DOI:
10.1039/D2BM00905F
(Paper)
Biomater. Sci., 2022,
10, 7004-7014
Sustained release of heme–albumin as a potential novel therapeutic approach for age-related macular degeneration†
Received
8th June 2022
, Accepted 18th October 2022
First published on 2nd November 2022
Abstract
Globally, age-related macular degeneration (AMD) is the third most common visual impairment. Most often attributed to cellular fatigue with aging, over expression of reactive oxygen species (ROS) causes ROS accumulation in the retina, leading to chronic inflammatory immune signaling, cellular and tissue damage, and eventual blindness. If left uncontrolled, the disease will progress from the dry form of AMD to more severe forms such as geographic atrophy or wet AMD, hallmarked by choroidal neovascularization. There is no cure for AMD and treatment options are limited. Treatment options for wet AMD require invasive ocular injections or implants, yet fail to address the disease progressing factors. To provide more complete treatment of AMD, the application of a novel anti-inflammatory heme-bound human serum albumin (heme–albumin) protein complex delivered by antioxidant ROS scavenging polydopamine (PDA) nanoparticles (NPs) for sustained treatment of AMD was investigated. Through the induction of heme oxygenase-1 (HO-1) by heme–albumin in retinal pigment epithelial (RPE) cells, anti-inflammatory protection may be provided through the generation of carbon monoxide (CO) and biliverdin during heme catabolism. Our results show that the novel protein complex has negligible cytotoxicity towards RPE cells (ARPE-19), reduces oxidative stress in both inflammatory and ROS in vitro models, and induces a statistically significant increase in HO-1 protein expression. When incorporated into PDA NPs, heme–albumin was sustainably released for up to 6 months, showing faster release at higher oxidative stress levels. Through its ability to react with ROS, heme–albumin loaded PDA NPs showed further reduction of oxidative stress with minimal cytotoxicity. Altogether, we demonstrate that heme–albumin loaded PDA NPs reduce oxidative stress in vitro and can provide sustained therapeutic delivery for AMD treatment.
1. Introduction
Age-related ocular disorders continue to increase in prevalence as the average global lifespan continues to grow, with the third leading cause of blindness relating to age-related macular degeneration (AMD).1 Today, the prevalence of AMD, globally, affects more than 196 million people with the projected number of people afflicted to increase to 288 million by 2040, with the United States alone accounting for over 11 million cases in patients over 60.2,3
Disease progression of AMD comes in two forms: early and late stage. Early or dry AMD is the most common form of the disease, accounting for almost 90% of cases and is hallmarked by intracellular or extracellular protein aggregation and toxic debris, resulting in lipofuscin and drusen formation, respectively, lipid oxidation, and chronic inflammation in the region.4–6 Advanced dry AMD or geographic atrophy (GA) results in degradation of the macular and retinal tissues, spreading to cover significant fractions of posterior segment tissues, and leading to partial or complete vision loss.7 There are currently no approved treatment strategies for dry AMD. Alternatively, late stage or wet AMD is the more advanced version of the disorder, found in 10% of cases, and is characterized by increased production of vascular endothelial growth factor (VEGF) leading to new vascularization in the choroid.4 The sub-retinal blood vessels breach the integrity of the Bruch's membrane into the retinal pigment epithelium (RPE), causing permanent damage to photoreceptor cells, RPE tearing and detachment, sub-retinal haemorrhage, fibrotic scarring, and permanent vision loss.8,9
Factors attributed to disease progression in both types of AMD include age, location and sunlight exposure, diet, smoking status, and demographics.10 Common among these contributors is their impact on generation of oxidative stress within the posterior segment of the eye and the damaging effects to ocular tissues.11 Metabolic imbalances between prooxidative and antioxidant regulation allow excess production of reactive oxygen species (ROS) which accumulate in the retina.12–14 ROS react with the surrounding proteins, lipids, and cellular DNA, causing damage and further generation of ROS, beginning a deteriorating cycle for the involved tissue.12,14 While the exact understanding of early AMD propagation is not confirmed, research has shown clear correlation supporting the relationship between oxidative injury to the RPE and surrounding tissues and cells, and the association of lipoproteins and parainflammatory immune responses within the retina.15,16
There is currently no clinically available cure for either dry or wet AMD. Early dry AMD treatments consist mostly of regular observation and documentation for early signs of neovascularization. More commonly, treatment of wet AMD includes frequent, up to monthly, intravitreal injections of anti-VEGF therapeutics to inhibit further growth factor binding and neovascularization.17 The invasive and costly injections are required for the rest of patients' lives and often experience low patient compliance and under treatment of the disease.18 Additionally, the injections increase the patient's risk for injection related complications such as endophthalmitis, rhegmatogenous retinal detachment, and increases in ocular inflammation and pressure.19 Intraocular implants, specifically Genentech's Susvimo®, is a newly approved extended port delivery system that is capable of providing 6 months of continued anti-VEGF application with minimally invasive refilling of the drug reservoir. Despite the improved injection frequency, the device requires surgical implantation and has shown a high incidence rate of endophthalmitis, generally 3 times higher than the rate for traditional intravitreal injection of anti-VEGF.20 Intravitreal injection of anti-VEGF treatments for patients with dry AMD are uncommon as they have not shown the same success at mitigation of disease progression as when used for wet AMD.6 Furthermore, links between extensive anti-VEGF use and exacerbation of retinal and macular atrophy have been seen in patients with dry AMD.21,22 Newly emerging investigative treatments for dry and wet AMD include cell-based therapies for regrowth of retinal cells, viral vector gene therapy, and complement cascade inhibition to mitigate inflammatory signalling and toxic complement by-products that are largely associated with disease progression.23,24
Utilization of the natural oxidative stress reduction pathways for treating inflammation and ROS remains an unexplored therapeutic approach for treating AMD. Within this response, heme oxygenase-1 (HO-1) has been found to be an essential protein in combating oxidative stress and inflammation.25 HO-1 maintains intracellular heme levels through the catabolism of the pro-oxidative protoporphyrin into the anti-inflammatory and antioxidant molecules biliverdin and carbon monoxide (CO).26 The generation of both CO and biliverdin provide essential mediation of the oxidative stress signals produced by immune cells. Pro-inflammatory cytokines activate expression of p38 mitogen-activated protein kinase (p38 MAPK), a transcription factor important in induction of HO-1, with α and β isoforms, that present pro-apoptotic or anti-apoptotic impacts, respectively.27,28 HO-1, and the production of CO, increases expression of the p38β isoform resulting in upregulation of anti-inflammatory IL-10 expression with simultaneous downregulation of pro-inflammatory cytokines.26,27 Additionally, conversion of biliverdin to bilirubin is catalysed via the biliverdin reductase (BVR) enzyme pathway. Bilirubin has shown promising antioxidant effects through ROS reduction, leading to its conversion back to biliverdin, providing a continual antioxidant protective loop.29–31
We therefore hypothesize that intentional and specific induction of HO-1 for therapeutic relief shows potential as a novel treatment for AMD. HO-1 has shown to be expressed in RPE cells and would be responsive during oxidative assault in AMD.32,33 To induce expression of the immune enzyme and generate the anti-inflammatory and antioxidant by-products, we propose the application of heme bound to human serum albumin (heme–albumin) to activate expression of HO-1. Additionally, HSA has shown antioxidant properties of its own as a major source of reactive free thiols that can scavenge ROS and its high affinity for dangerous free transition ions.34 Thus, as a synthesized protein complex, heme–albumin would be a dual threat therapeutic to combat the oxidative stress of AMD that leads to disease progression.
Direct application of heme–albumin, through intravitreal injection, would be inadequate to provide sustained, continued treatment against AMD due to rapid drug clearance from the vitreous.35 Therefore, polydopamine (PDA) nanoparticles (NPs) will be utilized for delivery of heme–albumin as an easily synthesized, and mucoadhesive drug delivery system for treatment of AMD. Recent studies in imaging, periodontal, and autoimmune disorder treatment applications, show that the self-polymerizing NPs are capable of ROS scavenging, providing additional antioxidant benefits in combating oxidative stress in AMD, without inducing cytotoxicity.36–38
We therefore hypothesize that heme–albumin, a novel protein therapeutic utilizing the naturally occurring heme molecule, delivered by ROS scavenging PDA NPs, will thoroughly and effectively combat both inflammation and ROS induced oxidative stress in retinal pigment epithelial cells, potentially providing a more complete defense against the perpetuating factors of both forms of AMD.
