
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExhaustive Baeyer–Villiger oxidation: a tailor-made post-polymerization modification to access challenging poly(vinyl acetate) copolymers†
Pengfei
Ma
 ab,
Christopher M.
Plummer
c,
Wenjun
Luo
ab,
Jiyan
Pang
a,
Yongming
Chen
db and
Le
Li
*ab
ab,
Christopher M.
Plummer
c,
Wenjun
Luo
ab,
Jiyan
Pang
a,
Yongming
Chen
db and
Le
Li
*ab
aSchool of Chemistry, Sun Yat-sen University, Guangzhou, 510275, P. R. China. E-mail: lile26@mail.sysu.edu.cn
bKey Laboratory for Polymeric Composite and Functional Materials of Ministry of Education, Sun Yat-sen University, Guangzhou 510275, P. R. China
cInternational Centre for Research on Innovative Biobased Materials (ICRI-BioM)—International Research Agenda, Lodz University of Technology, Zeromskiego 116, 90-924 Lodz, Poland
dSchool of Materials Science and Engineering, Sun Yat-sen University, Guangzhou 510275, P. R. China
First published on 6th September 2022
Abstract
The discovery of exhaustive (nearly quantitative) post-polymerization modifications (PPM) relies heavily on the efficiency of their corresponding small-molecule protocols. However, the direct translation of existing small-molecule protocols into PPM methods has never been guaranteed due to the intrinsic differences between small-molecule substrates and polymers. Herein, we introduce the direct optimization on polymers (DOP) as a complementary approach to developing exhaustive PPM reactions. As proof of the DOP concept, we present an exhaustive Baeyer–Villiger (BV) post-modification which cannot be accessed by conventional approaches. This user-friendly methodology provides general access to synthetically challenging copolymers of vinyl acetate and more activated monomers (MAMs) including both statistical and narrow-dispersed block copolymers. Furthermore, a scalable one-pot copolymerization/exhaustive BV post-modification procedure was developed to produce such materials showing improved performance over regular PVAc.
Introduction
Complementary to conventional polymerization approaches, post-polymerization modification (PPM) represents another appealing strategy for polymer synthesis.1 Polymer chemists continuously seek ideal PPM reactions where an exhaustive (nearly quantitative) functionalization can be achieved. The most common approach to access such transformations is the adaption of the corresponding small-molecule synthetic methodologies. The most notable examples include “click” reactions2 and multicomponent reactions.1c,3 However, the translation of small-molecule protocols to polymers is not straightforward. Indeed, the development of an exhaustive PPM reaction is even more technically challenging than that of its small-molecule prototype in many cases (Scheme 1A and Fig. S1†). An exhaustive PPM reaction requires both a high degree of mechanistic fidelity and sufficient functionalization rates to prevent the production of defects,4 chain scission or cross-linking,1b,5 since any side reaction will negatively alter the properties of the resultant polymers. Such stringent requirements greatly limit the reaction diversity6 of exhaustive PPM methods. An additional bottleneck for the development of PPM methods is the intrinsic difference between small-molecule substrates and polymers (e.g. reactivity, solubility, and packing/aggregation) which has been greatly overlooked (Scheme 1B). Indeed, the adaption of “mismatched” small-molecule protocols into PPMs was often fruitless. Therefore, we speculated that direct optimization on polymers (DOP) could constitute a new strategy and potentially accelerate the discovery of PPM reactions. In the DOP approach, polymer substrates will be used as model compounds for optimization, and the unique properties of polymers will be considered from the beginning of development.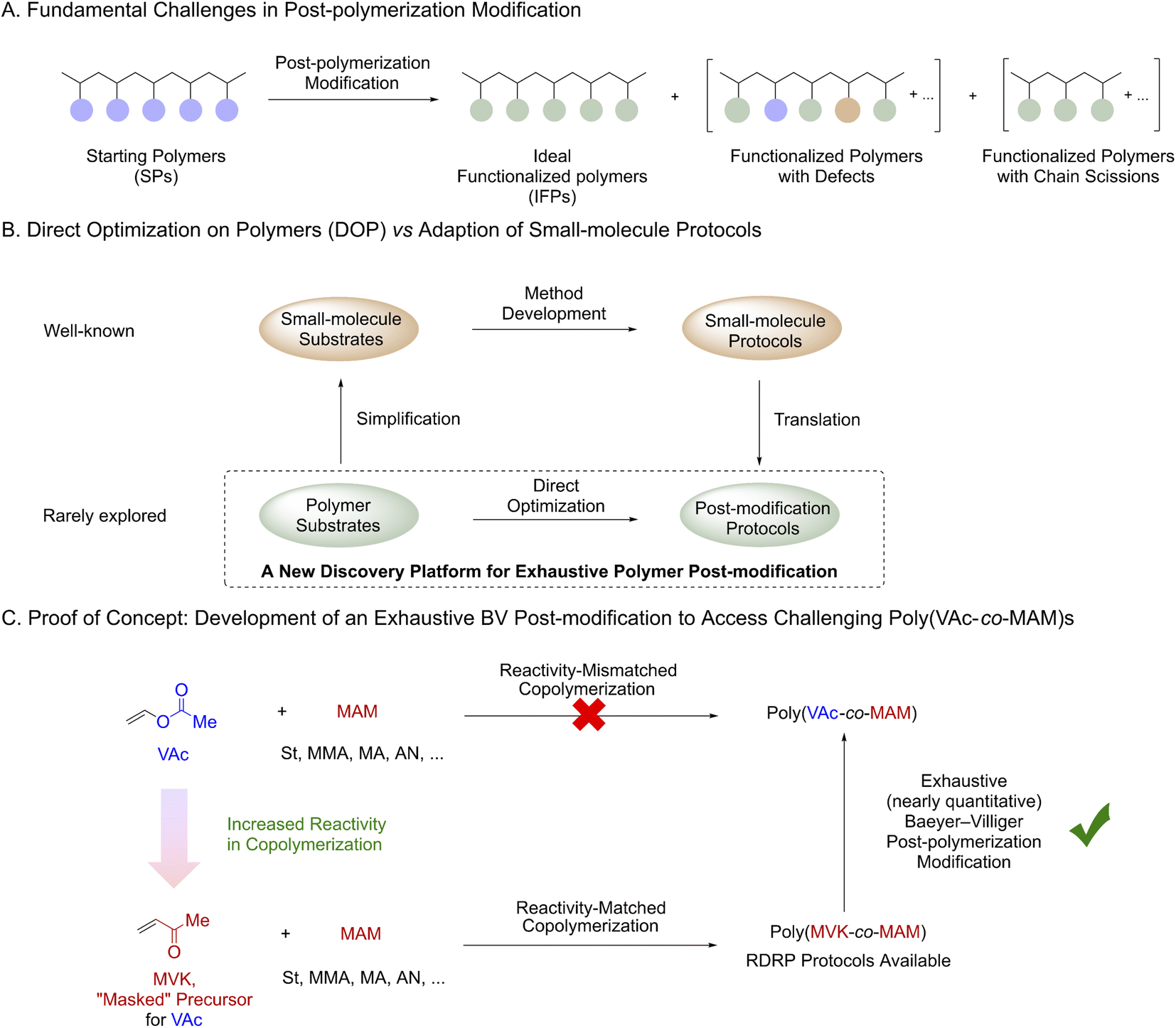 | ||
| Scheme 1 Accessing synthetically challenging poly(VAc-co-MAM)s with an exhaustive Baeyer–Villiger post-polymerization modification. | ||
To validate the effectiveness of the DOP approach, we aimed to develop an exhaustive Baeyer–Villiger (BV) post-modification of poly(methyl vinyl ketone) and its copolymers (Scheme 1C). Such a method can be applied to prepare challenging copolymers of vinyl acetate (VAc) and more activated monomers (MAMs),7 whose reactivities are mismatched in copolymerization.8 In our proposal, commercially available methyl vinyl ketone (MVK) would be used as a “masked-VAc” monomer since MVK readily copolymerizes with other MAMs8 and is also compatible with multiple reversible deactivation radical polymerization (RDRP) techniques.9
We were aware that significant challenges might occur during the development. Although small-molecule BV oxidation10 protocols were rich in literature, multiple side-reaction pathways such as aldol reaction/condensation,11 undesired oxidation12 and radical cleavage13 were known to be associated with BV oxidation. Such side reactions could introduce defects into the polymer backbone and negatively affect the molar mass and dispersity (Ð) of the polymers. In addition, uncertainty regarding the regioselectivity of the proposed BV post-modification must be considered since such selectivity for a polymer substrate has never been established. Apparently, the development of an exhaustive oxidative PPM reaction remains a challenging task. However, recent accomplishments14 in oxidative PPMs encouraged us to explore the proposed chemistry. For instance, Klausen14d and Ouchi14c individually developed highly efficient oxidative PPMs for their customized boron-containing polymers using organoboron oxidation chemistry.15 Herein, we report an exhaustive Baeyer–Villiger (BV) post-modification as the first example developed by the DOP approach. This BV post-modification provides a general method to prepare a wide range of synthetically challenging homopolymers, statistical, and block copolymers of VAc.
