
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Molecular mechanism of a large conformational change of the quinone cofactor in the semiquinone intermediate of bacterial copper amine oxidase†
Mitsuo
Shoji
 *ab,
Takeshi
Murakawa
c,
Shota
Nakanishi
d,
Mauro
Boero
e,
Yasuteru
Shigeta
a,
Hideyuki
Hayashi
f and
Toshihide
Okajima
df
*ab,
Takeshi
Murakawa
c,
Shota
Nakanishi
d,
Mauro
Boero
e,
Yasuteru
Shigeta
a,
Hideyuki
Hayashi
f and
Toshihide
Okajima
df
aCenter for Computational Sciences, University of Tsukuba, 1-1-1 Tennodai, Tsukuba 305-8577, Ibaraki, Japan. E-mail: mshoji@ccs.tsukuba.ac.jp
bJST-PRESTO, 4-1-8 Honcho, Kawaguchi 332-0012, Saitama, Japan
cDepartment of Biochemistry, Osaka Medical and Pharmaceutical University, 2-7 Daigakumachi, Takatsuki 569-8686, Osaka, Japan
dInstitute of Scientific and Industrial Research, Osaka University, 8-1 Mihogaoka, Ibaraki 567-0047, Osaka, Japan
eUniversity of Strasbourg, Institut de Physique et Chimie des Matériaux de Strasbourg, CNRS, UMR 7504, 23 rue du Loess, F-67034, France
fDepartment of Chemistry, Osaka Medical and Pharmaceutical University, 2-7 Daigakumachi, Takatsuki 569-8686, Osaka, Japan
First published on 23rd August 2022
Abstract
Copper amine oxidase from Arthrobacter globiformis (AGAO) catalyses the oxidative deamination of primary amines via a large conformational change of a topaquinone (TPQ) cofactor during the semiquinone formation step. This conformational change of TPQ occurs in the presence of strong hydrogen bonds and neighboring bulky amino acids, especially the conserved Asn381, which restricts TPQ conformational changes over the catalytic cycle. Whether such a semiquinone intermediate is catalytically active or inert has been a matter of debate in copper amine oxidases. Here, we show that the reaction rate of the Asn381Ala mutant decreases 160-fold, and the X-ray crystal structures of the mutant reveals a TPQ-flipped conformation in both the oxidized and reduced states, preceding semiquinone formation. Our hybrid quantum mechanics/molecular mechanics (QM/MM) simulations show that the TPQ conformational change is realized through the sequential steps of the TPQ ring-rotation and slide. We determine that the bulky side chain of Asn381 hinders the undesired TPQ ring-rotation in the oxidized form, favoring the TPQ ring-rotation in reduced TPQ by a further stabilization leading to the TPQ semiquinone form. The acquired conformational flexibility of TPQ semiquinone promotes a high reactivity of Cu(I) to O2, suggesting that the semiquinone form is catalytically active for the subsequent oxidative half-reaction in AGAO. The ingenious molecular mechanism exerted by TPQ to achieve the “state-specific” reaction sheds new light on a drastic environmental transformation around the catalytic center.
1 Introduction
A vast majority of enzyme-catalysed reactions proceed through multiple elementary processes realizing a series of catalytic intermediates.1,2 In every single process connecting the intermediates, various catalysed chemical events occur such as nucleophilic, electrophilic, and elimination reactions, and electron/proton transfer. These processes tend to induce conformational changes in the active-site residue(s)/cofactor, which in turn have the effect of enhancing the reactivity. Generally, conformational diversity and multiple states are key factors enhancing the catalytic functions;3 on the other hand, limiting the conformational changes is also important to achieve high catalytic activity.4 These enzymatic molecular mechanisms are further complicated by unclarified protonation and electronic states. All these uncertainties make enzymatic reactions very difficult to reveal. The catalytic intermediates can be detected spectrophotometrically, or by X-ray crystallography as freeze-trapped structures giving insightful information to unveil their reaction mechanisms, provided that they are transiently accumulated depending on the rate constants of the reaction steps. Nonetheless, both spectroscopic and structural characterization studies of transient conformational changes and unaccumulated intermediates remain elusive. To overcome this difficulty, computational approaches making use of hybrid quantum mechanics/molecular mechanics (QM/MM) and molecular dynamics (MD) simulations are nowadays a reliable tool to provide information not accessible to experimental probes. These well-assessed techniques allow for an unambiguous determination of reaction pathways, including conformational changes, and disclose the atomic-level structural details of the short-lived transition state of each elemental process.Copper amine oxidases (CAOs) catalyse the oxidative deamination of primary amines into their corresponding aldehydes and exert fundamental functions in a wealth of aerobic organisms from bacteria to yeast, plants, and mammals.5–7 CAOs in microorganisms have a nutritional role in catabolizing primary amines.7 In higher eukaryotes, CAOs in animals participate in the degradation of bio-active amines, and regulation of cell adhesion, cell death, and collagen cross-linking.7 CAOs in plants exert an active role in wound healing, cell growth, and biosynthesis of various compounds including some alkaloids and lignin.7 CAOs have a homodimer structure with a subunit molecular mass of 70–95 kDa.8–10 The active site is buried inside a large β-sandwich domain, containing one divalent copper ion (Cu(II)) and a redox-active organic cofactor, topaquinone (TPQ)11,12 originating from the post-translational modification of a specific tyrosine residue via a copper and oxygen-dependent autocatalytic reaction.13
The catalytic reaction of CAO is composed of two half-reactions, one reductive and the other oxidative, as shown in Fig. 1A.14,15 During the reductive half-reaction, an initial oxidized form of TPQ (TPQox) is converted into the substrate Schiff base (TPQssb) through the nucleophilic attack of a substrate amine on the O5 carbonyl group. TPQssb is further converted into the product Schiff base (TPQpsb) through stereospecific proton transfer. Then, TPQpsb is hydrolyzed to the corresponding aldehyde and aminoresorcinol (TPQamr). The latter is equilibrated with a semiquinone radical (TPQsq) plus monovalent Cu(I) that is formed by a single electron transfer from TPQamr to Cu(II).
 | ||
| Fig. 1 (A) The proposed catalytic cycle of AGAO.14 The stable and intermediate states are the oxidized form (TPQox), the substrate Schiff base (TPQssb), the product Schiff base (TPQpsb), aminoresorcinol (TPQamr), the semiquinone radical (TPQsq), and iminoquinone (TPQimq). Among all these, only TPQsq is detected in the on-copper conformation. (B) The conformational changes of TPQ in the aminoresorcinol and semiquinone radical states. TPQamr is located away from the Cu coordination site (off-copper), while TPQsq, colored in green, approaches the Cu site (on-copper). Two X-ray crystal structures of AGAO are superimposed (PDB ID: 3X3X, 3X3Z).14 | ||
Using CAO of the soil bacterium Arthrobacter globiformis (AGAO), we have performed transient kinetic experiments for the reductive half-reaction to spectrophotometrically detect the TPQssb, TPQpsb, TPQamr, and TPQsq intermediates.16,17 Furthermore, we have determined the X-ray crystal structures of all these intermediate states trapped in AGAO crystals. In the initial TPQox state, the TPQ ring is located away from Cu(II) (“off-copper” conformation) with the O4 of the TPQ ring forming a hydrogen bond (H-bond) with a highly conserved Tyr residue (Tyr284). This off-copper conformation is preserved in TPQssb, TPQpsb, and TPQamr. Interestingly, a large conformational change of the TPQ ring is observed in the reaction step going from TPQamr to TPQsq;14,15 through this process, the O4 of the TPQ ring is ligated axially to the Cu in TPQsq (“on-copper” conformation). For the conformational change of TPQamr, the TPQ ring needs to undertake three motions: sliding (rotation of 53° around the Cα–Cβ bond), tilting up (20° rigid body rotation centered on the Cα atom), and phenol ring rotation (rotation of 180° around the Cβ–Cγ bond).14 This structurally challenging mechanism is further complicated by the occurrence of a one-electron transfer from TPQamr to Cu(II) and a concomitant deprotonation of the O4 of TPQamr, which are all required for TPQsq formation. Since the TPQ ring in the off-copper conformation is surrounded by a number of amino acid residues including Asp298, Asn381, and Tyr384, it has so far been expected that the bulky TPQ ring does not have enough free space to undergo a rotation, and that the TPQ ring should first slide out from the off-copper position, and then revolve into the on-copper position.14 However, this pathway has never been detailed. Among the residues in proximity of the off-copper TPQ ring, Asn381 is highly conserved in CAOs, and the side chain carboxamide group is situated on the TPQ ring of TPQox.18,19 The spectroscopic study presented in ref. 18 for Hansenula polymorpha CAO (HPAO) suggests that the Asn residue, analogous to Asn381 in AGAO, prevents TPQox from taking a nonproductive orientation by suppressing the mobility of the cofactor. The Asn381 residue is located close to the TPQ ring in both off-copper and on-copper conformations. As a result, the side chain is likely to affect the TPQ conformational change from TPQamr to TPQsq (Fig. 1B). When the other active-site residues are mutated by site-directed mutagenesis, various effects concerning the conformational changes or thermal flexibility were reported in former studies.17,20,21 However, the specific contribution of Asn381 to this process is still unclear, as, to date, there are only static structural data on the active-site structure of AGAO.
Several former studies focused on the flexibilities of the TPQ cofactor in TPQox, and on-copper, off-copper active and off-copper flipped conformations were confirmed.8,9,22–24 These conformational flexibilities of TPQox represent an important feature in the early stage of the process after TPQ biogenesis, where TPQ in CAO is produced from a Tyr residue on the Cu site and TPQ has to change the conformation to the catalytic site. Conversely, for the catalytic intermediates, 3D structures in non-off-copper and on-copper conformations are very limited in TPQsq.14,25 To preserve the optimal catalytic activity of TPQ, accurate control of the TPQ conformation is essential, and a conformational change along with an alternation of TPQ contributes directly to the inactivation.18,23 We remark that TPQsq formation depends on the CAO types and the source organisms, and the formation of TPQsq is not always observed for all CAOs.14,25 AGAO, Pisum sativum CAO (PSAO) and Escherichia coli CAO (ECAO) undergo the TPQ conformational change during TPQsq formation, whereas bovine serum CAO (BSAO) and HPAO do not undergo any TPQ conformational change and TPQsq is not formed during the catalytic cycle.14,25 For AGAO, PSAO and ECAO, spectroscopy studies have shown that TPQsq is generated in equilibrium with TPQamr,26,27 and their TPQsq are essential intermediates in the catalytic cycle. The formation of TPQsq influences the reoxidization step by O2 to generate an iminoquinone intermediate (TPQimq) and hydrogen peroxide.14,27,28 The reaction steps in the oxidative-half reaction still remain an open issue, and may also vary depending on TPQsq formation. Give this scenario, it is crucial to address the questions of why and how TPQsq formation and the large conformational change of TPQ occur in the AGAO catalytic cycle.
In the present study, we investigated the conformational changes in the quinone cofactor in AGAO using a synergy of experimental and theoretical methods. The conformational changes of the Asn381Ala (N381A) mutant were determined by X-ray crystallography and kinetic analyses. Then QM/MM methods were employed to unravel the conformational change pathways and electron transfer mechanism in the TPQamr to TPQsq transition. The contributions of the active-site residues close to TPQ and Cu(II) along the TPQ conformation are also inspected within the same computational approach. The insight provided by this work evidences the active role of Asn381 in the exceptionally large conformational change in TPQamr and elucidates the electron transfer mechanism from TPQamr to Cu(II) which is strictly dependent on the conformation and deprotonation of the TPQ ring. The large conformational change in the quinone cofactor observed in AGAO is a clear and detailed example of the conformational control exerted by the active-site residues in promoting and enhancing multistep and multi-conformational enzymatic reactions.