2. Materials and methods
2.1. Materials
Hemin, dopamine hydrochloride, heme assay kit, fortified bovine calf serum, bovine serum albumin (BSA), 10% neutral buffered formalin, and Dulbecco's phosphate buffered saline (DPBS), were purchased from Sigma-Aldrich (St. Louis, MO). Human serum albumin (HSA), 25 mg mL−1 was obtained from Octapharma® (Lachen, Switzerland). Hollow fiber tangential flow filtration (TFF) modules (D01-S050-05-N, polysulfone membrane, 50 kDa pore size, 60 individual hollow fibers, 0.5 mm internal diameter, 190 cm2 total surface area) were purchased from Repligen (Rancho Dominguez, CA). TFF system tubing and peristaltic pump (Masterflex L/S® precision pump tubing, EW-96410-16 and Masterflex L/S® Digital Drive with Easy-Load® 3 pump head, EW-77921-65) were obtained from Cole-Palmer (Vernon Hills, IL). Tube connections were acquired from Nordson Medical (Loveland, CO). Phosphoric acid (H3PO4), sodium chloride (NaCl), sodium hydroxide (NaOH), sodium phosphate monobasic (NaH2PO4) and dibasic (Na2HPO4), Triton X-100, and 0.2 μm Titan3 sterile filters were obtained from Fisher Scientific Inc. (Hampton, NH), and Dulbecco's Modified Eagle Medium (DMEM) F-12 was purchased from Thermo Fisher Scientific (Waltham, MA). HyClone penicillin–streptomycin39–41 100× solution and trypsin 0.05% (1×) were obtained from Cytiva (Marlborough, MA). Human retinal pigment epithelial cells (ARPE-19 cells, CRL2302) were purchased from American Type Culture Collection (ATCC, Rockville, MD). Colorimetric (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) (MTS) assay, DCFDA/H2DCFDA Cellular ROS Assay Kit, and human heme oxygenase 1 ELISA kit were procured from Abcam (Cambridge, United Kingdom). Pierce™ BCA Protein Assay Kit, p38β MAPK antibody (p38-1145), IL-1β and phospho-p38 MAPK ELISA kit, 4′,6-diamidino-2-phenylindole (DAPI), α-mouse Alexa Fluor 568 secondary antibody, and human PCR primers (IL-6, IL-1β, and GAPDH,) along with SuperScript™ VILO™ cDNA Synthesis Kit were purchased from Thermo Fisher Scientific Inc (Waltham, MA). Additional primers for IL-1β, IL-6, caspase-3, and caspase-9 were purchased from Integrated DNA Technologies (Coralville, IA).
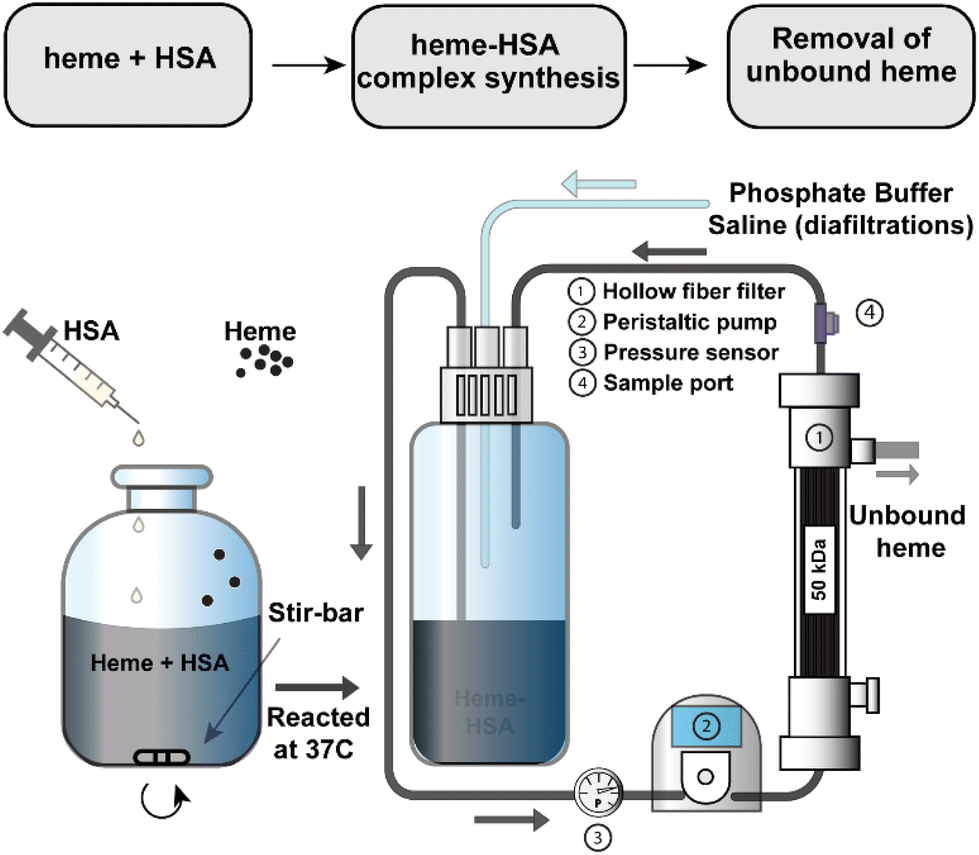
2.2. Heme–albumin synthesis and purification
A 3 mM solution of heme was solubilized in 100 mM NaOH and combined with a 1.5 mM solution of HSA. Heme stability in various non-aqueous conditions, including NaOH, has been verified in previous studies.39–41 The solution was mixed briefly before incubation at 37 °C for 1 hour after which the pH was adjusted to 7.4 with a phosphoric acid/NaCl solution and sterile filtered through a 0.2 μm filter. To remove any unbound heme, the heme–albumin protein complex was buffer exchanged over a 50 kDa TFF hollow fibre filter into phosphate buffered saline (PBS, pH 7.4). A schematic of the heme–albumin synthesis and buffer exchange process is shown in Fig. 1. Heme bound to albumin was quantified with a heme assay kit (n = 5), where 50 μL of a blank, 50 μL of heme calibrator, and 50 μL of diluted heme–albumin were added to individual wells in a 96-well clear plate. To each well, 200 μL of heme reagent was added. The contents of each well were mixed and left to react for 5 minutes at room temperature before the optical density was measured at 400 nm. Protein quantification was performed using a Dionex UltiMate 3000 UHPLC/HPLC system with an Acclaim SEC-1000 (4.6 × 300 mm) column with a 50 mM phosphate buffer mobile phase pH 7.4. Chromeleon 7 software was used to control and measure HPLC parameters such as flow rate (0.35 mL min−1), UV–visible absorbance detection (280 nm) and fluorescent light detection (excitation/emission (Ex/Em) at 285/333 nm). All samples were filtered through 0.2 μm syringe filters before analysis. The absorbance at 280 nm was used for total protein quantification of the heme–albumin complex, whereas confirmation of heme integration into albumin was determined by fluorescence quenching of heme at 285/333 nm compared to native albumin.
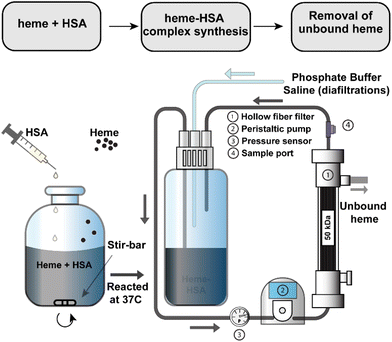 |
| | Fig. 1 Synthesis and purification schematic for heme–albumin (heme–HSA). The protein complex was synthesized under basic conditions before adjustment of the pH to 7.4 and buffer exchanged over a 50 kDa TFF hollow fiber filter into PBS pH = 7.4. | |
2.3. Circular dichroism of heme–albumin
The secondary structure of heme–albumin was investigated with a Jasco J-815 CD spectrometer (Easton, MD). The spectra was taken from 180–250 nm, 0.5 nm step, with a 0.1 cm quartz cuvette. Baseline correction was employed and sample concentrations of 0.1–0.2 mg mL−1 were used for analysis.
2.4. Heme–albumin loaded polydopamine nanoparticle synthesis
Dopamine hydrochloride precursor was reacted with a polymerization initiator solution composed of 45 mL deionized water (DI H2O) with 380 μL 1 N NaOH as published.42 The precursor was added to a 250 mL round bottom flask and initiator was added at a rate of 4 mL min−1 after which the self-polymerization reaction was left to proceed for 3 hours with constant stirring. The solution pH was adjusted to pH 7.4 before the addition of 100 mg of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 heme–albumin protein complex. After addition, protein loading was allowed to continue for 21 hours before centrifugation at 12100 rpm for 15 minutes and washing 3× times with DI H2O. The heme–albumin loaded PDA NPs were lyophilized at −88 °C and 0.002 mbar for 24 hours. Unloaded PDA NPs were centrifuged, washed, and lyophilized using the same procedure and were collected before protein addition.
:1 heme–albumin protein complex. After addition, protein loading was allowed to continue for 21 hours before centrifugation at 12100 rpm for 15 minutes and washing 3× times with DI H2O. The heme–albumin loaded PDA NPs were lyophilized at −88 °C and 0.002 mbar for 24 hours. Unloaded PDA NPs were centrifuged, washed, and lyophilized using the same procedure and were collected before protein addition.