Results and discussion
Initial attempts using classical BV protocols
Pioneering efforts16 to bring BV oxidation into the field of PPM suffered from a lack of reactivity and/or significant chain scission. Conventional BV oxidation conditions10 appeared to be destructive and incompatible with ketone-containing polymers. To obtain more mechanistic details, we decided to re-investigate this transformation using PMVK as a substrate.17 We first evaluated the influence of temperature and reaction time using a commercial PMVK sample (Fig. 1 and Table S2†). The BV oxidations of PMVK were conducted using a typical protocol of chloroform as a solvent and m-chloroperoxybenzoic acid (mCPBA) as an oxidant. Although higher conversions were obtained with prolonged periods at elevated temperatures, the molar mass of the polymers decreased significantly. Noticeably, our kinetic studies revealed that the BV post-modification proceeded rapidly during the early stage, but became sluggish after 24 h. Consistent with the earlier investigations,16 our results also suggested that the standard BV oxidation conditions were not efficient enough to complete the proposed post-modification of PMVK. Other oxidants such as hydrogen peroxide and peracetic acid were also evaluated but inferior results were obtained.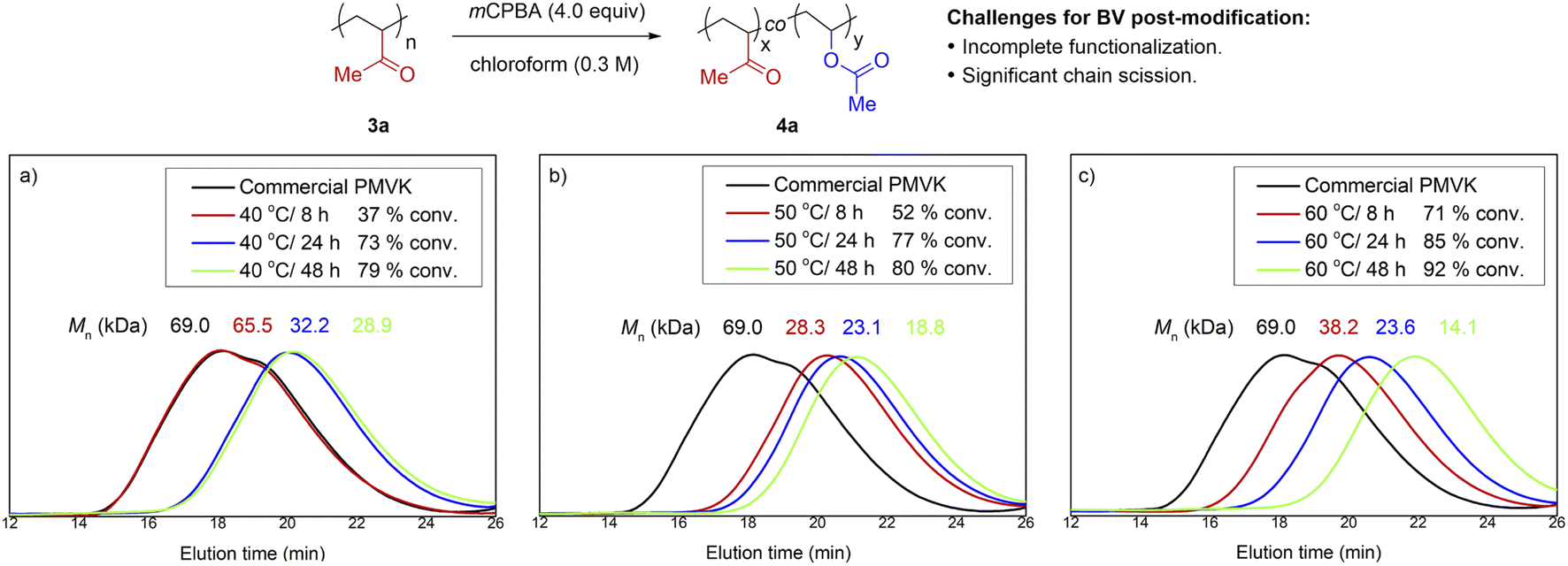 | ||
| Fig. 1 The influence of temperature and reaction time on the BV oxidation of PMVK in chloroform. SEC traces of commercial PMVK 3a before and after BV oxidation at (a) 40 °C, (b) 50 °C and (c) 60 °C with different reaction time. | ||
Unsuccessful attempts at the translation of small-molecule protocols to PMVK
A small-molecule model using 3-hexylundecan-2-one as a substrate was established at the outset of this study. A large number of high-yielding conditions from the literature were screened and modified (Table S1†). Unfortunately, the direct translation of small-molecule protocols to the post-modification of PMVK completely failed (Fig. 2 and Table S3†). The Mn of the polymers decreased significantly with the use of either acids or bases (Fig. 2a and b). In addition, the use of alternative oxidants such as peracetic acid (PAA), trifluoroperoxyacetic acid (TFPAA), and magnesium monoperoxyphthalate hexahydrate (MMPP) gave even worse results (Fig. 2c).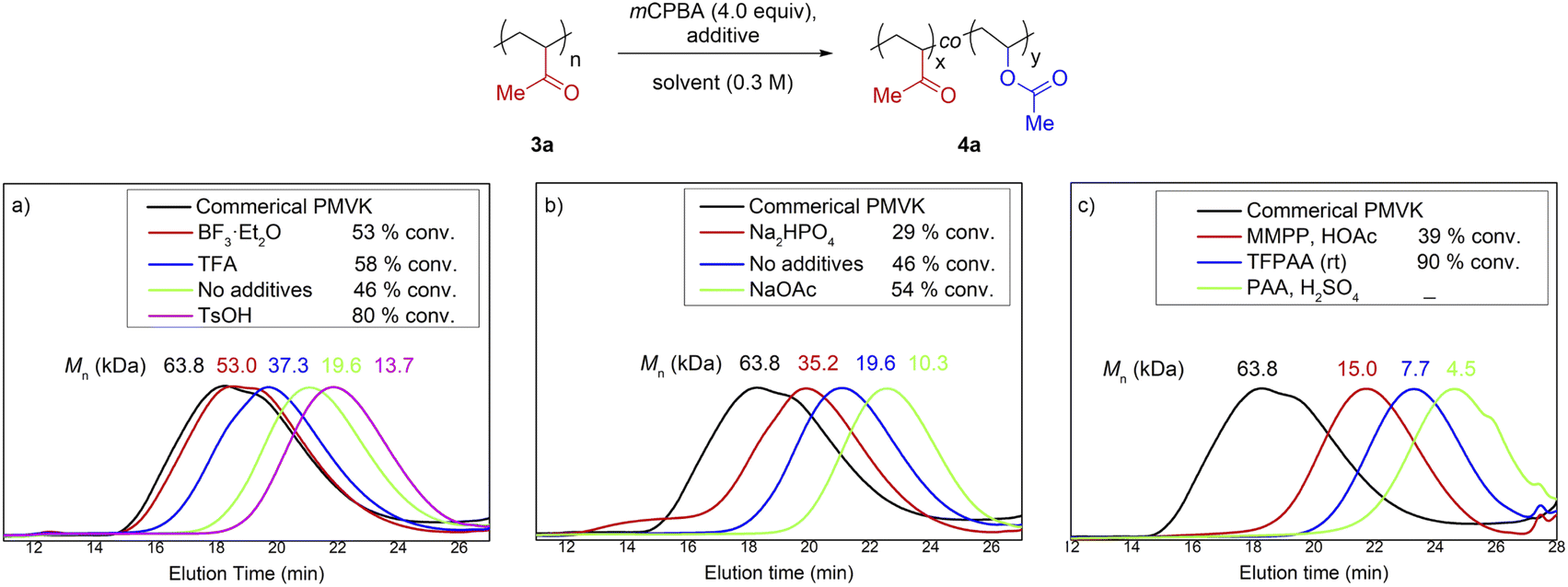 | ||
| Fig. 2 BV oxidation of PMVKs using high-yielding conditions optimized for the small-molecule model. SEC traces before and after BV oxidations: (a) acid catalyst, mCPBA as oxidant, (b) in the presence of inorganic base, mCPBA as oxidant, (c) MMPP and other peroxyacids as oxidant (see the detailed conditions in Table S3.†). | ||
The failure of the translation of the small-molecule model to PMVK suggests that the reactivity of a molecule with a single functional group such as 3-hexylundecan-2-one is quite different from that of PMVK. For a polymeric substrate, the electronic and steric interactions between functional groups can significantly affect its reactivity and possibly induce additional side reactions. Therefore, a small-molecule model would not always be able to guide the development of synthetic methods for polymers and other macromolecules.
We speculated that multiple side reactions could be responsible for the unsuccessful BV oxidation of PMVK. For example, acid-induced radical generation13 which is reported to be able to initiate even at low temperatures could cause significant chain scission. In addition, oxidative decomposition12 instigated by excessive oxidants, or Aldol-type condensations11 promoted by acid or base could also complicate the proposed PPM. A combination of a literature survey and our preliminary results suggested that a comprehensive evaluation of PMVK under BV conditions was required. Accordingly, we turned our focus to the DOP approach in the following studies.
Development of the BV oxidation of commercial PMVK homopolymers
We first identified that the combination of carbon-13 and proton nuclear magnetic resonance spectroscopy (13C and 1H NMR) was a reliable method to quantitatively characterize both PMVK and its product after BV oxidation. Errors relating to C–H decoupling of 13C NMR were evaluated and calibrated by 1H NMR prior to use.The effect of solvent was first investigated under a set of standard conditions (Table 1). Less polar solvents generally gave higher conversions while polar solvents were found to be problematic. Dichloromethane and chloroform, two common solvents for BV oxidation, were not suitable for the corresponding PPM due to both significant polymer degradation and only moderate reaction rates. Surprisingly, the optimal solvents were found to be halogen-substituted aromatic solvents such as fluorobenzene, chlorobenzene and 1,2,4-trichlorobenzene, which are rarely used in BV oxidation. Their improved performance may be partially attributed to the enhanced stability of these solvents to oxidative conditions. Among the solvents tested, 1,2,4-trichlorobenzene was found to be optimal in terms of both conversion and Mn. The BV post-modification proceeded smoothly in the first 8 h, but the Mn of the functionalized polymers declined with prolonged periods. We conjectured that the accumulation of m-chlorobenzoic acid was likely the cause of this phenomenon.
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Solvent | Conversionb (%) | M n,SEC (kDa) | Ð | Entry | Solvent | Conversionb (%) | M n,SEC (kDa) | Ð |
| a Standard conditions: PMVK 3a (41.7 mg, containing 0.60 mmol repeating units), mCPBA (2.40 mmol), and solvent (2.00 mL) stirred at 50 °C for 8 h. b Determined by 1H NMR of the crude products using 1,1,1,3-tetrachloropropane as an internal standard. c Determined by SEC (THF) analysis relative to polystyrene standards. d Reaction was run for 24 h instead of 8 h. The Mn/Ð of PMVK was 61.4 kDa/2.54. e Hexamethyldisiloxane (1.20 mmol) and dry mCPBA were employed. | |||||||||
| 1 | CHCl3 | 55 | 36.1 | 2.18 | 12 | DMF | 11 | 60.8 | 2.53 |
| 2 | CH2Cl2 | 44 | 41.9 | 2.27 | 13 | n-Hexane | 86 | 64.0 | 2.43 |
| 3 | 1,2-Dichloroethane | 52 | 61.9 | 2.50 | 14 | Toluene | 34 | 63.6 | 2.61 |
| 4 | EtOAc | 23 | 63.9 | 2.45 | 15 | (Trifluoromethyl)benzene | 80 | 63.7 | 2.44 |
| 5 | THF | 14 | 60.8 | 2.58 | 16 | Fluorobenzene | 71 | 69.8 | 2.61 |
| 6 | Et2O | 26 | 63.3 | 2.55 | 17 | Chlorobenzene | 75 | 71.3 | 2.57 |
| 7 | Dioxane | 12 | 62.1 | 2.56 | 18 | m-Dichlorobenzene | 79 | 67.7 | 2.49 |
| 8 | EtOH | 22 | 60.8 | 2.53 | 19 | o-Dichlorobenzene | 81 | 65.4 | 2.46 |
| 9 | (CF3)2CHOH | 16 | 55.2 | 2.49 | 20 | 1,2,4-Trichlorobenzene | 85 | 68.5 | 2.38 |
| 10 | MeCN | 16 | 54.1 | 2.43 | 21 | 1,2,4-Trichlorobenzened | >99 | 49.4 | 2.29 |
| 11 | N-Methylpyrrolidone | 12 | 59.6 | 2.58 | 22 | 1,2,4-Trichlorobenzened,e | >99 | 66.4 | 2.42 |

We speculated that a “precise” buffer could perhaps maintain the pH of the reaction mixture within a narrow window where polymer degradation would not occur. Indeed, even a subtle deviation from the “ideal” pH range could potentially induce chain scission. Initial efforts using various aqueous buffers were unsuccessful. Control experiments indicated that excessive water affected both the reaction rate and molar mass in a negative manner. Gratifyingly, an extensive screening (>700 experiments) of inorganic salts and organic additives revealed that hexamethyldisiloxane (TMS)2O (Tables S5–S7†) was able to buffer the system effectively. We conjectured that (TMS)2O slowly reacted with the m-chlorobenzoic acid accumulating during the oxidation to form a neutral trimethylsilyl m-chlorobenzoate species. Further suppression of chain scission by drying the commercial mCPBA reagent provided an optimal BV protocol for the exhaustive functionalization of PMVK. The optimal condition was obtained in Table 1, entry 22, Further experiments indicated that 4.0 equiv. of mCPBA was not required. Indeed, 1.2 equiv. of mCPBA was sufficient to complete the reaction, although a slower rate was observed.
Exhaustive BV oxidation of commercial PMVK homopolymers
With the first exhaustive functionalization condition in hand, we then conducted a BV oxidation of commercial PMVK on a preparative scale. In addition to 1H NMR and 13C NMR, the resulting polymer was fully characterized by size exclusion chromatography (SEC), Fourier transform infrared spectroscopy (FT-IR), differential scanning calorimetry (DSC), and thermogravimetric analysis (TGA).The 1H NMR and 13C NMR spectra of the polymer samples before and after oxidation are displayed in Fig. 3. The peak at δ = 4.87 ppm was assigned to the protons attached to carbon b′ of the post-modified polymer (Fig. 3a, bottom). Meanwhile, the peaks at δ = 2.3–2.7 ppm, corresponding to the protons attached to the carbon b of PMVK, disappeared after the oxidation (Fig. 3a, top). These results indicated that the BV oxidation completely transformed the PMVK homopolymer into a PVAc homopolymer. The 13C NMR spectra also provided additional evidence. In Fig. 3b, the carbonyl signal of the ketone of PMVK (δ = 210.2 ppm) disappeared completely after the oxidation, while a carbonyl signal corresponding to an ester appeared at 170.4 ppm. In addition, the conversion of PMVK to PVAc was further confirmed by FT-IR.
