
Substituent-dependent absorption and fluorescence properties of perylene bisimide radical anions and dianions†‡
Rebecca
Renner
a,
Matthias
Stolte
ab,
Julia
Heitmüller
 c,
Tobias
Brixner
bc,
Christoph
Lambert
ab and
Frank
Würthner
*ab
c,
Tobias
Brixner
bc,
Christoph
Lambert
ab and
Frank
Würthner
*ab
aUniversität Würzburg, Institut für Organische Chemie, Am Hubland, 97074 Würzburg, Germany. E-mail: wuerthner@uni-wuerzburg.de
bUniversität Würzburg, Center for Nanosystems Chemistry (CNC), Theodor-Boveri-Weg, 97074 Würzburg, Germany
cUniversität Würzburg, Institut für Physikalische und Theoretische Chemie, Am Hubland, 97074 Würzburg, Germany
First published on 5th August 2021
Abstract
Perylene-3,4:9,10-bis(dicarboximides) (PBIs) rank among the most important functional dyes and organic semiconductors, but only recently have their radical anions and dianions attracted interest for a variety of applications. Here, we systematically elucidate the functional properties (redox, absorption, and emission) of five PBI anions and dianions bearing different bay-substituents attached to the chromophore core. Cyclic voltammetry measurements reveal the influence of the substituents ranging from electron-withdrawing cyano to electron-donating phenoxy groups on the oxidation and reduction potentials that relate to the HOMO and LUMO levels ranging from −7.07 eV to −6.05 eV and −5.01 eV to −4.05 eV, respectively. Spectroelectrochemical studies reveal a significant number of intense absorption bands in the NIR-spectral range (750–1400 nm) for the radical anions, whereas the dianionic species are characterized by similar spectra to those for the neutral dyes, however being bathochromically shifted and with increased molar extinction coefficients of approximately 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1. The increase of the transition dipole moment is up to 56% and accompanied by an almost cyanine-like red-shifted (by 300 nm) absorption spectrum for the most electron-poor tetracyanotetrachloro PBI. Whilst the outstanding fluorescence properties of the neutral PBIs are lost for the radical anions, an appreciable near-infrared (NIR) fluorescence with a quantum yield of up to 18% is revealed for the dianions by utilizing a custom-built flow-cell spectroelectrochemistry setup. Time-dependent density functional theory calculations help to assign the absorption bands to the respective electronic transitions.
000 M−1 cm−1. The increase of the transition dipole moment is up to 56% and accompanied by an almost cyanine-like red-shifted (by 300 nm) absorption spectrum for the most electron-poor tetracyanotetrachloro PBI. Whilst the outstanding fluorescence properties of the neutral PBIs are lost for the radical anions, an appreciable near-infrared (NIR) fluorescence with a quantum yield of up to 18% is revealed for the dianions by utilizing a custom-built flow-cell spectroelectrochemistry setup. Time-dependent density functional theory calculations help to assign the absorption bands to the respective electronic transitions.
New conceptsDuring the last decade our group has made many contributions to the field of perylene bisimide dyes that constitute a most versatile class of absorbers, emitters and redox systems. Furthermore, they are widely utilized as building blocks for aggregates and functional materials. Most recently, new perspectives arose for perylene bisimides in electro- and photocatalysis. In this regard it is surprising that the properties of their radical anions and dianions remained rather unexplored despite their importance for many of these applications. After we were recently able to establish a custom-made setup for the controlled generation of specific redox states in our optical spectrometers, we could characterize not only the absorption but also the emission properties of the reduced species, as described in the current manuscript. We expect that this fundamental study will be most useful for everyone interested in radical anionic and dianionic species of perylene bisimide dyes, i.e. researchers interested in photoinduced electron transfer reactions, battery materials, photocatalysis, organic photovoltaics, etc. |
Introduction
About 100 years ago, perylene-3,4:9,10-bis(dicarboximide) (PBI) dyes firstly attracted attention as vat dyes and later on as industrial pigments.1–3 When used as vat dyes, the water-insoluble PBI pigment is reduced to the soluble leuco form, corresponding to an anionic species, which can be processed easily in textile or paper dying and other applications. Upon exposure of this reduced species to air the dye is re-oxidized, similar to indigo,4 giving the coloured fabric.5 Due to the straightforward adjustability of optical (absorption, emission) and electronic (ionization potential, electron affinity) properties of PBIs by substitution, a large variety of PBI derivatives has been developed in particular during the last decade.6,7 By this means, more sophisticated optoelectronic applications of PBIs emerged in n-channel organic field effect transistors (OFETs),8,9 ion batteries,10 electrocatalytic reduction of oxygen to hydrogen peroxide,11 organic light emitting diodes (OLEDs)12,13 and organic photovoltaics (OPVs).14,15 These applications, similar to the vat dying process, rely on the electron-deficient character of the PBI core and the formation of comparably stable delocalized radical anions and dianions upon the addition of electrons.16–18 Likewise, PBIs enjoy great popularity as acceptor components in molecular dyads due to the formation of stable and spectroscopically characteristic anionic species.19–21 More recently, the use of PBI radical anions and dianions generated by photoexcitation in the presence of triethylamine was further demonstrated as photosensitizers, catalysing the dehalogenation reaction of a broad variety of aryl halides, further revealing the versatile applicability of the robust reduced PBI species.22,23 Investigations by transient absorption (TA) spectroscopy proved the efficient photoinduced electron transfer from the excited doublet state of the PBI radical anion to electron acceptors.24,25 This characteristic of PBIs and related rylene bisimides might be further utilized in the development of new photosensitizers as catalysts for reactions, which would otherwise need highly negative potentials. In this regard Xie and coworkers just recently demonstrated the stepwise reduction of a PBI dye under irradiation, used for the injection of an electron into TiO2 nanoparticles coated with platinum as a co-catalyst, which could be used for the efficient reduction of water into hydrogen.26Chemical reduction of PBIs remains a field to be further explored, as reports are relatively scarce due to the ambient instability of most reduced PBIs. Accordingly, most anions and dianions reported in the literature are generated in situ electrochemically or chemically by reduction with sodium dithionite in deoxygenated water, affording stable solutions for months due to the delocalized aromatic character of these PBI dyes.16,18,27 Unfortunately, these species cannot be easily isolated from solution. The first report of an isolated and completely characterized, albeit zwitterionic radical anion that is stable under ambient conditions shows the feasibility of the generation of isolable PBI radical anions in general.28 This organic zwitterion can be considered as a PBI radical anion, which is stabilized by a σ-bonded imidazolium cation. Furthermore, it can be reduced or oxidized further by titration with tetrabutylammonium fluoride or nitrosyl tetrafluoroborate, respectively. In the same year, an extremely electron-poor PBI, having chlorine atoms attached in bay-positions as well as cyano groups in ortho-positions was isolated and characterized by single crystal X-ray diffraction.29 Reduction of this molecule with palladium on activated charcoal, hydrogen and sodium hydrogen carbonate gave the PBI dianion disodium salt, which was stable even under ambient conditions for months.29 In the following years, more examples of ambient stable radical anions and dianions have been reported, using more moderate reducing agents like potassium carbonate in DMF30,31 or tetraphenylphosphonium iodide.32
Usually, characterization of the reduced species remains scarce and only qualitative UV/Vis/NIR absorption spectra of the neutral as well as reduced species are reported, while insights into emission properties are missing. Recently, after Rybtchinski's first observation of fluorescence for the parent core-unsubstituted PBI dianion,27 we were able to disclose the first example of a fluorescence spectrum for the dianion of a tetraphenoxy-substituted PBI, utilizing a flow-cell setup that enabled the investigation of the emission as well as femtosecond and coherent two-dimensional (2D) electronic spectroscopy measurements.33 The latter technique indeed points to another major application of PBI dyes as acceptors in donor–acceptor dyads. Here, PBIs are the preferred acceptors due to the characteristic signature of the PBI radical anion bands in the NIR spectral range.34 However, whilst TA signatures can be easily assigned for those PBIs that have been characterized by spectroelectrochemistry, they are not available for the vast majority of core-substituted PBIs that could be of value for such studies due to the tunability of their redox properties. Because until now, a concise series of PBIs with a systematic variation of their redox properties has been missing, we herein investigate PBIs in the neutral as well as in their radical anionic and dianionic states for a consistent series of five PBI dyes bearing the 2,6-diisopropylphenyl imide (Dipp) substituent and different bay-substituents by electrochemical and spectroelectrochemical measurements. Furthermore, we also investigate the emission properties for the dianions of this series of PBIs. Within our series, the respective first reduction potential varies by about 1 eV and also comprises planar as well as heavily twisted chromophores. Furthermore, (time-dependent) density functional theory ((TD-)DFT) calculations have been performed to systematically assess the origin of the observed molecular properties and spectral features of the differently charged species.