2.5. Nanoparticle characterization
Lyophilized nanoparticles were resuspended in DI H2O at 1 mg mL−1 and characterized by a FEI Tecnai G2 Spirit transmission electron microscope (TEM) (Thermo Fisher, Waltham, MA) with 1% uranyl acetate. Scanning electron microscopy (SEM) of both the loaded and unloaded PDA NPs were conducted on a Thermo Fisher Scientific Apreo LoVac UXR (Waltham, MA). Lyophilized NP samples were fixed on carbon graphite tape before image capture. The hydrodynamic diameter and zeta potential of the nanoparticles was measured using a BI-200SM Goniometer and ZetaPals Zeta Potential Analyzer (Brookhaven Instruments Corp., Holtsville, NY). Dynamic light scattering (DLS) was performed at an angle of 90°and a wavelength of 637 nm. The hydrodynamic diameter was obtained by using the average values from the nonlinear least-squared algorithm in the instrument software. A total of n = 10 measurements were taken by ZetaPals and the value was averaged to determine the zeta potential of the nanoparticles.
2.6.
In vitro protein release
A 4 mL solution of heme–albumin loaded polydopamine nanoparticles at a nanoparticle concentration in solution of 1 mg mL−1 was incubated at 37 °C at varying concentrations of oxidative stressor: 0, 0.5, and 1 mM H2O2 in DPBS, for investigation of sustained release of heme–albumin. At time points 1 day, 4 days, 1 week, 2 weeks, 1 month, and monthly for up to 6 months, nanoparticles were collected by centrifugation at 20000 rpm for 20 minutes before retrieval of all the supernatant, and 4 mL of replacement oxidant solution was added to maintain sink conditions. Supernatants were stored at 4 °C and analysed by BCA protein quantification assay.
2.7. Cell culture
ARPE-19 cells were cultured with DMEM/F-12 media supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin 100× solution. Media was exchanged every 1–3 days and cells were passaged at 80–90% confluence. At passaging, cells were washed with 10 mL DPBS before addition of 3–5 mL of 0.05% trypsin and incubated at 37 °C for 5 minutes. Cells were visualized under a microscope to confirm non-adherence and trypsin was neutralized with the addition of 5–7 mL of media. Cells were pelleted at 130 g for 7 minutes before media was aspirated and resuspended for plating.
2.8. Cell viability assay
The cytotoxicity of heme–albumin, PDA NPs, and heme–albumin PDA NPs were investigated using ARPE-19 cells. The cells were seeded at 4000 cells per well in a clear 96-well plate.42 After 24 hours, to confirm cell adherence, varying concentrations of heme–albumin (10–2000 μg mL−1) or NP concentrations, both unloaded and loaded (10–200 μg mL−1) were applied to the wells and left to incubate for an additional 24 hours. Media contacting the therapeutic was removed and MTS stain was applied. The cells were stained for 3 hours before measurement of optical density at 490 nm with a Varioskan™ Lux multimode microplate reader using SkanIT software (Thermo Fisher Scientific, Waltham, MA).
2.9. Oxidative stress assay
The ability of heme–albumin, PDA NPs and heme–albumin loaded PDA NPs to reduce oxidative stress in both inflammatory and ROS models in ARPE-19 cells was investigated using published techniques with slight modifications.42 Cells were seeded at a density of 4 × 104 cells per well in a 96-well dark walled plate and left to adhere for 24 hours. Therapeutic agent heme–albumin, blank PDA NPs, or heme–albumin loaded PDA NPs were applied at varying concentrations (10–1000 μg mL−1 or 10–200 μg mL−1 for both nanoparticle groups, respectively) to the cells directly. In addition to the therapeutic, lipopolysaccharide (LPS) (10 μg mL−1) or H2O2 (100 μM) were applied to the wells. After 24 hours of incubation with therapeutic and stressing agent, the cells were washed with 100 μL 1× dilution buffer, stained for 45 minutes with 100 μL of 10 μM DCFDA dye and washed again with dilution buffer before fluorescence at Ex/Em = 485/535 nm was measured with the microplate reader.
2.10. ELISA quantification
The ability of heme–albumin to induce expression of HO-1, under baseline and stressed conditions, was quantified using a heme oxygenase 1 ELISA kit. Heme–albumin's impact on IL-1β and activated p38 MAPK in retinal cells were also measured. Briefly, ARPE-19 cells were seeded in 6-well plates at 1 × 106 cells per well (n = 6). Cells were allowed to adhere 24 hours before aspiration of media and treatment with either 500 μg mL−1 heme–albumin, 100 μM H2O2 or co-treatment with heme–albumin and 100 μM H2O2. Fresh media was used as a basal control. Incubation with treatment was continued for 24 hours before analysis. Cell lysis and ELISA were performed via manufacturer's recommendation.
2.11. PCR quantification
Investigation of heme–albumin's effect on baseline expression of pro-inflammatory cytokines, compared to control, or heme–albumin's impact on gene expression in oxidative challenge conditions was analysed in an identical manner as described above. 1 × 106 ARPE-19 cells per well were seeded in a 6-well plate (n = 6) and treated with 500 μg mL−1 of heme–albumin after 24 hours. Media was used as a baseline control. The samples were collected and isolated with Tryzol. After RNA quantification, the cDNA was made using SuperScript™ VILO™ cDNA Synthesis Kit. The total mass used to prepare the cDNA was 2500 ng. 50 ng of cDNA was used to perform the PCRs. Human IL-6, IL-1β, caspase-3, and caspase-9 were used as target genes for investigation of heme–albumin's impact on pro-apoptotic and inflammatory cytokine expression. Primer sequences for PCR are supplied in ESI.† GAPDH was used as a housekeeping gene.
2.12. p38β MAPK immunofluorescent staining
Changes in production of the p38β MAPK protein were measured by immunofluorescent staining. ARPE-19 cells were seeded in a 12-well plate before treatment with either 100 μM H2O2 or co-treatment with 100 μM H2O2 and 500 μg mL−1 of heme–albumin (n = 4). Cells without either treatment were used as a negative control. After 24 hours incubation with treatments, cells were fixed with 0.1% Triton X-100 for 10 minutes, blocked with 5% BSA for 1 h, and stained with p38 β MAPK primary antibody, in 1% BSA, overnight. Mouse IgG secondary antibody was added before DAPI staining.
2.13. Statistical analysis
All statistical analysis was performed using one-way ANOVA with Tukey honestly significant difference (HSD) test in JMP software. Results were considered statistically significant for p ≤ 0.05.
3. Results and discussion
3.1. Heme–albumin characterization
Heme and albumin concentrations were quantified separately after TFF purification of heme–albumin by heme assay kit and SEC-HPLC, respectively, to determine the working molar ratio of 2:1 heme:HSA. Heme binding to albumin was determined by SEC-HPLC and is presented in Fig. 2. Incorporation of heme into albumin quenches the native fluorescence of albumin. Measured at Ex/Em = 285/330 nm, unbound HSA has a natural fluorescence, due to the aromatic amino acids, tryptophan and tyrosine.43 When heme binds to the protein, the fluorescence is substantially quenched due to the interaction of the heme molecule and fluorescence contributing amino acids, verifying the formation of the heme–albumin protein complex. Heme binding to albumin resulted in a slight increase in molecular weight of heme–albumin compared to HSA, resulting in a small change in the protein elution time as seen in Fig. 2 with a small left shift of the heme–albumin peak compared to HSA. The secondary structure of heme–albumin compared to HSA was examined with CD and showed no significant deviation from the native HSA secondary structure. CD spectra was measured from 180–260 nm and presented as molar ellipticity shown in Fig. 2. Unfolding under basic conditions allows for heme incorporation into the hydrophobic binding sites within the protein that are entrapped within after pH adjustment back to physiological pH.
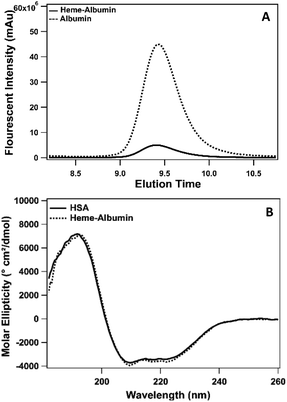 |
| | Fig. 2 (A) Size exclusion HPLC (SEC-HPLC) of albumin and heme–albumin monitored via fluorescence spectrometry. Incorporation of heme into albumin was detected by fluorescence quenching at Ex/Em = 285/330 (top). Unbound amino acids within HSA fluoresce at the measured wavelengths as shown by the dotted line in the figure. Binding of heme to the protein quenches the produced signal shown by the reduced fluorescent intensity. (B) CD spectra of heme–albumin and native HSA measured with a Jasco J-815 CD spectrophotometer (bottom). Unfolding and folding of HSA for formation of the heme–albumin does not alter the secondary structure of the protein complex. | |
3.2. Polydopamine nanoparticle characterization
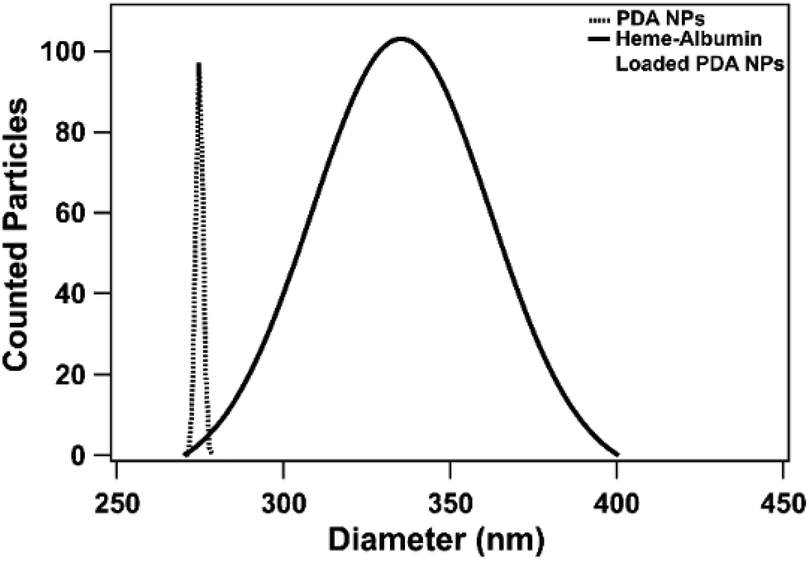
Both unloaded and heme–albumin loaded PDA NPs were characterized to determine the morphology and properties of the drug delivery system. Unloaded PDA NPs possessed a spherical morphology that remained when loaded with heme–albumin. Particles without protein had a mean hydrodynamic diameter of 189 nm and measured 296.4 ± 40.6 nm through TEM sizing. Both diameters are larger than previously reported nanoparticles with similar synthesis procedures but within the range of size suitable for use in drug delivery.42 Heme–albumin loaded PDA NPs possessed a hydrodynamic diameter of 229 nm and a measured diameter by TEM analysis of 307.5 ± 27.9 nm. TEM and SEM images of unloaded and heme–albumin loaded PDA NPs are shown in Fig. 4 and allow for confirmation of the spherical morphology of the different types of NPs. Protein loading of the heme–albumin loaded PDA NPs can be seen in the SEM images as the surface texture between unloaded and loaded PDA NPs has changed. DLS results are presented with a Gaussian distribution in Fig. 3. Zeta potential of both nanoparticle types was investigated and shown in Table 1.
