
Membrane-wrapped nanoparticles for nucleic acid delivery
Mackenzie A.
Scully
 a,
Eric H.
Sterin
a and
Emily S.
Day
*abc
a,
Eric H.
Sterin
a and
Emily S.
Day
*abc
aBiomedical Engineering, University of Delaware, Newark, DE 19716, USA. E-mail: emilyday@udel.edu
bMaterials Science and Engineering, University of Delaware, Newark, DE 19716, USA
cCenter for Translational Cancer Research, Helen F. Graham Cancer Center and Research Institute, Newark, DE 19713, USA
First published on 29th June 2022
Abstract
There is an unmet need for carriers that can deliver nucleic acids (NAs) to cancer cells and tumors to perpetuate gene regulation and manage disease progression. Membrane-wrapped nanoparticles (NPs) can be loaded with exogenously designed nucleic acid cargoes, such as plasmid deoxyribonucleic acid (pDNA), messenger ribonucleic acid (mRNA), small interfering RNA (siRNA), microRNA (miRNA), and immunostimulatory CpG oligodeoxynucleotides (CpG ODNs), to mitigate challenges presented by NAs’ undesirable negative charge, hydrophilicity, and relatively large size. By conjugating or encapsulating NAs within membrane-wrapped NPs, various physiological barriers can be overcome so that NAs experience increased blood circulation half-lives and enhanced accumulation in intended sites. This review discusses the status of membrane-wrapped NPs as NA delivery vehicles and their advancement in gene regulation for cancer management in vitro and in vivo. With continued development, membrane-wrapped NPs have great potential as future clinical tools to treat cancer and other diseases with a known genetic basis.
1. Introduction
1.1. Membrane-wrapped nanoparticles for cargo delivery and disease management
Coating nanoparticles (NPs) with cell-derived membranes is a “top-down” approach to creating bioinspired delivery vehicles that can hide from the immune system and deliver cargo to desired cells (Fig. 1). This method harnesses the unique combination of surface receptors, proteins, and phospholipids present on cellular membranes to enable effective biointerfacing.1,2 Natural cell membrane coatings are advantageous because they replicate the abundance, variety, and complexity of proteins that mediate cellular interactions with other cells and biomolecules in their microenvironment; “bottom-up” ligand conjugation strategies cannot achieve this level of complexity, which limits their immune evasion and targeting capabilities.3 Traditionally, NPs have been coated with poly(ethylene glycol) (PEG) to limit protein opsonization and clearance by the mononuclear phagocytic system (MPS), and/or coated with one or more targeting ligands (e.g., antibodies, peptides) to enable cell-specific binding. While this approach can increase NP circulation time and accumulation in target tissues, the level of improvement is modest and PEGylation has been associated with undesired immunological responses.4,5 Since it has been challenging to design stealth and actively targeted NPs with the desired capabilities by ligand conjugation techniques, researchers have turned to biomimicry as a promising biointerfacing strategy. The concept was introduced by Zhang and colleagues, who used red blood cell (RBC) membranes to camouflage poly(lactic-co-glycolic acid) (PLGA) NPs from immune clearance.6 They confirmed that CD47 “marker-of-self” proteins found on RBCs were successfully transferred in a “right-side-out” orientation onto the PLGA NP surface.1,2,6 Since CD47 acts as a “don't eat me” signal to inhibit phagocytosis by immune cells, the RBC-wrapped PLGA NPs exhibited 64% less macrophage engulfment in vitro than control NPs and their circulation half-life in mice was approximately double that of PEGylated NPs.6,7 These findings demonstrated the immense potential of membrane-wrapping as a biointerfacing strategy.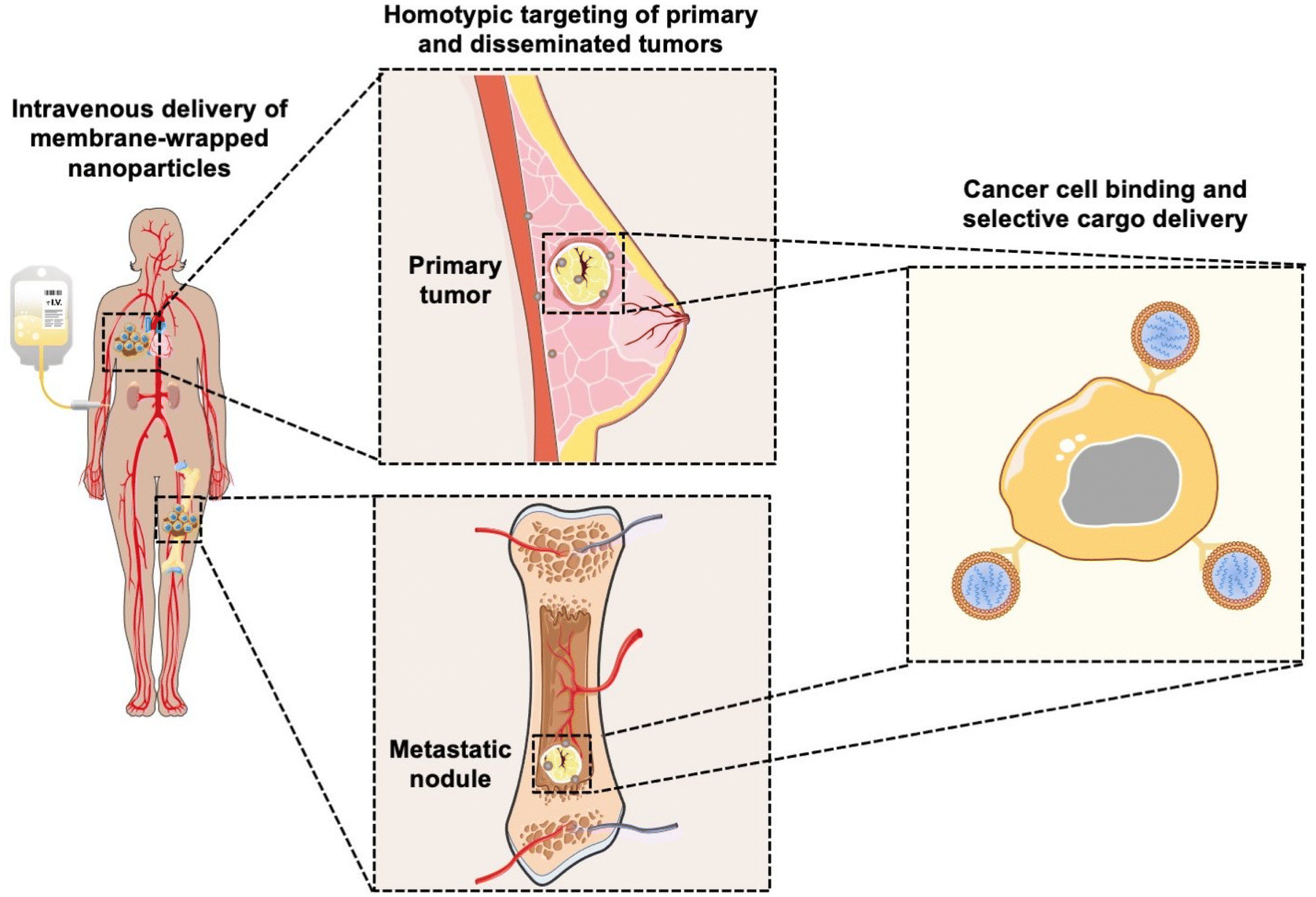 | ||
| Fig. 1 Scheme depicting the ability of membrane-wrapped NPs to accumulate in primary tumors and metastatic lesions and deliver their cargo to specific cells. Portions of this figure were produced using Servier Medical Art templates (https://smart.servier.com). Servier Medical Art by Servier is licensed under a Creative Commons Attribution 4.0 Unported License. | ||
Since the introduction of RBC membranes as NP coatings, the field of biomimicry has exploded with various membrane-wrapped NPs developed for use as standalone or combination therapies. Source cell membranes include RBCs, leukocytes, platelets, stem cells, cancer cells, immune cells, and bacteria.5 Each membrane type offers unique advantages. For example, cancer cell membranes impart NPs with the ability to target homotypic cells at either primary or metastatic tumor sites (Fig. 1) and leukocyte membranes allow NPs to detect circulating tumor cells (CTCs) in the bloodstream.8,9 These interactions are mediated by cell adhesion molecules expressed on the cell membrane exterior, such as TF-Ag (Thomsen-Friedenreich antigen), E-cadherin, or EpCAM (epithelial cell adhesion molecule), which are transferred onto the membrane-wrapped NPs and increase their ability to bind cancer cells throughout the body.10–13 This improved binding ultimately improves the delivery and efficacy of the cargo carried by the NPs. To create NPs with multiple biointerfacing capabilities, hybrid cell membrane-wrapped NPs have also been developed.14 Dehaini et al. fused RBC membranes with platelet membranes and then coated polymer NPs with the fused membrane vesicles.14 This retained the membrane markers and functionality that is characteristic to each cell type and allowed the NPs to perform increasingly complex tasks within biologically relevant contexts in vitro and in vivo.14 Overall, both singular and hybrid membrane wrapping approaches appear to provide enhanced in vivo circulation and disease site accumulation compared to conventional ligand conjugation strategies.15–17
Given their promising capabilities, membrane-wrapped NPs have been widely explored for drug delivery, immune modulation, disease detection, detoxification, imaging, and phototherapy singularly or in combination with other methods.1,5 The core–shell structure characteristic of membrane-wrapped NPs makes the platform versatile to target specific disease profiles and unlocks nearly endless potential therapeutic strategies. The NP core must be designed to provide the desired functionalities and ensure proper “right-side-out” orientation of the membrane around the NP such that the extracellular membrane components can interact with the outside microenvironment and other cells.3 Various synthetic materials have been used as NP cores, as detailed in a prior review.3 This versatility of core material allows a wide selection of hydrophobic or hydrophilic cargoes to be encapsulated within membrane-wrapped NPs, which enables improved pharmacokinetics and efficacy compared to freely delivered counterparts. Fig. 2 illustrates the fundamental synthesis steps for creating cell membrane-wrapped NPs including the various methods employed for cell membrane extraction and membrane-core fusion. Most research to date has focused on hydrophobic drug cargoes, but interest is growing in developing membrane-wrapped NPs for hydrophilic nucleic acid (NA) delivery. This review highlights recent progress in this emerging field, including a discussion of the barriers to systemic NA delivery, examples of membrane-wrapped NPs that have enabled successful NA delivery, and future avenues of investigation.
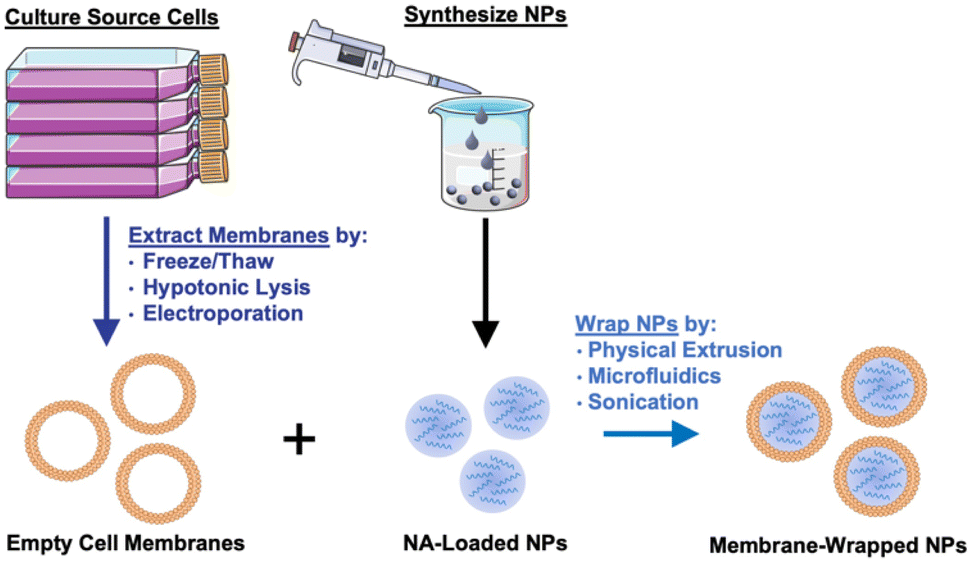 | ||
| Fig. 2 Illustration of the synthesis of membrane-wrapped NPs. Portions of this figure were produced using Servier Medical Art templates (https://smart.servier.com). Servier Medical Art by Servier is licensed under a Creative Commons Attribution 4.0 Unported License. | ||
1.2 Nucleic acids are beneficial therapeutics that face systemic delivery barriers
NAs are powerful tools that can modulate gene expression in target cells to enable the treatment of inherited and acquired genetic diseases.18 The most commonly explored NAs for disease management include plasmid deoxyribonucleic acid (pDNA), messenger ribonucleic acid (mRNA), small interfering RNA (siRNA), microRNA (miRNA), and immunostimulatory CpG oligodeoxynucleotides (CpG ODNs). While pDNA and mRNA increase the expression of the gene they encode, siRNA and miRNA utilize RNA interference (RNAi) to decrease the expression of desired genes. By comparison, CpGs stimulate immune cells to engage the body's immune response to attack diseased cells. Each NA's mechanism of action is described in more detail in section 2 and depicted in Fig. 3. While these distinct NAs all have immense therapeutic potential, they share similar characteristics that decrease their functionality when freely delivered. Namely, they are negatively charged, hydrophilic, and relatively large (on the order of 10–100 kDa).19 These characteristics limit their ability to overcome physiological barriers including opsonization and phagocytosis by cells of the MPS, degradation by endogenous nucleases, penetration across vascular barriers and into diseased tissue, and entry into target cells.20,21 Accordingly, freely delivered NAs exhibit short blood circulation half-lives and limited accumulation in target sites. Further, exogenously delivered synthetic NAs can activate an undesirable innate immune response and cytokine production through endosomal and cytoplasmic recognition pathways.22 To avoid these issues, researchers have considered local delivery methods, such as intratumoral injection of naked NAs, but this has proven unsuccessful and is not clinically feasible because many disease sites are difficult to reach and/or locate precisely and completely.23,24 Moreover, local delivery requires repeated administration since doses are quickly diluted at the injection site and this is not acceptable for long-term therapeutics.23 Hence, to be successful therapeutically, NAs require a carrier that can support effective and sustainable systemic delivery.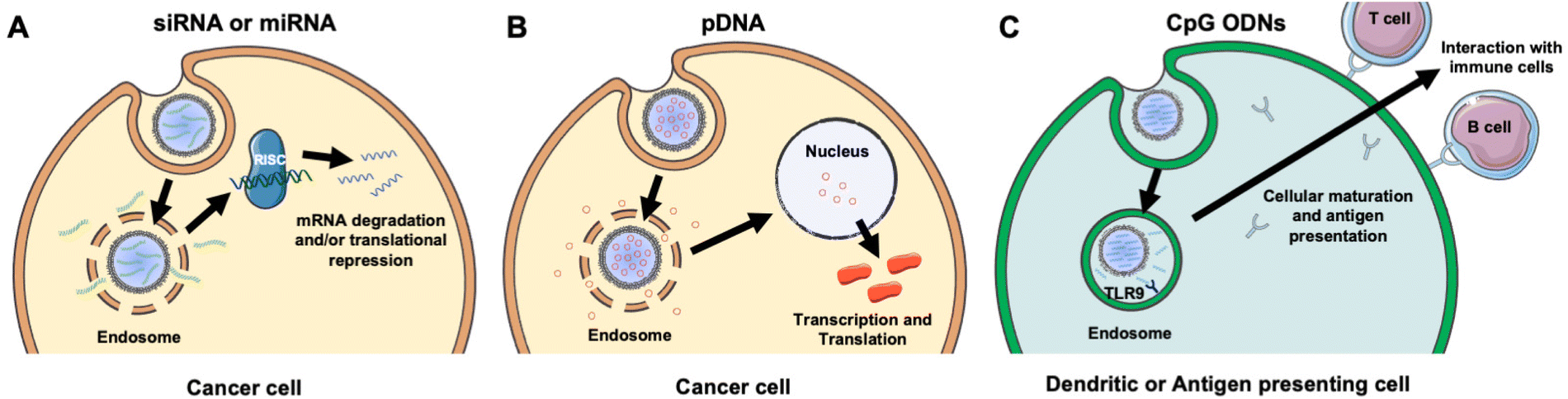 | ||
| Fig. 3 Scheme showing the intracellular fate and mechanism of action of different NA cargoes carried by membrane-wrapped NPs. (A) Upon endosome escape, the antisense strand of siRNA or miRNA duplexes is incorporated into the RNA induced silencing complex (RISC) and guides it to complementary mRNA that is then degraded or translationally repressed. (B) Following pDNA delivery to the nucleus, it is transcribed and translated to increase production of the encoded protein. (C) Upon NP entry into dendritic or antigen presenting cells, CpGs are recognized by TLR9 receptors within endosomes leading to cellular maturation and antigen presentation to stimulate an anti-tumor immune response. Portions of this figure were produced using Servier Medical Art templates (https://smart.servier.com). Servier Medical Art by Servier is licensed under a Creative Commons Attribution 4.0 Unported License. | ||
Assuming a NA nanocarrier can successfully bypass physiological barriers to arrive at the intended site, it must then enter the desired cells, escape endosomes, and deliver therapeutic payloads of the cargo to the correct cytoplasmic or nuclear location. mRNA and siRNA must engage RISC in the cytoplasm, while pDNA must be delivered to the nucleus. NPs loaded with NAs or other cargoes typically enter cells by clathrin-dependent endocytosis, caveolae-dependent endocytosis, flotillin-mediated endocytosis, or pinocytosis.25,26 To enable successful gene regulation, NPs carrying NAs must be designed to escape or rupture endosomes after uptake and subsequently release stable NAs into the cytoplasm to reach the correct intracellular target and perpetuate gene regulation.18,25 Endosomal escape is commonly achieved via pH-dependent degradation because endosomes are more acidic than the cytosol or extracellular environment.27 Designing NPs that can encapsulate hydrophilic NAs with satisfactory loading efficiency while also avoiding unintended burst release of the cargo prior to cellular delivery is challenging.19,25,26 However, the use of cellular membranes as an exterior coating on the NP surface can improve NA delivery by preventing immune clearance, facilitating NP endocytosis by specific cells, and protecting the NA cargo until it is released post endosomal escape.3,28 The following sections discuss research to date that has demonstrated the benefits of using membrane-wrapped NPs for siRNA, pDNA, and CpG delivery, with a particular emphasis on membrane-wrapped NPs for NA delivery to cancer cells and tumors.
2. Membrane-wrapped NPs for siRNA delivery
2.1. Singular membrane-wrapped NPs for tumor-targeted siRNA delivery
An attractive method to regulate gene expression in diseased cells is through the intracellular delivery of exogenous synthetic siRNA. Following cellular uptake and endosomal escape, siRNA is incorporated into the RNA-induced silencing complex (RISC), which retains the antisense siRNA strand. The RISC/antisense complex then selectively degrades complementary mRNA strands in the cell cytosol, reducing target protein production (Fig. 3A).23 A major advantage of RNAi is that siRNA molecules can target virtually any protein, including those that are considered undruggable by small molecules due to lack of an effective binding site.29 The diverse targeting provided by RNAi offers broad therapeutic impact.24 Numerous NPs have been developed to deliver siRNA into tumors to elicit RNAi, and this section summarizes recent progress in the use of membrane-wrapped NPs for tumor-specific siRNA delivery. Section 2.1 focuses on NPs that have a singular type of membrane as the coating, and section 2.2 discusses NPs with hybrid membrane coatings.Biomimetic siRNA nanocarriers have been developed using membranes derived from cell sources including cancer cells, mesenchymal stem cells, and platelets. Each of these membrane coatings enhance NP circulation time and tumor-specific delivery by harnessing the unique proteins present on the natural cell membrane that regulate immunogenicity and cell–cell interactions.27,30 For example, cancer cell membranes enable homotypic targeting, which is believed to be mediated by “self-recognition molecules” on the cell surface.8,9,28,31 Cancer cells adhere strongly to one another to form primary tumors and metastases, and cancer cell membrane-wrapped NPs can exploit this property to improve tumor-specific delivery of siRNA or other cargo. This was demonstrated in a study where PLGA NPs were co-loaded with doxorubicin (Dox) and siRNA targeting programmed death-ligand 1 (si.PD-L1) and coated with HeLa human cervical adenocarcinoma cell membranes (the resultant NPs were termed “CCMNPs”).31 Although not explicitly examined in the work, suppressing PD-L1 should sensitize cancer cells to death by natural killer and T cells. The CCMNPs were synthesized by a nanoprecipitation method in which 200 μL of 20 μM siRNA and 10 mg mL−1 Dox were added to 25 mg PLGA in 800 μL dichloromethane (DCM), which was further emulsified in 2% polyvinyl alcohol (PVA) 1500 (w/v). For dosing cells in vitro, CCMNPs or unwrapped control NPs were added to cells with media at a Dox concentration of 1.5 μg mL−1. In vitro flow cytometry and MTT studies showed that CCMNPs exhibited greater uptake and cytotoxic effects in homotypic HeLa cells than in heterotypic MDA-MB-231 human breast cancer cells, demonstrating the specificity afforded by the membrane coating. A preliminary western blot analysis qualitatively showed that CCMNPs decreased PD-L1 expression in HeLa cells 48 hours post-treatment relative to controls of no treatment, freely delivered Dox and siRNA, and unwrapped NPs, but the study lacked quantitative analysis and comparison to CCMNPs loaded with scrambled siRNA. It would be intriguing to compare the level of silencing achieved by CCMNPs to that achieved with commercial transfection agents to demonstrate the benefit of the biomimetic nanocarriers. Overall, this study provides proof-of-concept that cancer cell membrane-wrapped NPs can enhance the delivery of both hydrophobic (Dox) and hydrophilic (siRNA) payloads to targeted tumor cells.
Mesenchymal stem cells (MSCs) have also been used as a membrane source for NP coating because MSCs are recruited to the tumor microenvironment by growth factors and cytokines that are secreted during tumor angiogenesis and stroma formation.32–37 MSC membrane-coated NPs have remarkably long circulation and strong tumor targeting capabilities leading to increased cargo delivery to the tumor microenvironment, which includes several cell types beyond cancer cells alone.38 The potential of MSC-coated NPs to mediate tumor-specific siRNA delivery was demonstrated in 2018 by Mu et al., who coated iron oxide (Fe3O4) NPs with polydopamine (PDA) and siRNA (Fe3O4@PDA-siPlk1@MSCs NPs) to perform imaging-guided photothermal therapy (PTT) and siRNA delivery in human prostate cancer cells (DU145).36 The siRNA was designed to suppress Plk1, a proto-oncogene with high expression in tumor cells whose knockdown increases apoptosis and inhibits tumor cell growth. DU145 cells were treated with naked siPlk1, Lipofectamine 2000/siPlk1 polyplexes (Lipo2K/siPlk1), Fe3O4@PDA-siPlk1@MSCs NPs, or Fe3O4@PDA-siRNA@MSCs (wrapped NPs carrying a scrambled siRNA control) at equivalent doses of 10 nM siRNA for 24 hours prior to qRT-PCR analysis or for 72 hours prior to western blot analysis. The Fe3O4@PDA-siPlk1@MSCs and Lipo2K/siPlk1 achieved approximately 50% and 40% mRNA knockdown at 24 hours compared to controls, respectively, but the data lacked statistical significance. The 72-hour western blot data qualitatively confirmed this result, as only Fe3O4@PDA-siPlk1@MSCs and Lipo2K/siPlk1 had reduced Plk-1 protein band intensity versus controls. Based on the promising mRNA and protein knockdown observed in vitro, studies were performed in vivo to assess the therapeutic effect of Fe3O4@PDA-siPlk1@MSCs when intravenously administered to Balb/c nude mice bearing DU145 xenograft tumors. Biodistribution studies showed that Fe3O4@PDA-siRNA@MSCs exhibited greater tumor accumulation than unwrapped NPs, which improved their performance as magnetic resonance imaging probes. To examine treatment effect, mice were administered either PBS, or 100 μg of Fe3O4@PDA, Fe3O4@PDA-siRNA@MSCs, or Fe3O4@PDA-siPlk1@MSCs, then tumors were irradiated 24 hours later with an 808 nm laser (0.6 W cm−2) for 6 minutes. Tumor volume was monitored over 15 days post-therapy, and Fe3O4@PDA-siPlk1@MSCs + laser (which combines siPlk1 delivery with PTT) reduced tumor volume by about 60% versus pre-treatment values. By comparison, tumor volume decreased by ∼40% in mice treated with the scrambled control Fe3O4@PDA-siRNA@MSCs + laser and increased by ∼25% in mice treated with the non-wrapped control Fe3O4@PDA + laser. This demonstrates the benefit of the MSC coating for enhancing tumor delivery and PTT efficacy, as well as the benefit of the siPlk1 to enhance tumor reduction versus scrambled siRNA. TUNEL assays performed on excised tumors from the Fe3O4@PDA-siPlk1@MSCs + laser group showed many tumor cells underwent apoptosis, and hematoxylin and eosin (H&E) staining also revealed substantial changes to the tumor architecture versus PBS controls.
Platelet membranes are another advantageous membrane exterior because they shield NPs from immune system attack while facilitating binding to damaged blood vessels or specific pathogens.38,39 This supports their use to treat diseases ranging from atherosclerosis to bacterial infections.14 Excitingly, activated platelets have also been shown to target CTCs, which are created when primary tumors shed tumor cells off via hematogenous dissemination to distant organs during metastasis.40 Activated platelet membrane-wrapped NPs have been proven to target and adhere to CTC-associated micro-thrombi in the vasculature to deliver therapeutic cargoes and prevent the spread of tumor cells.40 Therefore, platelet membrane-wrapped NPs have great potential as siRNA delivery vehicles to mitigate tumor progression. Zhuang et al. explored this potential by coating zeolitic imidazolate framework-8 (ZIF-8) porous metal–organic framework (MOF) NPs with platelet membranes to enable targeted siRNA delivery in vivo (Fig. 4A).41 These P-MOF-siRNA NPs were synthesized using 500 nM siRNA, 20 mg mL−1 2-methylimidazole, and 1 mg mL−1 zinc nitrate hexahydrate and maintained sufficient siRNA loading due to the strong electrostatic interactions between the framework's metal nodes and the siRNA's backbone phosphates in addition to the physical placement within the NP.41 Not only were these P-MOF-siRNA NPs biomimetic, they were also pH-dependent, exhibiting minimal siRNA release when placed in pH 7.4 buffer but burst release within a few hours after exposure to acidic pH 5.0 (Fig. 4B). This pH-dependent property would allow the P-MOF-siRNA NPs to protect the siRNA during intravenous transport but facilitate siRNA release after internalization by tumor cells.
 | ||
| Fig. 4 Representative example of a membrane-wrapped siRNA nanocarrier. (A) Platelet membrane-coated siRNA-loaded MOFs (P-MOF-siRNA) were generated by mixing the siRNA payload with Zn2+ and 2-methylimidazole (mim), followed by coating with a natural cell membrane derived from platelets. (B) siRNA release from P-MOF-siRNA at pH 5.0 or pH 7.4 over time (n = 3, mean ± SD). (C) Flow cytometry analysis of siRNA uptake in SK-BR-3 cells 24 hours after incubation with free siRNA, P-MOF-siRNA, or R-MOF-siRNA. (D) Fluorescence microscopy images of siRNA localization in SK-BR-3 cells 1, 4, 8, and 24 hours post incubation with P-MOF-siRNA (scale bar = 20 μm; siRNA = green; nuclei = blue; endosomes = red). (E) Immunofluorescence analysis of survivin protein expression in SK-BR-3 cells treated with different siRNA nanocarriers for 48 hours (scale bar = 20 μm; survivin = purple; nuclei = blue). (F) PCR analysis of relative survivin mRNA expression in SK-BR-3 cells after incubation with various siRNA nanocarriers for 48 hours (n = 3, mean + SD). (G) Growth kinetics of subcutaneous SK-BR-3 tumors in nu/nu mice treated intravenously with P-MOF-siRNASur or R-MOF-siRNASur every 3 days for four total administrations (n = 5; mean ± SEM). (H) Survival of the mice in (G) over time (n = 5). From ref. 41 (J. Zhuang, et al., Target gene silencing in vivo by platelet membrane-coated metal–organic framework nanoparticles, Sci. Adv., 2020, 6). Reprinted with permission from the American Association for the Advancement of Science (AAAS), copyright 2020. | ||
For in vitro studies, the P-MOF-siRNA NPs were compared to RBC wrapped MOF-siRNA NPs (R-MOF-siRNA NPs) using SK-BR-3 human breast cancer cells that overexpress HER2 receptors as a model. Flow cytometry and confocal microscopy proved P-MOF-siRNA NPs had significantly reduced uptake in murine J774 macrophages and increased uptake in SK-BR-3 cancer cells compared to R-MOF-siRNA NPs (Fig. 4C). To understand the intracellular trafficking of the siRNA cargo, fluorescently labeled siRNA was delivered via P-MOF-siRNA NPs and its intracellular localization monitored over 24 hours. The siRNA payload was primarily at the cell periphery 1 hour after incubation, and within LysoTracker labeled endocytic compartments by 4 hours post incubation. By 8 hours, the siRNA fluorescence was visualized throughout the cell, indicating that the pH responsive properties of the MOF scaffold allowed the siRNA to reach the cytosol (Fig. 4D). Building on this observation, the gene silencing capabilities of P-MOF-siRNA NPs were examined using Survivin as a relevant target as it is an anti-apoptotic gene whose expression correlates with HER2 expression. SK-BR-3 cells were treated with 50 nM siRNA targeting Survivin freely (siRNASur), loaded within P-MOF-siRNA (P-MOF-siRNASur) or R-MOF-siRNA (R-MOF-siRNASur) NPs, or treated with platelet-wrapped empty MOFs (P-MOF) or scrambled siRNA MOF controls (P-MOF-siRNANC). The P-MOF-siRNASur reduced cell proliferation by ∼60% at 48 hours and by ∼40% at 72 hours compared to controls. Western Blot and immunofluorescence staining qualitatively confirmed the P-MOF-siRNASur inhibited Survivin protein expression compared to controls (Fig. 4E), while PCR analyses showed P-MOF-siRNASur NPs reduced Survivin mRNA expression by ∼80% after 48 hours compared to only ∼40% knockdown for R-MOF-siRNASur controls (Fig. 4F).
Notably, the P-MOF-siRNA NPs retained functionality in vivo when intravenously injected in nude mice bearing subcutaneous SK-BR-3 tumors.41 Biodistribution studies using DiD fluorophore-loaded MOF NPs found that P-MOF-siRNASur NPs had sixfold higher accumulation in tumors than R-MOF-siRNASur NPs. For therapeutic studies, mice received NPs intravenously at a dose of 2 nmol siRNA per injection every 3 days for a total of 4 administrations. Tumors in untreated mice grew 16-fold in volume by day 50 compared to approximately 8-fold by day 80 for P-MOF-siRNASur NPs (Fig. 4G). Moreover, untreated mice survived a median of 50 days whereas P-MOF-siRNASur NPs and R-MOF-siRNASur NPs extended median survival to 92 days and 66 days, respectively (Fig. 4H). Collectively, these studies show that P-MOF-siRNASur NPs could effectively deliver stable siRNA into the cytosol of cancer cells to successfully silence gene expression and hinder cancer progression in vitro and in vivo. It is important to note that siRNA was the only therapeutic agent in this formulation. Therefore, this study demonstrates that therapeutic amounts of siRNA without additional chemotherapy drugs can be delivered in vivo and withstand intravenous travel to regulate tumor growth and increase survival. This study serves as a promising benchmark for future gene regulation nanoformulations.
The full therapeutic potential of siRNA-mediated gene silencing can only be realized if a carrier can successfully load the siRNA, protect it during circulation, and deliver it into the cytosol of diseased cells. Further, the dose of siRNA delivered must be sufficient to cause substantial gene knockdown that results in lasting tumor regression without relapse. The above studies clearly prove that membrane coatings increase tumor specific delivery of siRNA nanocarriers to yield more potent tumor reduction within 48 hours after administration. However, it remains to be seen whether the membrane-wrapped NP designs and dosing strategies that have worked in preclinical animal models will remain effective when translated to human clinical use. Clinical application will require much larger NP volumes with consistent siRNA loading, and the concentration, frequency, and duration of dosing will need to be altered to achieve complete tumor eradication. If siRNA-mediated gene silencing cannot eliminate tumors as a standalone therapy, the studies described here indicate that the addition of PTT or other therapeutic strategies to RNAi mediated by membrane-wrapped NPs can increase tumor reduction in an additive or synergistic manner.
2.2 Hybrid cell membrane-wrapped NPs for siRNA delivery
The papers in section 2.1 demonstrate the potential of singular membrane-wrapped NPs for siRNA delivery, but scientists have recently started to fuse different cell membranes together to integrate the benefits of each membrane type. These hybrid membranes combine the functions of the original cell membranes, such as the half-life extension from RBCs with the active tumor targeting from cancer cells.42–45 Alternatively, they can offer new functions that are not provided via monotypic cell membrane coatings, such as presentation of tumor antigens to antigen-presenting cells through lymph node homing.5 Given these benefits, hybrid membrane coating strategies are likely to be widely explored in the field moving forward.One recent study explored the use of hybrid membrane-wrapped NPs for siRNA delivery. In this study, a lipoic acid (LA) and cross-linked peptide-lipoic acid micelle cross-linked nano-platform (LC) was coated with a fusion of membranes from bone marrow mesenchymal stem cells (BMSCs) and prostate cancer (PCa) cells.46 Prostrate cancer cell membranes were selected to increase the NPs’ targeting to primary tumors while BMSCs were selected to facilitate homing to bone marrow to treat any bone metastases. The therapeutic target for gene silencing was sterol regulatory element-binding protein (SREBP), as overexpressed SREBP leads to irregular lipid biosynthesis that promotes prostate cancer growth and bone metastases. Silencing SREBP should inhibit PI3K/AKT signaling and thereby increase cellular sensitivity to Docetaxel (DTX), a potent chemotherapy drug for treating bone metastatic castration-resistant prostate cancer (BmCRPC) that faces extreme issues of chemoresistance and adverse side effects when administered freely. With this hybrid membrane-wrapped micelle NP coloaded with siSREBP1 and DTX, the nanosystem labeled PB@LC/D/siR was used for targeted delivery of siRNA and DTX to the bone metastatic niche of BmCRPC in vitro and in vivo.46
The BMSC membranes were derived from 4-week-old Sprague-Dawley rats while PCa cell membranes were sourced from two human prostate cancer cell lines derived from the bone metastatic site, PC-3 and C4-2B. Confocal microscopy studies showed that PB@LCs loaded with Coumarin-6 as a model drug (PB@LC/C6) were colocalized with LysoTracker Red labeled lysosomes after 1 hour of incubation but showed dissociation after 4 hours as green fluorescence separated from red fluorescence. This indicates that PB@LC can escape lysosomes and serve as an effective siRNA delivery system. Real-time fluorescence quantitative PCR of PC-3 cells cultured in media mimicking the BmCRPC microenvironment was performed to measure mRNA expression of SREBP1 and its downstream target SCD1 after treatment with scrambled siRNA (siCon), freely delivered siSREBP1, unwrapped NPs (LC/D/siR), or the full system PB@LC/D/siR for 48 hours at a dose of 10 μg mL−1 siRNA. This showed PB@LC/D/siR suppressed SREBP1 and SCD1 mRNA by approximately 50–60% compared to controls, and this was confirmed qualitatively by Western Blot. Hence, PB@LC/D/siR have promise as RNAi therapeutics for BmCRPC.
To confirm the system worked in vivo, Balb/c nude mice were used to establish a BmCRPC model by injecting PC-3 cells into the long axis of the shinbone.46 Biodistribution studies using DiR fluorophore-loaded NPs showed that PB@LC/DiR exhibited increasing accumulation in bone tumor sites over 24 hours. To assess therapeutic effects, PB@LC/D/siR NPs (full system with DTX at 1 mg kg−1 and siSREBP1 at 0.25 mg kg−1) were injected every 3 days for a total of 4 treatments. Impressively, PB@LC/D/siR significantly reduced tumor volume, maintained mice body weight, and significantly prevented reduction of bone mineral density compared to saline, siCon, siSREBP1, DTX, and unwrapped controls. Importantly, PCR on ex vivo tumors showed PB@LC/D/siR caused a significant ∼70% reduction in mRNA expression of SREBP1 and SCD1 compared to siCon, siSREBP1, and unwrapped NPs, and qualitative western blot analyses affirmed this result. Hence, PB@LC/D/siR were able to yield effective RNAi therapy in BmCRPC in vitro and in vivo while maintaining low toxicity from DTX. Future siRNA delivery vehicles that build on the hybrid biomimetic membrane cloaking approach may dramatically improve the efficacy of RNAi as a treatment strategy for diverse disease types.
While hybrid membrane-wrapped NPs loaded with siRNA have immense potential, they face the same therapeutic challenges as singular membrane-wrapped siRNA nanocarriers. Fortunately, as the field of biomimicry progresses, membrane coatings are being innovated to combine the most advantageous elements of different cell types or add in non-native elements to the membrane coating to increase targeting or endosomal escape. Endosomal escape of siRNA (or miRNA) is critical to allow therapeutic payloads to complex with RISC and cause subsequent mRNA degradation and/or translational repression of target proteins. The development of hybrid membrane coatings shows promise that the field is advancing to customized and intricately designed membrane coatings that can overcome both extracellular and intracellular barriers to NA delivery.
3. Membrane-wrapped NPs for plasmid DNA delivery
The goal of RNAi-based cancer therapy is to suppress the expression of overabundant oncogenes, but an alternative treatment approach is to increase the expression of tumor suppressor genes. This can be facilitated by the intracellular delivery of pDNA, which is usually found naturally as double-stranded, circular DNA molecules in bacteria. Naturally occurring pDNA contains a variety of elements including an origin of replication, repeating units that regulate the number of pDNA copies, the encoded gene(s) of interest, and other components. Due to these characteristics, pDNA can effectively replicate within a host cell, creating proteins in the process. Therefore, pDNA found initial usage in biomanufacturing within bacteria to mass produce proteins,47 amino acid sequences,48 and viral vectors.49 More recently, pDNA has been utilized to elicit protein responses in vivo for applications including gene therapy,50 vaccines,51 and anti-cancer therapeutics.52To induce a therapeutic response, pDNA must be delivered into the nucleus so that it can be transcribed into mRNA (Fig. 3B). The requirement for nuclear localization poses an additional delivery challenge compared to RNA therapeutics that need only reach the cytoplasm. To reach the nucleus the pDNA, and other DNA nanostructures, must traverse through the nuclear pore complex (NPC), which allows diffusion-based transport of small molecules (less than 9 nm) but requires active transport for larger molecules such as pDNAs.53 This must be taken into consideration when designing pDNA delivery vehicles or other systems that need to reach the nucleus.53
pDNA is commonly delivered via non-viral vectors such as lipoplexes or polyplexes, which incorporate cationic molecules such as polyethylenimine (PEI) to electrostatically bind anionic NAs and deliver them to the cytoplasm and/or nucleus.18 The original proposed mechanism of polyplex endosomal escape was the “proton sponge” hypothesis that suggested the buffering capacity of polyamine carriers causes osmotic rupture of the endosomal membrane and subsequent escape of polyplexes into the cytosol.19 However, more recent studies have shown a lack of endosomal pH change within the endosomes of cells treated with PEI and that endosomal escape does not always cause total endosomal rupture, two discoveries that contradict the proton sponge hypothesis.19 While the mechanism of endosomal escape remains debated, it is clear that combining pDNA with PEI can significantly improve endosomal membrane disruption, cytosolic delivery, and transport to the nucleus to yield effective gene therapy. However, PEI has inherent cytotoxicity due to its highly positive charge and it lacks an innate ability to target specific cells.54 Cloaking PEI/pDNA polyplexes with cell-derived membranes seems to provide a means to overcome these limitations.
Han et al. recently reported the results of studies comparing unwrapped PEI/pDNA complexes encoding for luciferase (pLuc) to those wrapped with glioblastoma cell membranes (Fig. 5A).55 Surprisingly, when introduced to C6 glioblastoma cells cultured in serum-free medium, the unwrapped complexes yielded greater luciferase expression than their wrapped counterparts, despite flow cytometry indicating the wrapped polyplexes exhibited greater cell uptake. The authors concluded that uptake efficiency could not account for the lower luciferase expression and proposed that the membrane wrapping could be interfering with the endosomal escape properties of PEI. Interestingly, when the study was performed in cells cultured in serum-containing medium, the membrane-wrapped polyplexes exhibited greater cellular uptake and transfection efficiency than unwrapped polyplexes, likely because serum proteins would more readily adsorb onto the positively charged naked PEI/pDNA polyplexes, resulting in their aggregation and reduced efficacy. Uptake studies also validated the ability to achieve homotypic targeting with this system, as PEI/pLuc polyplexes coated with membranes derived from C6 cells, N2A cells, or L2 cells yielded the greatest increase in luciferase activity when administered to the homotypic cell line (e.g., N2A-wrapped polyplexes yielded the greatest luciferase expression in N2A cells) (Fig. 5B–D).
 | ||
Fig. 5 Representative example of a membrane-wrapped pDNA carrier. (A) TEM images of unwrapped PEI25k/pSV-Luc complexes and PEI25k/pSV-Luc/CM nanoparticles prepared at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:20 weight ratio. Scale bar indicates 100 nm. (B–D) Luciferase transfection efficiency of PEI25k/pSV-Luc/CM nanoparticles prepared with cell membranes from (B) C6, (C) N2A, and (D) L2 cells and transfected into different cell types to assess homotypic targeting and pDNA delivery. The data indicate the mean ± standard deviation of quadruplicated experiments. ***P < 0.001 compared with the other groups. *P < 0.05 compared with the other groups. **P < 0.01 compared with PEI25k/pSV-Luc/CM(C6), but no statistical significance compared with PEI25k/pSV-Luc/CM(N2A). (E) Analysis of tumor suppression enabled by pHSVtk delivery. The PEI25k/pHSVtk/CM nanoparticles and PEI25k/pHSVtk complexes were intratumorally injected into C6 glioblastoma tumors in rats. After 7 days, the brains were harvested and subjected to Nissl staining to quantify relative tumor size by measuring tumor areas in ImageJ. *P < 0.05 compared with the other groups (n = 6). (F) TUNEL assay of excised tumors from the study described in (E). Scale bar indicates 50 μm. Reproduced from ref. 55 (S. Han, et al., J. Controlled Release, 2021, 338) with permission from Elsevier BV, copyright 2021. :1:20 weight ratio. Scale bar indicates 100 nm. (B–D) Luciferase transfection efficiency of PEI25k/pSV-Luc/CM nanoparticles prepared with cell membranes from (B) C6, (C) N2A, and (D) L2 cells and transfected into different cell types to assess homotypic targeting and pDNA delivery. The data indicate the mean ± standard deviation of quadruplicated experiments. ***P < 0.001 compared with the other groups. *P < 0.05 compared with the other groups. **P < 0.01 compared with PEI25k/pSV-Luc/CM(C6), but no statistical significance compared with PEI25k/pSV-Luc/CM(N2A). (E) Analysis of tumor suppression enabled by pHSVtk delivery. The PEI25k/pHSVtk/CM nanoparticles and PEI25k/pHSVtk complexes were intratumorally injected into C6 glioblastoma tumors in rats. After 7 days, the brains were harvested and subjected to Nissl staining to quantify relative tumor size by measuring tumor areas in ImageJ. *P < 0.05 compared with the other groups (n = 6). (F) TUNEL assay of excised tumors from the study described in (E). Scale bar indicates 50 μm. Reproduced from ref. 55 (S. Han, et al., J. Controlled Release, 2021, 338) with permission from Elsevier BV, copyright 2021. | ||
The authors used confocal microscopy to evaluate the endosome escape properties of membrane-wrapped polyplexes and found they exhibit reduced escape compared to unwrapped polyplexes. The addition of chloroquine, a drug which induces endosomolysis, did not increase luciferase expression in cells exposed to unwrapped polyplexes but did increase expression in cells exposed to wrapped polyplexes. This suggests that unwrapped polyplexes are better able to escape endosomes than wrapped polyplexes, but the greater uptake and stability of the membrane-wrapped system will ultimately allow it to achieve greater transfection efficiency. To test transfection efficiency in vivo, membrane-wrapped polyplexes carrying pDNA encoding the suicide gene HSVtk were administered intratumorally to rats bearing intracranial C6 glioblastomas, and they substantially reduced tumor size by over 50% compared to unwrapped polyplexes (Fig. 5E) and increased tumor apoptosis based on TUNEL assay results (Fig. 5F).55 Future studies that build on this work should explore the therapeutic potential of pDNA carriers administered systemically rather than intratumorally.
The versatility of membrane wrapping for pDNA delivery was demonstrated by Liu et al., who developed three kinds of cell membrane-modified PEI/DNA capsules (CPDcs) utilizing adherent human embryonic kidney epithelial cells (293T), human uterus/cervix adenocarcinoma cells (HeLa), or human liver carcinoma epithelial cells (HepG2).54 Each of the CPDcs systems produced were labeled as 293T-CPDc, HeLa-CPDc, and HepG2-CPDc, respectively. Different PEI/DNA mass ratios and PEI molecular weights were tested for transfection efficiency and cytotoxicity to determine the optimal formulation. It was determined that the best quality ratio of PEI/DNA for CPDc preparation was 2.5/1 with 30k PEI (PEI30k/DNA) and 0.75/1 with 70 PEI (PEI70k/DNA). Corresponding membrane wrapped CPDcs were labeled as follows: 293T-CP30Dc, HepG2-CP30Dc, HeLa-CP30Dc, 293T-CP70Dc, HepG2-CP70Dc, and HeLa-CP70Dc. Of these formulations, 293T-CP30Dc exhibited the highest encapsulation efficiency of DNA at 72.3% at a mass ratio of 2/2.5/1 (CM/PEI/DNA) while 293T-CP70Dc had the highest encapsulation efficiency of 85.3% at a mass ratio of 2/0.75/1 after 15 extrusions. After the two polyplex capsules were optimized, in vitro gene transfection experiments were performed to determine therapeutic potential. The EGFP plasmid acted as a reporter gene to determine the transfection efficiency of 293T-CPDc in 293T cells via fluorescence microscopy and quantitative flow cytometry. The transfection efficiency of 293T-CP70Dc (∼75%) was greater than that of 293TCP30Dc (∼33%), which agreed with the study's trend of 293T-CP70Dc increasing cell uptake compared to 293T-CP30Dc. The 293T-CP70Dc also increased transfection efficiency from unwrapped PEI70k/DNA controls by 10%. When 293T-CP70Dc was tested in homologous 293T cells, HeLa cells, or HepG2 cells, they yielded 76.8% transfection efficiency in target 293T cells, which was significantly increased from only 39.2% and 37.5% in HepG2 and HeLa, respectively. Compared to unwrapped PEI70k/DNA controls in 293T cells, 293T-CP70Dc increased transfection efficiency by 9.3%. Therefore, the 293T cell membrane coating increased cell uptake in homologous cells thereby increasing the transfection efficiency of the polyplex.
To investigate if adding more cell-sourced surface moieties would enhance transfection efficiencies, the group doped the surface of the CP70Dc system with the cultured extracellular matrix (ECM) of 293T, HepG2, or HeLa cells and labeled them ECM-293T-CP70Dc, ECM-HepG2-CP70Dc, and ECM-HeLa-CP70Dc, respectively. The ECM addition was hypothesized to increase adhesion of the CPDc to cells to subsequently improve uptake and transfection efficiency. In fluorescence microscopy and flow cytometry studies, ECM-CP70Dc showed homologous targeting to their respective source cells, which was 10–20% higher than observed for CP70Dcs without ECM additions. In vitro gene transfection assays showed that ECM-CP70Dc increased EGFP expression by 20–40% in homologous cells compared to CP70Dcs without ECM additions, but this data lacked statistical significance.54 Despite lack of significance, it is evident that ECM doping shows promising benefits towards increasing homologous cell uptake and transfection efficiency.
In a final example, Kaneti et al. loaded PEI/pDNA polyplexes in MSC “nanoghosts” (NGs) by electroporation, which did not significantly alter their size or charge compared to non-electroporated NGs.35In vitro studies showed that MSC-NGs loaded with plasmids encoding for the hemopexin-like domain (pPEX) increased PEX DNA and mRNA by 40 and 51 times, respectively, compared to unwrapped pPEX. This led to final PEX protein levels that were more than 7 times greater for MSC-NGs compared to controls. The in vivo biodistribution and safety profile were studied in C57BL mice using pDNA encoding for GFP. The MSC-NGs distributed primarily to kidneys, livers, and lungs, with less accumulation in the spleen, and no significant changes in white blood cell counts were observed after one day, suggesting the MSC-NG formulation could be a safe pDNA delivery vehicle. To demonstrate the anti-cancer potential of the system, MSC-NG-pPEX were administered intravenously to mice with subcutaneous PC3 prostate cancer tumors, and a single dose yielded an impressive 76% inhibition in tumor growth after 2 weeks as compared to untreated animals and other controls (naked pDNA and empty MSC membranes). Similar data was shown against an A549 metastatic non-small cell lung cancer model (50% tumor inhibition after 1 week). These results affirm the immense potential of membrane-wrapped pDNA carriers in biomedical applications.
To date, pDNA as a therapeutic cargo has achieved moderate success in the literature. While complexing pDNA with PEI can elicit strong endosomal escape properties, the resultant polyplexes are too toxic for clinical use. The studies discussed here provide evidence that coating PEI/pDNA polyplexes with cell-derived membranes can offer a way to mitigate some of their toxicity and allow them to achieve efficient gene regulation in vitro and in vivo. Developing strategies to retain cell targeting while promoting endosomal escape after cellular uptake would greatly benefit this system. Further work is warranted to realize the full potential of these systems.
4. Membrane-wrapped NPs for immune adjuvant delivery
While most studies using membrane-wrapped NPs for NA delivery have focused on eliciting gene regulation via siRNA or pDNA delivery, newer research has developed these systems for delivery of immunostimulatory CpG ODNs (Fig. 3C). CpGs are short sections of DNA that include a cytosine nucleotide directly followed by a guanine nucleotide.56 They mimic the response initiated from pathogen-associated molecular pathways (PAMPs), or more plainly, they attempt to set off the same cascade of molecular signaling that occurs when there is an infection.57 CpG 1826 is a common motif used as an adjuvant that initiates an immune response. It has been specifically implicated in triggering the toll-like receptor 9 (TLR9) signaling pathway which causes antigen presenting cells to initiate their maturation process and begin to interact with T- and B-cells58 – this CpG-TLR9 relationship is a key link between the innate and adaptive immune responses. As TLR9 has been shown to traffic to endosomal compartments containing CpG ODNs, leading to CpG-TLR9 binding and immune stimulation, it is an optimal target for NPs that accumulate in endosomes following cellular uptake.58 Additionally, TLR9 has the narrowest expression profile of the toll-like receptor family, being expressed almost exclusively on plasmacytoid dendritic cells and B cells,59 making it an ideal candidate for immune stimulation while avoiding off-target effects. Theoretically, eliciting this response under the right conditions could generate a robust immune response against a chosen molecule.Kroll et al. utilized this technique to stimulate an immune response against cancer.60 Specifically, they loaded PLGA polymer cores with CpG 1826 and wrapped them in B16-F10 mouse melanoma cell membranes. Membrane wrapping increased the size of the particles and altered the charge to mimic that of the membrane vesicles, and no loss of adjuvant in the core was observed post-wrapping. Dye-labeled CpG-loaded NPs were incubated with bone marrow-derived dendritic cells (BMDCs), which rapidly internalized the particles, stimulating release of inflammatory cytokines. Notably, cytokine production was triggered by CpG-loaded NPs using roughly one tenth the concentration of free CpG. Additionally, the particles generated greater amounts of costimulatory markers in BMDCs than freely delivered CpG.60
As immune-stimulating particles, CpG-loaded membrane-wrapped NPs can be used as a treatment as well as a prophylactic cancer vaccine.60 The following groups were used to study the efficacy of the system: blank control, CpG NP without membrane (CpG-NP), empty cancer cell wrapped NP without CpG (CCNP), CCNP combined with free CpG (CCNP + fCpG), whole cell lysate and free CpG (WC + fCpG), and the CCNPs loaded with CpG (CpG-CCNP). The CpG-CCNP group significantly increased (almost doubled) the percentage of CD8+ cells that were positive for the gp100 antigen, a known epitope on the B16-F10 cell membranes. A similar increase was also seen for the TRP2 peptide, a secondary epitope. This immune response against certain epitopes on the cell membranes translated to successful vaccination in vivo. Mice were vaccinated with the groups described above and challenged 21 days later with B16-F10 tumor cells. Approximately 86% of mice vaccinated with CpG-CCNPs showed no tumor growth up to 150 days after challenge with tumor cells. Meanwhile, mice vaccinated with CCNPs + fCpG had a median survival of 40 days and all but one mouse in the control groups reached the experimental endpoint (death or a tumor size greater than 200 mm2) by day 48 after tumor challenge. These results show that successful cancer vaccination can be achieved by combined delivery of both the adjuvant (CpG) and antigen (cell membranes) in the same vehicle.
However, use of this system as a post-tumor inoculation treatment showed modest tumor growth inhibition. The CpG-CCNPs merely increased survival by a few days as compared to no treatment controls. The authors suggest that the aggressive nature of the B16-F10 tumor model and the strong immunosuppression in this disease state are possible explanations for the lack of efficacy of the CpG-CCNPs alone as a treatment.60 To boost cellular immunity, the CpG-CCNPs were paired with a checkpoint blockade cocktail of anti-CTLA4 and anti-PD1 antibodies. This formulation was able to keep 50% of the cohort below the tumor size threshold through day 48 post challenge whereas the checkpoint blockade cocktail alone was as effective as the CpG-CCNPs alone – confirming other studies that suggest this checkpoint blockade cocktail does not show significant efficacy in B16-F10 tumor models. The results suggest that the combination of membrane-wrapped NPs, adjuvant, and anti-immunosuppression through checkpoint blockades can act synergistically by regulating different facets of the immune system.
Two other groups have proven that CpG 1826-loaded PLGA NPs coated with cell-derived membranes provide robust anti-cancer effects.61,62 Johnson et al. used C1498 acute myeloid leukemia (AML) cell membranes as an NP coating,61 while Zhang et al. wrapped NPs with hybrid membranes fused from BALB/c-derived bone marrow dendritic cells and ID8 ovarian cancer cells.62 In a unique approach, Johnson et al. administered the AML cell membrane-wrapped NPs (ACMNPs) to mice that were initially challenged with C1498 cells on day 0 and subsequently received chemotherapy on days 1 through 5 to induce remission. ACMNP vaccination occurred on days 26, 33, and 40, followed by re-challenge with C1498 cells on day 163. Impressively, the AMCNPs extended median survival to 4.4 weeks post re-challenge, compared to 2.7 weeks for mice treated with whole cell lysate vaccine.61 Likewise, the hybrid dendritic cell/cancer cell membrane-wrapped NPs developed by Zhang et al. enabled successful treatment of allograft, patient-derived xenograft, and metastatic ovarian cancer tumor models in mice.62 In the subcutaneous and PDX models the membrane-wrapped NPs were administered post-tumor challenge, while the NPs were administered prior to tumor challenge in the metastatic model.62 This demonstrates that the system has potential as both a tumor treatment and vaccination strategy.
In summary, CpG delivery via membrane-wrapped NPs has significant potential as a vaccine adjuvant. In the study by Kroll et al., inoculation produced a robust immune response as measured by inflammatory markers and T cell differentiation, which drastically increased cancer cell rejection in vivo.60 Similar results were obtained in the studies by Johnson et al. and Zhang et al.,61,62 affirming the robustness of this approach. Notably, delivering CpG with membrane-wrapped NPs is advantageous as the targeting properties of the membrane coating can lead to a localized immunostimulatory response. Additionally, since the antigens on the cancer cell membranes remain in close proximity to the adjuvant CpG molecules, a more potent immune response is created. However, as this technique employs multiple modes of immune stimulation, potential adverse effects need to be fully elucidated prior to clinical translation. Overall, the combination of biomimetic, cell membrane-wrapping and immune stimulation should be further explored as a research topic.
5. Conclusions and future directions
Membrane-wrapped NPs show great promise as NA delivery vehicles for gene regulation and/or immunotherapy. As discussed in this review, membrane-wrapped NPs have been shown to successfully deliver siRNA, pDNA, and CpG ODNs into target cells and to outperform their unwrapped counterparts. These studies (summarized in Table 1) illustrate the fundamental benefits of membrane wrapping including homotypic targeting and increased cellular uptake – both of which are necessary for successful NA delivery. Homotypic targeting leads to greater accumulation of the NPs at the target site, which simultaneously increases cargo delivery to the cell type of interest and decreases off-target effects. The papers discussed in this review show that the increased cellular uptake and/or tumor delivery translates to greater NA efficacy in vitro and in vivo as compared to alternative state-of-the-art systems. In an intriguing observation, membrane wrapping was shown to decrease cellular uptake of PEI/pDNA complexes in vitro, but yield greater efficacy in vivo due to the enhanced circulation and homotypic targeting enabled by the membrane-expressed proteins.55 Membrane wrapping also reduced the short-term toxicity of the PEI complexes, but this needs to be more thoroughly investigated in future work.| Core material | Membrane coating | Nucleic acid cargo | Disease model studied | Hydrodynamic diameter (nm) | Zeta potential (mV) | Notable results | Ref. |
|---|---|---|---|---|---|---|---|
| a Depending on the size and charge of PEI/DNA core. | |||||||
| PLGA | Human cervical cancer cells (HeLa) | siRNA (siPD-L1) | Human cervical cancer (HeLa) in vitro | 110 | Not reported | Western Blot results indicating gene silencing | 31 |
| Iron oxide coated with polydopamine (Fe3O4@PDA) | Mesenchymal stem cells (MSCs) | siRNA (siPLK1) | Human prostate cancer (DU145) in vitro and in xenograft mouse models | 109 | −30.3 ± 1.3 | Reduced tumor volume by ∼60% | 36 |
| Zeolitic imidazolate framework-8 (ZIF-8) porous metal–organic framework (MOF) | Platelets | siRNA (siSurvivin) | Human breast cancer (SK-BR-3) in vitro and in subcutaneous tumors in mice | 175 | ∼−35 | Extended median survival 1.4× | 41 |
| Lipoic acid | Bone marrow mesenchymal stem cells (BMSCs) and prostate cancer cells (PC-3 and C4-2B) | siRNA (siSREBP) | Human prostate cancer (PC-3) in vitro and tumors implanted in long axis of shinbone of mice | 92.7 | −22.0 | ∼70% reduction in target mRNA expression of treated tumors | 46 |
| PEI/pDNA | Glioblastoma cells (C6) | Plasmid DNA encoding the suicide gene HSVtk | Rat glioblastoma (C6) intracranial tumors | 120 | −32 | Decreased tumor size by 50% when administered intratumorally | 55 |
| PEI/pDNA | Human liver carcinoma epithelial cells (HepG2), human embryonic kidney epithelial cells (293T), and human cervical cancer cells (HeLa) | Plasmid DNA encoding EGFP | Human liver carcinoma cells (HepG2), human embryonic kidney epithelial cells (293T), and Human cervical cancer cells (HeLa) in vitro | 220–225a | −8 to 15a | Increased transfection efficiency by 9.3% | 54 |
| PEI/pDNA | MSCs | Plasmid DNA encoding PEX | Prostate cancer cells (PC3) in vitro and mice with subcutaneous PC3 tumors | 204 ± 17 | −16 ± 2 | 76% inhibition in tumor growth | 35 |
| PLGA | Mouse melanoma cells (B16-F10) | CpG-1826 | Mouse melanoma (B16-F10) in vitro and in vivo | ∼150 | ∼−30 | 86% of vaccinated mice did not develop tumors when treated prior to cell inoculation | 60 |
| PLGA | Mouse acute myeloid leukemia cells (C1498) | CpG-1826 | Acute myeloid leukemia in vitro and in vivo | ∼165 | ∼−40 | Increased median survival in a post-remission vaccination and re-challenge model | 61 |
| PLGA | Mouse ovarian cancer cells (ID8) and dendritic cells | CpG-1826 | Ovarian cancer in vitro and in vivo | ∼70 | ∼−30 | Fused cell membrane NPs decreased growth of subcutaneous, PDX, and metastatic tumors in mice with greater efficacy than singular membrane-coated NPs | 62 |
Notably, membrane-wrapped NA carriers can be used not only for gene regulation but also for cancer vaccination by incorporating CpG ODNs as the payload.60–62 These systems use membrane-expressed proteins to support homotypic targeting and increased cell uptake, but also exploit the membrane as an antigen source. The simultaneous delivery of both tumor cell membrane antigens and CpG ODN adjuvants to dendritic cells leads to a potent anti-cancer immune response that is greater than what would be achieved through delivery of antigen or adjuvant alone.
Despite the immense promise of membrane-wrapped NA nanocarriers, there are several challenges – both basic and translational in nature – that need to be overcome before these NPs can yield clinical success. One area of research where minimal work has been completed is understanding the biomechanical and biochemical fate of these NPs after they have been administered to the body. As the integrity of the membrane wrapping relies on electrostatic interactions, questions arise surrounding its fate after being subjected to high shear and serum–protein interactions in blood vessels. Future work should investigate the structure and membrane-protein content of the NPs before and after exposure to physiologic environments to add knowledge to the field.
Also related to the membrane coating, the mechanisms of homotypic targeting and immune evasion are not fully understood. Future work should aim to determine the receptors that mediate homotypic binding and cell uptake or that facilitate immune tolerance, as this would help advance not just the field of membrane-coated NPs, but the field of nanomedicine more broadly.
To improve the efficacy of the systems reported to date, researchers could develop methods to minimize loss of cargo that occurs during the membrane wrapping process and/or control the rate of NA release from the NP to ensure cargo is not lost prematurely in circulation. In general, encapsulation/loading efficiencies and release rates are underreported, so adding this information to future publications will greatly benefit the field.
Moving forward, researchers could expand the breadth of membrane-wrapped NPs by incorporating cargo such as mRNA or CRISPR-Cas9 for gene editing strategies. To date, systems including polymeric NPs, lipid-based NPs, viral vectors, exosomes, and bioinert frameworks (i.e. silica, metal–organic, and gold NPs) have been used preclinically to deliver gene editing tools.63 The use of membrane coatings could elicit higher efficacy by directing more of the gene editing cargo to the target location. Moreover, developing methods to direct the intracellular localization of the NA cargo might also enhance gene regulation potency.
Finally, future work should evaluate membrane-wrapped NPs in increasingly complex animal models and focus on manufacturing scale-up and other aspects related to commercialization and clinical implementation. Moving from the lab bench to the clinical setting will raise numerous uncertainties that must be addressed. For example, membrane coatings could be either autogenic (derived from the specific patient) or allogenic (derived from another patient and/or from an immortalized cell line) – both have advantages and disadvantages regarding immunogenicity and manufacturability. Speculatively, autogenic cells would be less immunogenic but harder to culture in large amounts, while allogenic cells could have higher immunogenicity but easier manufacturability. Researchers will need to carefully consider the pros and cons of each membrane source when designing therapeutic NPs, and the best choice will likely depend on the specific disease application. Other issues related to clinical implementation of membrane-wrapped NPs include scale-up to the large volumes required for patient use and maintenance of high and consistent NA cargo loading. As NA nanocarriers without membrane coatings have already begun to enter the clinic, some of these manufacturing scale-up questions may be readily addressed.
In summary, membrane-wrapped NPs have great potential as NA delivery vehicles, but challenges to their clinical translation also exist. Once these issues are addressed, NA-loaded membrane-wrapped NPs are poised to transform medical practice.
Author contributions
Conceptualization—ESD; investigation—MAS, EHS; project administration—ESD; supervision—ESD; visualization—MAS, EHS; writing—original draft—MAS, EHS; writing—review & editing—MAS, EHS, ESD.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding was provided by grants from the National Science Foundation (DMR-1752009) and National Institutes of Health (R35GM119659 and 1P20GM139760-1). The content is solely the responsibility of the authors and does not necessarily reflect the views of the funding agencies.References
- R. H. Fang, Y. Jiang, J. C. Fang and L. Zhang, Biomaterials, 2018, 128, 69–83 CrossRef PubMed.
- A. V. Kroll, R. H. Fang and L. Zhang, Bioconjugate Chem., 2017, 28, 23–32 CrossRef CAS PubMed.
- J. C. Harris, M. A. Scully and E. S. Day, Cancers, 2019, 11, 1–19 CrossRef PubMed.
- J. J. F. Verhoef and T. J. Anchordoquy, Drug Delivery Transl. Res., 2013, 3, 499–503 CrossRef CAS PubMed.
- Y. Liao, Y. Zhang, N. T. Blum, J. Lin and P. Huang, Nanoscale Horiz., 2020, 5, 1293–1302 RSC.
- C.-M. J. Hu, L. Zhang, S. Aryal, C. Cheung, R. H. Fang and L. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 10980–10985 CrossRef CAS PubMed.
- C.-M. J. Hu, R. H. Fang, B. T. Luk, K. N. H. Chen, C. Carpenter and W. Gao, et al. , Nanoscale, 2013, 5, 2664–2668 RSC.
- H. Sun, J. Su, Q. Meng, Q. Yin, L. Chen and W. Gu, et al. , Adv. Mater., 2016, 28, 9581–9588 CrossRef CAS PubMed.
- J.-Y. Zhu, D.-W. Zheng, M.-K. Zhang, W.-Y. Yu, W.-X. Qiu and J.-J. Hu, et al. , Nano Lett., 2016, 16, 5895–5901 CrossRef CAS PubMed.
- S. K. Khaldoyanidi, V. V. Glinsky, L. Sikora, A. B. Glinskii, V. V. Mossine and T. P. Quinn, et al. , J. Biol. Chem., 2003, 278, 4127–4134 CrossRef CAS PubMed.
- Q. Zhao, M. Barclay, J. Hilkens, X. Guo, H. Barrow, J. M. Rhodes and L.-G. Yu, Mol. Cancer, 2010, 9, 154 CrossRef PubMed.
- V. V. Glinsky, G. V. Glinsky, K. Rittenhouse-Olson, M. E. Juflejt, O. V. Glinskii, S. L. Deutscher and T. P. Quinn, Cancer Res., 2001, 61, 4851–4857 CAS.
- M. Mareel, K. Vleminckx, S. Vermeulen, Y. Gao, L. Vakaet Jr., M. Bracke and F. van Roy, Prog. Histochem. Cytochem., 1992, 26, 95–106 CrossRef CAS PubMed.
- D. Dehaini, X. Wei, R. H. Fang, S. Masson, P. Angsantikul, B. T. Luk, Y. Zhang, M. Ying, Y. Jiang, A. V. Kroll, W. Gao and L. Zhang, Adv. Mater., 2017, 29, 1606209 CrossRef PubMed.
- Q. Yang, Y. Xiao, Y. Yin, G. Li and J. Peng, Mol. Pharm., 2019, 16, 3208–3220 CrossRef CAS PubMed.
- Y. Long, X. Wu, Z. Li, J. Fan, X. Hu and B. Liu, Biomater. Sci., 2020, 8, 5088–5105 RSC.
- Q. Sun, J. Wu, L. Jin, L. Hong, F. Wang, Z. Mao and M. Wu, J. Mater. Chem. B, 2020, 8, 7253–7263 RSC.
- H. J. Vaughan, J. J. Green and S. Y. Tzeng, Adv. Mater., 2020, 32, e1901081 CrossRef PubMed.
- M. Durymanov and J. Reineke, Front. Pharmacol., 2018, 9, 971 CrossRef PubMed.
- R. Kanasty, J. R. Dorkin, A. Vegas and D. Anderson, Nat. Mater., 2013, 12, 967–977 CrossRef CAS PubMed.
- H. Yin, R. L. Kanasty, A. A. Eltoukhy, A. J. Vegas, J. R. Dorkin and D. Anderson, Nat. Rev. Genet., 2014, 15, 541–555 CrossRef CAS PubMed.
- M. Chandler, M. B. Johnson, M. Panigaj and K. A. Afonin, Curr. Opin. Biotechnol., 2020, 63, 8–15 CrossRef CAS PubMed.
- K. A. Whitehead, R. Langer and D. G. Anderson, Nat. Rev. Drug Discovery, 2009, 8, 129–138 CrossRef CAS PubMed.
- H. J. Kim, A. Kim, K. Miyata and K. Kataoka, Adv. Drug Delivery Rev., 2016, 104, 61–77 CrossRef CAS PubMed.
- S. Prabha, G. Arya, R. Chandra, B. Ahmed and S. Nimesh, Artif. Cells, Nanomed., Biotechnol., 2016, 44, 83–91 CrossRef CAS PubMed.
- T. Sun, Y. S. Zhang, B. Pang, D. C. Hyun, M. Wang and Y. Xia, Angew. Chem., Int. Ed., 2014, 53, 12320–12364 CAS.
- R. H. Fang, A. V. Kroll, W. Gao and L. Zhang, Adv. Mater., 2018, 30, 1706759 CrossRef PubMed.
- D. M. Valcourt, J. Harris, R. S. Riley, M. Dang, J. Wang and E. S. Day, Nano Res., 2018, 11, 4999–5016 CrossRef CAS PubMed.
- S. Ganesh, M. L. Koser, W. A. Cyr, G. R. Chopda, J. Tao and X. Shui, et al. , Mol. Cancer Ther., 2016, 15, 2143–2154 CrossRef CAS PubMed.
- Z. He, Y. Zhang and N. Feng, Mater. Sci. Eng., C, 2019, 106, 110298 CrossRef PubMed.
- M. Chen, M. Chen and J. He, Artif. Cells, Nanomed., Biotechnol., 2019, 47, 1635–1641 CrossRef CAS PubMed.
- C. Xu, Q. Feng, H. Yang, G. Wang, L. Huang and Q. Bai, et al. , Adv. Sci., 2018, 5, 1800382 CrossRef PubMed.
- C. Gao, Z. Lin, Z. Wu, X. Lin and Q. He, ACS Appl. Mater. Interfaces, 2016, 8, 34252–34260 CrossRef CAS PubMed.
- J. Cao, J. Qi, X. Lin, Y. Xiong, F. He, W. Deng and G. Liu, Front. Bioeng. Biotechnol., 2021, 9, 707208 CrossRef PubMed.
- L. Kaneti, T. Bronshtein, N. M. Dayan, I. Kovregina, N. L. Khait and Y. Lupu-Haber, et al. , Nano Lett., 2016, 16, 1574–1582 CrossRef CAS PubMed.
- X. Mu, J. Li, S. Yan, H. Zhang, W. Zhang, F. Zhang and J. Jiang, ACS Biomater. Sci. Eng., 2018, 4, 3895–3905 CrossRef CAS PubMed.
- M. Zhang, F. Zhang, T. Liu, P. Shao, L. Duan and J. Yan, et al. , Int. J. Nanomed., 2020, 15, 10183–10197 CrossRef CAS PubMed.
- V. Vijayan, S. Uthaman and I. K. Park, Polymers, 2018, 10, 983 CrossRef PubMed.
- C.-M. J. Hu, R. H. Fang, K.-C. Wang, B. T. Luk, S. Thamphiwatana and D. Dahaini, et al. , Nature, 2015, 526, 118–121 CrossRef CAS PubMed.
- J. Li, Y. Ai, L. Wang, P. Bu, C. C. Sharkey and Q. Wu, et al. , Biomaterials, 2017, 76, 52–65 CrossRef PubMed.
- J. Zhuang, H. Gong, J. Zhou, Q. Zhang, W. Gao, R. H. Fang and L. Zhang, Sci. Adv., 2020, 6, eaaz6108 CrossRef CAS PubMed.
- D. Wang, H. Dong, M. Li, Y. Cao, F. Wang and K. Zhang, et al. , ACS Nano, 2018, 12, 5241–5252 CrossRef CAS PubMed.
- Q. Jiang, Y. Liu, R. Guo, X. Yao, S. Sung, Z. Pang and W. Yang, Biomaterials, 2019, 192, 292–308 CrossRef CAS PubMed.
- J. Xiong, M. Wu, J. Chen, Y. Liu, Y. Chen and G. Fan, et al. , ACS Nano, 2021, 15, 19756–19770 CrossRef CAS PubMed.
- H. Chen, J. Deng, X. Yao, Y. He, H. Li and Z. Jian, et al. , J. Nanobiotechnol., 2021, 19, 342 CrossRef CAS PubMed.
- J. Chen, Z. Wu, W. Dong, C. Xiao, Y. Zhang and S. Gao, et al. , Theranostics, 2020, 10, 1619–1632 CrossRef CAS PubMed.
- A. L. Demain and P. Vaishnav, Biotechnol. Adv., 2009, 27, 297–306 CrossRef CAS PubMed.
- K. L. Piers, M. H. Brown and R. E. W. Hancock, Gene, 1993, 134, 7–13 CrossRef CAS PubMed.
- R. M. Kotin, Hum. Mol. Genet., 2011, 20, R2–R6 CrossRef CAS PubMed.
- M. Manthorpe, F. Cornefert-Jensen, J. Hartikka, J. Felgner, A. Rundell, M. Margalith and V. Dwarki, Hum. Gene Ther., 1993, 4, 419–431 CrossRef CAS PubMed.
- E. F. Fynan, R. G. Webster, D. H. Fuller, J. R. Haynes, J. C. Santoro and H. L. Robinson, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 11478–11482 CrossRef CAS PubMed.
- D. Lew, S. E. Parker, T. Latimer, A. M. Abai, A. Kuwahara-Rundell and S. G. Doh, et al. , Hum. Gene Ther., 1995, 6, 553–564 CrossRef CAS PubMed.
- H. Li, L. W. C. Ho, L. K. C. Lee, S. Liu, C. K. W. Chan, X. Y. Tian and C. H. J. Choi, Nano Lett., 2022, 22, 3400–3409 CrossRef CAS PubMed.
- L. Liu, Y. Chen, C. Liu, Y. Yan, Z. Yang, X. Chen and G. Liu, Mol. Pharm., 2021, 18, 2803–2822 CrossRef CAS PubMed.
- S. Han, Y. Lee and M. B. Lee, J. Controlled Release, 2021, 338, 22–32 CrossRef CAS PubMed.
- G. J. Weiner, H. M. Liu, J. E. Wooldridge, C. E. Dahle and A. M. Krieg, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 10833 CrossRef CAS PubMed.
- T. Kawai and S. Akira, Nat. Immunol., 2010, 11, 373–384 CrossRef CAS PubMed.
- E. Latz, A. Schoenemeyer, A. Visintin, K. A. Fitzgerald, B. G. Monks and C. F. Knetter, et al. , Nat. Immunol., 2004, 5, 190–198 CrossRef CAS PubMed.
- Y.-C. Chuang, J.-C. Tseng, L.-R. Huang, C.-M. Huang, C.-Y. F. Huang and T.-H. Chuang, Front. Immunol., 2020, 11, 1075 CrossRef CAS PubMed.
- A. V. Kroll, R. H. Fang, Y. Jiang, J. Zhou, X. Wei, C. L. Yu, J. Gao, B. T. Luk, D. Dehaini, W. Gao and L. Zhang, Adv. Mater., 2017, 29, 1703969 CrossRef PubMed.
- D. T. Johnson, J. Zhou, A. V. Kroll, R. H. Fang, M. Yan and C. Xiao, et al. , Leukemia, 2021, 36, 994–1005 CrossRef PubMed.
- L. Zhang, W. Zhao, J. Huang, F. Li, J. Sheng, H. Song and Y. Chen, Front. Immunol., 2022, 13, 828263 CrossRef CAS PubMed.
- T. V. Mashel, Y. V. Tarakanchikova, A. R. Muslimov, M. V. Zyuzin, A. S. Timin, K. V. Lepik and B. Fehse, Biomaterials, 2020, 258, 120282 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |