
A multi-functional SiO32−-releasing hydrogel with bioinspired mechanical properties and biodegradability for vascularized skeletal muscle regeneration†
Pengcheng
Xu‡
 a,
Qiang
Yang‡
bc,
Lin
Zhang‡
a,
Kang
Wu
a,
Yanjie
Bai
d,
Saijilafu
a,
Huilin
Yang
a,
Huan
Zhou
*b,
Xiao
Lin
*a and
Lei
Yang
*ab
a,
Qiang
Yang‡
bc,
Lin
Zhang‡
a,
Kang
Wu
a,
Yanjie
Bai
d,
Saijilafu
a,
Huilin
Yang
a,
Huan
Zhou
*b,
Xiao
Lin
*a and
Lei
Yang
*ab
aInstitute of Orthopedics and Department of Orthopedics, The First Affiliated Hospital, Soochow University, Suzhou 215006, China. E-mail: ylei@hebut.edu.cn; xlin@suda.edu.cn
bCenter for Health Science and Engineering (CHSE), School of Health Sciences and Biomedical Engineering, Hebei University of Technology, Tianjin 300130, China. E-mail: zhouhuan@hebut.edu.cn
cDepartment of Minimally Invasive Spine Surgery, Tianjin Hospital, Tianjin 300211, China
dDepartment of Chemical Engineering, Hebei University of Technology, Tianjin 300130, China
First published on 15th April 2022
Abstract
Vascularized skeletal muscle regeneration remains a great medical need but significant challenge. Biomaterial strategies that can facilitate the regeneration of muscle fibers and blood vessels are unavailable. Herein, we report a new cell- and drug-free biomaterial-based strategy for the repair of severely injured skeletal muscles. A novel multi-functional silicate ion-releasing hydrogel (SRH) was developed by dissolving PVA and starch in Na2SiO3 solutions, followed by freeze–thawing treatment. The mechanical properties and degradation profile of the SRH could be easily adjusted by altering the amylose/amylopectin ratio of starch. The SRH efficiently releases silicate ions to create a favorable microenvironment for enhanced skeletal muscle repair, while the mechanical properties and biodegradability of SRHs is adjusted to match the muscle regeneration environment. Silicate ions released from the SRH simultaneously promote myoblast proliferation and myogenic differentiation, decrease oxidative stress, and enhance the angiogenesis of vascular endothelial cells in vitro. Silicate ions released from the SRH scaffold with bioinspired mechanical properties and biodegradability promote the de novo formation of muscle fibers and blood vessels while inhibiting tissue fibrosis, leading to enhanced vascularized muscle regeneration in vivo. With multiple biofunctions and mechanical/degradation tunability, the SRH platform bears great potential in the skeletal muscle tissue engineering and treatment of formidable clinical problems such as volumetric muscle loss and sarcopenia.
 Lei Yang | Lei lives in Tianjin, just outside of Beijing in China, and directs the School of Health Sciences and Biomedical Engineering at the Hebei University of Technology. He is a biomaterial engineer and entrepreneur focusing on developing revolutionary biomaterials to enable less invasive treatment and long-lasting solutions for patients with musculoskeletal diseases. He is the recipient of the Leaders in Innovation Fellowship and LIF Rising Star, Royal Academy of Engineering (UK), and the International Society of Orthopaedic Surgery and Traumatology (SICOT) Research Award. Lei obtained his PhD degree from the Brown University, USA, and his bachelor's and master's degrees in Materials Engineering from the Tsinghua University, China. He enjoys making aircraft models and taking care of a freshwater fish tank while not working. |
1. Introduction
Skeletal muscles are the basis of human movement and many physiological functions.1 Traumatic skeletal muscle defects result in the permanent loss of function,2 leading to economic and social pressures on millions of patients worldwide.3 Free functional muscle transfer, as the current standard clinical treatment for severe skeletal muscle injury, is often associated with poor prognosis and increased complications.4,5 However, new clinical strategies for the effective repair of severely injured skeletal muscles are still lacking.Highly metabolic skeletal muscles require a complex network of capillaries to support nutrient and metabolite exchange. Thus, vascularized muscle regeneration is critical for the effective repair of severely injured skeletal muscles. Biomaterial-based strategies have shown potential to improve de novo skeletal muscle formation,6,7 but regenerating vascularized skeletal muscles remains difficult6,8 owing to the limited endogenous myogenesis in high oxidant stress environments and inadequate blood vessel formation.9,10 Besides, cell- and drug-free biomaterial strategies that can facilitate the regeneration of muscle fibers and blood vessels have not yet been developed.11
Recent studies revealed that the delicate design of the microstructure and the physical properties of biomaterials are able to improve their bio-activity for skeletal muscle regeneration.12,13 However, these strategies still face the limitations of a complicated process and potential toxicity of functional additives (i.e., nanotubes or nanoparticles). Our recent studies demonstrated the potential of biofunctional ions in promoting soft tissue repair14–16 by modulating biological responses. Owing to their known angiogenic activity,17,18 silicate ions have been used to enhance vascularization in various tissues such as bone, adipose tissue, and skin.18–20 However, the therapeutic effect of silicate ions for promoting vascularized skeletal muscle regeneration remains unclear. Herein, we propose a new biomaterial-based strategy for skeletal muscle repair by the local delivery of silicate ions from a hydrogel scaffold and test its efficacy in promoting vascularized muscle regeneration.
Nevertheless, the biological role of silicate ions released from the hydrogel scaffold in mediating skeletal muscle regeneration and the influence of the degradation rate of the hydrogel scaffold on skeletal muscle repair remain unclear. Although the silicate 45S5 bioactive glass has been tested for the repair of skeletal muscle defects, beneficial effects were more ascribed to calcium ions in it.21 Based on a previous report on the beneficial effect of silica nanoparticles on myoblast fusion,22 we hypothesize that silicate ions with appropriate releasing kinetics from the hydrogel scaffold could create a favorable myogenic microenvironment that enhances the de novo formation of muscle fibers. Meanwhile, the in vivo therapeutic efficacy of silicate ions is believed to be regulated by the degradation rate of the hydrogel scaffold.23 To test the above hypothesis, we prepared a novel silicate ion-releasing hydrogel (SRH) platform by incorporating Na2SiO3 into hydrogels with degradation tunability. The myogenic effect of the SRH was evaluated, both in vitro and in vivo, in comparison with that of a chloride ion-releasing hydrogel (CRH) prepared by incorporating NaCl into the same hydrogel scaffold. The effect of the degradation rate of the SRH on skeletal muscle tissue repair was also investigated in vivo.
2. Materials and methods
2.1. Preparation of the SRH and other hydrogels
High-amylose corn starch (HAS, amylopectin ∼40%, as determined by the manufacturer) and waxy corn starch (WS, amylopectin ∼90%, as determined by the manufacturer) were purchased from Lihua Starch Co. Ltd (Qinhuangdao, China). Poly (vinyl alcohol) (PVA, degree of polymerization 1750 ± 50, degree of hydrolysis ≥99%), Na2SiO3·9H2O (analytical purity) and NaCl (analytical purity) were obtained from Sinopharm Chemical Reagent (China). All chemicals were used without further purification. Deionized water was used throughout the experiments. Silicate ion-releasing hydrogels (SRHs) were prepared by the following procedure. First, 4.21 wt% PVA and 8.42 wt% starch (mass ratios of HAS to WS are described in Table S1, ESI†) were added in deionized water and stirred in a water bath at 90 °C for 2 h. Subsequently, 3.16 wt% Na2SiO3·9H2O was added into the PVA/starch solution and stirred at 60 °C for 30 min. Finally, the mixed solution was poured into a mold of desired dimension and frozen at −20 °C for 10 h, and then thawed at room temperature for 2 h. The freezing/thawing process was repeated three times to form the SRH composite hydrogels. PVA (4.21 wt%), PVA/Na2SiO3 (PVA-Si, PVA 4.21 wt%, Na2SiO3·9H2O 3.16 wt%) and PVA/starch (PVA 4.21 wt%, starch 8.42 wt%) hydrogels were prepared according to the above method. Chloride ion-releasing hydrogels (CRH) were prepared by replacing Na2SiO3 with NaCl under the premise of the same molar mass of Na according to the above procedure.2.2. Scanning electron microscopy (SEM) analysis
For microstructure characterization, the prepared hydrogel samples were freeze-dried thoroughly in a freeze dryer and exposed the internal structure of the samples. After this, the samples were sputter-coated using an ion-sputter coating instrument (SC7620, Quorum, UK), and then the microstructure of the samples was observed using a scanning electron microscope (SEM, JSM-7100F, JEOL, Japan).2.3. Rheological tests
The rheological properties of PVA, PVA-Si, CRH and SRH hydrogels were studied using a rheometer (AR2000, TA Instruments, USA) equipped with a temperature control unit, in an oscillating mode using a plate–plate configuration (a plate diameter of 20 mm and a gap between the upper and lower plates of 1 mm). The samples were prepared with 20 mm in diameter and 1 mm in thickness. Frequency sweep tests were performed from 0.1 to 50 Hz at 37 °C and 1% strain.2.4. An in vitro degradation test and ion release measurement
For the degradation measurement of the hydrogels, hydrogels were completely dried and weighed (W0). All samples were immersed in deionized water at a concentration of 0.1 g ml−1 at 37 °C. The immersion solution was refreshed every other day. At predetermined time points, the solution with the hydrogel was centrifuged (2000 rpm for 5 min) and the supernatant was discarded. Afterward, the remaining hydrogels in the centrifuge tube were completely dried and weighed (Wi). The degradation status of hydrogels was then defined by the following equation:where W0 is the original dry weight of the hydrogel and Wi is the dry weight of the hydrogel after immersed for different time periods.
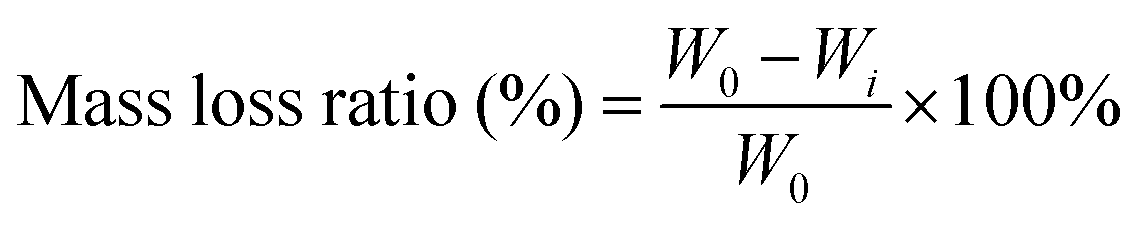
For characterizing the silicate release behavior, hydrogel samples were immersed in deionized water at 37 °C with a solid-to-liquid ratio of 0.1 g mL−1. The extract solution was collected and replenished every day up to 7 days. The concentration of silicate ions in extracts was measured by inductively coupled plasma-optical emission spectrometer (ICP-OES, Optima 8000/S10, PerkinElmer, USA).
2.5. Preparation of hydrogel extracts
Hydrogel extracts were prepared for in vitro cell experiments according to the ISO 10993–5 standard. Hydrogels were sterilized by UV light for 24 h and immersed in a high-glucose Dulbecco's modified Eagle's medium (DMEM) with 10 vol% fetal bovine serum (FBS) and 1 vol% penicillin–streptomycin solution at a concentration of 0.1 g ml−1 for 24 h. Then, the extracts were collected and filtrated through a 0.22 μm membrane filter (Millex-GP, Millipore, USA) to obtain a hydrogel extract medium for cell experiments. The regular cell culture medium (a high-glucose DMEM with 10 vol% fetal bovine serum and 1 vol% penicillin–streptomycin solution) was used as the control group. C2C12 myoblasts and HUVECs were purchased from the Stem Cell Bank of the Chinese Academy of Sciences.2.6. Cell proliferation and viability
C2C12 myoblasts (density: 3500 cells per well) or HUVECs (density: 4000 cells per well) were seeded in 96-well plates and filled with 100 μl of the regular cell culture medium. After 24 h, the cell culture medium was replaced with the hydrogel extract, and the control group was replaced with the fresh regular cell culture medium. After 1 and 3 days, the cell viability was measured using a Cell Counting Kit-8 (CCK-8, Dojindo Molecular Technologies, Inc., Japan) according to the method provided by the reagent manufacturer. Finally, the optical density (OD) value of each well was measured at 450 nm using a microplate reader (PowerWave X, BioTek Instruments, USA). The cell viability was calculated bywhere ODi is the OD value for the extract group and OD0 is the OD value for the control group.