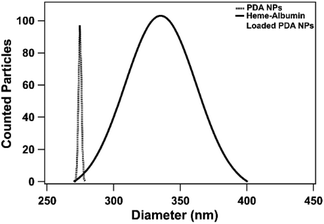 |
| | Fig. 3 Hydrodynamic diameter of heme–albumin loaded PDA NPs and unloaded PDA NPs measured with a BI-200SM Goniometer. Incorporation of heme–albumin is demonstrated by an increase in hydrodynamic diameter compared to unloaded particles. | |
 |
| | Fig. 4 TEM images of both unloaded (A) and heme–albumin loaded PDA NPs (B) are shown and confirm the spherical morphology of the NPs was maintained with the addition of the protein complex. SEM images of unloaded (C) and loaded (D) NPs show the surface coating of heme–albumin. | |
Table 1 Zeta potential of PDA NPs with and without incorporation of the heme–albumin protein complex
| |
Zeta potential (mV) |
Std deviation (mV) |
| Unloaded |
−31.10 |
±5.44 |
| Loaded |
−44.41 |
±3.42 |
3.3.
In vitro release of heme–albumin
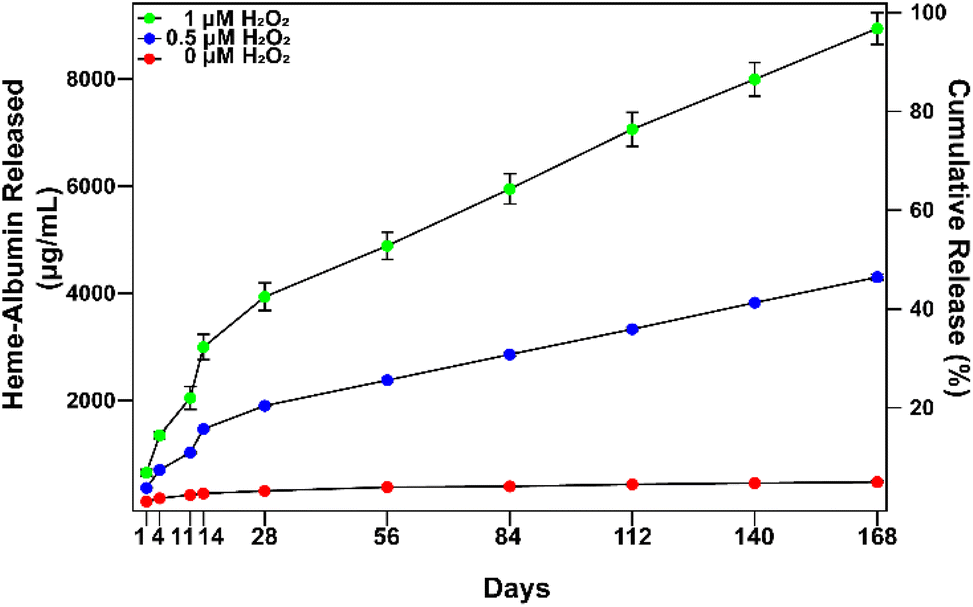
In vitro extended release of heme–albumin from PDA NPs was investigated at varying concentrations of oxidizing agent using 0, 0.5, and 1 mM H2O2 as a model for the oxidative stress conditions present in AMD. At time points of 1, 4, 11, 14, 28, 56, 84, 112, 140, and 168 days, NPs were collected and total protein in the supernatant was measured by the BCA assay. The results are presented in Fig. 5. At all concentrations of H2O2, heme–albumin loaded PDA NPs provided consistent and sustained release of heme–albumin. More protein was released at higher oxidative stress levels, as expected, providing tuneable therapeutic dosing for the changing oxidative stress conditions present in AMD. Cumulative release of the protein at 140 days at 0.5 and 1 mM H2O2 was 4301.6 ± 54.8 μg mL−1 and 8942.4 ± 295.6 μg mL−1, respectively. Compared to published uses of PDA NPs, where it has been demonstrated that PDA NPs provided up to 6 months of extended delivery of model protein BSA, our study confirms the ability of the PDA NPs to provide sustained and ROS-responsive release of new protein therapeutics such as the heme–albumin complex.42
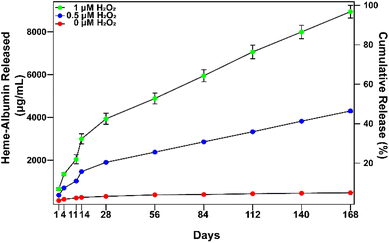 |
| | Fig. 5
In vitro release of heme–albumin from heme–albumin loaded PDA NPs incubated at 1 mg mL−1. Increasing oxidative stress with higher concentrations of H2O2 increased release rate, demonstrating ROS-responsive release of protein therapeutics from PDA NPs (n = 3). Error bars represent the standard deviation. | |
3.4. Cytotoxicity of heme–albumin, heme–albumin loaded PDA NPs, and blank NPs
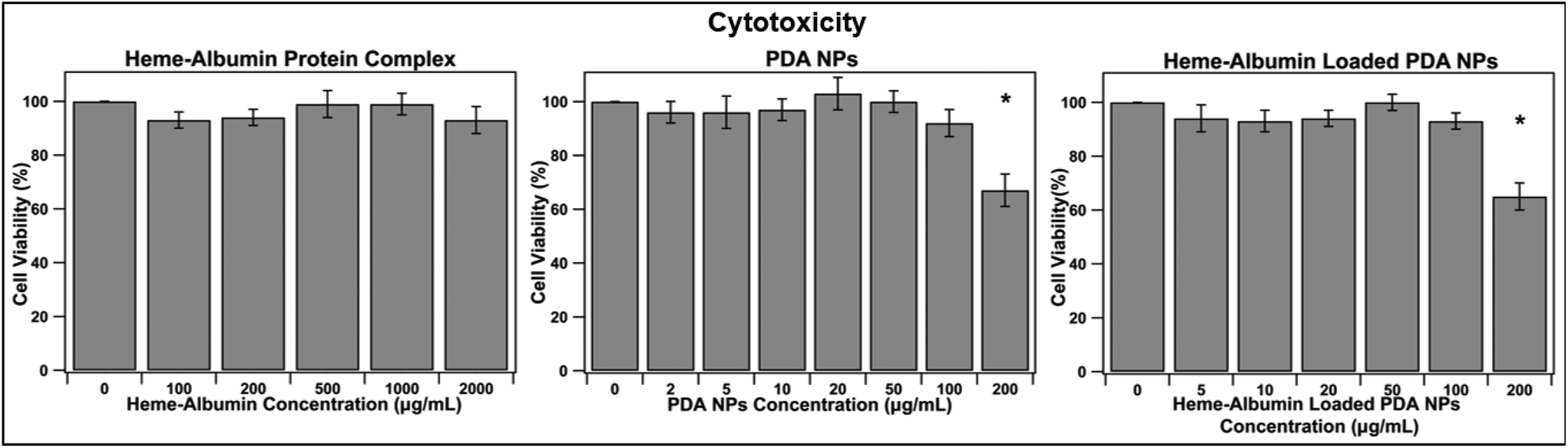
Biocompatibility is essential for determining the therapeutic use of any developed drug or drug delivery system. The synthesized heme–albumin protein complex, and both unloaded and loaded PDA NPs, were investigated with retinal pigment epithelial cells for understanding the interactions with one of the most impacted cell types present in AMD. The toxicity of heme–albumin as a therapeutic has yet to be determined and was investigated at varying concentrations. Between 100–2000 μg mL−1, heme–albumin showed no significant cytotoxicity to the dosed ARPE-19 cells after 24-hour incubation.
The LC50 of the therapeutic is >2000 μg mL−1, showing potential for a wide range of available therapeutic doses. Further, cytotoxicity of blank and heme–albumin loaded PDA NPs were evaluated using the same methods. ARPE-19 cells were incubated with varying concentrations of either blank, non-protein loaded NPs, or heme–albumin loaded PDA NPs, ranging from 2–200 μg mL−1, and stained with MTS dye 24 hours after initial dosing. As shown in Fig. 6, both loaded and unloaded PDA NPs were found to have minimal cytotoxicity (p < 0.05) to the retinal cells at concentrations less than 200 μg mL−1.
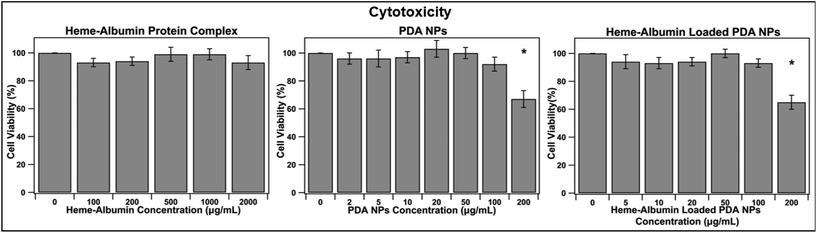 |
| | Fig. 6 Cytotoxicity analysis of heme–albumin, PDA NPs, and heme–albumin loaded PDA NPs. ARPE-19 cells were incubated with varying concentrations of therapeutic and cell viability was measured with the MTS assay (n = 6). Statistically significant differences for p < 0.05 is denoted with *. Heme–albumin showed no significant cytotoxicity to the ARPE-19 cells, and both unloaded and heme–albumin loaded PDA NPs only showed significant loss of cell viability at 200 μg mL−1 concentration. | |
3.5. Oxidative stress reduction in inflammatory model
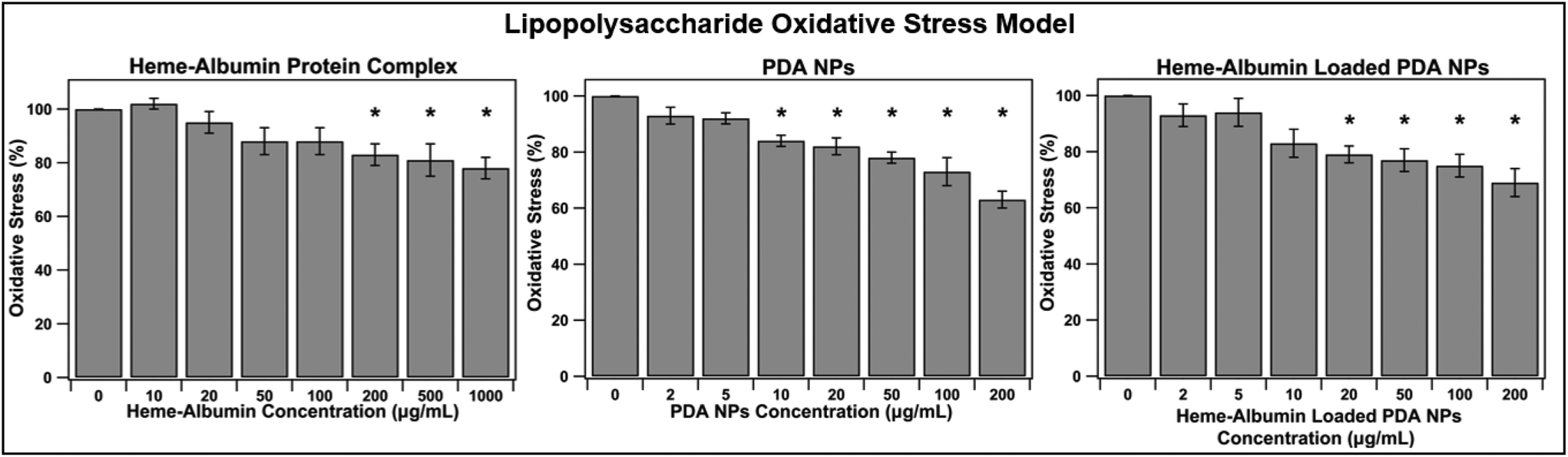
Chronic inflammation is a hallmark of dry AMD and plays a crucial role in disease progression to geographic atrophy and wet AMD. The constant oxidative injury that occurs with continued immune signalling leads to cellular fatigue and tissue damage within the retina and affected surrounding regions. The ability of heme–albumin to combat oxidative stress in an inflammatory model was investigated. A concentration of 10 μg mL−1 of LPS was applied to the ARPE-19 cells in conjunction with varying concentrations of the therapeutic groups. After 24 h, the cells were stained with 10 μM DCFDA stain for 45 minutes before fluorescence was measured at Ex/Em = 485/535 nm. The results are shown in Fig. 7. Heme–albumin was able to provide significant reduction in oxidative stress, when compared to the control at all therapeutic concentrations above 200 μg mL−1 to a maximum reduction of 22 ± 10% at 1000 μg mL−1. Both nanoparticle types, unloaded PDA NPs and heme–albumin loaded PDA NPs were able to significantly combat oxidative stress in LPS inflammatory model.
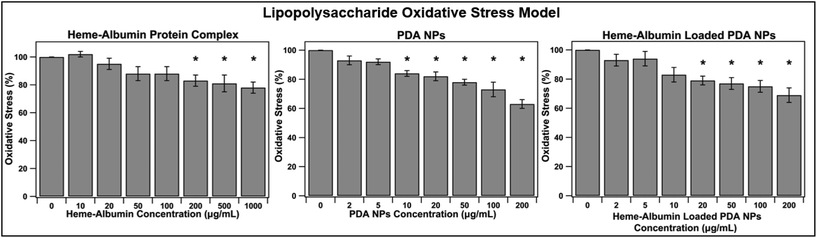 |
| | Fig. 7 Ability of treatment groups to reduce oxidative stress in a LPS model of inflammation (n = 6). Statistical significance at p < 0.05 is denoted with *. Heme–albumin reduced inflammatory oxidative stress by 22 ± 10% and heme–albumin loaded PDA NPs reduced stress at concentrations above 20 μg mL−1 (p < 0.05). Heme–albumin loaded PDA NPs reduced oxidative stress by 25 ± 10% at concentrations of 100 μg mL−1 compared to the untreated control. | |
3.6. Oxidative stress reduction in H2O2 ROS model
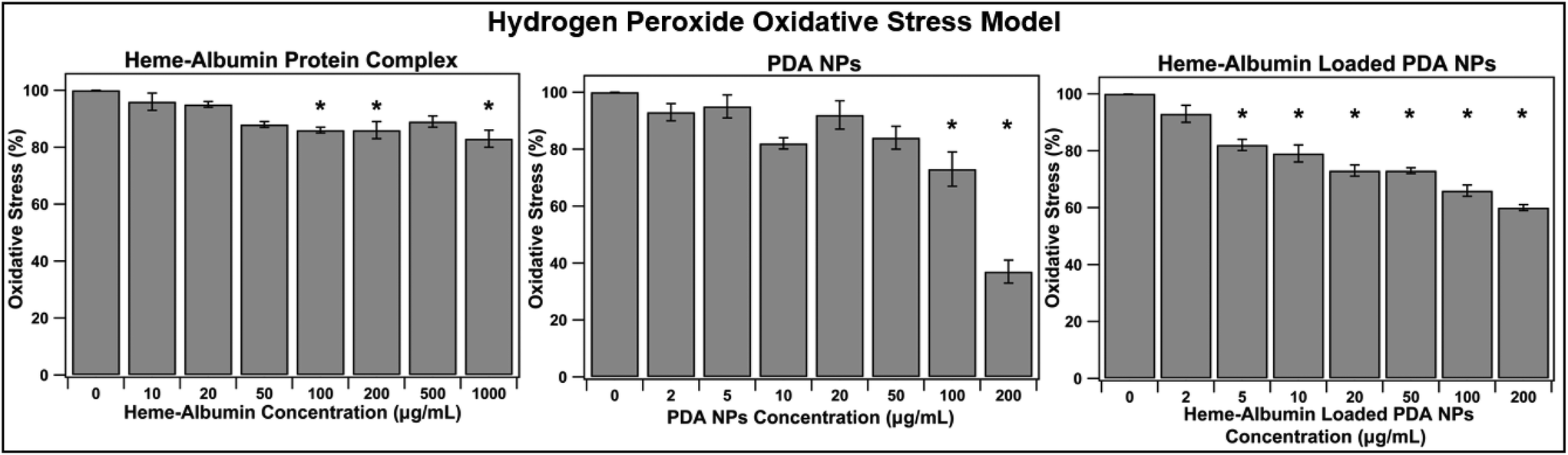
With aging and metabolic fatigue, the imbalance that occurs within the posterior segment of the eye results in accumulation of ROS, causing tissue damage and overexpression of inflammatory cytokines and growth factors. The ability of heme–albumin, unloaded PDA NPs, and heme–albumin loaded PDA NPs to reduce oxidative stress from ROS was therefore investigated. ARPE-19 cells were challenged with 100 μM H2O2 and simultaneously treated with varying concentrations of therapeutics. As shown in Fig. 8, heme–albumin, at a concentration of 1000 μg mL−1, reduced oxidative stress by 17 ± 3% (p = 0.005) as compared to control which experienced no oxidative challenge or therapeutic dosing.
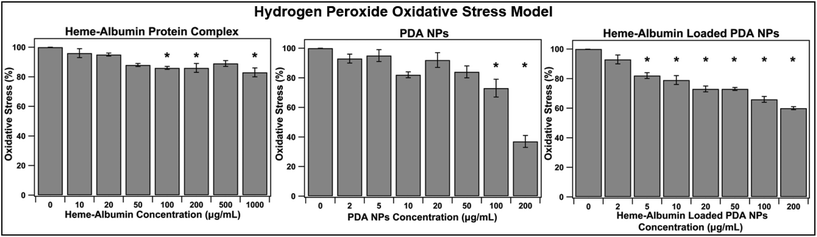 |
| | Fig. 8 Ability of treatment groups to reduce oxidative stress in H2O2 ROS model (n = 6). Significance of p < 0.05 is denoted with *. Heme–albumin was able reduce oxidative stress induced by 100 μM H2O2 by 17%. When co-treated with H2O2, heme–albumin loaded PDA NPs were able to reduce oxidative stress by 34 ± 6%. | |
Continued reduction in oxidative stress was achieved by heme–albumin loaded PDA NPs that showed a more significant decrease in oxidative stress compared to both other treatment groups with a peak reduction of 34 ± 6% (p < 0.0001) at concentrations with demonstrated minimal cytotoxicity.
3.7. Heme–oxygenase 1 expression is increased by heme–albumin
The proposed therapeutic activity of heme–albumin is provided by induction of the HO-1 enzyme that, through catabolism of heme, generates the anti-inflammatory and antioxidant components CO and biliverdin. Expression of HO-1 by exposure to 500 μg mL−1 heme–albumin was quantified by ELISA, at both baseline and stressed conditions, as depicted in Fig. 9. Compared to untreated control, there was a significant increase in the expression of HO-1 (p = 0.002) in RPE cells from 118.9 ± 14.9 pg mL−1 to 274.0 ± 44.3 pg mL−1 at baseline conditions. Further expression of HO-1 was shown under stressed conditions where, compared to the oxidatively challenged control, HO-1 expression increased from 135.9 ± 25.2 pg mL−1 to 381.1 ± 96.33 pg mL−1, validating the potential of the proposed treatment pathway for the therapeutic protein complex.
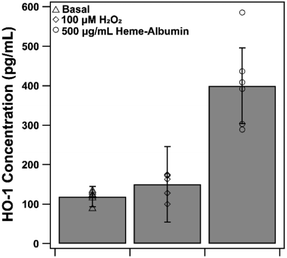 |
| | Fig. 9 HO-1 expression at basal, stressed and co-treatment with 500 μg mL−1 heme–albumin. Compared to basal and stressed ARPE-19 cells, application with heme–albumin showed a significant increase in HO-1 concentration of almost 3 times the protein expressed (*p < 0.05). | |
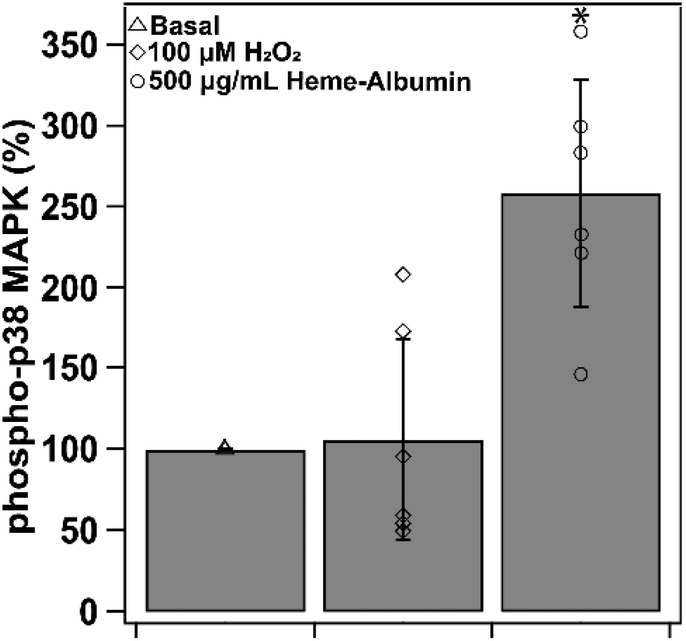
3.8. Heme–albumin increases phospho-p38 MAPK expression
Induction of HO-1 and production of CO by-product is theorized to provide anti-inflammatory relief through cytokine regulation through the p38 MAPK pathway. Heme–albumin's ability to increase expression of activated p38 MAPK was measured through ELISA and was shown to induce a statistically significant effect (p < 0.05) in ARPE-19 (Fig. 10). Compared to basal condition control, heme–albumin caused a 200% increase in activated protein expression.
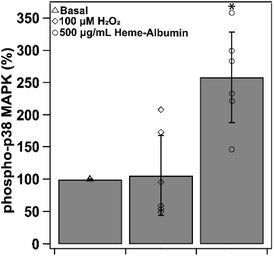 |
| | Fig. 10 Phospho-p38MAPK expression at basal, stressed and co-treatment with 500 μg mL−1 heme–albumin. Compared to basal control, application with heme–albumin showed a significant increase in phospho-p38 MAPK expression (*p < 0.05). | |
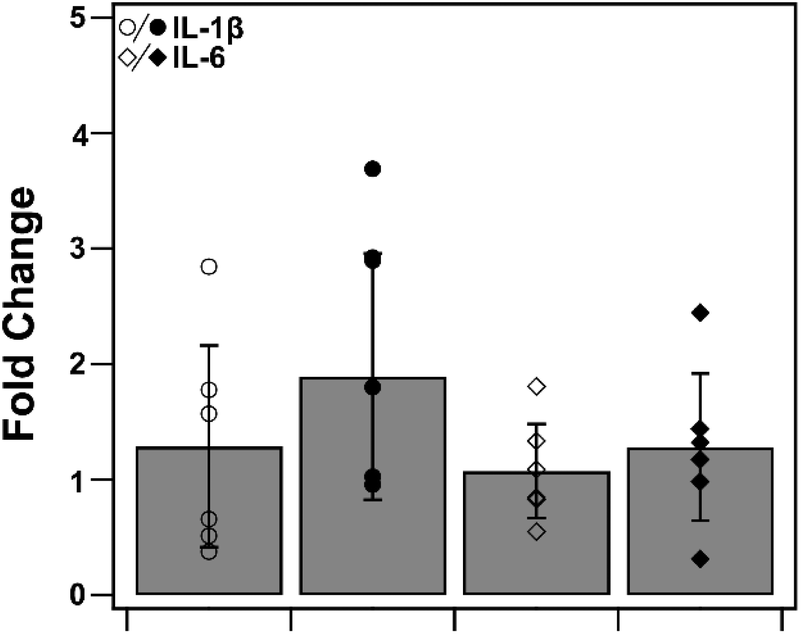
3.9. Heme–albumin does not induce pro-inflammatory cytokine response without activation of p38 MAPK
To determine heme–albumin's impact on inflammatory signalling in unstressed conditions, PCR analysis was conducted on pro-inflammatory cytokines, IL-1β and IL-6. Cytokines were measured and compared to untreated control. Results are shown in Fig. 11. Treatments of 500 μg mL−1 heme–albumin without oxidative challenge do not induce additional cytokine expression as compared to untreated control.