2 Materials and methods
2.1 Preparation, characterization and X-ray crystallography of the N381A mutant of AGAO
Details for site-directed mutagenesis (N381A), enzyme preparation, and kinetics analyses for TPQ biogenesis and enzyme catalysis are reported in the ESI.† A holo form of the N381A mutant was crystallized by a microdialysis method with the crystallization buffer, 1.05 M potassium-sodium tartrate in 25 mM HEPES buffer, pH 6.8, as described in a former work for the wild-type (WT) enzyme.17 For the determination of the crystal structures of the catalytic intermediates anaerobically reduced using 2-phenylethylamine (2-PEA), the holo-form crystals in the dialysis button were transferred into the new reservoir solution containing 45% (v/v) glycerol in an anaerobic glove box (SGV-65V glove box, AS ONE corporation) at 16 °C for 24 h. Then, the crystals were soaked in a new reservoir solution containing 45% (v/v) glycerol and 4 mM 2-PEA at 16 °C for 60 min and subsequently frozen by rapid cooling in liquid CF4. Before exposure to X-rays, these crystals were analysed by single-crystal microspectrophotometry at 100 K as previously reported.17 The X-ray diffraction data were collected at 100 K with synchrotron X-radiation (λ = 0.9 Å) using an MX-225HE detector (Rayonix, L.L.C.) in the BL44XU beamline station at the SPring-8 facility (Hyogo, Japan). The collected data were processed and scaled using Mosflm29 and Scala in CCP4,30 respectively. Molecular replacement for phase determination was done by Phaser31 using the WT AGAO structure (PDB ID: 1IU7)13 as a search model. The obtained initial structure was subjected to rigid-body refinement and was further refined with Phenix.32 The manual model building and its validation were performed with Coot.33 Ramachandran plots were calculated using MolProbity34 for structure validation. The details and statistics of crystallographic refinement are summarized in Table S2.† Atomic coordinates and structure factors of the holo form and the substrate-reduced form of N381A AGAO were deposited in the Protein Data Bank with the accession codes 7WIR and 7WIS, respectively.2.2 Computational details
The X-ray crystal structure of AGAO, determined with a resolution of 1.51 Å, was obtained from the Protein Data Bank (PDB ID: 3X3Z).14 This crystal structure corresponds to the TPQamr state in the off-copper conformation. An inhibitor Cl− anion coordinated to the Cu(II) in the original PDB was removed. Among all the available conformers, residues with the highest occupancy were selected and titratable residues were protonated according to their state in the corresponding neutron crystal structure (PDB ID: 6L9C).35 A dimer model was solvated into a water droplet with a 60 Å radius and the whole system was kept in a neutral charge state by replacing some of the water molecules with 36 Na+ ions. The whole system was equilibrated via classical MD within the Amberff99 force field framework.36 This equilibration process was achieved by an annealing MD at 250 K lasting for 10 ps to relax the solvent water molecules and all the added H atoms in AGAO. The coordinates of the heavy atoms determined by X-ray crystallography were kept fixed during this MD stage.After this annealing step, we moved to QM/MM simulations. The selected QM subsystem consists of side chains including the residues Tyr284, Asp298, Tyr384, Asn381, TPQ, His431, His433, and His592, plus the Cu(II) and water molecules in the active site (Fig. 2). For the electronic structure description of the QM region, we resorted to the density functional theory (DFT) in a spin-unrestricted scheme at the UB3LYP-D3/DZVP level. The remaining classical part of the system was treated at the same MM level, with the Amberff99 force field, as used in the equilibration step. The hybrid exchange-correlation functional B3LYP was complemented by Grimme's D3 dispersion correction.37 The basis set adopted to describe the electronic structure consists of valence double zeta plus polarized (DZVP) functions, specifically, LANL-2DZ for Cu and 6-31G* for the other atoms.38–40 This computational set-up has already been assessed in terms of the accuracy of the structures and energetics of the relevant enzymatic reactions.41–44 Geometry optimizations were performed for all the atoms within a 15 Å radius from the centre in the QM region. An electronic embedding scheme and link hydrogen atoms were adapted for the cut across covalent bonds at the QM/MM interface, and QM/MM non-bonded interactions were explicitly computed without introducing a cutoff distance for all the energy calculations. The sampling of the reaction pathways and the location of the transition states was done with the nudged elastic band (NEB) method.45–47 We used 13 images for the first rough searches, and in the high energy regions close to the barriers where transition states are expected to be located, we further refined the sampling by performing NEB calculations with an additional 13 images.
 | ||
| Fig. 2 The QM/MM model used in our simulations for the AGAO dimer. In the figure, the AGAO monomers, the Na+ ions, and the solvent water molecules are colored differently. The highlighted panel shows the details of the active site. Atoms in the QM region are shown in licorice representation with the main residues and water molecules labeled according to the discussion in the main text. | ||
The N381A mutant model was constructed by replacing the Asn381 residue with Ala. TPQox models for the WT AGAO and the N381A mutant were obtained by replacing the cofactor moieties in the reduced state (TPQamr) with those in the oxidized form (TPQox).
All the MD and QM/MM calculations were performed using the NWChem 6.8 program package.48 The molecular structures shown in the figures were drawn using the VMD program.49
3 Results and discussion
3.1 Characteristics of the N381A mutant
Asn381 is highly conserved among CAOs and is located at the closest position to the TPQ ring. For this reason, it plays a key role in the AGAO catalytic cycle. To evaluate the contribution of Asn381 to the TPQ conformational change and catalytic reactions, we replaced the Asn381 residue with Ala and conducted a kinetic and structural analysis. The WT and the N381A mutant of AGAO were expressed in E. coli and purified to homogeneity (Fig. S1†). The fundamental properties of the N381A mutant enzyme are summarized in Table 1 together with those of the WT for comparison. As with TPQ in the WT, TPQ was generated from the precursor Tyr residue in N381A. However, the rate of TPQ biogenesis was reduced to 1/180-fold that in the WT (Fig. S2†). A steady-state kinetic analysis of the overall catalytic reaction with 2-PEA showed a Km value for the N381A (1.9-fold) rather similar to that of the WT, but a very low kcat value (1/160-fold) for N381A compared with that of the WT enzyme (Table 1). To identify the reaction steps accounting for the significant decrease in the catalytic activity of the N381A mutant, changes in the UV-vis absorption spectra initiated by the addition of 2-PEA were measured under single-turnover (anaerobic) conditions with a stopped-flow spectrometer (Fig. S3†). During the reductive half-reaction of the WT enzyme with 2-PEA, the absorption peak at 480 nm of TPQox disappeared in about 4 ms. Then, the two peaks at 316 nm and 410 nm, assigned to TPQssb and TPQpsb, respectively, arose. Accompanied by a concomitant decay of these two peaks, new peaks assigned to TPQsq (peaks at 365, 438, and 465 nm) eventually appeared during about 200 ms as shown in Fig. S3B†.17 Conversely, for the N381A mutant, the TPQ-derived 495 nm absorption band gradually decreased within 100 ms upon anaerobic mixing with 2-PEA, and no new absorption peak above 300 nm was observed during the reaction (Fig. S3A†). The final spectrum detected in the measurement showed no absorption in the visible region, indicating that TPQamr is the final product and that the equilibrium between TPQamr and TPQsq significantly shifts toward the former. Thus, the absence of the detection of any intermediate state indicates that the initial TPQssb formation from TPQox is in the rate-limiting step in the reductive half-reaction of the N381A mutant enzyme with the substrate amine.| Rate of TPQ biogenesis (min−1) | Steady-state kinetic parameters | ||
|---|---|---|---|
| K m (μM) | k cat (s−1) | ||
| a Values in parentheses indicate the ratio with the WT AGAO. | |||
| WT | 2.300 × 10−1 ± 3.4 × 10−3 (1) | 4.80 ± 0.43 (1) | 98.2 ± 5.4 (1) |
| N381A | 1.3000 × 10−3 ± 5.7 × 10−6 (1/180) | 8.90 ± 0.35 (1.9) | 0.610 ± 0.012 (1/160) |
3.2 X-ray crystallographic analysis of the N381A mutant
The X-ray crystallographic structures were determined for the TPQox and TPQamr forms in the N381A mutant, hereafter indicated as N381Aholo and N381Aholo/PEA, respectively (Table S1†). In the single-crystal UV-vis absorption spectra, N381Aholo was characterized by a peak centered at 480 nm corresponding to TPQox, and N381Aholo/PEA exhibited a spectrum similar to that of TPQamr formed during the reaction with 2-PEA (Fig. S4†), confirming that the TPQamr intermediate has been successfully freeze-trapped after 2-PEA soaking. As a noticeable feature, the electron density of the active-site structure of N381Aholo shows that TPQ can take two conformations as depicted in Fig. 3A. The occupancies of the two conformers, a and b, of TPQ are 0.47 and 0.53, respectively (Fig. S5†). The conformer a of TPQ has essentially the same conformation as the WT TPQ (normal form), whereas the conformer b is rotated by about 170° around the Cβ–Cγ axis (flipped form). For both conformers, the C4 hydroxyl group of TPQ forms H-bonds with the highly conserved Tyr residue (Tyr284 in AGAO) with a distance of 2.7 Å and 2.5 Å for the normal and flipped forms, respectively. The conformer b has the O5 carbonyl of TPQ not directed toward the substrate-binding site, indicating an inactive conformation. | ||
| Fig. 3 X-ray structures of the holo N381A crystal in the TPQox and TPQamr conformations. The TPQ rings are flipped in both states. (A) The TPQ ring-flipped conformation of TPQN381Aox is colored by element, in which the non-ring-flipped conformation is colored in cyan. (B) The N381A structure of TPQamr is superimposed on the WT structure (PDBID: 3X3Z),14 colored in green for a direct comparison. | ||
Concerning the structure of N381Aholo/PEA, based on single-crystal micro-spectrophotometry and the electron density maps, we modelled TPQamr for the cofactor at the position of the amino acid residue 382. In the structure, the electron density corresponding to the product phenylacetaldehyde was observed at the active site. A remarkable feature is a fact that TPQamr presents a flipped form with rotations of about 20° around the Cα–Cβ bond and about 150° around the Cβ–Cγ bond with respect to the off-copper conformation of the WT structure14 (Fig. 3B). Details of the active site and TPQamr in the N381A mutant are shown in Fig. S6.† The O4 atom of the phenol of TPQamr in the N381A mutant approached Asp298, or rather moved away from Cu. This TPQamr still does not reach an on-copper conformation. This is consistent with the evidence that the final conformer identified by the spectral change of the reductive half-reaction of the N381A mutant enzyme with 2-PEA is assigned to TPQamr but not to TPQsq. It is evident that the N381A mutant enzyme lacks the ability for the cofactor to retain an appropriate conformation in both TPQox and the substrate-reduced forms (TPQamr/TPQsq) for the catalytic reaction. In particular, the alternative position of TPQamr in the N381A mutant suggests that specific and selective TPQ conformational regulations are required for the WT-AGAO catalytic reactions, especially for TPQsq formation. Hence, by resorting to detailed QM/MM simulations, presented in the following paragraphs, we aim to elucidate the conformational change pathway from TPQamr to TPQsq associated with electron transfer to the Cu centre, focusing on the N381A mutant lacking the WT catalytic ability because of its peculiar conformation.
3.3 Relation between TPQ protonation and TPQsq formation
We started our computational study by inspecting the possible protonation states of AGAO in the TPQamr and TPQsq states. We recall that the characteristic UV-vis absorption spectrum with maxima at around 440 and 470 nm14 originates from TPQsq. This spectrum is quite different from the broad and peak less spectra of TPQamr, which can be obtained as an intermediary state in solution upon anaerobic substrate reduction. The crystal structure was also determined in the presence of NaCl or NaBr by anaerobic soaking with the substrate.14 The halide ions are bound to the axial position of the Cu active site and act as anionic inhibitors. The characteristic absorption peaks of the TPQsq state are ascribed to an electron transfer from TPQamr to Cu(II), leading to the formation of Cu(I). Electronic structure calculations for the different protonation states of TPQamr have shown that the protonation of both the O2 and O4 sites of TPQ is required to prevent oxidation by the Cu(II) centre. Indeed, the energy differences for a single electron transfer from the protonated and deprotonated forms at the O4 site in TPQamr, corresponding to 1h and 1 in Fig. 4, turned out to be 18.8 eV (433 kcal mol−1) and −32.9 eV (−758 kcal mol−1), respectively. From this energetics, we can infer that a single electron transfer from TPQamr to Cu(II) is more favoured in 1 and is strictly related to the deprotonation of TPQamr. Furthermore, after a structural optimization of these conformations, the Cu coordinating water molecules, Wax and Weq, are released by the formation of Cu(I), and this also holds for the off-copper conformation. We also found that the dissociation of the water ligands (Wax and Weq) is energetically unfavourable by more than 25 kcal mol−1 in the Cu(II) state. These results suggest that the TPQ coordination to the Cu site must occur after the formation of a TPQ semiquinone radical and Cu(I). Complementary calculations have shown that O4-TPQamr deprotonation is energetically more favourable than the deprotonation of O2-TPQamr by ΔE = 12.3 kcal mol−1. Based on a synthetic model compound (aminophenol), the experimentally determined pKa values are 9.59 and 11.62 for O4 and O2,50 respectively, which are consistent with the present QM/MM results. The overall picture provided by these analyses is that TPQamr is fully protonated in TPQamr, and that TPQsq is formed upon an electron transfer to the Cu centre when TPQamr is deprotonated at the O4 site. Moreover, the simulations are fully consistent with the experimental detection of TPQamr when the O4 of TPQamr is protonated in the off-copper conformation.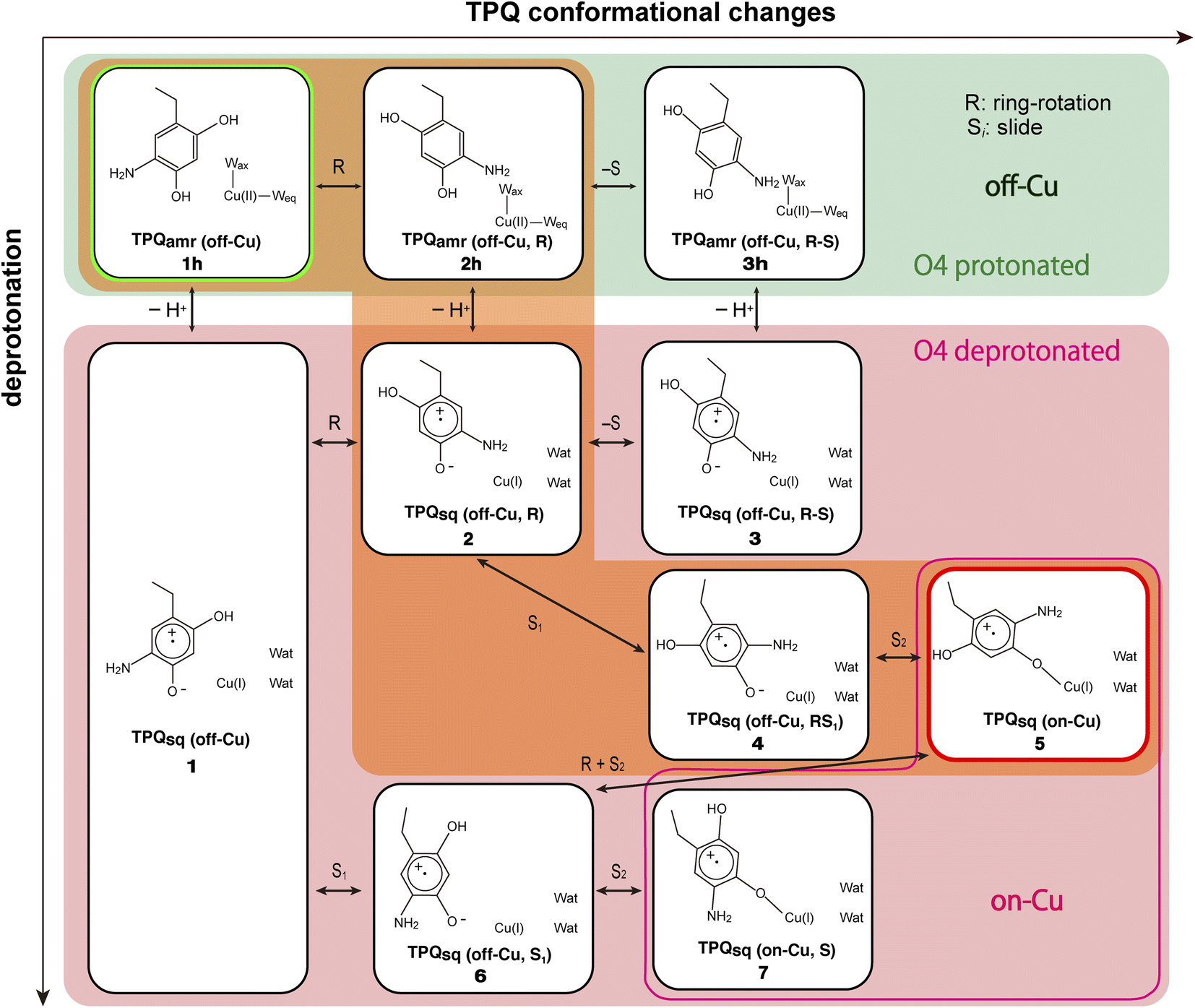 | ||
| Fig. 4 A comprehensive view of the intermediate states on which the present work is focused for the TPQamr to TPQsq transition. The letter “h” added in the labeling as in 1h–3h stands for the protonated state on O4-TPQ. The orange background highlights the main reaction pathway, whereas the green background refers to the O4-TPQ protonated state, and the red one refers to the deprotonated one. | ||
3.4 Viable transition pathways of the TPQ conformational change
This paragraph is focused on the possible reaction pathways from the off-copper TPQamr to the on-copper TPQsq. The TPQ conformational change involves two main motions, (i) slide and (ii) ring-rotation. The slide motion of the TPQ ring takes place when a rotation of about 50° occurs around the Cα–Cβ bond along with tilting up in a rigid-body motion of ∼20° with respect to the centre represented by the Cα atom.14 The ring-rotation of this same TPQ, instead, occurs by a rotation of 180° around the Cβ–Cγ bond. By considering the ordered sequence of the two main motions and the two possible rotational directions of the TPQ ring-rotation, clockwise and counter-clockwise, four alternative pathways can be identified:(I) Clockwise-ring-rotation and slide.
(II) Counter-clockwise-ring-rotation and slide.
(III) Slide and clockwise-ring-rotation.
(IV) Slide and counter-clockwise-ring-rotation.
In a former study,14 we suggested that the TPQ ring needs to slide out from the off-copper position to perform a rotation and that pathway (III) is preferable to the alternative ones.14 Yet, a thorough and deeper analysis was not possible at that time. We filled this gap here. Specifically, we could observe that unless a conformational change of the surrounding residues occurs (Fig. 1B and S7†) these pathways are hindered to various extents. More precisely, in pathway (I), the side chains of Asn381 and Tyr384 prevent TPQamr ring-rotation. In pathway (II), the main chain of TPQ and the side chain of Val282 blocks the 2-hydroxyl group of TPQamr and the 5-amino group of TPQamr, respectively. Concerning pathway (III), the main chain of Phe407 and the side chain of Tyr384 prevent ring-rotation. Finally, in pathway (IV), the side chains of Asn381 and His433 come in close contact with the TPQ moiety, thus hindering any further motion. Among these four pathways, the steric hindrance in pathway (II) seems inevitable because the hindrance during the first counter-clockwise-ring-rotation occurs within the TPQamr residue (Fig. S8A†). The steric hindrance in pathway (IV) for the counter-clockwise-ring-rotation is also unavoidable because TPQsq and the side chain of His433 are both tightly coordinated to Cu(I) (Fig. S8B†). For these reasons, we focus here on the pathways (I) and (III).
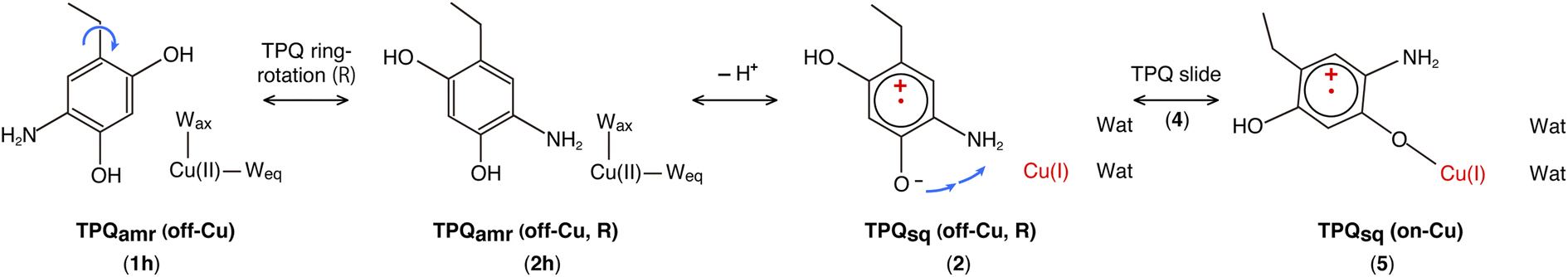
Our QM/MM simulations allowed us to identify several intermediate states with specific conformations and protonation states. All the intermediate states are sketched and shown in Fig. 4, where the protonated states of the O4-TPQ are labelled by adding a second letter “h”. 1h and 5 denote the conformations assumed upon off-copper and non-ring-rotation of one of TPQamr and the on-copper one of TPQsq, respectively. These correspond to the AGAO structures determined by X-ray crystallography.14 States 2h and 2 have TPQ rings that are rotated with respect to 1h and 1, respectively, and take the off-copper conformation. In the middle of the slide step in pathway (I) (2 → 5 in Fig. 4), a stable intermediate state, labelled as 4, was found. In this state, the rotation angles around the Cα–Cβ bond of TPQ along 2 → 4 and 4 → 5 are 16.1° and 37.3°, respectively.
On these grounds, the TPQ slide movement can be seen as a combination of two subsequent steps, an initial slide motion (S1) followed by a second one (S2). We could also identify a state labelled as 6 generated by the slide motion from state 1 without rotating the TPQsq ring corresponding to the slide motion S1. The state labelled as 7 is defined as the one in which the ring takes the on-copper conformation but is turned over with respect to structure 5 and can be formed from 6via the S2 slide.
Another important issue is the moment in which the deprotonation of O4-TPQ occurs. There are three possibilities for this process to happen: the first one (A) is before the TPQ conformational change; the second one (B) is during the TPQ conformational change; and the third one (C) is after the TPQ conformational change. We remind that the deprotonation of O4-TPQamr induces the change Cu(II) → Cu(I) and that the ligand exchange at the Cu(II) site is energetically demanding. This result suggests that pathway (C) can be ruled out since the TPQ on-copper conformation does not proceed because of the presence of Wax. Similarly, in the first slide movement of the TPQ conformational change, such as for instances (III) and (IV), O4-TPQ must be displaced close to the Cu axial position in the presence of Wax. This means that the first ring-rotation motion of TPQamr is favoured before the TPQamr deprotonation in the case of (B). Hence, the pathways to be considered can be summarized as:
(IA): deprotonation, TPQsq clockwise-ring-rotation and slide (1h → 1 → 2 → 4 → 5).
(IB): clockwise-ring-rotation of TPQamr, deprotonation, and TPQsq slide (1h → 2h → 2 → 4 → 5).
(IIIA): deprotonation, TPQsq slide, and clockwise-ring-rotation (1h → 1 → 6 → 5).
and are sketched and shown in Fig. 4.
States 3h and 3 in Fig. 4 can also be formed as alternative TPQ conformational states originating from 2h and 2 by slide motions with rotations of −88.4° and −85.1°around the Cα–Cβ bond, respectively. The conformation indicated as 3h corresponds to the TPQamr conformation observed in the X-ray crystal structure of the N381A mutant (N381Aholo/PEA). Reaction steps 2h → 3h and 2 → 3 correspond to the negative slide motion (−S) in Fig. 4. All the states in the O4-TPQ protonated form take an electronic structure typical of TPQamr, whereas all the states in the deprotonated form present the electronic structure of TPQsq. The X-ray crystal structures of the WT TPQamr (PDB ID:3X3Z), N381Aholo/PEA (this work), and WT TPQsq (PDB ID: 3X3X) are well reproduced by states 1h, 3hNA, and 5, whose root-mean-square-deviation (RMSD) of the atoms inside the QM region are 0.291, 0.392, and 0.516 Å, respectively (see Table S3†).
The energies of the TPQ O4-protonated states in the WT and N381A mutant were evaluated with respect to the energies in states 1h and 1hNA, respectively. For O4-deprotonated states in the WT, the energies were calculated by taking as a reference the energy of state 5. This corresponds to the observed deprotonation equilibrium between TPQamr and TPQsq. The validity and advantage of adopting these reference energy values, closely related to the assignment of pKa, are discussed in Section 3.17. For the N381A mutant, the relative energy in deprotonated state XNA (ΔE(XNA)) was converted by referring to the energy differences in the WT. The energy correction was expressed as ΔE(XNA) = E(XNA) − E(1hNA) + E(1h) − E(5), where E(XNA) represents the total energy in state XNA.
3.5 Ring-rotation of TPQamr in the protonated form (1h → 2h)
The initial step of pathway (IB) is the ring-rotation of TPQamr at the off-copper position. This originates from a clockwise rotation around the Cβ–Cγ bond (1h → 2h). The calculated energy profile for the WT along with the optimized structures are shown in Fig. 5. In the initial TPQamr state (1h), the TPQamr ring forms H-bonds with Asp298, Tyr284, and Wax with typical (non-hydrogen) distances of 3.3, 2.7, and 2.9 Å, respectively; the most relevant atomic distances are summarized in Table S1.† An NEB calculation was used to sample the reaction path and locate the transition state of the TPQamr ring-rotation (TS(1h,2h)). The relative energy of the TS(1h,2h) with respect to 1h is ΔE(TS(1h,2h)) = 23.3 kcal mol−1. The side chains of Asn381 and Tyr384 in TS(1h,2h) are located in the proximity of TPQamr at the distances of 2.7 and 2.7 Å, to be compared with the distances of 4.2 and 4.6 Å in 1h. These same side chains move away giving room to TPQamr rotation. The 5-amino group of TPQamr forms new H-bonds with Oδ and Nδ of Asn381 with distances of 2.7 and 2.8 Å, respectively (Table S1†). We remark that the phenol group of Tyr384, pushed by TPQamr, can form one H-bond with Oδ1 of Asp298 (Table S1†). These interactions contribute to reducing the energy barrier for TPQamr ring-rotation (Fig. 4).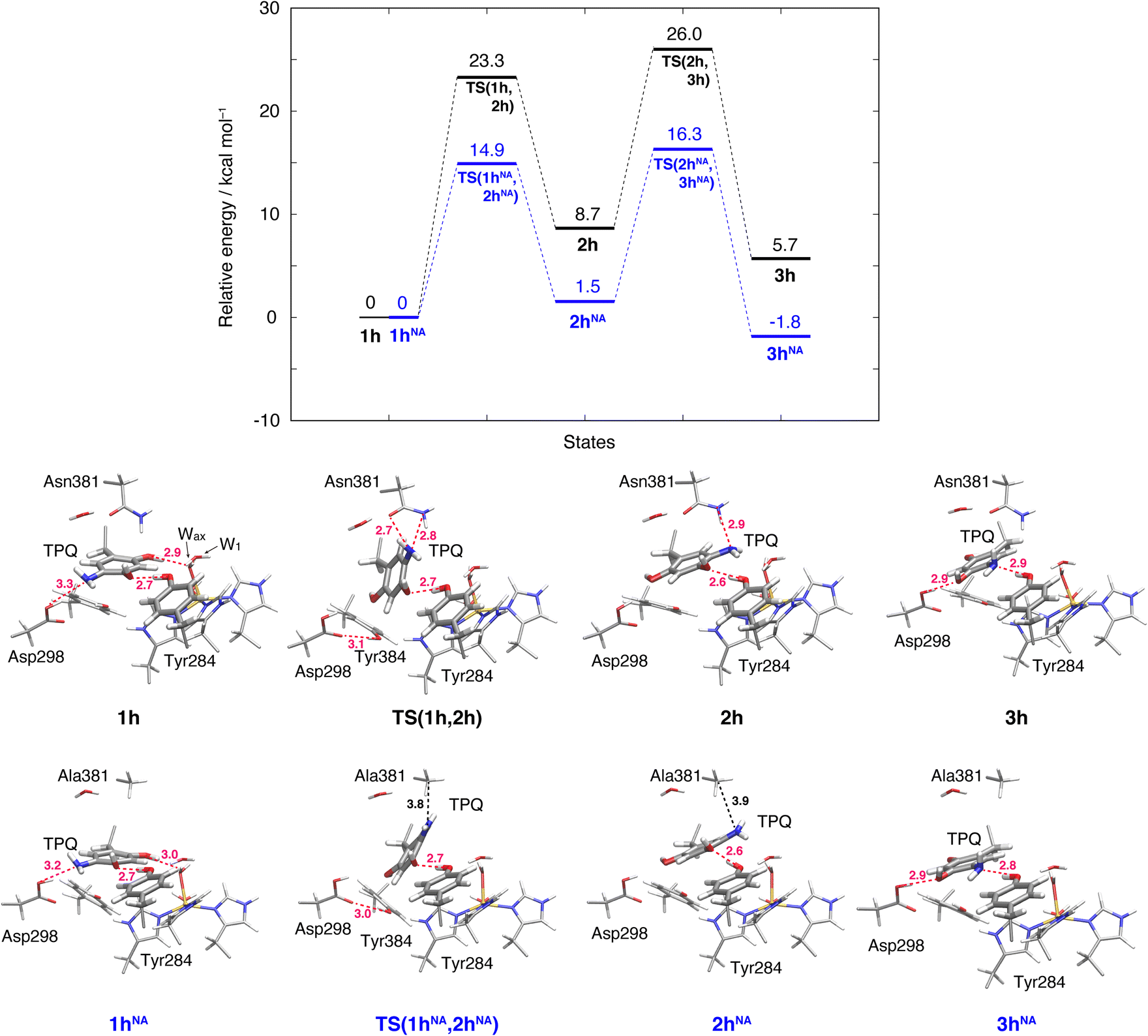 | ||
| Fig. 5 Relative energies associated with the TPQ conformational change in the protonated state. Results for the WT and the N381A mutant (NA) are shown as black and blue lines, respectively. Molecular structures of the intermediate and transition states are reported with the most important distances between heavy atoms. | ||
To evaluate the effects of the carboxamide side-chain of Asn381, we also considered the N381A mutant and inspected the conformational change on the off-copper and protonated TPQamr state (1hNA) to the ring-rotated form (2hNA) within our QM/MM computational approach (Fig. 5). For the N381A mutant model, no steric hindrance by the small side chain of Ala381 was observed, and the activation barrier of the 1hNA → 2hNA transition was ΔE(TS(1hNA,2hNA)) = 14.9 kcal mol−1 relative to 1hNA. The carboxamide group of Asn381 was expected to hinder TPQ rotation. However, as shown by our simulations, TPQamr can rotate by overcoming a rather modest barrier formed by the carboxamide group, calculated as ΔE(TS(1h,2h)) − ΔE(TS(1hNA,2hNA)) = 8.4 kcal mol−1, through forming a specific H-bond between the carboxamide group of Asn381 and the amino group of the TPQ ring in the WT.
The ring-rotated state (2h) is rather unstable with respect to the initial state of TPQamr (1h) by ΔE(2h) = 8.7 kcal mol−1 for the WT (Fig. 5). As Wax in the hydration shell of the Cu centre is close to the 5-amino group of TPQamr, the ring-rotation of TPQ around the Cβ–Cγ bond cannot be completed (compare 1h and 2h in the lower panels of Fig. 5). The dihedral angle ΔA(Cα, Cβ, Cγ, N5) = 142.7° is clearly lower than the expected 180° value for a full rotation. The conformation of the carboxamide group of Asn381 in 2h of the WT does not completely revert to the original position in 1h. These interactions keep the 5-amino group trapped between the carboxamide group of Asn381 and Wax at distances of 2.9 and 3.3 Å, respectively, through H-bonds (Table S1†). In the N381A mutant, the state corresponding to 2hNA was stabilized with respect to the WT state 2h (ΔE(2hNA) = 1.5 kcal mol−1) (Fig. 5). 2hNA maintains a short H-bond of 2.6 Å between TPQamr and Tyr284. The molecular structures of the WT AGAO (2h) and N381A mutant (2hNA) are practically identical apart from the geometry of the mutated carboxamide group of Asn381 and the 5-amino group of TPQamr. The destabilization of 2h can be ascribed to the interaction of TPQamr with Asn381 that has a closer position to TPQamr than in 2hNA.
3.6 Alternative TPQamr ring-rotated state in the protonated form (3h)
The TPQamr ring-rotated state of AGAO can also take an alternative conformation, labelled as 3h, in AGAO. Such a conformation of TPQamr was experimentally found by X-ray crystallography in the TPQamr form of the N381A mutant (Fig. S6†). The energy profile leading to 3h was sampled for both the WT and N381A mutant. State 3h can be formed from 2h by a slide motion of −88.4° of TPQamr, and this occurs in the opposite direction with respect to the on-copper formation. Pathway 2h → 3h can be summarized as a negative slide motion (−S) (see Fig. 4). In 3h, TPQamr forms short H-bonds (2.9 Å) with Asp298 and Tyr284 (3h in Fig. 5). In spite of this H-bond stabilized 3h structure, compared to 2h, the system is still energetically located above 1h by ΔE(3h) = 5.7 kcal mol−1 (Fig. 5). Our NEB sampling of the reaction path indicates that the barrier for the 2h → 3h transition (ΔE(TS(2h,3h)) = 26.0 kcal mol−1) is higher than the one for 1h → 2h. Thus, we can infer that the formation of 3h is slower than that of 2h in the WT. In the N381A mutant, 3hNA becomes more stable than the initial TPQamr state (1hNA) (ΔE(3hNA) = −1.8 kcal mol−1) and an NEB calculation showed a relatively low barrier of ΔE(TS(2hNA,3hNA)) = 16.3 kcal mol−1, which is likely to allow for rapid conversion of 2hNA into 3hNA in TPQamr.3.7 TPQsq ring-rotation in the deprotonated form (1 → 2)
The TPQsq ring-rotation of the deprotonated form is a crucial process in pathway (IA). Upon the deprotonation of O4-TPQamr in 1h, TPQamr and Cu(II) are spontaneously converted to TPQsq and Cu(I) by single-electron transfer, and the two water molecules coordinated to the Cu(I) centre are released as discussed in Section 3.3. The hydrating water molecules can move rather easily and have enough free space to escape. For this reason, we removed redundant H2O in all the deprotonated states defined in the present study (1–7). This allows for easier evaluation and comparison of the relative energies of different structures in the deprotonated state.The NEB energy profile and the main intermediate states are shown in Fig. 6 and the structure of the transition state is reported in Fig. S9 of the ESI.† In a way analogous to step 1h → 2h in the protonated form, the TPQsq ring-rotation 1 → 2 step occurs by drifting of the side chains of Asn381 and Tyr384, thereby giving room to the formation of a specific H-bond between TPQ and Asn381 in TS(1,2). As 1 and 2 have higher relative energies, ΔE(1) = 8.0 kcal mol−1 and ΔE(2) = 12.3 kcal mol−1, the NEB estimated barrier is ΔE(TS(1,2)) = 30.8 kcal mol−1. In 2, the 5-amino group of TPQsq is close to the Cu(I) site (4.6 Å), to be compared with a distance of 5.1 Å in 2h, and TPQ forms H-bonds with Asn381, Tyr284, and W1 with distances of 3.1, 2.6 and 2.7 Å, respectively. Superimposed structures of 2vs.2h and 1vs.1h shown in Fig. S10† show the small changes affecting these H-bonds. In the N381A mutant, states 1NA, 2NA and the transition state TS(1NA,2NA) are all higher in energy than the corresponding WT states by 4 kcal mol−1 (Fig. 6). These energy profiles clearly suggest that TPQ ring-rotation is unfavourable in the deprotonated state of TPQsq compared to the protonated state of TPQamr in both the WT and N381A mutant.
 | ||
| Fig. 6 Relative energies associated with the TPQ conformational change over 1–5 states in the deprotonated form. Results for the WT and the N381A mutant (NA) are shown as black and blue lines, respectively. Molecular structures of the intermediate states are reported with the most important distances between heavy atoms. | ||
3.8 TPQsq slide in the deprotonated form (3 → 2)
Within our simulation protocol, we inspected the conformational change and energetics of the deprotonated TPQsq from 3 to 2 (Fig. 6). States 3 and 3NA turned out to be located at the relative energies of 5.8 and 2.3 kcal mol−1, respectively. The 2 and 2NA structures have higher relative energy, namely 12.3 and 5.7 kcal mol−1, respectively. In the WT, the transition state energy for the TPQsq slide motion along the reaction path from 3 to 2 was ΔE(TS(3,2)) = 21.9 kcal mol−1. This is lower than the energies of transition states in the protonated state (ΔE(TS(1h,2h)) = 23.3 and ΔE(TS(2h,3h)) = 26.0 kcal mol−1). On the other hand, in the N381A mutant, the corresponding energy was ΔE(TS(3NA,2NA)) = 19.0 kcal mol−1. This energy is higher than those of transition states in the protonated state (ΔE(TS(1hNA,2hNA)) = 14.9 and ΔE(TS(2hNA,3hNA)) = 16.3 kcal mol−1).3.9 Slide motion of the deprotonated form after the ring-rotation (2 → 4 → 5)
The steps of 2 → 4 → 5 correspond to the final slide motion in both pathways (IA) and (IB). Through this pathway, state 2 with the ring-rotated TPQsq undergoes two subsequent slide movements, S1 (2 → 4) and S2 (4 → 5), and eventually forms state 5 representing the on-copper conformation (Fig. 4). This pathway is highlighted in orange in Fig. 4. During the TPQsq slide occurring after the realization of state 2, O4-TPQsq approaches the Cu(I), as evidenced by a significant reduction of the distance from 6.3 Å to 2.1 Å in the on-copper state of 5. A stable intermediate state 4 is present before 5 (d(O4, Cu) = 4.1 Å). A H-bond of 2.5 Å is formed between O4-TPQsq and Tyr284 in state 4 (see Table S2†), but is absent in 5 (Fig. 6). The relative energy of state 4, ΔE(4) = 3.5 kcal mol−1, indicates higher stability with respect to the previous states 1–3. On-copper state 5 is the most stable state (ΔE(5) = 0 kcal mol−1) in the deprotonated form (Fig. 6 and Table S2†). Along the reaction path obtained from an NEB calculation, we could not locate any additional transient state for the slide motion S1 (2 → 4), while a TS(4,5) of 10.0 kcal mol−1 (Fig. 6) has to be overcome to perform the slide motion S2 (4 → 5). In the transition state (TS(4,5)), TPQsq is located in the proximity of His431 and characterized by distances of 3.4 Å between O4-TPQsq and Cε-His431 and 3.4 Å between C4-TPQsq and Cε-His431.In on-copper state 5, the amino group of TPQsq is at typical H-bond distances from the main chain carbonyl of Thr403 (N5-TPQsq, O-Thr403: 3.3 Å) and the Sδ atom of Met602 (N5-TPQsq, Sδ-Met602: 3.3 Å) (Table S2†). The 2-OH group of TPQsq interacts with the carboxamide group of Asn381 via two H-bonds, specifically O2-TPQsq with Nδ-Asn381 (3.0 Å), and O2-TPQsq with Oδ-Asn381 (2.96 Å). The short H-bond between Tyr284 and TPQsq changes from a direct interaction to an indirect H-bond mediated by the bridging water molecule W1 at a distance of 2.9 Å from O4-TPQsq in 5. These H-bond interactions are also preserved in the X-ray crystal structure (PDB ID: 3X3X) and contribute to stabilizing states 5 (WT) and 5NA (N381A mutant). We observed that the H-bond between O2-TPQsq and Asn381 is lost in 5NA, and as a result, the distance between O2-TPQsq and Cβ-Ala381 increases to 4.1 Å. The H-bond interaction between O2 of TPQsq and Asn381 in the WT can be formed only in state 5. Similar to the WT, we did not find any energy barrier for the TPQsq slide motion S1 (2NA → 4NA) in the N381A mutant, while a transition state energetically located at 11.1 kcal mol−1 characterizes the slide motion S2 from 4NA to 5NA (Fig. 6). As with the WT, the on-copper state in the N381A mutant is most stable in TPQsq (ΔE(5NA) = −0.4 kcal mol−1) compared to other deprotonated states such as 4NA (ΔE(4NA) = 0.7 kcal mol−1) and 3NA (ΔE(3NA) = 2.2 kcal mol−1). From these results, we can infer that in both the WT and the N381A mutant, pathway 2 → 4 → 5 is favoured and represents a viable reaction channel (Fig. 4 and 6). A feature worthy of note is that TPQsq of 5NA is less stable than TPQamr of 3hNA in the N381A mutant due to the stabilization of 3hNA as described in Section 3.6. These relative energies show that the energetic driving force required to generate TPQsq from TPQamr is reduced by ΔΔE(5NA, 3hNA) − ΔΔE(5, 3h) = 7.1 kcal mol−1 in the N381A mutant.
3.10 Slide motion of TPQsq after the deprotonation in the 1 → 6 pathway
This TPQsq slide motion (1 → 6) (Fig. 4) is the first conformational change occurring along pathway (IIIA). The energy profiles are shown in Fig. 7. During the TPQsq slide, the H-bond between TPQsq and Tyr284 is preserved. In state 6, after the slide motion, two H-bonds are formed, one of 2.5 Å between O4-TPQsq and O-Tyr284, and the second one of 2.8 Å between N5-TPQsq and O-Tyr284 (6 in Fig. 7 and Table S2†). The H-bond between the carboxy group of Asp298 and the amino group of TPQsq formed in 1 is lost in 6 (3.6 and 6.4 Å in 1 and 6, respectively). The relative energy of the transition state is ΔE(TS(1,6)) = 19.8 kcal mol−1, and the slide state 6 is more destabilized compared to state 1 (ΔE(6) = 12.9 kcal mol−1) (Fig. 7).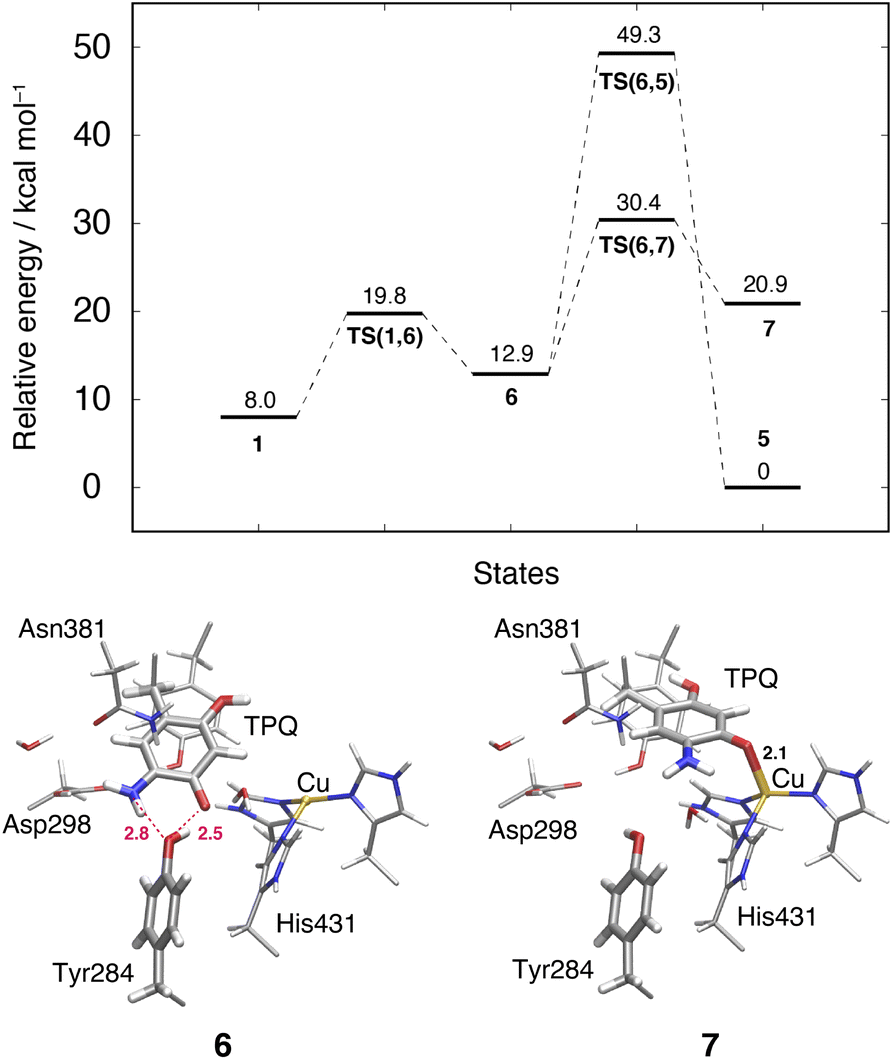 | ||
| Fig. 7 Relative energies of alternative TPQ conformational changes in the deprotonated state, including the TPQ slide motion (1 → 6 → 7) and the rotation ( → 5). The conformations of the intermediate states are reported along with the most relevant distances between heavy atoms. | ||
3.11 Subsequent TPQsq ring-rotation in the deprotonated form (6 → 5)
The ring-rotation of TPQsq (6 → 5) after the slide motion (1 → 6) is the subsequent conformational change along pathway (IIIA), whose energy profile is shown in Fig. 7. State 6 of TPQsq converts to 5 upon a rotation of the TPQsq ring around the Cβ–Cγ bond, corresponding to a change from 101.2° to −72.5° in terms of the dihedral angle Cα–Cβ–Cγ–Cδ2, thus bringing TPQsq closer not only to the side chains of Asn381 and Tyr384, but also to the main chains of Val406 and Phe407 and the side chain of His433. Compared to the atoms in the side chains, those in the main chain are more rigid and less likely to make room for the ring-rotation of the bulky TPQsq. In fact, we could observe that no H-bonds are formed between TPQsq and Asn381 during the 6 → 5 transition, and TPQsq needs to be close to the side chains of Tyr384 (O-Tyr384, O2-TPQsq; 2.6 Å) and Val406 (Cα-Val406, N5-TPQsq; 3.1 Å) at the transition state TS(6,5). The motion of His433 also induces dissociation of the coordinated imidazole side chain from the Cu(I). Therefore, the energy of the transition state for the TPQsq ring-rotation becomes rather high, ΔE(TS(6,5)) = 49.3 kcal mol−1 (Fig. 7). Due to this large barrier, the reaction channel 6 → 5 is not easily realizable.3.12 TPQ additional slide in the deprotonated form (6 → 7)
An additional slide motion of TPQsq can take place, bringing the system from state 6 to 7, in which the TPQ ring has the on-copper but ring-rotated conformation (Fig. 4 and 7). The outcome of our calculations indicated that state 7 is energetically unstable, with ΔE(7) = 20.9 kcal mol−1 (Fig. 7). From a structural standpoint, in 7, the O4-TPQsq can coordinate to the Cu(I) with a distance of 2.1 Å. Yet, TPQsq forms new H-bonds with the main chain of Asp383 and the phenol group of Tyr284, more precisely, N-Asp383, O2-TPQsq (3.5 Å) and O-Tyr284, N5-TPQsq (3.2 Å). In the transition state, the O4 atom of TPQsq is located at a position suitable to overcome the phenol group of Tyr284 (O-Tyr284, O4-TPQsq equal to 2.5 Å). The relative energy of the transition state for this slide, corresponding to the second TPQsq-slide (S2) from 4 to 5, is ΔE(TS(6,7)) = 30.4 kcal mol−1. This value is not excessively high, but because of the higher energy of state 7 compared to state 5 (Fig. 7), step 6 → 7 does not seem to be a viable reaction channel in the AGAO catalytic cycle.Taken together, the results presented in Sections 3.11 and 3.12 indicate that there is no low barrier route in pathway (III) with slide and clockwise-ring-rotation steps. The relative energy of the transition state (TS(6,5)) for the TPQsq ring-rotation after the slide motion is too high for the reaction to proceed, and the high relative energy of state 7 that is defined as another on-copper form (Fig. 4 and 7) after the complete TPQsq slide indicates the intrinsic instability of the system. Furthermore, attempts to find the 7 → 5 NEB pathways showed these were very unstable and are not converged to a low-barrier pathway. We can then rule out pathway (III) for the conformational change process from off-copper TPQamr to on-copper TPQsq (1h → 5).