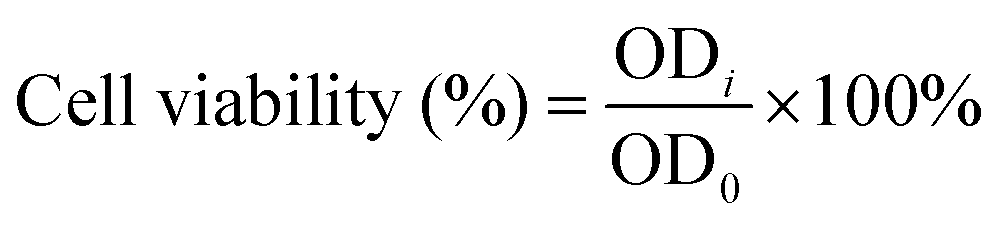
For the live/dead staining assay, C2C12 myoblasts or HUVECs were seeded in 24-well plates at a density of 8000 cells per well. As mentioned above, the cells were replaced with hydrogel extracts after adherence and continued to be cultured for 1 and 3 days. The cells were then stained using a LIVE/DEAD viability kit (Thermo Fisher Scientific, USA) and imaged using a fluorescence microscope (Carl Zeiss, Zeiss Ax overt 200M, Germany).
2.7. Mature myotube actin filament staining
C2C12 myoblasts were seeded in 24-well plates at a density of 5 × 104 cells per well. Cells were cultured in the regular cell culture medium until a confluent. Once the cells attained 90% confluence, the medium was switched to the regular differentiation medium (rDM, the high-glucose DMEM with 2 vol% horse serum and 1 vol% penicillin–streptomycin solution) or the hydrogel extract-based differentiation medium (hDM, obtained by extracting the hydrogel with the rDM) for the fusion of myoblasts into myotubes. Mature myotubes were formed after 6 days of culture in the rDM or hDM. The nucleus and filamentous actin (F-actin) of the cells were observed by DAPI/phalloidin staining. Briefly, the cells were fixed for 30 min on ice in 4% paraformaldehyde (Beyotime, China), washed three times with PBS, and permeabilized in 0.1% Triton-X100 (Beyotime, China) for 15 min. Then, the cells were stained with rhodamine–phalloidin (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100, Everbright Inc, USA) and diamidino-2-phenylinodole (DAPI, 1:500, Beyotime, China) for 30 min at room temperature in the dark.
:100, Everbright Inc, USA) and diamidino-2-phenylinodole (DAPI, 1:500, Beyotime, China) for 30 min at room temperature in the dark.
2.8. MHC immunofluorescence
To determine the muscle-specific marker expression, immunofluorescence staining for the muscle specific marker myosin heavy chain (MHC) was performed. Briefly, the cells were fixed using 4% of paraformaldehyde (Beyotime, China) for 30 min. The fixed cells were permeabilized with 0.1% Triton X-100 (Beyotime, China) for 15 min and blocked with 2% bovine serum albumin (BSA) (Sigma, USA) for 1 h at 37 °C. For staining, the cells were incubated with the following primary antibodies: mouse-anti-myosin heavy chain (1:10, Novus Biologicals, MAB4470, USA) for overnight at 4 °C and followed by incubation with fluorescence-labeled secondary antibodies (1:500, Alexa Fluor 488 goat anti-mouse IgG, ab150113) for 2 h at 37 °C. Nuclei were labeled using DAPI (1:500, Beyotime, China). The stained cells were visualized using a fluorescence microscope and analyzed using the ImageJ software. The average myotube diameter and the fusion index were quantified using the ImageJ software.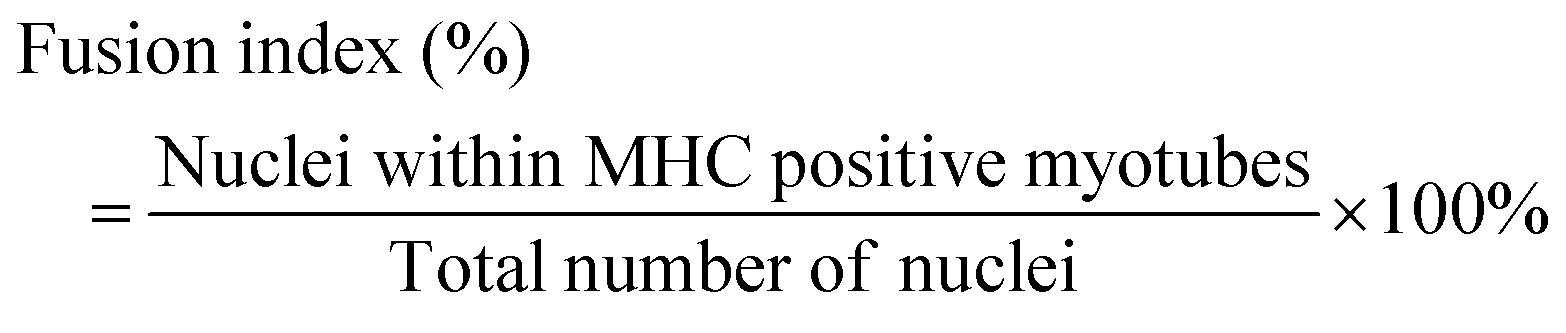
2.9. Intracellular ROS production detection
The intracellular ROS level of C2C12 myoblasts under H2O2 treatment was measured using a ROS Assay kit (Beyotime, China). C2C12 myoblasts were seeded in 24-well plates at a density of 2 × 104 cells per well. After cell adherence, the medium was replaced with hydrogel extracts or the fresh DMEM, and 250 μM H2O2 was added at the same time to induce oxidative stress, which is a well-established method to induce oxidative stress in C2C12 myoblasts.24 After continued culture for 24 h, the medium was removed and incubated 2′,7′-dichlorofluorescin diacetate (DCFH-DA) diluted with the serum-free medium (1:1000) in a 37 °C incubator for 30 minutes. Intracellular ROS can oxidize non-fluorescent DCFH to produce fluorescent DCF. Then, the cells were washed with a serum-free medium to fully remove DCFH-DA that did not enter the cells. Finally, the images were captured by using fluorescence microscopy and quantified using ImageJ software. All values were expressed as a ratio relative to the fluorescence intensity of cells treated without H2O2.
2.10. Scratch wound healing assay
HUVECs were seeded in a 6-well plate at a density of 2 × 105 cells per well. The cells were adherent and attained full confluence. Then, a 200 μL sterile pipette tip was used to make a straight wound in the cell layer, and the medium was replaced with the fresh hydrogel extract medium containing 1% FBS. Images of the HUVEC migration process were observed using a light microscope (Carl Zeiss, Germany) at 0, 12 and 24 h after the cells were scratched. The ImageJ software was used to quantify the distance between two scratch edges (wound distance) at each time point.2.11. Transwell assay
The Transwell assay was performed within a 24 well Transwell chamber (pore size, 8 μm; corning, USA). HUVECs (5 × 104 cells) were seeded on the upper chamber and added with the serum-free DMEM. Then, the upper chamber was partially immersed in the lower chamber containing 600 μL of different hydrogel extract media. After 12 h of incubation, a cotton swab was used to gently wipe away the non-migrated HUVECs on the upper surface of the membrane. HUVECs that migrated to the lower surface of the membrane were fixed with 4% paraformaldehyde and stained with 0.2% crystal violet, then observed and photographed under an inverted microscope. ImageJ software was used to count the number of migrated HUVECs.2.12. Tube formation assay
According to the manufacturer's instructions, HUVECs (1 × 104) were seeded onto a 48-well plate coated with 200 μL of Matrigel (Corning, USA) and cultured in different hydrogel extracts and the regular cell culture medium. After 12 h, the cells were photographed and the tube formation was analyzed using the ImageJ software.2.13. qRT-PCR
The expression levels of myogenesis specific genes (MyoD, myogenin (Myog) and MHC) and angiogenesis specific genes (vascular endothelial growth factor (VEGF) and kinase insert domain containing receptor (KDR)) were measured by qRT-PCR. Briefly, the total RNA was extracted using an extraction kit (Cell Total RNA Isolation Kit V2, Vazyme Company, Nanjing, China) according to the manufacturer's instructions. To measure the concentration and purity of the isolated RNA, a spectrophotometer (FLX800T, Biotek, USA) was used. Then, the total RNA was converted to cDNA using a reverse transcription system (Toyobo Co., Ltd, Japan). The synthesized cDNA was used to conduct the quantitative RT-PCR using a real-time PCR detection system (Bio-Rad, USA), and the gene expression was normalized using glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and calculated using the 2−ΔΔCT method. In addition, gene levels of cells cultured in the regular medium were set as one-fold. All primer sequences are provided in Table S2 (ESI†).2.14. In vivo implantation of hydrogels into the rat tibialis anterior (TA) muscle defect model
The animal study was approved by the Ethics Committee of Soochow University (Approval No. SUDA20200407A01). Twenty-four (n = 4 per each group) adult male Sprague–Dawley (SD) rats (8–10 weeks old; 200–220 g; Experimental Animal Centre of Soochow University, Suzhou, China) were randomly divided into six groups: (1) Untreated, (2) CRH, (3) PVA-Si hydrogel, (4) H4W0 hydrogel, (5) Na2SiO3 injection, and (6) H3W1 hydrogel. Before the surgery, rats were weighted and anesthetized using 1% pentobarbital sodium intraperitoneal injection (40 mg kg−1). The muscle defect model was prepared by the resection of the TA muscle according to a previous study.25 Briefly, a longitudinal incision was made parallel to the tibia on the outside of the right calf to expose the tibial anterior muscle. Then, a surgical defect (∼7 × 5 × 2 mm) was created in the middle third of the TA muscle using a scalpel. For hydrogel treatment groups, hydrogels were implanted into the defect immediately after hemostasis. For the untreated group, the injury was left without any treatment. For the Na2SiO3 injection group, 100 μL of the Na2SiO3 solution (0.5 mM) was injected intramuscularly into the defect site immediately after hemostasis and weekly after the surgery (totally four injections were applied). The fascia and skin were closed using vicryl (6-0) and nylon (3-0) interrupted sutures, respectively. After surgery, meloxicam (2 mg kg day−1) was subcutaneously administered for 3 days. Animals were sacrificed at day 28 post-surgery by euthanasia.2.15. Muscle morphology and weight
Immediately after euthanasia, each TA muscle was dissected from the animal with Achilles tendon intact and fat and skin removed. Once images were taken with the camera, the weight (g) of the TA muscle was measured. The TA muscle weight (% of Contralateral) was calculated byFollowing the measurements, TA muscles were fixed in 10% neutral formalin for histological analysis.
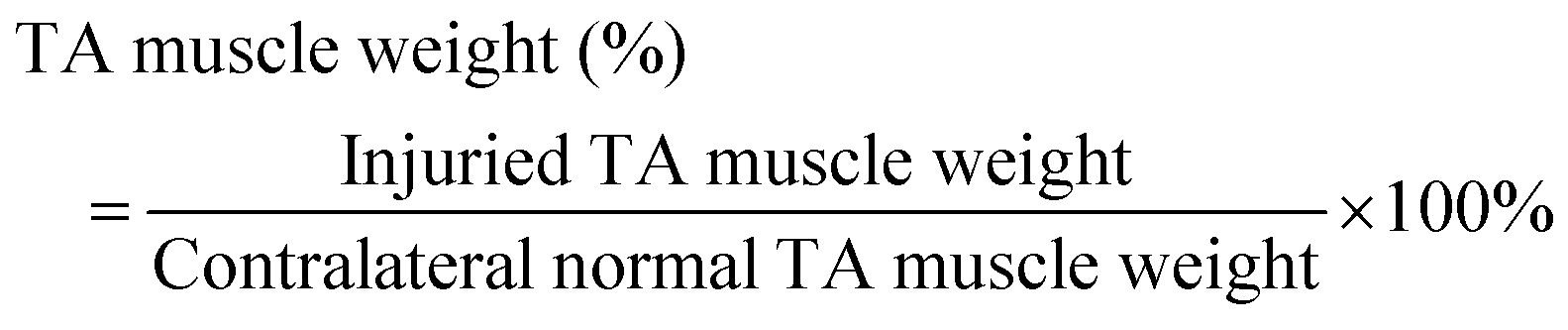
2.16. Histology and immunohistochemistry staining
After fixation in 10% neutral formalin for 48 h, the TA muscle specimens were dehydrated using an alcohol gradient. Then, the specimen was embedded in a paraffin block using a paraffin-embedding machine. The paraffin block was serially sliced at a thickness of 6 μm using a microtome, and the sections were subjected to hematoxylin and eosin (H&E) staining and Masson trichrome (MT) staining. For immunohistochemistry, sections were dewaxed and the antigen was repaired with citrate buffer. Samples were then treated with 0.1% Triton X-100 for 15 min and 2% BSA for 1 h. Sections were incubated overnight with CD31 (1:50, Abcam, Cambridge, UK) primary antibody at 4 °C. After incubation with primary antibodies, tissue sections were incubated with biotin-conjugated secondary antibodies and avidin-biotin enzyme reagents for 30 min. Chromogen 3,30-diaminobenzidine (DAB) tetrahydrochloride was used for colour development, followed by counterstaining with haematoxylin. Images were captured at 20× magnification using a Zeiss Axiocam microscope and analyzed using the ImageJ software as described previously. Briefly, for H&E-stained sections, the number of the centronucleated muscle fibers was manually measured. For MT-stained sections, the number of blue pixels was found using the ImageJ software and then normalized by area. The percentage of the fibrotic area was calculated by dividing the blue-dyed area by the total area. Finally, for CD31-stained sections, the number of brown pixels in the injured area was counted to calculate the area of CD31+ vessels, representing the degree of vascularization.
2.17. Statistical analysis
Data are expressed as mean ± standard deviation. For the multiple comparison test statistical evaluation, one way-ANOVA followed by Tukey's post-test was conducted using the GraphPad Prism 7 software (GraphPad Software, San Diego, CA, USA). To compare the differences between two groups, two-tailed unpaired Student's t-tests were performed. p Values <0.05 were considered statistically significant.3. Results and discussion
3.1. SRH preparation and characterization
We developed a new type of biodegradable silicate ion-releasing hydrogel (SRH) by incorporating Na2SiO3 into a poly(vinyl alcohol) (PVA)/starch network (Fig. 1). Na2SiO3 provided silicate ions and the PVA/starch hydrogel served as the ion-carrier (with high biocompatibility26) for controlled release. We adjusted the release kinetics and degradation rate of the SRH to test the hypothesis that silicate ions with appropriate releasing kinetics from the hydrogel scaffold possess myogenic and angiogenic effects and investigate the influence of the degradation rate of delivery scaffolds in their therapeutic efficacy in vivo. Based on our recent studies,27,28 we hypothesized that, by tuning the amylose/amylopectin ratio of starch, the material properties of the PVA/starch hydrogel could be regulated. Therefore, we prepared a library of different SRHs in which the starch was composed of different amylose/amylopectin ratios, and characterized their mechanical, ion releasing and degradation properties. The high-amylose corn starch (HAS, ∼40% amylopectin) and waxy corn starch (WS, ∼90% amylopectin) were used to adjust the amylose/amylopectin ratio. SRHs with increasing HAS/WS ratios were designated as H0W4, H1W3, H2W2, H3W1, and H4W0 (see Table S1, ESI† for detailed compositions); these hydrogels changed from a viscous liquid to a robust gel (Movie S1, ESI†), confirming the substantial effect of the starch type on the physical characteristics of the SRHs. Scanning electron microscopic characterization showed that all SRHs had a homogenous network (Fig. 2A and Fig. S1, ESI†). To confirm the myogenic and angiogenic activity of silicate ions rather than the PVA/starch scaffold, a CRH was prepared and set as the control material for in vitro and in vivo tests. The composition of the CRH was identical to that of H3W1, except for the replacement of Na2SiO3 with NaCl.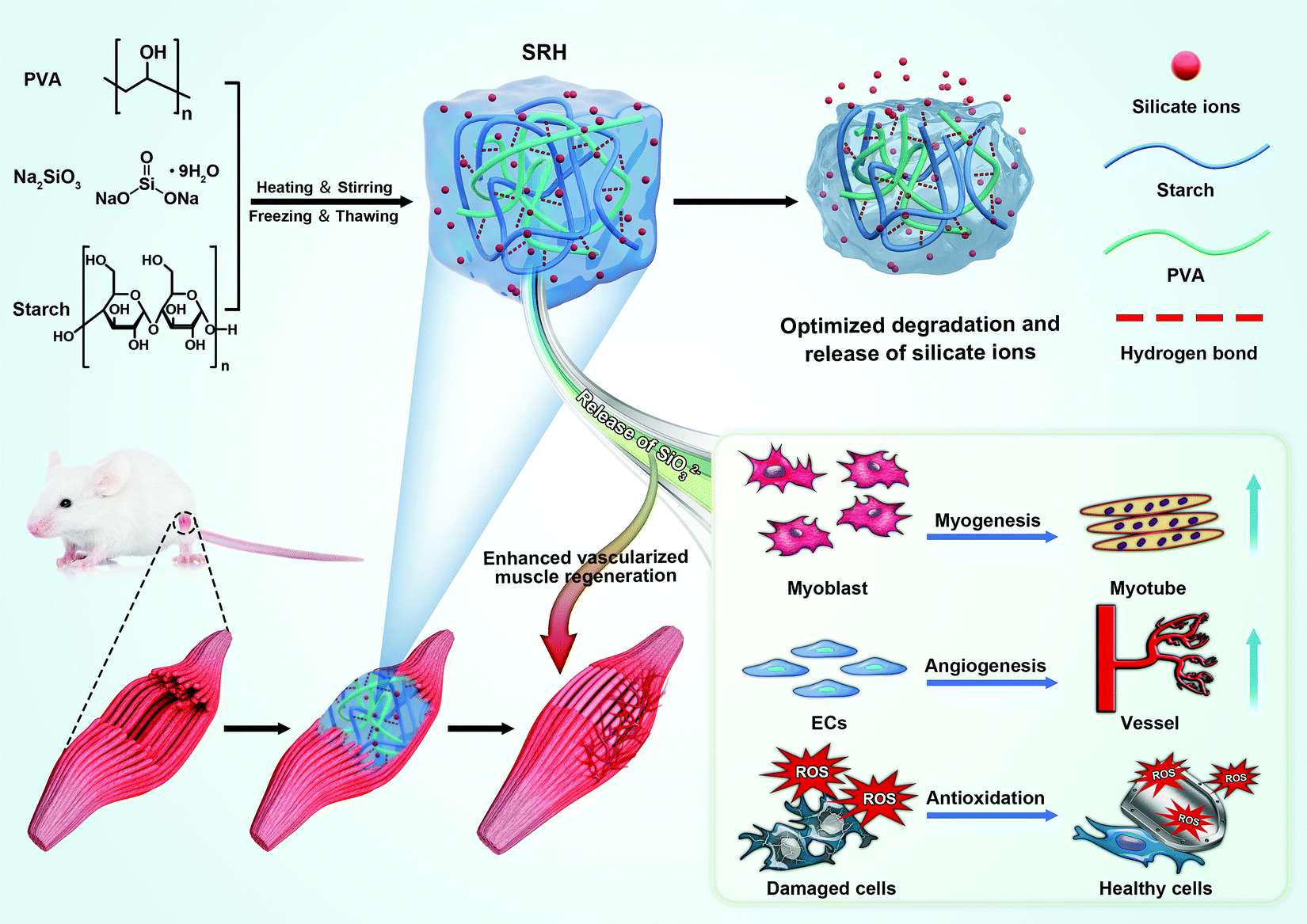 | ||
| Fig. 1 Schematic illustration of the fabrication process, properties and multiple functions of the silicate ion-releasing hydrogel (SRH) scaffold, and its application in skeletal muscle injury repair for enhancing vascularized muscle regeneration. | ||
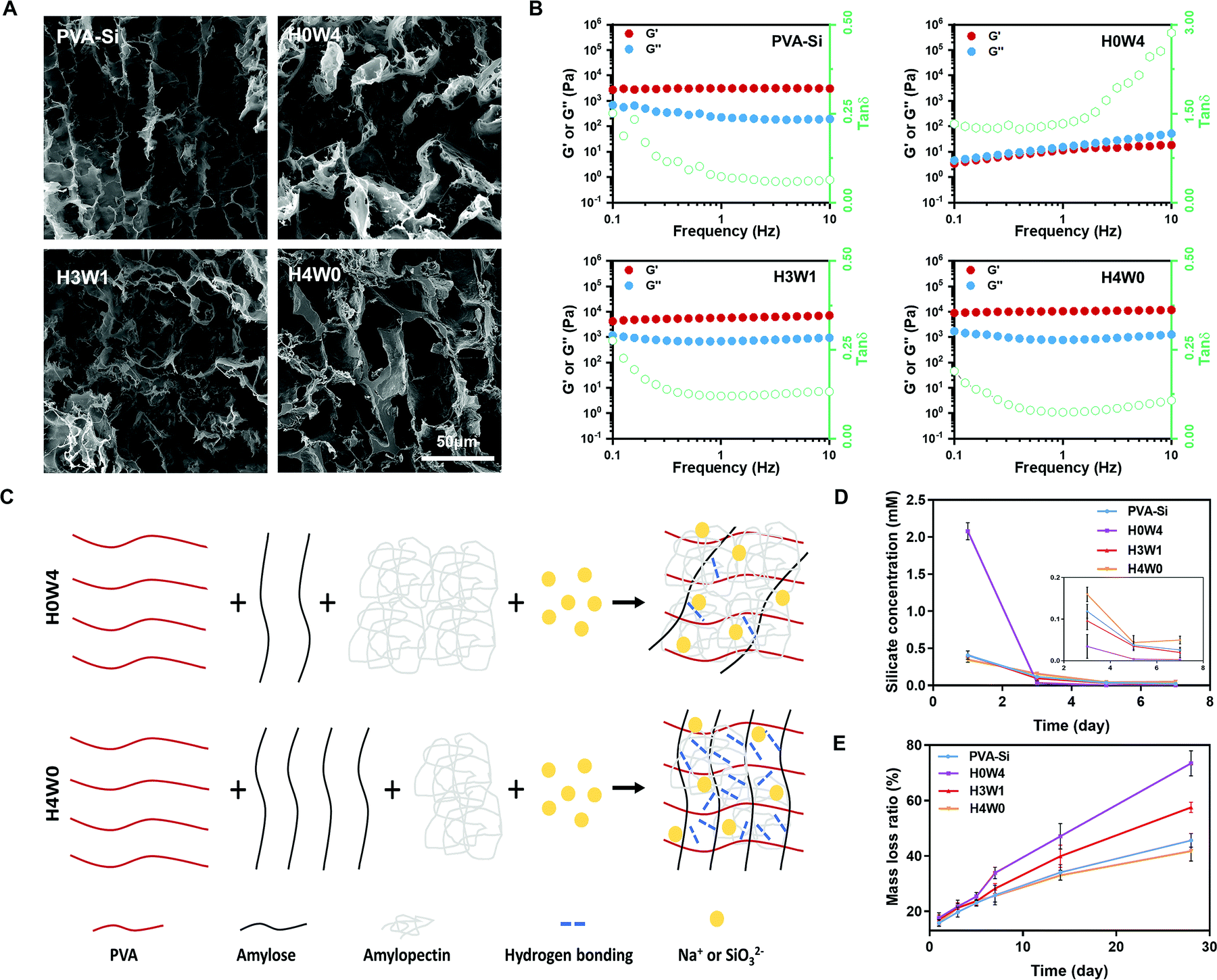 | ||
| Fig. 2 Microstructure, mechanical and degradation properties of hydrogels. (A) SEM images of PVA-Si and SRHs. (B) Dependence of G′, G′′ and the G′′/G′ ratio (loss factor) of PVA-Si and SRHs on the frequency of oscillation. (C) A schematic illustrating the network of H0W4 and H4W0. (D) Silicate concentrations of immersion solution after soaked with various silicate-containing hydrogels for different time periods, and the inset shows the silicate concentrations of each group on the 3rd, 5th, and 7th day. (E) The variation of the mass loss ratio of hydrogels with time during soaking in deionized water for 28 days. | ||
To be used in skeletal muscle repair, the SRH should also possess muscle-like stiffness and the capability to withstand cyclic muscle deformation. Therefore, we first characterized the mechanical properties of SRHs by rheological and tensile/compressive tests. The rheological properties of the hydrogels are shown in Fig. 2B and Fig. S2 (ESI†). The rheological properties of PVA-Si were similar to those of PVA, both exhibiting solid-like and elastic gel characteristics. The addition of HAS (H4W0) to the SRH network did not change the loss modulus (G′′) of the hydrogel but significantly increased its storage modulus (G′) and therefore reduced the G′′/G′ ratio, indicating an enhancement of the elasticity of the hydrogel. Interestingly, the addition of WS (H0W4) substantially altered the rheological properties of the hydrogel, which exhibited a higher G′′ than G′, and both were strongly frequency-dependent, behaving as a viscoelastic liquid. The G′ of H0W4 was three orders of magnitude lower than that of H4W0. Fourier transform infrared spectroscopy analysis (Fig. S3 and Text S1, ESI†) demonstrated that no chemical reactions occurred in SRHs, and the different effects of HAS and WS were attributed to physical interactions in the network. Considering the widely reported plasticization effect of inorganic salts on starch/PVA-based networks,29,30 Na2SiO3 should also act as a plasticizer in the SRH network, inhibiting intramolecular hydrogen bonding and the crystallization of PVA and starch (Fig. S3, S4 and Text S1, ESI†), and enhancing the interaction between the PVA chain and starch molecules to affect the mechanical properties of the SRH. As illustrated in Fig. 2C, amylopectin formed limited interactions with the PVA chain because of its highly branched and inflexible structure (with side chains much shorter than the main chain), leading to a significant softening effect.31 By contrast, amylose, which has a linear chain structure, easily formed a strong network with PVA chains via hydrogen bonding and chain entanglement,32 resulting in a strong network with high mechanical properties. Based on this mechanism, we effectively regulated the mechanical properties of the SRH by tuning the amylose/amylopectin ratio in the presence of a certain amount of Na2SiO3 (Fig. 2B and Fig. S2, ESI†). As a result, the storage moduli of PVA-Si, H3W1, and H4W0 were all at levels that supported the growth of myogenic cells.33
Next, we characterized the compressive and tensile behaviors of different SRHs (Fig. S5, ESI†). The addition of Na2SiO3 to pure PVA did not significantly alter its modulus, but substantially decreased its tensile strength and extensibility. For SRHs, both the compressive modulus (H2W2:1.07 ± 0.28 kPa; H3W1:3.08 ± 1.38 kPa; H4W0:14.60 ± 2.46 kPa) and tensile modulus (H2W2:3.64 ± 1.41 kPa; H3W1:19.66 ± 2.55 kPa; H4W0:90.11 ± 10.62 kPa) increased gradually as the HAS/WS ratio increased. The tensile moduli of H3W1, PVA-Si, and H4W0 were similar to the elastic modulus of natural murine skeletal muscles (ranging from 12 to 52 kPa)34 and at a level that supported myogenic cell survival and fusion.33 In addition to the modulus, the type of starch could also substantially affect the tensile strength and extensibility of the SRH. Notably, despite its lower modulus, H3W1 showed the comparable tensile strength and higher extensibility compared with H4W0, which could be explained by the hybrid network structure of the SRH constructed with a hydrogen bonding-mediated PVA/amylose network and ionic interaction-mediated amylopectin/ion complexes. Specifically, H3W1 should be synergistically strengthened by hydrogen bonding, which would preserve the elasticity and ionic crosslinking that effectively dissipate the crack energy under loading, resulting in simultaneous improvements in the tensile strength, extensibility, and toughness of hybrid hydrogels.14,35 Overall, the above results indicated that the mechanical properties of SRHs could be finely tuned by adjusting the amylose/amylopectin ratio. Among investigated compositions, PVA-Si, H3W1, and H4W0 exhibited mechanical characteristics suitable for skeletal muscle tissue engineering.12
The biological activities of silicate ions relied on their release kinetics from the SRH,36 which was tested by immersing samples in deionized water for up to 7 days and the critical time period for the delivery of therapy to enhance muscle regeneration.37 All hydrogels showed an efficient release of silicate ions on the first day (Fig. 2D) when minimal degradation occurred, indicating the ease of silicate ion transport through the hydrogel network. However, H0W4 showed a typical burst release of silicate ions, probably because of its high fluidity and degradability (as shown in the following degradation test). By contrast, silicate ions were continuously released from PVA-Si, H3W1, and H4W0 hydrogels with similar release behaviors. The potential toxicity induced by the release of silicate ions was also evaluated. As shown by viability test results in myoblasts, the silicate ions released from PVA-Si, H3W1, and H4W0 on the first day were all at a concentration that enhanced the viability of myoblasts (Fig. S6 and Text S1, ESI†), and the concentrations on days 5 and 7 (0.03–0.05 mM) were within the effective range to promote vascularization.38
In order to predict the in vivo degradation profiles of the SRHs, we next conducted an in vitro degradation test on hydrogels for up to 4 weeks (Fig. 2E), which is the normal duration of physiologic muscle healing.39 Overall, all the tested SRHs were biodegradable. The degradation rate of H4W0 was slightly lower than that of PVA-Si, probably because of the higher hydrogen bonding strength and the crosslinking density of H4W0, which hinders water uptake and debonding.40 The degradation rate of the SRH increased as the waxy starch content increased, which could be attributed to a decrease in the hydrogen-bonding density and the high swelling power of the waxy starch.31,41 Notably, in addition to its higher shear modulus, tensile strength, and extensibility, H3W1 also exhibited better degradability than PVA-Si, indicating the ability to independently tune the degradation behaviors of the SRH while maintaining appropriate mechanical properties.
Interestingly, we also found that the addition of Na2SiO3 significantly inhibited the growth of Staphylococcus aureus (the most common infectious pathogen in musculoskeletal traumatology)42 on the hydrogel surface (Fig. S7, ESI†) as the bacterial density decreased from 1115.5 ± 126.3 to 3.73 ± 2.6 counts mm−2. The pH values of the SRH extracts at day 1 were within 8–8.5, and gradually decreased with the immersion time (Fig. S8, ESI†). As reported before, a microenvironment with an alkaline pH inhibits the growth of most bacteria,43 probably by reducing bacterial ATP levels.44 Thus, the SRH might inhibit the adhesion of S. aureus by creating a basic microenvironment at the vicinity of the hydrogel surface. The bacterial inhibitory effect of the SRH could avoid the implant-related bacterial infection in the treatment of skeletal muscle trauma. In addition, owing to the presence of free ions in the network, the SRHs showed relatively high conductivity (Fig. S9, ESI†) compared to recently reported conductive hydrogels,13 enabling the delivery of electrical signals during healing that facilitates muscle regeneration by promoting the growth, differentiation, and communication of skeletal muscle cells.45,46
Taken together, we developed a new type of SRHs that simultaneously exhibited muscle-like mechanical properties, tunable degradability, continuous release of silicate ions, high cell compatibility, and efficient bacterial inhibition effects, thereby providing a solid basis for applications in skeletal muscle injury repair. Next, we performed in vitro experiments to determine whether silicate ions with appropriate releasing kinetics from the hydrogel scaffold mediate vascularized muscle regeneration by investigating the effects of silicate-enriched SRH extracts on the growth and function of myogenic and angiogenic cells. H3W1, which exhibited an appropriate releasing profile similar to those of PVA-Si and H4W0, was selected for the in vitro study. To determine the bio-effects of silicate ions (rather than the leachable of PVA or starch), the CRH was used as the control material for the in vitro study, which was prepared by replacing the Na2SiO3 of H3W1 with NaCl. Notably, the CRH exhibited similar rheological properties, tensile/compressive moduli, degradation profiles, and conductivity as those of the H3W1 hydrogel (Fig. S10, ESI†), besides being a perfect control material for the in vivo study.
3.2. Silicate ions from the SRH promoted the proliferation and myogenic differentiation of C2C12 myoblasts
C2C12 myoblasts were derived from mouse satellite cells and have been widely used for the in vitro evaluation of myogenic potential. Therefore, we investigated the biological effects of SRH extracts on the proliferation and myogenic differentiation of C2C12 myoblasts. Cell Counting Kit 8 (CCK8) assays (Fig. 3A) demonstrated that the SRH extracts significantly promoted the proliferation of C2C12 myoblasts compared with the cell culture medium (control group). By contrast, the cell proliferation in the CRH extract group was comparable to that in the control group. Furthermore, live/dead staining assays (Fig. 3B) also showed a higher density of viable C2C12 myoblasts in the SRH group than those in the CRH and control groups.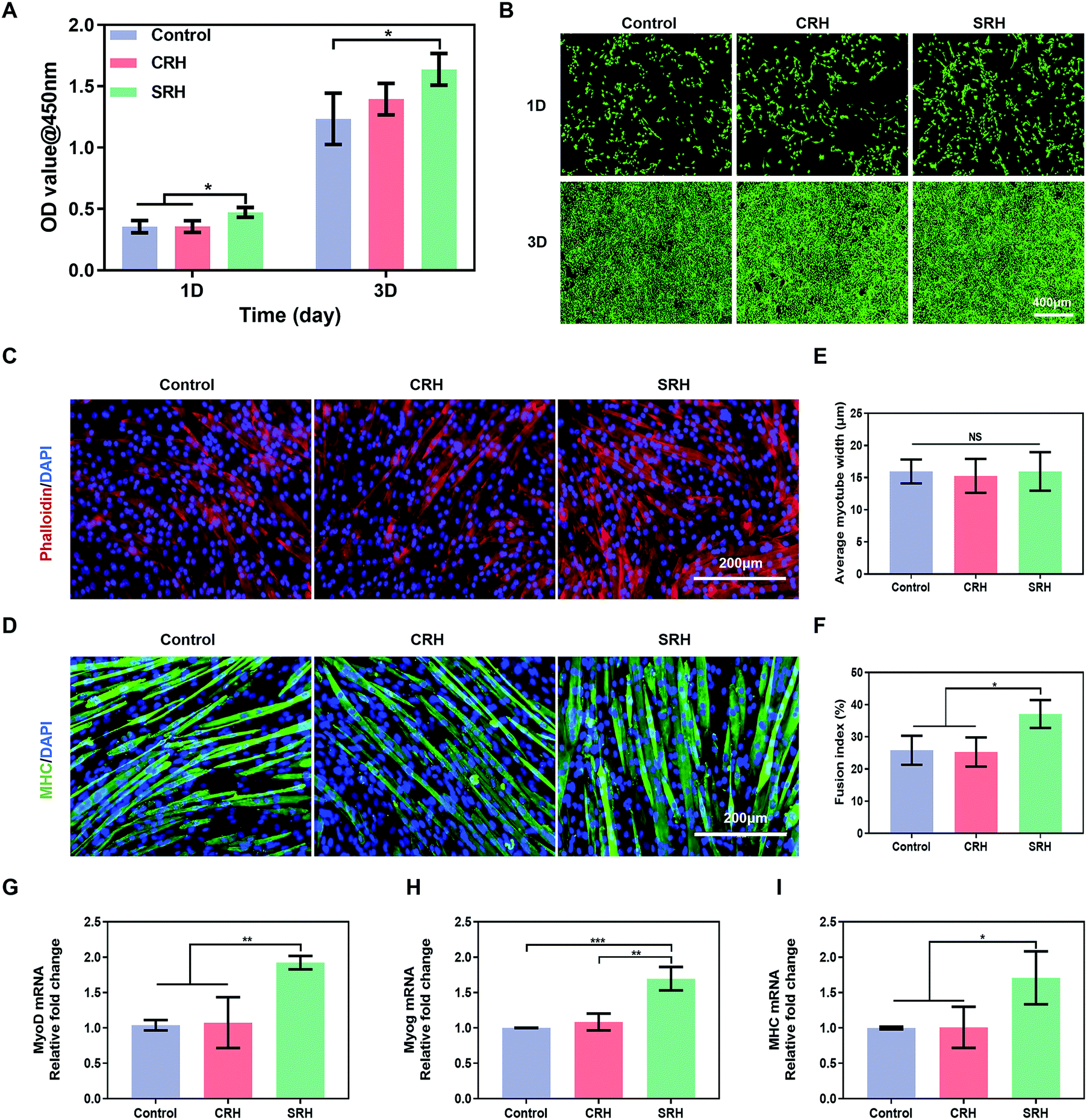 | ||
| Fig. 3 Silicate ions from the SRH promotes the proliferation and myogenic differentiation of C2C12 myoblasts. (A) Optical density values (@450 nm) of the CCK-8 assay on C2C12 myoblasts cultured in the hydrogel extracts or regular cell culture media for 1 and 3 days. (B) Fluorescence micrographs of live/dead stained C2C12 myoblasts after being cultured with the hydrogel extracts or regular cell culture media for 1 and 3 days. (C and D) Actin filament staining (Phalloidin: red, DAPI: blue) and immunofluorescence staining of the myogenic maker myosin heavy chain (MHC) (MHC: green, DAPI: blue) of myotubes formed by culturing C2C12 myoblasts with the rDM or hDM for 6 days. (E and F) Quantification of the average myotube width and fusion index after culturing C2C12 myoblasts with the rDM or hDM for 6 days. (G–I) Quantitative real-time PCR analysis of the myogenic gene expression of myotube formed by culturing C2C12 myoblasts with the rDM or hDM for 6 days. All data are expressed as mean ± S.D (n = 3). The p-values by one-way ANOVA are indicated. *p < 0.05, **p < 0.01, ***p < 0.001. rDM: regular differentiation medium. hDM: hydrogel extract-based differentiation medium. | ||
Subsequently, we evaluated the effects of hydrogel extracts on the myogenic differentiation of C2C12 myoblasts. C2C12 myoblasts were cultured in the regular differentiation medium (rDM) or the hydrogel extract-based differentiation medium (hDM) to evaluate myotube formation. Fig. 3C and D represent actin filament staining and MHC immunofluorescence staining images of the mature myotubes. All groups showed obvious myotube formation after culturing for 6 days in the rDM or hDM. To quantitatively analyze the effects of SRH extracts on myoblast differentiation, we measured the width and fusion index of the multinucleated fused myotubes. There were no significant differences in the average diameter of myotubes in each group (Fig. 3E). However, the fusion index in the SRH group (37.1% ± 4.3%) was significantly higher than those in the control (25.9% ± 4.5%) and CRH (25.3% ± 4.5%) groups (Fig. 3F).
The expression levels of myogenic-specific MyoD, Myog, and MHC genes, which are expressed at the early, late, and fully differentiated stages of myogenic development, respectively,47 were further evaluated (Fig. 3G–I). The expression level of MyoD in the SRH group was approximately 1.9-fold higher than those in the control and CRH groups. MyoD is a regulator of myogenesis from initiation to maturity, which not only promotes myogenic differentiation but also accelerates myoblast proliferation.48 Therefore, the previously observed promotion of C2C12 myoblast proliferation by the SRH may be related to the upregulation of MyoD expression. The expression of the muscle-specific transcription factor Myog was also improved in the SRH group, partially because of the upregulation of MyoD, which acts as an upstream regulator of markers for Myog.49MHC is a muscle filament protein that determines the metabolism and contraction properties of skeletal muscle fibers and is essential for myotube development.50 Notably, the expression levels of MHC genes in the SRH group were significantly higher than those in the CRH and control groups, indicating that silicate ions released by the SRH could promote the maturation of myotubes.
Taken together, these results demonstrated that cell- and drug-free SRH extracts could promote the proliferation and myogenic differentiation of C2C12 myoblasts, directly contributing to the enhanced regeneration of myofibers in vivo. It was reported that the expression of insulin-like growth factor 1 (IGF-1) from the osteoblast was significantly increased after incubation with 45S5 bioactive glass extracts.51 IGF-1 effectively mediates the growth of skeletal muscle tissues.52 In addition, silicate ions could promote in situ bone regeneration by activating the mammalian target of the rapamycin (mTOR) signaling pathway in the nervous system.19 The activation of the mTOR signaling pathway promoted myogenic differentiation through upregulating key myogenic determinant genes.53 Therefore, the myogenic effect of SRH extracts might be related to the stimulation of IGF-1 secretion and the activation of mTOR signaling.
Loss of vascular components, early inflammatory responses, and transfer of the remaining muscle tissue to a more oxidized phenotype synergistically lead to a highly oxidative microenvironment with the overproduction of reactive oxygen species (ROS).9,10,54 Excessive ROS production in the early post-traumatic period can increase proteolysis and skeletal muscle catabolism,55 disturb the normal healing process following skeletal muscle injury,56 and even lead to irreversible damage of skeletal muscle cells.57 Consequently, we next investigated the antioxidant properties of the SRH extracts to evaluate their ability to attenuate the damaging effects of oxidative stress during the early healing stages after an injury.
3.3. Silicate ions from the SRH protected C2C12 myoblasts from oxidative stress
We further investigated the protective effects of SRH extracts on the viability and myogenic differentiation of myoblasts when exposed to H2O2-induced oxidative stress. First, we evaluated the inhibitory effects of SRH extracts on the H2O2-induced expression of intracellular ROS using the redox-active dye 2′,7′-dichlorodihydrofluorescein diacetate (Fig. 4A and B). Compared with those of cells in the H2O2-free culture medium, the intracellular ROS levels of C2C12 myoblasts in the H2O2-containing culture medium increased by approximately 4.7-fold. Notably, the ROS levels in cells exposed to H2O2-containing SRH extracts were significantly decreased (∼1.9-fold decrease) compared with those in cells grown in the H2O2-containing culture medium, indicating that SRH extracts could reverse the overproduction of ROS in myogenic cells in an oxidative microenvironment. The antioxidative effects of silicate ions may be related to their ability to enhance the expression of superoxide dismutase-1 in cells that convert ROS into oxygen and water, as reported in previous studies on other cell lines.58,59 Next, we directly measured the C2C12 myoblast viability under oxidative stress conditions (Fig. 4C). After incubation in the H2O2-containing culture medium for 1 day, the viability of C2C12 myoblasts was significantly reduced to 68.33% ± 4.12% compared with that of cells in the H2O2-free control group, whereas this reduction was significantly attenuated in H2O2-containing SRH extracts (80.24% ± 4.34% compared with that of cells in the H2O2-free control group) compared with that in the H2O2-containing culture medium group. Moreover, the viability of cells in the H2O2-containing SRH extract group (85.19% ± 4.30% that of cells in the H2O2-free control group) was significantly higher than that of cells in the H2O2-containing regular cell culture medium group (74.67% ± 3.46% that of cells in the H2O2-free control group) after culturing for 3 days. The protective effects of SRH extracts on the viability of C2C12 myoblasts against H2O2 were further confirmed using live/dead staining. After 3 days of culture in the H2O2-containing culture medium, many dead cells were detected. In comparison, the toxic effects of H2O2 were attenuated by silicate ion-enriched SRH extracts, which elicited an increase in the density of living cells and a decrease in that of dead cells (Fig. 4D). These results suggested that SRHs have protective effects on the viability of myoblasts in high oxidative stress environments, probably by reducing the cellular production of ROS.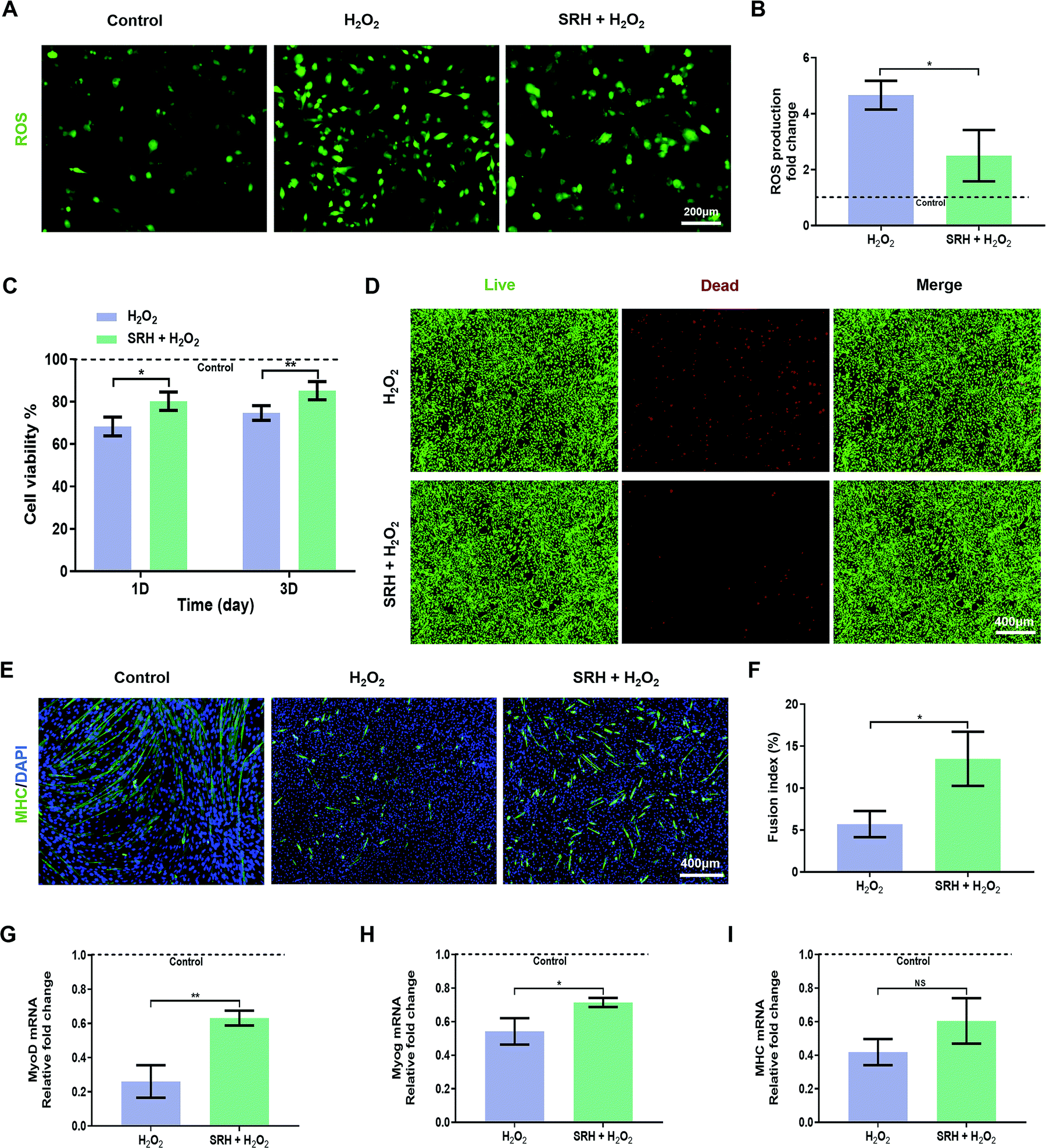 | ||
| Fig. 4 Silicate ions from the SRH protect C2C12 myoblasts against oxidative damage. (A) The ROS levels of C2C12 myoblasts after being cultured with the H2O2 added hydrogel extracts or regular cell culture media for 24 h assayed by a DCFH probe (ROS: green) and (B) quantitatively analyzed using ImageJ. (C) Viability of C2C12 myoblasts after being cultured with H2O2-containing hydrogel extracts or H2O2-containing regular cell culture media for 1 and 3 days versus a control group (H2O2-free regular cell culture media, viability set as 100%). (D) Fluorescence micrographs of live/dead stained C2C12 myoblasts after being cultured with H2O2-containing hydrogel extracts or H2O2-containing regular cell culture media for 3 days. (E) Immunofluorescence staining of the myogenic maker MHC (MHC: green, DAPI: blue) after cultured with the H2O2-containing hDM or rDM for 6 days and (F) the quantification of fusion index. (G–I) Quantitative real-time PCR analysis of myogenic gene expression in myotubes after being cultured with the H2O2-containing hDM or H2O2-containing rDM for 6 days versus a control group (H2O2-free regular cell culture media, gene expression set as 1). All data are expressed as mean ± S.D (n = 3). The p-values by two-tailed unpaired Student's t-test are indicated. *p < 0.05, **p < 0.01. rDM: regular differentiation medium. hDM: hydrogel-extract-based differentiation medium. | ||
High ROS levels also affect the efficiency of myogenic differentiation in myogenic cells by inducing the activation of the nuclear factor-κB pathway, resulting in decreases in MyoD expression and myogenesis capability.60 Therefore, we investigated the effects of SRH extracts on the myogenic differentiation of C2C12 myoblasts under high oxidative stress induced by H2O2. To quantify the morphological changes in myotubes, MHC immunofluorescence staining was performed on differentiated myoblasts (Fig. 4E). After culturing in the H2O2-containing rDM for 6 days, only a small number of scattered MHC-positive myotubes were formed, and the lengths and diameters of myotubes were significantly reduced compared with those in the normal control group. Although the myotubes generated in the H2O2-containing hDM were also atrophied compared with those in the normal control group, both the densities and lengths of the myotubes were substantially higher than those in the H2O2-containing rDM group. Quantitative analysis of the fusion indexes of the two groups (Fig. 4F) further confirmed that SRH extracts could attenuate the damaging effects of oxidative stress on the myotube formation capability of myogenic cells. The expression levels of myogenic differentiation-related genes in C2C12 myoblasts were further quantified (Fig. 4G–I). The expression levels of MyoD, Myog, and MHC genes were significantly decreased in H2O2-containing groups compared with those in the H2O2-free control group, highlighting the inhibitory effects of excessive oxidative stress on myogenic differentiation by SRH extracts. Notably, these results also demonstrated the effects of SRH extracts on the myogenic differentiation of C2C12 myoblasts in the presence of H2O2. Specifically, the expression levels of MyoD and Myog genes in the H2O2-containing hDM group were significantly increased by approximately 2.4- and 1.3-fold, respectively, compared with those in the H2O2-containing rDM group. The MHC gene expression in the H2O2-containing hDM group also increased by approximately 1.4-fold compared with that in the H2O2-containing rDM group, although the difference was not statistically significant. These results demonstrated that the SRHs effectively alleviated intracellular ROS production in myoblasts in an oxidative microenvironment generated by H2O2, thereby enhancing the viability and myogenic differentiation of myogenic cells and eventually enhancing muscle regeneration in the unavoidable oxidative stress microenvironment.
3.4. Silicate ions from the SRH promoted the migration, tube formation, and angiogenic differentiation of human umbilical vein endothelial cells (HUVECs)
HUVECs, which are highly proliferative and capable of forming capillaries,61 are commonly used to study angiogenesis, which is critical for vascularized muscle regeneration. CCK8 and live/dead staining assays revealed that the viability of HUVECs was similar in SRH, CRH, and control groups (Fig. S11, ESI†). The ability of HUVECs to initiate angiogenesis in hydrogel extracts was investigated using various assays. First, we evaluated the migration of HUVECs, which is important for vessel formation. Wound healing assays (Fig. 5A) demonstrated that the silicate ions released from the SRHs significantly enhanced HUVEC migration, with a marked decrease in the wound distance compared with those in the CRH and control groups at 24 h after wounding (Fig. 5D). Furthermore, the invasion ability of HUVECs, which was assessed using Transwell invasion chamber assays (Fig. 5B), was obviously increased in SRH extracts compared with those in CRH extracts and the control medium (Fig. 5E).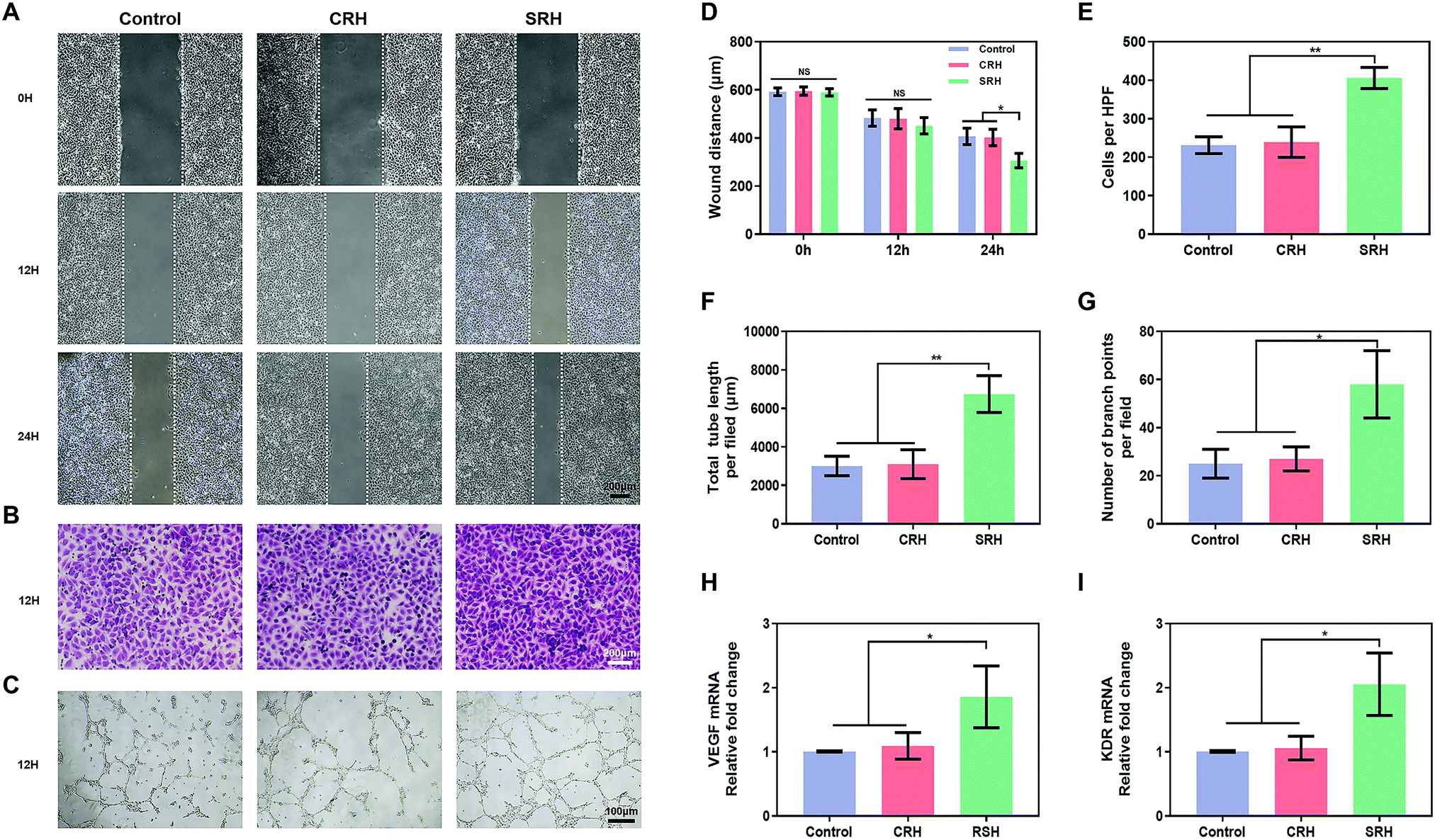 | ||
| Fig. 5 Silicate ions from the SRH promote the in vitro migration, tube formation and angiogenic differentiation of HUVECs. (A) Representative microscopic photographs of HUVEC migration when cultured with hydrogel extracts or regular cell culture media for 0, 12, and 24 h. (B) Transwell images of HUVECs cultured with hydrogel extracts or regular cell culture media for 24 h, and the cells that migrated to the lower chamber were stained with crystal violet. (C) Typical images of HUVECs cultured on the Matrigel basement membrane matrix in the presence of hydrogel extracts or regular cell culture media for 12 h. (D) Quantification of the migration rate of different groups in the wound healing assay. (E) Migratory cell number of HUVECs per high-power field (HPF) was calculated by ImageJ in the Transwell migration assay. (F and G) Quantitation of the tube length and branch points in the tube formation assay. (H and I) Relative mRNA expression of VEGF and KDR in HUVECs after cultured with hydrogel extracts or regular cell culture media for 5 days. All data are expressed as mean ± S.D (n = 3). The p-values by one-way ANOVA are indicated. *p < 0.05, **p < 0.01. | ||
Next, we used tube formation assays in Matrigel to evaluate the ability of HUVECs to organize into tube-like structures, which is the first visual indication of establishing the capillary lumen and the new vascular network. As shown in Fig. 5C, a low-extensive network with incomplete tubes was formed by HUVECs in the control and CRH groups at 12 h. By contrast, HUVECs in the SRH group exhibited more complex tube formation and enhanced the tube density compared with those in the other two groups. Quantitative analyses of the total tube length and the number of branch points (Fig. 5F and G) confirmed that the silicate-rich SRH extracts improved the tube formation ability of HUVECs. The above results indicated that silicate ions derived from SRHs could enhance the migration of HUVECs and promote the formation of capillary structures, consistent with previous findings in other silicate-containing biomaterials.20,62
Vascular endothelial growth factor (VEGF) plays important roles in angiogenesis by binding to its receptor kinase insert domain-containing receptor (KDR) on the cell membrane to directly stimulate EC proliferation, migration, and angiogenesis.63 Therefore, to further clarify the mechanisms mediating the pro-angiogenic effects of SRH extracts, the expression levels of VEGF and KDR genes in HUVECs were quantified using qRT-PCR (Fig. 5H and I). After 5 days of incubation, the expression levels of VEGF and KDR in HUVECs in the SRH group were markedly increased compared with those in CRH and control groups. The enhanced production of the key pro-angiogenic factor VEGF could activate EC behaviors, such as migration and tube formation, and ultimately improve the blood vessel growth. By contrast, as one of the most important VEGF receptors in the angiogenesis pathway, KDR is directly involved in cell migration and vascular invasion.64 These findings supposed that the SRHs could upregulate VEGF and KDR in HUVECs by releasing silicate ions, thereby promoting angiogenesis.
Taken together, the findings of these in vitro assays demonstrated that the silicate ions released by SRHs at an appropriate concentration could simultaneously enhance the proliferation and myogenic differentiation of C2C12 myoblasts in both normal and high oxidative stress environments, as well as promote migration, tube formation, VEGF expression, and KDR expression in HUVECs.
3.5. Local release of silicate ions from the SRH with bioinspired mechanical properties and biodegradability improved vascularized muscle regeneration
We next tested the ability of SRHs to enhance vascularized skeletal muscle regeneration after severe injury in vivo using a rat TA muscle defect model. Considering their similar ion release profiles and different degradation behaviors, we evaluated PVA-Si, H3W1, and H4W0 in this experiment. The CRH was used as a control material to evaluate the effects of silicate ions. In addition, treatment by weekly injection of Na2SiO3 was also performed as a control to evaluate the necessity for a hydrogel scaffold to achieve the continuous delivery of silicate ions (Fig. 6A).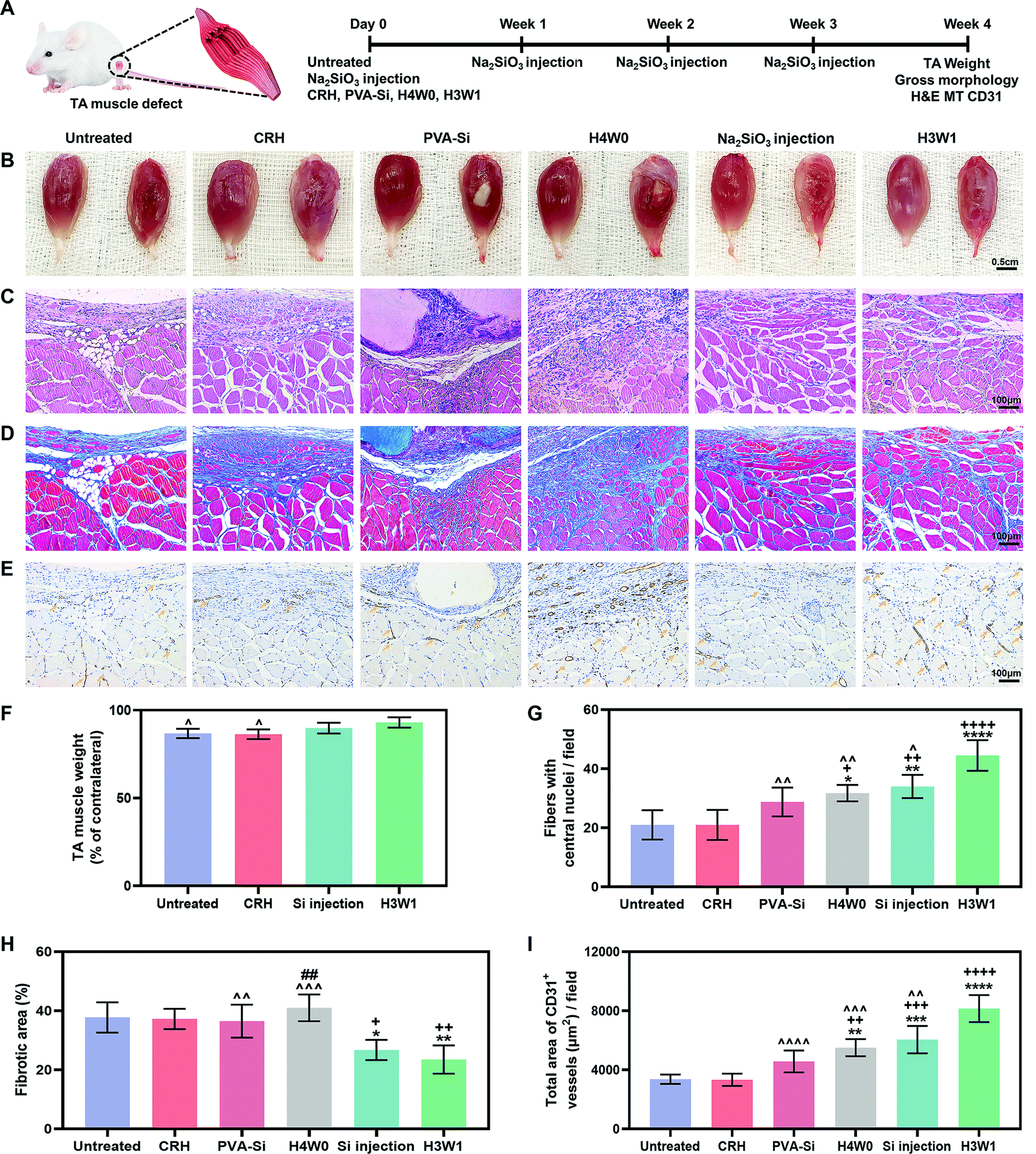 | ||
| Fig. 6 SRH improves vascularized muscle regeneration in the rat tibialis anterior (TA) muscle defect model. (A) Schematic illustration of the experimental design. (B) Gross appearance of TA muscles of different groups that harvested at 4 weeks after the operation. Right muscle: the TA muscle of the right leg (injured TA). Left muscle: the TA muscle of the left leg (contralateral normal TA). (C) Hematoxylin and eosin staining of the regenerated tissues of defect sites at 4 weeks after the operation. (D) Masson's trichome staining of the regenerated tissues of defect sites at 4 weeks after the operation. (E) Immunohistochemical staining of CD31 expression demonstrated the vascular-specific markers (yellow arrow) in the regenerated skeletal muscle tissue of the defect site at 4 weeks after the operation. (F) The weight of the TA muscle of different groups at 4 weeks after the operation (normalized with the weight of the contralateral normal TA muscle). (G) Quantitative number of centronucleated myofibers in the regenerated tissue at 4 weeks after the operation. (H) Quantitative of the fibrotic area in the de novo formed tissue region at 4 weeks after the operation. (I) Quantification of the total area of CD31-positive vessels per field. All data are expressed as mean ± S.D (n = 4, ^p < 0.05, ^^p < 0.01, ^^^p < 0.001 and ^^^^p < 0.0001 versus the H3W1 group; +p < 0.05, ++p < 0.01, +++p < 0.001 and ++++p < 0.0001 versus the untreated group; *p < 0.05, **p< 0.01, ***p < 0.001 and ****p < 0.0001 versus the CRH group; ##p < 0.01 versus the Si injection group). The p-values by one-way ANOVA are indicated. | ||
At 4 weeks post-surgery, the images of the gross morphology revealed no signs of infection or severe inflammation in all groups (Fig. 6B), and the residual implant material was still visible in the PVA-Si and H4W0 groups. H3W1 and CRH were completely degraded within 4 weeks, consistent with the normal duration of physiologic muscle healing (∼4 weeks). Obvious signs of muscle damage were observed in the untreated and CRH groups, with clear depressions on the surface where the TA was removed. By contrast, irregular new tissue filling was observed at the defect site in the Na2SiO3 injection group. Compared with the other groups, sites treated with H3W1 showed reduced fibrosis and smooth filling of muscle defects, although the results were still different from the ordered striations of normal muscles. We also quantified TA weights (Fig. 6F), except in the PVA-Si and H4W0 groups, based on the significant influence of residual implants on the muscle weight. The TA weight after treatment with H3W1 was similar to that of the contralateral normal muscle and was significantly increased compared with those in the untreated and CRH groups, indicating the effectiveness of H3W1 in promoting the restoration of muscle morphology and weight.
Hematoxylin and eosin (H&E) staining, Masson's trichrome (MT) staining, and immunohistochemical CD31 staining were then performed to systematically evaluate myogenesis, fibrosis level, and angiogenesis. The H&E staining results (Fig. 6C) clearly demonstrated the complete degradation of H3W1 and CRH within 4 weeks. By contrast, PVA-Si maintained a uniform white opaque appearance and clear boundary with the surrounding tissues, whereas H4W0 showed partial degradation with the infiltration of tissues. The higher in vivo degradation rate of H4W0 than PVA-Si was probably related to the accelerated hydrolysis of starch by glycosidase (amylase), which is present in body fluids.65 Inflammatory cells accompanied by fibroblast-like cells were observed in the vicinity of the PVA-Si hydrogel with collapsed muscle fibers and a small number of new myofibers. Although the bottom of the H4W0 hydrogel was fused with the muscle, large numbers of non-myogenic cells were observed at the degradation site to wrap the immature small new myofibers. Excessive infiltration of fibroblasts and adipose tissue was observed in the defect site of the CRH group, similar to that in the untreated group, indicating that the CRH hydrogel could not effectively enhance the restoration of skeletal muscle defects. Interestingly, newly regenerated myofibers with central nuclei were found in the defect sites of the H3W1 and Na2SiO3 injection groups, and the new myofibers in the H3W1 group were denser and more regular in shape than those in the Na2SiO3 injection group. Quantification analysis (Fig. 6G) demonstrated that the H3W1 group contained the highest number of new myofibers. At the same time, the numbers of new myofibers in the H4W0 and Si injection groups were significantly higher than those in the untreated and CRH treatment groups.
MT staining (Fig. 6D) showed extensive fibrosis and adipose tissue deposition in the untreated and CRH-treated groups. Intense collagen staining was also observed in the defect sites of the PVA-Si and H4W0 groups, which may be attributed to the high stiffness of the hydrogels66 and the persistent inflammatory reaction around the slowly degradable hydrogel. By contrast, only a small amount of fibrotic tissue was deposited around the new muscle fibers in the Na2SiO3 injection and H3W1 groups. The above observations were confirmed by quantification of the fibrotic area (Fig. 6H); according to the statistical analysis results, the fibrotic area percentages in the Na2SiO3 injection and H3W1 groups were significantly lower than those in other groups.
Immunohistochemical CD31 staining (Fig. 6E) demonstrated low vascularization in the untreated and CRH groups, with the most staining observed at the junction of the fibrous scar and muscle tissues. By comparison, all three silicate-containing hydrogels, as well as Na2SiO3 injection, enhanced CD31+ vessel formation. Fewer CD31+ vessels were found in the PVA-Si group, and these vessels were mainly distributed at the muscle/hydrogel interface. This may be related to the slow degradation of PVA-Si, which causes an inflammatory microenvironment, thereby impeding EC infiltration and vessel formation. Although most of the CD31+ vessels in the H4W0 group were located in the fibrotic area, considerable vascular infiltration was also observed around the underlying muscle fibers. Injection of Na2SiO3 also resulted in the formation of a vascular network around the new muscle fibers. Notably, mature vascular-like structures with visible lumens were identified in muscle defects treated with H3W1, indicating that H3W1 significantly promoted vascularization in regenerated skeletal muscles. The area of the CD31+ vessels in defect was also quantified (Fig. 6I). Compared with those in the untreated and CRH groups, H4W0, Na2SiO3 injection, and H3W1 groups all showed significant increases in the CD31+ vessel area. Similar trends were observed in the PVA-Si treatment group, although the differences were not significant. More importantly, the area of CD31+ vessels was significantly higher in the H3W1 group than the other groups.
The substantially enhanced vascularized skeletal muscle regeneration and reduced fibrotic tissue formation in the H3W1 group compared with the CRH group clearly demonstrated the therapeutic effects of silicate ions on skeletal muscle repair because the composition only differed in the type of anion (SiO32− for H3W1 and Cl− for CRH) coupled with the hydrogels showed comparable mechanical properties, degradation profiles, and conductivities. Moreover, the significantly improved restoration of skeletal muscle injury in the H3W1 group compared with that in the Na2SiO3 injection group suggested the necessity of a hydrogel scaffold for the application of the “ion therapy” scenario, serving as a temporary matrix in the early healing stages and not only supplying a source for continuous release of functional ions but also providing the physical support and exerting local mechanical signals mimicking muscle-specific microenvironments to further enhance regeneration. Importantly, our in vivo results demonstrated that the scaffold degradation rate greatly affected the tissue repair rate and the quality of skeletal muscles. Although similar ion-release profiles were observed, H3W1, which was completely degraded within the duration of muscle healing, showed significantly improved therapeutic ability compared with PVA-Si and H4W0. The beneficial effect of the appropriate degradation rate is potentially due to the prevention of rapid degradation-induced scar tissue filling or the slow degradation-induced prolonged immune responses.67
Overall, our findings demonstrated that silicate ions delivered to an injury site from the SRH scaffold with bioinspired mechanical properties and biodegradability could simultaneously enhance the de novo formation of muscle fibers and blood vessels and inhibit tissue fibrosis. A recent study demonstrated the therapeutic effects of silicon-containing 45S5 bioactive glass in the repair of skeletal muscle defects.21 However, it is a challenge to determine the contributing ions to its bioactivity due to its complex ionic composition. The difference between silicate ions from Na2SiO3 and those released from bioactive glasses should be determined in the future to evaluate the potential of silicon-containing bioactive glasses for muscle regeneration. To the best of our knowledge, this is the first report on the therapeutic ability of silicate ions to repair skeletal muscle defects. The observed therapeutic effects of silicate ions could be attributed to their synergistic effects on improving angiogenesis and myogenesis and attenuating oxidative stress damage, thereby promote vascularized skeletal muscle regeneration in vivo. The therapeutic effects of silicate ions can also be realized by promoting cell–cell communication. For example, VEGF expression, which was upregulated in SRH extracts, has been shown to promote the proliferation and migration of myoblasts and inhibit apoptosis.68 The role of silicate ions in modulating cell–cell interactions during the healing process of skeletal muscles will be investigated in the future.
The emergence of ion-releasing hydrogels may facilitate the establishment of novel strategies for tissue engineering and repair, enabling the development of cell- and drug-free clinical approaches for the repair of diverse soft tissue injuries, including skin wounds and cartilage defects. Optimization of the mechanical properties and biodegradability to match the tissue regeneration microenvironment is required when designing these novel hydrogels as temporal substrates to guide tissue regeneration.
4. Conclusions
Using a facile and environmentally friendly method, we developed a novel multi-functional silicate ion-releasing PVA/starch hydrogel platform for promoting vascularized skeletal muscle regeneration. The mechanical properties and degradation profile of the hydrogel could be easily tuned by adjusting the amylose/amylopectin ratio of starch. In vitro studies revealed that silicate ions released from the hydrogel scaffold could create a favorable regenerative microenvironment that promotes myoblast proliferation and myogenic differentiation, decreases oxidative stress, and enhances angiogenesis. An in vivo study demonstrated that the degradation rate of the scaffold is key to the therapeutic efficacy of silicate ions. Silicate ions delivered to a muscle injury site from the hydrogel scaffold with bioinspired mechanical properties and biodegradability simultaneously enhanced the de novo formation of muscle fibers and blood vessels and inhibited tissue fibrosis, facilitating vascularized muscle regeneration. These simple but important findings could be extrapolated to a broad area of skeletal muscle tissue engineering and used as guidance for the design of bioactive materials to treat formidable clinical problems such as volumetric muscle loss and sarcopenia.Author contributions
Pengcheng Xu, Qiang Yang and Lin Zhang: experimental design and execution, writing and revision; Kang Wu: material testing and data analysis; Saijilafu: revision and data interpretation; Yanjie Bai and Huilin Yang: revision and grant acquisition; Lei Yang, Xiao Lin and Huan Zhou: conceptualization, data interpretation, revision, supervision, and grant acquisition.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors are grateful for the financial support from the National Natural Science Foundation of China (No. 82025025, 31801585, 32171321 and 81622032), the National Key Research and Development Program of China (No. 2020YFC1107401), the Suzhou Science and Technology Project (No. SYS2019022), the China Postdoctoral Science Foundation (No. 2020T130459), the expert workstation program of Yunnan Province (202205AF150025) and the Priority Academic Program Development of Jiangsu High Education Institutions (PAPD).References
- S. Ostrovidov, V. Hosseini, S. Ahadian, T. Fujie, S. P. Parthiban, M. Ramalingam, H. Bae, H. Kaji and A. Khademhosseini, Tissue Eng., Part B, 2014, 20, 403–436 CrossRef PubMed.
- B. T. Corona, J. C. Wenke and C. L. Ward, Cells Tissues Organs, 2016, 202, 180–188 CrossRef PubMed.
- T. Vos, A. A. Abajobir, K. H. Abate, C. Abbafati, K. M. Abbas, F. Abd-Allah, R. S. Abdulkader, A. M. Abdulle, T. A. Abebo and S. F. Abera, Lancet, 2017, 390, 1211–1259 CrossRef.
- C.-H. Lin, Y.-T. Lin, J.-T. Yeh and C.-T. Chen, Plast. Reconstr. Surg., 2007, 119, 2118–2126 CrossRef CAS PubMed.
- S.-H. Lin, D. C.-C. Chuang, Y. Hattori and H.-C. Chen, J. Reconstr. Microsurg., 2004, 20, 227–235 CrossRef PubMed.
- M. M. Smoak and A. G. Mikos, Mater. Today Bio, 2020, 7, 100069 CrossRef CAS PubMed.
- M. M. Carleton and M. V. Sefton, J. Biomed. Mater. Res., Part A, 2021, 109, 2720–2739 CrossRef PubMed.
- D. Gholobova, L. Terrie, M. Gerard, H. Declercq and L. Thorrez, Biomaterials, 2019, 235, 119708 CrossRef PubMed.
- S. A. Dos Santos, A. J. Serra, T. G. Stancker, M. C. B. Simões, M. A. dos Santos Vieira, E. C. Leal-Junior, M. Prokic, A. Vasconsuelo, S. S. Santos and P. D. T. C. de Carvalho, Oxid. Med. Cell. Longevity, 2017, 2017, 5273403 Search PubMed.
- J. Gilbert-Honick, S. R. Iyer, S. M. Somers, R. M. Lovering, K. Wagner, H.-Q. Mao and W. L. Grayson, Biomaterials, 2018, 164, 70–79 CrossRef CAS PubMed.
- W. Kim, C. H. Jang and G. Kim, Chem. Eng. J., 2021, 419, 129570 CrossRef CAS.
- T. Hu, M. Shi, X. Zhao, Y. Liang, L. Bi, Z. Zhang, S. Liu, B. Chen, X. Duan and B. Guo, Chem. Eng. J., 2022, 428, 131017 CrossRef CAS.
- J. Ge, Y. Li, M. Wang, C. Gao, S. Yang and B. Lei, Chem. Eng. J., 2021, 425, 130333 CrossRef CAS.
- X. Lin, Y. Mao, P. Li, Y. Bai, T. Chen, K. Wu, D. Chen, H. Yang and L. Yang, Adv. Sci., 2021, 8, 2004627 CrossRef CAS PubMed.
- Y. Mao, M. Pan, H. Yang, X. Lin and L. Yang, Front. Mater. Sci., 2020, 14, 232–241 CrossRef.
- J. Yin, P. Xu, K. Wu, H. Zhou, X. Lin, L. Tan, H. Yang, K. Yang and L. Yang, Acta Metall. Sin. (Engl. Lett.), 2021, 35, 853–866 CrossRef.
- L. Ma, Y. Zhou, Z. Zhang, Y. Liu, D. Zhai, H. Zhuang, Q. Li, J. Yuye, C. Wu and J. Chang, Sci. Adv., 2020, 6, eabb1311 CrossRef CAS PubMed.
- X. Wang, L. Gao, Y. Han, M. Xing, C. Zhao, J. Peng and J. Chang, Adv. Sci., 2018, 5, 1800776 CrossRef PubMed.
- Y. Ma, K. Jiao, Q. Wan, J. Li, M. Liu, Z. Zhang, W. Qin, K. Wang, Y. Wang, F. R. Tay and L. Niu, Bioact. Mater., 2022, 9, 475–490 CrossRef CAS PubMed.
- K. Dashnyam, J. O. Buitrago, T. Bold, N. Mandakhbayar, R. A. Perez, J. C. Knowles, J.-H. Lee and H.-W. Kim, Biomater. Sci., 2019, 7, 5221–5231 RSC.
- W. Jia, H. Hu, A. Li, H. Deng and Q. Fu, Acta Biomater., 2019, 103, 306–317 CrossRef PubMed.
- O. Lambert, S. Poussard, M. Decossas, O. L. Bihan, S. Mornet and G. Naudin, Int. J. Nanomed., 2015, 10, 1479 CrossRef PubMed.
- L. Chang, Y. Li, M. Li, S. Liu, J. Han, G. Zhao, C. Ji, Y. Lyu, G. M. Genin and B. Bai, Chem. Eng. J., 2021, 420, 130398 CrossRef CAS.
- F. Li, Y. Li, Y. Tang, B. Lin, X. Kong, O. A. Oladele and Y. Yin, Mol. Biol. Rep., 2014, 41, 7715–7722 CrossRef CAS PubMed.
- J. Ge, K. Liu, W. Niu, M. Chen, M. Wang, Y. Xue, C. Gao, P. X. Ma and B. Lei, Biomaterials, 2018, 175, 19–29 CrossRef CAS PubMed.
- S. M. Andrabi, P. Singh, S. Majumder and A. Kumar, Chem. Eng. J., 2021, 423, 130219 CrossRef CAS.
- X. Lin, Y. Liu, A. Bai, H. Cai, Y. Bai, W. Jiang, H. Yang, X. Wang, L. Yang and N. Sun, Nat. Biomed. Eng., 2019, 3, 632–643 CrossRef CAS PubMed.
- Y. Mao, P. Li, J. Yin, Y. Bai, H. Zhou, X. Lin, H. Yang and L. Yang, J. Mater. Sci. Technol., 2021, 63, 228–235 CrossRef CAS.
- X. Jiang, T. Jiang, L. Gan, X. Zhang, H. Dai and X. Zhang, Carbohydr. Polym., 2012, 90, 1677–1684 CrossRef CAS PubMed.
- J. Lu, J. Gu, O. Hu, Y. Fu, D. Ye, X. Zhang, Y. Zheng, L. Hou, H. Liu and X. Jiang, J. Mater. Chem. A, 2021, 9, 18406–18420 RSC.
- N. Ben Ammar, T. Saied, M. Barbouche, F. Hosni, A. Hamzaoui and M. Sen, Polym. Bull., 2017, 75, 3825–3841 CrossRef.
- P. Chen, Y. Zhang, Q. Qiao, X. Tao and F. Xie, Food Hydrocolloids, 2020, 111, 106134 Search PubMed.
- E. Sleep, B. D. Cosgrove, M. T. Mcclendon, A. T. Preslar, C. H. Chen, M. H. Sangji, C. M. R. Pérez, R. D. Haynes, T. J. Meade and H. M. Blau, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E7919 CrossRef CAS PubMed.
- P. M. Gilbert, K. L. Havenstrite, K. E. G. Magnusson, A. Sacco, N. A. Leonardi, P. Kraft, N. K. Nguyen, S. Thrun, M. P. Lutolf and H. M. Blau, Science, 2010, 329, 1078–1081 CrossRef CAS PubMed.
- S. Gu, G. Cheng, T. Yang, X. Ren and G. Gao, Macromol. Mater. Eng., 2017, 302, 1700402 CrossRef.
- S. Bose, G. Fielding, S. Tarafder and A. Bandyopadhyay, Trends Biotechnol., 2013, 31, 594–605 CrossRef CAS PubMed.
- C. A. Cezar, P. Arany, S. A. Vermillion, B. R. Seo, H. H. Vandenburgh and D. J. Mooney, Adv. Healthcare Mater., 2017, 6, 1700202 CrossRef.
- H. Yu, J. Peng, Y. Xu, J. Chang and H. Li, ACS Appl. Mater. Interfaces, 2016, 8, 703–715 CrossRef CAS PubMed.
- S. Kenji, L. Yong, F. William, F. Kazumasa and B. Neil, Muscle Nerve, 2003, 28, 365–372 CrossRef PubMed.
- S. B. Qasim, S. Husain, H. Ying, M. Pogorielov, V. Deineka, M. Lyndin, A. Rawlinson and I. U. Rehman, Polym. Degrad. Stab., 2017, 136, 31–38 CrossRef CAS.
- X. Zhang, D. Guo, J. Xue, S. Yanniotis and I. Mandala, Food Funct., 2017, 8, 3792–3802 RSC.
- M. Morgenstern, C. Erichsen, M. Militz, Z. Xie, J. Peng, J. Stannard, W. J. Metsemakers, D. Schaefer, V. Alt and K. Søballe, J. Orthop. Res., 2021, 39, 136–146 CrossRef CAS PubMed.
- E. Padan, E. Bibi, M. Ito and T. A. Krulwich, Biochim. Biophys. Acta, Biomembr., 2005, 2, 67–88 CrossRef PubMed.
- J. Tan, D. Wang, H. Cao, Y. Qiao, H. Zhu and X. Liu, ACS Appl. Mater. Interfaces, 2018, 49, 42018–42029 CrossRef PubMed.
- H. Jo, M. Sim, S. Kim, S. Yang, Y. Yoo, J.-H. Park, T. H. Yoon, M.-G. Kim and J. Y. Lee, Acta Biomater., 2017, 48, 100–109 CrossRef CAS PubMed.
- H. Ding, M. Zhong, Y. J. Kim, P. Pholpabu, A. Balasubramanian, C. M. Hui, H. He, H. Yang, K. Matyjaszewski and C. J. Bettinger, ACS Nano, 2014, 8, 4348–4357 CrossRef CAS PubMed.
- S. Jana, S. K. L. Levengood and M. Zhang, Adv. Mater., 2016, 28, 10588–10612 CrossRef CAS PubMed.
- J. P. K. Hyatt, G. E. McCall, E. M. Kander, H. Zhong, R. R. Roy and K. A. Huey, Muscle Nerve, 2008, 38, 861–866 CrossRef CAS PubMed.
- C. Bentzinger, X. W. Yu and M. A. Rudnicki, CSH Perspect. Biol., 2012, 4, a008342 Search PubMed.
- M. J. McClure, K. Garg, D. G. Simpson, J. J. Ryan, S. A. Sell, G. L. Bowlin and J. J. Ericksen, J. Tissue Eng. Regener. Med., 2013, 10, E239–E249 CrossRef PubMed.
- P. Valerio, M. M. Pereira, A. M. Goes and M. F. Leite, Biomaterials, 2004, 15, 2941–2948 CrossRef PubMed.
- J. Menetrey, C. Kasemkijwattana, C. S. Day, P. Bosch, M. Vogt, F. H. Fu, M. S. Moreland and J. Huard, J. Bone Jt. Surg., Br. Vol., 2000, 1, 131–137 CrossRef.
- A. Matsumoto, A. Pasut, M. Matsumoto, R. Yamashita, J. Fung, E. Monteleone, A. Saghatelian, K. I. Nakayama, J. G. Clohessy and P. P. Pandolfi, Nature, 2017, 541, 228–232 CrossRef CAS PubMed.
- T. Chao, D. M. Burmeister, B. T. Corona and S. M. Greising, J. Appl. Physiol., 2019, 126, 1541–1549 CrossRef CAS PubMed.
- J. O. Ogunbileje, D. N. Herndon, A. J. Murton and C. Porter, J. Burn. Care Res., 2018, 39, 100–108 Search PubMed.
- P. Seale, B. Bjork, W. Yang, S. Kajimura, S. Chin, S. Kuang, A. Scime, S. Devarakonda, H. M. Conroe and H. Erdjument-Bromage, Nature, 2008, 454, 961–967 CrossRef CAS PubMed.
- C. M. Hee, O. J. Rong, N. D. Yang and T. Reshma, Oxid. Med. Cell. Longevity, 2015, 2016, 1–13 Search PubMed.
- A. Ilyas, T. Odatsu, A. Shah, F. Monte, H. K. W. Kim, P. Kramer, P. B. Aswath and V. G. Varanasi, Adv. Healthcare Mater., 2016, 5, 2199–2213 CrossRef CAS PubMed.
- M. Felipe, C. Tugba, R. Daniel, I. Fareed, H. V. Kojouharov, B. M. Chen, H. K. W. Kim, P. B. Aswath and V. G. Varanasi, J. Tissue Eng. Regener. Med., 2018, 12, 2203 CrossRef PubMed.
- E. Ardite, J. A. Barbera, J. Roca and J. Fernándezcheca, Am. J. Pathol., 2004, 165, 719–728 CrossRef CAS.
- M. Šalandová, I. A. van Hengel, I. Apachitei, A. A. Zadpoor, B. C. van der Eerden and L. E. Fratila-Apachitei, Adv. Healthcare Mater., 2021, 10, 2002254 CrossRef PubMed.
- C. Yang, H. Ma, Z. Wang, M. R. Younis, C. Liu, C. Wu, Y. Luo and P. Huang, Adv. Sci., 2021, 8, 2100894 CrossRef CAS PubMed.
- F. G. Rocha, C. A. Sundback, N. J. Krebs, J. K. Leach, D. J. Mooney, S. W. Ashley, J. P. Vacanti and E. E. Whang, Biomaterials, 2008, 29, 2884–2890 CrossRef CAS PubMed.
- P. Carmeliet and D. Collen, Ann. N. Y. Acad. Sci., 2010, 902, 249–264 CrossRef PubMed.
- C. Elvira, J. Mano, J. San Roman and R. Reis, Biomaterials, 2002, 23, 1955–1966 CrossRef CAS PubMed.
- J. S. Silver, K. A. Günay, A. A. Cutler, T. O. Vogler and K. S. Anseth, Sci. Adv., 2021, 7, 4501 CrossRef PubMed.
- C. A. Cezar and D. J. Mooney, Adv. Drug Delivery Rev., 2015, 84, 188–197 CrossRef CAS PubMed.
- A. Germani, A. Di Carlo, A. Mangoni, S. Straino, C. Giacinti, P. Turrini, P. Biglioli and M. C. Capogrossi, Am. J. Pathol., 2003, 163, 1417–1428 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tb00388k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |