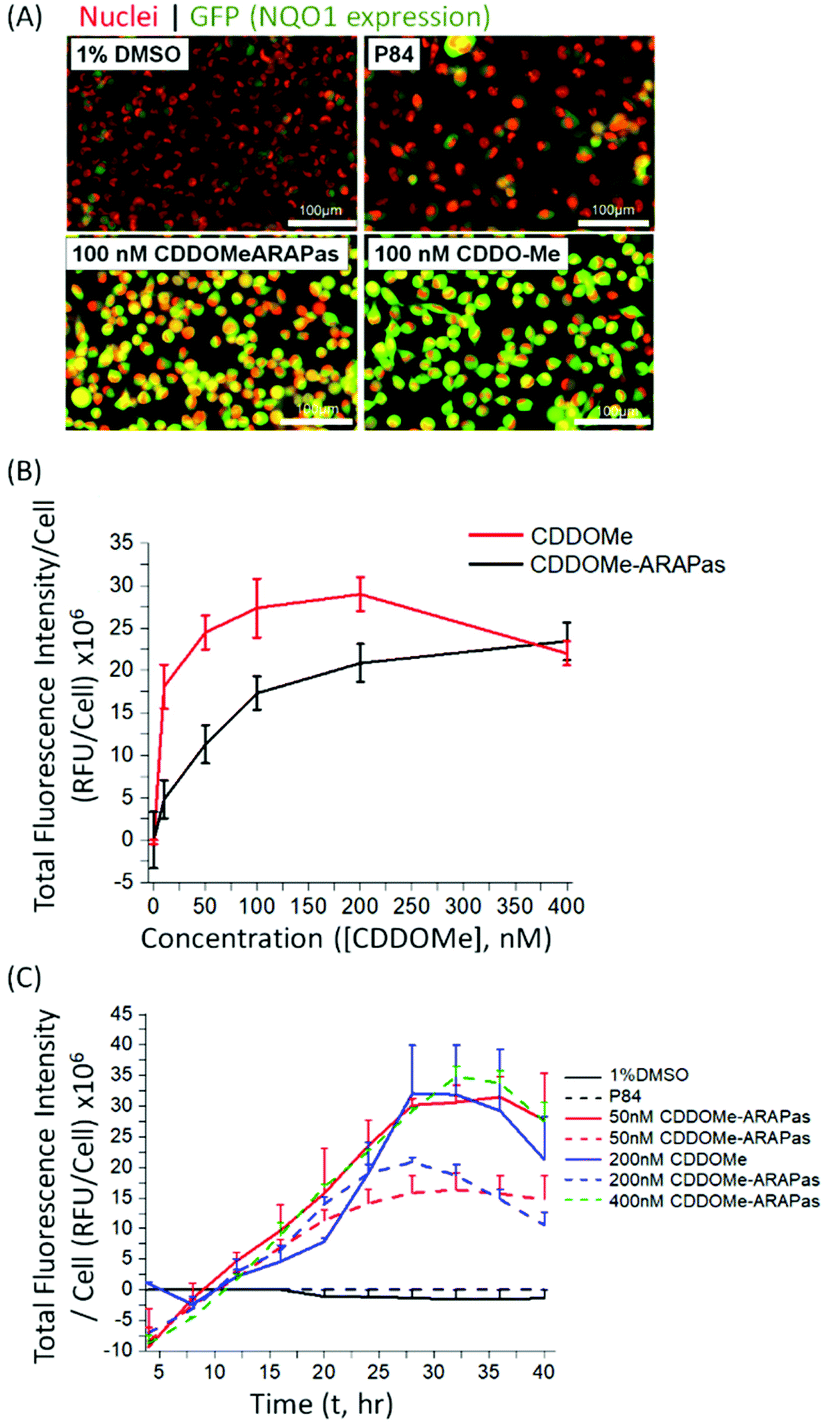 | ||
| Fig. 3 Activation of Nrf2 by CDDOMe-ARAPas. H1299 cells were treated with either 1% DMSO, P84 polymer (at equivalent concentrations as present in most concentrated NP preparations), CDDOMe (10–400 nM), or CDDOMe-ARAPas (10–400 nM) for 24–40 hours (37 °C, 5% CO2). (A) Representative images of live cell GFP-NQO1 fluorescence as a proxy of Nrf2 activation in H1299 cells after 24 hours as collected by the Cytation 5 plate reader. Shown here is treatment with 1% DMSO vehicle, or P84 polymer at concentrations equivalent to that found in the most concentrated nanoparticle preparations, 100 nM CDDOMe-ARAPas and 100 nM CDDOMe. Scale bar is 100 μm. (B) Dose-dependent activation of NQO1 transcription by CDDOMe (red solid line) and CDDOMe-ARAPas (black solid line). GFP fluorescence intensity per cell was quantified with an average of 200 cells counted per measurement. Data represents the means ± 1 S.D. of N = 4–5 independent biological experiments, conducted in sextuplicate. (C) Time-dependent activation of NQO1 transcription by CDDOMe and CDDOMe-ARAPas. H1299 cells were treated with either 1% DMSO, P84 polymer (at equivalent concentrations as present in most concentrated NP preparations), CDDOMe (solid lines, 50–200 nM), or CDDOMe-ARAPas (dashed lines, 50–400 nM) for up to 40 hours. GFP fluorescence intensity per cell was quantified with an average of 200 cells counted per measurement. Data represents the means ± SEM, N = 2–4 independent biological experiments. | ||
We then examined the activation of Nrf2 by CDDO-Me and CDDOMe-ARAPas over the course of 40 hours, with kinetic live cell imaging, at varying concentrations (Fig. 3C). Herein we found that GFP expression is sustained between 24–40 h. We additionally recapitulated the trend observed in Fig. 3B, wherein higher concentrations of CDDOMe-ARAPas are required (up to 400 nM) to sustain GFP expression at similar levels to un-encapsulated CDDO-Me. Taken together, this data indicates firstly, that CDDOMe is released in a delayed manner by CDDOMe-ARAPas and secondly, is capable of activating NRf2 in vitro.
Toxicity of CDDOMe-ARAPas
We also assayed the effect of both CDDOMe and CDDOMe-ARAPas upon macrophage viability with a MTT assay (Fig. 4A) and an Annexin V/Apoptosis flow cytometry assay (Fig. 4B and C). Naïve macrophages were incubated with increasing concentrations of either CDDOMe or CDDOMe-ARAPas at 0–2000 nM over 24 hours and in the case of MTT, then incubated with MTT reagent for a further 4 hours. To determine the EC50 we fitted the MTT data with a growth/sigmoidal curve with a dose response function where the upper asymptote was fixed at 100%. The calculated EC50 were 274 (95% CI 220–328) and 378 (95% CI 348–408), for CDDOMe and CDDOMe-ARAPas respectively (Fig. 4A). The lower EC50 for CDDOMe compared to CDDOMe-ARAPas supports a slower release of CDDOMe from the ARAPas, consistent with data in Fig. 3. Analysis of the flow cytometry AnnV/apoptosis data (Fig. 4B and C) determined that the total viable cell number following either CDDOMe or CDDOMe-ARAPas treatment was reduced in a dose dependent manner.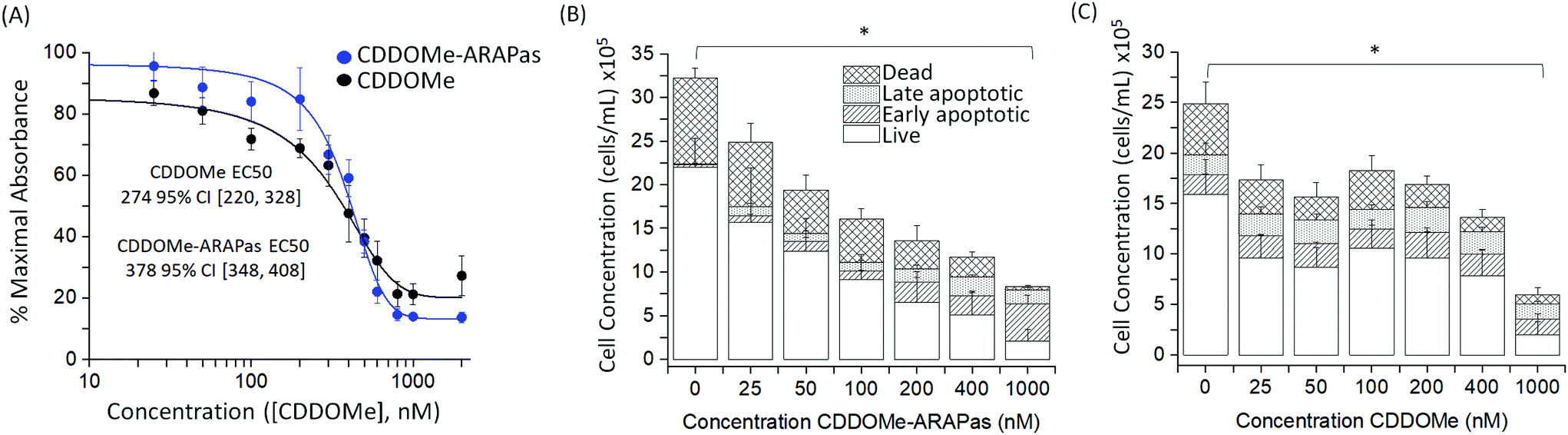 | ||
| Fig. 4 Toxicity of CDDOMe and CDDOMe-ARAPas. (A) MTT of RAW 264.7 macrophages treated with CDDOMe or CDDOMe-ARAPas. Murine macrophages were incubated with CDDOMe or CDDOMe-ARAPas in a 0–2000 nM dose range. Data represents N = 3–5 independent experiments ± SEM with 8 replicates per experiment. Data were fitted with a growth/sigmoidal curve with a dose response function in OriginPro 2018b software where the upper asymptote was fixed at 100 and the lower asymptote unfixed. The R square (COD) value for the CDDOMe-ARAPas and CDDOMe fitted curve were 0.98 and 0.99 respectively. The EC50 was derived from the fitted curves and were 274 95% CI [220, 328] and 378 95% CI [348, 408] for CDDOMe and CDDOMe-ARAPas respectively. (B) and (C) Quantification of viable, early apoptotic, late apoptotic and dead RAW 264.7 macrophages following treatment with CDDOMe or CDDOMe-ARAPas. Macrophages were incubated for 24 hours with 25–1000 nM (B) CDDOMe-ARAPas or (C) CDDOMe and then cells were analyzed by the Muse Annexin V and Dead Cell Assay Kit. *p < 0.05 compared to control (0 μM CDDOMe-(ARAPas)). Data presented as means ± SEM (N = 3–5 independent experiments in triplicate). Data were analyzed using multivariate analysis of variance (MANOVA) to compare the effect of treatment with either CDDOMe or CDDOMe-ARAPas as well as concentration on total live cell population and then total dead cell population (where early and late apoptotic and dead cell populations were grouped together). Based on this analysis, we found a significant difference due to concentration, Wilk's Lambda λ = 0.302, (F(12,62) = 4.24, P < 0.001), with a large effect size estimate ωMult2 = 0.637. No difference was found due to treatment between CDDOMe or CDDOMe-ARAPas. A separate univariate analysis (two-way ANOVA) revealed that the significant effect was upon the live cell population (including both CDDOMe and ARAPas) (F(6,32) = 5.72, P < 0.001), but not the dead cell population. A one-way ANOVA was conducted to examine the effect of concentration of either CDDOMe or CDDOMe-ARAPas on total live cell population. The one-way ANOVA for CDDOMe was significant (F(6, 14) = 3.21, P = 0.034) and for CDDOMe-ARAPas (F(6, 18) = 3, P = 0.033). Tukey post-hoc tests found a significant difference between the live cell population at 0 and 1000 nM treatment in both CDDOMe (P = 0.04952) and CDDOMe-ARAPas (P = 0.02475). | ||
We used multivariate ANOVA (MANOVA) to compare the effect of treatment (CDDOMe or CDDOMe-ARAPas) and concentration on total live, and total dead cell populations (where early and late apoptotic and dead cell populations were grouped together). This revealed a significant difference due to concentration, Wilk's Lambda λ = 0.302, (F(12, 62) = 4.24, P < 0.001), with a large effect size estimate ωMult2 = 0.64 on the live cell population. No difference was found when comparing the effect due to treatment with either CDDOMe or CDDOMe-ARAPas. A separate univariate analysis (2-way ANOVA) revealed that the significant effect of dose was upon the live cell population (F(6, 32) = 5.72, P < 0.001), but not the dead cell population (F(6, 32) = 0.993, P = 0.45). Overall, this indicates that treatment with CDDOMe or CDDOMe-ARAPas significantly reduced the total viable cells without affecting total dead cells (Fig. 4B and C). Taken together, these results suggest that lower concentrations of CDDOMe and CDDOMe-ARAPas reduced viable cell number without significant toxicity, which suggests inhibition of proliferation. Previous work by Khoo and co-workers with a related compound, CDDO-imidazole, found that 100 nM did not significantly affect viability in HepG2 cells, although 1000 nM led to a loss of ∼94% of absorbance in the MTT assay.40 They also additionally found that 100 nM CDDO-imidazole did not result in toxicity in RAW 264.7 cells. This is in line with our results, albeit in the murine macrophage cell line (RAW 264.7 cells), and an analog of this chemical. In support of the suggestion that CDDOMe may affect macrophage proliferation, in another study, Probst and colleagues41 found that an analog of CDDO-Me inhibited growth in 8 different cancer cell lines with an IC50 ranging from 159 to 363 nM.