3.13 TPQox ring-rotation in the oxidized form
To inspect the TPQox rotation, we constructed the oxidized states in the WT and in the N381A mutant by replacing the TPQamr moiety with TPQox. The 180°-flipped conformation of TPQox was found stable in both the WT and N381A mutant. The two conformers a and b observed in the X-ray structures of N381Aholo were reproduced by our N381A mutant models (OXNA, OXTNA) within an RMSD tolerance of 0.577 and 0.610 Å, respectively (Table S4†). The role played by the residue Asn381 was investigated within our QM/MM approach and the resulting energy profiles and intermediate states for the WT and N381A mutant are shown in Fig. 8.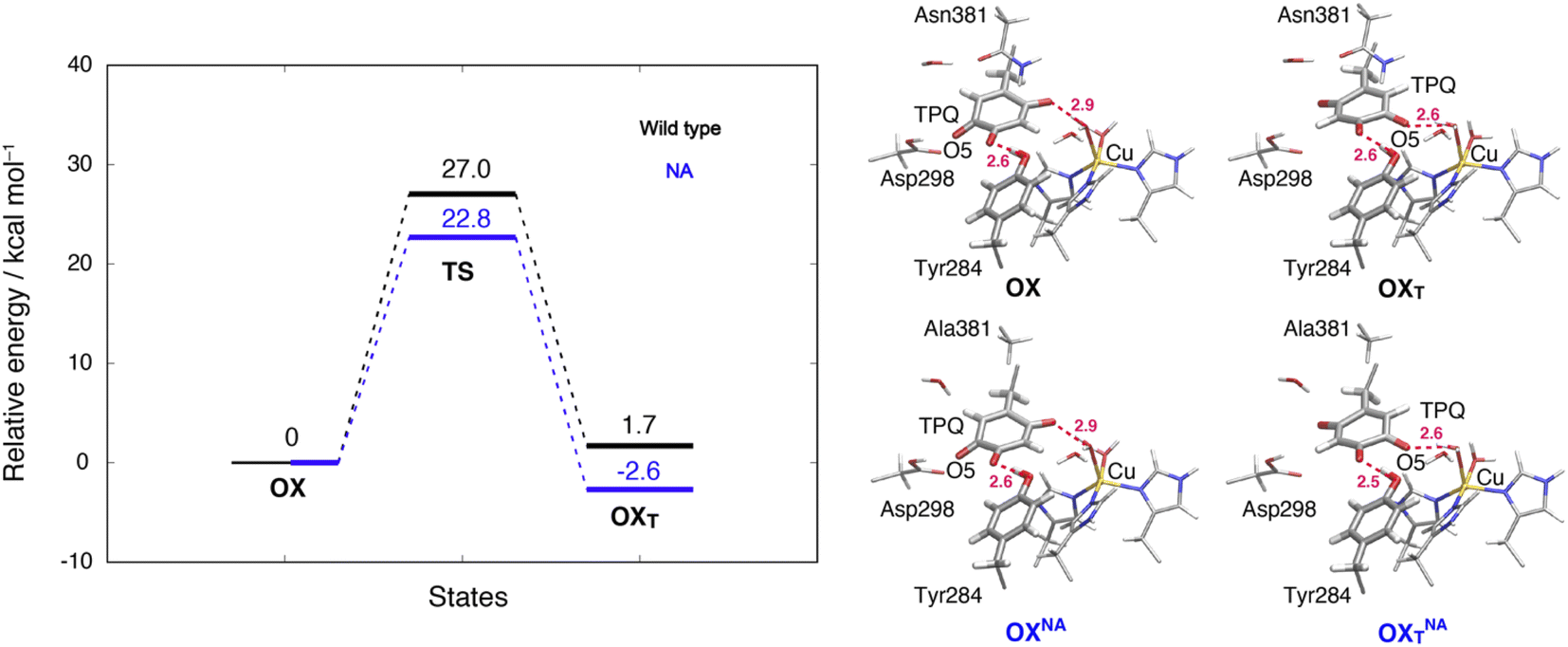 | ||
| Fig. 8 Relative energies and related conformations for TPQ rotation in the oxidized form. Results for the WT and the N381A mutant are shown in black and blue, respectively. Most relevant distances between the heavy atoms are explicitly indicated. | ||
We noticed that the ring-rotated state of TPQox (OXT) is slightly unstable with respect to the state in the unrotated conformation (ΔE(OXT) = 1.7 kcal mol−1) for the WT, while in the N381A mutant, OXTNA is more stable than OXNA by ΔE(OXTNA) = −2.6 kcal mol−1. The energy barrier of the N381A mutant for the TPQox ring rotation (ΔE(OXTNA) = 22.8 kcal mol−1) is lower by 4.2 kcal mol−1 than that of the WT, suggesting that the process can occur at room temperature. These features are consistent with the X-ray crystal structure of N381Aholo where the rotated conformation of TPQox could be observed only for the N381A mutant. The H-bond between O4-TPQox and the side-chain OH group belonging to Tyr284 is preserved during the TPQ rotation, with length variations in the range 2.5–2.7 Å in the N381A mutant (see Table S2†). In a way analogous to TPQamr, the TPQox group is stabilized upon TPQox rotation by forming an H-bond of 2.8 Å with the carboxamide group of Asn381 (O5-TPQox, Nδ-Asn381) in the TS(OX,OXT) of the WT (Table S2†). During TPQox rotation, the methyl group of Ala381 retains its position in the N381A mutant, indicating that steric hindrance between TPQ and Asn381 is significantly reduced because of the small size of the side chain. From these results, we can infer that the rotated conformation of TPQ (OXTNA) is more favourable in the N381A mutant than in the WT. Thus, the residue Asn381 contributes to the stabilization of the unrotated TPQox conformation and limits the unproductive rotation of the TPQox plane in the oxidized form, whereas the TPQ ring rotation and sliding during TPQsq formation are not hindered or suppressed by the side chain of Asn381. Furthermore, Asn381 contributes to preventing an overstabilization of the rotated intermediate states in TPQamr such as 3hNA.
3.14 Comparative experimental and QM/MM analyses of TPQamr in the N381A mutant and WT
The X-ray crystal structure of N381Aholo/PEA provided evidence for the unusually flipped conformation of TPQamr, with respect to the off-copper structure of the WT,14 as shown in Fig. 3B. The energy profile in Fig. 5 showed that 3hNA is more stable than 1hNA and 2hNA, and that the highest barrier connecting them is 16.3 kcal mol−1, which is sufficiently low to be overcome at room temperature. The stable intermediate of TPQamr switches from 1hNA to 3hNA in the N381A mutant, contrary to what has been observed in the protonated TPQamr of the WT, where the relative energy of 1h is significantly lower than that of 2h and 3h (Fig. 5). Among the TPQsq states in Fig. 6, product TPQsq states (5, 5NA) are most stable, but 3hNA is more stable than 5NA. These energy profiles in Fig. 5 and 6 are qualitatively consistent with the fact that only the TPQamr state could be observed in the N381A mutant by using UV-vis spectra and X-ray crystallography.3.15 Role of Asn381
In principle, the rather high degree of rigidity of the carboxamide group of Asn381 would jeopardize the rotation of TPQamr. Nonetheless, we could show that this is not necessarily true. In fact, the Asn381 carboxamide group can be displaced and give room to TPQamr to rotate in the WT, since the energy contributions of the group to the rotation barriers do not exceed 10 kcal mol−1, as assessed by the decrease in the TS energy levels by the N381 mutation (Fig. 5). The energy profile of the N381A mutant indicates that the ring-rotated state 3hNA of TPQamr is more stable than the non-ring-rotated one 1hNA (ΔE = −1.8 kcal mol−1, Fig. 5). This indicates that the intermediate state during the catalytic reaction of the N381A mutant is trapped at TPQamr (3hNA) suppressing the accumulation of TPQsq during the reductive half-reaction. The net effect is that Asn381 destabilizes the nonproductive TPQ ring-rotated conformations of both TPQox and TPQamr states, thus promoting only the desired catalytic reaction. We can also remark that the ring-rotation of TPQ in the off-copper conformation is allowed by Asn381.3.16 Role of TPQsq in the AGAO catalytic cycle
For AGAO, a fast oxidation of Cu(I)-TPQsqvia a direct reaction with O2 was detected from kinetics analysis of the transient absorption spectra.14,27 Nonetheless, to react with O2, a conformational change from the on-copper to an off-copper of TPQsq was required.25 The computed energy profile in the deprotonated form (Fig. 6) suggests that the on-copper conformation (5) is easily converted into a transient off-copper conformation (4) in TPQsq, and a direct coordination of O2 to the Cu(I) axial position becomes possible in the oxidative half-reaction. The kinetics of TPQsq oxidation in AGAO could not be explained in terms of an outer-sphere mechanism,14,27 in which TPQamr reacts with O2 without forming TPQsq and Cu(I). All these results support the notion that TPQsq with a large conformational change and Cu(II) are exploited in AGAO.Conversely, for BSAO, HPAO-1 and HPAO-2, no TPQ conformational change has been evidenced and TPQsq is not formed during the catalytic cycle. The catalytic rates of Co(II)-substituted CAOs are similar to the wild-type copper containing one for these CAOs,51–53 while AGAO, PSAO and ECAO significantly reduce their catalytic activities to 2.2%, 4.7% and 12%, respectively, upon Co(II)-substitution.12,54,55 These results suggest that the former CAOs undergo oxidative half-reactions via an outer-sphere mechanism, which is different from the inner-sphere mechanism expected in the latter CAOs.
3.17 Deprotonation of TPQamr in pathway (I)
The deprotonation process that could occur in pathway (I) remains elusive in our QM/MM-based simulations. To encompass the deprotonation issue, we can resort to experimental data that is provided by the pH dependency of the equilibrium between TPQamr and TPQsq. When AGAO is anaerobically reduced by high-affinity substrates such as 2-PEA, the pH dependency shows that two ionizable groups characterized by pKa1 = 5.96 and pKa2 = 7.74 are involved in the equilibrium shift.14 The pKa1 and pKa2 are ascribed to 5-NH2 of TPQamr and O4-TPQsq,14 on the basis of the pKas of model compounds, 5.88 (ref. 50) and 6.39 (ref. 56), respectively, although ambiguity still remains for the difference between the experimental pKa value of O4-TPQamr and that of the model compound (9.59).14The 5-NH2 group and O2-TPQamr interact with the side-chain carboxyl group of Asp298 and the Cu(II)-coordinated Wax, respectively, in the off-copper conformation (1h) (Table S2†). The ring-rotation to convert 1h to 2h changes the environments of both 5- and 2-groups simultaneously: The 5-NH2 group interacts with Wax, Tyr284, and Asn381 through hydrogen bonds, whereas the O2-TPQamr lacks a hydrogen bond and comes closer to the Asp298 side chain (Table S2†). Thus, the electrostatic environments of TPQamr in 2h would significantly perturb the electronic state of TPQamr. It presumably results in the decrease of the pKa of O4-TPQamr, although the O4-TPQamr forms a hydrogen bond with Tyr284 (Table S2†). We predicted that the pKa of O4-TPQamr in the off-copper conformation (1h) is similar to that indicated by the TPQamr model compound (pKa = 9.59)50 and that after the ring-rotation, the pKa of O4-TPQamr (2h) reduces to the experimentally determined value (pKa = 7.74), which is comparable to that of the O4-TPQsq (pKa = 6.39) found in the TPQsq model compound56 as described previously.14 The present QM/MM calculation also reveals that the ring-rotation promotes the deprotonation of O4-TPQamr. We can infer that the deprotonation of 2h (2h → 2) is more favourable by 4.4 kcal mol−1 in comparison with that of 1h (1h → 1) (Fig. 5 and 6).
For state 3h, it is expected that its deprotonation to 3 is preferable in energy if the conformational change of the −S movement from 2h to 3h becomes a lower energy barrier process. The present energy barrier (26.0 kcal mol−1) of 2h → 3h suggests an unfavourable pathway (Fig. 5, see Section 3.6). However, the deprotonation of 3h (3h → 3) is more favoured by 7.9 kcal mol−1 than that of 1h (1h → 1) (Fig. 5 and 6). The formation of 2h represents the most direct pathway. However, the alternative and more indirect pathway, visiting also the conformations 3h and 3 is still viable along the route of 1h → 2h → 3h → 3 → 2 → 4 → 5 (Fig. 4), although this reaction channel is characterized by a slower kinetics in the 2h → 3h transition. These energy profiles suggest a possibility that the pathway via3h may be utilized with different substrates or in other CAOs.
The assignment of pKa2 = 7.74 to O4-TPQsq provides an important basis for discussing the overall reaction pathway. States 1h and 5, which are the most stable configurations in TPQamr and TPQsq, respectively, are the main species detected at pH values around 7.74 ref. 14. Thus, at pH 7.74, we can directly compare the energy profiles of TPQ O4-protonated states (Fig. 5) and TPQ O4-unprotonated states (Fig. 6 and 7), by adjusting the relative energies of 1h and 5, since 1h and 5 + H+ have the same chemical potential at this pH. At other pH values, a similar comparison can be done by offsetting Fig. 6 and 7 upward by 1.36 (7.74 − pH) kcal mol−1 relative to Fig. 5. Based on the profiles overlaid in this way we can deduce the plausible overall reaction pathway in the WT as described in Section 4.
4 Conclusions
The possible reaction pathways characterized by the TPQ conformational change during the TPQamr to TPQsq transition were investigated for the AGAO system providing an atomistic insight into the detailed process and a thorough analysis of the associated energetics. From the molecular structures of the surrounding residues and TPQamr, and deprotonation time of TPQamr, we have narrowed down the most viable reaction pathways into three, all of them including the clockwise-ring-rotation of TPQ, i.e., (IA) 1h → 1 → 2 → 4 → 5, (IB) 1h → 2h → 2 → 4 → 5, and (IIIA) 1h → 1 → 6 → 5.The first and third pathways (IA and IIIA) have higher energy barriers of more than 30 kcal mol−1 for the 1 → 2 and 6 → 5 transitions. Therefore, the most favourable pathway is (IB): the TPQamr → TPQsq reaction in the WT AGAO proceeds through the TPQamr ring-rotation, deprotonation of O4H-TPQamr, and TPQsq slide. This reaction mechanism is summarized in Fig. 9. The reaction pathway via3h (1h → 2h → 3h → 3 → 2 → 4 → 5) can be regarded as a more indirect route in the WT AGAO compared to the direct pathway (IB), and this pathway via3h is not preferable, at least under the condition of the present theoretical model. In the N381A mutant AGAO, the main reaction pathway is the indirect route via3hNA: 1hNA → 2hNA (⇄ 3hNA) → 2NA → 4NA → 5NA. The most stable state is 3hNA and the highest energy barrier along this pathway is the 3hNA → 2hNA step with an energy barrier of 18.1 kcal mol−1 (ΔE(2hNA, 3hNA) − ΔE(3hNA)). The actual TPQamr ring-rotation state (3hNA) was determined from the X-ray crystal structure of the N381A mutant.
 | ||
| Fig. 9 Reaction mechanism of the TPQamr → TPQsq transition obtained in the present study. | ||
The large conformational change of the TPQamr ring in the WT AGAO can only be permitted before TPQamr deprotonation, and this conformational change is essential for stable formation of TPQsq in AGAO. On the other hand, for TPQox, the TPQ ring flip partially occurs in the N381A mutant, but it is absent in the WT. The TPQ ring-flipped conformation in TPQox (OXTNA) and the related energetics are determined by a synergy of the X-ray crystal structure and QM/MM calculation for the N381A mutant.
This seems reasonable because the unusually large conformational change taking place in the TPQ ring-rotation and sliding motions in TPQox would reduce the probability of the nucleophilic attack of the substrate amines toward the O5 carbonyl of TPQox being directed to the substrate-binding pocket. This provides a clear picture of the ingenious role played by Asn381 in directing the reaction pathway starting either from TPQox or from TPQamr.
Pathway (IB) is also consistent with the results of transient kinetics experiments of the AGAO catalytic reaction.14 Our previous study has demonstrated that the rate constant of the TPQamr → TPQsq step (k+4 = 39 s−1 at 4 °C) is much lower than the large value that is predicted for electron transfer from a donor to an acceptor apart from a short distance (the distances from O4 and O2-TPQamr to the Cu centre = 6.7 and 4.8 Å, respectively, in AGAO and PDB ID: 3X3Z). In fact, temperature-jump relaxation studies have shown that the rate constant of the electron transfer (kET) is 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s−1 for PSAO.57 In addition, the dependence of k±4 on solvent viscosity in AGAO suggested the presence of a large conformational change.14 Therefore, we have predicted that the electron transfer is gated by the conformational change. The present study clearly showed that the TPQ ring-rotation at the off-copper position, followed by TPQ deprotonation, is a relevant change for electron transfer from TPQamr to TPQsq. Interestingly, the active site of PSAO9 contains two dissociable residues Lys296 and Glu412 located close to TPQ, and the former is hydrogen-bonded with O4-TPQ (Fig. S11†). These dissociable side chains may significantly reduce the pKa of O4-TPQamr in the off-copper position, facilitating deprotonation. Furthermore, in PSAO, a less bulky residue, Asn389, is located under the TPQ ring at the position corresponding to Tyr384 that restricts the ring-rotation in AGAO, while the highly conserved Asn residue (corresponding to Asn381 in AGAO), Asn386, is located on the TPQ ring (Fig. S11†). This difference is expected to enable TPQ ring-rotation through a low energy barrier. As a result, PSAO mainly has the ring-rotated conformation of TPQox in the X-ray crystal structure9 (Fig. S11†). Despite the unreactive conformation, the distinct features of the active-site structure may facilitate the deprotonation of O4-TPQamr in PSAO, resulting in very fast electron transfer and efficient TPQsq formation.
000 s−1 for PSAO.57 In addition, the dependence of k±4 on solvent viscosity in AGAO suggested the presence of a large conformational change.14 Therefore, we have predicted that the electron transfer is gated by the conformational change. The present study clearly showed that the TPQ ring-rotation at the off-copper position, followed by TPQ deprotonation, is a relevant change for electron transfer from TPQamr to TPQsq. Interestingly, the active site of PSAO9 contains two dissociable residues Lys296 and Glu412 located close to TPQ, and the former is hydrogen-bonded with O4-TPQ (Fig. S11†). These dissociable side chains may significantly reduce the pKa of O4-TPQamr in the off-copper position, facilitating deprotonation. Furthermore, in PSAO, a less bulky residue, Asn389, is located under the TPQ ring at the position corresponding to Tyr384 that restricts the ring-rotation in AGAO, while the highly conserved Asn residue (corresponding to Asn381 in AGAO), Asn386, is located on the TPQ ring (Fig. S11†). This difference is expected to enable TPQ ring-rotation through a low energy barrier. As a result, PSAO mainly has the ring-rotated conformation of TPQox in the X-ray crystal structure9 (Fig. S11†). Despite the unreactive conformation, the distinct features of the active-site structure may facilitate the deprotonation of O4-TPQamr in PSAO, resulting in very fast electron transfer and efficient TPQsq formation.
The reaction mechanism of AGAO elucidated here implies that the amino acid residues in the active site, Asn381 and Tyr384, as well as the TPQ cofactor are remarkably dynamical and flexible. Nonetheless, they can reorient and rearrange depending on the multistep reaction channel and play a major role in promoting the catalytic reaction. Besides AGAO, other enzymes such as dihydrofolate reductase, flavin-dependent N-hydroxylase, cytochrome c oxidase, and electron transfer flavoprotein undergo large conformational changes during their own catalytic reactions.3,58–60 Their structural changes are utilized in their specific reaction steps to stabilize intermediates, to eject the spent NADP+, and to trigger proton transfer and rapid electron transfer. From a general standpoint, important conformational changes are a general paradigm rather ubiquitous for realizing efficient biological functions.
We stress the fact that the present study aims at providing insights into the reaction pathways of TPQsq formation in AGAO, along with accurate evaluations of their energy profiles. This paves the route to forthcoming studies of the structural flexibilities for the whole AGAO. We are confident that the present work will stimulate additional studies exploiting the joint use of molecular dynamics simulations and experimental reaction kinetics.
Data availability
Crystallographic data of the N381A AGAO have been deposited in the Protein Data Bank under IDs 7WIR (holo, TPQoxN381A) and 7WIS (substrate-reduced, TPQamrN381A). The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
M. S., T. M., M. B., Y. S., H. H., and T. O. participated in research design. M. S. conducted the theoretical calculations, and T. M. and T. O. conducted the experiments. All authors contributed to perform the data analysis and to write the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by (i) JST-PRESTO, Japan (grant number JPMJPR19G6), (ii) JSPS KAKENHI (grant numbers: 16KT0055, 19K05694, 20H05088, 20H05448, 20H05453, and 22H04916), (iii) the Cooperative Research Program of the “Network Joint Research Center for Materials and Devices”, and (iv) the MEXT Quantum Leap Flagship Program (MEXT Q-LEAP) (grant number JPMXS0120330644). X-ray diffraction data of the protein crystals were obtained using synchrotron beamline station BL44XU at SPring-8, under the Cooperative Research Program of the Institute for Protein Research, Osaka University (Proposal No.: 2006A6809, 2006B6809, 2007A6904, and 2007B6904). Numerical calculations were carried out under the support of the (i) Multidisciplinary Cooperative Research Program in CCS, University of Tsukuba and (ii) HPCI system research project (project ID: hp210115) using the supercomputer Fugaku provided by RIKEN. M. B. thanks the HPC Mesocenter at the University of Strasbourg funded by the Equipex Equip@Meso project and the CPER Alsacalcul/Big Data, and the Grand Equipement National de Calcul Intensif (GENCI) under allocation DARI-A0100906092.References
- R. N. Perham, Annu. Rev. Biochem., 2000, 69, 961–1004 CrossRef CAS PubMed.
- G. G. Hammes, Biochemistry, 2002, 41, 8221–8228 CrossRef CAS PubMed.
- A. Ramanathan, A. Savol, V. Burger, C. S. Chennubhotla and P. K. Agarwal, Acc. Chem. Res., 2014, 47, 149–156 CrossRef CAS PubMed.
- D. E. Koshland Jr, Nat. Med., 1998, 4, 1112–1114 CrossRef CAS PubMed.
- M. J. McPherson, M. R. Parsons and C. M. Wilmot, Handbook of metalloproteins, John Wiley & Sons, Ltd, 2001, vol 2, 1245–1257 Search PubMed.
- D. L. Wertz and J. P. Klinman, Handbook of metalloproteins, John Wiley & Sons, Ltd, 2001, vol 2, pp. 1258–1271 Search PubMed.
- T. Okajima and K. Tanizawa, Mechanism of TPQ Biogenesis in Prokaryotic Copper Amine Oxidase, CRC Press, Boca Raton, FL, 2009, 103–118 Search PubMed.
- M. R. Parsons, M. A. Convery, C. M. Wilmot, K. D. S. Yadav, V. Blakeley, A. S. Corner, S. E. V. Phillips, M. J. McPherson and P. F. Knowles, Structure, 1995, 3, 1171–1184 CrossRef CAS PubMed.
- V. Kumar, D. M. Dooley, H. C. Freeman, J. M. Guss, I. Harvey, M. A. McGuirl, M. C. J. Wilce and V. M. Zubak, Structure, 1996, 4, 943–955 CrossRef CAS PubMed.
- M. C. J. Wilce, D. M. Dooley, H. C. Freeman, J. M. Guss, H. Matsunami, W. S. McIntire, C. E. Ruggiero, K. Tanizawa and H. Yamaguchi, Biochemistry, 1997, 36, 16116–16133 CrossRef CAS PubMed.
- S. M. Janes, D. Mu, D. Wemmer, A. J. Smith, S. Kaur, D. Maltby, A. L. Burlingame and J. P. Klinman, Science, 1990, 248, 981–987 CrossRef CAS PubMed.
- S. Kishishita, T. Okajima, M. Kim, H. Yamaguchi, S. Hirota, S. Suzuki, S. Kuroda, K. Tanizawa and M. Mure, J. Am. Chem. Soc., 2003, 125, 1041–1055 CrossRef CAS PubMed.
- R. Matsuzaki, T. Fukui, H. Sato, Y. Ozaki and K. Tanizawa, FEBS Lett., 1994, 351, 360–364 CrossRef CAS PubMed.
- T. Murakawa, A. Hamaguchi, S. Nakanishi, M. Kataoka, T. Nakai, Y. Kawano, H. Yamaguchi, H. Hayashi, K. Tanizawa and T. Okajima, J. Biol. Chem., 2015, 290, 23094–23109 CrossRef CAS.
- T. Murakawa, S. Baba, Y. Kawano, H. Hayashi, T. Yano, T. Kumasaka, M. Yamamoto, K. Tanizawa and T. Okajima, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 135–140 CrossRef CAS.
- T. Murakawa, T. Okajima, S. Kuroda, M. Taki, Y. Yamamoto, H. Hayashi and K. Tanizawa, Biochem. Biophys. Res. Commun., 2006, 342, 414–423 CrossRef CAS PubMed.
- Y. C. Chiu, T. Okajima, T. Murakawa, M. Uchida, M. Taki, S. Hirota, M. Kim, H. Yamaguchi, Y. Kawano, N. Kamiya, S. Kuroda, H. Hayashi, Y. Yamamoto and K. Tanizawa, Biochemistry, 2006, 45, 4105–4120 CrossRef CAS PubMed.
- B. Schwartz, E. L. Green, J. Sanders-Loehr and J. P. Klinman, Biochemistry, 1998, 37, 16591–16600 CrossRef CAS PubMed.
- T. Murakawa, H. Hayashi, T. Sunami, K. Kurihara, T. Tamada, R. Kuroki, M. Suzuki, K. Tanizawa and T. Okajima, Acta Crystallogr. D, 2013, 69, 2483–2494 CrossRef CAS PubMed.
- H. Matsunami, T. Okajima, S. Hirota, H. Yamaguchi, H. Hori, S. Kuroda and K. Tanizawa, Biochemistry, 2004, 43, 2178–2187 CrossRef CAS PubMed.
- R. H. Moore, M. A. Spies, M. B. Culpepper, T. Murakawa, S. Hirota, T. Okajima, K. Tanizawa and M. Mure, J. Am. Chem. Soc., 2007, 129, 11524–11534 CrossRef CAS PubMed.
- L. Rongbao, J. P. Klinman and F. S. Mathews, Structure, 1998, 6, 293–307 CrossRef.
- M. Mure, Acc. Chem. Res., 2004, 37, 131–139 CrossRef CAS PubMed.
- J. Finney, H.-J. Moon, T. Ronnebaum, M. Lantz and M. Mure, Arch. Biochem. Biophys., 2014, 546, 19–32 CrossRef CAS PubMed.
- B. J. Johnson, E. T. Yukl, V. J. Klema, J. P. Klinman and C. M. Wilmot, J. Biochem., 2013, 288, 28409–28417 CAS.
- A. Mukherjee, V. V. Smirnov, M. P. Lanci, D. E. Brown, E. M. Shepard, D. M. Dooley and J. P. Roth, J. Am. Chem. Sci., 2008, 130, 9459–9473 CrossRef CAS.
- E. M. Shepard, K. M. Okonski and D. M. Dooley, Biochemistry, 2008, 47, 13907–13920 CrossRef CAS.
- V. J. Klema and C. M. Wilmot, Int. J. Mol. Sci., 2012, 13, 5375–5405 CrossRef CAS PubMed.
- A. G. W. Leslie, Joint CCP4 EESF-EACMB Newsletter on Protein Crystallography, SERC Daresbury Laboratory, 1992 Search PubMed.
- M. D. Winn, C. C. Ballard, K. D. Cowtan, E. J. Dodson, P. Emsley, P. R. Evans, R. M. Keegan, E. B. Krissinel, A. G. W. Leslie, A. McCoy, S. J. Mc-Nicholas, G. N. Murshudov, N. S. Pannu, E. A. Potterton, H. R. Powell, R. J. Read, A. Vagin and K. S. Wilson, Acta Crystallogr. D, 2011, 67, 235–242 CrossRef CAS PubMed.
- A. J. McCoy, R. W. Grosse-Kunstleve, P. D. Adams, M. D. Winn, L. C. Storoni and R. J. Read, J. Appl. Crystallogr., 2007, 40, 658–674 CrossRef CAS PubMed.
- P. D. Adams, P. V. Afonine, G. Bunkóczi, V. B. Chen, I. W. Davis, N. Echols, J. J. Headd, L.-W. Hung, G. J. Kapral, R. W. Grosse-Kunstleve, A. J. McCoy, N. W. Moriarty, R. Oeffner, R. J. Read, D. C. Richardson, J. S. Richardson, T. C. Terwilliger and P. H. Zwart, Acta Crystallogr. D, 2010, 66, 213–221 CrossRef CAS PubMed.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Acta Crystallogr. D, 2010, 66, 486–501 CrossRef CAS PubMed.
- V. B. Chen, W. B. Arendall, J. J. Headd, D. A. Keedy, R. M. Immormino, G. J. Kapral, L. W. Murray, J. S. Richardson and D. C. Richardson, Acta Crystallogr. D, 2010, 66, 12–21 CrossRef CAS PubMed.
- T. Murakawa, K. Kurihara, M. Shoji, C. Shibazaki, T. Sunami, T. Tamada, N. Yano, T. Yamada, K. Kusaka, M. Suzuki, Y. Shigeta, R. Kuroki, H. Hayashi, T. Yano, K. Tanizawa, M. Adachi and T. Okajima, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 10818–10824 CrossRef CAS.
- J. Wang, P. Cieplak and P. A. Kollman, J. Comput. Chem., 2000, 21, 1049–1074 CrossRef CAS.
- S. Grimme, S. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270 CrossRef CAS.
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299 CrossRef CAS.
- M. Shoji, T. Murakawa, M. Boero, Y. Shigeta, H. Hayashi and T. Okajima, RSC Adv., 2020, 10, 38631–38639 RSC.
- M. Shoji, Y. Abe, M. Boero, Y. Shigeta and Y. Nishiya, Phys. Chem. Chem. Phys., 2020, 22, 16552–16561 RSC.
- S. Yamasaki, M. Shoji, M. Kayanuma, Y. Sladek, D. K. Inaoka, Y. Matsuo, T. Shiba, L. Young, A. L. Moore, K. Kita and Y. Shigeta, Biochim. Biophys. Acta, Bioenerg., 2021, 1862, 148356–148359 CrossRef CAS PubMed.
- M. Shoji, N. Watanabe, Y. Hori, K. Furuya, M. Umemura, M. Boero and Y. Shigeta, Astrobiology, 2022, 22, 1129–1142 CrossRef CAS PubMed.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef CAS.
- R. Elber and M. Karplus, Chem. Phys. Lett., 1987, 139, 375–380 CrossRef CAS.
- M. Valiev, E. J. Bylaska, N. Govind, K. Kowaiski, T. P. Straatsma, H. J. J. van Dam, D. Wang, J. Nieplocha, E. Apra, T. L. Windus and W. A. de Jong, Comput. Phys. Commun., 2010, 181, 1477 CrossRef CAS.
- W. Humphery, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef.
- M. Mure and J. P. Klinman, J. Am. Chem. Soc., 1993, 115, 7117–7127 CrossRef CAS.
- A. Bellelli, L. Morpurgo, B. Mondovi and E. Agostinelli, Eur. J. Biochem., 2010, 267, 3264–3269 CrossRef.
- S. A. Mills, Y. Goto, Q. Su, J. Plastino and J. Klinman, Biochemistry, 2002, 41, 10577–10584 CrossRef CAS.
- C. M. Chang, V. J. Klema, B. J. Johnson, M. Mure, J. P. Klinman and C. M. Wilmot, Biochemistry, 2010, 49, 2540–2550 CrossRef CAS PubMed.
- S. A. Mills, D. E. Brown, K. Dang, D. Sommer, A. Bitsimis, J. Ngugen and D. M. Dooley, J. Biol. Inorg Chem., 2012, 17, 507–515 CrossRef CAS PubMed.
- M. A. Smith, P. Pirrat, A. R. Pearson, C. R. P. Kurtis, C. H. Trinh, T. G. Gaule, P. F. Knowles, S. E. V. Philips and M. J. McPherson, Biochemistry, 2010, 49, 1268–1280 CrossRef CAS.
- R. H. Bisby, S. A. Johnson, A. W. Parker and S. M. Tavender, Laser Chem., 1999, 19, 201–208 CrossRef CAS.
- P. N. Turowski, M. A. McGuirl and D. M. Dooley, J. Biol. Chem., 1992, 268, 17680–17682 CrossRef.
- J. W. Setser, J. R. Heemstra Jr, C. T. Walsh and C. L. Drennan, Biochemistry, 2014, 53, 6063–6077 CrossRef CAS.
- L. Qin, J. Liu, D. A. Mills, D. A. Proshlyakov, C. Hiser and S. Ferguson-Miller, Biochemistry, 2009, 48, 5121–5130 CrossRef CAS PubMed.
- H. S. Toogood, D. Leys and N. S. Scrutton, FEBS J., 2007, 274, 5481–5504 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Key atomic distances, details of the crystallographic refinement, molecular structures of the transition states, UV-vis spectra and cartesian coordinates of all the states are provided. See https://doi.org/10.1039/d2sc01356h |
| This journal is © The Royal Society of Chemistry 2022 |