
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reverse sequence polymerization-induced self-assembly in aqueous media: a counter-intuitive approach to sterically-stabilized diblock copolymer nano-objects†
Nicholas J. W.
Penfold
 a,
Thomas J.
Neal
*a,
Corentin
Plait
a,
Andrew E.
Leigh
a,
Gwen
Chimonides
a,
Mark J.
Smallridge
b and
Steven P.
Armes
*a
a,
Thomas J.
Neal
*a,
Corentin
Plait
a,
Andrew E.
Leigh
a,
Gwen
Chimonides
a,
Mark J.
Smallridge
b and
Steven P.
Armes
*a
aDainton Building, Department of Chemistry, The University of Sheffield, Brook Hill, Sheffield, South Yorkshire S3 7HF, UK. E-mail: s.p.armes@sheffield.ac.uk; tneal@ed.ac.uk
bGEO Specialty Chemicals, Hythe, Southampton, Hampshire SO45 3ZG, UK
First published on 11th October 2022
Abstract
Polymerization-induced self-assembly (PISA) is a powerful platform technology for the efficient synthesis of block copolymer nanoparticles in many types of solvents, including water. In PISA, a soluble precursor block is used to grow a second insoluble block, which leads to in situ self-assembly of the block copolymer chains. Thus, in the case of aqueous PISA, the water-soluble block is always prepared first because this confers steric stabilization. Herein, we challenge this paradigm by demonstrating that amphiphilic diblock copolymer chains can be prepared in water by preparing the hydrophobic block first via reversible addition–fragmentation chain transfer (RAFT) polymerization. This counter-intuitive reverse sequence PISA formulation utilizes an ionic RAFT agent to conduct the RAFT aqueous dispersion polymerization of 2-hydroxypropyl methacrylate (HPMA), which results in the formation of charge-stabilized PHPMA latex particles of ∼500 nm diameter. Initial attempts to chain-extend these hydrophobic PHPMA chains with water-miscible monomers such as glycerol monomethacrylate (GMA) were unsuccessful, with only uncontrolled free radical polymerization being observed in the aqueous phase. However, using a water-immiscible monomer such as isopropylideneglycerol methacrylate (IPGMA) enabled the synthesis of charge-stabilized PHPMA-PIPGMA latex particles. Subsequent acid hydrolysis of the PIPGMA block led to the in situ formation of sterically-stabilized PHPMA-PGMA diblock copolymer spheres. Alternatively, dissolution of the precursor PHPMA latex in a methanol/water binary mixture enables RAFT solution polymerization of water-miscible monomers such as GMA or N,N′-dimethylacrylamide (DMAC) to be achieved with good control. The resulting amphiphilic diblock copolymer chains then undergo self-assembly in aqueous solution after removal of the methanol co-solvent. Finally, this reverse sequence PISA protocol can also be applied to other vinyl monomers such as 2-methoxyethyl methacrylate (MOEMA) or diacetone acrylamide (DAAM), which significantly broadens its scope.
Introduction
Over the past decade it has become clear that polymerization-induced self-assembly (PISA) is a powerful and versatile platform technology for the convenient synthesis of a wide range of block copolymer nano-objects in the form of concentrated colloidal dispersions.1–11 Typically, PISA involves growing a polymer chain from one end of a precursor chain using an appropriate monomer in a suitable solvent. The second chain becomes insoluble when it reaches a certain critical mean degree of polymerization: this induces micellar nucleation and leads to the formation of sterically-stabilized diblock copolymer nanoparticles.12–15 Many vinyl monomers have been used to prepare such nanoparticles and most of these studies have employed reversible addition–fragmentation chain transfer (RAFT) polymerization.16–19 This is no doubt because this technique is highly tolerant of monomer functionality.19,20It is well-established that PISA can be conducted in many different types of solvents.21–31 In the case of water, PISA can be performed using either water-miscible or water-immiscible monomers via either aqueous dispersion polymerization or aqueous emulsion polymerization, respectively.32–67 For such aqueous PISA formulations, it is seemingly essential that the hydrophilic block must be prepared first to ensure that colloidal stability is retained via a steric stabilization mechanism. Recently, we have challenged this paradigm by demonstrating that the hydrophobic water-insoluble block can be prepared before generating the hydrophilic block: this new approach is termed reverse sequence PISA.68 Herein, we report on our new experiments based on this counter-intuitive strategy. More specifically, we conduct the RAFT aqueous dispersion of 2-hydroxypropyl methacrylate (HPMA) and the RAFT aqueous emulsion polymerization of 2-methoxyethyl methacrylate (MOEMA)15 using a previously reported cationic morpholine-functional RAFT agent known as MPETTC (Scheme 1).69 At low pH, the morpholine end-groups become protonated, which leads to the formation of a cationic precursor latex that remains colloidally stable via a charge stabilization mechanism. Two methods were subsequently explored to chain-extend the latex and hence introduce an appropriate steric stabilizer block. The first method involved dissolution of the hydrophobic precursor latex with a suitable water-miscible co-solvent, with subsequent polymerization of a hydrophilic monomer to produce the steric stabilizer block via RAFT solution polymerization. The second method involved extending the hydrophobic chains via RAFT seeded emulsion polymerization using a second water-immiscible monomer, isopropylideneglycerol monomethacrylate (IPGMA). Again, the resulting diblock copolymer latex remains colloidally stable owing to charge stabilization. Subsequently, the acetone protecting groups on the PIPGMA chains can be removed via acid hydrolysis to generate an amphiphilic PHPMA-PGMA diblock copolymer that self-assembles in situ to form sterically-stabilized diblock copolymer nano-objects. In addition, the RAFT aqueous dispersion polymerization of diacetone acrylamide (DAAM) using a carboxylic acid-based RAFT agent at pH 7 is briefly explored.
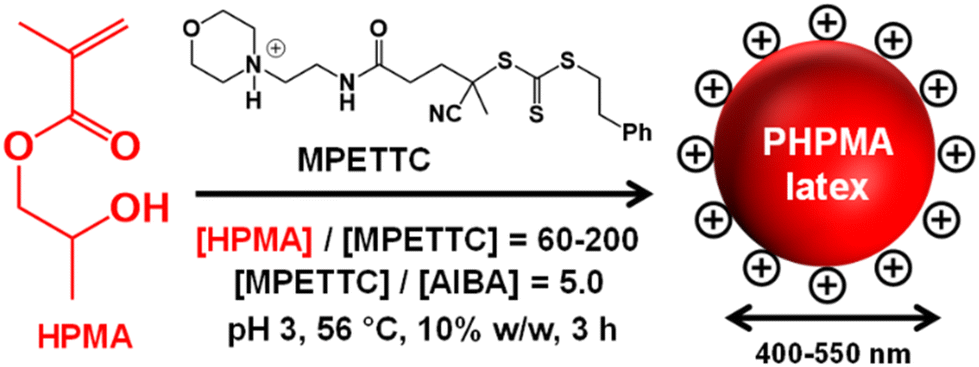 | ||
| Scheme 1 Schematic cartoon of the RAFT aqueous dispersion polymerization of HPMA using a cationic morpholine-functionalized RAFT agent (MPETTC) and an azo initiator (AIBA) at pH 3 to yield a charge-stabilized precursor PHPMA latex at 56 °C. | ||
Results and discussion
Synthesis of latex precursors via RAFT aqueous dispersion polymerization
Kinetic data were obtained for the RAFT aqueous dispersion polymerization of HPMA at 56 °C by periodic sampling of the reaction mixture followed by 1H NMR spectroscopy studies and DMF GPC analysis. This scoping experiment was performed at pH 3 by targeting a mean degree of polymerization (DP) of 140 for the PHPMA chains at 10% w/w solids. The conversion vs. time curve and corresponding semi-logarithmic plot are shown in Fig. S1.† Initially, a relatively slow RAFT solution polymerization was observed, with only 43% conversion being achieved after 1.5 h at 56 °C. Thereafter, a gradual increase in turbidity indicated micellar nucleation; this coincides with a dramatic increase in the rate of polymerization, with more than 99% conversion being obtained within 3 h. Similar observations have been reported for conventional aqueous PISA syntheses.70–72 Visual inspection of the final latex dispersion indicated a milky-white, free-flowing fluid (see Fig. 1A).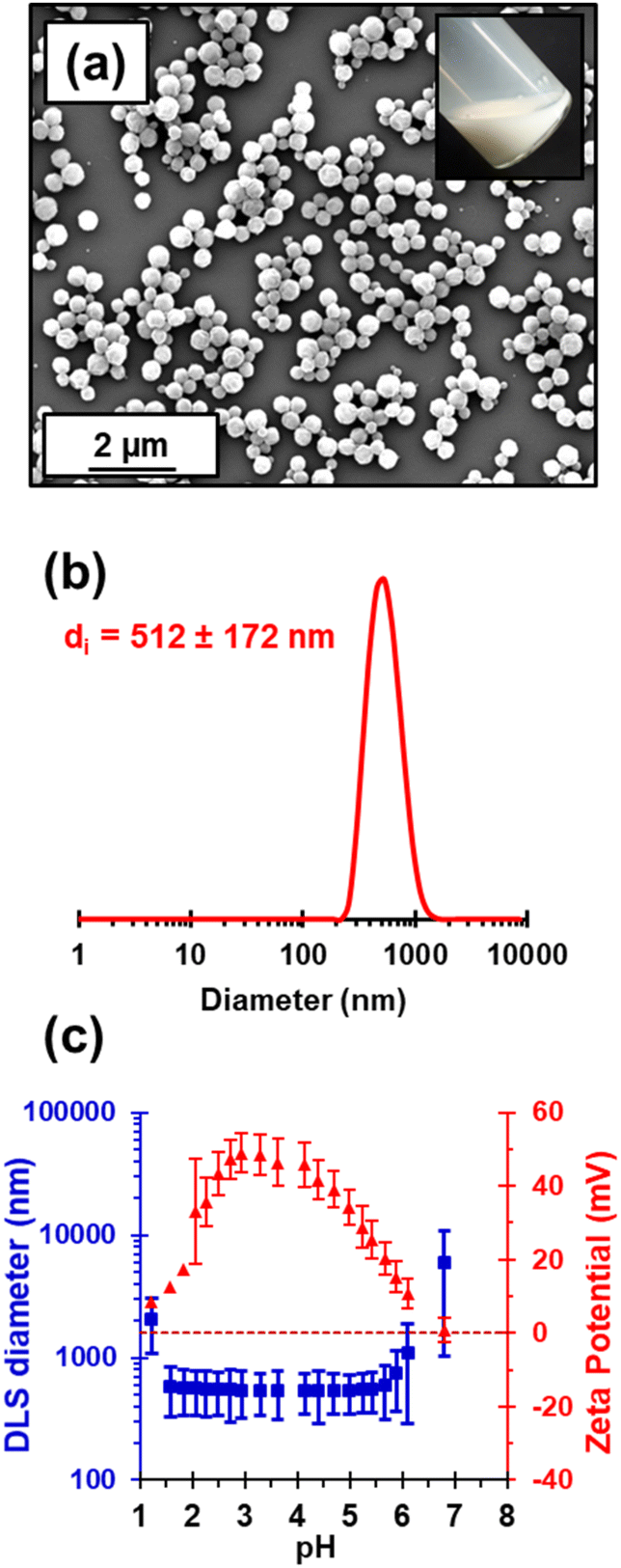 | ||
| Fig. 1 (a) SEM image recorded for a dried PHPMA140 latex, indicating its spherical morphology (digital photograph of a 10% w/w aqueous dispersion; see inset). (b) Intensity-average particle size distribution obtained by dynamic light scattering analysis of a 0.1% w/w PHPMA140 latex at pH 3. (c) Variation in DLS diameter and zeta potential with pH for this charge-stabilized PHPMA140 latex [a 0.1% w/w aqueous dispersion in 1 mM KCl was initially prepared at pH 3 and the solution pH was adjusted using KOH or HCl]. | ||
Importantly, the evolution in Mn is linear with monomer conversion and the relatively narrow molecular weight distributions (Mw/Mn < 1.15) confirm that this polymerization proceeds with good RAFT control.73–77 Scanning electron microscopy (SEM) images revealed a polydisperse spherical morphology with a number-average SEM diameter of 432 ± 78 nm (see Fig. 1a). This is consistent with dynamic light scattering (DLS) studies, which indicate a z-average diameter of 512 ± 172 nm (see Fig. 1b). Such latex particles are much larger than those usually obtained for conventional aqueous PISA syntheses of sterically-stabilized diblock copolymer nanoparticles, for which mean diameters of less than 100 nm are typically obtained when targeting spheres.56,60,78,79 In the present study, the weakly hydrophobic nature of the PHPMA chains results in their partial plasticization by water80–82 and hence a somewhat larger than expected latex diameter. Indeed, higher magnification SEM images suggest that some degree of dehydration of the PHPMA latex particle occurs under the ultrahigh vacuum conditions required for this technique (see Fig. S2†). It is well-known that ionic RAFT end-groups can have a dramatic effect on the electrophoretic footprint39,50,69,83,84 and colloidal stability85 of block copolymer nano-objects. Accordingly, the pH-responsive behavior of this PHPMA140 latex was examined using DLS and aqueous electrophoresis (see Fig. 1C). Zeta potentials of approximately +40 to +50 mV were observed between pH 2.5 and pH 4.5 owing to protonation of the morpholine-based RAFT groups located at the latex surface. Above pH 4.5, these tertiary amine groups gradually become deprotonated, leading to an isoelectric point at around pH 7. Below pH 2.5, the excess acid acts as a salt and screens the cationic surface charge, leading to lower zeta potentials. DLS studies indicate a colloid stability window from pH 1.8 to pH 5.6; the latex particles retain a sufficiently high zeta potential over this pH range to remain stable with respect to aggregation.