CDDOMe-ARAPas inhibit inflammation in classically activated macrophages
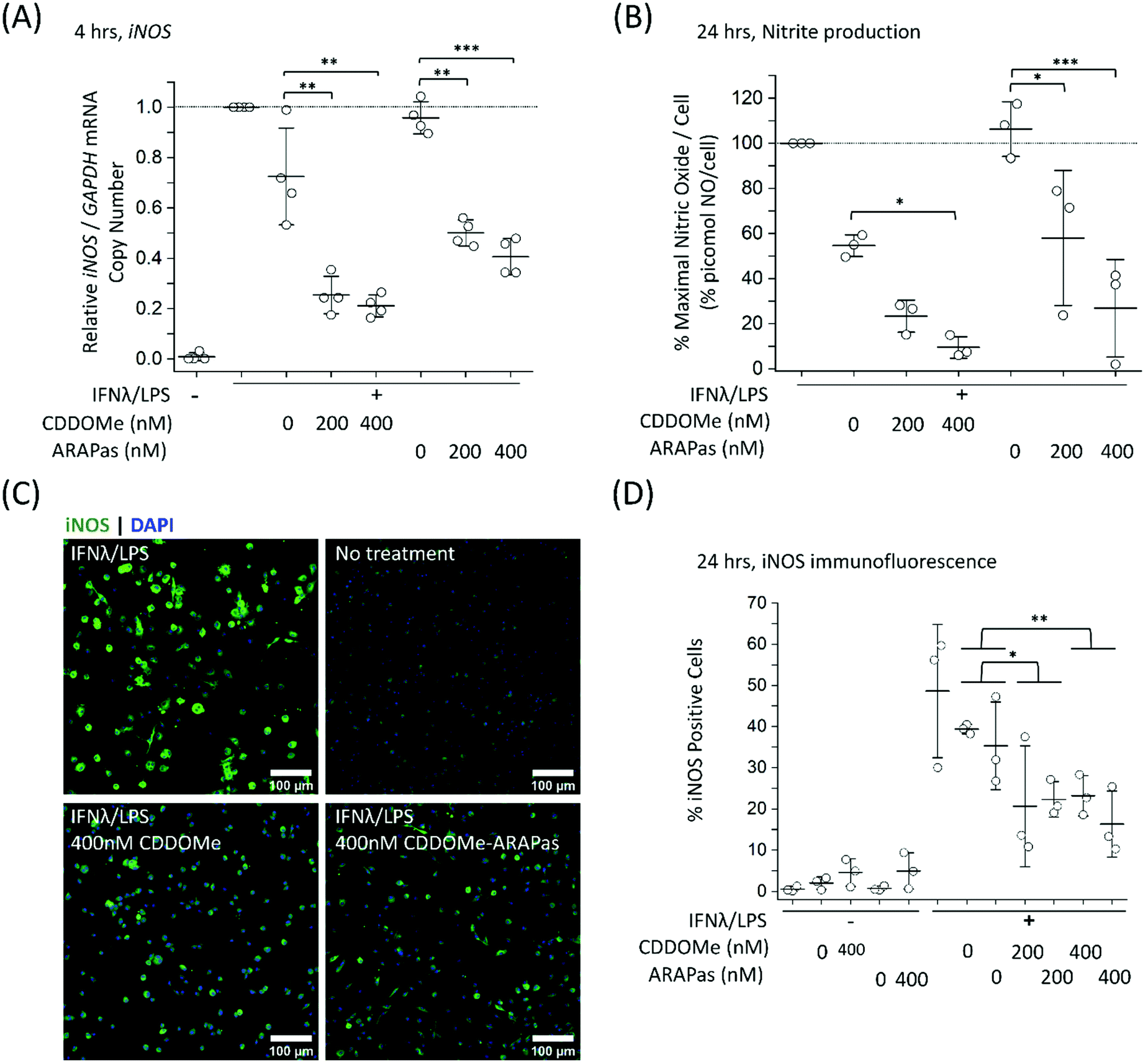
Previous studies by Kobayashi and co-workers reported that Nrf2 negatively regulates iNOS expression as well as that of pro-inflammatory cytokine genes, including IL6 and IL1b.20 This results in inhibition of LPS-induced expression of these genes in murine BMDMs. Moreover, synthetic triterpenoids such as CDDOMe were initially identified as potent inhibitors of iNOS protein expression in murine macrophages.42 Thus, we sought to investigate if CDDOMe and, more importantly, the corresponding CDDOMe-ARAPas could negatively regulate iNOS in both RAW macrophages (Fig. 5) and primary murine BMDMs (Fig. 6). We also examined whether treatment could inhibit transcription of IL1b (Fig. 7). Firstly, we examined iNOS mRNA levels with digital droplet PCR (ddPCR) in classically stimulated (IFNλ/LPS) murine RAW 264.7 macrophages in the presence or absence of CDDOMe or CDDOMe-ARAPas after 4 h of incubation (Fig. 5A). iNOS mRNA levels were normalized to GAPDH mRNA copy number and are represented as relative to the iNOS/GAPDH mRNA copy number which was produced solely due to IFNλ/LPS stimulation. A factorial ANOVA was conducted to determine the effect of either treatment with CDDOMe-ARAPas or CDDOMe alone and dose on relative iNOS/GAPDH mRNA levels in RAW macrophages. The model was significant (F(5, 14) = 43.13, P < 0.0001), and treatment (F(1, 14) = 102.8, P < 0.0001) and dose (F(2, 14) = 49.1, P < 0.0001) were both found to be significant. Thus, the effect size of treatment was ωp2 = 0.8, and for dose ωp2 = 0.8. Moreover, the interaction of dose and treatment with ARAPas vs. CDDOMe alone was significant (F(2, 14) = 9.67, P = 0.002). Additionally, post hoc Tukey tests found that there were significant decreases in relative iNOS/GAPDH mRNA levels between vehicle and both 200 and 400 nM of either CDDOMe alone or CDDOMe-ARAPas (Fig. 5A). Secondly, we examined the production of nitric oxide by measuring nitrite in the media, as an index of iNOS protein activity (Fig. 5B). Amount of NO was normalized to total cell count (picomol NO per cell), and then expressed as a percentage of maximal picomol NO per cell produced solely due to IFNλ/LPS stimulation. We conducted a factorial ANOVA analyzing the effects of treatment with either CDDOMe-ARAPas or CDDOMe alone as well as dose upon % maximal picomol of NO per cell. Overall, this model was significant (F(5, 15) = 8.04, P = 0.0007). Analysis of the main effects showed that there was no statistically significant effect comparing treatment between CDDOMe vs. CDDOMe-ARAPas (F(1, 15) = 0.005, P = 0.95), however dose did have a statistically significant effect upon % maximal picomol of NO per cell (F(2, 15) = 18.63, P < 0.0001) with an effect size of ωp2 = 0.6. Moreover, a post-hoc Tukey test found a significant decrease in % maximal picomol of NO per cell between the vehicle and 400 nM for CDDOMe-ARAPas and CDDOMe (Fig. 5B). Finally, we quantified iNOS protein expression via immunofluorescence (Fig. 5C and D). Classically stimulated murine RAW 264.7 macrophages were fixed, permeabilized and stained for iNOS protein expression, which was quantified as percentage (%) Positive Cells. Images were thresholded, and cells that exhibited fluorescence above this threshold were counted as positive, and expressed as a percentage of the total cell number (Fig. 5D). We conducted a 2-way ANOVA to analyze the effect of treatment (with either CDDOMe-ARAPas or CDDOMe) or dose on % Positive Cells. The 2-way ANOVA model was significant (F(5, 12) = 14.62, P < 0.0001). Simple main effects analysis showed that both treatment (CDDOMe vs. CDDOMe-ARAPas) (F(1, 12) = 40.51, P < 0.0001) and dose had a statistically significant effect upon % Positive Cells (F(2, 12) = 9.47, P = 0.003) with an effect size of ωp2 = 0.7 and ωp2 = 0.5, respectively. Additionally, the interaction between the two independent variables was significant (F(2, 12) = 5.59, P = 0.02) with an effect size of ωp2 = 0.3. Overall, a post-hoc Tukey test found a significant decrease on % Positive Cells between 0 nM and 400 nM (P = 0.002). Thus, overall our data support that CDDOMe and CDDOMe-ARAPas inhibit iNOS protein expression. This is completely in line with previous results reported by Khoo and co-workers using an analog of CDDO-Me, CDDO-Imidazole.40 These authors reported that co-treatment with 100 nM CDDO-Im and IFNλ/LPS in RAW 264.7 macrophages significantly inhibited the induction of iNOS mRNA expression at 6 and 16 h.40 Moreover, it significantly decreased iNOS protein expression following 20hr co-incubation with IFNλ/LPS.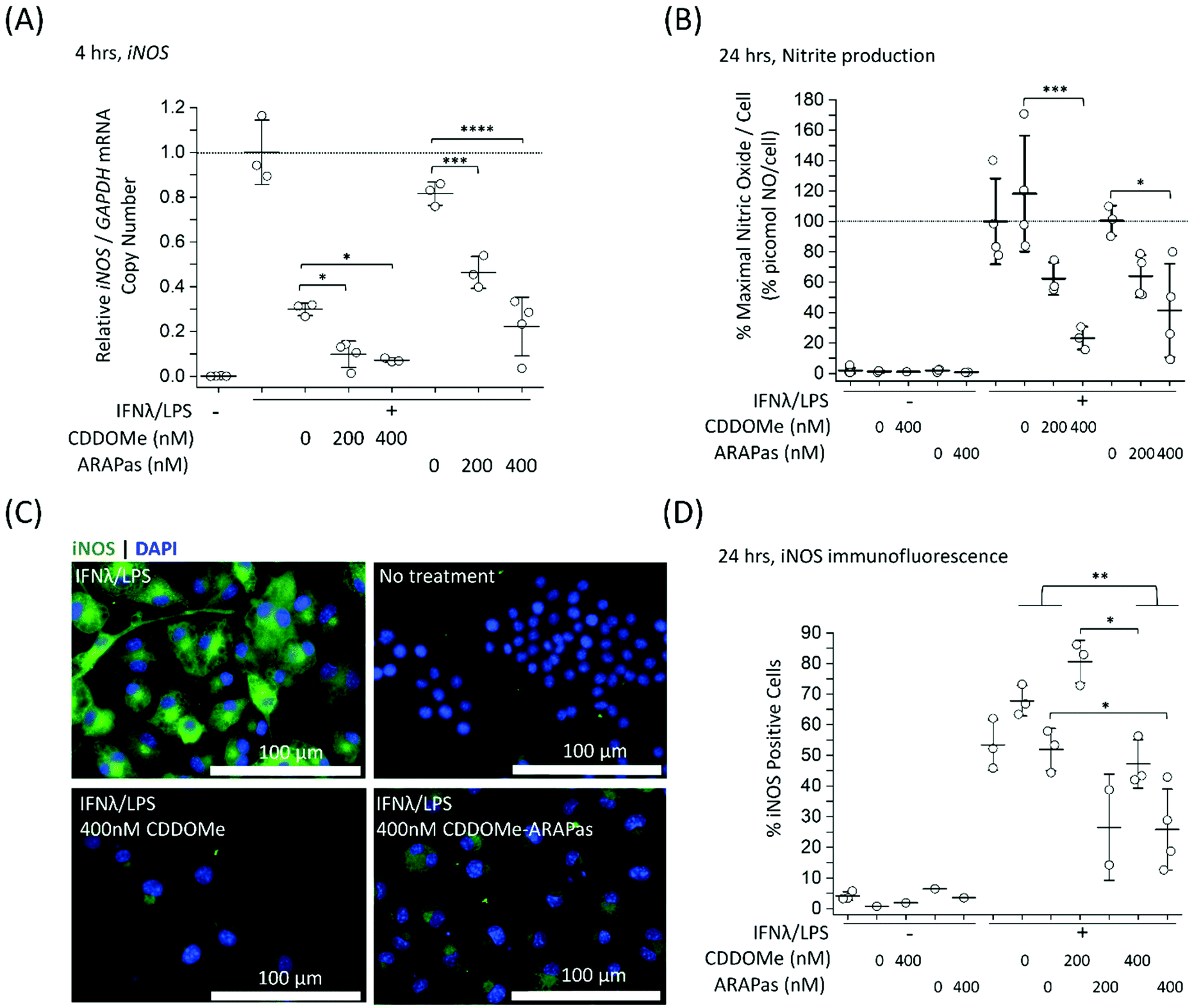 | ||
| Fig. 5 Inhibition of iNOS transcription, iNOS protein activity and expression in classically stimulated RAW 264.7 macrophages. RAW 264.7 macrophages were classically stimulated with IFNλ (10 ng mL−1, 7 h) followed by treatment with LPS (100 ng mL−1, 4–24 h) in the presence or absence of treatments and their respective vehicles. The media was then collected and cells were either washed and scraped into sterile HBSS and pelleted for RNA extraction, or fixed (2% PFA), permeabilized (0.3% Triton X-100 in 10 mM PBS) and then stained with anti-iNOS antibody followed by an appropriate secondary antibody. Secondary antibody alone and no antibody controls were included for all experiments. (A) CDDOMe and CDDOMe-ARAPas inhibit iNOS mRNA expression after 4 h incubation. Data represents the mean of N = 3–4 independent biological replicates, ±1 S.D. A factorial ANOVA was conducted to determine the effect of either CDDOMe or CDDOMe-ARAPas treatment and dose upon relative iNOS mRNA levels normalized to GAPDH copy number (*P < 0.05, ***P < 0.001, ****P < 0.0001). (B) CDDOMe and CDDOMe-ARAPas inhibit nitrite production after 24 h incubation. Nitrite in the media was reduced with acidic iodide reducing agent, and the resulting NO produced was detected with a Sievers NO analyzer. Cells were counted via DAPI staining, and NO normalized to cell count. Picomol NO/Cell is expressed as a percentage of maximal picomol/NO produced due to IFNλ/LPS stimulation. Data represents the mean of 3–4 independent biological replicates, ±1 S.D. A factorial ANOVA was conducted to determine the effect of either CDDOMe or CDDOMe-ARAPas treatment and dose upon % maximal picomol NO per cell (*P < 0.05, ***P < 0.001). (C) Representative images of immunofluorescence of iNOS-stained murine RAW macrophages after 24 h incubation. Fixed and permeabilized cells were stained with an anti-iNOS antibody followed by a secondary antibody conjugated to Alexa Fluor 488 dye, and then counter-stained with DAPI for nuclear detection. Scale bar is 100 μm. (D) CDDOMe and CDDOMe-ARAPas inhibit iNOS protein expression in classically stimulated macrophages after 24 h incubation. Fluorescence intensity in the 488 channel was thresholded and then positive cells that expressed fluorescence above this threshold were taken as a percentage of total cell count. Data represents the mean of N = 1–3 biological replicates, with 3 biological replicates for all IFNλ/LPS treated conditions, conducted in sextuplicate, ±1 S.D. A 2-way ANOVA was conducted to determine the effect of either CDDOMe or CDDOMe-ARAPas treatment and dose upon % Positive Cells (*P < 0.05, **P < 0.01). | ||
 | ||
| Fig. 6 Inhibition of iNOS transcription, iNOS protein activity and expression in classically stimulated murine bone marrow derived macrophages (BMDMs). BMDMs were classically stimulated with IFNλ (10 ng mL−1, 7 h) followed by treatment with LPS (100 ng mL−1, 4–24 h) in the presence or absence of treatments and their respective vehicles. The media was then collected and cells were either washed and scraped into sterile HBSS and pelleted for RNA extraction, or fixed (2% PFA), permeabilized (0.3% Triton X-100 in 10 mM PBS), and then stained with anti-iNOS antibody followed by an appropriate secondary antibody. Secondary antibody alone and no antibody controls were included for all experiments. (A) CDDOMe and CDDOMe-ARAPas inhibit iNOS mRNA expression after 4 h incubation. Data represents the mean of N = 4 independent biological replicates, ±1 S.D. A factorial ANOVA was conducted to determine the effect of either CDDOMe or CDDOMe-ARAPas treatment and dose upon relative iNOS mRNA expression normalized to GAPDH copy number (**P < 0.01, ***P < 0.001). (B) CDDOMe and CDDOMe-ARAPas inhibit nitrite production after 24 h incubation. Nitrite in the media was reduced with acidic iodide reducing agent, and the resulting NO produced was detected with a Sievers NO analyzer. Cells were counted via DAPI staining, and NO normalized to cell count. Data represents the mean of 3 independent biological replicates, ±1 S.D. A factorial ANOVA was conducted to determine the effect of either CDDOMe or CDDOMe-ARAPas treatment and dose upon % maximal picomol NO per cell (*P < 0.05, ***P < 0.001). (C) Representative images of immunofluorescence of iNOS-stained BMDMs after 24 h stimulation. Fixed and permeabilized cells were stained with an anti-iNOS antibody followed by a secondary antibody conjugated to Alexa Fluor 488 dye, and then counter-stained with DAPI for nuclear detection. Scale bar is 100 μm. (D) CDDOMe and CDDOMe-ARAPas inhibit iNOS protein expression in classically stimulated BMDMs after 24 h incubation. Fluorescence intensity in the 488 channel was thresholded and then positive cells that expressed fluorescence above this threshold were taken as a percentage of total cell count. Data represents the mean of N = 3 biological replicates, conducted in 6-plicate, ±1 S.D. A 2-way ANOVA was conducted to determine the effect of either CDDOMe or CDDOMe-ARAPas treatment and dose upon % Positive Cells (*P < 0.05, **P < 0.01). | ||
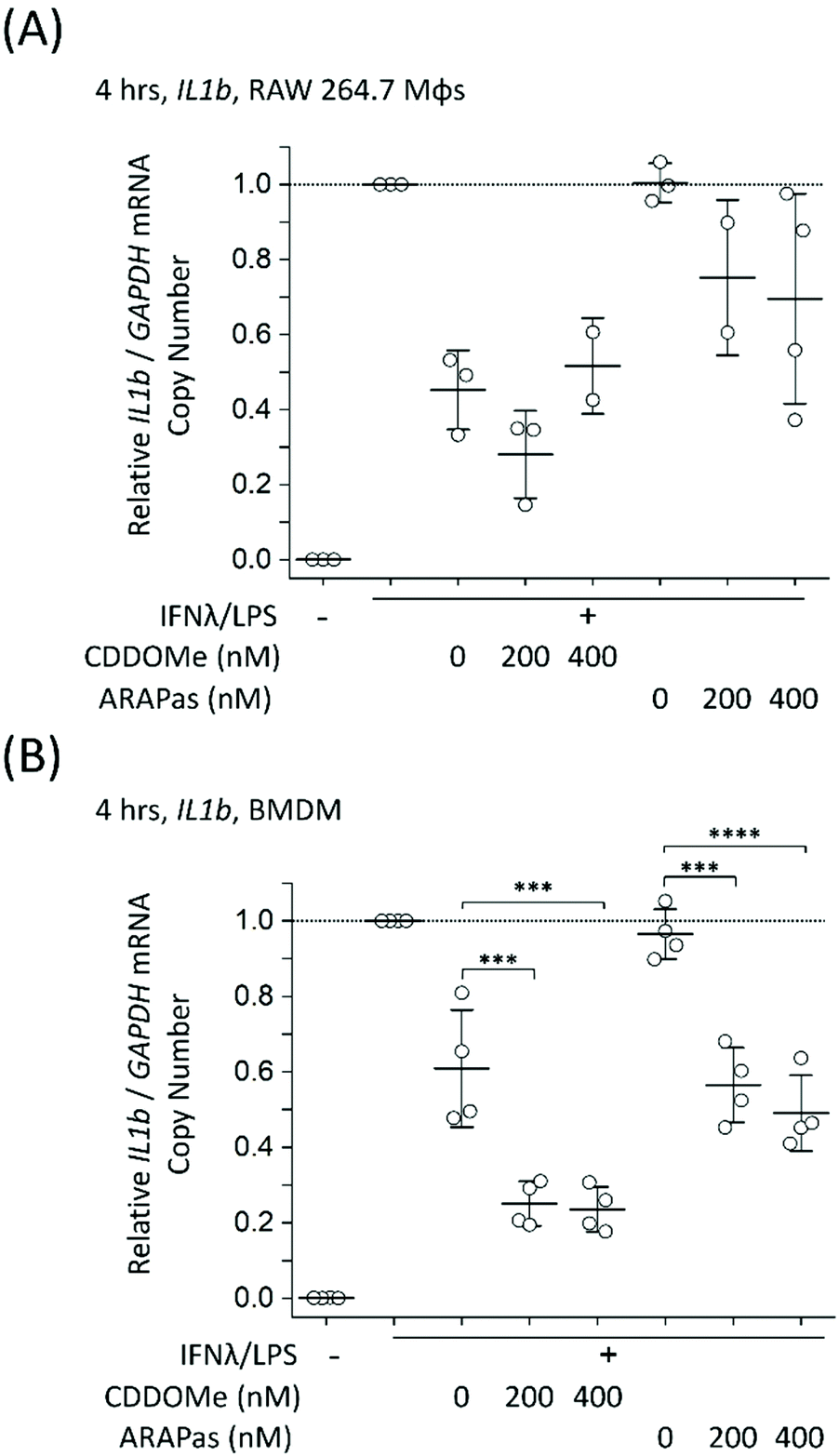 | ||
| Fig. 7 CDDOMe and CDDOMe-ARAPas treatment and IL1b mRNA trasncription in classically stimulated (A) RAW 264.7 macrophages and (B) murine bone marrow derived macrophages (BMDMs). Cells were classically stimulated with IFNλ (10 ng mL−1, 7 h) followed by treatment with LPS (100 ng mL−1, 4 h) in the presence or absence of treatments and their respective vehicles. The media was then collected and cells were pelleted for RNA extraction. Data represents the mean of N = 2–4 independent biological replicates, ±1 S.D. A factorial ANOVA was conducted to determine the effect of either CDDOMe or CDDOMe-ARAPas treatment and dose upon relative iNOS/GAPDH mRNA copy number (***P < 0.001, ****P < 0.0001). | ||
Although RAW264.7 macrophages are useful compared to primary cells due to both ease of cell propagation and homogenous genetic background resulting in reduced experimental variability; they are originally derived from a virus-induced leukemic tumor developing in BALB/C Abelson mice.43 This origin may result in a distinct inflammatory response compared to that of in vivo macrophages. Indeed, recent reports have demonstrated significant differences in the transcriptomic profile following LPS stimulation between murine BMDMs and RAW264.7 cells.44 Moreover, at least one report indicates that commercially available RAW264.7 macrophages from ATCC can still express ecotropic and polytropic murine leukemia virus,45 which may additionally alter inflammatory signalling. In light of this, we additionally examined the capacity of both CDDOMe and CDDOMe-ARAPas to inhibit iNOS mRNA levels, iNOS-derived nitrite production and iNOS protein expression in classically stimulated murine BMDMs. Successful isolation of BMDMs was confirmed via presence of positive immunofluorescence staining with two markers for macrophages: cd11b and cd68. Representative images of this characterization are shown in ESIFig. 1.† Firstly, we examined iNOS mRNA levels via ddPCR following classical stimulation (IFNλ/LPS) in the presence or absence of CDDOMe or CDDOMe-ARAPas after 4hr (Fig. 6A). A factorial ANOVA was conducted to determine the effect of either treatment with CDDOMe-ARAPas or CDDOMe alone and dose on relative iNOS/GAPDH mRNA levels in BMDMs. The model was significant (F(5, 18) = 13.62, P < 0.0001), and treatment (F(1, 18) = 12.24, P = 0.003) and dose (F(2, 18) = 27.83, P < 0.0001) were both significant. The effect size of treatment was ωp2 = 0.3, and for dose ωp2 = 0.7. However, the interaction of dose and treatment with ARAPas and CDDOMe alone was not significant (F(2, 18) = 0.09, P = 0.92). Additionally, post hoc Tukey tests found that there were significant decreases in relative iNOS/GAPDH mRNA levels between vehicle and both 200 and 400 nM of either CDDOMe alone or CDDOMe-ARAPas (Fig. 6A). Secondly, we again examined the production of nitric oxide in the media, as an index of iNOS protein activity (Fig. 6B). Here, for each independent biological repeat, the response in picomol NO per cell due to classical stimulation without any vehicle was set as the maximal response (100%), and the other values within an independent biological repeat were calculated as a percentage of this. We conducted a factorial ANOVA analyzing the effects of treatment with either CDDOMe-ARAPas or CDDOMe alone as well as dose upon % of maximal picomol of NO per cell. Overall, this model was significant (F(5, 12) = 17.26, P < 0.0001), and analysis of the main effects showed that there was a statistically significant effect comparing treatment between CDDOMe vs. CDDOMe-ARAPas (F(1, 12) = 29.1, P = 0.0001), with an effect size of ωp2 = 0.6. Additionally, dose did have a statistically significant effect upon % of maximal picomol of NO per cell produced (F(2, 12) = 26.86, P < 0.0001) with an effect size of ωp2 = 0.7. Moreover, post-hoc Tukey tests found a significant decrease in % of maximal picomol of NO per cell between the vehicle and both 200 and 400 nM for CDDOMe-ARAPas and between the vehicle and 400 nM CDDOMe (Fig. 6B). Finally, we examined iNOS protein expression in murine BMDMs classically stimulated for 24 h, via immunofluorescence as previously described for RAW264.7 macrophages (Fig. 6C and D). We conducted a 2-way ANOVA to analyze the effect of treatment (with either CDDOMe-ARAPas or CDDOMe) or dose on percentage (%) Positive Cells. The 2-way ANOVA model was significant (F(5, 12) = 3.38, P = 0.039). Simple main effects analysis showed that while treatments between CDDOMe or CDDOMe-ARAPas was not significant (F(1, 12) = 0.59, P = 0.46), the effect due to dose was (F(2, 12) = 7.76, P = 0.007). Moreover, we found that the effect size for dose was ωp2 = 0.4. A post-hoc Tukey test found a significant difference between both 200 and 400 nM and vehicle (P = 0.02, P = 0.01, respectively). Overall, this data indicates that treatment with CDDOMe and CDDOMe-ARAPas results in inhibition of iNOS protein expression.
To further examine the role of CDDOMe and CDDOMe-ARAPas in inhibition of inflammatory signalling in classically stimulated macrophages, we quantified IL1b mRNA expression via ddPCR in both RAW 264.7 macrophages and BMDMs (Fig. 7). Data is expressed relative to the IL1b/GAPDH mRNA copy number that is induced by classical stimulation alone. Firstly, CDDOMe or CDDOMe-ARAPas did not decrease relative IL1b/GAPDH mRNA in RAW 264.7 macrophages (Fig. 7A). This stood in direct contrast to our results with BMDMs. A factorial ANOVA was conducted to determine the effect of treatment (CDDOMe-ARAPas or CDDOMe alone) and dose on relative IL1b mRNA levels in BMDMs (Fig. 7B). The model was significant (F(5, 18) = 31.57, P < 0.0001), and both treatment (F(1, 18) = 62.17, P < 0.0001) and dose (F(2, 18) = 47.28, P < 0.0001) were significant. The effect size of treatment was ωp2 = 0.7, and for dose ωp2 = 0.8. However, the interaction of dose and treatment with ARAPas vs. CDDOMe alone was not significant (F(2, 18) = 0.57, P = 0.58). Post hoc Tukey tests found that there were highly significant decreases (P < 0.001) in relative IL1b/GAPDH mRNA levels between vehicle and both 200 and 400 nM of either CDDOMe alone or CDDOMe-ARAPas. Taken together, this highlights the important differences that exist between the inflammatory response of RAW264.7 macrophages and primary BMDMs.
In further studies we looked at the 24 h time point for both IL1b and iNOS mRNA expression in classically stimulated RAW macrophages but found no significant differences (ESIFig. 2A and B†). Additionally we examined IL6 mRNA expression in classically stimulated RAW macrophages following 4 h incubation with CDDOMe or CDDOMe-ARAPas, and found that, in line with the lack of activation of IL1b mRNA expression, there was also no significant difference in IL6 mRNA expression (ESIFig. 2C†).
Notably, the literature shows a reduction in IL1b mRNA in primary macrophages. Kobayashi and colleagues found that IL1b was negatively regulated by the Nrf2 transcription factor in murine BMDMs.20 Dayalan Naidu and co-workers reported that another synthetic CDDO analog inhibited Il1B mRNA expression in LPS-stimulated (1 ng mL−1) primary murine peritoneal macrophages at 4 h,46 with this dependent on the Cys 151 present in the Keap1. In another study, Zheng and co-workers reported that in LPS-stimulated (100 ng mL−1) RAW 264.7 macrophages, pre-treated for 1 h with CDDO-Me (500 nM), IL-1β protein production following 8 and 24hr incubation periods was inhibited, however no statistical tests on this data were reported.47In vivo, CDDO Me administration has reduced IL1-β protein in mesenteric adipose tissue of high fat diet fed mice,48 and mRNA expression of IL-1β, IL-6 and TNF-α in the liver and epididymal fat of diet-induced diabetic mice.49 Together with our data, these studies support that pharmacological Nrf2 activation can decrease IL1B expression in primary murine macrophages. Reductions both in vitro and in vivo may however be influenced by the stimulatory conditions employed, and the cell/tissue type, which may explain the lack of inhibition in RAW 264.7 macrophages.
CDDOMe-ARAPas localize to atherosclerotic plaque
Athero-prone mice (LDLr−/− and ApoE−/−) were high fat diet fed for 15 weeks in order to induce atherosclerotic plaque generation. To assess whether CDDOMe-ARAPas accumulated in atherosclerotic lesions, these mice were intravenously injected with 3 mg kg−1 of DiD-CDDOMe-ARAPas (25 μg mL−1 DiD dye). This was an equivalent dose of 125 μg kg−1 of DiD fluorescent dye. 24 h following injection these animals were sacrificed and the hearts, aortae, and a number of organs (liver, kidney, lung, gut, spleen) were excised for histology, and plasma collected. Fig. 8A shows representative fluorescence images of the aortic sinus region of ApoE−/− and LDLr−/− animals injected with 3 mg kg−1 of DiD-CDDOMe-ARAPas, whilst representative images of vehicle-injected animals are shown in ESIFig. 3.† We quantified thresholded fluorescence intensity normalized to lesion area (RFU/lesion area, Fig. 8B) in the atherosclerotic plaque of the aortic sinus of both LDLr−/− and ApoE−/− mice. We conducted a 2-way ANOVA to determine the effect of both the genotype and treatment (DiD-CDDOMe-ARAPas vs vehicle) on RFU/lesion area. The 2-way ANOVA model was significant (F(3, 11) = 7.41, P = 0.0055). Notably, we found that whilst genotype did not significantly affect RFU/lesion area (F(1, 11) = 0.23, P = 0.64), treatment with CDDOMe-ARAPas did (F(1, 11) = 21.3, P = 0.0008), with an effect size of ωp2 = 0.57. Additionally the interaction between the variables was not significant (F(1, 11) = 0.04, P = 0.84). Overall, our data confirms that CDDOMe-ARAPas localize in atherosclerotic plaque.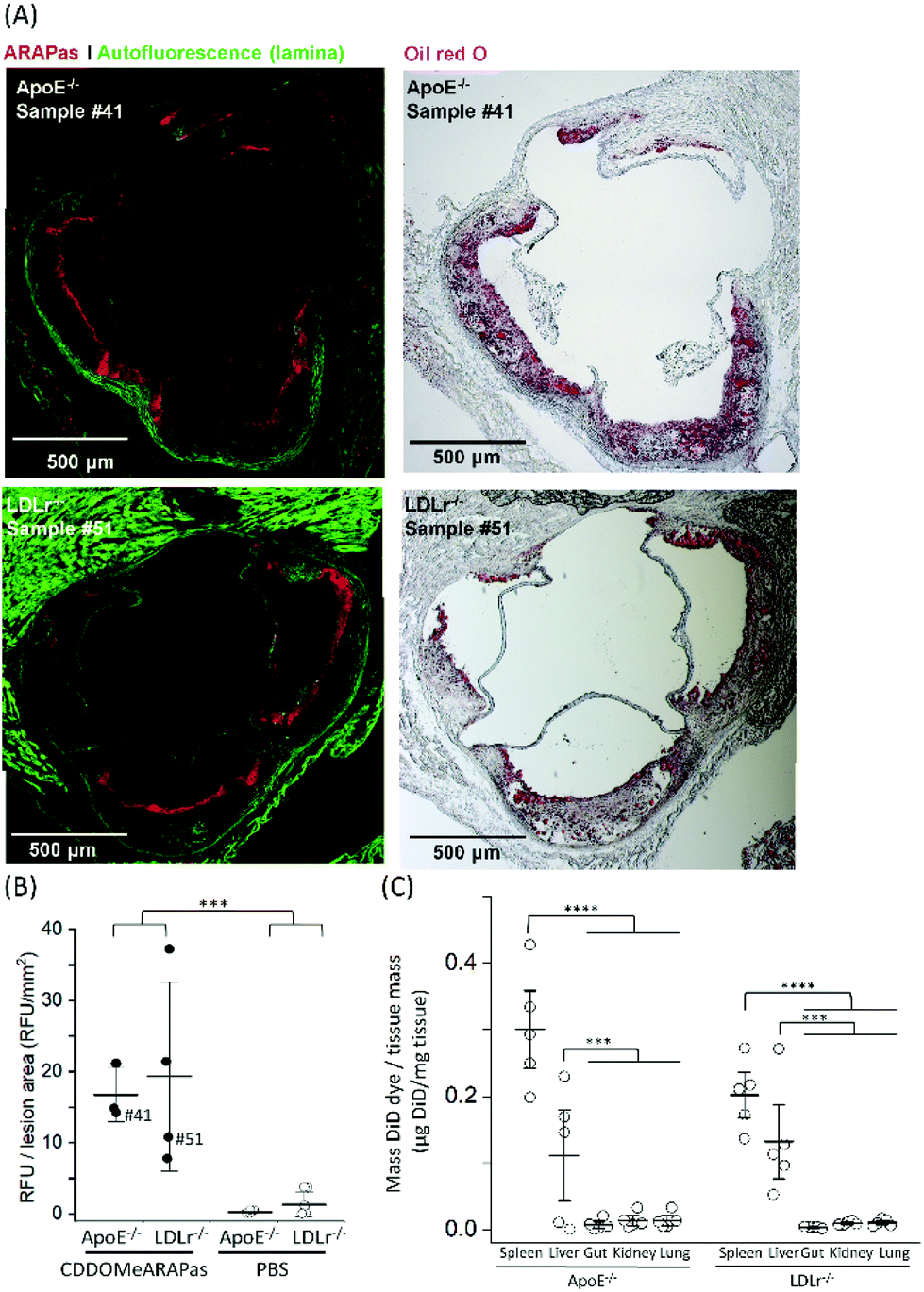 | ||
| Fig. 8 CDDOMe-ARAPas localize to atherosclerotic plaque in athero-prone mice. LDLr−/− and ApoE−/− mice were fed a high fat diet for 15 weeks and then intravenously injected with 3 mg kg−1 of DiD-CDDOMe-ARAPas (5 mL kg−1) or an equivalent volume of 10 mM PBS. 24 hours later their organs and plasma were collected. (A) Representative images of localization of DiD-CDDOMe-ARAPas in atherosclerotic plaque of both ApoE−/− and LDLr−/− mice. Tissue sections were imaged by fluorescence microscopy (left panels) and then the same tissue sections were stained with Oil Red O (right panels) to confirm the visualization of atherosclerotic plaque. Scale bar is 500 μm. (B) CDDOMe-ARAPas selectively localize to atherosclerotic plaque. Aortic root sections were analyzed with fluorescence microscopy to determine localization of DiD-CDDOMeARAPas in atherosclerotic plaque. DiD fluorescence intensity was thresholded and normalized to lesion area (determined by auto-fluorescence in the 488 channel). Data represents the mean of 3–5 independent biological replicates, ±1 S.D. A factorial ANOVA was conducted to determine the effect of CDDOMe-ARAPas treatment and genotype upon RFU/lesion area (***P < 0.001). (C) Biodistribution of CDDOMe-ARAPas in athero-prone mice. Organs were weighed and homogenized in 5% Triton-X-100 in 10 mM PBS, and subsequently extracted with isopropanol. Fluorescence intensity was recorded and μg of DiD determined with a standard curve and normalized to tissue mass. Data represents the mean of 3–5 independent biological replicates, with fluorescence measurements conducted in triplicate, ±1 S.D. Sham-injected genotype-matched animals were also measured and for each organ, the basal μg DiD per mg tissue was deducted. A factorial ANOVA was conducted to determine the effect of genotype and organs upon μg DiD per mg tissue (***P < 0.001, ****P < 0.0001). | ||
Nanoparticles take advantage of a dysfunctional endothelium with larger inter-endothelial junctions to accumulate in sites of vascular inflammation.14,50 Work by Beldman and co-workers14,51 support a paradigm where intravenously injected nanoparticles chiefly enter atherosclerotic plaque through para-cellular means by crossing the luminal endothelial junctions. Early atherosclerotic plaque have been shown to be associated with more severe endothelial barrier disruption, whilst advanced stage disease progressions are associated with improved endothelial barrier permeability.14 Beldman and colleagues examined the junctional integrity of atherosclerotic lesions in both early (6 weeks old) and advanced (12 weeks old) plaque, and chiefly found that early atherosclerotic endothelial junctions could have spaces reaching up to 3μm in width, with slight but significant endothelial normalization for older more advanced lesions. Thus, previous pre-clinical studies have successfully integrated nanoparticles into sites of atherosclerotic plaque, some of which were able to inhibit plaque formation and decreased the number of atherosclerotic lesions in mouse models of atherosclerosis.16,50,52–55Following para-cellular transport, engulfment by plaque-associated macrophages is expected, which allows for nanoparticle retention in atherosclerotic plaque. This is likely to reflect phagocytosis due to both the presence of the protein corona that plays a role in nanoparticle-cell interactions,56 and nanoparticle size being >200 nm.15 In the present study, we have not investigated the role of plaque-resident macrophages in the in vivo accumulation of DiD-CDDOMe-ARAPas. However, overall, the atherosclerotic plaque accumulation of DiD-CDDOMe-ARAPas in our study is consistent with the literature, which shows that nanoparticles can accumulate selectively in atherosclerotic plaque primarily due to the enhanced permeation and retention (EPR) effect.14,50 The cellular mechanisms that influence nanoparticle uptake into atherosclerotic lesions is a continuing area of study.
In addition, we also examined the bio-distribution of the DiD-CDDOMe-ARAPas at 24 h in each of the genotypes (Fig. 8C). To analyze their biodistribution, the organs were homogenized and the lysate extracted with isopropanol and DiD fluorescence measured with a fluorescence spectrometer. The average fluorescent signal from sham animals (injected with PBS vehicle) was subtracted from the signal in CDDOMe-ARAPas treated animals. We conducted a 2-way ANOVA to examine the influence of genotype and organs upon μg DiD per mg tissue. Overall, the 2-way ANOVA model was significant (F(9, 40) = 19.5, P < 0.0001), and it revealed that the effect of genotype was not significant (F(1, 40) = 1.44, P = 0.24), whilst organs did influence μg DiD per mg tissue (F(4, 40) = 41.6, P < 0.0001), with an effect size of ωp2 = 0.45. Moreover, post hoc Tukey tests revealed that μg DiD per mg tissue was significantly different particularly in the liver (P < 0.0001), and spleen (P < 0.001) compared to the other organs (kidney, lung, gut).
