
Bile acid linked β-glucan nanoparticles for liver specific oral delivery of biologics
Ayreen S.
Chowdhury†
ab,
Renu
Geetha Bai†
ac,
Tamanna
Islam
a,
Muhammad
Abir
d,
Mahesh
Narayan
 e,
Zehedina
Khatun
*a and
Md
Nurunnabi
*adf
e,
Zehedina
Khatun
*a and
Md
Nurunnabi
*adf
aDepartment of Pharmaceutical Sciences, School of Pharmacy, University of Texas at El Paso, El Paso, TX 79902, USA. E-mail: mnurunnabi@utep.edu; zkhatun1@utep.edu; Tel: +915-747-8335 Tel: +915-747-8098
bDepartment of Bioscience, School of Science and Technology, Nottingham Trent university, Nottingham, NG11 8NS, UK
cSchool of Natural Sciences and Health, Tallinn University, Tallinn, 10120, Estonia
dAerospace Center (cSETR), University of Texas at El Paso, El Paso, TX 79965, USA
eDepartment of Chemistry and Biochemistry, University of Texas at El Paso, El paso, TX 79965, USA
fBiomedical Engineering, College of Engineering, University of Texas at El Paso, El Paso, TX 79965, USA
First published on 8th April 2022
Abstract
Oral delivery remains one of the most convenient routes for drug administration compared to intravenous, intramuscular, and via suppositories. However, due to the risk of degradation, and proteolysis of molecules in the acidic gastric medium, as well as the difficulty of transporting large molecules through the intestinal membrane, more than half of the therapeutic molecules are prohibited for oral administration. Moreover, most of the large molecules and biological therapeutics are not available in oral dosage form due to their instability in the stomach and inability of intestinal absorption. To achieve expected bioavailability, an orally administered therapeutic molecule must be protected within the stomach, and transportation facilitated via the small intestine. In this project, we have introduced a hybrid carrier, composed of Taurocholic Acid (TA) and β-Glucan (TAG), that is shown to be effective for the simultaneous protection of the biologics in acidic buffer and simulated gastric juice as well as facilitate enhanced absorption and transportation via the small intestine. In this project, we have used an eGFP encoded plasmid as a model biologic to prepare particles mediated with TAG. TAG show the potential of enhancing transfection and expression of eGFP as we have observed two fold higher expression in the cell upon coincubation for 4 h. In vivo studies on orally dosed mice showed that eGFP expression in the liver was significantly higher in TAG containing particles compared to particles without TAG. The findings suggest that the TAG carrier is capable of not only preserving biologics but also transporting them more efficiently to the liver. As a result, this strategy can be employed for a variety of liver-targeted therapeutic delivery to treat a variety of liver diseases.
1. Introduction
Oral medications are preferred over other routes of administration due to their numerous benefits, including patient compliance, convenience, cost effectiveness, and non-invasiveness.1–4 Besides systemic absorption via oral delivery, the oral route is also considered as the most effective route of drug administration to treat local diseases in the gastrointestinal tract.4–8 However, oral formulations have several problems, particularly with peptides and proteins, including poor stability in the gastric environment and low/no bioavailability.9–11 The acidic condition in the stomach facilitates proteolyzing or degrading of the biologics and mucus membrane to prevent drug penetration and absorption. To overcome these barriers, formulations of various approaches have been considered and investigated including mucoadhesive gel, polymeric particles, enteric coated granules and biocompatible materials based oral devices.4,12–15 Several steps have been taken to develop oral dosage forms of these pharmaceutical compounds that target select transport proteins and are transferred via transporters to improve oral absorption and bioavailability.16Previously, we have studied and reported bile acid transporter mediated oral delivery of anticancer drugs, imaging agents, and gene encoded Glucagon-like peptide (GLP)-1.17–19 Our findings demonstrated that the bile acid transporter, which is over-expressed in the small intestine, enhances the absorption and transportation of both small and large molecules. Interestingly, followed by absorption bile-acid-rich vehicles were found to accumulate within the liver. We also investigated the potential of β-glucan as an oral drug delivery vehicle.20 We observed that β-glucan in association with the GRGD peptide not only protects the antigen from harsh gastric conditions but also enhances the absorption of antigens.
Over the last few decades, β-glucan has been well-studied for various drugs and vaccine delivery.21–26 Recently, we have investigated how β-glucan interacts with lipid molecules in the presence of water and that resulted in the formulation of nano/microparticles.27 In this study, we have hypothesized that the use of β-glucan, an acid-resistant polysaccharide, can protect a sophisticated biological molecule from the hostile intra-gastric environment. On top of protection, β-glucan also works as an intestinal drug transporter and enhancer by binding to the dectin-1 receptor on dendritic cells.28–31 We have also hypothesized that the incorporation of a bile-acid moiety can further facilitate the absorption and transportation of the biological payloads at higher rates. Therefore, we have synthesized a vehicle composed of Taurocholic Acid (TA), a hydrophilic bile acid, and β-Glucan (TAG) to test this hypothesis. This vehicle is expected to demonstrate higher transportation, intestinal absorption, and transportation profiles via the synergistic interaction of the individual components. β-Glucan is employed to fabricate nanoparticles; loaded vaccines will be shielded from stomach acid and enzymes; and β-glucan can induce immunological responses.
Over 95% of bile acid–drug conjugates reabsorb in the small intestine during recirculation.32 Therefore, the incorporation of bile acid derivatives as a part of the oral drug delivery vehicle demonstrated excellent liver accumulation. TAG vehicles are expected to be transported via bile acid transporters, found in the apical compartment of ileum enterocytes of the small intestine. Once the TAG is absorbed and transported to the systemic circulation, it is anticipated that they deposit within the liver through the basolateral compartment of hepatocytes. In the basolateral compartment of liver cells, sodium taurocholate cotransporting polypeptide reabsorbs bile acid–drug conjugates from the hepatic portal vein, completing one enterohepatic circulation cycle.
To provide a proof-of-concept, we have prepared a multi-layered particle composed of TAG and eGFP (enhanced green fluorescent protein), a model plasmid. The 1H-NMR spectrum confirms the chemical conjugation between TA and β-glucan via the formation of an amide bond. The plasmid (eGFP) was condensed with cationic bPEI in varying ratios followed by wrapping by TAG. Characterization data reveal that particle sizes range from 20 to 30 nm, and 100 to 200 nm in diameter, for bPEI/eGFP (polyplex) and TAG/polyplex, respectively. Stability studies conducted under a variety of conditions, including acidic conditions, demonstrate that the TAG coated polyplex has better stability in the acid buffer than the free polyplex. In vitro studies with EaHY cell lines with TAG/polyplex show 2× higher expression of eGFP than polyplex, an indication of the potential of TAG for cellular uptake induction. Both polyplex and TAG/polyplex were orally administered to healthy mice to establish pharmacokinetic and biodistribution profiles of eGFP. The results show that TAG/polyplex is absorbed via the small intestine and deposited within the liver. Furthermore, the expression of eGFP within the liver tissues even indicated their specificity toward liver cells. Non-specific accumulations were limited to kidney tissue which is indicative of the excretion/elimination mechanism of the particles via kidney filtration. In summary, our platform can be used for the delivery of oral biologics to liver-specific diseases including liver cancer, fibrosis hepatitis, and fatty liver disease.
2. Materials and methods
2.1. Materials
β-Glucan extracted from barley (low viscosity) (MW: 166![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da) was purchased from Megazyme (Island). 4-Nitrophenylchloroformate (4-NPC) (>98%), 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide (EDC) (>98%), and triethylamine (TEA) (>99%) were purchased from TCI (Japan). Branched polyethylenimine (BPEI), taurocholic acid (TA), enhanced green fluorescence protein (eGFP) plasmid, ethylenediamine (EDA), and N-hydroxy-succinimide (>98%) (NHS) were purchased from Sigma-Aldrich (St Louis, MO).
000 Da) was purchased from Megazyme (Island). 4-Nitrophenylchloroformate (4-NPC) (>98%), 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide (EDC) (>98%), and triethylamine (TEA) (>99%) were purchased from TCI (Japan). Branched polyethylenimine (BPEI), taurocholic acid (TA), enhanced green fluorescence protein (eGFP) plasmid, ethylenediamine (EDA), and N-hydroxy-succinimide (>98%) (NHS) were purchased from Sigma-Aldrich (St Louis, MO).
2.2. Synthesis and characterization of TAG
:acetonitrile (50:50, V/V). The aqueous phase was collected and lyophilized using a freeze dryer.