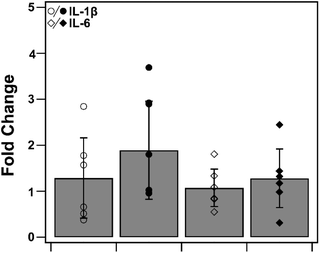 |
| | Fig. 11 PCR quantification of pro-inflammatory cytokines, IL-1β and IL-6. Treatment of ARPE-19 cells with heme–albumin did not induce significant additional pro-inflammatory protein expression compared to control. | |
3.10. Treatment of heme–albumin in oxidative stress conditions reduces pro-apoptotic and IL-6 gene expression
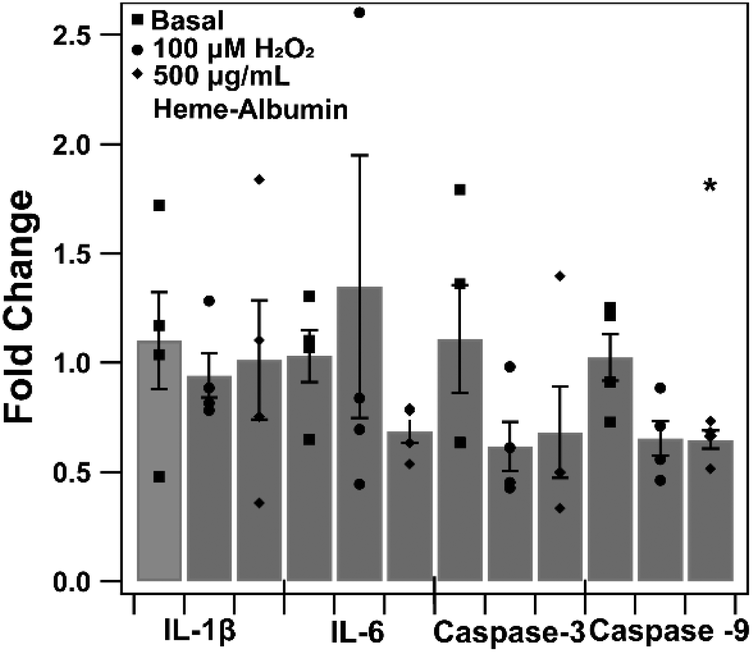
The impact of heme–albumin on pro-inflammatory and pro-apoptotic gene expression in oxidative stress conditions was determined and results are presented in Fig. 12. In both interleukins, there was not a statistically significant expression of Il-1β or IL-6 nor was there a statistical difference in pro-apoptotic caspase-3 gene expression. Lastly, the pro-apoptotic gene caspase-9 did show a statistically significant difference (p = 0.0468) compared to untreated control, showing that heme–albumin is able to reduce pro-apoptotic mRNA expression in oxidative stress conditions.
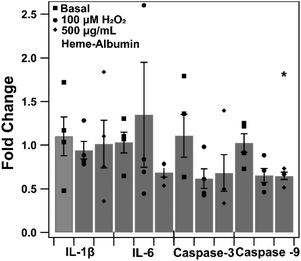 |
| | Fig. 12 qtPCR of pro-inflammatory and apoptotic gene expression in oxidative conditions with 500 μg mL−1 heme–albumin treatment. A statistically significant reduction in caspase-9 gene expression (p = 0.0468) was measured. | |
3.11. Heme–albumin impacts IL-1β expression with oxidative challenge
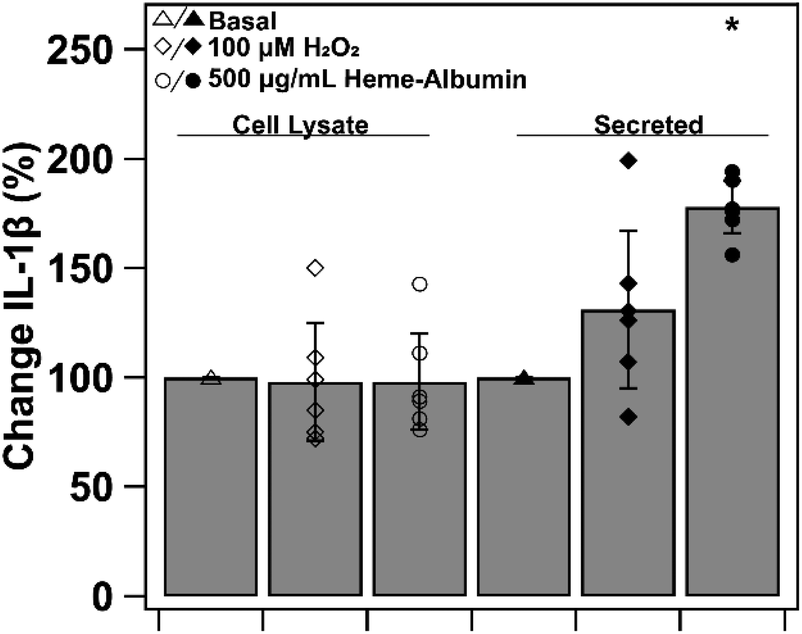
To investigate heme–albumin's impact on inflammatory signalling, ELISA quantification of expressed protein was conducted on pro-inflammatory cytokine, IL-1β. Cytokine expression of basal, oxidatively stressed and co-treatment of oxidative stress and heme–albumin in both cell lysate and media was evaluated. Results are shown in Fig. 13 as percent expression compared to control. Compared to basal control in cell lysate, neither 100 μM H2O2 challenge or co-treatment with oxidative challenge or heme–albumin caused a statistically significant change in IL-1β expression. Secretion of IL-1β did show a statistically significant difference (p < 0.05) compared to untreated control and is theorized to be related to the increase in protein expression of p38 MAPK.44,45
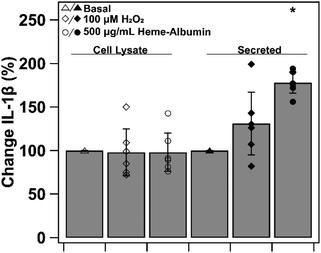 |
| | Fig. 13 IL-1β expression in ARPE-19 cell lysate (open markers) and secreted media (closed markers) treated with basal, oxidatively challenged, or co-treatment with heme–albumin and oxidative challenged conditions. Treatment with heme–albumin induced a statistically significant (p < 0.05) increase in secreted IL-1β, compared to untreated control, relating to the relationship between IL-1β and p38 MAPK activation. | |
3.12. Heme–albumin induces p38β MAPK isoform expression
Induction of HO-1 by heme–albumin for anti-inflammatory and antioxidant relief of oxidative stress activated further downstream expression of p38 MAPK. Heme–albumin's ability to increase total p38 MAPK expression was demonstrated by immunofluorescence staining of ARPE-19 cells. This is shown in Fig. 14.
 |
| | Fig. 14 Increased expression of p38 MAPK by co-treatment of 500 μg mL−1 heme–albumin with 100 μM H2O2 compared to treatment with 100 μM H2O2 or basal conditions alone is shown by immunofluorescence staining. ARPE-19 cells were treated with each treatment group for 24 hours before fixation, permeabilization, and staining with p38 MAPK antibody. | |
4. Conclusion
The pathogenesis of AMD is complex and is exacerbated largely by the confounding oxidative stress of chronic inflammation and ROS affecting the central vision, causing tissue damage and inevitable blindness if left untreated. Treatment for dry AMD with antioxidant and glucocorticoid steroid therapies have shown potential benefits but long-term steroid use is often unfavourable due to their undesirable side effects, including cataract.46 The current standard treatment for the ocular disorder involves up to monthly intravitreal injections that can result in poor patient compliance and treatment management, often leading to further vision loss. To improve treatment options, a novel heme–albumin protein complex delivered by sustained release from polydopamine nanoparticles was investigated to address the inflammatory aspect underlying both forms of AMD. As a free molecule, heme is a low molecular weight and hydrophobic molecule that readily entraps itself in lipophilic cell membranes and lipoproteins, but when bound to human serum albumin (HSA), an important serum protein capable of carrying a variety of types of cargo, heme's hydrophobicity and cytotoxicity are mitigated. The synthesis of both heme–albumin protein complex and loaded PDA nanoparticles was performed to address inflammation and ROS present in AMD. Morphology and zeta potential of the heme–albumin loaded PDA NPs and unloaded PDA NPs were characterized and the in vitro release of heme–albumin at varying concentrations of H2O2 from protein loaded PDA NPs were quantified over a 6-month period, demonstrating sustained release for several months. Higher concentrations of H2O2 induced release of heme–albumin, providing increased therapeutic potential against deleterious levels of ROS. Heme–albumin showed no significant cytotoxicity to ARPE-19 cells up to 2000 μg mL−1 whereas both unloaded and heme–albumin loaded PDA NPs showed minimal cytotoxicity up to concentrations of 200 μg mL−1. Heme–albumin was able to reduce oxidative stress in both inflammatory and ROS models with reductions of up to 22% and 17%, respectively (p = 0.005). Additionally, the novel protein therapeutic caused a significant difference in the expression of HO-1, an essential enzyme in the immune stress response which is vital for therapeutic action. With the oxidative stress elevated by AMD development, HO-1 would be highly expressed to provide immune support. Delivery of heme–albumin loaded PDA NPs could therefore potentially provide sustained delivery of heme–albumin to further induce the anti-inflammatory and antioxidant benefits of HO-1. Incorporated into the delivery system, heme–albumin loaded PDA NPs showed a maximum reduction of oxidative stress of 25% and 34% (p < 0.001) for inflammatory and ROS models, respectively. Compared to traditional, frequent intravitreal administration of anti-VEGF and oral administration of high dose antioxidants for treatment of wet and dry AMD, heme–albumin loaded PDA NPs have potential for reduced injection frequency and a dual method of combating the causes of oxidative assault for innovative treatment of AMD. Future studies will explore safety and efficacy of this therapeutic approach in in vivo models of AMD.