Next, the target PHPMA DP was systematically varied from 60 to 200 (see Table S1†). High HPMA conversions (>99%) were achieved in all cases and relatively narrow, unimodal molecular weight distributions (see Fig. 2a) were indicated by DMF GPC analysis. Furthermore, the linear relationship between Mn and the target PHPMA DP (combined with dispersities of less than 1.13) indicate that good RAFT control was achieved for such surfactant-free aqueous PISA syntheses (see Fig. 2b). Each of these eight latexes was diluted to 0.1% w/w (using 1 mM KCl adjusted to pH 3) to afford dispersions for DLS and aqueous electrophoresis studies (see Fig. 2C). Unexpectedly, the latex with the lowest target DP (PHPMA60) proved to be unstable with respect to dilution and became solubilized, as judged by its relatively low scattered light intensity. However, with the benefit of hindsight this is perhaps not surprising: it is well known that similarly short PHPMA blocks can become water-dispersible when conjugated to hydrophilic blocks.81 In contrast, each of the other seven PHPMA latexes proved to be stable to dilution. Zeta potentials of approximately +50 mV were determined for this latex series regardless of the target PHPMA DP (see Fig. 2C). Similarly, there was surprisingly little change in z-average diameter when varying the PHPMA DP from 80 to 160. These observations suggest that at least some (perhaps most) of the cationic morpholine RAFT end-groups remain buried within the interior of the latex particles, rather than being exclusively located at their surface. DLS studies also indicated that PHPMA latexes with DPs greater than 160 were relatively polydisperse and contained some macroscopic precipitate. This suggests that a DP of 160 may represent a realistic upper limit for such aqueous PISA syntheses, at least under the stated conditions. Nevertheless, in principle this should be sufficient to access sphere and worm morphologies via subsequent chain extension experiments using appropriate water-soluble methacrylic monomers.86,87 Many RAFT agents contain a carboxylic acid group.88,89 In part, this is because such functionality usually ensures that these compounds exist as solids at ambient temperature, which reduces their malodor. In the context of the present study, we wanted to examine whether carboxylic acid-based RAFT agents might offer the opportunity to conduct the surfactant-free RAFT aqueous dispersion polymerization of HPMA at pH 7, i.e. under conditions where ionization should confer negative surface charge via anionic carboxylate groups. Accordingly, we attempted the synthesis of PHPMA90 latexes using a carboxylic acid-functionalized trithiocarbonate-based RAFT agent known as PETTC (see Scheme S1† and Experimental section for further details).90,91 Indeed, these syntheses yielded free-flowing, milky-white dispersions. 1H NMR spectroscopy and DMF GPC analysis indicated high conversion (>99%) with the HPMA polymerization proceeding under RAFT control (Mn = 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400, Mw/Mn = 1.12). Furthermore, SEM and DLS studies indicated polydisperse, anionic spherical latexes with a zeta potential of −52 ± 7 mV (see Fig. S3†). Interestingly, such latexes were somewhat smaller (z-average diameter = 213 ± 29 nm) than the corresponding cationic latexes. We hypothesize that conducting these experiments at higher temperatures (70 °C vs. 56 °C) resulted in somewhat less hydrated PHPMA latexes and therefore smaller particles. In the remainder of this manuscript, we focus mainly on chain extension experiments conducted using cationic PHPMA latexes prepared using MPETTC. We have chosen to include the synthesis and characterization details for the anionic PHPMA latexes prepared using PETTC because such precursor particles exhibit complementary pH-sensitive behavior and hence may be of interest for specific applications.
400, Mw/Mn = 1.12). Furthermore, SEM and DLS studies indicated polydisperse, anionic spherical latexes with a zeta potential of −52 ± 7 mV (see Fig. S3†). Interestingly, such latexes were somewhat smaller (z-average diameter = 213 ± 29 nm) than the corresponding cationic latexes. We hypothesize that conducting these experiments at higher temperatures (70 °C vs. 56 °C) resulted in somewhat less hydrated PHPMA latexes and therefore smaller particles. In the remainder of this manuscript, we focus mainly on chain extension experiments conducted using cationic PHPMA latexes prepared using MPETTC. We have chosen to include the synthesis and characterization details for the anionic PHPMA latexes prepared using PETTC because such precursor particles exhibit complementary pH-sensitive behavior and hence may be of interest for specific applications.
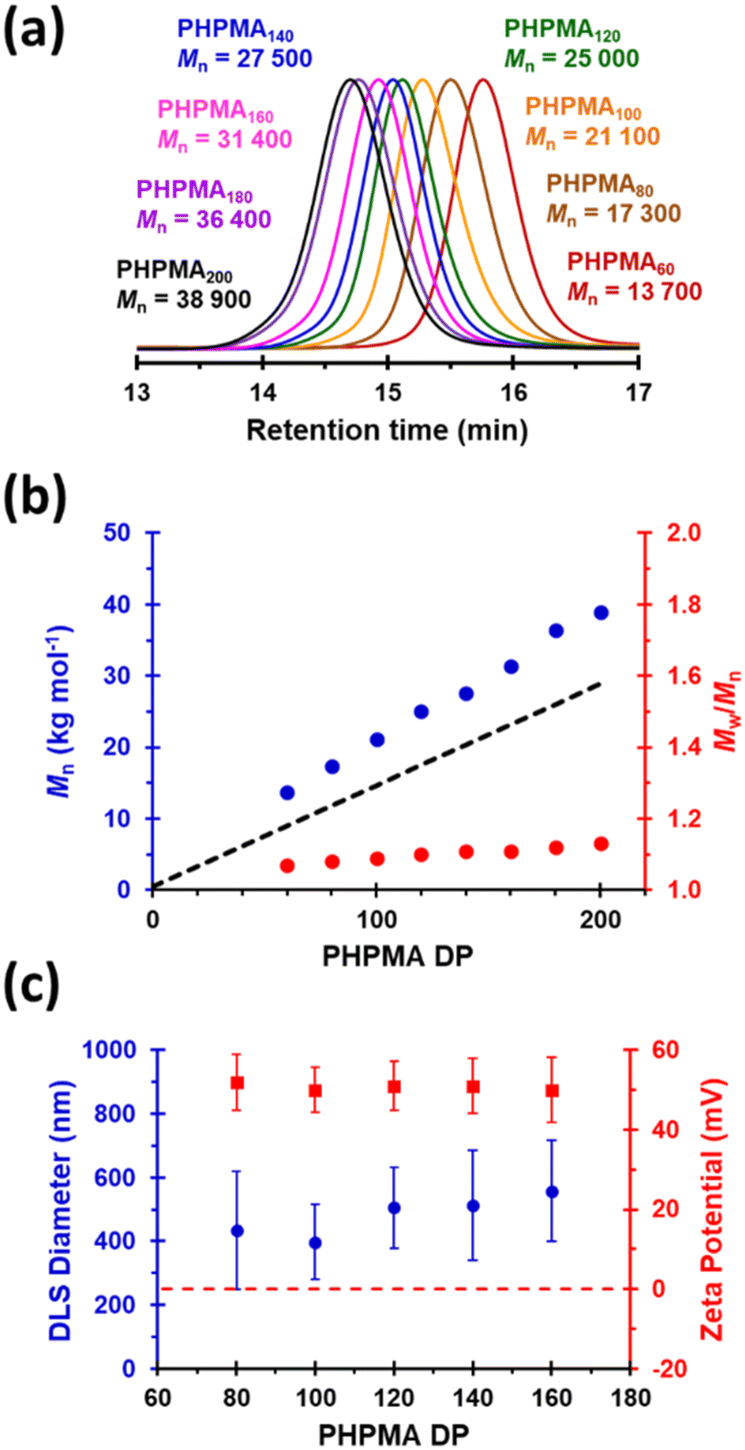 | ||
| Fig. 2 (a) Normalized DMF GPC curves (refractive index detector; calibrated against a series of poly(methyl methacrylate) standards) recorded for eight PHPMA latexes prepared using MPETTC RAFT agent where the PHPMA DP was systematically varied from 60 to 200. (b) Linear evolution in number-average molecular weight (Mn) and dispersity (Mw/Mn) with increasing target PHPMA DP for the same series of eight PHPMA latexes (the dashed line indicates the theoretical Mn). (c) Variation in z-average diameter and zeta potential with target PHPMA DP for the same series of eight PHPMA latexes. | ||
Synthesis of PHPMA-PGMA diblock copolymer nanoparticles
Next, chain extension of the cationic PHPMA140 precursor was attempted in aqueous media using glycerol monomethacrylate (GMA) targeting PHPMA140-PGMA56 diblock copolymer nano-objects. In an initial scoping experiment, GMA monomer was simply added to the as-synthesized aqueous PHPMA140 latex (prepared using MPETTC) and allowed to polymerize for 16 h at 56 °C without any additional AIBA initiator. Unfortunately, DMF GPC analysis (using refractive index and UV detectors in tandem) indicated that only uncontrolled free radical solution polymerization of the GMA had occurred, with the production of water-soluble PGMA homopolymer chains rather than the desired diblock copolymer nano-objects (see Fig. S4†). With the benefit of hindsight, this is because the aqueous phase contained unreacted initiator and there is no physical reason for the water-miscible GMA monomer to enter the PHPMA latex particles (which contain the buried RAFT chain-ends). In view of these negative results, the PHPMA latex synthesis was repeated in the absence of any azo initiator using a blue LED source (λmax = 395 nm, 0.37 mW cm−2).92–97 This photoinitiated RAFT protocol97–100 (see ESI; pages S15 and S16†) afforded colloidally stable PHPMA145 latex particles at 10% solids (>99% conversion) and DMF GPC analysis indicated good RAFT control (Mn = 26900; Mw/Mn = 1.10). However, subsequent photoinitiation of GMA in the presence of this PHPMA latex again led to the formation of a substantial fraction of polydisperse water-soluble PGMA chains that contained no RAFT chain-ends (see Fig. S5†). Presumably, this is because photoinitiation generates water-soluble radicals, which then polymerize the GMA monomer in the aqueous continuous phase via uncontrolled free radical polymerization, rather than within the latex particles.
To circumvent this problem, we added sufficient methanol (32% v/v) to the aqueous precursor latex to form soluble PHPMA chains, which enables their efficient chain extension to be achieved via RAFT solution polymerization of GMA in an aqueous methanol milieu in the absence of any additional AIBA initiator. In such syntheses, GMA conversions of 98–99% were routinely obtained. Moreover, DMF GPC analysis indicated efficient chain extension of the PHPMA140 precursor with a Mn of 46300 and Mw/Mn of 1.15 being obtained when targeting PHPMA160-PGMA90 (see Fig. S6† and Fig. 3a); synthetic details for related diblock copolymers are summarized in Table S2.† The methanolic co-solvent was then removed from the reaction mixture under reduced pressure and the resulting aqueous solution was freeze-dried to obtain a copolymer powder. Redispersion of this copolymer in ice-cold water at 10% w/w and pH 7, followed by warming to 20 °C led to in situ self-assembly of the amphiphilic PHPMA-PGMA diblock copolymer chains to afford either spherical (see Fig. 3b and c) or worm-like nanoparticles (see Fig. 3d), depending on the precise diblock copolymer composition.82 Furthermore, the resulting worm-like nanoparticles displayed thermoresponsive behavior, as evidenced by temperature-dependent rheology experiments (see Fig. 3e). At room temperature, the worm-like nanoparticles form a percolating 3D network to produce a free-standing gel.67 However, when cooled, these worm-like nanoparticles undergo a morphological transition to form non-interacting spherical nanoparticles, thus producing a free-flowing liquid. Similar thermoresponsive behavior for PGMA-PHPMA nano-objects prepared by conventional PISA has been widely reported.43,45,72,80,101 Although the above RAFT solution polymerization protocol was undoubtedly successful, we wished to examine whether wholly aqueous reverse sequence PISA formulations were feasible.
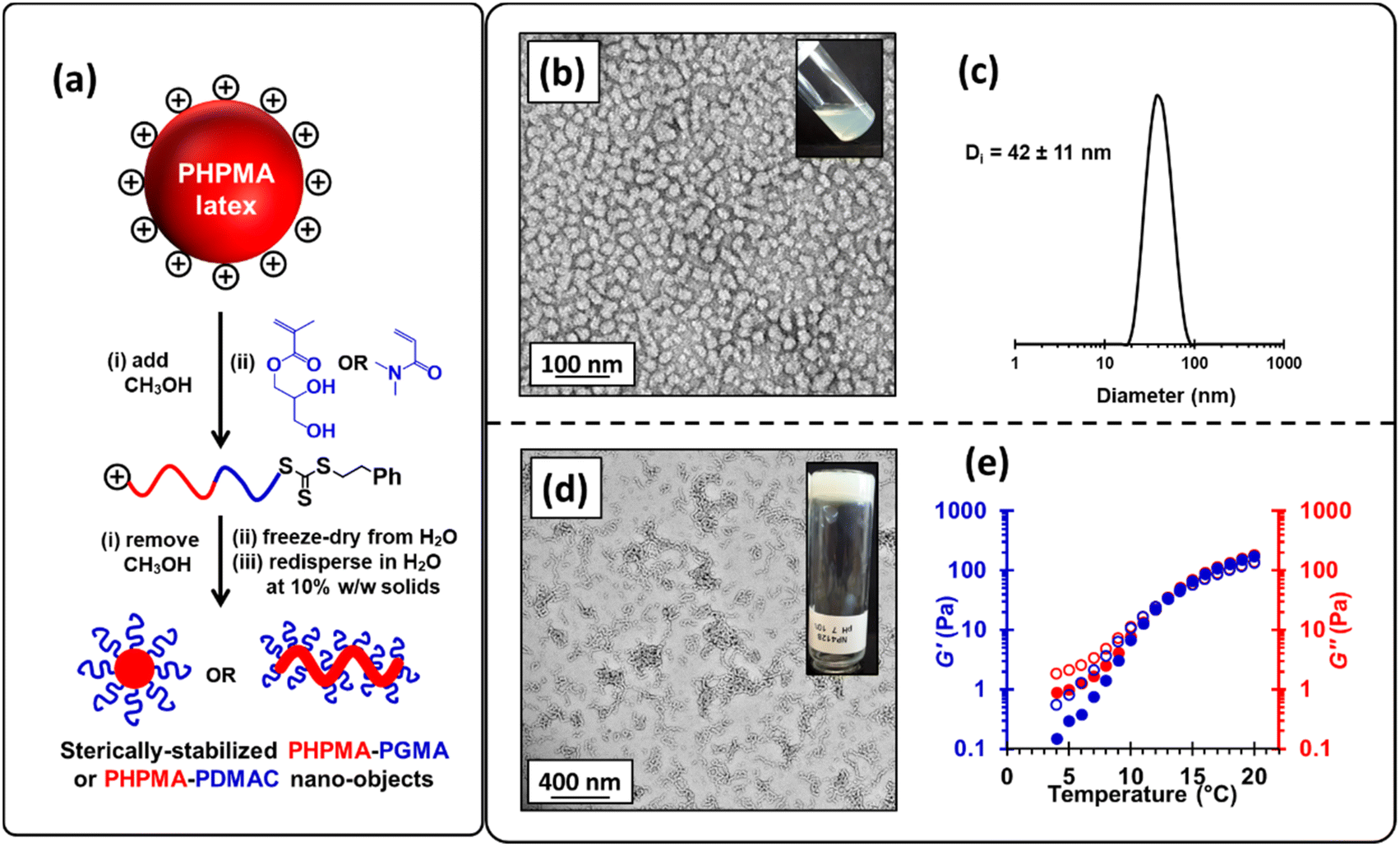 | ||
| Fig. 3 (a) Schematic representation of a reverse sequence PISA synthesis. A charge-stabilized cationic PHPMA latex precursor (prepared by surfactant-free RAFT aqueous dispersion polymerization of HPMA using MPETTC) is first dissolved by adding sufficient methanol co-solvent. The soluble PHPMA chains are then chain-extended via RAFT solution polymerization of either GMA or DMAC to produce PHPMA-PGMA or PHPMA-PDMAC diblock copolymers in a water/methanol mixture. Methanol co-solvent is then removed under vacuum prior to freeze-drying from water. Subsequent redispersion of the amphiphilic diblock copolymer chains in initially cold water at 10% w/w solids induces their self-assembly to yield sterically-stabilized nanoparticles. (b) TEM image recorded after drying a 0.1% w/w aqueous dispersion of PHPMA160-PGMA90 nanoparticles. These nanoparticles formed a free-flowing liquid at 10% w/w solids (see inset digital photograph). (c) DLS particle size distribution data obtained for a 0.1% w/w aqueous dispersion of PHPMA160-PGMA90 nanoparticles at pH 7 and 20 °C. (d) TEM image recorded after drying a dilute aqueous dispersion of PHPMA160-PGMA55 nanoparticles at 20 °C. The inset shows a digital photograph of the free-standing 10% w/w worm gel that is formed at 20 °C. (e) Variation in storage (G′, filled circles) and loss (G′′ open circles) moduli with temperature for a 10% w/w aqueous dispersion of PHPMA160-PGMA55 nanoparticles at pH 7. The heating sweep (red data) was performed prior to the cooling sweep (blue data). A thermal equilibration time of 5 min was allowed for each 1 °C increment and data were recorded at 1% applied strain and 1 rad s−1. | ||
Recently, we reported that poly(isopropylideneglycerol monomethacrylate) (PIPGMA) particles prepared via RAFT aqueous emulsion polymerization of IPGMA can be readily converted into water-soluble PGMA chains via acid hydrolysis.102 Accordingly, water-miscible GMA was replaced with water-immiscible IPGMA monomer (see Fig. 4a). Thus, a precursor cationic PHPMA140 latex (prepared at 10% w/w solids using MPETTC) was chain-extended via seeded RAFT aqueous emulsion polymerization of IPGMA using a one-pot protocol, targeting a final diblock composition of PHPMA140-PIPGMA55 at 14% w/w solids. Unlike GMA, the water-immiscible IPGMA monomer is preferentially located within the hydrophobic PHPMA latex cores, where it can be polymerized with good RAFT control to afford charge-stabilized PHPMA140-PIPGMA55 particles within 2 h. TEM and DLS confirmed the presence of large, spherical latex particles with a modest increase in z-average diameter compared to that of the precursor PHPMA140 latex (see Fig. S7†). Subsequent acid hydrolysis of the PIPGMA block using HCl (pH ∼ 1.2) at 70 °C afforded a 13.2% w/w aqueous dispersion. 1H NMR spectroscopy and TEM studies (see Fig. 4) indicate that the charge-stabilized PHPMA140-PIPGMA55 latex is transformed into sterically-stabilized PHPMA140-PGMA55 spherical nanoparticles. This is an example of reverse sequence PISA because the hydrophobic structure-directing block is synthesized first, rather than last.68
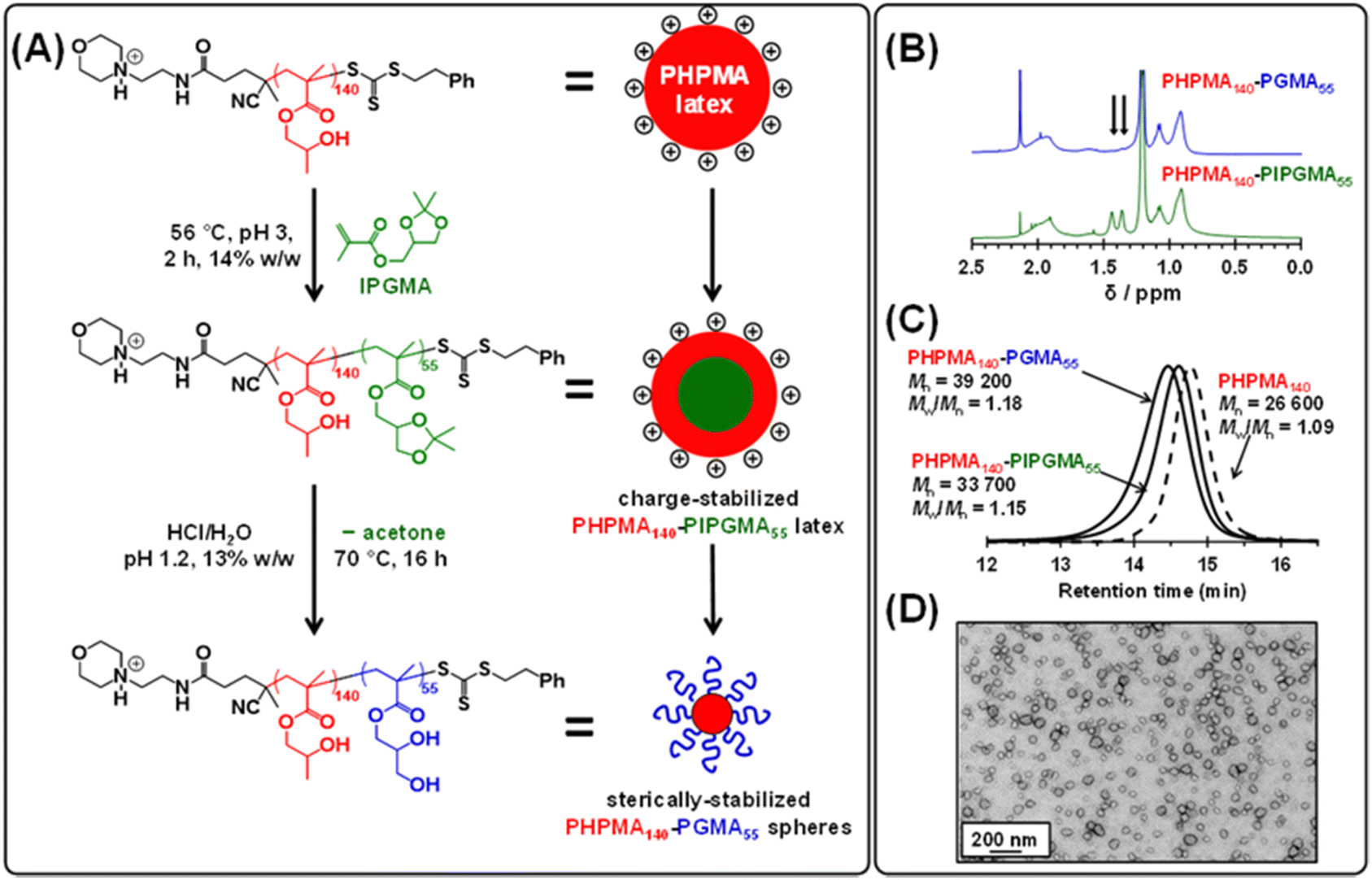 | ||
| Fig. 4 (A) Reaction scheme for the surfactant-free one-pot synthesis of sterically-stabilized PHPMA140-PGMA55 spherical nanoparticles. A charge-stabilized cationic PHPMA140 precursor latex is chain-extended via RAFT emulsion polymerization of IPGMA to afford a charge-stabilized PHPMA140-PIPGMA55 diblock copolymer latex. Subsequent acid hydrolysis at pH 1.2 affords sterically-stabilized PHPMA140-PGMA55 spheres. (B) 1H NMR spectra recorded for PHPMA140-PIPGMA55 and PHPMA140-PGMA55 diblock copolymers in d7-DMF. Hydrolysis of PHPMA140-PIPGMA55 to form PHPMA140-PGMA55 is confirmed by the disappearance of the six methyl protons (black arrows) at 1.36 ppm and 1.44 ppm associated with the acetone protecting group. (C) DMF GPC curves recorded for the PHPMA140, PHPMA140-PIPGMA55 and PHPMA140-PGMA55. Molecular weight data are expressed relative to a series of near-monodisperse poly(methyl methacrylate) calibration standards. (D) TEM image obtained after drying a 0.1% w/w dispersion of sterically-stabilized PHPMA140-PGMA55 nanoparticles at pH 1.2. | ||
Chain extension of a PHPMA latex with DMAC to afford PHPMA-PDMAC nano-objects
So far, we have demonstrated that PHPMA latexes can be chain-extended using a wholly aqueous formulation when employing a water-immiscible monomer (i.e. IPGMA). An alternative strategy is molecular dissolution of the PHPMA latex using a water-miscible co-solvent. In principle, this provides an opportunity to prepare new PHPMA-based diblock copolymers, where the water-soluble block is, for example, poly(N,N′-dimethylacrylamide) (PDMAC). Notably, well-defined PDMAC-PHPMA diblock copolymers cannot be prepared using a conventional PISA formulation. This is because the hydrophilic PDMAC precursor cannot be efficiently chain-extended using a methacrylic monomer such as HPMA since fragmentation of the radical intermediate preferentially forms the methacrylic tertiary radical rather than the acrylamide-based secondary radical, thus resulting in a mixture of PDMAC and PHPMA homopolymers.103–105 This is a well-known limitation of RAFT chemistry: methacrylates must be polymerized before acrylamides for efficient radical cross-over.106 Similar synthetic problems have been recently reported by Tan and co-workers when attempting to chain-extend a hydrophilic acrylic block with HPMA.107Accordingly, we added sufficient methanol co-solvent to dissolve an aqueous dispersion of cationic PHPMA140 latex particles to prepare a range of PHPMA-PDMAC diblock copolymers in the resulting water/methanol binary mixture (see Fig. S8† and Fig. 3a). Representative kinetic data for the RAFT solution polymerization of DMAC using a PHPMA140 precursor in a 68:32 v/v water/methanol mixture at 56 °C are shown in Fig. S8.† When targeting a PDMAC DP of 160, more than 90% DMAC conversion was achieved within 4 h with good RAFT control, as demonstrated by the linear evolution of Mn with monomer conversion and narrow dispersities. Informed by such kinetic experiments, a precursor PHPMA140 latex (Mn = 29300, Mw/Mn = 1.10) was molecularly dissolved via methanol addition and the resulting PHPMA140 chains were then chain-extended to prepare a PHPMA140-PDMAC160 diblock copolymer (Mn = 39800, Mw/Mn = 1.17). DMF GPC analysis indicated a relatively high blocking efficiency for the PHPMA chains. The methanol co-solvent was then removed under reduced pressure, followed by freeze-drying to afford a PHPMA140-PDMAC160 diblock copolymer powder. Subsequent redispersion of these amphiphilic copolymer chains in initially ice-cold water (to ensure molecular dissolution of the PHPMA chains)82 at 10% w/w solids resulted in the formation of spherical nanoparticles (z-average diameter = 41 ± 12 nm; see Fig. 5), with the hydrophilic PDMAC block conferring steric stabilization.82 Similarly, adjusting the diblock copolymer composition to target PHPMA160-PDMAC40 (Mn = 34500, Mw/Mn = 1.13) resulted in the formation of a free-flowing liquid at 5 °C. At its native solution pH of 4.7, this 10% w/w copolymer dispersion underwent a thermoreversible sol–gel transition on heating to form a soft, free-standing gel at 20 °C with a bulk modulus, G′, of 97 Pa (see Fig. S9†).80
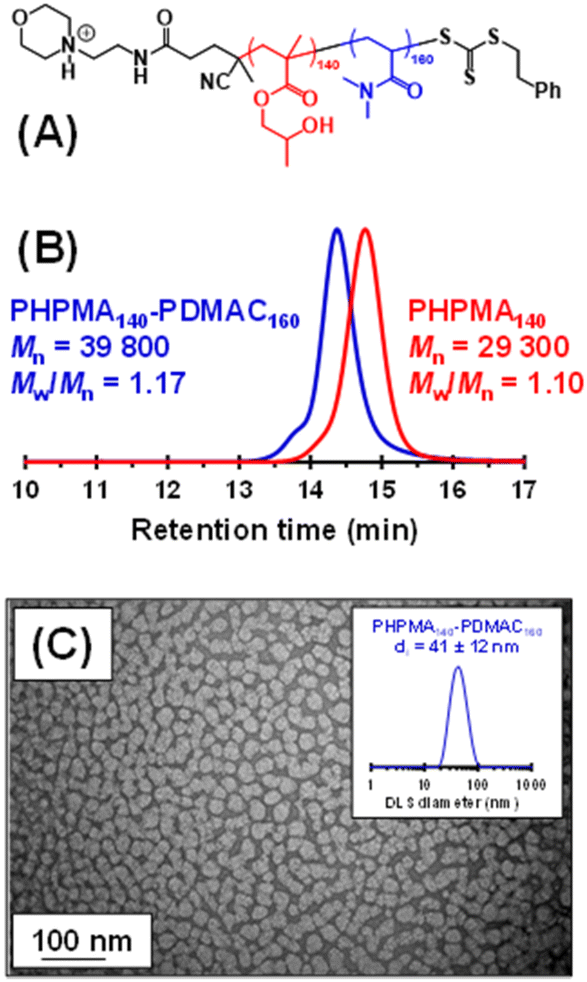 | ||
| Fig. 5 (A) Chemical structure of the PHPMA140-PDMAC160 diblock copolymer. (B) DMF GPC curves recorded for the PHPMA140 precursor and the final PHPMA140-PDMAC160 diblock copolymer. Molecular weight data are expressed relative to a series of near-monodisperse poly(methyl methacrylate) calibration standards. (C) TEM images recorded after drying a 0.1% w/w dispersion of PHPMA140-PDMAC160 nanoparticles, indicating a pseudo-spherical morphology. Intensity-average particle size distribution obtained by dynamic light scattering studies of a 0.1% w/w dispersion of cationic PHPMA140 latex particles at pH 3. | ||
Precursor latex based on an acrylamide-based monomer
We also wished to examine whether other water-miscible monomers could be used to form charge-stabilized precursor latexes. In principle, diacetone acrylamide (DAAM) should be a suitable monomer.108–113 In order to prepare such PDAAM latexes, an appropriate RAFT agent was selected and the solution pH was adjusted to ensure colloidal stabilization. Accordingly, a PDAAM99 latex was prepared at 10% w/w solids via RAFT aqueous dispersion polymerization of DAAM using a carboxylic acid-functionalized PETTC RAFT agent at pH 7 to confer anionic surface charge and hence charge stabilization. More than 99% DAAM conversion was achieved and GPC analysis indicated an Mn of 22600 and an Mw/Mn of 1.18 (see Scheme S2†). DLS studies of this precursor PDAAM latex gave a z-average diameter of 71 nm and a polydispersity index of 0.12.
Chain extension was achieved via RAFT solution polymerization of DMAC in a 61:39 w/w water/methanol mixture to afford a well-defined PDAAM99-PDMAC40 diblock copolymer (Mn = 25400, Mw/Mn = 1.21; see Scheme S2†). This proof-of-concept experiment confirms that this new reverse sequence PISA route is applicable to acrylamide-based monomers as well as methacrylic monomers, which significantly broadens the scope of this strategy.
Synthesis of latex precursors via RAFT aqueous emulsion polymerization
So far, we have focused on water-miscible monomers for the synthesis of the charge-stabilized precursor latex. However, RAFT PISA of water-immiscible monomers has been widely reported in the PISA literature.57,58,114–126 Interestingly, many RAFT aqueous emulsion polymerization formulations only yield kinetically-trapped spheres.78,114,124,127–129 Recently, we reported the synthesis of sterically-stabilized spheres, worms or vesicles via RAFT aqueous emulsion polymerization of MOEMA by systematic variation of the target DP of the PMOEMA block while employing a relatively short steric stabilizer block. This was rationalized in terms of the relatively high aqueous solubility of the MOEMA monomer (19.6 g dm−3 at 70 °C) compared to many other water-immiscible vinyl monomers. In view of this prior study, we selected MOEMA for the reverse sequence aqueous PISA formulations described herein.More specifically, MOEMA was polymerized using MPETTC at pH 3 to ensure protonation of this RAFT agent's morpholine group (Fig. 6a). Initial scoping experiments indicated that stable PMOEMA latexes could be consistently obtained between 5.0 and 7.5% w/w solids when conducting the RAFT aqueous emulsion polymerization of MOEMA at 56 °C. Further experiments confirmed that minimal latex coagulation was observed for syntheses conducted at up to 10% w/w solids but extensive coagulation was observed at either 15 or 20% w/w solids (see Fig. S10†). Kinetic data were obtained for the RAFT emulsion polymerization of MOEMA when targeting a PMOEMA DP of 60 and 7.5% w/w solids at 56 °C. In this experiment, aliquots were periodically extracted from the reaction mixture and analysed by 1H NMR spectroscopy and GPC (Fig. 7).
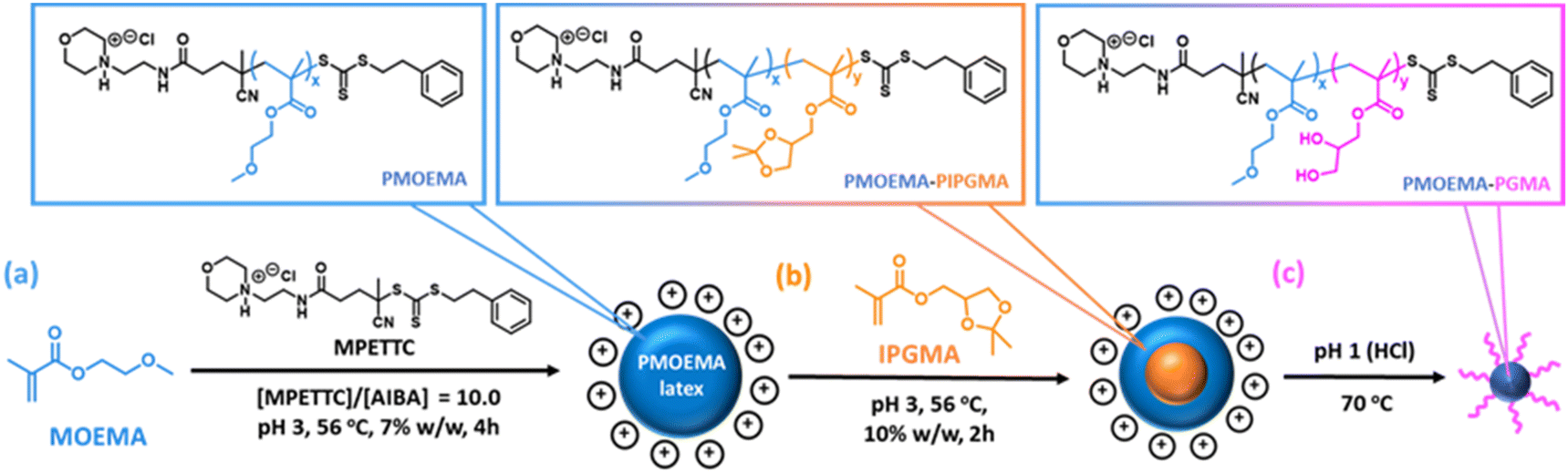 | ||
| Fig. 6 Schematic representation of a reverse sequence aqueous PISA formulation based on the RAFT aqueous emulsion polymerization of 2-methoxyethyl methacrylate (MOEMA). (a) Formation of the cationic PMOEMA latex precursor. (b) Chain extension of the PMOEMA chains with isopropylideneglycerol monomethacrylate (IPGMA) to form a PIPGMA-PMOEMA core–shell latex. (c) Deprotection of the hydrophobic PIPGMA block via acid hydrolysis to produce hydrophilic PGMA chains and hence amphiphilic PMOEMA-PGMA diblock copolymer nanoparticles. | ||
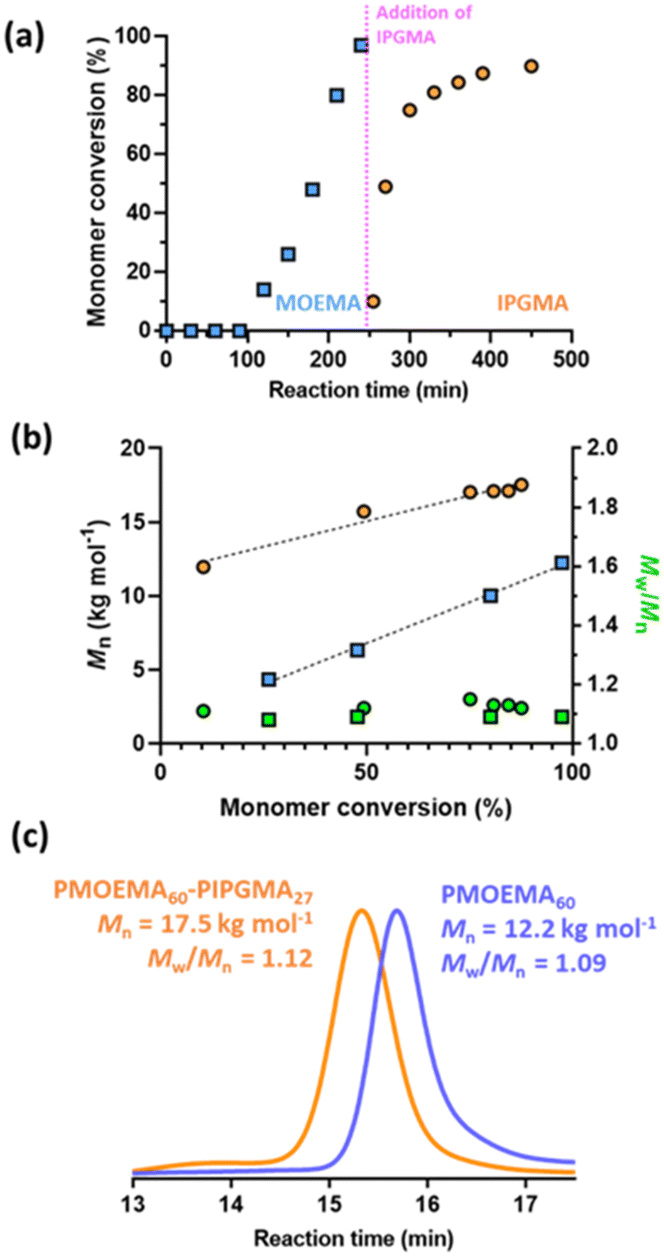 | ||
| Fig. 7 Kinetic data obtained for the RAFT aqueous emulsion polymerization of MOEMA using a cationic MPETTC RAFT agent at 56 °C and the subsequent extension of the PMOEMA chains with IPGMA: (a) monomer conversion vs. time curves; (b) evolution in number-average molecular weight (Mn) and dispersity (Mw/Mn) with monomer conversion. Polymerization conditions: [MOEMA]/[MPETTC] molar ratio = 60, [IPGMA]/[PMOEMA60] = 27, [MPETTC]/[AIBA] molar ratio = 5.0, targeting 7.5% w/w solids at pH 3. (c) DMF GPC curves recorded for PMOEMA60 and PMOEMA60-PIPGMA27. Molecular weight data are expressed relative to a series of near-monodisperse poly(methyl methacrylate) calibration standards. | ||
The MOEMA conversion remained relatively low for the first 100 min. This slow initial stage is most likely related to the relatively low concentration of this monomer within the aqueous phase, although induction periods for RAFT polymerizations appear to be a more general phenomenon.130 Nevertheless, micellar nucleation occurs at around 10% conversion, which corresponds to an instantaneous PMOEMA DP of just 6. Monomer then diffuses from the monomer droplet reservoirs into these nascent particles, leading to a relatively high local concentration and hence the observed rapid rate acceleration (Fig. 7a). A final MOEMA conversion of more than 99% is achieved within the following 140 min, giving a total reaction time of 4 h at 56 °C. The evolution in the molecular weight distribution for this first-stage polymerization was assessed by GPC. The number-average molecular weight (Mn) increased linearly with monomer conversion, indicating that this MOEMA polymerization proceeds with good RAFT control (Fig. 7b). Moreover, dispersities remained low (Mw/Mn < 1.20) throughout the polymerization.
A series of PMOEMAx latexes were prepared targeting various DPs (x = 20, 30, 40, 50, 60 or 70) to examine the effect of this parameter on the final particle size. 1H NMR studies indicated that more than 99% MOEMA conversion could be obtained in each case.
As expected, GPC analysis confirmed the expected progressive increase in PMOEMA Mn when targeting higher x values, with minimal increase in the final Mw/Mn (Fig. 8a). Each PMOEMA latex was then diluted to 0.1% w/w and analyzed by DLS to determine its z-average diameter. A monotonic increase in particle size was observed with increasing DP, with a z-average diameter of more than 200 nm being obtained when targeting PMOEMA70 (Fig. 8b).
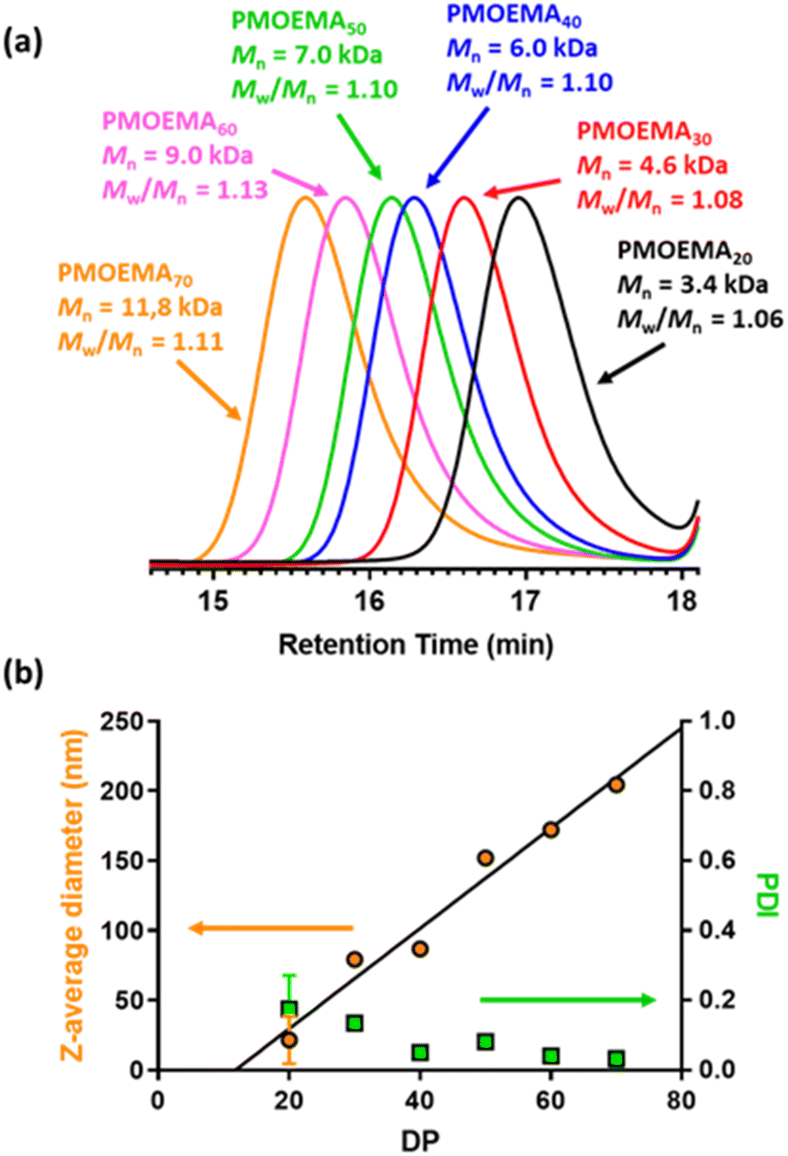 | ||
| Fig. 8 (a) Normalized DMF GPC curves (refractive index detector; calibrated using a series of poly(methyl methacrylate) standards) recorded for six cationic PMOEMA latexes where the PMOEMA DP was systematically varied from 20 to 70. (b) Variation in z-average diameter and DLS polydispersity (PDI) vs. PMOEMA Mn for the same series of six PMOEMA latexes. | ||
The initial PMOEMA seed latex was then chain-extended with IPGMA to produce a diblock copolymer latex. Empirically, we found that a one-pot synthesis protocol prevented multimodal molecular weight distributions. Thus, IPGMA is added to the reaction mixture just as the initial MOEMA polymerization approaches full conversion (with no further AIBA initiator being required). The water-immiscible IPGMA monomer diffuses through the aqueous phase and the second-stage polymerization takes place within the IPGMA-swollen PMOEMA latex particles. IPGMA has a significantly lower aqueous solubility than MOEMA,102 so PIPGMA is expected to be more hydrophobic than PMOEMA. Hence the PIPGMA block should form the latex core while the PMOEMA block forms the shell, as depicted schematically in Fig. 6.
Kinetic data were also obtained for the IPGMA polymerization. In this case, no induction period was observed and approximately 87% conversion can be achieved within 210 min at 56 °C (see Fig. 7a). GPC analysis confirmed that the Mn for the diblock copolymer chains increased linearly with IPGMA conversion and Mw/Mn values remained below 1.20. Moreover, a relatively high blocking efficiency was observed for the PMOEMA precursor chains. Thus, good RAFT control is also achieved for the second-stage IPGMA polymerization.
The pH-responsive behavior of the PMOEMA-PIPGMA latex particles was assessed by DLS and aqueous electrophoresis (Fig. S11†). Zeta potentials ranging from +10 to +15 mV were observed between pH 2 and pH 5 because the morpholine-based RAFT end-groups are protonated under such conditions and thus confer cationic surface charge. Above pH 5, these tertiary amine groups gradually became deprotonated, leading to an isoelectric point at around pH 7. DLS studies confirmed that the latex particles retained their colloidal stability between pH 2.0 and pH 5.8. However, the near-neutral latex particles began to flocculate above the latter pH, with macroscopic precipitation being observed at pH 6.5.
Deprotection of PMOEMA-PIPGMA latex to form PMOEMA-PGMA nanoparticles
As reported by Jesson et al., PIPGMA can be readily converted into PGMA via acid hydrolysis.102 Because PGMA is hydrophilic, acid hydrolysis of the hydrophobic PIPGMA block should lead to the in situ formation of an amphiphilic PMOEMA-PGMA diblock copolymer. Accordingly, acid hydrolysis of the PIPGMA block was performed at pH 1: 1H NMR spectroscopy studies confirmed that essentially all of the IPGMA repeat units were converted into GMA repeat units within 3 h at 70 °C (Fig. S12†). This drives a dramatic change in copolymer morphology: the double-hydrophobic PMOEMA-PIPGMA chains within the relatively large charge-stabilized latex particles rearrange to form much smaller sterically-stabilized diblock copolymer nanoparticles with PGMA now acting as the steric stabilizer. Indeed, the initial milky-white dispersion becomes a relatively transparent dispersion (see inset digital photographs in Fig. 9). Moreover, DLS studies indicated that the z-average diameter is significantly reduced from 145 nm for the PMOEMA40-PIPGMA27 precursor latex to just 23 nm for the final PMOEMA40-PGMA27 nanoparticles (Fig. 9a). TEM images recorded before and after hydrolysis followed by dialysis (Fig. 9b & c) confirmed that the precursor latex particles are converted into much finer spherical nanoparticles after hydrolysis. Furthermore, there is minimal difference between the GPC traces recorded for the initial PMOEMA40-PIPGMA27 PMOEMA40-PGMA27 and final PMOEMA40-PGMA27 diblock copolymer chains (Fig. S13†). The resulting aqueous dispersion of PMOEMA-PGMA nanoparticles was subsequently dialyzed against deionized water overnight to remove small molecule impurities (excess acid, traces of acetone, spent initiator etc.). After dialysis, the final pH of the aqueous dispersion was essentially that of deionized water (pH ∼ 6). In summary, a rather efficient reverse sequence PISA formulation has been developed based on RAFT aqueous emulsion polymerization.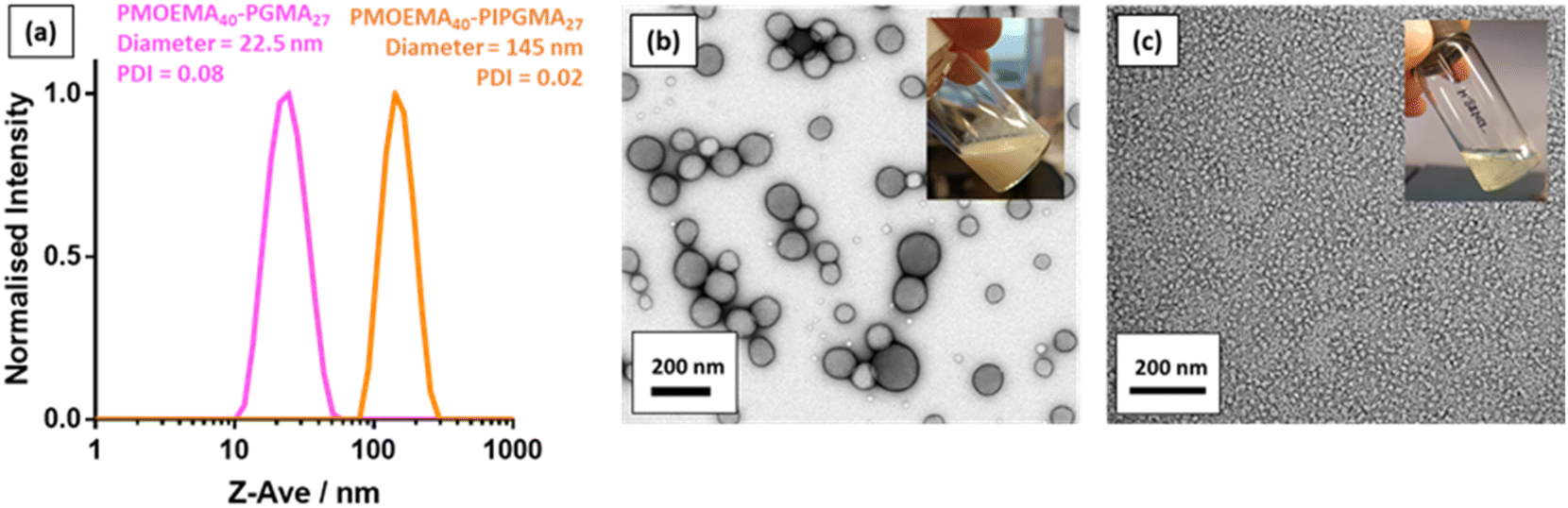 | ||
| Fig. 9 (a) Normalized DLS intensity-average particle size distributions recorded at 20 °C for 0.1% w/w aqueous dispersions of (i) charge-stabilized PMOEMA40-PIPGMA27 latex particles and (ii) sterically-stabilized PMOEMA40-PGMA27 nanoparticles. (b) TEM image recorded after drying the PMOEMA40-PIPGMA27 latex, which formed a free-flowing turbid dispersion at 8.2% w/w solids (see inset digital photograph). (c) TEM image recorded after drying an aqueous dispersion of PMOEMA40-PGMA27 nanoparticles (after dialysis against deionized water). These nanoparticles afford a relatively transparent free-flowing dispersion at 7.3% w/w solids (see inset digital photograph). | ||
DLS and aqueous electrophoresis were used to assess the pH-responsive behavior of the nanoparticles obtained after deprotection of the PIPGMA block (Fig. S11c†). At pH 2, the sterically-stabilized PMOEMA40-PGMA27 nanoparticles exhibited a zeta potential of +10 mV owing to the protonated morpholine-based RAFT end-groups. An isoelectric point was observed at pH 5 and the zeta potential became −10 mV at pH 8.
This weakly anionic character is attributed to the presence of a small number of methacrylic acid (MAA) repeat units within the PGMA stabilizer block owing to partial hydrolysis of methacrylic ester bonds. Thus, acetone deprotection of the IPGMA residues via acid hydrolysis is not perfectly selective. Further investigations indicated that approximately one MAA unit was produced per diblock copolymer chain. It was hypothesized that performing the hydrolysis at pH 2 might prevent ester hydrolysis. Unfortunately, these milder conditions required much longer reaction times to achieve complete deprotection but did not prevent this unwanted side-reaction. Unlike the precursor PMOEMA40-PIPGMA27 latex particles, the PMOEMA40-PGMA27 nanoparticles remained colloidally stable across the entire pH range studied owing to the steric stabilization conferred by the hydrophilic PGMA block.
Effect of added salt on the copolymer morphology
Varying amounts of NaCl were added to a 7.3% w/w aqueous dispersion of PMOEMA40-PGMA27 spherical nanoparticles to produce salt concentrations of 50, 100 or 150 mM, respectively. In the presence of 50 mM NaCl, the dispersion became significantly more turbid and viscous but remained a free-flowing fluid. In contrast, a free-standing gel was obtained in the presence of either 100 or 150 mM NaCl (Fig. 10). According to the PISA literature, this suggests a salt-induced sphere-to-worm morphology transition.131 | ||
| Fig. 10 Digital photograph (left) illustrating the effect of addition of 50–150 mM NaCl to an aqueous dispersion of PMOEMA40-PGMA27 nanoparticles at pH 6 to produce a soft free-standing gel at 20 °C. The corresponding TEM image (right) confirms the anisotropic worm-like particles that are formed in the presence of 150 mM NaCl. The inset image shows a shear-induced polarized light image of the corresponding 7.3% w/w worm dispersion recorded at a maximum shear rate of 1.0 s−1. The Maltese cross motif indicates birefringence arising from the in situ worm alignment that occurs under such conditions.132 | ||
TEM studies confirmed that a sphere-to-worm transition occurred in the presence of sufficient NaCl (Fig. 10). The added salt screens the anionic charge arising from the MAA repeat unit(s) within the otherwise neutral PGMA chains, which enables the amphiphilic PMOEMA40-PGMA27 chains to self-assemble to form worms. In contrast, mutual repulsion between the weakly anionic spheres formed in the absence of any added salt prevents their stochastic 1D fusion to form worms.39 It is well-known that highly anisotropic worms can form a 3D percolating network via multiple inter-worm contacts, which enables the formation of a free-standing gel at relatively low copolymer concentration.67 Moreover, shear-induced polarized light imaging (SIPLI) studies133 of the free-standing gel recorded at 20 °C under constant shear produced a distinctive Maltese cross motif (see inset within TEM image shown in Fig. 10), which is characteristic of the shear-induced alignment of anisotropic worms.45
Conclusions
We report new surfactant-free aqueous PISA formulations that enable the structure-directing hydrophobic block to be synthesized first when targeting amphiphilic diblock copolymers. This counter-intuitive reverse sequence approach requires an ionic RAFT agent to confer charge stabilization on the precursor latex, which can be prepared via RAFT aqueous dispersion polymerization of 2-hydroxypropyl methacrylate (HPMA). Unfortunately, attempted chain extension of this PHPMA latex precursor using water-miscible monomers only led to their uncontrolled free radical polymerization in the aqueous phase. However, chain extension utilizing a water-immiscible monomer, isopropylideneglycerol monomethacrylate (IPGMA), afforded a charge-stabilized PHPMA-PIPGMA diblock copolymer latex; this is because the water-immiscible IPGMA monomer preferentially partitions into the latex core. Subsequent acid hydrolysis afforded the desired sterically-stabilized PHPMA-PGMA nano-objects. Alternatively, addition of methanol to solubilize the precursor PHPMA latex enables efficient chain extension via RAFT solution polymerization in water/methanol binary mixtures. This protocol enables the formation of both PHPMA-PGMA diblock copolymer nanoparticles and also new copolymer examples that cannot be accessed via conventional aqueous PISA. Moreover, this counter-intuitive reverse sequence PISA approach is also applicable to acrylamide-based monomers. Finally, charge-stabilized poly(2-methoxyethyl methacrylate) (PMOEMA) latexes were prepared via RAFT aqueous emulsion polymerization. Subsequent chain extension using IPGMA leads to a charge-stabilized PMOEMA-PIPGMA diblock copolymer latex of approximately 150 nm diameter. Finally, acid-catalyzed removal of the acetone protecting groups from the PIPGMA block affords amphiphilic PMOEMA-PGMA diblock copolymer chains that undergo in situ self-assembly to form sterically-stabilized diblock copolymer nanoparticles. Given the excellent control over molecular weight distribution, relatively high atom economy and minimal levels of residual monomer, we envisage that such new reverse sequence PISA routes are likely to become a useful addition to the synthetic polymer chemist's toolbox for the rational synthesis of functional block copolymer nanoparticles.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. P. A. acknowledges a four-year EPSRC Established Career Particle Technology Fellowship (EP/R003009) which provided post-doctoral support for T. J. N. GEO Specialty Chemicals (Hythe, UK) is thanked for supplying the HPMA, IPGMA and GMA monomers used in this study.References
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 CrossRef CAS PubMed.