2.3. Preparation of formulations
We have followed the previously reported and optimized methods for polyplex and layer-by-layer wrapping.19 eGFP and bPEI were separately diluted with saline (PBS) at 0.1 mg mL−1. To prepare the polyplex, an eGFP encoded plasmid DNA (pDNA) was initially condensed with branched polyethyleneimine (bPEI) at a N/P ratio of 5:1 to form a stable gene complex, viz. the polyplex (PP). Equal volumes of bPEI and eGFP were simultaneously introduced into a round bottom flask with constant stirring. The electrostatic interaction between the cationic bPEI and anionic eGFP facilitates the formation of particles upon stirring (at room temperature). The pre-synthesized TAG was dissolved in PBS at a concentration of 1 mg mL−1. Then an equal volume of the TAG solution and PP solution were simultaneously introduced into a round bottom flask with concurrent stirring with a ratio of 1/1 (w/w). The TAG wraps around the surface of PP and forms TAG/polyplex nanoparticles via a layer-by-layer coating strategy. Both PP and TAG/polyplex were characterized for their morphology and stability.
2.4. Characterization of the formulations
Morphological characterization of PP and BTCA-PP was performed using a transmission electron microscope/TEM (ELS-8000, Photal, Osaka, Japan).33 Particle sizes and morphology of the nanocomplex were estimated by dynamic laser scattering (DLS) (Malvern Panalytical, United Kingdom). Particle charge was measured using a zeta analyzer (JEOL, Japan). The chemical conjugation and amide bond formation between β-glucan and TA was evaluated by using 1H-NMR.20For TEM analysis, the formulations were dispersed in water and sonicated in a water bath for 30 seconds before dropping on the TEM grid.34 After 1 min of adding solution to the grid, excess water was eliminated using dry paper tissue and allowed to further dry in air under a fume-hood. The hydrodynamic volume and size changes, in various buffers, and the duration of the nanocomplex were measured and the charge of the particle was also evaluated.
2.5. Stability analysis
We measured the stability of the formulations in simulated gastric juice and different acidic buffers (pH 3 and 5) and investigated changes in their size distribution and surface charges.20 To evaluate the stability of the nanoparticles (polyplex and TAG/polyplex), the material is dispersed in 1 mg mL−1 simulated gastric juice (Sigma Aldrich) and their size and zeta potential are measured by DLS. To further investigate their stability in an “empty” stomach, the formulations were dissolved in an acidic buffer at a concentration of 1 mg mL−1. Size distribution and zeta potential were measured at different time points.2.6. In vitro gene expression study
Human umbilical vein cell line EaHy926 was purchased from ATCC and cultured according to the instructions provided (CRL-2922™, ATCCA). The cells were transferred to a 8-well chamber and incubated for another 24 h prior to adding the formulation. 200 μL of solutions (1 μg mL−1 equivalent to eGFP) of both polyplex and TAG/polyplex was added to each well and incubated for 4 h to facilitate internalization of the particles and expression of eGFP.35 The cells were then imaged using a confocal microscope with a green filter to capture comparative expression levels.2.7. Biodistribution of the eGFP
C57BL/6 was purchased from Charles River Lab (Waltham, MA) and housed at the University of Texas at El Paso (UTEP) vivarium. The protocol (#A-201910-1) for the animal studies was evaluated and approved by UTEP's Institutional Animal Care and Use Program (IACUP) committee. Polyplex and TAG/polyplex were dissolved in PBS at a concentration of 100 mg mL−1 (equivalent to eGFP) and 100 μL of the solution was administered via oral gavage (equivalent to 10 μg of eGFP). The animals were sacrificed within 24 h of post administration and the organs were harvested for quantitative and qualitative analysis of eGFP expression in various organs.36 The tissues were fixed with 10% paraformaldehyde for 30 min and transferred into paraffin block, followed by transferring and storing at −80 °C. Regarding microscopic imaging, the tissue block was cut at 10 μm thickness and embedded on glass slides. The tissue embedded glass slides were imaged using a confocal microscope with a green filter to capture the degree of eGFP expression. Regarding quantitative analysis, the tissue was dissolved in PBS and homogenized. The homogenized tissues were centrifuged, and the precipitated tissues were discarded. The supernatant was used to measure the intensity of eGFP using a microplate reader. Tissues from untreated healthy mice were used as a negative control and subtracted from the reading to correct for auto fluorescence.193. Results and discussion
The chemical conjugation of TA with BG was achieved using different amine catalysts and confirmed by 1H-NMR analysis. Fig. 1 shows the 1H-NMR of BG (A), TA (B), and TAG (C), respectively. The NMR peak, chemical shift, and J-coupling were analyzed by the MestReNova (version 14.2.3) software. The values of the chemical shift obtained for each of the different proton contents in the samples from the 1H-NMR (400 MHz, DMSO) experiment are listed below:The chemical shift for BG: δ 4.68 (s, 3H), 3.34 (d, J = 15.1 Hz, 1H). The chemical shift for TA: δ 7.67 (s, 1H), 4.32 (d, J = 4.4 Hz, 1H), 4.12 (d, J = 3.5 Hz, 1H), 3.99 (dd, J = 27.6, 5.3 Hz, 1H), 3.78 (s, 1H), 3.61 (s, 1H), 3.25 (dq, J = 15.1, 6.6 Hz, 2H), 3.18 (s, 2H), 2.90 (s, 1H), 2.74 (s, 1H), 2.21 (dt, J = 23.4, 11.8 Hz, 1H), 2.08–1.90 (m, 1H), 1.64 (d, J = 12.6 Hz, 2H), 1.42 (s, 3H), 1.37 (d, J = 13.5 Hz, 1H), 1.25 (s, 1H), 1.15 (q, J = 7.0 Hz, 1H), 0.92 (d, J = 6.4 Hz, 3H), 0.84 (s, 1H), 0.81 (s, 3H), 0.59 (s, 3H). The chemical shift for TAG: δ 5.43 (s, 3H), 5.30 (s, 1H), 4.69 (s, 7H), 4.35 (s, 4H), 3.80 (s, 3H), 3.70 (s, 1H), 3.56 (s, 3H), 3.13 (s, 2H), 3.06 (s, 1H). Based on the analysis, it can be concluded that BG can effectively interact with TA to form TAG via preferentially creating the –OCO–NH–C2H4–NH–O– linkage. The following reaction presented in Scheme 1 potentially represents the trajectory towards the formation of TAG.
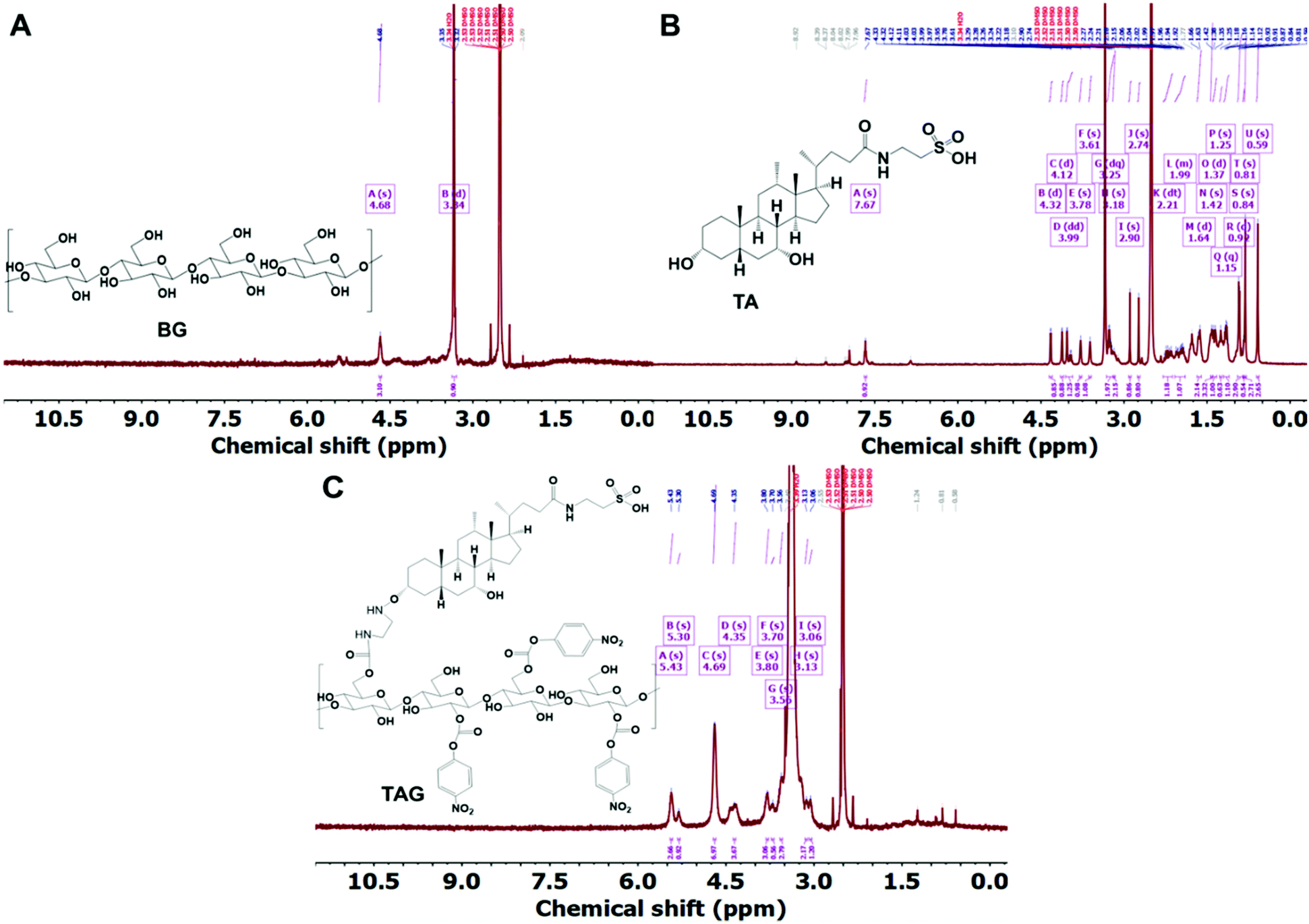 | ||
| Fig. 1 A, B, and C represent the 1H-NMR plots of BG, TA, and TAG, respectively. The peak-assignment confirms the chemical conjugation between BG and TA via amide bond formation. | ||
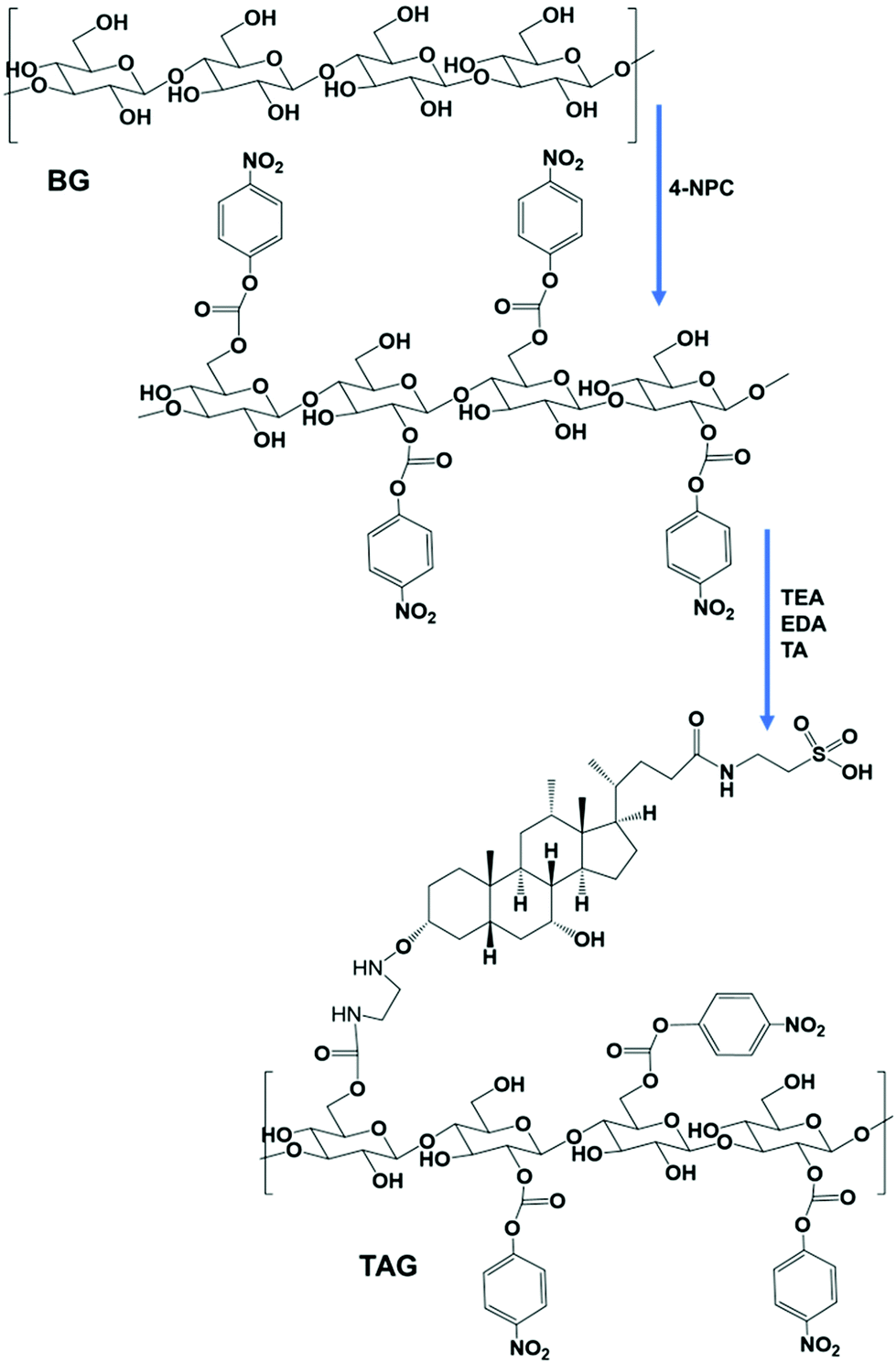 | ||
| Scheme 1 The scheme shows the chemical conjugation process and bond formation between β-glucan and TA. The feed molar ratio of β-glucan and TA was 1:10. The chemical conjugation and amide bond formation between β-glucan and TA were confirmed by 1H-NMR. | ||
Fig. 2A outlines the steps of polyplex and TAG/polyplex formation. Polyplex is formed by the assembly of bPEI and plasmid DNA, whereas TAG is formed by the combination of β-glucan and TA. During the formation of polyplex, we observe that the polyplex of 1:3 and 1:5 (N:P ratios) is saturated as the size was very much stable, based on the size distribution data (Fig. 2B). The zeta potential data also reveal that the surface charge of 1:2 polyplex is cationic and higher than that of 1:3 polyplex. The measured average charge of bPEI was 37.1 mV, whereas the charge of TAG was −78.8 and −151 mV for 1 mg ml−1 and 0.5 mg mL−1 respectively. The size and charge analysis were performed by various EGFP:bPEI ratios. From the analysis the optimal lowest size was observed for the N:P ratio of 1. However, depending on the increase in the N:P ratio the charge of the system changed from negative to positive values as shown in Fig. 2B. As the N:P ratio increases the size and charge of the polyplex showed a more stable profile in Fig. 2C. Therefore, based on the size distribution and zeta potential value, we selected polyplex (1:2 N:P ratio) for the next step.