Results and discussion
Molecular design and chemical reduction
Five PBIs have been chosen for the investigation of their molecular properties in their neutral as well as reduced states. Three of them (PBI–Cl, PBI–H, and PBI–OPh) are commonly applied as organic semiconductors35–38 or fluorescent dye derivatives,39 whilst the two others complement the series based on a comparably very high reduction potential (PBI–ClCN, see ESI‡)29 and a fixed helical twist (PBI–Ph)40, which seem to be important for the successful application of these PBIs as non-fullerene acceptors in OPV (Fig. 1a).41,42 For all derivatives, the bulky 2,6-diisopropylphenyl (Dipp) has been chosen as the imide residue to prevent aggregation and ensure a high solubility.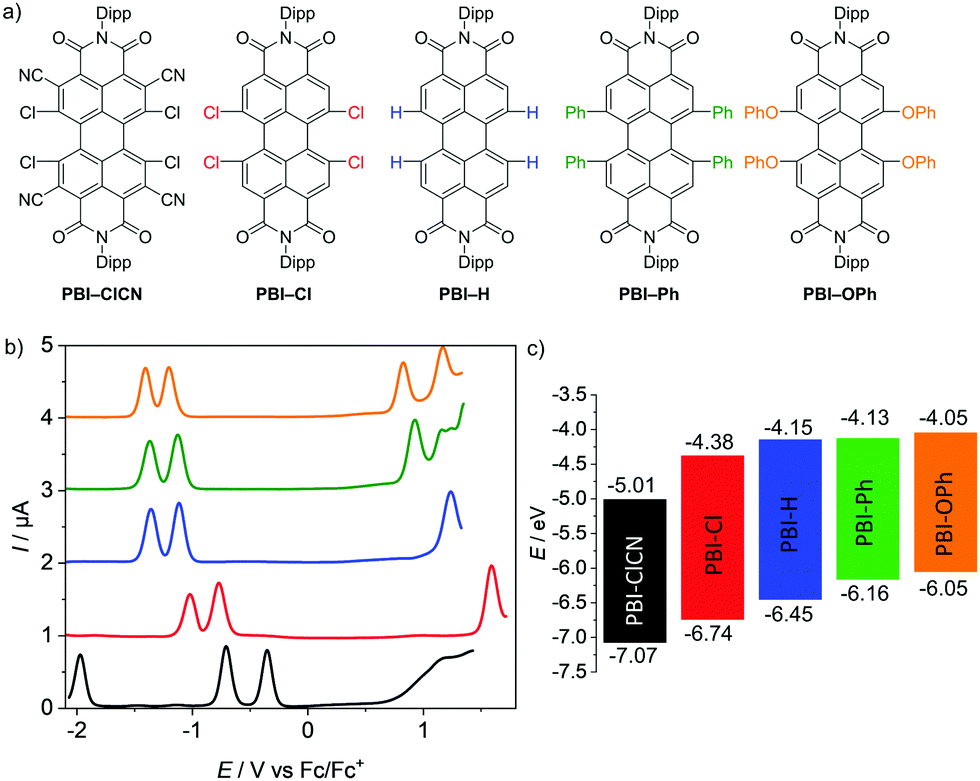 | ||
| Fig. 1 (a) Molecular structures of the PBIs with bulky 2,6-diisopropylphenyl (Dipp) imide substituents investigated by electrochemical reduction. (b) Square wave voltammograms of PBI–ClCN (black), PBI–Cl (red), PBI–H (blue), PBI–Ph (green) and PBI–OPh (orange). Measurements were performed using DCM solutions (c0 = 2 × 10−4 M) at room temperature, using TBAHFP (0.1 M) as electrolyte (scan rate 100 mV s−1, SW amplitude 25 mV, SW frequency 15 Hz). (c) Schematic diagram of the electrical band gaps of the investigated PBIs (energy level of Fc/Fc+ with respect to the vacuum level = −5.15 eV).15 | ||
Generally, either chemical reduction, photoinduced electron transfer or electrochemical reduction can be used to obtain the reduced species of PBIs. Advantageous of the former method would be the straightforward measurement of the desired species by a variety of techniques without the need for elaborate setups due to different measurement geometries and accordingly we first pursued the reduction with KC8 in THF.43 As the stoichiometric addition of the reducing agent did not give the desired dianions, an excess of up to four equivalents of the reducing agent was used. The reaction could be monitored by the naked eye, due to prominent changes of the coloured PBI solutions. Upon the addition of 18-crown-6 to the solution, the reaction was significantly accelerated. All the obtained products could be dissolved in 1,2-difluorobenzene for the measurement of UV/Vis/NIR absorption spectra. However even under inert conditions, the oxidation of the generated dianions into the radical anions was observed (Fig. S7, ESI‡). The increased stability of the radical anions compared to the dianions is quite remarkable and has already been observed for other organic radical anions.43,44 As the stability of the dianions for hours in solution would be necessary for the full characterization of their optical properties, this reduction method did not provide the needed species with sufficient selectivity. Furthermore, the spectra could only be measured in the rather exotic solvent 1,2-difluorobenzene due to an even more pronounced instability in other solvents. As more common solvents like dichloromethane or chloroform are desirable to be more comparable to the literature reported TA spectra, we focused our attention on the electrochemical reduction to spectroscopically characterize the radical anions and dianions of all PBIs.