Plasma chemistry profile in vivo
A number of plasma protein markers including aspartate transaminase (AST), alanine transaminase (ALT), and albumin (ALB) for liver toxicity, blood urea nitrogen (BUN) and creatinine for kidney toxicity, and finally cholesterol, low density lipoprotein, and triglycerides (TRIG) for a lipid profile, were measured. The data were analyzed using multivariate analysis of variance (MANOVA) to compare the effect of treatment with either CDDOMe-ARAPas or PBS and genotype (C57bl6, LDLr−/− vs ApoE−/−) upon these markers (ESITable 2†). The MANOVA revealed a significant difference due to treatment, Wilk's Lambda λ = 0.337, (F(8, 13) = 3.192, P = 0.031), and genotype, Wilk's λ = 0.042 (F(16, 26) = 6.26, P < 0.001), with a significant interaction between treatment and genotype, Wilk's λ = 0.33 (F(8, 13) = 3.252, P = 0.029). The effect size of treatment upon ALT was ωp2 = 0.53, and upon AST ωp2 = 0.67. Pairwise comparisons confirm that ApoE−/− and LDLr−/− have significantly increased levels of plasma cholesterol and LDL compared to those levels in age-matched normal chow-fed C57bl/6 mice, whilst triglyceride levels are only significantly increased in LDLr−/− mice compared to both C57bl/6 (P = 0.001), and ApoE−/(P < 0.0001). Regarding effects upon liver and kidney toxicity markers, pairwise comparisons revealed that CDDOMe-ARAPas treatment significantly increased ALT (P = 0.006) and AST (P < 0.0001) in LDLr−/− mice, but without significant changes observed in ApoE−/− mice. In ApoE−/− mice, pairwise comparisons show a significant decrease in BUN (P = 0.028) and increase in creatinine (P = 0.026). No other marker showed significant changes between treatments, or within genotypes. An increase in serum levels of AST and ALT was also observed in patients treated with CDDO-Me.57 In this study the increase in serum levels of transaminases was transient. Even though the increase in serum levels of these enzymes is associated with hepatotoxic effects, Lewis and colleagues suggested that Nrf2 activation increases serum levels of AST and ALT by increasing their gene expression in both hepatic and extra-hepatic tissues.57 The authors showed that serum levels of AST and ALT are lower in Nrf2-null mice and higher in Keap1-knockdown mice. Since the CDDO-Me-ARAPas accumulated significantly in the liver, it is possible that hepatic Nrf2 activation could drive AST and ALT gene expression. It would be important to assess Nrf2 activation status in the liver and assess whether the observed increased in AST and ALT is transient.CDDOMe-ARAPas locally activate Nrf2 in atherosclerotic plaque
Finally, we examined whether CDDOMe and CDDOMe-ARAPas activate canonical Nrf2 regulated genes (GCLC and NQO1) in both murine macrophages and in high fat diet fed LDLr−/− mice. Firstly, we examined the transcription of GCLC in classically stimulated RAW macrophages (Fig. 9A), murine BMDMs (Fig. 9B) and in the aortic arch of high fat diet fed LDLr−/− mice (Fig. 9C). IFNλ/LPS treatment markedly decreased GCLC mRNA levels in both RAW macrophages and BMDMs, in line with previous literature.58 Overall, we found that CDDOMe or CDDOMe-ARAPas (200–400 nM) increased GCLC mRNA levels in both RAW 264.7 macrophages (Fig. 9A) and in BMDMs (Fig. 9B) after IFNλ/LPS treatment. A factorial ANOVA revealed an overall significant model (F(5, 10) = 14.22, P = 0.0003), and simple main effects analysis revealed that the difference between treatments was not significant (F(1, 10) = 0.24, P = 0.64), and dose had a significant effect on relative GCLC/GAPDH mRNA levels (F(2, 10) = 30.76, P < 0.0001) with an effect size of ωp2 = 0.8. Overall, GCLC mRNA levels were increased by CDDOMe (1.7–1.9 fold) and by CDDOMe-ARAPas (3–4.5 fold) (Fig. 9A). Post hoc Tukey pairwise comparisons revealed that this increase was significant for CDDOMe-ARAPas at both 200 and 400 nM (Fig. 9A). Regarding GCLC/GAPDH mRNA levels in classically stimulated BMDMs, a factorial ANOVA was conducted to determine the effect of treatment type (CDDOMe-ARAPas or CDDOMe) and dose on relative GCLC/GAPDH mRNA levels in BMDMs (Fig. 9B). The model was significant (F(5, 18) = 5.71, P = 0.002). While dose had a significant effect (F(2, 18) = 14.15, P = 0.0002), there was no differences between CDDOMe and CDDOMe-ARAPas (F(1, 18) = 0.06, P = 0.81). The effect size of dose was ωp2 = 0.5. Post hoc Tukey tests found that overall there was a significant increase between treatment with 200 nM and 400 nM compared to 0 nM, with GCLC/GAPDH mRNA levels being increased by CDDOMe by ∼2.7 fold and by CDDOMe-ARAPas by ∼3.6–3.8-fold.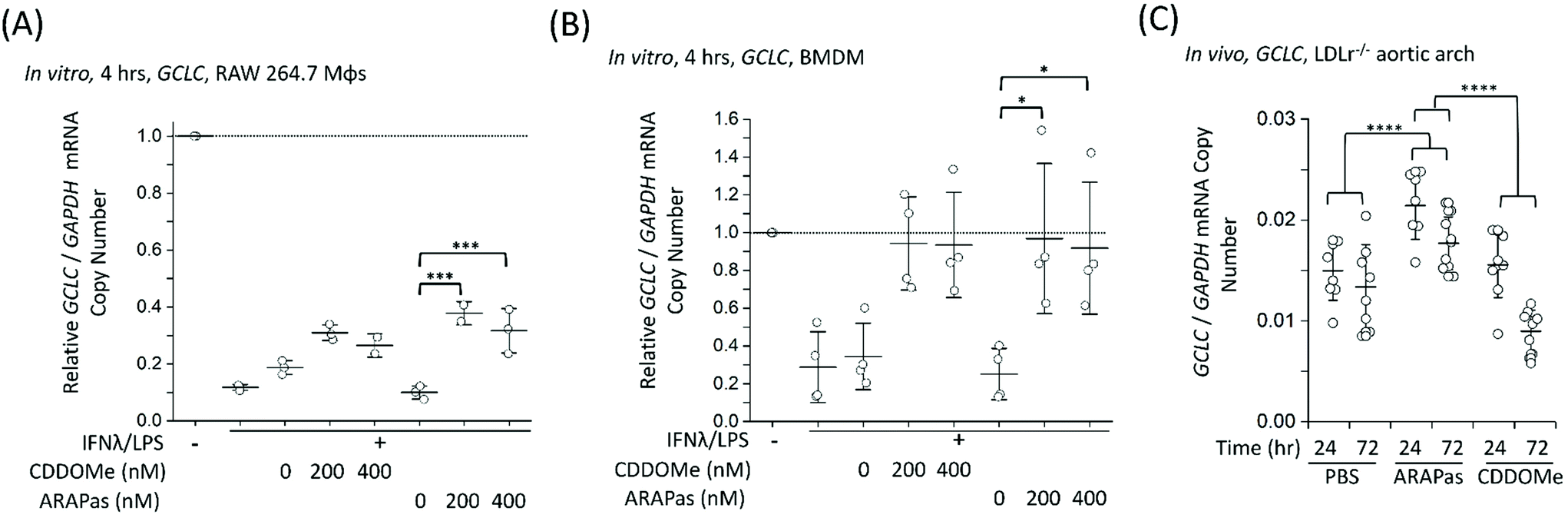 | ||
| Fig. 9 Activation of Nrf2-regulated genes in vitro and in vivo by CDDOMe-ARAPas. GCLC mRNA level were recorded in either classically stimulated murine RAW 264.7 macrophages, BMDMs or in aortic arch homogenates of high fat diet fed LDLr−/− mice. RAW 264.7 macrophages and BMDMs were classically stimulated with IFNλ (10 ng mL−1, 7 h) followed by treatment with LPS (100 ng mL−1, 4 h) in the presence or absence of treatments and their respective vehicles. Cells were then pelleted for RNA extraction. 4–6 weeks old LDLr−/− mice were high fat diet fed for 8 weeks (till 12–14 weeks old) and then either injected intravenously with 1 mg kg−1 of CDDOMe-ARAPas or 10 mM PBS, or intraperitoneally with 1 mg kg−1 CDDOMe dissolved in DMSO. Aortic arches were collected at either 24 h or 72 h post injection of CDDOMe-ARAPas, 10 mM PBS, or CDDOMe. (A) Activation of GCLC mRNA expression in classically activated murine RAW macrophages following 4 h incubation. Data represents the mean of 2–3 independent biological replicates, ±1 S.D. A factorial ANOVA was conducted to determine the effect of classical stimulation, treatment and dose upon relative GCLC/GAPDH mRNA copy number (***P < 0.001). (B) Activation of GCLC mRNA expression in classically activated murine BMDMs following 4 h incubation. Data represents the mean of 4 independent biological replicates, ±1 S.D. A factorial ANOVA was conducted to determine the effect of classical stimulation, treatment and dose upon relative GCLC/GAPDH mRNA copy number (*P < 0.05). (C) Activation of GCLC mRNA expression in LDLr−/− aortic arch homegenates. Data represents the mean of 8–10 independent biological replicates for each condition, ±1 S.D. A factorial ANOVA was conducted to determine the effect of treatment with either PBS, CDDOMe or CDDOMe-ARAPas and time upon GCLC mRNA expression normalized to GAPDH copy number (****P < 0.0001). | ||
We next wanted to confirm whether selective delivery of CDDOMe-ARAPas to atherosclerotic plaque (as shown in Fig. 8) led to selective increases in mRNA expression of the Nrf2-regulated genes GCLC and NQO1. LDLr−/− mice were high fat diet fed over 8 weeks and then intravenously injected with either 10 mM PBS, or 1 mg kg−1 CDDOMe-ARAPas (equivalent to ∼3 mg kg−1 nanoparticle mass). HPLC-UV-VIS was used to determine the concentration of CDDOMe in ARAPas suspensions, and the volume injected was adjusted to ensure a dose of 1 mg kg−1. To compare to untargeted CDDOMe, we intra-peritoneally injected an equimolar dose of CDDOMe (1 mg kg−1), dissolved in DMSO. Typically, the doses cited in the literature for CDDOMe treatment range from 3–100 mg kg−1,59–61 with a majority employing 10 mg kg−1. We chose to use a lower dose than previously reported to better parse out whether encapsulation of CDDOMe in ARAPas, resulting in localized delivery to atherosclerotic plaque, would result in localized Nrf2 activation that is absent in un-targeted CDDOMe. 24 and 72 hours following injection, the aortic arch (a site of preferential atherosclerotic plaque formation in mice) was harvested, homogenized, and mRNA extracted and purified. GCLC (Fig. 9C) mRNA in aortic arch homogenates was detected utilizing ddPCR. Firstly, we conducted a 2-way ANOVA to determine whether treatment (PBS, CDDOMe-ARAPas, CDDOMe alone) and time (24 vs. 72 h) influenced GCLC mRNA expression normalized to GAPDH copy number. The model was significant (F(5, 45) = 15.5, P < 0.0001), and simple main effects analysis revealed that both treatment (F(2, 45) = 25, P < 0.0001) and time (F(1, 45) = 21, P < 0.0001) significantly influenced GCLC mRNA expression, although the interaction was non-significant (F(2, 45) = 3, P = 0.07). Overall, the effect size for treatment was ωp2 = 0.48, and for time ωp2 = 0.28. Post-hoc Tukey tests showed that there was a significant increase in GCLC/GAPDH mRNA expression for CDDOMe-ARAPas vs. PBS (P < 0.0001) and for CDDOMe-ARAPas vs. CDDOMe alone (P < 0.0001) (Fig. 9C). On the other hand, there was a non-significant change in GCLC/GAPDH mRNA levels for CDDOMe alone vs. PBS (P = 0.13).
Next we examined whether CDDOMe and CDDOMe-ARAPas activate another canonical Nrf2-regulated gene, NQO1 in both classically stimulated RAW murine macrophages (ESIFig. 4A†) and in high fat diet fed LDLr−/− mice (ESIFig. 4B†). We conducted a factorial ANOVA on whether treatment with CDDOMe or CDDOMe-ARAPas, and dose influenced relative NQO1/GAPDH mRNA levels in RAW macrophages (ESIFig. 4A†). This model was significant (F(5, 12) = 4.38, P = 0.02), and revealed that both treatment type (CDDOMe vs. CDDOMe-ARAPas) (F(1, 12) = 10.1, P = 0.008) and dose were significant (F(2, 12) = 6.17, P = 0.014), with effect sizes of ωp2 = 0.3 and ωp2 = 0.4, respectively. However, the interaction of these two variables was not significant (F(2, 12) = 1.1, P = 0.36). Post hoc Tukey tests showed that NQO1 mRNA expression was significantly increased by the 400 nM dose of CDDOMe or CDDOMe-ARAPas (ESIFig. 4A†). In vivo, we also conducted a 2-way ANOVA with treatment (PBS, CDDOMe-ARAPas, CDDOMe alone) and time (24 vs 72 h). The model was significant (F(5, 47) = 14, P < 0.0001), and simple main effects analysis revealed that both treatment (F(1, 47) = 16.5, P < 0.0001) and time (F(2, 47) = 26.2, P < 0.0001) significantly influenced NQO1 mRNA expression, although the interaction was not significant (F(2, 47) = 0.42, P = 0.66). Overall, the effect size for treatment was ωp2 = 0.37, and for time ωp2 = 0.32. Post-hoc Tukey tests showed that there was an increase in mRNA expression for CDDOMe-ARAPas vs. PBS (P < 0.00969) and for CDDOMe-ARAPas vs. CDDOMe alone (P < 0.0001) (ESIFig. 4B†).
Khoo and co-workers previously found that an analog of CDDO-Me, CDDO-imidazole, was able to dose-dependently increase HO1 expression in naïve RAW 264.7 macrophages, as well as significantly increase HO1 expression above that of the HO1 already expressed in IFNλ/LPS treated RAW 264.7 macrophages.40 Thus, we also investigated the effect of CDDOMe and CDDOMe-ARAPas injection in vivo on HO1 and SOD1 mRNA expression at 24 and 72 hours post-injection (ESIFig. 5†). However, we did not find a change to the expression of these mRNA for any treatment in vivo.
In the literature, several compounds known as Nrf2-inducers (tBHQ,22 Ebselen,23 CDDO-Me analogue dh404,24,59 and oleanolic acid25) have augmented endogenous antioxidant systems and limited inflammation thereby preventing atherosclerosis development or progression in diabetes-aggravated atherosclerosis.59 In particular, Tan and colleagues using a related synthetic triterpenoid analog, dh404, examined mRNA levels of NQO1, Glutathione S Transferase (GSH-S-T) and Glutathione peroxidase 1 (GPx1) in the kidney cortex following 5 weeks of treatment. In their study, only the 20 mg kg−1 dose (not 3 or 10) had increased GSH-S-T, and GPx1 mRNA in diabetic animal kidney cortex relative to the vehicle.59 Overall, the results presented in Fig. 9C and ESI Fig. 4B† support that selective delivery of CDDOMe-ARAPas to atherosclerotic plaque results in a targeted increase in mRNA expression of Nrf2-regulated genes in the aortic arch, which is absent in equi-molar untargeted CDDOMe. However, we do not know what cells are responsible for the increase in mRNA of Nrf2 transcriptional targets. In light of the reported anti-inflammatory and athero-protective nature of pharmacological Nrf2 induction, we speculate that localized delivery will allow for greater targeted effect with lower doses. A barrier for the clinical use of redox-modulatory drugs is their adequate access to sites of redox dysfunction such as atherosclerotic plaque, thus our study begins to address this important challenge.
Conclusions
Herein we report the successful encapsulation of the potent Nrf2 activator drug, CDDO-methyl into polymeric nanoparticles via flash nanoprecipitation (FNP)29 to generate CDDOMe Antioxidant Response Activating nanoParticles (CDDOMe-ARAPas). FNP was chosen as our nanoparticle synthesis method as it is highly translational, being amenable to large-scale (kg day−1) manufacturing whilst robustly maintaining nanoparticle characteristics and homogeneity.30 We describe the physiochemical characteristics of our nanoparticles as well as their in vitro release of CDDO-Me, resulting in potent Nrf2 activation. We additionally demonstrate their internalization by naïve murine macrophages and inhibition of pro-inflammatory iNOS and IL1b induction by classically activated macrophages. We go on to show that these nanoparticles selectively accumulate in atherosclerotic plaque in two widely used genotypes of athero-prone mice (ApoE−/− and LDLr−/−). Finally, we show that the CDDOMe-ARAPas successfully activate the expression of Nrf2-regulated genes both in vitro in murine macrophages and in vivo in the aortic arch. Moreover, equi-molar doses of un-encapsulated CDDOMe fail to induce expression under the same conditions in vivo. It was beyond the scope of this study to assess whether intra-plaque delivered Nrf2 activator drugs such as CDDO-Me, prevent atherosclerotic progression, which will be assessed in future work. Overall, these studies demonstrate the successful intra-plaque delivery of antioxidant-based therapeutics employing a highly translational nanoparticle synthesis technique. Our studies support the paradigm that targeted delivery of redox-active therapeutics is superior to systemic delivery for modulation of the intra-plaque environment.20Author contributions
Sophie Maiocchi: Conceptualization, methodology, formal analysis, investigation, data curation, writing – original draft, visualization. Ana Cartaya: Investigation, visualization. Sydney Thai: Investigation, formal analysis, data curation. Adam Akerman: Methodology, resource. Edward Bahnson: Conceptualization, methodology, formal analysis, resources, writing – review & editing, supervision, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Institutes of Health, National Heart Lung and Blood Institute [K01HL145354] to E. M. B. S. M. is supported by the Leon and Bertha Golberg Fellowship. A. E. C. is supported by National Heart Lung and Blood Institute [F31HL156427]. We gratefully acknowledge Vicky Madden and Kristen White for assistance with conventional TEM. The Microscopy Services Laboratory, Department of Pathology and Laboratory Medicine, is supported in part by P30 CA016086 Cancer Center Core Support Grant to the UNC Lineberger Comprehensive Cancer Center. We also gratefully acknowledge Marina Sokolsky, Olesia Gololobova and Jacob Ramsey of the Nanomedicines Characterization Core Facility (NCore) at the Center for Nanotechnology in Drug Delivery (CNDD) at UNC School of Pharmacy for their assistance with Nanosight Nanotracking Analysis and HPLC UV-VIS analysis of nanoparticle samples. Additionally, we thank Ling Wang in the Animal Histopathology and Laboratory Medicine Core for expert technical assistance with analysis of plasma chemistry. The Animal Histopathology and Laboratory Medicine core is supported in part by an NCI Center Core Support Grant (5P30CA016080-42). Microscopy was performed at the UNC Neuroscience Microscopy Core supported, in part, by funding from the NIH-NINDS Neuroscience Center Support Grant P30 NS045892 and the NIH-NICHD Intellectual and Developmental Disabilities Research Center Support Grant U54 HD079124.Notes and references
- World Health Organization, Cardiovascular Diseases (CVDs) Fact Sheet, World Health Organization, 2021, https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) Search PubMed.
- E. J. Benjamin, M. J. Blaha, S. E. Chiuve, M. Cushman, S. R. Das, R. Deo, S. D. de Ferranti, J. Floyd, M. Fornage, C. Gillespie, C. R. Isasi, M. C. Jiménez, L. C. Jordan, S. E. Judd, D. Lackland, J. H. Lichtman, L. Lisabeth, S. Liu, C. T. Longenecker, R. H. Mackey, K. Matsushita, D. Mozaffarian, M. E. Mussolino, K. Nasir, R. W. Neumar, L. Palaniappan, D. K. Pandey, R. R. Thiagarajan, M. J. Reeves, M. Ritchey, C. J. Rodriguez, G. A. Roth, W. D. Rosamond, C. Sasson, A. Towfighi, C. W. Tsao, M. B. Turner, S. S. Virani, J. H. Voeks, J. Z. Willey, J. T. Wilkins, J. H. Wu, H. M. Alger, S. S. Wong and P. Muntner, Circulation, 2017, 135, e146–e603 CrossRef PubMed.
- P. Libby, J. E. Buring, L. Badimon, G. K. Hansson, J. Deanfield, M. S. Bittencourt, L. Tokgözoğlu and E. F. Lewis, Nat. Rev. Dis. Primers, 2019, 5, 56 CrossRef PubMed.
- M. Bäck, A. Yurdagul, I. Tabas, K. Öörni and P. T. Kovanen, Nat. Rev. Cardiol., 2019, 16, 389–406 Search PubMed.
- G. K. Hansson and A. Hermansson, Nat. Immunol., 2011, 12, 204–212 CrossRef CAS PubMed.
- C. Weber and H. Noels, Nat. Med., 2011, 17, 1410–1422 CrossRef CAS PubMed.
- D. Harrison, K. K. Griendling, U. Landmesser, B. Hornig and H. Drexler, Am. J. Cardiol., 2003, 91, 7–11 CrossRef.
- U. Förstermann, N. Xia and H. Li, Circ. Res., 2017, 120, 713–735 CrossRef PubMed.
- K. J. Bubb, G. R. Drummond and G. A. Figtree, Cardiovasc. Res., 2020, 116, 532–544 CrossRef CAS PubMed.
- E. S. M. Bahnson, H. A. Kassam, T. J. Moyer, W. Jiang, C. E. Morgan, J. M. Vercammen, Q. Jiang, M. E. Flynn, S. I. Stupp and M. R. Kibbe, Antioxid. Redox Signaling, 2016, 24, 401–418 CrossRef CAS PubMed.
- A. Cartaya, S. Maiocchi and E. M. Bahnson, Curr. Pathobiol. Rep., 2019, 7, 47–60 CrossRef CAS PubMed.
- A. M. Flores, J. Ye, K. U. Jarr, N. Hosseini-Nassab, B. R. Smith and N. J. Leeper, Arterioscler., Thromb., Vasc. Biol., 2019, 39, 635–646 CrossRef CAS PubMed.
- N. E. Buglak, E. V. Batrakova, R. Mota and E. S. M. Bahnson, Oxid. Med. Cell. Longevity, 2018, 2018, 2468457 Search PubMed.
- T. J. Beldman, T. S. Malinova, E. Desclos, A. E. Grootemaat, A. L. S. Misiak, S. Van Der Velden, C. P. A. A. Van Roomen, L. Beckers, H. A. Van Veen, P. M. Krawczyk, R. A. Hoebe, J. C. Sluimer, A. E. Neele, M. P. J. De Winther, N. N. Van Der Wel, E. Lutgens, W. J. M. Mulder, S. Huveneers and E. Kluza, ACS Nano, 2019, 13, 13759–13774 CrossRef CAS PubMed.
- H. Hillaireau and P. Couvreur, Cell. Mol. Life Sci., 2009, 66, 2873–2896 CrossRef CAS PubMed.
- S. Chono, Y. Tauchi, Y. Deguchi and K. Morimoto, J. Drug Targeting, 2005, 13, 267–276 CrossRef CAS PubMed.
- J. Mimura and K. Itoh, Free Radicals Biol. Med., 2015, 88, 221–232 CrossRef CAS PubMed.
- A. K. Ruotsalainen, M. Inkala, M. E. Partanen, J. P. Lappalainen, E. Kansanen, P. I. Mäkinen, S. E. Heinonen, H. M. Laitinen, J. Heikkilä, T. Vatanen, S. Hörkkö, M. Yamamoto, S. Ylä-Herttuala, M. Jauhiainen and A. L. Levonen, Cardiovasc. Res., 2013, 98, 107–115 CrossRef CAS PubMed.
- H. K. Jyrkkänen, E. Kansanen, M. Inkala, A. M. Kivela, H. Hurttila, S. E. Heinonen, G. Goldsteins, S. Jauhiainen, S. Tiainen, H. Makkonen, O. Oskolkova, T. Afonyushkin, J. Koistinaho, M. Yamamoto, V. N. Bochkov, S. Ylä-Herttuala and A. L. Levonen, Circ. Res., 2008, 103, e1–e9 CrossRef PubMed.
- E. H. Kobayashi, T. Suzuki, R. Funayama, T. Nagashima, M. Hayashi, H. Sekine, N. Tanaka, T. Moriguchi, H. Motohashi, K. Nakayama and M. Yamamoto, Nat. Commun., 2016, 7, 11624 CrossRef CAS PubMed.
- A. L. Levonen, M. Inkala, T. Heikura, S. Jauhiainen, H. K. Jyrkkänen, E. Kansanen, K. Määttä, E. Romppanen, P. Turunen, J. Rutanen and S. Ylä-Herttuala, Arterioscler., Thromb., Vasc. Biol., 2007, 27, 741–747 CrossRef CAS PubMed.
- I. Lazaro, L. Lopez-Sanz, S. Bernal, A. Oguiza, C. Recio, A. Melgar, L. Jimenez-Castilla, J. Egido, J. Madrigal-Matute and C. Gomez-Guerrero, Front. Pharmacol., 2018, 9, 819 CrossRef PubMed.
- P. Chew, D. Y. C. Yuen, N. Stefanovic, J. Pete, M. T. Coughlan, K. A. Jandeleit-Dahm, M. C. Thomas, F. Rosenfeldt, M. E. Cooper and J. B. De Haan, Diabetes, 2010, 59, 3198–3207 CrossRef CAS PubMed.
- S. M. Tan and J. B. de Haan, Redox Rep., 2014, 19, 107–117 CrossRef CAS PubMed.
- M. Gamede, L. Mabuza, P. Ngubane and A. Khathi, Molecules, 2019, 24(2), 340 CrossRef PubMed.
- Y. Y. Wang, Y. X. Yang, H. Zhe, Z. X. He and S. F. Zhou, Drug Des., Dev. Ther., 2014, 8, 2075–2088 Search PubMed.
- N. D. Camp, R. G. James, D. W. Dawson, F. Yan, J. M. Davison, S. A. Houck, X. Tang, N. Zheng, M. B. Major and R. T. Moon, J. Biol. Chem., 2012, 287, 6539–6550 CrossRef CAS PubMed.
- A. A. Cohen, N. Geva-Zatorsky, E. Eden, M. Frenkel-Morgenstern, I. Issaeva, A. Sigal, R. Milo, C. Cohen-Saidon, Y. Liron, Z. Kam, L. Cohen, T. Danon, N. Perzov and U. Alon, Science, 2008, 322, 1511–1516 CrossRef CAS PubMed.
- C. E. Markwalter, R. F. Pagels, B. K. Wilson, K. D. Ristroph and R. K. Prud'homme, J. Visualized Exp., 2019, 2019 Search PubMed.
- J. Feng, C. E. Markwalter, C. Tian, M. Armstrong and R. K. Prud'homme, J. Transl. Med., 2019, 17, 200 CrossRef PubMed.
- M. Danaei, M. Dehghankhold, S. Ataei, F. Hasanzadeh Davarani, R. Javanmard, A. Dokhani, S. Khorasani and M. R. Mozafari, Pharmaceutics, 2018, 10(2), 57 CrossRef PubMed.
- Y. Zhao, M. Huo, Z. Xu, Y. Wang and L. Huang, Biomaterials, 2015, 68, 54–66 CrossRef CAS PubMed.
- M. Ogawa, R. Uchino, A. Kawai, M. Kosugi and Y. Magata, Nucl. Med. Biol., 2015, 42, 299–304 CrossRef CAS PubMed.
- V. Schäfer, H. von Briesen, R. Andreesen, A. M. Steffan, C. Royer, S. Tröster, J. Kreuter and H. Rübsamen-Waigmann, Pharm. Res., 1992, 9, 541–546 CrossRef PubMed.
- S. S. Yu, C. M. Lau, S. N. Thomas, W. G. Jerome, D. J. Maron, J. H. Dickerson, J. A. Hubbell and T. D. Giorgio, Int. J. Nanomed., 2012, 7, 799–813 CrossRef CAS PubMed.
- E. J. Cho, H. Holback, K. C. Liu, S. A. Abouelmagd, J. Park and Y. Yeo, Mol. Pharm., 2013, 10, 2093–2110 CrossRef CAS PubMed.
- S. M. D’Addio, A. A. Bukari, M. Dawoud, H. Bunjes, C. Rinaldi and R. K. Prud’homme, Philos. Trans. R. Soc., A, 2016, 374, 2072 CrossRef PubMed.
- S. A. Abouelmagd, B. Sun, A. C. Chang, Y. J. Ku and Y. Yeo, Mol. Pharm., 2015, 12, 997–1003 CrossRef CAS PubMed.
- A. T. Dinkova-Kostova and P. Talalay, Arch. Biochem. Biophys., 2010, 501, 116–123 CrossRef CAS PubMed.
- N. K. H. Khoo, L. Li, S. R. Salvatore, F. J. Schopfer and B. A. Freeman, Sci. Rep., 2018, 8, 2295 CrossRef PubMed.
- B. L. Probst, I. Trevino, L. McCauley, R. Bumeister, I. Dulubova, W. C. Wigley and D. A. Ferguson, PLoS One, 2015, 10, e0122942 CrossRef PubMed.
- F. G. Favaloro Jr., T. Honda, Y. Honda, G. W. Gribble, N. Suh, R. Risingsong and M. B. Sporn, J. Med. Chem., 2002, 45, 4801–4805 CrossRef PubMed.
- W. C. Raschke, S. Baird, P. Ralph and I. Nakoinz, Cell, 1978, 15, 261–267 CrossRef CAS PubMed.
- A. Das, C. S. Yang, S. Arifuzzaman, S. Kim, S. Y. Kim, K. H. Jung, Y. S. Lee and Y. G. Chai, Front. Immunol., 2018, 9, 22 CrossRef PubMed.
- J. W. Hartley, L. H. Evans, K. Y. Green, Z. Naghashfar, A. R. Macias, P. M. Zerfas and J. M. Ward, Retrovirology, 2008, 5, 1 CrossRef PubMed.
- S. Dayalan Naidu, A. Muramatsu, R. Saito, S. Asami, T. Honda, T. Hosoya, K. Itoh, M. Yamamoto, T. Suzuki and A. T. Dinkova-Kostova, Sci. Rep., 2018, 8, 8037 CrossRef PubMed.
- S. Zheng, Y. R. Santosh Laxmi, E. David, A. T. Dinkova-Kostova, K. H. Shiavoni, Y. Ren, Y. Zheng, I. Trevino, R. Bumeister, I. Ojima, W. C. Wigley, J. B. Bliska, D. F. Mierke and T. Honda, J. Med. Chem., 2012, 55, 4837–4846 CrossRef CAS PubMed.
- C. H. Dinh, A. Szabo, Y. Yu, D. Camer, H. Wang and X. F. Huang, ScientificWorldJournal, 2015, 2015, 549352 Search PubMed.
- P. K. Saha, V. T. Reddy, M. Konopleva, M. Andreeff and L. Chan, J. Biol. Chem., 2010, 285, 40581–40592 CrossRef CAS PubMed.
- M. Durymanov, T. Kamaletdinova, S. E. Lehmann and J. Reineke, J. Controlled Release, 2017, 261, 10–22 CrossRef CAS PubMed.
- T. J. Beldman, M. L. Senders, A. Alaarg, C. Pérez-Medina, J. Tang, Y. Zhao, F. Fay, J. Deichmöller, B. Born, E. Desclos, N. N. van der Wel, R. A. Hoebe, F. Kohen, E. Kartvelishvily, M. Neeman, T. Reiner, C. Calcagno, Z. A. Fayad, M. P. J. de Winther, E. Lutgens, W. J. M. Mulder and E. Kluza, ACS Nano, 2017, 11, 5785–5799 CrossRef CAS PubMed.
- R. Duivenvoorden, J. Tang, D. P. Cormode, A. J. Mieszawska, D. Izquierdo-Garcia, C. Ozcan, M. J. Otten, N. Zaidi, M. E. Lobatto, S. M. Van Rijs, B. Priem, E. L. Kuan, C. Martel, B. Hewing, H. Sager, M. Nahrendorf, G. J. Randolph, E. S. G. Stroes, V. Fuster, E. A. Fisher, Z. A. Fayad and W. J. M. Mulder, Nat. Commun., 2014, 6, 239 Search PubMed.
- M. Zhang, J. He, C. Jiang, W. Zhang, Y. Yang, Z. Wang and J. Liu, Int. J. Nanomed., 2017, 12, 533–558 CrossRef CAS PubMed.
- M. E. Lobatto, Z. A. Fayad, S. Silvera, E. Vucic, C. Calcagno, V. Mani, S. D. Dickson, K. Nicolay, M. Banciu, R. M. Schiffelers, J. M. Metselaar, L. Van Bloois, H. S. Wu, J. T. Fallon, J. H. Rudd, V. Fuster, E. A. Fisher, G. Storm and W. J. M. Mulder, Mol. Pharm., 2010, 7, 2020–2029 CrossRef CAS PubMed.
- F. M. van der Valk, D. F. van Wijk, M. E. Lobatto, H. J. Verberne, G. Storm, M. C. M. Willems, D. A. Legemate, A. J. Nederveen, C. Calcagno, V. Mani, S. Ramachandran, M. P. M. Paridaans, M. J. Otten, G. M. Dallinga-Thie, Z. A. Fayad, M. Nieuwdorp, D. M. Schulte, J. M. Metselaar, W. J. M. Mulder and E. S. Stroes, Nanomedicine, 2015, 11, 1039–1046 CrossRef CAS PubMed.
- X. Cheng, X. Tian, A. Wu, J. Li, J. Tian, Y. Chong, Z. Chai, Y. Zhao, C. Chen and C. Ge, ACS Appl. Mater. Interfaces, 2015, 7, 20568–20575 CrossRef CAS PubMed.
- J. H. Lewis, M. Jadoul, G. A. Block, M. P. Chin, D. A. Ferguson, A. Goldsberry, C. J. Meyer, M. O'Grady, P. E. Pergola, S. A. Reisman, W. C. Wigley and G. M. Chertow, Clin. Transl. Sci., 2021, 14, 299–309 CrossRef CAS PubMed.
- H. Zhang, H. Liu, L. Zhou, J. Yuen and H. J. Forman, Free Radicals Biol. Med., 2017, 113, 304–310 CrossRef CAS PubMed.
- S. M. Tan, A. Sharma, N. Stefanovic, D. Y. C. Yuen, T. C. Karagiannis, C. Meyer, K. W. Ward, M. E. Cooper and J. B. De Haan, Diabetes, 2014, 63, 3091–3103 CrossRef PubMed.
- A. Sharma, L. Rizky, N. Stefanovic, M. Tate, R. H. Ritchie, K. W. Ward and J. B. de Haan, Cardiovasc. Diabetol., 2017, 16, 33 CrossRef PubMed.
- M. Chin, C. Y. Lee, J. C. Chuang, R. Bumeister, W. C. Wigley, S. T. Sonis, K. W. Ward and C. Meyer, Am. J. Physiol.: Renal, Fluid Electrolyte Physiol., 2013, 304, F1438–F1446 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1bm01421h |
| This journal is © The Royal Society of Chemistry 2022 |