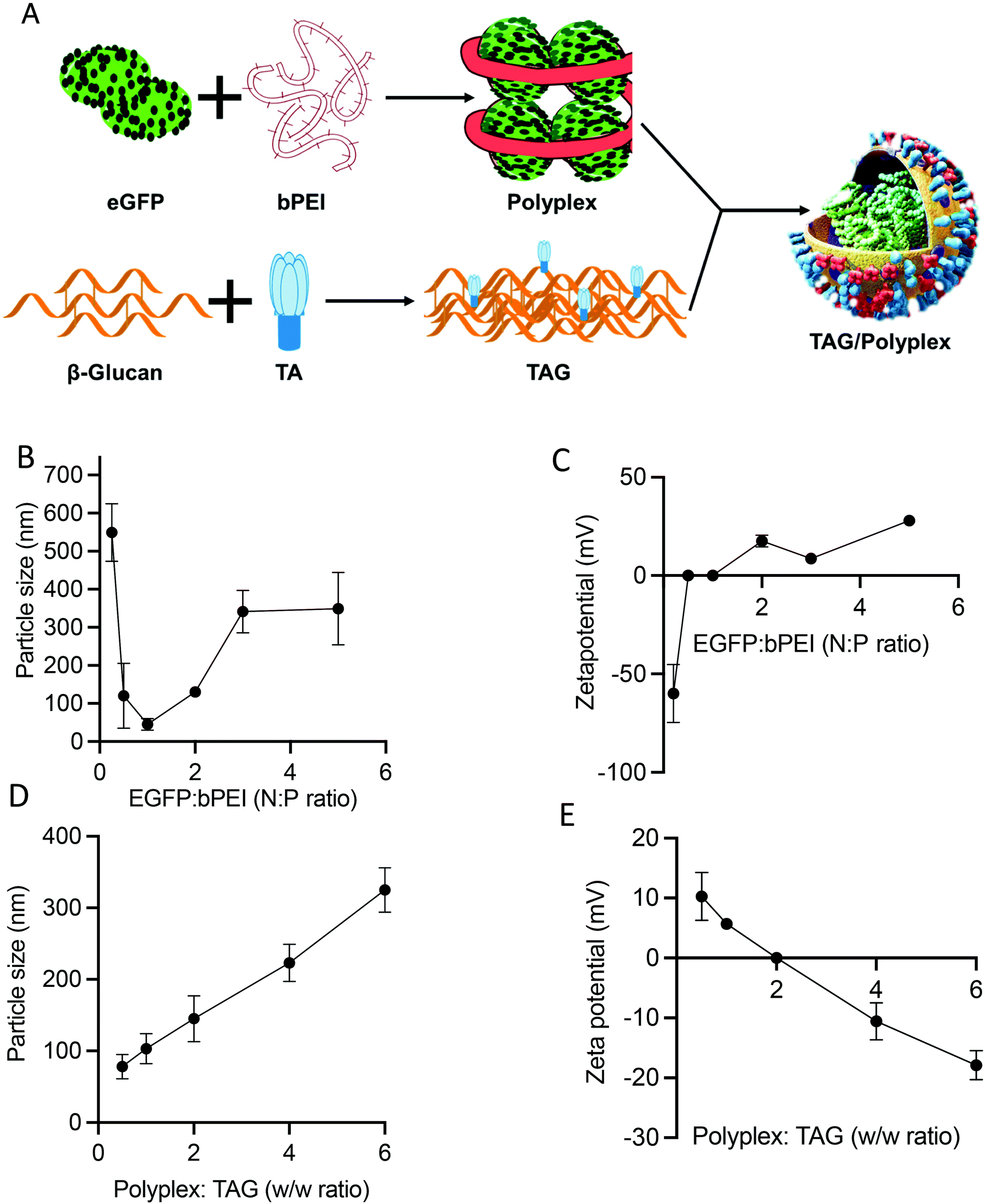 | ||
| Fig. 2 The scheme represents how the TAG/polyplex complex was formed (A). Particle size (B) and zeta potential (C) of polyplex. Particle size (D) and zeta potential (E) of TAG/polyplex. An EGFP:bPEI ratio of 1:2 was considered to prepare TAG/polyplex particles. | ||
During the process of TAG/polyplex complex preparation, polyplex acts as the solid core where TAG is the shield covering and protecting the biologic containing inner core. The particle size and charge of the TAG/polyplex were evaluated using DLS and zeta analyses. The particle size analysis of the TAG/polyplex is displayed in Fig. 2D and the zeta potential analysis is shown in Fig. 2E. As the w/w ratio increases, the particle size of TAG/polyplex increases and the zeta values shifted from positive to negative. The TAG creates a layer on the outer surface of the polyplex via electrostatic interactions where β-glucan is expected to provide protection from the acidic environment in the lumen of the stomach. TA is expected to be exposed on the outermost surface of the particles to be able to interact with bile acid transporters of the intestine. Therefore, polyplex wrapping by TAG not only results in a size increment but also impacts surface charge. The TAG/polyplex (1:0.5 and 1:1 w/w) particles exhibited positive surface charges of 10 and 6 mV, respectively.
The TEM images reveal the morphological features of the “as-prepared” polyplex and TAG/polyplex (Fig. 3). The polyplex showed a homogeneous distribution of the complex in the size range of 10–50 nm diameter. Nevertheless, after wrapping with negatively charged TAG the size increases to 147.5 ± 1.3 nm in diameter. The size and morphology results indicate that the polyplex is completely wrapped by TAG as an outer shell, which causes an increase in particle size. Furthermore, the TEM analysis displayed the formation of a shield (thin coating) of TAG covering the comparatively compact polyplex core. The larger size of the TAG/polyplex particles also indicates that the individual nanoparticle may consist of multiple polyplexes loaded into it. The coating of TAG conjugates over the polyplex was initiated to cover the cationic surface, thus protecting the pDNA from gastrointestinal degradation and finally facilitating intestinal absorption and transportation of the entrapped eGFP via transporters across the small intestine.
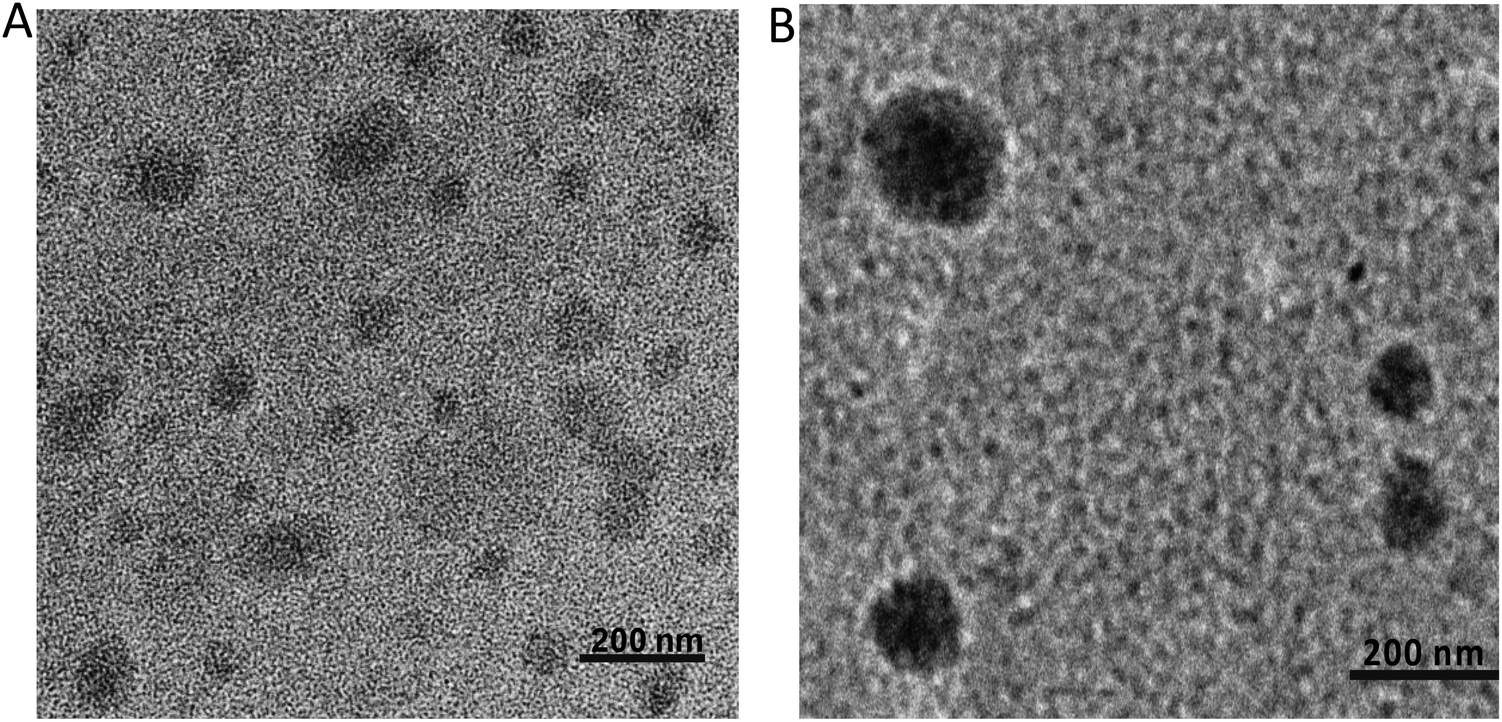 | ||
| Fig. 3 Size and morphology were measured using transmission electron microscopy (TEM) for polyplex (A) and TAG/polyplex (B). Scale bars represent 200 nm. | ||
The stability of the polyplex and TAG/polyplex was tested for 0–12 h duration in simulated gastric juice. Simulated gastric juice contains pepsin on top of hydrochloric acid, sodium chloride and water. Upon incubation the formulation interacts with the pepsin as well as free protons. Polyplex revealed a tremendous increment in size from 147.5 nm to 467.4 nm, attributed to the possible interaction between the cationic polyplex and the proton concentration in the acidic stomach. Another possible reason for size increment is the potential aggregation of the complex, which may have played a vital role in size increment. By contrast, the size distribution of TAG/polyplex formulation remained almost constant without many changes in the size of the particles (Fig. 4A), which showcases the stable nature of TAG/polyplex under stomach simulated conditions. This finding provides a proof of our hypothesis that TAG wrapping can potentially protect the entrapped polyplex as well as eGFP from being aggregated, dissociated or proteolyzed. The zeta potential profile of polyplex and TAG/polyplex also showed similar trends. The negative zeta potential of the polyplex slowly increased which is directly proportional to the incubation period, whereas the zeta potential value of the TAG/polyplex particle remained stable without much difference in the duration of incubation, as displayed in Fig. 4B.