Cyclic and square wave voltammetry
The redox properties of the PBI series have been investigated by cyclic voltammetry (CV) and square wave (SW) voltammetry under identical experimental conditions in dichloromethane (DCM) using tetrabutylammonium hexafluorophosphate (TBAHFP) as the electrolyte. The SW voltammograms as well as a schematic diagram of the electrical band gaps are depicted in Fig. 1b and c and the half-wave potentials and the calculated HOMO and LUMO potentials are listed in Table 1 (Fig. S4–S6, ESI‡). At least two reversible reduction waves are observed for all five derivatives. Values range from −0.14 V for the first reduction and −0.48 V for the second reduction of the strongest electron acceptor PBI–ClCN, which are in line with the reported values for a derivative thereof,29 to −1.10 V and −1.29 V for the first and second reduction waves, respectively, for the weakest electron acceptor PBI–OPh.45 The other derivatives follow the expected sequence by exhibiting their first reduction wave at −0.77 V for PBI–Cl, −1.00 V for PBI–H and −1.02 V for PBI–Ph, respectively. The potentials of the second reduction wave follow the same trend with values of −1.02 V, −1.22 V and −1.25 V for the latter three PBIs, respectively. Interestingly, the first and second reduction potentials of the planar PBI–H and the heavily twisted PBI–Ph are very similar, indicating the comparably small influence of a directly attached aryl unit compared to stronger electron withdrawing groups like halogens or electron donating aryloxy groups on the electronic properties of the molecule. Integration of the first and second reduction peaks in the SW spectra proved that the ratio of the electronic quantity of the reduction processes from PBI to PBI•− and PBI•− to PBI2− is one, substantiating the nature of the proposed reduced species. A third reversible reduction wave at −1.73 V could only be observed for PBI–ClCN, accentuating the electron deficiency of this derivative due to the additional cyano groups in the ortho-position. The effect of the substituents on the redox potentials is even more pronounced for their respective oxidation processes.45 Due to the highly electron-deficient character of PBI–ClCN, no oxidation could be observed within the available potential range of the used electrolyte system, whereas one oxidation process is observed at +1.59 V and +1.30 V for PBI–Cl and PBI–H, respectively. Due to the shift of the oxidation waves to lower potentials, two oxidation processes are observable for the other two PBIs at +1.01 V and +1.19 V for PBI–Ph and +0.90 V and +1.21 V for PBI–OPh. The values are summarized in Table 1. The HOMO and LUMO energy levels have been calculated from the half-wave potentials of the first oxidation and reduction processes, which were obtained by the CV measurements with the energy level of the ferrocene/ferrocenium redox couple set to −5.15 eV vs. vacuum.15 They cover a broad range of almost 1 eV within this series between −7.07 eV and −6.05 eV for the HOMOs and −5.01 eV and −4.05 eV for the LUMOs for PBI–ClCN and PBI–OPh (Fig. 1c), respectively. Notably, PBI–ClCN ranks according to this analysis among the most electron-deficient π-cores, comparable to tetracyanoquinodimethane (TCNQ).46,47| E Red11/2 [V] | E Red21/2 [V] | E Red31/2 [V] | E Ox11/2 [V] | E Ox21/2 [V] | E HOMO [eV] | E LUMO [eV] | ΔECVgap [eV] | ΔEOptgap [eV] | |
|---|---|---|---|---|---|---|---|---|---|
| a Not observed. b Calculated according to the known literature procedure using the experimentally determined redox potentials (EHOMO = −[EOx11/2 + 5.15 eV] and ELUMO = −[ERed11/2 + 5.15 eV]) and the energy level of Fc/Fc+ with respect to the vacuum level (−5.15 eV).15 c Calculated according to EHOMO = ELUMO − E(λmax). d Optical band gap higher than electronic band gap probably due to distortion of the band shape due to the overlap of different transitions. Elaborate calculations taking correction factors into account can be found in Table S1 and Table S2 in the ESI. | |||||||||
| PBI–ClCN | −0.14 | −0.48 | −1.73 | —a | —a | −7.07c | −5.01 | 2.06 | 2.13d |
| PBI–Cl | −0.77 | −1.02 | —a | +1.59 | —a | −6.74 | −4.38 | 2.36 | 2.21 |
| PBI–H | −1.00 | −1.22 | —a | +1.30 | —a | −6.45 | −4.15 | 2.30 | 2.26 |
| PBI–Ph | −1.02 | −1.25 | —a | +1.01 | +1.19 | −6.16 | −4.13 | 2.03 | 1.82 |
| PBI–OPh | −1.10 | −1.29 | —a | +0.90 | +1.21 | −6.05 | −4.05 | 2.00 | 1.99 |
UV/Vis/NIR absorption properties in neutral and reduced states
The UV/Vis/NIR absorption spectra of the neutral, radical anionic and dianionic PBIs in DCM could be obtained by electrochemical reduction in situ with a custom built spectroelectrochemical cell and are depicted in Fig. 2 and the derived data are collected in Table 2 (for details see the ESI‡ and Fig. S8–S18, Table S3). The optical band gap calculated with the help of the absorption spectra of the neutral species and extrapolation by a linear curve (Tauc's plot)48 is in all cases, except PBI–ClCN, lower in energy than the electrical band gap, which is expected since the threshold for the absorption of photons is smaller than the threshold for the creation of an electron–hole pair.49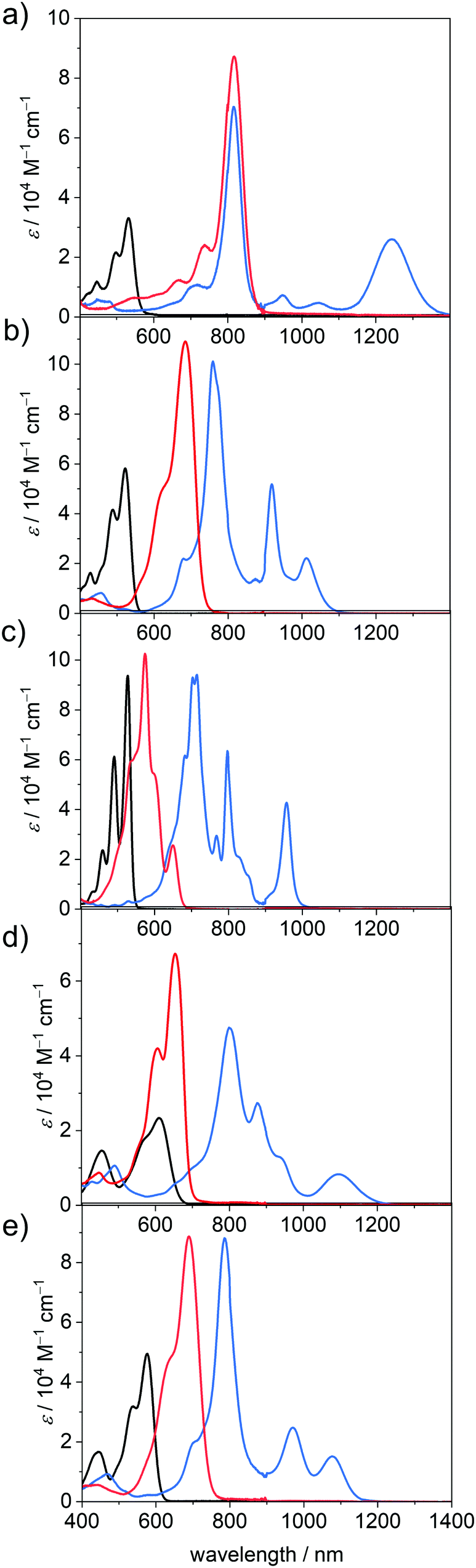 | ||
| Fig. 2 UV/Vis/NIR absorption spectra of the neutral (black), radical anionic (blue) and dianionic (red) PBI species of (a) PBI–ClCN, (b) PBI–Cl, (c) PBI–H, (d) PBI–Ph, and (e) PBI–OPh measured in DCM (c0 = 4 × 10−4 M) at room temperature using TBAHFP (0.1 M) as electrolyte. | ||
| λ max [nm] | ε max [M−1 cm−1] | μ eg [D] | λ em [nm] | Δ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/char_e0e1.gif) Stokes [cm−1] Stokes [cm−1] |
Φ f [%] | τ F [ns] | |
|---|---|---|---|---|---|---|---|
| a Fluorescence spectra of PBI2− have been measured in front-face geometry (22.5°) and all spectra are corrected against the photomultiplier sensitivity and the lamp intensity. b Fluorescence quantum yields of the neutral PBIs were determined using the dilution method (OD < 0.05) and N,N′-bis(2,6-diisopropylphenyl)-perylene-3,4:9,10-bis(dicarboximide) (ΦF = 1.00 in CHCl3) or N,N′-bis(2,6-diisopropylphenyl)- 1,6,7,12-tetraphenoxy-perylene-3,4:9,10-bis(dicarboximide) (for PBI–OPh, ΦF = 0.96 in CHCl3)39 as reference. Fluorescence quantum yields of PBI2− were determined relative to PBI by excitation at 385 nm (PBI–Cl), 415 nm (PBI–H), 490 nm (PBI–Ph) and 398 nm (PBI–OPh). c Fluorescence lifetimes were determined with EPL picosecond pulsed diode lasers for time-correlated single photon counting (λex = 530 nm (PBI–ClCN), λex = 550 nm (PBI–Cl, PBI–H, PBI–OPh), λex = 560 nm (PBI–Ph), λex = 725 nm (PBI–Cl2−), λex = 662 nm (PBI–H2−), λex = 725 nm (PBI–Cl2−), λex = 677 nm (PBI–Ph2−), λex = 735 nm (PBI–OPh2−)). d Significant overestimation due to multiple transitions present in the S0–S1 absorption band. e Not observed. | |||||||
| PBI–ClCN | 531 | 33900 |
—d | 575 | 1510 | 0.1 | 0.15, 2.10 |
| PBI–ClCN2− | 817 | 87200 |
—d | —e | —e | —e | —e |
| PBI–Cl | 522 | 58200 |
8.7 | 549 | 940 | 57 | 5.06 |
| PBI–Cl2− | 684 | 109100 |
12.6 | 722 | 770 | 3 | 2.04 |
| PBI–H | 527 | 93700 |
8.8 | 534 | 210 | 99 | 3.72 |
| PBI–H2− | 574 | 102600 |
3.5 | 662 | 300 | 18 | 5.92 |
| PBI–Ph | 609 | 23400 |
6.4 | 677 | 1650 | 46 | 10.87 |
| PBI–Ph2− | 653 | 67300 |
10.0 | 676 | 520 | 15 | 4.36 |
| PBI–OPh | 576 | 49600 |
8.7 | 608 | 915 | 96 | 6.01 |
| PBI–OPh2− | 689 | 88700 |
12.4 | 738 | 960 | 17 | 3.93 |
In accordance with earlier results for these and related PBIs,1 the unsubstituted planar PBI–H exhibits an intense absorption band of the S0-S1 transition with vibronic progression with an absorption maximum (λmax) at 527 nm and a high molar extinction coefficient (εmax) of 93700 M−1 cm−1 in DCM. Upon substitution in the bay-position, as is the case for all other derivatives within this series, the spectra become more broadened and show a decrease in εmax and the vibronic progression of the S0–S1 transition is less clearly visible. Furthermore, the S0–S2 transition, which is symmetry forbidden for the planar chromophore,50 becomes allowed and can be observed as a second absorption maximum at about 440 nm. This effect is most pronounced for PBI–Ph, which contains the most twisted chromophore.40 The attachment of electron withdrawing halogen atoms in the bay-position does not exhibit a large influence on the absolute position of the absorption maximum (PBI–Cl 522 nm; PBI–ClCN 531 nm), whereas the more electron donating groups like tetraaryl (PBI–Ph 609 nm) or tetraaryloxy (PBI–OPh 576 nm) afford a significant redshift, reflecting electronic interactions, i.e., electron donation from the bay-substituents to the PBI core.51
Under the influence of an electrochemical potential between −0.1 and −1.2 V, significant spectral changes occur for all PBI dyes in DCM. All radical anions exhibit multiple characteristic spectral features in the UV/Vis/NIR region, each with a prominent and broad absorption band at 957 nm or above (PBI–H), which is commonly assigned to the D0-D1 transition (vide infra).52 In the case of PBI–ClCN this NIR transition appears at 1243 nm, followed by PBI–Ph (1097 nm), PBI–OPh (1076 nm) and PBI–Cl (1011 nm) (Fig. 2). The absorption spectra in the UV/Vis/NIR region of all five radical anions exhibit at least three clearly distinguishable transitions. The most prominent absorption bands are also bathochromically shifted with respect to those of the neutral PBIs and are found for all derivatives between 715 nm for the unsubstituted PBI–H and 817 nm for PBI–ClCN. Whereas the electron withdrawing groups exert only a minor influence on the wavelength of the lowest energy absorption band in the neutral state, they affect those of all absorption bands in the radical anionic state in a similarly pronounced way as the electron donating substituents. Furthermore, different from PBI–H with an almost unchanged εmax value of 94100 M−1 cm−1 upon reduction to the radical anion, a significant increase between 174% (PBI–Cl, 101100 M−1 cm−1) up to 212% (PBI–ClCN, 70300 M−1 cm−1) in εmax is found for the main absorption band of all the bay-substituted derivatives. Additionally, at least one further transition can be observed between the absorption maximum for the most intense band and the lowest energy absorption band of the radical anions. For the unsubstituted derivative PBI–H, even more distinguishable absorption features than for the other PBIs are found, which originate from the more rigid and planar π-scaffold, similar to the neutral species. These intense and characteristic absorption bands make the here investigated PBI derivatives particularly valuable for the investigation of photoinduced electron transfer processes in molecular dyads as well as solid state materials.52
Upon further reduction at a bias between −0.4 and −1.3 V, the absorption spectra of all PBI dianions simplify to one prominent absorption band that is located between 650 and 900 nm. This hypsochromic shift with respect to the NIR bands of the radical anion species is accompanied by an additional increase in band intensity. Thus, the values for εmax increase further for all the derivatives to more than 100000 M−1 cm−1 in the case of PBI–Cl (684 nm) and PBI–H (574 nm) and to more than twice the intensity of the respective neutral species in the cases of PBI–Ph (67300 M−1 cm−1 at 653 nm) and PBI–ClCN (87200 M−1 cm−1 at 817 nm). Surprisingly, only the dianion of the parent PBI–H displays an optical spectrum of rather different shape with a transition at 649 nm of lower intensity (25600 M−1 cm−1) followed by a strong absorbing and structured absorption spectrum at about 574 nm. The increase in tinctorial strength for these PBI dianions is indeed remarkable. Integration of the respective isolated longest wavelength S0–S1 absorption bands of the neutral and the dianionic species shows again an increase in the transition dipole moments μeg from 8.7 to 12.6 D (PBI–Cl), 6.4 to 10.0 D (PBI–Ph) and 8.7 to 12.4 D (PBI–OPh) for all derivatives except PBI–H, where it decreases from 8.8 to 3.5 D (Table 2). Some of these values might be overestimated due to the presence of multiple transitions in the respective S0-S1 absorption band (vide infra).
Fluorescence properties in the neutral and dianionic states
The fluorescence spectra, fluorescence quantum yields (Φf) and fluorescence lifetimes (τF) of the five neutral PBIs and four dianionic PBIs could be obtained within an SEC flow-cell setup (Fig. 3, Table 2 and Fig. S8b, S20–S22, ESI‡). As expected, all the radical anions showed no emission. The Φf of the dianions were determined relative to the respective neutral species within one SEC run, while the fluorescence decay curves were recorded by time-correlated single-photon counting (TCSPC) in front-face geometry (22.5°). All emission spectra of the neutral and dianionic PBIs show mirror-like shape with respect to their corresponding absorption spectra, with the exception of PBI–Ph2−, which exhibits an unexpected narrow fluorescence band with a well-defined vibronic progression. All the bay-substituted PBIs show almost equal (PBI–OPh2−) or even decreased Stokes shifts (Δ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) Stokes) in their dianionic states, while the unsubstituted PBI–H exhibited a slight increase from 210 cm−1 (PBI) to 300 cm−1 (PBI2−), which is still smaller than the ΔStokes of the other derivatives due to the more rigid framework and lack of substituents. The same holds true for τF, which is increased again only for PBI–H from 3.72 ns (PBI) to 5.92 ns (PBI2−), while it is decreased in all other derivatives upon reduction.
Stokes) in their dianionic states, while the unsubstituted PBI–H exhibited a slight increase from 210 cm−1 (PBI) to 300 cm−1 (PBI2−), which is still smaller than the ΔStokes of the other derivatives due to the more rigid framework and lack of substituents. The same holds true for τF, which is increased again only for PBI–H from 3.72 ns (PBI) to 5.92 ns (PBI2−), while it is decreased in all other derivatives upon reduction.
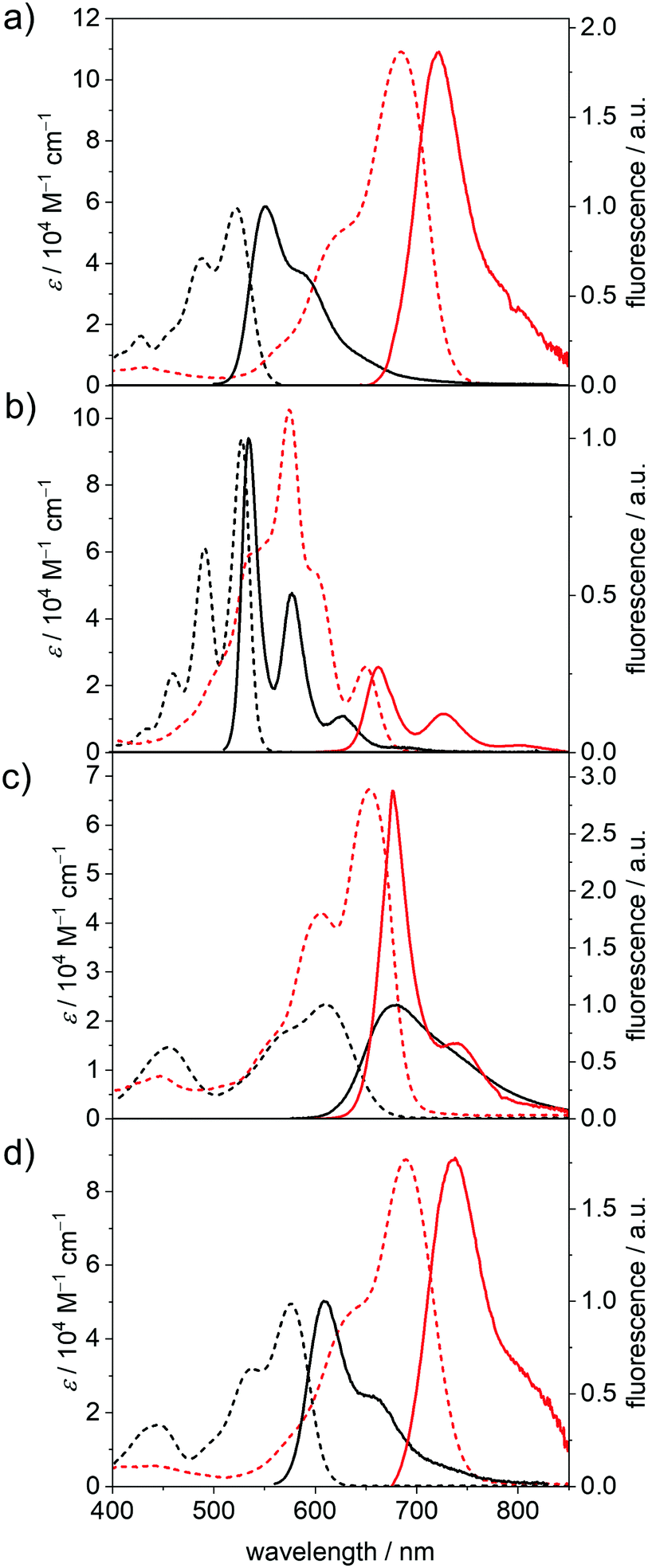 | ||
| Fig. 3 UV/Vis/NIR absorption spectra (dashed lines) as well as the fluorescence spectra (solid lines) of the neutral (black) and dianionic (red) (a) PBI–Cl, (b) PBI–H, (c) PBI–Ph and (d) PBI–OPh measured in DCM (c0 = 4 × 10−4 M for absorption, c0 = 1 × 10−5 M for fluorescence of PBI and c0 = 1 × 10−4 M for fluorescence of PBI2−) at room temperature using TBAHFP (0.1 M) as electrolyte. | ||
The Φf of all dianions are significantly reduced as expected for the emission due to the decreased band gap53 but are still appreciably high with 18% at 662 nm (PBI–H2−) and 17% at 738 nm (PBI–OPh2−) compared to, e.g., higher rylene dyes emitting in the region between 650 and 950 nm.54–56 For PBI–Cl2− an emission maximum at 722 nm, with reduced ΔStokes of 770 cm−1 and the most pronounced decrease of the quantum yield within this series is noted from 57% (PBI) to 3% (PBI2−). PBI–Ph exhibits a large ΔStokes of 1650 cm−1 affording λem of 677 nm. This indicates the participation of the core-substituents in the lowest excited state upon photoexcitation and the following structural relaxation.20 Interestingly, the Φf of the arylated dye does not decrease as significantly upon reduction as the other derivatives, from 46% of the neutral PBI–Ph to 15% for PBI–Ph2−. Together with the structured fluorescence spectrum such high quantum yield corroborates the rigidification effect impaired by the four phenyl units that are interlocked based on the crystallographic evidence from the related chromophores.57 The neutral PBI–ClCN exhibits only very weak fluorescence upon excitation at 490 nm, with a λem at 575 nm and a Φf of merely 0.1%. Because this is much lower than the reported quantum yield of 11% of the neutral congener with electron-withdrawing fluoroalkyl chains in the imide position we attribute the fluorescence quenching to a photoinduced electron transfer from the Dipp substituent into the highly electron-deficient core.58 Similar processes have been reported for the more electron-rich imide substituents to the parent electron-poor PBI.20,59
The decrease of Φf upon reduction for all the derivatives can be ascribed to an increase of the rate of non-radiative decay processes, as expected due to the smaller band gap for the dianionic state.60 The trend observed in τF corresponds to the trend obtained for the natural lifetimes τ0 by analysis according to the Strickler–Berg relationship.61 For PBI–H, τ0 is slightly increased in the dianion (3.64 ns to 3.75 ns), while for the other three derivatives PBI–Cl (5.14 ns to 4.50 ns), PBI–Ph (17.5 ns to 5.08 ns) and PBI–OPh (6.89 ns to 5.22 ns) τ0 is decreased in the measured reduced state of the molecules due to the increase of the transition dipole moment and oscillator strength. The radiative decay rates kr obtained by this analysis nevertheless decrease in all the derivatives upon reduction, while the non-radiative decay rates knr are increased, confirming the experimental results of the decreasing quantum yields in the PBI dianions.
Theoretical investigations
In order to further elucidate the trends observed by optical spectroscopy upon electrochemical reduction, (time-dependent) density functional theory ((TD-)DFT) calculations were performed. For simplicity, the Dipp imide residues were replaced by methyl groups, as their impact on the molecular properties of the chromophore is negligible.62The geometry-optimized structures of all derivatives with the exception of PBI–H exhibit a large twist of the two naphthalene subunits between 31.0° (PBI–OPh) and 39.5° (PBI–ClCN). This results in a distortion of the PBI π-MO system. In all derivatives the LUMO of the neutral PBI, the SOMO of the radical anion (PBI•−) and the HOMO of the dianion (PBI2−) are visually indistinguishable and thus in Fig. 4 (Fig. S24 and 25, ESI‡) only the MOs of the neutral PBI are shown.
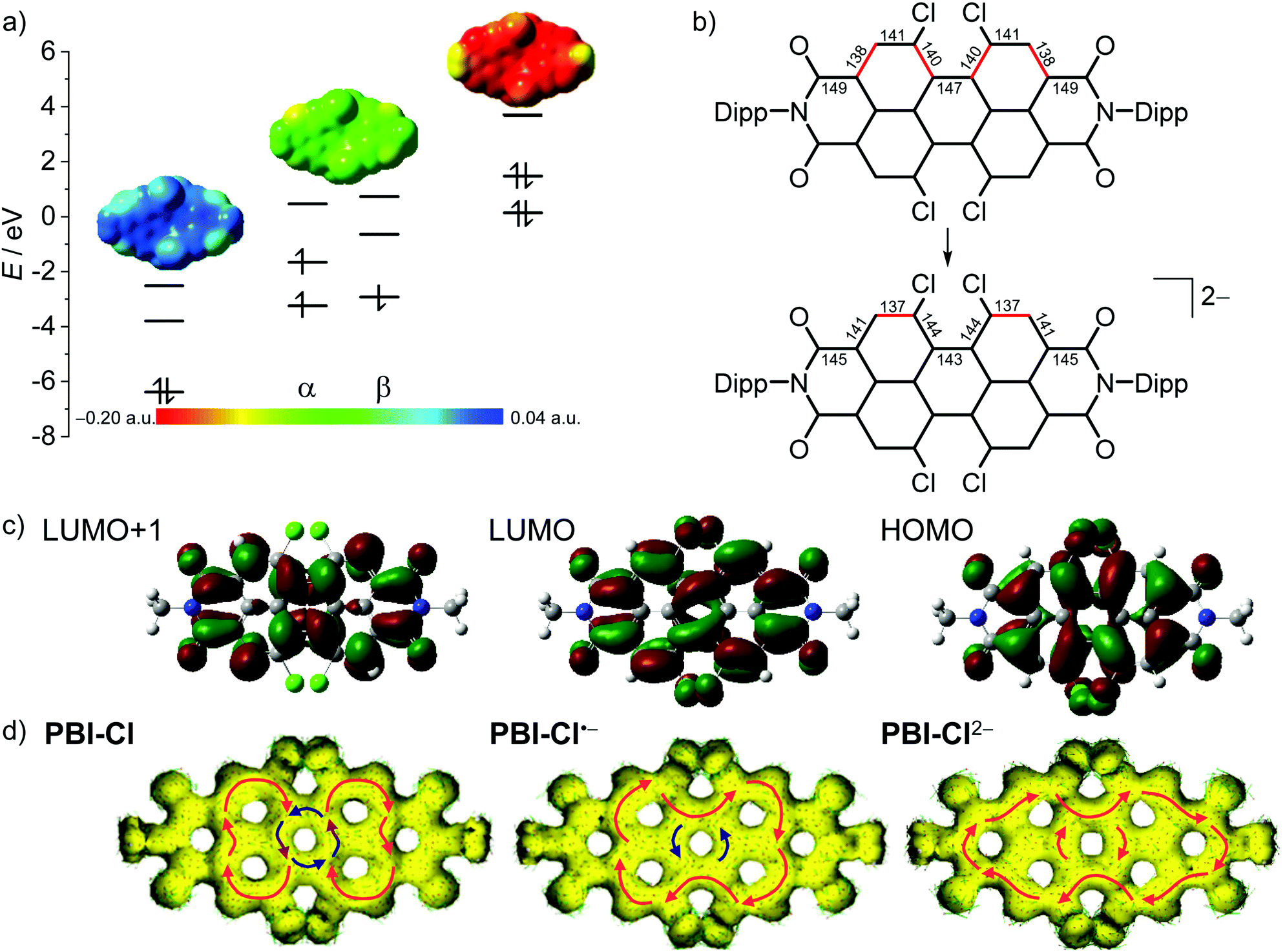 | ||
| Fig. 4 (a) The HOMO, LUMO and LUMO+1 levels as obtained from DFT calculations of PBI–Cl (left), the corresponding α- and β-spin MO levels of PBI–Cl•− (middle) and HOMO−1, HOMO and LUMO levels of PBI–Cl2− (right), as well as the electrostatic potential maps of the respective derivative (top, isovalue 0.020 a.u.). (b) Calculated bond length for perylene carbon–carbon bonds in neutral and dianionic states in pm (for details, see Table S4, ESI‡). Bonds drawn in red indicate a high double-bond character. (c) The HOMO, LUMO and LUMO+1 (right to left) of PBI–Cl. The corresponding orbitals of PBI–Cl•− and PBI–Cl2− are visually indistinguishable. Orbitals and electrostatic potential maps were calculated using DFT (B3LYP/6-31g(d)). (d) Calculated AICD isosurface plots of PBI–Cl, PBI–Cl•− and PBI–Cl2−. Clockwise ring current is depicted with red arrows and counterclockwise ring current with blue arrows (isosurface value 0.025). | ||
As depicted in Fig. 4b for the tetrachlorinated PBI–Cl there are significant changes in bond length upon reduction from the neutral to the dianionic state. Whilst the changes in bond length are comparably minor for the inner bonds of the perylene (<3 pm), the conjugated outer perimeter experiences a significant alteration where bonds either get shorter by up to 3.8 pm from 148.5 pm to 144.7 pm or longer by up to 3.4 pm from 138.0 pm to 141.4 pm. This change in bond length relates to a reversal of single versus double bond character, which is in accordance with the bonding character between the involved atoms in the LUMO (Fig. 4c). As a consequence, the PBI skeleton of PBI–Cl (and likewise the other derivatives) is contracted by about 66 pm along the long molecular axis upon twofold reduction and expanded by about 57 pm along the short axis. It is interesting to relate these bond length alterations to the carbon–carbon bond stretching vibration that occurs upon photoexcitation of PBI into the S1 state, i.e., promotion of an electron from the HOMO into the LUMO, and that causes the characteristic vibronic progression in the absorption and fluorescence bands of PBIs. The most striking change of bond length concerns the bonds connecting the two naphthalene subunits, e.g., in the tetrachlorinated PBI–Cl from 146.8 pm for the neutral PBI via 144.8 pm for the radical anion to 143.1 pm in the dianion (Table S4, ESI‡). This decreased bond length in the dianion for the central connections between the naphthalene subunits as well as the bond length alterations for the peripheral C–C carbon chain is in accordance with calculations by Iron, Rybtchinski and co-workers for the unsubstituted PBI16,27 and our experimental results by single-crystal X-ray analysis for a derivative of PBI–ClCN.29 This corroborates a stiffening of the π-scaffold for the central ring and planarization of the PBI core with a reduction of the twist angle from 35.4° to 31° in PBI–Cl due to the additional electron density in the system (Table S4, ESI‡).
The transitions to the excited states obtained by TD-DFT calculations are in good accordance with the experimental results concerning the number and intensities of the observed transitions (Fig. S23, Table S5–S19, ESI‡). While the calculated transition energies agree reasonably well with the neutral PBIs, e.g., PBI–H with 488 and 527 nm for the calculated and experimental spectra, respectively, they deviate in some examples of the radical anions and dianions quite severely. Nevertheless, the bathochromic shift of the radical anion spectra visible in all derivatives, as well as the subsequent hypsochromic shift of the most prominent transitions of the dianions is substantiated by the theoretical results. The calculated energies for the allowed optical transitions could be correlated to the respective experimental values well enough by shifting the whole spectrum, so that the transitions with the highest oscillator strength exhibit the same energy as the absorption maximum in the experimental absorption spectrum, allowing for an assignment of the individual absorption bands (Fig. S23, ESI‡) with which some further conclusions could be made (vide infra).
According to the calculations, for the neutral state of the most electron-deficient PBI–ClCN, two transitions are quite close in energy around 570 nm and thus no clear separation thereof can be observed in the experimental spectra (Table S5, ESI‡). In the other three bay-substituted derivatives, the bands of the experimental spectra could be assigned to the S0–S1 transition as well as the S0–S2 transition at slightly higher energies. In the planar unsubstituted PBI–H, only the S0–S1 transition exhibits a noteworthy oscillator strength, corresponding to former reports and the symmetry forbiddance of the S0–S2 transition.63 Due to the spin-separated energy levels in the radical anionic state, the assignment of the transitions is more complex. In the bay-chlorinated PBIs PBI–ClCN and PBI–Cl the SOMO–LUMO transition corresponds to the lowest-energy transition in the NIR region and thus can be assigned to D0–D1 transition. The same holds true for PBI–OPh. In the unsubstituted PBI–H, this transition causes the absorption band at 649 nm in the calculated spectrum, while the most bathochromically shifted absorption band corresponds to a transition of the electron from the highest occupied β-spin orbital to the lowest unoccupied β-spin orbital. In the tetraphenyl derivative PBI–Ph, the SOMO–LUMO transition corresponds to the absorption maximum at 679 nm, while for the lowest-energy absorption band a similar transition of the electron in the β-MO as in PBI–H is found. The predictions for optically allowed transitions of PBI radical anions are probably less accurate than the predictions for the neutral and dianionic species due to the radical nature of the PBIs and the neglect of solvent effects.16
In the calculated transitions of the dianions, the HOMO–LUMO transition thereof corresponds to the lowest-energy transition in all derivatives except for PBI–Cl and PBI–OPh. For these two derivatives, the lowest-energy transition is not well described by the simple HOMO–LUMO transition but rather by a HOMO–LUMO+1 transition, which suggests that the excitation energy is actually not equal to the HOMO–LUMO orbital energy difference. Thus, even though the calculations are of good assistance in the interpretation of the experimental spectra, the reality might be more complex. In accordance with the experimental UV/Vis/NIR spectrum of PBI–H2−, the calculations predict a significantly smaller oscillator strength of 0.0938 for the S0-S1 transition than for the S0-S2 transition with 0.8905 (see Fig. 2c and Table S13, ESI‡). The intensity of the absorption bands relates to the oscillator strength or the transition dipole moment.
Unfortunately, the experimental determination of the transition dipole moments (μeg) of some derivatives, especially in their reduced states, is not straightforward for this series of dyes due to a significant overlap of different transitions, thus overestimating the values significantly (vide supra). However, the values for μeg of neutral PBI–Cl, PBI–H, PBI–Ph and PBI–OPh could be determined by integration of the absorption band corresponding to the S0–S1 transition of the experimental spectra to be 8.7 D, 8.8 D, 6.4 D and 8.7 D, which fits reasonably well with the values obtained from TD-DFT calculations of 7.6 D, 8.8 D, 6.6 D and 8.1 D, respectively (Table S20, ESI‡). Theoretical determination of μeg from TD-DFT calculations for all species shows a decrease of the transition dipole moment for the lowest energy transition upon the first reduction of the PBIs with the exception of the directly arylated PBI–Ph, where μeg of the radical anion is increased compared to the neutral PBI. Upon further reduction to the dianion, a further increase of μeg is noted for all PBI dyes with values reaching above 9.0 D for most derivatives. Similar to the previous results (vide supra), the calculations predict μeg of all five neutral PBIs as well as their dianionic states reasonably well (Table S20, ESI‡).