Author contributions
MM Allyn was the main contributor responsible for data curation, formal analysis, visualization, and writing original manuscript draft and revisions. Additionally, MM Allyn played a role in methodology development and conceptualization of project direction. Author MA Rincon-Benavides contributed secondarily to data curation and formal analysis. HL Chandler performed experimental design and data analysis for ELISA and immunofluorescent staining. Author N Higuita-Castro contributed to methodology to assist and guide author MA Rincon-Benavides. Authors AF Palmer and KE Swindle-Reilly were primary contributors to investigation conceptualization and methodology along with review and editing of manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Funding for this research was provided by the Young Investigator Student Fellowship Award from Prevent Blindness Ohio, The Ohio Lions Eye Research Foundation Disaster Relief Grant, The Ohio State University Institute for Materials Research Kickstart Facility Grant, and the Ohio State University College of Engineering.
References
-
World Health Organisation, Global Data on Visual Impairment 2010, 2010, p. 17 Search PubMed
 .
.
- The Lancet Global Health: Vision loss could be treated in one billion people worldwide, unlocking human potential and accelerating global development | Michigan Medicine, https://www.uofmhealth.org/news/archive/202102/lancet-global-health-vision-loss-could-be-treated-one, (accessed 9 November 2021).
- Age-Related Macular Degeneration (AMD) Data and Statistics | National Eye Institute, https://www.nei.nih.gov/learn-about-eye-health/outreach-campaigns-and-resources/eye-health-data-and-statistics/age-related-macular-degeneration-amd-data-and-statistics, (accessed 31 August 2021).
- J. Ambati and B. J. Fowler, Neuron, 2012, 75, 26–39 CrossRef CAS PubMed .
- N. M. Schultz, S. Bhardwaj, C. Barclay, L. Gaspar and J. Schwartz, Clin. Ther., 2021, 43, 1792–1818 CrossRef PubMed .
- R. P. Danis, J. A. Lavine and A. Domalpally, Clin. Ophthalmol., 2015, 9, 2159 CrossRef CAS PubMed .
- R. Mahmoudzadeh, J. W. Hinkle, J. Hsu and S. J. Garg, Curr. Opin. Ophthalmol., 2021, 32, 294–300 CrossRef PubMed .
- A. Gheorghe, L. Mahdi and O. Musat, Rom. J. Ophthalmol., 2015, 59, 74 Search PubMed .
- H. E. Grossniklaus and W. R. Green, Am. J. Ophthalmol., 2004, 137, 496–503 CrossRef PubMed .
- T. J. Heesterbeek, L. Lor Es-Motta, C. B. Hoyng, Y. T. E. Lechanteur and A. I. Den Hollander, Ophthalmic Physiol. Opt., 2020, 40, 140–170 CrossRef PubMed .
- L. F. Hernández-Zimbrón, R. Zamora-Alvarado, L. Ochoa-De La Paz, R. Velez-Montoya, E. Zenteno, R. Gulias-Cañizo, H. Quiroz-Mercado and R. Gonzalez-Salinas, Oxid. Med. Cell. Longevity, 2018, 8374647 Search PubMed .
- S. G. Jarrett and M. E. Boulton, Mol. Aspects Med., 2012, 33, 399–417 CrossRef CAS PubMed .
- L. J. Rohowetz, J. G. Kraus and P. Koulen, Int. J. Mol. Sci., 2018, 19(11), 3362 CrossRef PubMed .
- M. Nita and A. Grzybowski, Oxid. Med. Cell. Longevity, 2016, 2016 DOI:10.1155/2016/3164734 .
- P. S. Mettu, A. R. Wielgus, S. S. Ong and S. W. Cousins, Mol. Aspects Med., 2012, 33, 376–398 CrossRef CAS PubMed .
- D. V. Telegina, O. S. Kozhevnikova and N. G. Kolosova, Biochemistry, 2018, 83, 1009–1017 CAS .
- A. G. Smith and P. K. Kaiser, Expert Opin. Emerging Drugs, 2014, 19, 157–164 CrossRef CAS PubMed .
- J. G. Garweg, J. J. Zirpel, C. Gerhardt and I. B. Pfister, Graefe's Arch. Clin. Exp. Ophthalmol., 2018, 256, 823–831 CrossRef PubMed .
- K. G. Falavarjani, Q. D. Nguyen, K. Ghasemi Falavarjani and Q. D. Nguyen, Eye, 2013, 27, 787–794 CrossRef PubMed .
- A. Sharma, N. Parachuri, N. Kumar, B. D. Kuppermann and F. Bandello, Eye, 2021, 1–2 Search PubMed .
- M. Gemenetzi, A. J. Lotery and P. J. Patel, Eye, 2016, 31, 1–9 CrossRef PubMed .
- H. J. Cho, S. G. Yoo, H. S. Kim, J. H. Kim, C. G. Kim, T. G. Lee and J. W. Kim, Am. J. Ophthalmol., 2015, 159, 285–292 CrossRef CAS PubMed .
- T. A. Cabral de Guimaraes, M. Daich Varela, M. Georgiou and M. Michaelides, Br. J. Ophthalmol., 2022, 106, 297–304 CrossRef PubMed .
- A. Sadaka and G. P. Giuliari, Clin. Ophthalmol., 2012, 6, 1325–1333 Search PubMed .
- A. A. Waza, Z. Hamid, S. Ali, S. A. Bhat and M. A. Bhat, Inflammation Res., 2018, 67, 579–588 CrossRef CAS PubMed .
- R. Gozzelino, V. Jeney and M. P. Soares, Annu. Rev. Pharmacol. Toxicol., 2010, 50, 323–354 CrossRef CAS PubMed .
- G. Silva, A. Cunha, I. P. Grégoire, M. P. Seldon and M. P. Soares, J. Immunol., 2006, 177, 1894–1903 CrossRef CAS PubMed .
- A. D. Bachstetter and L. J. Van Eldik, Aging Dis., 2010, 1, 199 Search PubMed .
- C. Mancuso, Free Radicals Biol. Med., 2021, 172, 521–529 CrossRef CAS PubMed .
- W. Chen, G. J. Maghzal, A. Ayer, C. Suarna, L. L. Dunn and R. Stocker, Free Radicals Biol. Med., 2018, 115, 156–165 CrossRef CAS PubMed .
- B. Wegiel and L. E. Otterbein, Front. Pharmacol., 2012, 3 MAR, 47 Search PubMed .
- R. Krishnan Kutty, C. N. Nagineni, G. Kutty, J. J. Hooks, G. J. Chader and B. Wiggert, J. Cell Physiol., 1994, 159, 371–378 CrossRef PubMed .
- J. Kuesap, B. Li, S. Satarug, K. Takeda, I. Numata, K. Na-Bangchang and S. Shibahara, Biochem. Biophys. Res. Commun., 2008, 367, 413–419 CrossRef CAS PubMed .
- M. Taverna, A. L. Marie, J. P. Mira and B. Guidet, Ann. Intensive Care, 2013, 3, 1–7 CrossRef PubMed .
- M. M. Allyn, R. H. Luo, E. B. Hellwarth and K. E. Swindle-Reilly, Front. Med., 2021, 8, 787644 CrossRef PubMed .
- H. Zhao, Z. Zeng, L. Liu, J. Chen, H. Zhou, L. Huang, J. Huang, H. Xu, Y. Xu, Z. Chen, Y. Wu, W. Guo, J. H. Wang, J. Wang and Z. Liu, Nanoscale, 2018, 10, 6981–6991 RSC .
- X. Bao, J. Zhao, J. Sun, M. Hu and X. Yang, ACS Nano, 2018, 12, 8882–8892 CrossRef CAS PubMed .
- S. Jia, S. Dong, H. Liu, H. Yu, Z. Chen, S. Wang, W. Li, R. Peng, F. Li, Q. Jiang and J. Liu, Biomater. Sci., 2022, 10, 3309 RSC .
- T. Kostka, J. Fohrer, C. Guigas, K. Briviba, N. Seiwert, J. Fahrer, P. Steinberg and M. T. Empl, Arch Toxicol, 2020, 94, 3911–3927 CrossRef CAS PubMed .
- R. J. Wong, H. J. Vreman, S. Schulz, F. S. Kalish, N. W. Pierce and D. K. Stevenson, J. Perinatol., 2011, 31, S35–S41 CrossRef CAS PubMed .
- F. Vallelian, C. A. Schaer, J. W. Deuel, G. Ingoglia, R. Humar, P. W. Buehler and D. J. Schaer, Pharmacol. Res. Perspect., 2018, 6, 2 Search PubMed .
- P. Jiang, A. Choi and K. E. Swindle-Reilly, Nanoscale, 2020, 12, 1–33 RSC .
- M. Anraku, R. Shintomo, K. Taguchi, U. Kragh-Hansen, T. Kai, T. Maruyama and M. Otagiri, Life Sci., 2015, 134, 36–41 CrossRef CAS PubMed .
- J. J. Baldassare, Y. Bi and C. J. Bellone, J. Immunol., 1999, 162, 5367–5373 CAS .
- J. Taylor, D. L. Dewitt, J. Saklatvala, S. H. Ridley, S. J. Sarsfield, J. C. Lee, H. F. Bigg and T. E. Cawston, J. Immunol., 1997, 158, 3165–3173 Search PubMed .
- R. S. Sulaiman, M. Kadmiel and J. A. Cidlowski, Steroids, 2018, 133, 60–66 CrossRef CAS PubMed .
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *abf
*abf