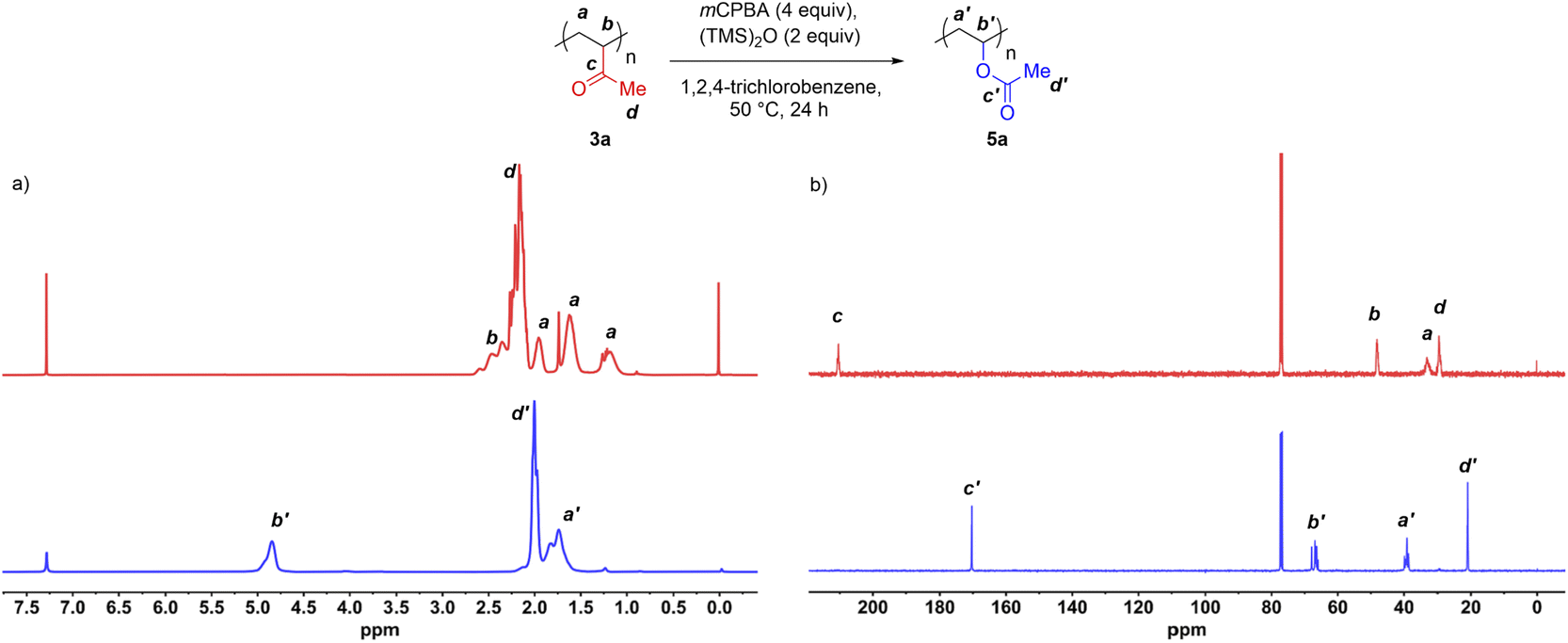 | ||
| Fig. 3 Exhaustive BV oxidation of commercial PMVK homopolymers. Comparison of (a) 1H NMR spectra and (b) 13C NMR spectra before and after exhaustive BV oxidation of commercial PMVK 3a. The NMR spectra were recorded using CDCl3 as a solvent. | ||
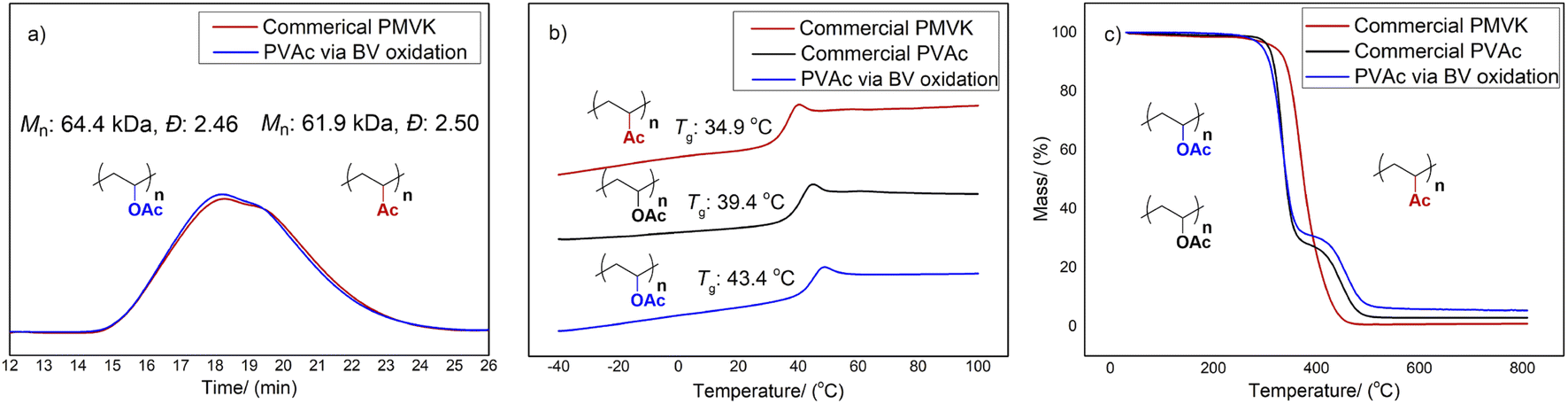
The Mn and Ð of the resulting PVAc polymer 5a were obtained by SEC (Fig. 4a). A slightly higher Mn and a similar Ð were obtained after functionalization. The SEC traces indicated that our protocol successfully suppressed chain scission and cross-linking without affecting the molar mass and Ð. Meanwhile, glass transition temperature (Tg) data was collected using differential scanning calorimetry (DSC) (Fig. 4b). The Tg of PVAc 5a obtained by the exhaustive BV oxidation was close to the Tg of the commercial PVAc sample. In contrast, the Tg of PMVK 3a was slightly lower. Furthermore, the thermogravimetric analysis (TGA) of this sample after oxidation displayed a two-staged curve, which is analogous to that of PVAc (Fig. 4c). The initial degradation stage possibly relates to the elimination reaction of the side groups while the second stage may relate to the degradation of the polymer backbone. As a comparison, the TGA of the original PMVK sample exhibited a one-stage curve.
 | ||
| Fig. 4 Characterization of PVAc 5a prepared by exhaustive BV post-modification of PMVK 3a. Comparison of (a) SEC, (b) DSC, and (c) TGA data of PMVK 3a, commercial PVAc and PVAc 5a obtained by BV oxidation. | ||
Exhaustive BV oxidation of statistical copolymers of MVK and various monomers
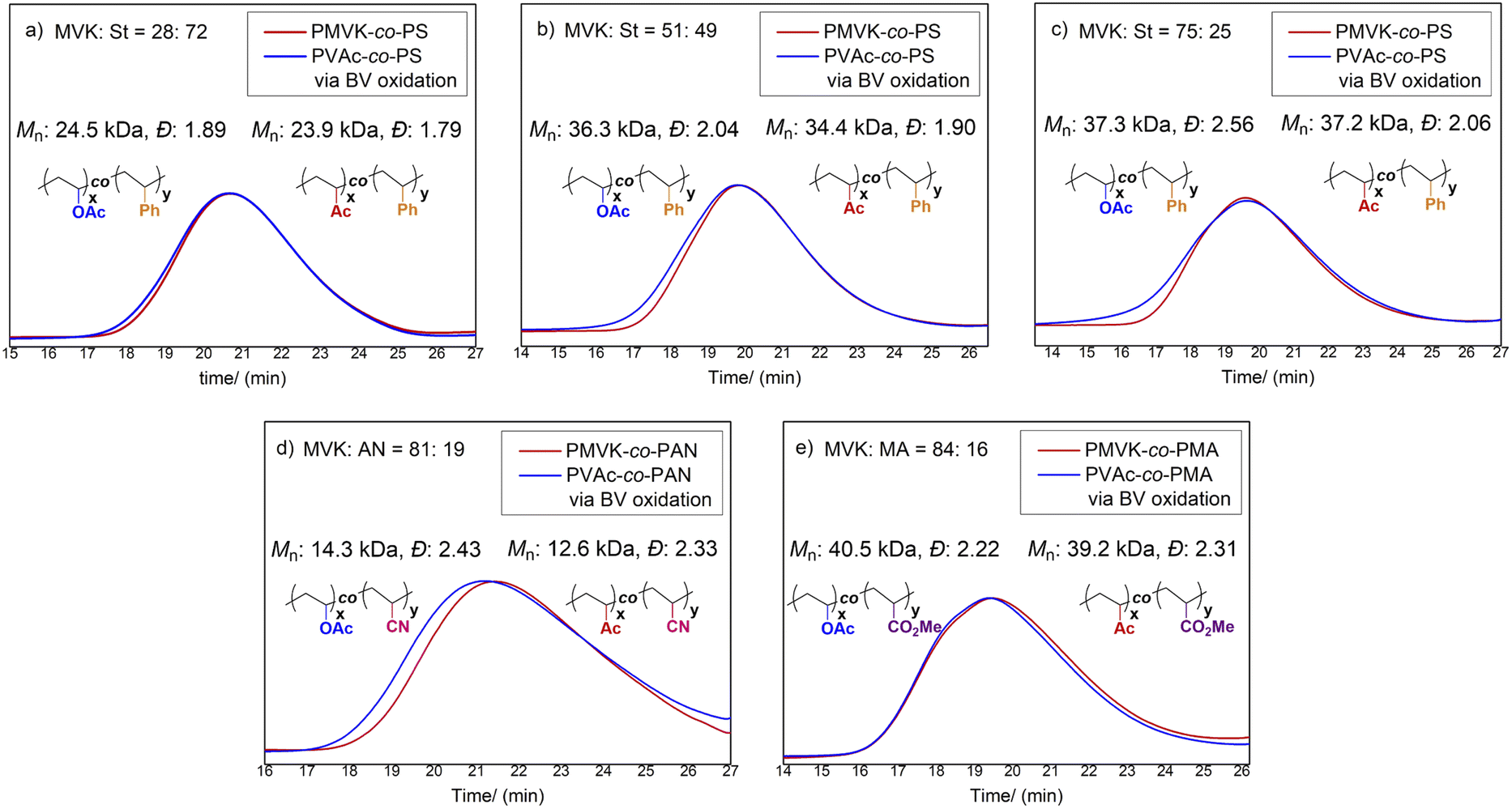
Although PVAc-derived copolymers are highly valuable, many PVAc copolymers, in particular those involving more activated monomers (MAMs), are rarely accessible by conventional copolymerization approaches.8a Indeed, copolymerization cannot occur effectively since VAc and MAMs have disparate reactivity ratios.8 Encouraged by our success using PMVK homopolymers, we expected that our exhaustive BV post-modification would provide a general solution for the synthesis of poly(VAc-co-MAM)s.Firstly, we prepared a number of PMVK-co-PSs with differing MVK/St feed ratios (20/80, 50/50, and 80/20) under AIBN-initiated free radical polymerization. The reactivity ratios of MVK and St monomers (rMVK = 0.29, rSt = 0.35)8a indicate that PMVK-co-PS copolymers favor a statistical distribution. As expected, almost identical MVK/St ratios (28/72, 51/49 and 75/25, respectively) were obtained in the resulting copolymers. Subsequently, the prepared PMVK-co-PS copolymers were functionalized using the BV protocol developed for the PMVK homopolymers. Unexpectedly, it was observed that the rates of BV oxidation for PMVK-co-PS copolymers were higher than of the PMVK homopolymers. The enhanced rates appeared to be proportional to the percentages of styrene units within the PMVK-co-PSs. Following the same trend, the rates of chain scission and cross-linking also increased. Gratifyingly, with careful tuning of the reaction temperature, a set of exhaustive modification conditions for PMVK-co-PS were achieved at 45 °C (MVK/St = 75/25), 40 °C (MVK/St = 51/49), and 35 °C (MVK/St = 28/72), respectively (Table S10†). Under these optimized conditions, the Mn and Ð were well controlled (Fig. 5). The reactivity differences between the PMVK homopolymers and PMVK-co-PS copolymers can be tentatively attributed to the fact that phenyl groups are less electron-withdrawing than acetyl and acetoxy groups. Mechanistically, the reaction rate of BV oxidation can be affected by the electronic properties of the migrating group. As a comparison, the direct copolymerization of VAc and St was also conducted. It was found that only 1% of the VAc monomer copolymerized while 94% conversion of St was obtained. Accordingly, the resulting copolymer was predominately composed of St. Besides PVAc-co-PS, PVAc copolymers with other MAMs such as acrylonitrile (AN), and methyl acrylate (MA) were also successfully prepared. Under the optimal BV conditions of 50 °C and 24 hours, PMVK-co-PAN and PMVK-co-PMA copolymers were fully converted to their corresponding PVAc-co-PAN and PVAc-co-PMA copolymers, respectively. No obvious chain scission or cross-linking were observed in the SEC traces (Fig. 5). All of the prepared statistical copolymers were thoroughly characterized. The “comonomer” ratios in the functionalized copolymers were consistent with the ratios of the starting copolymers (Tables S10 and S12†). Analysis of NMR spectral data (Fig. S4–S8†) confirmed that nearly all MVK units were converted to VAc units. FT-IR, DSC, and TGA data (Tables S11 and S13†) further supported the NMR spectra result.
 | ||
| Fig. 5 Synthesis of statistical copolymers of VAc and various monomers. SEC traces before and after the exhaustive BV oxidation of MVK statistical copolymers: (a) MVK/St (28/72) statistical copolymer 6a, (b) MVK/St (51/49) statistical copolymer 6b, (c) MVK/St (75/25) statistical copolymer 6c, (d) MVK/AN (81/19) statistical copolymer 6d and (e) MVK/MA (84/16) statistical copolymer 6e. | ||
Exhaustive BV oxidation of block copolymers of MVK and various monomers
In addition to the synthesis of statistical copolymers, the synthesis of block copolymers of VAc and MAMs remains challenging as well.18 Encouraged by our success in the synthesis of statistical poly(VAc-co-MAM)s, we anticipated that our BV post-modification strategy could be expanded to the synthesis of PVAc block copolymers with MAMs. Firstly, we prepared a number of PMVKs and poly(MAM-b-MVK)s via RDRP techniques. Although the atom transfer radical polymerization (ATRP) of vinyl ketone monomers16a is presently inaccessible, Wooley's RAFT protocol9f offered us an effective method to synthesize narrow-dispersed PMVKs and PMVK block copolymers. Unfortunately, none of our established BV protocols for the statistical copolymers were compatible with the post-modification of narrow-dispersed PMVK homopolymers. Significant degradation of the polymer architecture occurred in all cases (Table S8†). We speculated that the degradation was most likely caused by the end-group of the RAFT polymers. Indeed, this hypothesis was later verified by the fact that a RAFT-derived polymer without a sulfur-based end group19 performed well using our BV protocol (Table S8†).Previous studies indicated that sulfonic acid might be generated by the oxidation of the sulfur-based end-groups in the RAFT polymers.20 Although TMS2O was unable to buffer the more acidic –SO3H group, we speculated that another additive might neutralize both m-chlorobenzoic acid and the additional sulfonic acid. Significant efforts at using common bases as a buffer met with little success since the pH of the reaction mixture changed dynamically. Indeed, a fluctuating pH, no matter if higher or lower, could be detrimental to the PPM. Gratifyingly, t-butyl carbamate and urea were later identified as the most effective additives after a laborious screening (Tables 2 and S9†). With a modified procedure using 0.2 equiv. of urea as an additive, the PMVK homopolymer prepared by RAFT was nearly quantitatively transformed into a PVAc polymer without obvious chain scission or cross-linking. The structural integrity of the resulting PVAc polymer was verified by 1H and 13C NMR (Fig. S3†), and FT-IR. Furthermore, the MALDI-TOF MS (matrix-assisted laser desorption/ionization time-of-flight mass spectroscopy) spectrum indicated a spacing of 86.1 which is consistent with the molar mass of a Vac repeating unit (Fig. S13†).
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Additive | Conversionb (%) | M n (kDa) | Ð | Entry | Additive | Conversionb (%) | M n c (kDa) | Ð |
| a Standard conditions: RAFT homopolymer of MVK 3b (41.7 mg, containing 0.60 mmol repeating units), mCPBA (2.40 mmol), additive (0.12 mmol), and solvent (2.00 mL) stirred at 50 °C for 24 h unless noted otherwise. b Determined by 1H NMR of the crude products using 1,1,1,3-tetrachloropropane as an internal standard. c Determined by SEC (THF) analysis relative to polystyrene standards. d Hexamethyldisiloxane (1.20 mmol) and dry mCPBA were employed. e mCPBA was used without drying. | |||||||||
| 1d | Hexamethyldisiloxane | n.a. | 2.1 | 1.63 | 9 | 1,3-Diphenylurea | 69 | 4.3 | 1.39 |
| 2e | n.a. | >99 | 4.4 | 1.43 | 10 | p-Toluenesulfonamide | 98 | 3.3 | 1.35 |
| 3 | Triethylamine | 90 | 10.7 | 1.30 | 11 | Urea | >99 | 13.5 | 1.15 |
| 4 | N,N-Diisopropylethylamine | 88 | 10.0 | 1.32 | 12 | Thiourea | 98 | 11.8 | 1.21 |
| 5 | 2,6-Lutidine | 61 | 13.1 | 1.18 | 13 | Ammonium carbamate | 87 | 6.9 | 1.37 |
| 6 | NH4HCO3 | 96 | 9.4 | 1.29 | 14 | Methyl carbamate | 98 | 3.6 | 1.38 |
| 7 | NH4OAc | 83 | 6.9 | 1.34 | 15 | t-Butyl carbamate | >99 | 13.0 | 1.16 |
| 8 | Benzamide | 99 | 10.7 | 1.25 | |||||

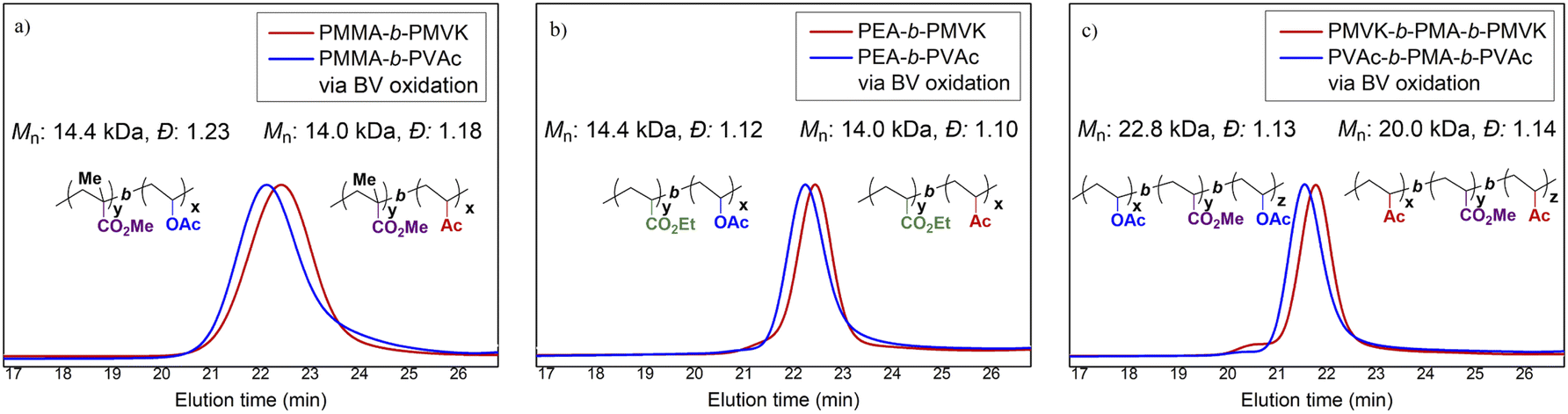
The modified BV protocol using the urea additive was further applied to the synthesis of PVAc block copolymers. Two di-block copolymers, poly(methyl methacrylate)-block-poly(methyl vinyl ketone) (PMMA-b-PMVK, Mn = 14.0 kDa, PMMA/PMVK = 7.7/6.3) and poly(ethyl acrylate)-block-poly(methyl vinyl ketone) (PEA-b-PMVK, Mn = 13.9 kDa, PEA/PMVK = 8.6/5.3), and one “ABA”-type tri-block copolymer, PMVK-b-PMA-b-PMVK (Mn = 20.0 kDa, PMVK/PMA/PMVK = 5.2/10.3/4.5), were synthesized by sequential RAFT polymerization. Subsequently, these three PMVK block copolymers were completely functionalized by the urea-modified BV protocol, with PMMA-b-PVAc, PEA-b-PVAc, and PVAc-b-PMA-b-PVAc being obtained without apparent degradation (Fig. 6 and Fig. S9–S11†). Notably, all of these copolymers were prepared using nearly identical conditions, and thereby the careful tuning of conditions for different MAMs is not required. This is distinct from the methods using switchable RAFT agent7b and dual RDRP agents21 where the choice of polymerization parameters and RDRP techniques were important.18b
 | ||
| Fig. 6 Synthesis of block copolymers of VAc and various monomers. SEC traces of (a) PMMA-b-PMVK di-block copolymer 7a, (b) PEA-b-PMVK di-block copolymer 7b and (c) PMVK-b-PMA-b-PMVK tri-block copolymer 7c before and after exhaustive BV oxidation. | ||
Scalable one-pot synthesis of PVAc-co-PS and its improved performance over PVAc adhesive
To streamline the preparation of poly(VAc-co-MAM)s, we developed a one-pot copolymerization/exhaustive BV post-modification procedure (Fig. 7, top). This convenient and scalable process allows us to synthesize PVAc-co-PS on a gram scale from MVK and St monomers directly without the purification of the PMVK-co-PS precursor. Noticeably, the use of 1,2,4-trichlorobenzene as a solvent for copolymerization was crucial for this process. Although the comprehensive properties of PVAc-co-PS 10 are yet to be investigated, this copolymer has exhibited an improved performance over the standard PVAc adhesive in our preliminary tests, especially when applied in aqueous conditions at elevated temperatures. The adhesive strength of PVAc-co-PS was maintained after 12 h of heating at 80 °C in water while that of regular PVAc was lost in 10 min under identical conditions (Fig. 7, bottom).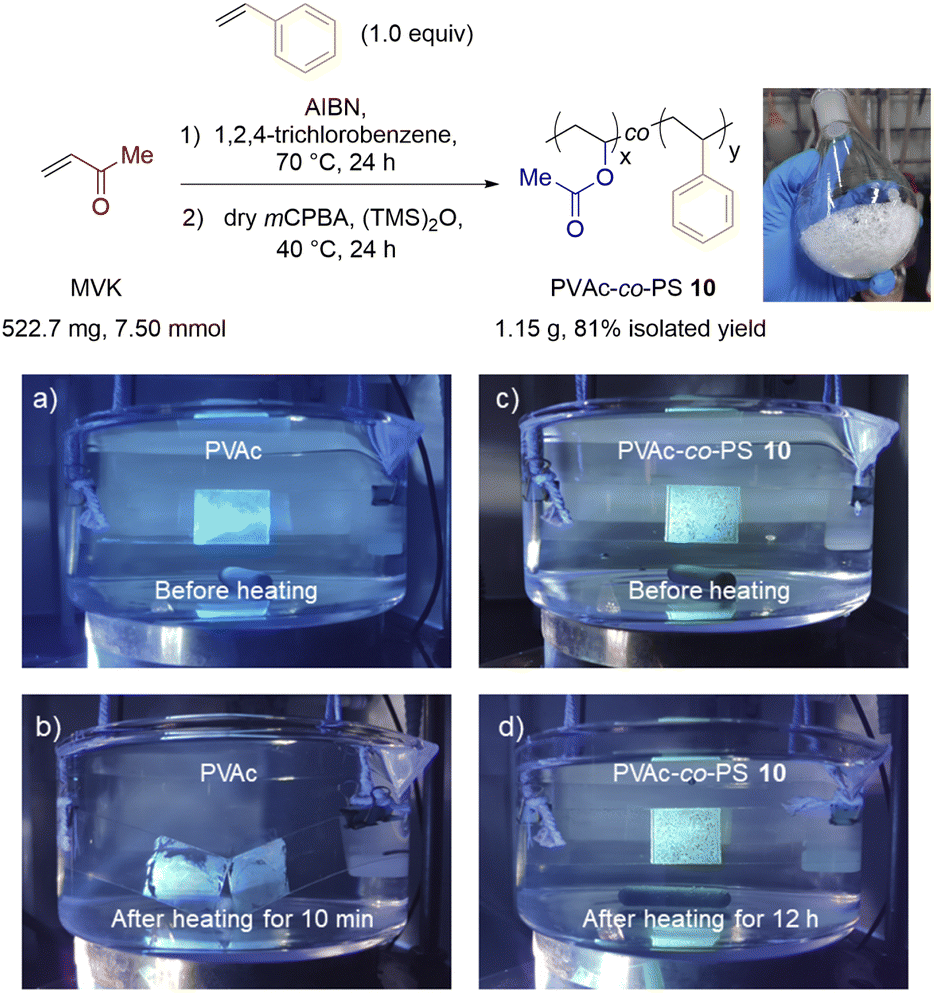 | ||
| Fig. 7 One-pot synthesis of PVAc-co-PS 10 and its adhesive performance. Images of glass slide lap joints bonded with PVAc (a) before and (b) after stirring in 80 °C water for 10 min, or PVAc-co-PS 10 (c) before and (d) after stirring in 80 °C water for 12 h. | ||
Conclusions
Baeyer–Villiger oxidation, a historically important transformation in small-molecule organic chemistry, has now emerged to be an efficient PPM reaction for PMVK and its copolymers. Initial unsuccessful attempts on a small-molecule model led us to develop and re-investigate the BV oxidation directly on PMVK polymers. After the elucidation of multiple factors affecting reaction kinetics and side reactions, a set of unique BV oxidation conditions have been developed and are able to completely functionalize PMVK and its copolymers without obvious chain scission or cross-linking.With this unique BV post-modification method in hand, we successfully prepared a broad array of poly(VAc-co-MAM) statistical copolymers that are rarely accessed by direct copolymerization. In addition, a number of PVAc di-block and tri-block copolymers with narrow molecular weight distributions were synthesized using this post-modification method. A gram-scale one-pot process was additionally developed to demonstrate the potential of a scalable production of such copolymers. The material properties and potential applications of the prepared poly(VAc-co-MAM) copolymers will be reported in due course.
In addition, we recognize that re-investigating a reaction using polymer substrates could be a valuable approach for the development of other PPM reactions, particularly when the direct translation of small-molecule protocols has failed. By applying this approach, the post-modifications of other commodity polymers such as poly(acrylic acid) and polyketones are currently being investigated in our laboratory.
Data availability
All supporting data can be found in the ESI.†Author contributions
L. L. conceived the project; L. L. and P. M. designed the research and analyzed the data; P. M. conducted the experimental work; L. L discussed and wrote the manuscript together with P. M., C. M. P., W. L., J. P., and Y. C.Conflicts of interest
Sun Yat-sen University has filed a patent application.Acknowledgements
Financial support from the National Natural Science Foundation of China (No. 21502241), the Natural Science Foundation of Guangdong Province (No. 2016A030313290), and Sun Yat-sen University is gratefully acknowledged. L.L. thanks Prof. Daniel Seidel (University of Florida), Prof. Seth B. Herzon (Yale University), Prof. Julian Zhu (Université de Montréal), Prof. Yongguang Jia (South China University of Technology), Prof. Hanchu Huang (Sun Yat-sen University), Prof. Zichen Li (Peking University) and Pyh Li for helpful discussions and support.Notes and references
- Selected reviews in recent literature: (a) C. M. Plummer, L. Li and Y. Chen, Polym. Chem., 2020, 11, 6862–6872 RSC; (b) J. B. Williamson, S. E. Lewis, R. R. Johnson III, I. M. Manning and F. A. Leibfarth, Angew. Chem., Int. Ed., 2019, 58, 8654–8668 CrossRef CAS PubMed; (c) E. Blasco, M. B. Sims, A. S. Goldmann, B. S. Sumerlin and C. Barner-Kowollik, Macromolecules, 2017, 50, 5215–5252 CrossRef CAS; (d) J. Romulus, J. T. Henssler and M. Weck, Macromolecules, 2014, 47, 5437–5449 CrossRef CAS; (e) K. A. Günay, P. Theato and H.-A. Klok, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1–28 CrossRef; (f) M. A. Gauthier, M. I. Gibson and H.-A. Klok, Angew. Chem., Int. Ed., 2009, 48, 48–58 CrossRef CAS PubMed; (g) N. K. Boaen and M. A. Hillmyer, Chem. Soc. Rev., 2005, 34, 267–275 RSC.
- Selected reviews on click chemistry: (a) J. Dong, L. Krasnova, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2014, 53, 9430–9448 CrossRef CAS PubMed; (b) J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249–1262 RSC; (c) W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2007, 28, 15–54 CrossRef CAS; (d) H. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- Selected reviews on multicomponent reactions: (a) M. A. R. Meier, R. Hu and B. Tang, Macromol. Rapid Commun., 2021, 42, 2100104 CrossRef CAS PubMed; (b) R. Kakuchi, Polym. J., 2019, 51, 945–953 CrossRef CAS.
- (a) J. Lee, A. J. Kalin, T. Yuan, M. Al-Hashimi and L. Fang, Chem. Sci., 2017, 8, 2503–2521 RSC; (b) R. Kakuchi and P. Theato, Post-polymerization Modifications via Active Esters, Functional Polymers by Post-Polymerization Modification: Concepts, Guidelines, and Applications, ed. P. Theato and H.-A. Klok, Wiley-VCH, Weinheim, 1st edn, 2013, pp. 45–64 Search PubMed.
- K. Pahnke, J. Brandt, G. Gryn’ova, C. Y. Lin, O. Altintas, F. G. Schmidt, A. Lederer, M. L. Coote and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2016, 55, 1514–1518 CrossRef CAS.
- Selected examples in recent literature: (a) P. Jung, A. D. Ziegler, J. Blankenburg and H. Frey, Angew. Chem., Int. Ed., 2019, 58, 12883–12886 CrossRef CAS PubMed; (b) Y. M. Wu and T. M. Swager, J. Am. Chem. Soc., 2019, 141, 12498–12501 CrossRef CAS; (c) C. Jeon, D. W. Kim, S. Chang, J. G. Kim and M. Seo, ACS Macro Lett., 2019, 8, 1172–1178 CrossRef CAS; (d) M. B. Larsen, S. Wang and M. A. Hillmyer, J. Am. Chem. Soc., 2018, 140, 11911–11915 CrossRef CAS PubMed.
- (a) D. J. Keddie, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 2012, 45, 5321–5342 CrossRef CAS; (b) M. Benaglia, J. Chiefari, Y. K. Chong, G. Moad, E. Rizzardo and S. H. Thang, J. Am. Chem. Soc., 2009, 131, 6914–6915 CrossRef CAS PubMed.
- (a) G. Odian, Principles of Polymerization, John Wiley & Sons, New York, 4th edn, 2004 Search PubMed; (b) F. R. Mayo, F. M. Lewis and C. Walling, J. Am. Chem. Soc., 1948, 70, 1529–1533 CrossRef CAS; (c) F. R. Mayo, C. Walling, F. M. Lewis and W. F. Hulse, J. Am. Chem. Soc., 1948, 70, 1523–1525 CrossRef CAS; (d) F. R. Mayo and F. M. Lewis, J. Am. Chem. Soc., 1944, 66, 1594–1601 CrossRef CAS.
- (a) I. Lee, E. H. Discekici, A. Anastasaki, J. R. de Alaniz and C. J. Hawker, Polym. Chem., 2017, 8, 3351–3356 RSC; (b) J. A. Reeves, M. L. Allegrezza and D. Konkolewicz, Macromol. Rapid Commun., 2017, 38, 1600623 CrossRef; (c) Y. Zhao, X. Liu, Y. Liu, Z. Wu, X. Zhao and X. Fu, Chem. Commun., 2016, 52, 12092–12095 RSC; (d) J. A. M. Hepperle, H. Luftmann and A. Studer, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2150–2160 CrossRef CAS; (e) K. Uehara, C. B. Wagner, T. Vogler, H. Luftmann and A. Studer, Angew. Chem., Int. Ed., 2010, 49, 3073–3076 CrossRef CAS PubMed; (f) C. Cheng, G. Sun, E. Khoshdel and K. L. Wooley, J. Am. Chem. Soc., 2007, 129, 10086–10087 CrossRef CAS PubMed.
- Selected reviews on Baeyer–Villiger oxidation: (a) L. Zhou, L. Lin, X. Liu and X. Feng, Baeyer–Villiger (BV) Oxidation/Rearrangement in Organic Synthesis, Molecular Rearrangements in Organic Synthesis, ed. C. M. Rojas, John Wiley & Sons, Hoboken, 1st edn, 2016, pp. 35–57 Search PubMed; (b) G. J. ten Brink, I. W. C. E. Arends and R. A. Sheldon, Chem. Rev., 2004, 104, 4105–4123 CrossRef PubMed; (c) M. Renz and B. Meunier, Eur. J. Org. Chem., 1999, 4, 737–750 CrossRef; (d) G. R. Krow, Org. React., 1993, 43, 251–798 CAS.
- (a) J. W. Krumpfer, E. Giebel, E. Frank, A. Muller, L. Ackermann, C. N. Tironi, G. Mourgas, J. Unold, M. Klapper, M. R. Buchmeiser and K. Mullen, Chem. Mater., 2017, 29, 780–788 CrossRef CAS; (b) J. N. Hay, Makromol. Chem., 1963, 67, 31–41 CrossRef CAS; (c) C. S. Marvel and C. L. Levesque, J. Am. Chem. Soc., 1938, 60, 280–284 CrossRef CAS.
- (a) M. C. Celina, Polym. Degrad. Stab., 2013, 98, 2419–2429 CrossRef CAS; (b) T. Q. Nguyen, Polymer Degradation and Stabilization, Handbook of Polymer Reaction Engineering, ed. T. Meyer and J. Keurentjes, Wiley-VCH, Weinheim, 2005, pp. 757–831 Search PubMed.
- B. Schweitzer-Chaput, T. Kurten and M. Klussmann, Angew. Chem., Int. Ed., 2015, 54, 11848–11851 CrossRef CAS PubMed.
- Selected examples on oxidative post-polymerization modifications: (a) L. Chen, K. G. Malollari, A. Uliana and J. F. Hartwig, J. Am. Chem. Soc., 2021, 143, 4531–4535 CrossRef CAS PubMed; (b) L. Chen, K. G. Malollari, A. Uliana, D. Sanchez, P. B. Messersmith and J. F. Hartwig, Chem, 2021, 7, 137–145 CrossRef CAS; (c) T. Nishikawa and M. Ouchi, Angew. Chem., Int. Ed., 2019, 58, 12435–12439 CrossRef PubMed; (d) H. L. van de Wouw, J. Y. Lee, E. C. Awuyah and R. S. Klausen, Angew. Chem., Int. Ed., 2018, 57, 1673–1677 CrossRef CAS; (e) Y. Dong, Z. Wang and C. Li, Nat. Commun., 2017, 8, 277 CrossRef PubMed; (f) A. Bunescu, S. Lee, Q. Li and J. F. Hartwig, ACS Cent. Sci., 2017, 3, 895–903 CrossRef CAS PubMed; (g) N. K. Boaen and M. A. Hillmyer, Macromolecules, 2003, 36, 7027–7034 CrossRef CAS.
- (a) T. Chinnusamy, K. Feeney, C. G. Watson, D. Leonori and V. K. Aggarwal, Oxidation of Carbon–Boron Bonds, Comprehensive Organic Synthesis II, ed. P. Knochel, Elsevier, Amsterdam, 2nd edn, 2014, pp. 692–718 Search PubMed; (b) H. C. Brown, C. Synder, B. C. Subba Rao and G. Zweifel, Tetrahedron, 1986, 42, 5505–5510 CrossRef CAS; (c) G. Zweifel and H. C. Brown, Org. React., 1963, 13, 1–54 CAS.
- (a) A. Mittal, S. Sivaram and D. Baskaran, Macromolecules, 2006, 39, 5555–5558 CrossRef CAS; (b) N. Kosaka, T. Hiyama and K. Nozaki, Macromolecules, 2004, 37, 4484–4487 CrossRef CAS; (c) A. W. P. Jarvie, N. Overton and C. B. S. Pourçain, J. Chem. Soc., Perkin Trans. 1, 1999, 115, 2171–2176 RSC.
- Our preliminary studies indicated that BV oxidations of other poly(vinyl ketones) were less regioselective than that of PMVK.
- Selected reviews in recent literature: (a) S. Pearson, C. S. Thomas, R. Guerrero-Santos and F. D'Agosto, Polym. Chem., 2017, 8, 4916–4946 RSC; (b) S. Harrisson, X. Liu, J. Ollagnier, O. Coutelier, J. Marty and M. Destarac, Polymers, 2014, 6, 1437–1488 CrossRef; (c) L. E. N. Allan, M. R. Perry and M. P. Shaver, Prog. Polym. Sci., 2012, 37, 127–156 CrossRef CAS; (d) H. Tang, M. Radosz and Y. Shen, in Controlled/“Living” Radical Polymerization of Vinyl Acetate, in Controlled/Living Radical Polymerization: Progress in ATRP, ed. K. Matyjaszewski, ACS Symposium Series, Washington, DC, 2009, 1023, pp. 139–157 Search PubMed; (e) C. Barner-Kowollik, Handbook of RAFT Polymerization, Wiley-VCH, Weinheim, 2008 Search PubMed.
- (a) E. H. Discekici, S. L. Shankel, A. Anastasaki, B. Oschmann, I. Lee, J. Niu, A. J. McGrath, P. G. Clark, D. Laitar, J. Read de Alaniz, C. J. Hawker and D. J. Lunn, Chem. Commun., 2017, 53, 1888–1891 RSC; (b) H. Willcock and R. K. O'Reilly, Polym. Chem., 2010, 1, 149–157 RSC; (c) M. Chen, G. Moad and E. Rizzardo, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6704–6714 CrossRef CAS; (d) S. Perrier, P. Takolpuckdee and C. A. Mars, Macromolecules, 2005, 38, 2033–2036 CrossRef CAS.
- L. Feng, K. A. Cavicchi, B. C. Katzenmeyer and C. Wesdemiotis, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 5100–5108 CrossRef CAS.
- (a) R. Nicolay, Y. Kwak and K. Matyjaszewski, Chem. Commun., 2008, 42, 5336–5338 RSC; (b) Y. Tong, Y. Dong, F. Du and Z. Li, Macromolecules, 2008, 41, 7339–7346 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc03492a |
| This journal is © The Royal Society of Chemistry 2022 |