The differences in oscillator strength of the first two transitions of PBI–H2− compared to the bay-substituted PBIs can be rationalized by investigation of the transition density (Fig. S27, ESI‡). It can be seen that the transition density of the S0–S1 transition of PBI–H2− is polarized along the short molecular axis and resembles the transition densities of the S0–S2 transitions of the four substituted PBI dianions. In contrast, the S0–S2 transition density of PBI–H2−, which is polarized along the long molecular axis (N,N′) exhibits a high oscillator strength in the absorption spectrum, which is comparable to the S0–S1 transitions of the substituted PBI dianions; thus the two differently polarized transitions are interchanged in the planar PBI dianion compared to the bay-substituted contorted systems.64
To further explore the aromaticity of the different reduced species, anisotropy of the induced current-density (AICD) calculations was performed for PBI–Cl and PBI–H as well as their respective reduced species.65,66 In both the neutral molecules, clockwise diamagnetic ring currents can be observed that are localized in the two naphthalene subunits, suggesting a Clar-type localization of aromaticity, while the central PBI ring exhibits a counterclockwise paramagnetic ring current, indicating anti-aromaticity (Fig. 4d, Fig. S28, ESI‡). Upon reduction, the clockwise ring current expands over the whole PBI core along the outer perimeter and the counterclockwise current in the centre vanishes. This reorganization of the π-electron delocalization has been elucidated before in a theoretical study by Iron and co-workers to explain the high stability of PBI dianions in water due to their increased aromatic character.16 These results from the AICD calculations match quite well to our earlier observations, i.e., the contraction of the bonds connecting the two naphthalene subunits due to increased conjugation and the increase of the absorption coefficients and bathochromic shifts of the absorption bands observed for PBI dianions.
Conclusion
In this article, the optical properties for a series of five perylene bisimide dyes bearing 2,6-diisopropylphenyl groups in the imide position but different bay-substituents on the π-core have been investigated upon their reduction into radical anionic and dianionic states in custom-built spectroelectrochemical cells for UV/Vis/NIR absorption and emission studies. Their first reduction potentials range almost over 1 V from −0.14 V for the most electron-deficient PBI–ClCN to −1.10 V for the most electron-rich PBI–OPh. Significant bathochromic shifts of all the absorption spectra upon the first reduction with the most red-shifted maximum at 1243 nm (PBI–ClCN) were observed. Upon further reduction, the absorption maxima shift again back hypsochromically compared to the radical anionic spectra. The molar extinction coefficients εmax increase in all derivatives upon reduction to approximately 100000 M−1 cm−1, with absorption maxima between 649 nm (PBI–H) and 817 nm (PBI–ClCN) for the dianions. While fluorescence data of neutral PBI dyes are abundantly reported, we could study and quantify the fluorescence properties (Φf, τF, ΔStokes) of the dianions of the whole series in a custom-built flow-cell setup. While the fluorescence quantum yield was reduced for all PBI dianions, due to an increased influence of non-radiative decay pathways, values of up to 18% (662 nm) and 17% (738 nm) were retained for PBI–H and PBI–OPh, respectively. DFT and TD-DFT calculations assist in the elucidation of the origin of the experimentally observed absorption bands as well as structural changes of all PBIs and their reduced species. They showed a localization of the MOs on the perylene core rather than on the substituents for the relevant energetic states. Furthermore, the experimental spectra could be brought into line with the calculated absorption spectra. Overall, based on our comprehensive library of five PBIs, electronic and optical properties of their radical anions and their dianions could be characterized in a systematic manner. This information should be of great value for the analysis of PBI-bearing molecular dyads and PBI-based solid-state materials by transient absorption spectroscopy and for the utilization of these dyes as sensitizers for photocatalysis.
Author contributions
Rebecca Renner: investigation, methodology, and visualization. Matthias Stolte: methodology, investigation, and writing. Julia Heitmüller: methodology, writing. Tobias Brixner: writing, supervision, and funding acquisition. Christoph Lambert: writing. Frank Würthner: conceptualization, writing, supervision, and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Dr David Bialas for his assistance with DFT and TD-DFT calculations. We are grateful for the financial support from the Deutsche Forschungsgemeinschaft (Grant Wu 317/20-2).Notes and references
- C. Huang, S. Barlow and S. R. Marder, J. Org. Chem., 2011, 76, 2386–2407 CrossRef CAS.
- K. Hunger and W. Herbst, Pigments, Organic in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2000 Search PubMed.
- W. Herbst and K. Hunger, Industrial Organic Pigments: Production, Properties, Applications, Wiley-VCH, Weinheim, 3rd edn, 2004 Search PubMed.
- E. Steingruber, Indigo and Indigo Colorants in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2004 Search PubMed.
- M. Greene, High Performance Pigments, Wiley-VCH, Weinheim, 2009, ch. 16 Search PubMed.
- A. Nowak-Król and F. Würthner, Org. Chem. Front., 2019, 6, 1272–1318 RSC.
- F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962–1052 CrossRef.
- R. Schmidt, J. H. Oh, Y.-S. Sun, M. Deppisch, A.-M. Krause, K. Radacki, H. Braunschweig, M. Könemann, P. Erk, Z. Bao and F. Würthner, J. Am. Chem. Soc., 2009, 131, 6215–6228 CrossRef CAS.
- B. A. Jones, A. Facchetti, M. R. Wasielewski and T. J. Marks, J. Am. Chem. Soc., 2007, 129, 15259–15278 CrossRef CAS PubMed.
- H.-G. Wang, S. Yuan, D.-I. Ma, X.-I. Huang, F.-I. Meng and X.-B. Zhang, Adv. Energy Mater., 2014, 4, 1301651 CrossRef.
- M. Warczak, M. Gryszel, M. Jakesova, V. Đerek and E. D. Głowacki, Chem. Commun., 2018, 54, 1960–1963 RSC.
- S. V. Dayneko, M. Rahmati, M. Pahlevani and G. C. Welch, J. Mater. Chem. C, 2020, 8, 2314–2319 RSC.
- G. Li, Y. Zhao, J. Li, J. Cao, J. Zhu, X. W. Sun and Q. Zhang, J. Org. Chem., 2015, 80, 196–203 CrossRef CAS.
- X. Zhan, Z.-A. Tan, B. Domercq, Z. An, X. Zhang, S. Barlow, Y. Li, D. Zhu, B. Kippelen and S. R. Marder, J. Am. Chem. Soc., 2007, 129, 7246–7247 CrossRef CAS.
- A. Nowak-Król, K. Shoyama, M. Stolte and F. Würthner, Chem. Commun., 2018, 54, 13763–13772 RSC.
- M. A. Iron, R. Cohen and B. Rybtchinski, J. Phys. Chem., 2011, 115, 2047–2056 CrossRef CAS.
- V. V. Roznyatovskiy, D. M. Gradner, S. W. Eaton and M. R. Wasielewski, Org. Lett., 2014, 16, 696–699 CrossRef CAS.
- R. O. Marcon and S. Brochsztain, Langmuir, 2007, 23, 11972–11976 CrossRef CAS.
- B. Rybtchinski, L. E. Sinks and M. R. Wasielewski, J. Am. Chem. Soc., 2004, 126, 12268–12269 CrossRef CAS PubMed.
- W. Kim, T. Kim, S. Kang, Y. Hong, F. Würthner and D. Kim, Angew. Chem., Int. Ed., 2020, 132, 8571–8578 CrossRef PubMed.
- M. S. Rodríguez-Morgande, T. Torres, C. Atienza-Castellanos and D. M. Guldi, J. Am. Chem. Soc., 2006, 128, 15145–15154 CrossRef.
- I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Science, 2014, 346, 725–728 CrossRef CAS PubMed.