- N. J. W. Penfold, J. Yeow, C. Boyer and S. P. Armes, ACS Macro Lett., 2019, 8, 1029–1054 CrossRef CAS PubMed.
- H. Phan, V. Taresco, J. Penelle and B. Couturaud, Biomater. Sci., 2021, 9, 38–50 RSC.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef CAS PubMed.
- F. D'Agosto, J. Rieger and M. Lansalot, Angew. Chem., Int. Ed., 2020, 59, 8368–8392 CrossRef.
- B. Charleux, G. Delaittre, J. Rieger and F. D'Agosto, Macromolecules, 2012, 45, 6753–6765 CrossRef CAS.
- A. B. Lowe, Polymer, 2016, 106, 161–181 CrossRef CAS.
- M. J. Derry, L. A. Fielding and S. P. Armes, Prog. Polym. Sci., 2016, 52, 1–18 CrossRef CAS.
- C. Liu, C. Y. Hong and C. Y. Pan, Polym. Chem., 2020, 11, 3673–3689 RSC.
- J. Yeow and C. Boyer, Adv. Sci., 2017, 4, 1700137 CrossRef.
- W. J. Zhang, C. Y. Hong and C. Y. Pan, Macromol. Rapid Commun., 2019, 40, e1800279 CrossRef PubMed.
- M. J. Derry, L. A. Fielding, N. J. Warren, C. J. Mable, A. J. Smith, O. O. Mykhaylyk and S. P. Armes, Chem. Sci., 2016, 7, 5078–5090 RSC.
- R. Takahashi, S. Miwa, F. H. Sobotta, J. H. Lee, S. Fujii, N. Ohta, J. C. Brendel and K. Sakurai, Polym. Chem., 2020, 11, 1514–1524 RSC.
- A. Czajka and S. P. Armes, Chem. Sci., 2020, 11, 11443–11454 RSC.
- E. E. Brotherton, F. L. Hatton, A. A. Cockram, M. J. Derry, A. Czajka, E. J. Cornel, P. D. Topham, O. O. Mykhaylyk and S. P. Armes, J. Am. Chem. Soc., 2019, 141, 13664–13675 CrossRef CAS PubMed.
- J. Chiefari, J. Jeffery, R. T. A. Mayadunne, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1999, 32, 7700–7702 CrossRef CAS.
- R. T. A. Mayadunne, E. Rizzardo, J. Chiefari, Y. K. Chong, G. Moad and S. H. Thang, Macromolecules, 1999, 32, 6977–6980 CrossRef CAS.
- G. Moad, Y. K. Chong, A. Postma, E. Rizzardo and S. H. Thang, Polymer, 2005, 46, 8458–8468 CrossRef CAS.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- D. J. Keddie, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 2012, 45, 5321–5342 CrossRef CAS.
- B. R. Parker, M. J. Derry, Y. Ning and S. P. Armes, Langmuir, 2020, 36, 3730–3736 CrossRef CAS PubMed.
- D. Zehm, L. P. D. Ratcliffe and S. P. Armes, Macromolecules, 2013, 46, 128–139 CrossRef CAS.
- M. J. Derry, T. Smith, P. S. O'Hora and S. P. Armes, ACS Appl. Mater. Interfaces, 2019, 11, 33364–33369 CrossRef CAS PubMed.
- C. György, S. J. Hunter, C. Girou, M. J. Derry and S. P. Armes, Polym. Chem., 2020, 11, 4579–4590 RSC.
- E. R. Jones, M. Semsarilar, P. Wyman, M. Boerakker and S. P. Armes, Polym. Chem., 2016, 7, 851–859 RSC.
- M. Semsarilar, N. J. W. Penfold, E. R. Jones and S. P. Armes, Polym. Chem., 2015, 6, 1751–1757 RSC.
- M. Semsarilar, V. Ladmiral, A. Blanazs and S. P. Armes, Polym. Chem., 2014, 5, 3466–3475 RSC.
- L. A. Fielding, M. J. Derry, V. Ladmiral, J. Rosselgong, A. M. Rodrigues, L. P. D. Ratcliffe, S. Sugihara and S. P. Armes, Chem. Sci., 2013, 4, 2081–2087 RSC.
- L. P. D. Ratcliffe, M. J. Derry, A. Ianiro, R. Tuinier and S. P. Armes, Angew. Chem., 2019, 131, 19140–19146 CrossRef.
- M. J. Rymaruk, S. J. Hunter, C. T. O’Brien, S. L. Brown, C. N. Williams and S. P. Armes, Macromolecules, 2019, 52, 2822–2832 CrossRef CAS.
- Q. Zhang and S. Zhu, ACS Macro Lett., 2015, 4, 755–758 CrossRef CAS PubMed.
- V. J. Cunningham, M. J. Derry, L. A. Fielding, O. M. Musa and S. P. Armes, Macromolecules, 2016, 49, 4520–4533 CrossRef CAS PubMed.
- Y. Ning, L. Han, M. J. Derry, F. C. Meldrum and S. P. Armes, J. Am. Chem. Soc., 2019, 141, 2557–2567 CrossRef CAS PubMed.
- C. J. Mable, N. J. Warren, K. L. Thompson, O. O. Mykhaylyk and S. P. Armes, Chem. Sci., 2015, 6, 6179–6188 RSC.
- K. L. Thompson, C. J. Mable, A. Cockram, N. J. Warren, V. J. Cunningham, E. R. Jones, R. Verber and S. P. Armes, Soft Matter, 2014, 10, 8615–8626 RSC.
- C. A. Figg, A. Simula, K. A. Gebre, B. S. Tucker, D. M. Haddleton and B. S. Sumerlin, Chem. Sci., 2015, 6, 1230–1236 RSC.
- G. Mellot, J. M. Guigner, L. Bouteiller, F. Stoffelbach and J. Rieger, Angew. Chem., Int. Ed., 2019, 58, 3173–3177 CrossRef CAS.
- D. E. Mitchell, J. R. Lovett, S. P. Armes and M. I. Gibson, Angew. Chem., Int. Ed., 2016, 55, 2801–2804 CrossRef CAS PubMed.
- J. R. Lovett, N. J. Warren, L. P. D. Ratcliffe, M. K. Kocik and S. P. Armes, Angew. Chem., Int. Ed., 2015, 54, 1279–1283 CrossRef CAS.
- X. Wang, S. Man, J. Zheng and Z. An, ACS Macro Lett., 2018, 7, 1461–1467 CrossRef CAS.
- L. D. Blackman, S. Varlas, M. C. Arno, A. Fayter, M. I. Gibson and R. K. O'Reilly, ACS Macro Lett., 2017, 6, 1263–1267 CrossRef CAS.
- L. D. Blackman, S. Varlas, M. C. Arno, Z. H. Houston, N. L. Fletcher, K. J. Thurecht, M. Hasan, M. I. Gibson and R. K. O'Reilly, ACS Cent. Sci., 2018, 4, 718–723 CrossRef CAS PubMed.
- I. Canton, N. J. Warren, A. Chahal, K. Amps, A. Wood, R. Weightman, E. Wang, H. Moore and S. P. Armes, ACS Cent. Sci., 2016, 2, 65–74 CrossRef CAS.
- R. Deng, M. J. Derry, C. J. Mable, Y. Ning and S. P. Armes, J. Am. Chem. Soc., 2017, 139, 7616–7623 CrossRef CAS PubMed.
- N. J. Warren, M. J. Derry, O. O. Mykhaylyk, J. R. Lovett, L. P. D. Ratcliffe, V. Ladmiral, A. Blanazs, L. A. Fielding and S. P. Armes, Macromolecules, 2018, 51, 8357–8371 CrossRef CAS PubMed.
- X. Wang, J. Zhou, X. Lv, B. Zhang and Z. An, Macromolecules, 2017, 50, 7222–7232 CrossRef CAS.
- C. J. Mable, M. J. Derry, K. L. Thompson, L. A. Fielding, O. O. Mykhaylyk and S. P. Armes, Macromolecules, 2017, 50, 4465–4473 CrossRef CAS PubMed.
- C. A. Figg, R. N. Carmean, K. C. Bentz, S. Mukherjee, D. A. Savin and B. S. Sumerlin, Macromolecules, 2017, 50, 935–943 CrossRef CAS.
- S. J. Byard, M. Williams, B. E. McKenzie, A. Blanazs and S. P. Armes, Macromolecules, 2017, 50, 1482–1493 CrossRef CAS PubMed.
- J. R. Lovett, N. J. Warren, S. P. Armes, M. J. Smallridge and R. B. Cracknell, Macromolecules, 2016, 49, 1016–1025 CrossRef CAS PubMed.
- N. J. Warren, J. Rosselgong, J. Madsen and S. P. Armes, Biomacromolecules, 2015, 16, 2514–2521 CrossRef CAS.
- S. Sugihara, A. H. Ma'Radzi, S. Ida, S. Irie, T. Kikukawa and Y. Maeda, Polymer, 2015, 76, 17–24 CrossRef CAS.
- P. Galanopoulo, P. Y. Dugas, M. Lansalot and F. D'Agosto, Polym. Chem., 2020, 11, 3922–3930 CAS.
- M. Chenal, J. Rieger, C. Vechambre, J.-M. Chenal, L. Chazeau, C. Creton and L. Bouteiller, Macromol. Rapid Commun., 2013, 1524–1529 CrossRef CAS.
- C. J. Mable, R. R. Gibson, S. Prevost, B. E. McKenzie, O. O. Mykhaylyk and S. P. Armes, J. Am. Chem. Soc., 2015, 137, 16098–16108 CAS.
- W. Zhang, F. D'Agosto, P. Y. Dugas, J. Rieger and B. Charleux, Polymer, 2013, 54, 2011–2019 CrossRef CAS.
- A. A. Cockram, T. J. Neal, M. J. Derry, O. O. Mykhaylyk, N. S. J. Williams, M. W. Murray, S. N. Emmett and S. P. Armes, Macromolecules, 2017, 50, 796–802 CrossRef CAS PubMed.
- F. L. Hatton, J. R. Lovett and S. P. Armes, Polym. Chem., 2017, 8, 4856–4868 RSC.
- T. R. Guimarães, Y. L. Bong, S. W. Thompson, G. Moad, S. Perrier and P. B. Zetterlund, Polym. Chem., 2021, 12, 122–133 RSC.
- C. György, J. R. Lovett, N. J. W. Penfold and S. P. Armes, Macromol. Rapid Commun., 2019, 40, 1–7 CrossRef.
- S. Y. Khor, N. P. Truong, J. F. Quinn, M. R. Whittaker and T. P. Davis, ACS Macro Lett., 2017, 6, 1013–1019 CrossRef CAS PubMed.
- N. J. Warren, O. O. Mykhaylyk, A. J. Ryan, M. Williams, T. Doussineau, P. Dugourd, R. Antoine, G. Portale and S. P. Armes, J. Am. Chem. Soc., 2015, 137, 1929–1937 CrossRef CAS PubMed.
- D. L. Beattie, O. O. Mykhaylyk and S. P. Armes, Chem. Sci., 2020, 11, 10821–10834 RSC.
- M. Sponchioni, C. T. O'Brien, C. Borchers, E. Wang, M. N. Rivolta, N. J. W. Penfold, I. Canton and S. P. Armes, Chem. Sci., 2020, 11, 232–240 RSC.
- S. J. Byard, C. T. O'Brien, M. J. Derry, M. Williams, O. O. Mykhaylyk, A. Blanazs and S. P. Armes, Chem. Sci., 2020, 11, 396–402 RSC.
- C. J. Mable, I. Canton, O. O. Mykhaylyk, B. Ustbas Gul, P. Chambon, E. Themistou and S. P. Armes, Chem. Sci., 2019, 10, 4811–4821 RSC.
- J. R. Lovett, M. J. Derry, P. Yang, F. L. Hatton, N. J. Warren, P. W. Fowler and S. P. Armes, Chem. Sci., 2018, 9, 7138–7144 RSC.
- T. J. Neal, N. J. W. Penfold and S. P. Armes, Angew. Chem., Int. Ed., 2022, 61, e202207376 CrossRef CAS PubMed.
- N. J. W. Penfold, J. R. Lovett, P. Verstraete, J. Smets and S. P. Armes, Polym. Chem., 2017, 8, 272–282 RSC.
- S. Sugihara, A. Blanazs, S. P. Armes, A. J. Ryan and A. L. Lewis, J. Am. Chem. Soc., 2011, 133, 15707–15713 CrossRef CAS PubMed.
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2011, 133, 16581–16587 CrossRef CAS PubMed.
- N. J. Warren, O. O. Mykhaylyk, D. Mahmood, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 1023–1033 CrossRef CAS PubMed.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2009, 62, 1402–1472 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2012, 65, 985–1076 CrossRef CAS.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS.
- V. J. Cunningham, A. M. Alswieleh, K. L. Thompson, M. Williams, G. J. Leggett, S. P. Armes and O. M. Musa, Macromolecules, 2014, 47, 5613–5623 CrossRef CAS.
- S. Boisse, J. Rieger, G. Pembouong, P. Beaunier and B. Charleux, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3346–3354 CrossRef CAS.
- A. Blanazs, R. Verber, O. O. Mykhaylyk, A. J. Ryan, J. Z. Heath, C. W. Douglas and S. P. Armes, J. Am. Chem. Soc., 2012, 134, 9741–9748 CrossRef CAS PubMed.
- J. Madsen, S. P. Armes, K. Bertal, S. MacNeil and A. L. Lewis, Biomacromolecules, 2009, 10, 1875–1887 CrossRef CAS PubMed.
- M. K. Kocik, O. O. Mykhaylyk and S. P. Armes, Soft Matter, 2014, 10, 3984–3992 RSC.
- N. J. W. Penfold, J. R. Lovett, N. J. Warren, P. Verstraete, J. Smets and S. P. Armes, Polym. Chem., 2016, 7, 79–88 RSC.
- J. Z. Du, H. Willcock, J. P. Patterson, I. Portman and R. K. O'Reilly, Small, 2011, 7, 2070–2080 CrossRef CAS PubMed.
- R. R. Gibson, S. P. Armes, O. M. Musa and A. Fernyhough, Polym. Chem., 2019, 10, 1312–1323 RSC.
- D. L. Beattie, O. O. Mykhaylyk, A. J. Ryan and S. P. Armes, Macromolecules, 2021, 17, 5602–5612 CAS.
- D. L. Beattie, O. O. Mykhaylyk, A. J. Ryan and S. P. Armes, Macromolecules, 2012, 45, 5099–5107 CrossRef.
- A. Lubnin, K. O'Malley, D. Hanshumaker and J. Lai, Eur. Polym. J., 2010, 46, 1563–1575 CrossRef CAS.
- J. T. Lai, D. Filla and R. Shea, Macromolecules, 2002, 35, 6754–6756 CrossRef CAS.
- M. Semsarilar, V. Ladmiral, A. Blanazs and S. P. Armes, Langmuir, 2012, 28, 914–922 CrossRef CAS PubMed.
- M. Semsarilar, E. R. Jones, A. Blanazs and S. P. Armes, Adv. Mater., 2012, 24, 3378–3382 CrossRef CAS PubMed.
- J. Yeow and C. Boyer, Adv. Sci., 2017, 4, 1700137 CrossRef PubMed.
- J. Yeow, O. R. Sugita and C. Boyer, ACS Macro Lett., 2016, 5, 558–564 CrossRef CAS PubMed.
- Y. J. Ma, P. Gao, Y. Ding, L. L. Huang, L. Wang, X. H. Lu and Y. L. Cai, Macromolecules, 2019, 52, 1033–1041 CrossRef.
- P. Gao, H. Cao, Y. Ding, M. Cai, Z. Cui, X. Lu and Y. Cai, ACS Macro Lett., 2016, 5, 1327–1331 CrossRef CAS PubMed.
- K. Ferji, P. Venturini, F. Cleymand, C. Chassenieux and J.-L. Six, Polym. Chem., 2018, 9, 2868–2872 RSC.
- M. Chen, M. Zhong and J. A. Johnson, Chem. Rev., 2016, 116, 10167–10211 CrossRef CAS PubMed.
- H. Wang, Q. Li, J. Dai, F. Du, H. Zheng and R. Bai, Macromolecules, 2013, 46, 2576–2582 CAS.
- K. Jung, C. Boyer and P. B. Zetterlund, Polym. Chem., 2017, 8, 3965–3970 CAS.
- W. Smulders, R. G. Gilbert and M. J. Monteiro, Macromolecules, 2003, 36, 4309–4318 CrossRef CAS.
- K. A. Simon, N. J. Warren, B. Mosadegh, M. R. Mohammady, G. M. Whitesides and S. P. Armes, Biomacromolecules, 2015, 16, 3952–3958 CrossRef CAS PubMed.
- C. P. Jesson, V. J. Cunningham, M. J. Smallridge and S. P. Armes, Macromolecules, 2018, 51, 3221–3232 CrossRef CAS PubMed.
- D. J. Keddie, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 2012, 45, 5321–5342 CrossRef CAS.
- J. Chiefari, R. T. A. Mayadunne, C. L. Moad, G. Moad, E. Rizzardo, A. Postma and S. H. Thang, Macromolecules, 2003, 36, 2273–2283 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2006, 59, 669–692 CrossRef CAS.
- C. P. Easterling, Y. Xia, J. Zhao, G. E. Fanucci and B. S. Sumerlin, ACS Macro Lett., 2019, 8, 1461–1466 CrossRef CAS PubMed.
- Y. X. Zhang, L. L. Yu, X. C. Dai, L. Zhang and J. B. Tan, ACS Macro Lett., 2019, 8, 1102–1109 CrossRef CAS PubMed.
- J. L. de la Haye, I. Martin-Fabiani, M. Schulz, J. L. Keddie, F. D'Agosto and M. Lansalot, Macromolecules, 2017, 50, 9315–9328 CrossRef.
- S. Parkinson, N. S. Hondow, J. S. Conteh, R. A. Bourne and N. J. Warren, React. Chem. Eng., 2019, 4, 852–861 RSC.
- G. Mellot, P. Beaunier, J. M. Guigner, L. Bouteiller, J. Rieger and F. Stoffelbach, Macromol. Rapid Commun., 2019, 40, e1800315 CrossRef PubMed.
- J. He, Q. Xu, J. Tan, L. Zhang, J. Tan and L. Zhang, Macromol. Rapid Commun., 2019, 40, e1800296 CrossRef PubMed.
- X. Wang, J. M. Zhou, X. Q. Lv, B. H. Zhang and Z. S. An, Macromolecules, 2017, 50, 7222–7232 CrossRef CAS.
- X. Wang, C. A. Figg, X. Q. Lv, Y. Q. Yang, B. S. Sumerlin and Z. S. An, ACS Macro Lett., 2017, 6, 337–342 CAS.
- N. P. Truong, M. V. Dussert, M. R. Whittaker, J. F. Quinn and T. P. Davis, Polym. Chem., 2015, 6, 3865–3874 RSC.
- S. Boissie, J. Rieger, K. Belal, A. Di-Cicco, P. Beanier, M. H. Li and B. Charleux, Chem. Commun., 2010, 46, 1950–1952 RSC.
- S. J. Hunter, N. J. W. Penfold, E. R. Jones, T. Zinn, O. O. Mykhaylyk and S. P. Armes, Macromolecules, 2022, 55, 3051–3062 CrossRef CAS PubMed.
- S. J. Hunter, J. R. Lovett, O. O. Mykhaylyk, E. R. Jones and S. P. Armes, Polym. Chem., 2021, 12, 3629–3639 RSC.
- B. Akpinar, L. A. Fielding, V. J. Cunningham, Y. Ning, O. O. Mykhaylyk, P. W. Fowler and S. P. Armes, Macromolecules, 2016, 49, 5160–5171 CrossRef CAS PubMed.
- C. J. Ferguson, R. J. Hughes, D. Nguyen, B. T. T. Pham, R. G. Gilbert, A. K. Serelis, C. H. Such and B. S. Hawkett, Macromolecules, 2005, 38, 2191–2204 CrossRef CAS.
- C. J. Ferguson, R. J. Hughes, B. T. T. Pham, B. S. Hawkett, R. G. Gilbert, A. K. Serelis and C. H. Such, Macromolecules, 2002, 35, 9243–9245 CrossRef CAS.
- F. Stoffelbach, L. Tibiletti, J. Rieger and B. Charleux, Macromolecules, 2008, 41, 7850–7856 CrossRef CAS.
- J. Rieger, G. Osterwinter, C. Bui, F. Stoffelbnach and B. Charleux, Macromolecules, 2009, 42, 5518–5525 CAS.
- D. Alaimo, C. Jérôme, B. Charleux, F. Stoffelbach, C. Bui and J. Rieger, Macromolecules, 2008, 41, 4065–4068 CrossRef.
- W. Zhang, F. D'Agosto, O. Boyron, J. Rieger and B. Charleux, Macromolecules, 2011, 44, 7584–7593 CrossRef CAS.
- M. Khan, T. R. Guimarães, R. P. Kuchel, G. Moad, S. Perrier and P. B. Zetterlund, Angew. Chem., Int. Ed., 2021, 60, 23281–23288 CrossRef CAS PubMed.
- T. R. Guimarães, M. Khan, R. P. Kuchel, I. C. Morrow, H. Minami, G. Moad, S. Perrier and P. B. Zetterlund, Macromolecules, 2019, 52, 2965–2974 CrossRef.
- J. Rieger, W. Zhang, F. Stoffelbach and B. Charleux, Macromolecules, 2010, 43, 6302–6310 CrossRef CAS.
- I. Chaduc, A. Crepet, O. Boyron, B. Charleux, F. D'Agosto and M. Lansalot, Macromolecules, 2013, 46, 6013–6023 CrossRef CAS.
- I. Chaduc, M. Girod, R. Antoine, B. Charleux, F. D'Agosto and M. Lansalot, Macromolecules, 2012, 45, 5881–5893 CrossRef CAS.
- K. G. E. Bradford, L. M. Petit, R. Whitfield, A. Anastasaki, C. Barner-Kowollik and D. Konkolewicz, J. Am. Chem. Soc., 2021, 143, 17769–17777 CrossRef CAS PubMed.
- S. M. North and S. P. Armes, Polym. Chem., 2020, 11, 2147–2156 RSC.
- O. O. Mykhaylyk, N. J. Warren, A. J. Parnell, G. Pfeifer and J. Laeuger, J. Polym. Sci., Part B: Polym. Phys., 2016, 54, 2151–2170 CrossRef CAS.
- A. Blanazs, R. Verber, O. O. Mykhaylyk, A. J. Ryan, J. Z. Heath, C. W. I. Douglas and S. P. Armes, J. Am. Chem. Soc., 2012, 134, 9741–9748 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2py01064j |
| This journal is © The Royal Society of Chemistry 2022 |