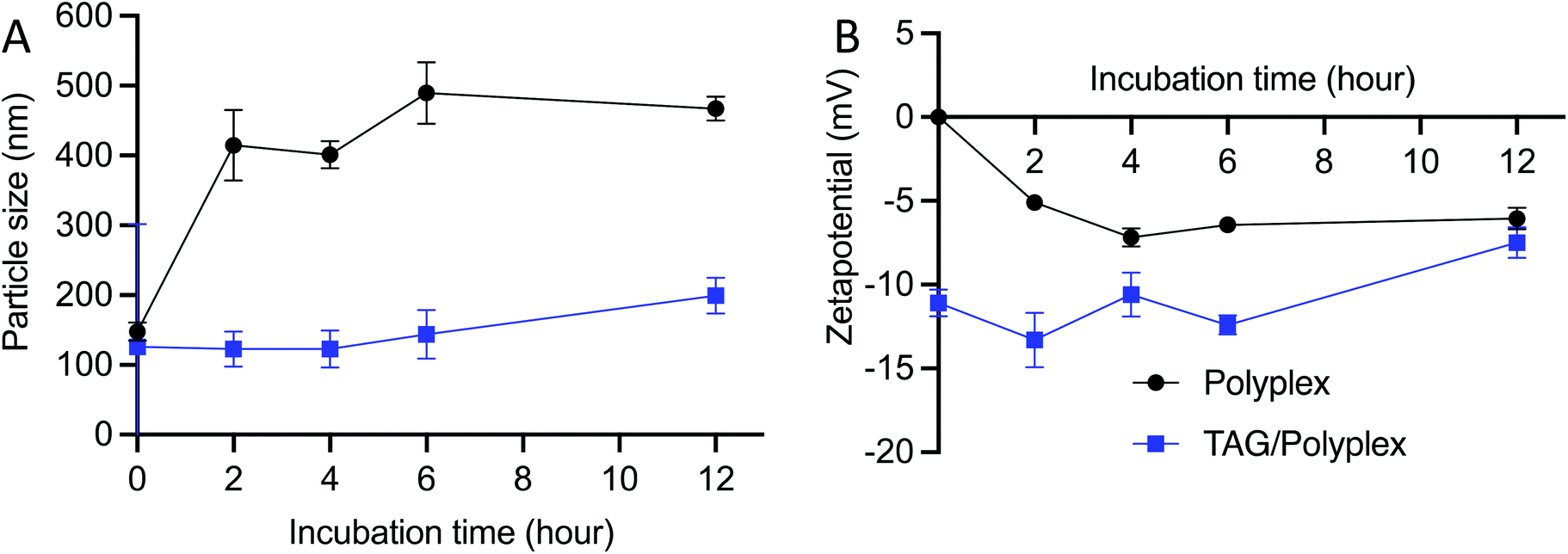 | ||
| Fig. 4 Stability of the particles in the simulated gastric juice. The size distribution (A) and zeta potential (B) stability profile, for 12 h of incubation, shows that the TAG/polyplex particles are more stable compared to that of polyplex within the gastric juice. | ||
To evaluate the stability profile of the oral biologic in the stomach, size distribution and zeta potential of the particles were analyzed by dissolving the formulations (polyplex and TAG/polyplex) in acidic pH buffer (pH 3 and pH 5) for a duration of 16 h. Fig. 5A depicts the changes in particle size changes of polyplex and TAG/polyplex in pH 3 and pH 5 buffers. In pH 3 buffer, the size of the polyplex fluctuated up to 4 h, and then stabilized in size until 8 h. This was then followed by a gradual decrease in size until 16 h. In pH 5, polyplex gradually increased in size up to 8 h and then showed a decrease in size. The size of TAG/polyplex in pH 3 buffer also showed an increase in size <400 nm due to possible aggregation of the complex till the 4 h. Then, due to charge stabilization it showed a decrease in size afterwards. In the case of polyplex in pH 3and 5 and TAG/polyplex in pH 3 the change in size after 8 h showed a similar stable size profile showing a decrease in size. However, TAG/polyplex in the pH 5 buffer showed a different profile. For TAG/polyplex in pH 5, the size ∼140 nm at 0 h has increased to >300 nm due to the charge interactions and aggregation process, however, after 4 h it showed a stable profile with almost the same size until 16 h. The findings demonstrate the ability of TAG to maintain the physical stability of the particle for a duration that is enough for them to pass through the stomach and reach out to the intestine.
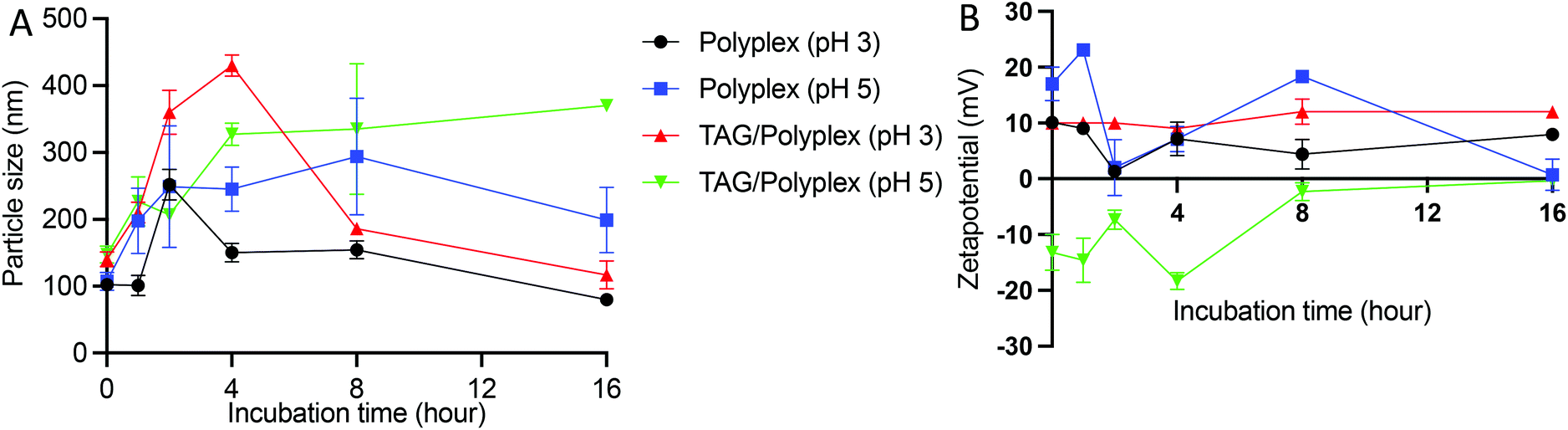 | ||
| Fig. 5 Stability profile of polyplex and TAG/polyplex in various pH buffers to mimic stomach acid. Both particle size (A) and zeta potential (B) show that the TAG containing particles are relatively more stable than polyplex. | ||
As shown in Fig. 5B, in the pH 3 buffer, the presence of protons (H+ ions) did not interfere with the original positive charge of the polyplex and thereforethe charge remained almost stable for the entire incubation duration. However, in the pH 5 buffer the polyplex showed fluctuations in charge (from +15 to −15 mV) due to the interaction of protons in the buffer until 8 h and then gradually increased and stabilized to a neutral charge during the tested duration. In the case of TAG/polyplex, in pH 3 buffer, the system showed a very stable zeta profile without many significant changes in values. Whereas, in the pH 5 buffer the charge of the TAG/polyplex also showed some variations (from a negative charge to an almost neutral charge) until 8 h and then displayed stable charge and, thus, zeta values.
The transport and update of the oral delivery system in the liver and small intestine are regulated by a bile acid uptake protein such as sodium/taurocholate co-transporting polypeptides (NTCPs) and apical sodium dependent bile acid transporter (ASBT) receptors.36–38 The quantitative cellular uptake and transfection efficiency of the polyplex and TAG/polyplex was tested on EaHY cells upon co-incubation for 4 h. The TAG modified polyplex (TAG/polyplex) exhibited higher expressions of enhanced green fluorescence in the treated cells compared to the polyplex. The green fluorescence intensity within EaHY cells correlates with its cellular uptake and expression of eGFP. The presence of bile acid on the TAG/polyplex particle enables increased cellular uptake and cell internalization, thereby enhancing transfection efficiency of TAG/polyplex (Fig. 6A). The relative quantitative fluorescence intensity (RFU) is displayed in Fig. 6B, where TAG/polyplex showed ∼350 RFU, which is almost 2-fold higher fluorescence intensity than that of polyplex treated cells at ∼170 RFU. This facilitates the quantitative analysis of the cellular uptake comparison between polyplex and TAG/polyplex.
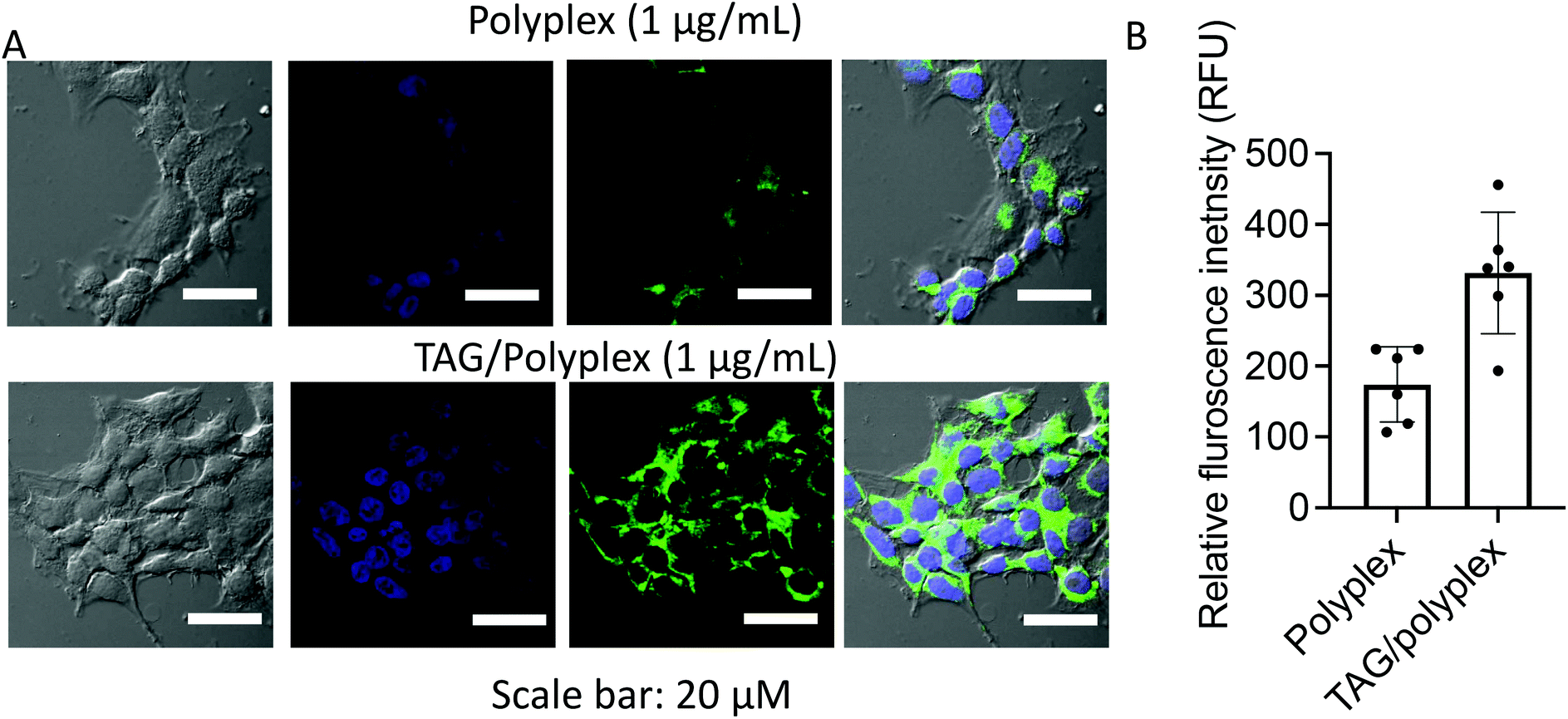 | ||
| Fig. 6 Cellular uptake and expression of eGFP at 4 h of post-incubation (A), and quantitative analysis of eGFP expression to each cell (B). Data are presented as mean ± STD, where n = 6. | ||
The TAG/polyplex was designed to improve intestinal absorption, cellular uptake, and transfection efficiency of the entrapped eGFP via specific intestinal receptors. The in vivo studies were performed, on mice models, to evaluate the oral absorption profile and biodistribution of eGFP and compared between TAG/polyplex and polyplex. A total of 5 treatment groups were used in the study with each group containing 5 mice. After 24 h of administration via oral gavage, the animals were sacrificed and dissected to collect the organs – small intestine (jejunum, duodenum, ileum), lungs, liver, heart, kidneys, and spleen to evaluate organ specific accumulation and expression of eGFP. As shown in Fig. 7A, the biodistribution of the entrapped eGFP in polyplex and TAG/polyplex corresponds to the green fluorescence in the images, whereas the buffer did not exhibit any fluorescence (negative control). The microscopic images show a significant enhancement in the fluorescence intensity of TAG/polyplex compared to the polyplex within the duodenum region. By contrast, a lighter green expression was observed in the duodenum and jejunum for polyplex. This result supports the notion of improved cellular uptake by the transporter-based targeting mechanism and transfection efficiency of the TAG containing carriers in 24 h. ImageJ was used to perform quantitative analysis of the microscopic images by picking a small region of interest (ROI) and measuring the mean intensity inside the ROI.39,40 Both images and quantitative analysis based on the distribution profile showed excellent expression of eGFP for TAG/polyplex in the liver. Whereas in the kidneys both polyplex and TAG/polyplex showed similar fluorescence intensities. The highest fluorescence intensity was observed in the duodenum followed by the liver and kidneys which support the efficient transfection of the gene complex through TAG/polyplex-mediated transport through the intestinal receptors. The enterohepatic circulation of bile acids enables TAG to absorb and transport through the small intestine and accumulate in the liver, resulting in a strong fluorescence intensity in liver tissues. Furthermore, the fluorescence exhibited by the animals treated with polyplex depicted the nonspecific transfection of eGFP, which could be due to the electrostatic interactions of polyplex with a negatively charged glycocalyx in mucin, it degrades or destabilizes the gene complex.
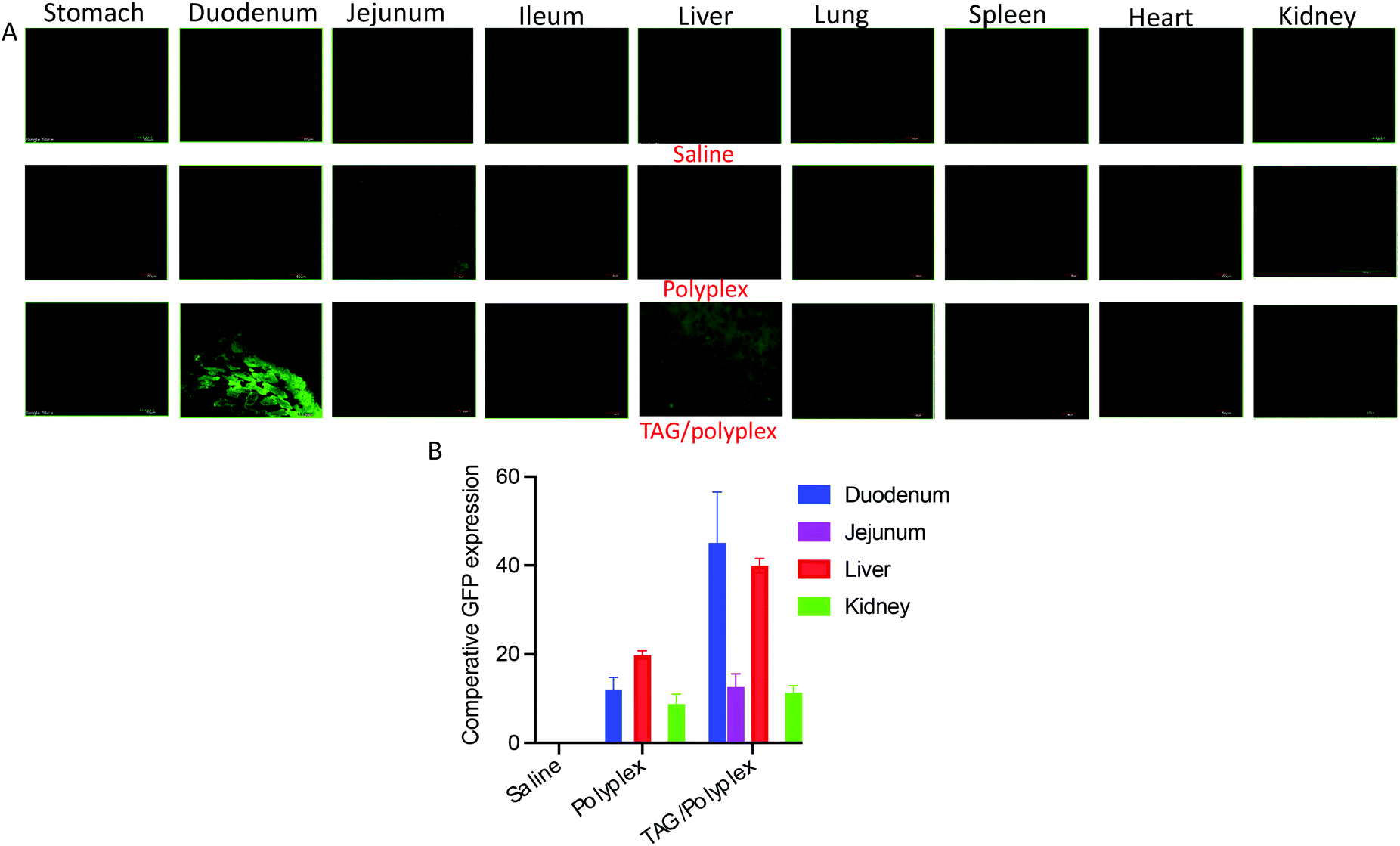 | ||
| Fig. 7 Biodistribution and quantitative expression of eGFP in different tissues/organs of orally administered polyplex and TAG/polyplex. TAG containing particles show duodenum-specific transportation and liver accumulation. Liver accumulation of eGFP for TAG-mediated particles was 3× higher than polyplex. | ||
From the in vivo studies, we underscore that the uptake of the gene complex occurred in the small intestine through enterocytes by the synergy of the bile acid transporter and dectin-1 receptor.41,42 Following that, the transfection efficacy of the orally administered TAG/polyplex in comparison with the cationic polyplex, expression of the gene was evaluated in different tissues. A relative quantitative biodistribution analysis (Fig. 7B) was performed in the stomach, duodenum, jejunum, ileum, heart, liver, lungs, spleen, and kidneys. The relative fluorescence analysis profile suggests that polyplex without TAG did not demonstrate any specific absorption profile in the mice tissues. However, nonspecific absorption is observed in the case of polyplex, which may result from their positive charge. The electrostatic interactions between negatively charged surface glycolipids and glycoproteins, and positively charged polyplex eventually result in nonspecific transportation via the intestine. Compared with polyplex, TAG/polyplex showed as much as two fold higher transfection efficiency in the liver, which indicates the ability of TAG to entrap and carry the biological payload to the liver.
4. Conclusion
In the current study, TA and β-glucan were synthesized using biocompatible amide bonds, which are also stable under an acidic environment. Various independent characterization approaches were used to validate the accuracy of the chemical conjugation and nanoparticle formulation via electrostatic interactions. Moreover, these nanoparticles remain stable for a long duration under acidic buffer conditions without compromising their integrity. Both in vitro and in vivo studies depicted that the TAG, a hybrid vehicle composed of bile acid and β-glucan, has the potential of shielding the biological molecules from the harsh environment of the stomach, as well as accelerating oral absorption and transportation to the systemic circulation. More importantly, the in vivo biodistribution profile demonstrate that the orally administered vehicle has target specificity for the liver. This demonstrates the potential and efficacy of this newly devised and precise method of administering oral large molecules, including, biologics, proteins, and peptides. We anticipate that, with further development, this promising nonviral gene delivery strategy can be used for the oral delivery of therapeutic genes and vaccines to treat various liver diseases.Conflicts of interest
There are no conflicts to delcare.Acknowledgements
We acknowledge funding by the Cancer Prevention Research Institute of Texas (CPRIT) through the Texas Regional Excellence in Cancer Award (TREC) under Award No. PR210153. Research reported in this publication was also supported by the Research Centers at Minority Institutions grant funded by the National Institute on Minority Health and Health Disparities of the National Institutes of Health under Award No. U54MD007592. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.References
- M. R. Patel, R. B. Patel and S. D. Thakore, Nanoemulsion in drug delivery. Applications of nanocomposite materials in drug delivery, Elsevier, 2018, pp. 667–700 Search PubMed.
- M. S. Alqahtani, M. Kazi, M. A. Alsenaidy and M. Z. Ahmad, Advances in oral drug delivery, Front. Pharmacol., 2021, 62 Search PubMed.
- B. Homayun, X. Lin and H.-J. Choi, Challenges and recent progress in oral drug delivery systems for biopharmaceuticals, Pharmaceutics, 2019, 11, 129 CrossRef CAS PubMed.
- S. Attili-Qadri, N. Karra, A. Nemirovski, O. Schwob, Y. Talmon and T. Nassar, et al., Oral delivery system prolongs blood circulation of docetaxel nanocapsules via lymphatic absorption, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 17498–17503 CrossRef CAS PubMed.
- S. Hua, Advances in oral drug delivery for regional targeting in the gastrointestinal tract-influence of physiological, pathophysiological and pharmaceutical factors, Front. Pharmacol., 2020, 11, 524 CrossRef CAS PubMed.
- V. Sankar, V. Hearnden, K. Hull, D. V. Juras, M. Greenberg and A. Kerr, et al., Local drug delivery for oral mucosal diseases: challenges and opportunities, Oral Dis., 2011, 17, 73–84 CrossRef PubMed.
- C. Paderni, D. Compilato, L. I. Giannola and G. Campisi, Oral local drug delivery and new perspectives in oral drug formulation, Oral Surg., Oral Med., Oral Pathol. Oral Radiol., 2012, 114, e25–e34 CrossRef PubMed.
- K. H. Clitherow, C. Murdoch, S. G. Spain, A. M. Handler, H. E. Colley and M. B. Stie, et al., Mucoadhesive electrospun patch delivery of lidocaine to the oral mucosa and investigation of spatial distribution in a tissue using MALDI-mass spectrometry imaging, Mol. Pharm., 2019, 16, 3948–3956 CrossRef CAS PubMed.
- Q. Zhu, Z. Chen, P. K. Paul, Y. Lu, W. Wu and J. Qi, Oral delivery of proteins and peptides: Challenges, status quo and future perspectives, Acta Pharm. Sin. B, 2021, 11, 2416–2448 CrossRef CAS PubMed.
- J. Renukuntla, A. D. Vadlapudi, A. Patel, S. H. Boddu and A. K. Mitra, Approaches for enhancing oral bioavailability of peptides and proteins, Int. J. Pharm., 2013, 447, 75–93 CrossRef CAS PubMed.
- S. Verma, U. K. Goand, A. Husain, R. A. Katekar, R. Garg and J. R. Gayen, Challenges of peptide and protein drug delivery by oral route: Current strategies to improve the bioavailability, Drug Dev. Res., 2021, 82, 927–944 CrossRef CAS PubMed.
- J. Zhang, P. Xu, A. Q. Vo and M. A. Repka, Oral drug delivery systems using core–shell structure additive manufacturing technologies: a proof-of-concept study, J. Pharm. Pharmacol., 2021, 73, 152–160 CrossRef PubMed.
- T. D. Brown, K. A. Whitehead and S. Mitragotri, Materials for oral delivery of proteins and peptides, Nat. Rev. Mater., 2020, 5, 127–148 CrossRef.
- L. Vaut, J. J. Juszczyk, K. Kamguyan, K. E. Jensen, G. Tosello and A. Boisen, 3D printing of reservoir devices for oral drug delivery: from concept to functionality through design improvement for enhanced mucoadhesion, ACS Biomater. Sci. Eng., 2020, 6, 2478–2486 CrossRef CAS PubMed.
- L. M. Ensign, R. Cone and J. Hanes, Oral drug delivery with polymeric nanoparticles: the gastrointestinal mucus barriers, Adv. Drug Delivery Rev., 2012, 64, 557–570 CrossRef CAS PubMed.
- S. Shahriar, J. Mondal, M. N. Hasan, V. Revuri, D. Y. Lee and Y.-K. Lee, Electrospinning nanofibers for therapeutics delivery, Nanomaterials, 2019, 9, 532 CrossRef CAS PubMed.
- Z. Khatun, M. Nurunnabi, G. R. Reeck, K. J. Cho and Y.-K. Lee, Oral delivery of taurocholic acid linked heparin–docetaxel conjugates for cancer therapy, J. Controlled Release, 2013, 170, 74–82 CrossRef CAS PubMed.
- Z. Khatun, M. Nurunnabi, K. J. Cho and Y.-K. Lee, Oral delivery of near-infrared quantum dot loaded micelles for noninvasive biomedical imaging, ACS Appl. Mater. Interfaces, 2012, 4, 3880–3887 CrossRef CAS PubMed.
- M. Nurunnabi, S.-A. Lee, V. Revuri, Y. H. Hwang, S. H. Kang and M. Lee, et al., Oral delivery of a therapeutic gene encoding glucagon-like peptide 1 to treat high fat diet-induced diabetes, J. Controlled Release, 2017, 268, 305–313 CrossRef CAS PubMed.
- D.-Y. Lee, M. Nurunnabi, S. H. Kang, M. Nafiujjaman, K. M. Huh and Y.-K. Lee, et al., Oral gavage delivery of PR8 Antigen with β-glucan-conjugated GRGDS carrier to enhance M-cell targeting ability and induce immunity, Biomacromolecules, 2017, 18, 1172–1179 CrossRef CAS PubMed.
- K. Lee, Y. Kwon, J. Hwang, Y. Choi, K. Kim and H.-J. Koo, et al., Synthesis and functionalization of β-glucan particles for the effective delivery of doxorubicin molecules, ACS Omega, 2019, 4, 668–674 CrossRef CAS.
- Y. Su, L. Chen, F. Yang and P. C. Cheung, Beta-d-glucan-based drug delivery system and its potential application in targeting tumor associated macrophages, Carbohydr. Polym., 2021, 253, 117258 CrossRef CAS PubMed.
- R. De Smet, L. Allais and C. A. Cuvelier, Recent advances in oral vaccine development: Yeast-derived β-glucan particles, Hum. Vaccines Immunother., 2014, 10, 1309–1318 CrossRef CAS PubMed.
- V. Vetvicka, L. Vannucci and P. Sima, β–glucan as a new tool in vaccine development, Scand. J. Immunol., 2020, 91, e12833 Search PubMed.
- J. Huang, C. Wu, S. Tang, P. Zhou, J. Deng and Z. Zhang, et al., Chiral Active β-Glucan Nanoparticles for Synergistic Delivery of Doxorubicin and Immune Potentiation, Int. J. Nanomed., 2020, 15, 5083 CrossRef CAS PubMed.
- E. Stuyven, F. Verdonck, I. Van Hoek, S. Daminet, L. Duchateau and J. P. Remon, et al., Oral administration of β-1, 3/1, 6-glucan to dogs temporally changes total and antigen-specific IgA and IgM, Clin. Vaccine Immunol., 2010, 17, 281–285 CrossRef CAS PubMed.
- T. Islam, M. N. Huda, M. A. Ahsan, H. Afrin, C. Joseph J Salazar and M. Nurunnabi, Theoretical and experimental insights into the possible interfacial interactions between β-Glucan and fat molecules in aqueous media, J. Phys. Chem. B, 2021, 125, 13730–13743 CrossRef CAS PubMed.
- H. Huang, G. R. Ostroff, C. K. Lee, C. A. Specht and S. M. Levitz, Robust stimulation of humoral and cellular immune responses following vaccination with antigen-loaded β-glucan particles, mBio, 2010, 1, e00164–e00110 CrossRef PubMed.
- P. R. Taylor, G. D. Brown, D. M. Reid, J. A. Willment, L. Martinez-Pomares and S. Gordon, et al., The β-glucan receptor, dectin-1, is predominantly expressed on the surface of cells of the monocyte/macrophage and neutrophil lineages, J. Immunol., 2002, 169, 3876–3882 CrossRef CAS PubMed.
- G. D. Brown, P. R. Taylor, D. M. Reid, J. A. Willment, D. L. Williams and L. Martinez-Pomares, et al., Dectin-1 is a major β-glucan receptor on macrophages, J. Exp. Med., 2002, 196, 407–412 CrossRef CAS PubMed.
- M. J. Elder, S. J. Webster, R. Chee, D. L. Williams, J. Hill Gaston and J. C. Goodall, β-Glucan size controls dectin-1-mediated immune responses in human dendritic cells by regulating IL-1β production, Front. Immunol., 2017, 8, 791 CrossRef PubMed.
- D. B. Njoku, H. V. Chitilian and K. Kronish, Hepatic physiology, pathophysiology, and anesthetic considerations, in Miller's Anesthesia, 2020, pp. 420–443 Search PubMed.
- M. Nurunnabi, M. Nafiujjaman, S.-J. Lee, I.-K. Park, K. M. Huh and Y.-K. Lee, Preparation of ultra-thin hexagonal boron nitride nanoplates for cancer cell imaging and neurotransmitter sensing, Chem. Commun., 2016, 52, 6146–6149 RSC.
- Z. Khatun, M. Nurunnabi, M. Nafiujjaman, G. R. Reeck, H. A. Khan and K. J. Cho, et al., A hyaluronic acid nanogel for photo–chemo theranostics of lung cancer with simultaneous light-responsive controlled release of doxorubicin, Nanoscale, 2015, 7, 10680–10689 RSC.
- M. A. Ehrmann, C. H. Scheyhing and R. F. Vogel, In vitro stability and expression of green fluorescent protein under high pressure conditions, Lett. Appl. Microbiol., 2001, 32, 230–234 CrossRef CAS PubMed.
- H. Li, H. Wei, Y. Wang, H. Tang and Y. Wang, Enhanced green fluorescent protein transgenic expression in vivo is not biologically inert, J. Proteome Res., 2013, 12, 3801–3808 CrossRef CAS PubMed.
- K. S. Kim, K. Suzuki, H. Cho and Y. H. Bae, Selected Factors Affecting Oral Bioavailability of Nanoparticles Surface-Conjugated with Glycocholic Acid via Intestinal Lymphatic Pathway, Mol. Pharm., 2020, 17, 4346–4353 CrossRef CAS PubMed.
- F. Deng and Y. H. Bae, Bile acid transporter-mediated oral drug delivery, J. Controlled Release, 2020, 327, 100–116 CrossRef CAS PubMed.
- E. C. Jensen, Quantitative analysis of histological staining and fluorescence using ImageJ, Anat. Rec., 2013, 296, 378–381 CrossRef PubMed.
- C. T. Rueden, J. Schindelin, M. C. Hiner, B. E. DeZonia, A. E. Walter and E. T. Arena, et al., ImageJ2: ImageJ for the next generation of scientific image data, BMC Bioinf., 2017, 18, 1–26 CrossRef PubMed.
- I. D. Iliev, Dectin-1 exerts dual control in the gut, Cell Host Microbe, 2015, 18, 139–141 CrossRef CAS PubMed.
- S. Cohen-Kedar, L. Baram, H. Elad, E. Brazowski, H. Guzner-Gur and I. Dotan, Human intestinal epithelial cells respond to β–glucans via Dectin–1 and Syk, Eur. J. Immunol., 2014, 44, 3729–3740 CrossRef CAS PubMed.
Footnote |
| † Equal contribution. |
| This journal is © The Royal Society of Chemistry 2022 |