- H.-X. Gong, Z. Cao, M.-H. Li, S.-H. Liao and M.-J. Lin, Org. Chem. Front., 2018, 5, 2296–2302 RSC.
- C. J. Zeman, S. Kim, F. Zhang and K. S. Schanze, J. Am. Chem. Soc., 2020, 142, 2204–2207 CrossRef CAS.
- C. Rosso, G. Filippini and M. Prato, Eur. J. Org. Chem., 2021, 1193–1200 CrossRef CAS.
- Y. Xu, J. Zheng, J. O. Lindner, X. Wen, N. Jiang, Z. Hu, L. Liu, F. Huang, F. Würthner and Z. Xie, Angew. Chem., Int. Ed., 2020, 59, 10363–10367 CrossRef CAS.
- E. Shirman, A. Ustinov, N. Ben-Shitrit, H. Weissman, M. A. Iron, R. Cohen and B. Rybtchinski, J. Phys. Chem. B, 2008, 112, 8855–8858 CrossRef CAS.
- D. Schmidt, D. Bialas and F. Würthner, Angew. Chem., Int. Ed., 2015, 54, 3611–3614 CrossRef CAS PubMed.
- S. Seifert, D. Schmidt and F. Würthner, Chem. Sci., 2015, 6, 1663–1667 RSC.
- Q. Li, H. Guo, X. Yang, S. Zhang and H. Zhang, Tetrahedron, 2017, 73, 6632–6636 CrossRef CAS.
- E. He, J. Wang, H. Liu, Z. He, H. Zhao, W. Bao, R. Zhang and H. Zhang, J. Mater. Sci., 2016, 51, 9229–9238 CrossRef CAS.
- Y. Kumar, S. Kumar, D. Bansal and P. Mukhopadhyay, Org. Lett., 2019, 21, 2185–2188 CrossRef CAS.
- J. Heitmüller, K. Eckstein, R. Renner, M. Stolte, T. Hertel, F. Würthner and T. Brixner, Spectrochim. Acta, Part A, 2021, 253, 119567 CrossRef.
- P. E. Hartnett, C. M. Mauck, M. A. Harris, R. M. Young, Y.-L. Wu, T. J. Marks and M. R. Wasielewski, J. Am. Chem. Soc., 2017, 139, 749–756 CrossRef CAS PubMed.
- L. H. Slooff, E. E. Bende, A. R. Burgers, T. Budel, M. Pravettoni, R. P. Kenny, E. D. Dunlop and A. Büchtemann, Phys. Status Solidi RRL, 2008, 2, 257–259 CrossRef CAS.
- Z. Chem, M. G. Debije, T. Debaerdemaeker, P. Osswald and F. Würthner, ChemPhysChem, 2004, 5, 137–140 CrossRef.
- R. Kinderman, L. H. Slooff, A. R. Burgers, N. J. Bakker, A. Büchtemann, R. Danz and J. A. M. van Rosmalen, J. Sol. Energy Eng., 2007, 129, 277–282 CrossRef CAS.
- B. E. Hardin, E. T. Hoke, P. B. Armstrong, J.-H. Yum, P. Comte, T. Torres, J. M. J. Fréchet, M. K. Nazeeruddin, M. Grätzel and M. D. McGehee, Nat. Photonics, 2009, 3, 406–411 CrossRef CAS.
- G. Seybold and G. Wagenblast, Dyes Pigm., 1989, 11(4), 303–317 CrossRef.
- B. Pagoaga, L. Giraudet and N. Hoffmann, Eur. J. Org. Chem., 2014, 5178–5195 CrossRef CAS.
- Y. H. Cai, L. J. Huo, X. B. Sun, D. H. Wei, M. S. Tang and Y. M. Sun, Adv. Energy Mater., 2015, 5, 1500032 CrossRef.
- Y. H. Cai, X. Y. Guo, X. B. Sun, D. H. Wei, M. M. Yu, L. J. Huo and Y. M. Sun, Sci. China Mater., 2016, 59, 427–434 CrossRef CAS.
- B. A. R. Günther, S. Höfener, R. Eichelmann, U. Zschieschang, H. Wadepohl, H. Klauk and L. H. Gade, Org. Lett., 2020, 22, 2298–2302 CrossRef PubMed.
- L. Ji, M. Haehnel, I. Krummenacher, P. Biegger, F. L. Geyer, O. Tverskoy, M. Schaffroth, J. Han, A. Dreuw, T. B. Marder and U. H. F. Bunz, Angew. Chem., Int. Ed., 2016, 55, 10498–10501 CrossRef CAS.
- P. Leowanawat, A. Nowak-Król and F. Würthner, Org. Chem. Front., 2016, 3, 537–544 RSC.
- S. Kumar, J. Shukla, Y. Kumar and P. Mukhopadhyay, Org. Chem. Front., 2018, 5, 2254–2276 RSC.
- M. Kozaki, K. Sugimura, H. Ohnishi and K. Okada, Org. Lett., 2006, 8, 5235–5238 CrossRef CAS PubMed.
- A. A. Haidry, J. Puskelova, T. Plecenik, P. Durina, J. Gregus, M. Truchly, T. Roch, M. Zahoran, M. Vargova, P. Kus, A. Plecenik and G. Plesch, Appl. Surf. Sci., 2012, 259, 270–275 CrossRef CAS.
- H. Fritzsche, J. Non-Cryst. Solids, 1971, 6, 49–71 CrossRef CAS.
- K. Nagarajan, A. R. Mallia, V. S. Reddy and M. Hariharan, J. Phys. Chem. C, 2016, 120, 8443–8450 CrossRef CAS.
- Z. Chen, U. Baumeister, C. Tschierske and F. Würthner, Chem. – Eur. J., 2007, 13, 450–465 CrossRef CAS.
- D. Gosztola, M. P. Niemczyk, W. Svec, A. S. Lukas and M. R. Wasielewski, J. Phys. Chem. A, 2000, 104, 6545–6551 CrossRef CAS.
- M. Bixon, J. Jortner, J. Cortes, H. Heitele and M. E. Michel-Beyerle, J. Phys. Chem., 1994, 98, 7289–7299 CrossRef CAS.
- L. Chen, C. Li and K. Müllen, J. Mater. Chem. C, 2014, 2, 1938–1956 RSC.
- F. Nolde, J. Qu, C. Kohl, N. G. Pschirer, E. Reuther and K. Müllen, Chem. – Eur. J., 2005, 11, 3959–3967 CrossRef CAS.
- F. O. Holtrup, G. R. J. Müller, H. Quante, S. De Feyter, F. C. De Schryver and K. Müllen, Chem. – Eur. J., 1997, 3, 219–225 CrossRef CAS PubMed.
- R. Renner, B. Mahlmeister, O. Anhalt, M. Stolte and F. Würthner, Chem. – Eur. J., 2021 DOI:10.1002/chem.202101877.
- S. Seifert, Doctoral dissertation, Julius-Maximilians-Universität Würzburg, 2017 Search PubMed.
- E. H. A. Beckers, S. C. J. Meskers, A. P. H. J. Schenning, Z. Chen, F. Würthner and R. A. J. Janssen, J. Phys. Chem. A, 2004, 108, 6933–6937 CrossRef CAS.
- R. Englman and J. Jortner, Mol. Phys., 1970, 18, 145–164 CrossRef CAS.
- S. J. Stricker and R. A. Berg, J. Chem. Phys., 1962, 37, 814–822 CrossRef.
- F. Würthner, Chem. Commun., 2004, 1564–1579 RSC.
- Z. Chen, U. Baumeister, C. Tschierske and F. Würthner, Chem. – Eur. J., 2007, 15, 450–465 CrossRef.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS.
- R. Herges and D. Geuenich, J. Phys. Chem. A, 2001, 105, 3214–3220 CrossRef CAS.
- D. Geuenich, K. Hess, F. Köhler and R. Herges, Chem. Rev., 2005, 105, 3758–3772 CrossRef CAS.
Footnotes |
| † Dedicated to Prof. Seth Marder on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available: General and synthetic methods; additional electrochemical and optical measurements as well as additional theoretical calculations. See DOI: 10.1039/d1mh01019k |
| This journal is © The Royal Society of Chemistry 2022 |