
Axially chiral bay-tetraarylated perylene bisimide dyes as non-fullerene acceptors in organic solar cells†‡
Bernhard
Mahlmeister
a,
Rebecca
Renner
b,
Olga
Anhalt
b,
Matthias
Stolte
ab and
Frank
Würthner
 *ab
*ab
aCenter for Nanosystems Chemistry (CNC), Universität Würzburg, Theodor-Boveri-Weg, 97074 Würzburg, Germany. E-mail: wuerthner@uni-wuerzburg.de
bInstitut für Organische Chemie, Universität Würzburg, Am Hubland, 97074 Würzburg, Germany
First published on 7th October 2021
Abstract
A series of twisted perylene bisimide (PBI) dyes bearing different imide substituents as well as various aryl substituents in 1,6,7,12 bay positions inducing conformationally stable axial chirality are investigated as non-fullerene acceptors (NFAs) in organic solar cells (OSCs). In a comprehensive study, both imide and aryl substituent variations are tested within a simple inverted bulk heterojunction (BHJ) architecture with donor polymer PCE-10. The results are rationalized by single crystal structure analyses revealing the importance of the sterical demand imparted by the substituents in both positions on the packing arrangements. Comparative studies of racemic and enantiopure samples of these axially chiral dyes showed that the OSC performance is only affected by the enantiopurity if the chromophore is not well shielded. In that case, intimate π–π-stacking is disfavored for enantiopure compounds, thereby improving the OSC efficiency significantly by up to 24%. For a more shielded PBI bearing bulky 2,6-diisopropylphenyl (Dipp) substituents at imide and 2-naphthyl at bay positions, a maximum power conversion efficiency (PCEmax) of 4.3% is achieved even for the racemate.
Introduction
Perylene bisimides (PBIs) opened up the field of non-fullerene acceptors (NFAs) in organic solar cells (OSCs) with the pioneering work of Tang in the very beginning of 1986.1 The embedding of a simple PBI derivative into a p–n junction not only demonstrated its utilization as a stable and electron-poor n-type semiconductor but also initialized the quest for vacuum-processable organic functional materials that are still of major interest when it comes to commercial large scale applications.2 PBIs show outstanding photostability3 and chemical customizability which allows tailoring of desired material properties.4 Thus, they found application in many parts of OSCs besides NFAs such as interlayer dopants,5 cathode interlayers6 and acceptor polymers.7 Even if the focus on current NFA research has moved towards solution-processable A–D–A structural motifs such as ITIC8,9 or Y6,10 PBIs still hold significant importance in this field.11,12 Todays PBI-based NFAs are typically designed as twisted or star-shaped arrangements of multiple PBIs attached to each other by single or annulated bonding in bay, ortho or imide position.13–18 This twisted geometry prevents strong aggregation of the planar PBI chromophores, enables good solubility and allows the formation of an amorphous, three dimensional packing arrangement.19,20 However, good solubility as well as twisted geometry can also be achieved by twisting the PBI chromophore itself. This can be realized by the sterical congestion of four aryl bay substituents directly attached to the PBI π-core resulting in a twist of the naphthalene subplanes against each other.21 As a result, the LUMO levels are lowered, a bathochromic shift of the absorption maximum is induced and the solubility is highly increased.22 This is of particular interest for solution-processed organic solar cells (OSCs) as the solubilizing alkyl chains can be omitted which do not contribute affirmatively to charge transport properties anyway. The highly twisted geometry itself and the steric demand of the four aryl substituents are effective tools to prevent the formation of large crystalline domains within the bulk-heterojunction (BHJ).23,24 This was exploited by Sun an co-workers who reached appreciable high efficiencies of up to 4.1% in a BHJ OSC using a bay-tetraphenyl PBI with cyclohexyl residues in imide position (1d, Fig. 1a) combined with donor polymer PCE-10.25 The surprisingly simple synthesis to accomplish the fourfold arylation in the crowded PBI bay area was pioneered in 2006 by Zhu and co-workers26 followed by the Hoffmann group in 201027 who exploited different Suzuki cross-coupling routes. Applying this coupling chemistry, we recently synthesized a series of tetraarylated PBIs (Fig. 1a) with aryl substituents ranging from phenyl and para-trifluoromethyl phenyl over 1- and 2-naphthyl up to 1-pyrenyl for which for the first time we obtained the structural proof on its chirally twisted geometry via single-crystal structure analyses.22 Based on early predictions on the formation of highly stable atropo-enantiomers for such tetraphenyl PBIs,28 first experimental investigation of tetraarylated PBIs chirality was done in 201429 but did not find a way into a solid-state application so far unlike the closely related ortho-π-extended PBI double-[7]heterohelicenes utilizing the chiral core twist for a circular polarized light detection device.30 Even if Sun and co-workers utilized the twisted geometry to design well-soluble and potent NFAs,24,25 the impact of enantiopurity as well as the imide substituents remained so far unexplored. Indeed, there is only little investigation on morphological effects that are expected to significantly alter thin-film device performances while using either enantiopure species or their racemic mixtures. Within the few reported examples no clear trends are yet provided by corresponding OSC and organic thin-film transistor (OTFT) devices. The impact of enantiopurity ranges from zero up to an increase in orders of magnitudes of the respective figures of merit.31,32 Among the important family of branched alkyl chains used in organic electronics,33 probably the one of the most investigated chiral residues is the branched 2-ethylhexyl chain. It is used in OSC donor polymers31 or diketopyrrolopyrroles (DPPs) for OSCs34 as well as OTFTs35 and single-crystal field-effect transistors (SCFETs).36 Smaller and more rigid chiral residues such as 2-methylbenzyl introduced on one or two sides of naphthalene diimides (NDIs) have also recently been investigated in thin-film devices.37,38 Interestingly, even if key parameters such as charge carrier mobility or π–π-stacking are assessed independently, the interpretation of their interplay within a final device is not always straightforward.38 Also, the proportion of the chiral moieties on molecular size is not solely decisive as demonstrated by Itoh and co-workers by investigating a C60 fullerene derivative with a small chiral methyl unit that changed the OSC performance significantly by a factor of two using either enantiopure species or their diastereomeric mixtures.39 Besides this point-chirality mediated by side chains, the π-scaffold can show inherent chirality itself. Thus, the investigation on packing effects upon enantiopurity are of major interest within the growing field of contorted polycyclic aromatic hydrocarbons (PAHs) for organic electronics and spintronics.40 Investigating carbon helicenes, the groups of Crassous41 and Fuchter42 thoroughly investigated their materials as NFAs in OSCs or as p-type semiconductor in OTFTs, respectively. Whereas the enantiopure NFA showed superior morphology, it was the racemic mixture of the 1-aza[6]helicenes enabling a favorable charge transport pathway in the OTFT devices. The chirality-functional–property relationship was also recently investigated within a phototransistor based on a polymer blend of chiral PBIs. Superior performance again was recorded for devices of the racemic mixture.43 However, similar investigations on twisted nanoribbons,44 chiral peropyrenes45 or perylene dyes46 are not yet reported. | ||
| Fig. 1 (a) Chemical structures of the P-atropo-enantiomer of PBI NFAs 1–5 (top) as well as donor polymer PCE-10 and solvent additive 1-chloronaphthalene (CN) (bottom). (b) Schematic diagram of HOMO and LUMO energy levels of PBI NFAs22 as well as PCE-10.47 Literature reported PCE-10 energy levels have been adjusted with respect to Fc/Fc+ of −5.15 eV. (c) Normalized UV/Vis/NIR absorption spectra of PBIs as well as PCE-10 neat films on quartz spin-casted from a chloroform solution (10 mg ml−1). | ||
Herein, we study the impact of both the aryl as well as imide substituents within two series of the recently reported twisted PBIs22 as NFAs (Fig. 1a) in simple solution-processed OSCs. Device efficiencies are correlated with molecular packing deduced from single crystal X-ray structure analysis. Following our systematic study we finally designed the new NFA 2e reaching highest efficiencies of up to 4.3% PCEmax in a simple inverted BHJ architecture. As these materials show inherent axial chirality, atropo-enantiomers of 1b and 1e could be successfully separated to investigate the impact of enantiopurity on device efficiency and its dependency on solvent additive 1-chloronaphthalene (CN). Single crystal X-ray analyses of enantiopure crystals then revealed major differences imparted by the flexibility of imide substituents and their ability to form supramolecular assemblies in the solid state guided by the sterical congestion of bay-substituents. This favourable interplay led to the formation of the hitherto highest PBI twist-angle of 38.3° of enantiopure 1b. Circular dichroism (CD) spectroscopy not only proved the resemblance of chromophore packing in amorphous thin films with the single crystal structures but also identified a unique enhancement in Cotton effect of this X-cross chromophoric dimer arrangement of enantiopure PBIs which is further supported by TD-DFT calculations.
Results and discussion
Twisted PBIs designed as non-fullerene acceptors
To obtain comparable insights into their NFA applicability, all tetraarylated PBIs were initially tested using the donor polymer PCE-1047 in an inverted BHJ device architecture of ITO/ZnO/PBI:PCE-10/MoO3/Al (Fig. 2a). OSCs were fabricated and characterized under inert conditions and AM1.5 G irradiation. The optimized active layer blend consists of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 weight ratio of donor and acceptor material and was spin cast from a 15 mg mL−1 chlorobenzene (CB) solution. The NFAs were screened along two series varying either imide or bay aryl-residues using 2% of CN as solvent additive (Fig. 2, Table 1 and Fig. S2, ESI‡). Within the series of 1a–1e, phenyl as smallest aryl moiety was kept constant in bay-position and the imide substituents were varied. 1b bearing flexible n-butyl chains reached a moderate maximum power conversion efficiency (PCEmax) of 3.31%. The overall high open circuit voltage (VOC) of 0.84–0.88 V for these dyes indicated a low energy loss at around 0.6 V confirming PCE-10 as matching donor part. The obtained fill factors (FF) of about 40% are typical for single-core PBIs but lack behind some other oligomeric PBI-based NFAs that reach up to 80% due to the presence of enlarged π-surfaces with better intermolecular contacts. Accordingly, for these oligomers three-dimensional chromophore arrangement is achieved without scarificing close edge-to-edge or edge-to-face PBI interactions to guarantee balanced charge carrier mobilities within the BHJ.12,13,48 The obtained moderate short-circuit current densities (JSC) of 9–11 mA cm−2 indicated a considerable PBI contribution to the generated overall photocurrent as corroborated by UV/Vis/NIR and external quantum efficiency (EQE) measurements showing a complementary donor–acceptor absorbance over most of the visible range (Fig. 1c, 2c and Fig. S2, see the ESI‡). A proper film formation was observed by atomic force microscope (AFM) revealing a very smooth and homogeneous surface with a root-mean-squared roughness (Rq) of less than 1 nm that holds true for all of the properly working devices of 1b–1e (Fig. S4, ESI‡). In contrast, the low solubility of methyl functionalized 1a hampers the formation of a homogeneous active layer due to strong phase separation (Fig. S4, ESI‡) leading to a drop in all J–V characteristics and thus a low PCEmax of only 0.73%. By increasing size and stiffness of the imide substituents from 3-pentyl (1c) to cyclohexyl (1d), the device performance increases gradually from 3.53% to 3.72% and reaches even 4.07% PCEmax for the 2,6-diisopropylphenyl (Dipp) substituted 1e. The shielding of the PBI core by size and rigidity of the aryl as well as imide substitutes is in line with the NFA performance within this series. We like to note that Sun and co-workers accomplished a higher PCEmax of 4.1% for 1d and the same donor polymer PCE-10 after optimization.25
:1 weight ratio of donor and acceptor material and was spin cast from a 15 mg mL−1 chlorobenzene (CB) solution. The NFAs were screened along two series varying either imide or bay aryl-residues using 2% of CN as solvent additive (Fig. 2, Table 1 and Fig. S2, ESI‡). Within the series of 1a–1e, phenyl as smallest aryl moiety was kept constant in bay-position and the imide substituents were varied. 1b bearing flexible n-butyl chains reached a moderate maximum power conversion efficiency (PCEmax) of 3.31%. The overall high open circuit voltage (VOC) of 0.84–0.88 V for these dyes indicated a low energy loss at around 0.6 V confirming PCE-10 as matching donor part. The obtained fill factors (FF) of about 40% are typical for single-core PBIs but lack behind some other oligomeric PBI-based NFAs that reach up to 80% due to the presence of enlarged π-surfaces with better intermolecular contacts. Accordingly, for these oligomers three-dimensional chromophore arrangement is achieved without scarificing close edge-to-edge or edge-to-face PBI interactions to guarantee balanced charge carrier mobilities within the BHJ.12,13,48 The obtained moderate short-circuit current densities (JSC) of 9–11 mA cm−2 indicated a considerable PBI contribution to the generated overall photocurrent as corroborated by UV/Vis/NIR and external quantum efficiency (EQE) measurements showing a complementary donor–acceptor absorbance over most of the visible range (Fig. 1c, 2c and Fig. S2, see the ESI‡). A proper film formation was observed by atomic force microscope (AFM) revealing a very smooth and homogeneous surface with a root-mean-squared roughness (Rq) of less than 1 nm that holds true for all of the properly working devices of 1b–1e (Fig. S4, ESI‡). In contrast, the low solubility of methyl functionalized 1a hampers the formation of a homogeneous active layer due to strong phase separation (Fig. S4, ESI‡) leading to a drop in all J–V characteristics and thus a low PCEmax of only 0.73%. By increasing size and stiffness of the imide substituents from 3-pentyl (1c) to cyclohexyl (1d), the device performance increases gradually from 3.53% to 3.72% and reaches even 4.07% PCEmax for the 2,6-diisopropylphenyl (Dipp) substituted 1e. The shielding of the PBI core by size and rigidity of the aryl as well as imide substitutes is in line with the NFA performance within this series. We like to note that Sun and co-workers accomplished a higher PCEmax of 4.1% for 1d and the same donor polymer PCE-10 after optimization.25
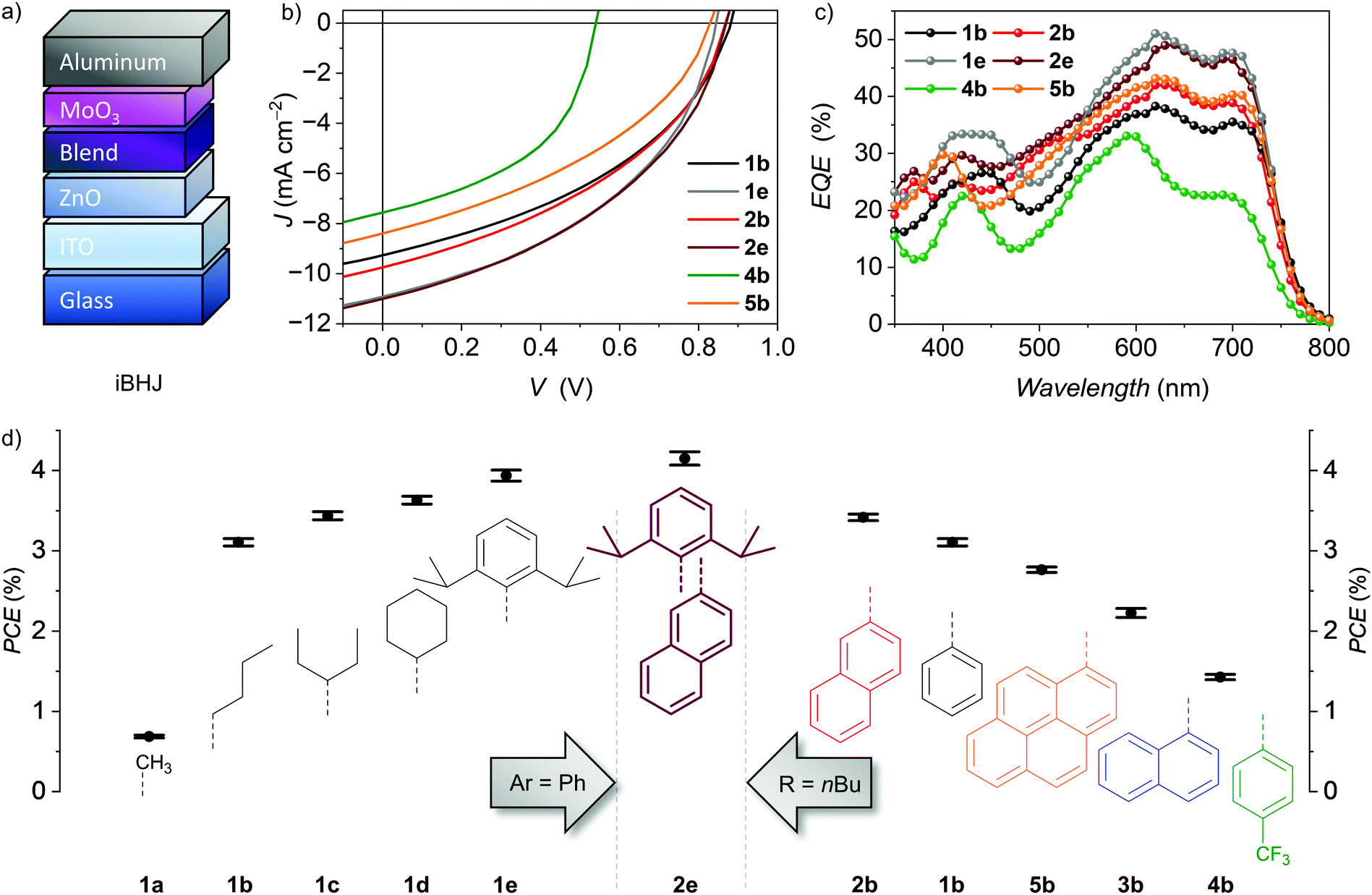 | ||
| Fig. 2 (a) Schematic device architecture of solution-processed inverted BHJ solar cells in ITO/ZnO/PBI:PCE-10/MoO3/Al configuration. (b) J–V characteristics of a selection of the best PCE-10 based devices using racemic mixtures of PBIs as NFAs. The total blend concentration was 15 mg ml−1 (16 mg ml−1 for 2e) in chlorobenzene (CB) with 1:1 donor/acceptor weight ratio for all devices (1.0:1.1 for 2e). 2% of 1-chloronaphthalene (CN) was used as solvent additive. (c) EQE spectra of the corresponding best devices displayed in (b). (d) OSC efficiencies of racemic PBIs used as NFAs along screening of imide (left: 1a–1e) and aryl substituents (right: 1b–5b) under optimized conditions of at least five independent devices. Error bars represent mean deviation. The solar cells were fabricated and characterized under inert conditions and AM1.5 G irradiation. | ||
:1 (1.0:1.1 for 2e) weight ratio with use of 2% of 1-chloronaphthalene (CN) as solvent additive. The cells in ITO/ZnO/PBI:PCE-10/MoO3/Al configuration were fabricated and characterized under inert conditions and AM1.5 G irradiation
| NFA (rac) | R, Ar | V OC (V) | J SC (mA cm−2) | FF (%) | PCEa (%) | PCEmax (%) |
|---|---|---|---|---|---|---|
| a Average of at least five up to ten independent devices. | ||||||
| 1a | Me, Ph | 0.60 ± 0.01 | −3.30 ± 0.11 | 35 ± 1 | 0.69 ± 0.02 | 0.73 |
| 1b | nBu, Ph | 0.84 ± 0.01 | −8.50 ± 0.24 | 44 ± 1 | 3.12 ± 0.11 | 3.31 |
| 1c | 3-Pent, Ph | 0.84 ± 0.01 | −9.66 ± 0.20 | 42 ± 1 | 3.40 ± 0.08 | 3.53 |
| 1d | Cy, Ph | 0.88 ± 0.01 | −9.93 ± 0.14 | 42 ± 1 | 3.63 ± 0.06 | 3.72 |
| 1e | Dipp, Ph | 0.84 ± 0.01 | −10.77 ± 0.32 | 44 ± 1 | 3.94 ± 0.10 | 4.07 |
| 2b | nBu, 2-Naph | 0.88 ± 0.01 | −9.60 ± 0.21 | 41 ± 1 | 3.45 ± 0.03 | 3.48 |
| 3b | nBu, 1-Naph | 0.76 ± 0.01 | −7.42 ± 0.27 | 40 ± 1 | 2.23 ± 0.08 | 2.35 |
| 4b | nBu, PhCF3 | 0.60 ± 0.01 | −5.49 ± 0.14 | 44 ± 1 | 1.43 ± 0.04 | 1.55 |
| 5b | nBu, 1-Pyr | 0.80 ± 0.01 | −8.77 ± 0.12 | 40 ± 1 | 2.76 ± 0.05 | 2.83 |
| 2e | Dipp, 2-Naph | 0.84 ± 0.01 | −11.17 ± 0.36 | 45 ± 1 | 4.15 ± 0.09 | 4.28 |
Due to the variation of aryl substituents, both steric and electronic factors are affected within the second series. Thus, n-butyl as the most flexible imide substituent was kept constant within the series 1b–5b, while the four aryl substituents where varied. Upon the attachment of electron-withdrawing para-trifluoromethyl units to the phenyl residues of 4b the device efficiency PCEmax decreased significantly from 3.31% (1b) to 1.55%. The pronounced drop in VOC to only 0.60 V is in line with its LUMO level lowered by 0.1 eV (Fig. 1b).22 The concomitant loss in JSC down to 5.49 mA cm−2 can be attributed to a significantly reduced donor contribution to the extracted photocurrent at 700 nm as the EQE (Fig. 2c and Fig. S2, ESI‡) reveals. However, as also the BHJ surface roughness is drastically increased (Rq = 13.3 nm, Fig. S4, ESI‡) it is probable to consider both electronic and morphological factors for the low performance. Enlarging the phenyl substituent to its next homologue, the PBI isomers bearing 1-naphthyl (3b) or 2-naphthyl (2b) residues are obtained. Even if both NFAs show the same LUMO levels and exhibit comparable homogeneous thin-films the more laterally extended 2b is highly superior in all OPV characteristics. Most prominent is the loss in VOC from 0.88 V to 0.76 V for 3b. Accordingly, while 2b reaches a PCEmax of 3.48%, for 3b only 2.35% are achieved. Our recent studies on conformational isomers in solution revealed low rotation barriers in 3b for its 1-naphthyl aryl residues and the presence of up to 14 stereoisomers in solution, while for 2b this number is reduced to four, due to the intimate π-stacking of the 2-naphthyl substituents.22 Presumably, the multitude of present species of 3b also in its amorphous spin-cast thin film will lead to a broadening of the density of states and hampers a uniform chromophore arrangement as well as defined interface with the donor polymer. Appending 1-pyrenyl instead of 1-naphthyl, 5b shows again slightly elevated PCEmax of up to 2.83% due to an increased VOC of 0.80 V and higher PBI contribution to the photocurrent revealed by EQE (Fig. 2c and Fig. S2, ESI‡). This can be correlated with the broadened 5b absorption due to the charge transfer (CT) character between electron-rich pyrene and electron-poor PBI within 5b. The electron donating character of the pyrene bay-substituents results in a localization of HOMO and LUMO on the pyrene units and PBI core, respectively.22
Combining the information from both the imide (1a–1e) as well as the aryl (1b–5b) substituent variation we successfully designed a new derivative 2e (for details see the ESI‡) consisting of the aryl (2-naphthyl) and imide groups (Dipp) that gave the highest PCEs within each respective series. By this systematic approach we were able to enhance the JSC as well as FF of the simple bulk-heterojunction solar cells further (Fig. 2b, Table 1 and Fig. S2, ESI‡). The optimized blend ratio of 1.0:1.1 resulted in the highest JSC of 11.2 mA cm−2 with EQE of up to 49% at 640 nm (Fig. 2c). An appreciable high FF of 45% was obtained to finally result in a PCEmax of up to 4.28%. This should result from the fusion of the discussed attributes such as its high degree of chromophore shielding while maintaining the conformational stability guided by the π-stacked 2-naphthyl units22 overall resulting in a homogeneous film forming of the PCE-10 blend. To the best of our knowledge this is the highest reported PCE value of a single-core PBI based NFA without an extension of the π-system or embedding of heteroatoms.
Molecular packing
To shed light into influence of the chromophore arrangement by the variation of the imide and aryl substituents on device performance, a comprehensive analysis on the most relevant single-crystal structures from racemic mixtures of 1b, 2b, 1e and 2e was performed. Single crystals of suitable size and quality could be obtained by slow-evaporation method out of either chloroform (1b) or chlorobenzene (1e, 2b, 2e) solution under optimized conditions using methanol or n-hexane as co-solvent, respectively. They reveal an anisotropic arrangement of alternating P- and M-atropo-enantiomers within their respective unit cells (Fig. 3). At this point it should be noted that the activation barriers for the conversion between P- and M-atropo-enantiomers is rather high (ΔG‡ ≈ 120 kJ mol−1).22 Accordingly P- and M-atropo-enantiomers will not interconvert under our procession conditions. All crystal structures depicted here contain solvent molecules as well as the small disorder of the n-butyl chains which is not depicted for clarity. Within this series, the flexible n-butyl residues of rac-1b allow the closest chromophore interactions between PBI headland region leading to a unique slip-stacked packing arrangement of alternating P- and M-atropo-enantiomers forming two isolated double strands within the unit cell of the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group (Fig. S6, ESI‡). Even as the π–π distances between two stacked imide units of adjacent chromophores within one double strand can come as close as 3.1 Å, a large lateral displacement up to 10.7 Å (center-to-center distance) is introduced by the sterical congestion of the bay-substituents preventing the chromophores from efficient excitonic Coulomb coupling (−140 cm−1 and −156 cm−1) along the double strand (see the Table S2, ESI‡).49
space group (Fig. S6, ESI‡). Even as the π–π distances between two stacked imide units of adjacent chromophores within one double strand can come as close as 3.1 Å, a large lateral displacement up to 10.7 Å (center-to-center distance) is introduced by the sterical congestion of the bay-substituents preventing the chromophores from efficient excitonic Coulomb coupling (−140 cm−1 and −156 cm−1) along the double strand (see the Table S2, ESI‡).49
 | ||
| Fig. 3 Molecular packing of PBIs rac-1b (a), rac-2b22 (b), rac-1e (c) and rac-2e (d) in their respective single crystal structures. PBI atropo-enantiomers are colored in blue (P) and red (M). The ellipsoids are set to 50% probability (C: gray). Disorder of the imide residues, hydrogen atoms as well as solvent molecules are omitted for clarity. Distances of closest π–π interactions (blue), the distance between the planes of adjacent chromophores (black) as well as twist angle between two chromophores (red) and distances between the centroids of the perylene cores (green) are indicated as well. | ||
As already mentioned the sterical congestion of the aryl substituents leads to stable axial chirality due to the core twist of the PBI. Considering the PBI naphthalene subunits as individual planes, twist angles of about 32° are measured between them for rac-1b. This is within the range of our previously reported twisted PBIs (1d, 2b)22 that is still below 37° as reported for the most contorted bay-tetrabromo PBIs due to the high van-der-Waals radii of its halogen bay substituents.21 In contrast to the spherical heavy atoms, two adjacent aryl moieties align to each other mostly co-parallel. Their intramolecular π–π distance can be given by the distance between the respective centroids that is similar for all derivatives and ranges between 3.2–3.5 Å. The rac-2b single crystal structure was reported previously by us.22 By increasing the PBI lateral extension going from phenyl (rac-1b) to 2-naphthyl (rac-2b), the crystal symmetry and the packing motif of alternating P- and M-atropo-enantiomers is preserved but now the enlarged aryl substituents incur the guiding of chromophore arrangement and confine the number of interacting chromophores down to a number of two (Fig. 3b). The closest π–π distance within the slip-stacked dimer arrangement is increased by 0.5 Å up to 3.6 Å but the chromophore centers come slightly closer down to 10.0 Å. Apart from this entity further adjacent chromophores stay more distant compared to the chromophores in rac-1b crystal. Thus, the distance between planes of P- and M-atropo-enantiomers of the same unit cell is increased from 6.0 Å to 7.2 Å and the side-on neighbours of same helicity in b-axis direction are moved away from 12.6 Å to 14.5 Å. In summary, the strain imparted by the n-butyl-promoted slip-stacked arrangement combined with the steric demand of the 2-naphthyl units directly contacting the chromophore leads to this considerably higher core twist of up to 36.6°. It is noteworthy that our previous work on these twisted PBIs revealed a multitude of conformational isomers of 3b in solution due to various possibilities for the arrangement of the aryl residues during the reaction that are similar in energy. This conformational variability was not as pronounced for 2b which might explain to the tremendous OSC performance differences for these two constitutional isomers (vide supra).
By changing the imide substituent from flexible n-butyl to the stiff and bulky Dipp moieties, the isolation of the chromophore cores within the crystal lattice significantly increases. The single crystal structure of rac-1e (Fig. 3c) reveals that intimate π–π-stacking between adjacent chromophores is no longer possible and that the center-to-center distance is increased to 8.5 Å due to the steric demand of the Dipp residues. In contrast to the close arrangement promoted by the flexible n-butyl chain, the strain on the isolated PBI core of rac-1e is reduced, yielding a twist angle of 32.2°. The maximum in chromophore isolation was obtained for rac-2e (Fig. 3d). In contrast to the previous structures, 2e has two pairs of enantiomers instead of one that are embedded within its unit cell of P21/c space group. The distance between adjacent P- and M-atropo-enantiomers in a slip-stack arrangement is large revealing center-to-center distances of 13.5 Å or 9.7 Å between the planes of the respective chromophores. The two molecules at the exterior of the unit cell are rotated by 73° towards the inner pair of enantiomers that are evenly distant by 16.8 Å. It becomes apparent that the steric demand of the Dipp residue of the outmost molecules promotes the side-on orientation of two chromophores of the same helicity whereas its 2-naphthyl units guide the distance within its heterochiral arrangement of the elementary cell. No chromophore interactions for rac-2e are possible which further reduces the strain on its π-scaffold leading to the smallest twist angle of only 30.9° within this series. While for each racemate both P- and M-atropo-enantiomers are alternatingly arranged within the respective unit cell, only the flexible n-butyl chains of 1b and 2b allow for some minor face-to-face π-interactions between them but only at the expense of a huge longitudinal displacement and accordingly large center-to-center distance.
Impact of enantiopurity on organic solar cell performance
As the steric demand of both substituents highly affects the molecular packing of alternating P- and M-atropo-enantiomers in the solid state of the racemate, we expected a pronounced difference for the packing structure for enantiopure compounds. For the bay-phenyl derivatives 1b (n-butyl) and 1e (Dipp), the P- and M-atropo-enantiomers could be fully separated in sufficient quantity as well as purity using a semipreparative HPLC device equipped with a chiral stationary phase, showing perfect mirror images of the respective CD spectra (vide infra and see the Fig. S7, ESI‡). In contrast, a sufficient separation of the most potent NFA 2e was not possible as a consequence of the effective shielding of the twisted core structure by the large 2-naphthyl and Dipp substituents that prevent sufficient chiral interactions with the stationary phase. OSCs of racemic mixtures as well as isolated P- and M-atropo-enantiomers of 1b and 1e (Fig. 4a) as NFAs were fabricated either without solvent additive or with 2% of CN (Fig. 4b, Table 2 and see the Fig. S3, ESI‡). The binary blends of n-butyl substituted 1b show a significant increase in PCE by about 24% from 2.5% up to 3.1% upon the change from the racemic mixture to the enantiopure P- or M-atropo-enantiomers averaged over at least ten devices. The reduced efficiency of rac-1b based devices compared to enantiopure P-1b or M-1b (Fig. 4b and c) solely derives from a reduced short-circuit current density, while VOC (0.84 V) and FF (40–41%) are almost unaffected. JSC of the binary blend without additive decreases by more than 18% from up to 9.0 (P/M) down to 7.5 mA cm−2 (rac). The corresponding EQE shows its maximum of 40% at 620 nm and drops down to 34% upon the change from enantiopure to the racemic NFA even while the overall thin-film absorbance remains the same for all three active layers (Fig. 4d). It is apparent that the extracted photocurrent of rac-1b is reduced equally within the entire spectral region and cannot be attributed to either the NFA region between 400–650 nm or the one of the donor polymer PCE-10 with its maximum in absorbance at 700 nm. Thus, both channel 1 (hole) and channel 2 (electron) contribution to the photocurrent50 are affected by the different packing of the enantiopure and racemic chiral NFAs. | ||
| Fig. 4 (a) Schematic illustration of P- (blue) and M- (red) atropo-enantiomers of chiral PBI 1. (b) J–V characteristics of best OSC devices with inverted configuration of ITO/PCE-10:PBI/MoO3/Al of 1b without solvent additive (solid) and with 2% CN (dashed) as P (blue), M (red) or racemic (black) PBI species. The solar cells were fabricated and characterized under inert conditions and AM1.5 G irradiation. (c) Mean PCEs of 1b (left) and 1e (right) based OSCs with use of no solvent additive (orange spheres) or 2% CN (green spheres) of 10 (1b) or 5 (1e) independent devices. Error bars represent mean deviation. (d) EQE (symbols) and UV/Vis/NIR absorption (solid line) spectra of corresponding P (blue), M (red) or racemic (black) 1b based devices without solvent additive. | ||
:1 weight ratio with optional use of 1-chloronaphthalene (CN) as solvent additive. The cells in ITO/ZnO/PBI:PCE-10/MoO3/Al configuration were fabricated and characterized under inert conditions and AM1.5 G irradiation
| NFA | CN (%) | V OC (V) | J SC (mA cm−2) | FF (%) | PCEa (%) | PCEmax (%) |
|---|---|---|---|---|---|---|
| a Average of at least ten (1b) or five (1e) independent devices. | ||||||
| P-1b | — | 0.84 ± 0.01 | −8.96 ± 0.25 | 41 ± 1 | 3.11 ± 0.08 | 3.25 |
| M-1b | — | 0.84 ± 0.01 | −8.91 ± 0.17 | 40 ± 1 | 2.99 ± 0.06 | 3.15 |
| rac-1b | — | 0.84 ± 0.01 | −7.50 ± 0.09 | 40 ± 1 | 2.52 ± 0.05 | 2.65 |
| P-1b | 2 | 0.84 ± 0.01 | −9.52 ± 0.17 | 44 ± 1 | 3.48 ± 0.07 | 3.68 |
| M-1b | 2 | 0.84 ± 0.01 | −9.51 ± 0.18 | 43 ± 1 | 3.45 ± 0.07 | 3.59 |
| rac-1b | 2 | 0.84 ± 0.01 | −8.50 ± 0.24 | 44 ± 1 | 3.12 ± 0.11 | 3.31 |
| P-1e | — | 0.84 ± 0.01 | −11.45 ± 0.31 | 39 ± 1 | 3.79 ± 0.08 | 3.88 |
| M-1e | — | 0.84 ± 0.01 | −11.35 ± 0.27 | 41 ± 1 | 3.86 ± 0.09 | 4.02 |
| rac-1e | — | 0.84 ± 0.01 | −10.66 ± 0.31 | 43 ± 1 | 3.82 ± 0.10 | 4.05 |
| P-1e | 2 | 0.84 ± 0.01 | −11.07 ± 0.25 | 42 ± 1 | 3.91 ± 0.09 | 4.02 |
| M-1e | 2 | 0.84 ± 0.01 | −11.35 ± 0.24 | 42 ± 1 | 3.97 ± 0.08 | 4.09 |
| rac-1e | 2 | 0.84 ± 0.01 | −10.77 ± 0.32 | 44 ± 1 | 3.94 ± 0.10 | 4.07 |
Space-charge limited current (SCLC) mobilities were evaluated according to literature known procedure51 and revealed an overall predominance in hole mobility over electron mobility by two orders of magnitude. Hole mobilities of 1.5 and 1.4 × 10−3 cm2 V−1 s−1 were found for P- and M-1b, slightly higher than the 1.1 × 10−3 cm2 V−1 s−1 for the corresponding rac-1b based device in ITO/PEDOT:PSS/PCE-10:1b/MoO3/Al architecture (Fig. S8 and Table S3, ESI‡). Interestingly, the electron mobility measured in a configuration of ITO/ZnO/PCE-10:1b/BCP/Al revealed elevated values of 13.9 × 10−6 cm2 V−1 s−1 for the rac-1b based device compared to slightly lower mobilities of 9.4 and 9.7 × 10−6 cm2 V−1 s−1 for the devices of the respective P- and M-atropo-enantiomers. This might show an improved forming of crystalline domains of the donor polymer in the presence of enantiopure P/M-1b PBIs whereas the elevated electron mobility of rac-1b is indicative for a more effective LUMO overlap of the chromophores in their racemic mixture even if the SCLC differences cannot solely explain the significant differences in overall PCE.
By applying 2% of the solvent additive CN on the 1b based blends, device efficiencies show an overall increase up to 3.5% (P, M) or 3.1% (rac) in PCE, respectively. Thus, the relative difference between enantiopure and racemic material shrinks down to about 11%, but is still clearly discriminable. This indicates a partial reduction on the impact of enantiopurity in molecular arrangement by the solvent additive. The overall rise in PCE observed in our studies for 1b-based devices by CN solvent additive originates from an equal increase of JSC and FF. This result slightly differs from observations reported for 1d-based OSCs by Sun and co-workers25 who solely recorded an increase in FF. They attributed this improvement to the prolonged dwell time of the material in solution due to the higher boiling point of the CN with respect to the solvent. However, AFM analysis did not reveal significant changes on the overall smooth and homogeneous active layers which matches with our results for all blends of P/M/rac-1b with a Rq between 0.5–0.7 nm (Fig. S4 and S5, ESI‡). In contrast to 1b, the Dipp-shielded 1e-based OSCs do not show a PCE dependency on enantiopurity and likewise the impact of CN solvent additive is rather modest (Fig. 4, Table 2). Overall higher PCE values of about 3.8–3.9% are recorded for the binary blends of P/M/rac-1e that only slightly increase to 3.9–4.0% when applying 2% of CN due to a subtle increase of FF by 1–3% to a maximum of 44%. Thus, for both, 1b and 1e, the additive promotes the device efficiency but with significantly less effect on 1e regardless of its enantiopurity. It is reasonable to suggest the CN guiding the PBI arrangement for 1b as it only shows little effect on the more shielded and isolated PBIs of 1e. Thus, it can be followed that the CN additive promotes the donor/acceptor interface or blend miscibility rather than improving the molecular order of the NFAs within their domains.
Chiral packing in single crystals and thin films
The variation in PCE of OSCs of racemic and enantiopure NFA 1b is solely attributed to changes in JSC (Fig. 4b, Table 2) which cannot be easily explained by differences in thin-film UV/Vis/NIR absorption or film morphology as these do not show significant alterations. To approach these morphological disparities in sub-nanometer scale on the molecular level we took the efforts to obtain the single-crystal structures of enantiopure P-1b and M-1e revealing tremendous differences in molecular arrangement. The single crystal structures were obtained by slow evaporation method likewise the racemates from a chloroform or chlorobenzene solution, respectively, using methanol as co-solvent. Similar to its racemic mixture, the flexible n-butyl chain of P-2b enables face-to-face π-stacking but now guides towards a unique twisted π-stacked dimer formation (Fig. 5a). The chromophores in this X-cross arrangement52 are stacked perfectly on top of each other showing a relative twist angle of 73° with respect to the N,N′-axes with a close center-to-center distance of about 4.9 Å. Most interestingly, the two π-stacked phenyl substituents of each chromophore additionally stack on top of the neighbouring naphthalene plane of the PBI within the dimer, therefore locking the molecular arrangement in a distinct conformation. Both molecules are distinguishable by a different torsional twist imparted by the sterical congestion of the phenyl substituents. Whereas the bottom PBI shows a twist angle of 34.2°, the topmost chromophore is twisted by 38.4°, supported by the helical stacking of its adjacent phenyl residues with the underlying naphthalene subunit of the bottom molecule. Within this three-part helix, short π–π-contacts (between centroids) of 3.3 Å are determined. Thus, this supramolecular arrangement of P-1b guides its chromophore into the most twisted configuration of a PBI π-scaffold reported so far in literature. This unique spherical dimeric entity arranges as isolated units in the large unit cell of P6522 space group consisting of 12 chromophores, six times larger than the close and unidirected packing in the racemic crystal of 1b. The enantiopure species is assumed to allow for more isotropic growth which might contribute to the superior OSC performance of enantiopure 1b over its racemic species thereof. The comparison of single-crystal structures can also be correlated to the SCLC mobilities showing increased electron mobilities for the racemic 1b with both a more intimate face-to-face π-stacking as well as a closer molecular network within its double strand compared to enantiopure P/M-1b (vide supra, or see the Fig. S6, ESI‡). | ||
| Fig. 5 Molecular packing of PBIs P-1b (a) and M-1e (b) within their respective single crystal structures (top), top view onto dimeric chromophore arrangements and side view (bottom) as well as rotated side view (P-1b). PBI atropo-enantiomers are colored in blue (P) and red (M). Distances between centroids of adjacent perylene cores (green), distances between planes of adjacent chromophores (light blue) and closest π–π distances between centroids (grey) are indicated as well as twist angles of single chromophores (black) and twist angles between adjacent chromophores (red). The ellipsoids are set to 50% probability (C: gray). Disorder of the imide residues, hydrogen atoms as well as solvent molecules are omitted for clarity. | ||
Similar to the single crystal structure of the racemate of 1e (Fig. 3c), the Dipp-substituted enantiopure M-1e reveals isolated chromophores with relatively low twist angles of the chiral chromophores of only 28.4° within its single crystal (Fig. 5b). The closest neighbour within its unit cell of P21 space group resembles to the dimeric arrangement of P-1b with a twist angle of 95° but with the lack of any face-to-face π-stacking as the center-to-center distance of 10.8 Å reaches more than twice the value of P-1b. This is also significantly higher than the center-to-center distance of 8.5 Å found in the corresponding racemic crystal of 1e. The long molecular axis of the chromophores are tilted by 25.2° towards each other and show a long-ranged edge to face orientation. Thus, no face-to-face π-stacking is present and the PBIs of enantiopure M-1e can be considered as isolated chromophores contrasting the closed dimeric species of enantiopure 1b. Accordingly, as no close chromophore–chromophore contacts are present, no significant difference in PCE is observable for rac-1e (4.0%) in comparison to P/M-1e (3.9%, vide supra, Fig. 3 and Table 2).
To assess the resemblance of molecular arrangement in both, single crystal structures and amorphous thin films, the chiroptical properties of the neat thin-films as well as BHJ with PCE-10 were investigated and compared with corresponding spectra in solution with equal optical density (Fig. 6 and Fig. S7, ESI‡). Tetraarylated PBIs show overall broad and structureless absorption spectra in dichloromethane (DCM) solution. Both, spectral shape and position is mainly preserved also in the amorphous thin films of 1b as well as of 1e (Fig. 6, top) with no significant difference to the respective solution spectra. Absorption maxima of 1b are located at 603 and 420 nm for the S0–S1 and S0–S2 transition, respectively. For 1e the main bands in the thin-film spectra are slightly blue-shifted by 7 nm from 610 to 603 nm whereas the S0–S2 maxima of both, solution and thin film are located at 454 nm. Only the thin-film spectra of 1b show a slightly increased S0–S2 band compared to the main transition, indicative for molecular interactions (vide infra). Despite of these overall resembling UV/Vis absorption properties, the CD thin-film spectra of P/M-1b show a unique behavior contrasting all other remaining CD spectra in DCM solution (1b, 1e) as well as the thin film of 1e. Thus, the CD amplitude of the P/M-1b thin-film S0–S1 transition is increased by a factor of two at 614 nm whereas the CD signal is of lower amplitude at about 540 nm (Fig. 6, bottom). To our understanding, this can be attributed to the helically twisted and π-stacked dimer entity of enantiopure 1b found in its single crystal structure giving rise to an excitonic Cotton effect. In contrast, 1e only shows isolated chromophores with no supramolecular interaction in solution and single crystal as well as thin-film and the respective active layer of the OSCs (Fig. S7, ESI‡). To rule out artefacts by anisotropy,30,53 AFM images were recorded to show a homogeneous surface without oriented crystalline domains visible (Fig. S5, ESI‡). By using the atomic coordinates obtained from single crystal structure of either monomeric or dimeric P-1b, theoretical calculations on wB97/def2SVP level of theory were performed to simulate the arising Cotton effect of the dimer structure of P-1b due to its chromophore coupling (Fig. S9, ESI‡).
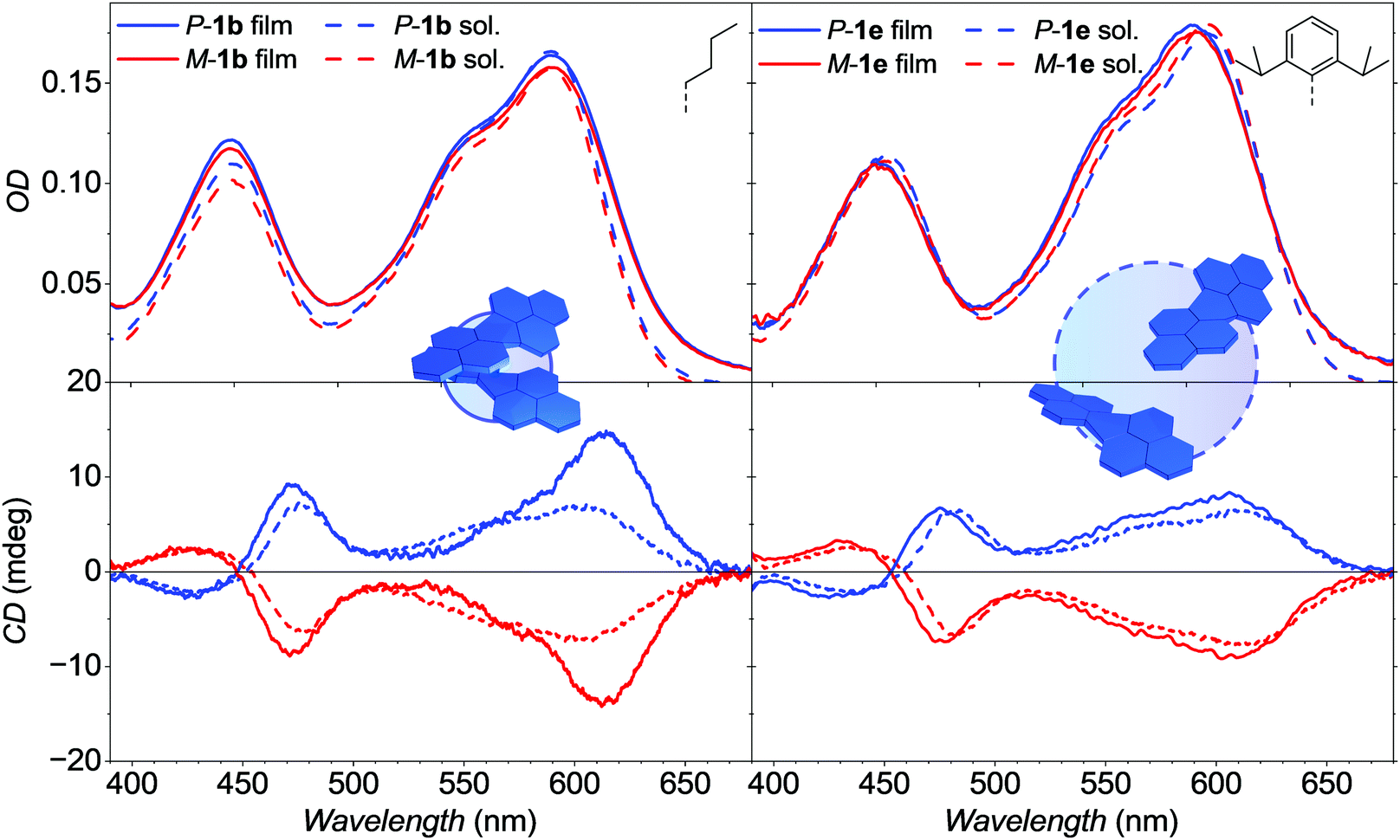 | ||
| Fig. 6 UV/Vis/NIR absorption (top) and CD (bottom) spectra of P- (blue) and M- (red) atropo-enantiomers of 1b (left) and 1e (right) in thin film (solid lines) and DCM solutions (dashed lines) at room temperature. Neat films were spin-cast from CHCl3 solution (10 mg ml−1) onto quartz substrates. The schematic insets represent the dimeric chromophore arrangement of enantiopure P-1b and P-1e species found in the respective single-crystal structures. | ||
It can be reasoned that the isolated chromophores of 1e (4.05%) and 2e (4.28%) show the highest performance as NFAs followed by the isolated dimers of enantiopure P/M-1b (3.68%) whereas rac-1b (3.31%) allows intimate face-to-face π-interactions that diminish the OSC performance. The higher hole mobility of the isolated dimeric entities of enantiopure P/M-1b blends as well as the reduced dependency on the solvent additive for P/M-1b and P/M/rac-1e based devices suggests a more favorable formation of crystalline domains of the donor polymer PCE-10 in the presence of the respective acceptor PBIs. Accordingly, the PBI enantiopurity only shows an impact on the OSC performance as long as the chromophores come into close vicinity and are not highly shielded by imide or bay substituents.
Conclusions
In summary, we investigated two series of conformationally stable and heavily twisted bay-tetraarylated PBI dyes as NFAs in BHJ devices with donor polymer PCE-10. In a comprehensive study on the variation of both imide (1a–1e) and aryl (1b–5b) substituents, we determined 2,6-diisopropylphenyl (imide) and 2-naphthyl (aryl) as the best substituents for PBI-based NFAs. Thus, the corresponding derivative 2e inheriting both substituents reached a peak performance of 4.3% PCEmax that is to the best of our knowledge the highest OSC efficiency of a single core PBI without lateral π-extension or embedding of heteroatoms. Our OSC results were correlated with chromophore packing deduced by single crystal structure analysis of the PBIs in racemic mixtures. Interestingly, the more isolated chromophores 1e and 2e with almost no π–π-interactions gave the highest device performance over 4% PCEmax. This reasoning was also corroborated by a comparison of devices built from either racemic or enantiopure PBIs. The effect was more pronounced for n-butyl-substituted PBI 1b that assembles as pure enantiomers into isolated dimers rather than a close slip-stacked double strand arrangement of the racemate as deduced from the respective single crystals. Thus, enantiopure PBIs of 1b afforded higher PCEs. This contrasts with Dipp-substituted PBI 1e for which π–π-contacts are already prohibited by the bulky imide substituents and no difference in device efficiency is observed. Therefore, we conclude that isolated and shielded chromophores such as 1e and 2e are potent NFA candidates regardless of their enantiomeric purity. Instead, PBIs bearing small and flexible alkyl chains such as n-butyl chains which allow for close π–π-interactions that are detrimental for efficient OSCs show a dependency on enantiopurity because of their higher propensity for the formation of slip-stacked arrangements.Our study opens up several perspectives for future NFA material design: (1) utilization of the PBI core twist can be considered also as a viable strategy for multi-chromophore systems that constitute the currently most potent PBI-based NFAs.12 (2) In contrast, by enhancing close π–π-interactions in the solid state as indicated for the still highly soluble P-1b, chromophoric coupling might introduce an excitonic Cotton effect. Thus, our materials with their appreciable semiconducting properties might be useful for detecting circularly polarized light in a phototransistor device. Recent progress in this field encourages research on enhancing chiroptical response towards the near-infrared spectral region.30,54 (3) Lastly, the loose solid-state packing of this highly absorbing and electron-deficient material will also allow for sublimation which might enable the fabrication of entirely vacuum-deposited devices.
Author contributions
Bernhard Mahlmeister: investigation, methodology, visualization, writing. Rebecca Renner: investigation, methodology. Olga Anhalt: investigation. Matthias Stolte: methodology, visualization, writing. Frank Würthner: conceptualization, writing, supervision, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the Bavarian State Ministry for Science and the Arts for the research program “Solar Technologies Go Hybrid”. The CPL/CD hybrid spectrometer was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Projektnummer 444286426.Notes and references
- C. W. Tang, Appl. Phys. Lett., 1986, 48, 183–185 CrossRef CAS
.
- A. Venkateswararao and K.-T. Wong, Bull. Chem. Soc. Jpn., 2021, 94, 812–838 CrossRef CAS
- F. Würthner, Chem. Commun., 2004, 1564–1579 RSC
- A. Nowak-Król and F. Würthner, Org. Chem. Front., 2019, 6, 1272–1318 RSC
- X. Wen, A. Nowak-Król, O. Nagler, F. Kraus, N. Zhu, N. Zheng, M. Müller, D. Schmidt, Z. Xie and F. Würthner, Angew. Chem., Int. Ed., 2019, 131, 13185–13189 CrossRef
- Z.-G. Zhang, B. Qi, Z. Jin, D. Qi, Y. Li and J. Wang, Energy Environ. Sci., 2014, 7, 1966–1973 RSC
- Q. Shi, J. Wu, X. Wu, A. Peng and H. Huang, Chem. – Eur. J., 2020, 26, 12510–12522 CrossRef CAS
- Y. Lin, J. Wang, Z.-G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2015, 27, 1170–1174 CrossRef CAS PubMed
- Y. Lin, F. Zhao, Q. He, L. Huo, Y. Wu, T. C. Parker, W. Ma, Y. Sun, C. Wang, D. Zhu, A. J. Heeger, S. R. Marder and X. Zhan, J. Am. Chem. Soc., 2016, 138(14), 4955–4961 CrossRef CAS
- A. Armin, W. Li, O. J. Sandberg, Z. Xiao, L. Ding, J. Nelson, D. Neher, K. Vandewal, S. Shoaee, T. Wang, H. Ade, T. Heumüller, C. Brabec and P. Meredith, Adv. Energy Mater., 2021, 11, 2003570 CrossRef CAS
- A. Nowak-Król, K. Shoyama, M. Stolte and F. Würthner, Chem. Commun., 2018, 54, 13763–13772 RSC
- V. Sharma, J. D. B. Koenig and G. C. Welch, J. Mater. Chem. A, 2021, 9, 6775–6789 RSC
- S. Chen, D. Meng, J. Huang, N. Liang, Y. Li, F. Liu, H. Yan and Z. Wang, CCS Chem., 2021, 3, 78–84 CrossRef CAS
- R. Xin, Z. Cheng, D. Meng, Z. Ren, J. Wei, Z. Wang, S. Yan and S. Yan, ACS Omega, 2020, 5(29), 18449–18457 CrossRef CAS PubMed
- T. Huang, H. Chen, J. Feng, A. Zhang, J. Wei, F. He and Z. Wang, ACS Mater. Lett., 2019, 1(4), 404–409 CrossRef CAS
- Z. Ma, H. Fu, D. Meng, W. Jiang, Y. Sun and Z. Wang, Chem. – Asian J., 2018, 13, 918–923 CrossRef CAS
- Y. Lin, Y. Wang, J. Wang, J. Hou, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2014, 26, 5137–5142 CrossRef CAS PubMed
- Y. Zhong, M. T. Trinh, R. Chen, G. E. Purdum, P. P. Khlyabich, M. Sezen, S. Oh, H. Zhu, B. Fowler, B. Zhang, W. Wang, C.-Y. Nam, M. Y. Sfeir, C. T. Black, M. L. Steigerwald, Y.-L. Loo, F. Ng, X.-Y. Zhu and C. Nuckolls, Nat. Commun., 2015, 6, 8242–8249 CrossRef CAS
- T. Ye, R. Singh, H.-J. Butt, G. Floudas and P. E. Keivanidis, ACS Appl. Mater. Interfaces, 2013, 5, 11844–11857 CrossRef CAS
- R. Singh, J. Lee, M. Kim, P. E. Keivanidis and K. Cho, J. Mater. Chem. A, 2017, 5, 210–220 RSC
- R. Schmidt, J. H. Oh, Y.-S. Sun, M. Deppisch, A.-M. Krause, K. Radacki, H. Braunschweig, M. Könemann, P. Erk, Z. Bao and F. Würthner, J. Am. Chem. Soc., 2009, 131, 6215–6228 CrossRef CAS PubMed
- R. Renner, B. Mahlmeister, O. Anhalt, M. Stolte and F. Würthner, Chem. – Eur. J., 2021, 27, 11997–12006 CrossRef CAS
- K. Fujimoto, M. Takahashi, S. Izawa and M. Hiramoto, Materials, 2020, 13(9), 2148–2178 CrossRef CAS
- X. G. Y. Cai, X. Sun, D. Wei, M. Yu, L. Huo and Y. Sun, Sci. China Mater., 2016, 59, 427–434 CrossRef
- Y. Cai, L. Huo, X. Sun, D. Wei, M. Tang and Y. Sun, Adv. Energy Mater., 2015, 5, 1500032 CrossRef
- W. Qiu, S. Chen, X. Sun, Y. Liu and D. Zhu, Org. Lett., 2006, 8, 867–870 CrossRef CAS PubMed
- M. Queste, C. Cadiou, B. Pagoaga, L. Giraudet and N. Hoffmann, New J. Chem., 2010, 34, 2537–2545 RSC
- P. Osswald and F. Würthner, J. Am. Chem. Soc., 2007, 129(46), 14319–14326 CrossRef CAS
- B. Pagoaga, L. Giraudet and N. Hoffmann, Eur. J. Org. Chem., 2014, 5178–5195 CrossRef CAS
- L. Zhang, I. Song, J. Ahn, M. Han, M. Linares, M. Surin, H.-J. Zhang, J. H. Oh and J. Lin, Nat. Commun., 2021, 12, 142–150 CrossRef CAS
- T. Ikai, R. Kojima, S. Katori, T. Yamamoto, T. Kuwabara, K. Maeda, K. Takahashi and S. Kanoh, Polymer, 2015, 56, 171–177 CrossRef CAS
- M. Chen, X. Jiao, J. Li, W. Wu, H. Xin, C. R. McNeill and X. Gao, Langmuir, 2019, 35, 6188–6195 CrossRef CAS
- F. Zhang, Y. Hu, T. Schuettfort, C.-A. Di, X. Gao, C. R. McNeill, L. Thomsen, S. C. B. Mannsfeld, W. Yuan, H. Sirringhaus and D. Zhu, J. Am. Chem. Soc., 2013, 135(6), 2338–2349 CrossRef CAS
- R. B. Zerdan, N. T. Shewmon, Y. Zhu, J. P. Mudrick, K. J. Chesney, J. Xue and R. K. Castellano, Adv. Funct. Mater., 2014, 24, 5993–6004 CrossRef CAS
- J. Liu, Y. Zhang, H. Phan, A. Sharenko, P. Moonsin, B. Walker, V. Promarak and T.-Q. Nguyen, Adv. Mater., 2013, 25, 3645–3650 CrossRef CAS
- T. He, P. Leowanawat, C. Burschka, V. Stepanenko, M. Stolte and F. Würthner, Adv. Mater., 2018, 30, 1804032 CrossRef
- M. Chen, J. Li, X. Jiao, X. Yang, W. Wu, C. R. McNeill and X. Gao, J. Mater. Chem. C, 2019, 7, 2659–2665 RSC
- S.-K. Jung, J. H. Heo, B. M. Oh, J. B. Lee, S.-H. Park, W. Yoon, Y. Song, H. Yun, J. H. Kim, S. H. Im and O. P. Kwon, Adv. Funct. Mater., 2020, 30, 1905951 CrossRef CAS
- K. Sugawara, N. Nakamura, Y. Yamane, S. Hayase, T. Nokami and T. Itoh, Green Energy Environ., 2016, 1, 149–155 CrossRef
- S.-H. Yang, R. Naaman, Y. Paltiel and S. S. P. Parkin, Nat. Rev. Phys., 2021, 3, 328–343 CrossRef
- P. Josse, L. Favereau, C. Shen, S. Dabos-Seignon, P. Blanchard, C. Cabanetos and J. Crassous, Chem. – Eur. J., 2017, 23, 6277–6281 CrossRef CAS
- Y. Yang, B. Rice, X. Shi, J. R. Brandt, R. Correa da Costa, G. J. Hedley, D.-M. Smilgies, J. M. Frost, I. D. W. Samuel, A. Otero-de-la-Roza, E. R. Johnson, K. E. Jelfs, J. Nelson, A. J. Campbell and M. J. Fuchter, ACS Nano, 2017, 11, 8329–8338 CrossRef CAS PubMed
- I. Song, J. Ahn, X. Shang and J. H. Oh, ACS Appl. Mater. Interfaces, 2020, 12(44), 49926–49934 CrossRef CAS
- S. Ma, J. Gu, C. Lin, Z. Luo, Y. Zhu and J. Wang, J. Am. Chem. Soc., 2020, 142(39), 16887–16893 CrossRef CAS PubMed
- W. Yang, G. Longhi, S. Abbate, A. Lucotti, M. Tommasini, C. Villani, V. J. Catalano, A. O. Lykhin, S. A. Varganov and W. A. Chalifoux, J. Am. Chem. Soc., 2017, 139, 13102–13109 CrossRef CAS PubMed
- G. Gao, X. Zhang, D. Meng, A. Zhang, Y. Liu, W. Jiang, Y. Sun and Z. Wang, RSC Adv., 2016, 6, 14027–14033 RSC
- S.-H. Liao, H.-J. Jhuo, Y.-S. Cheng and S.-A. Chen, Adv. Mater., 2013, 25, 4766–4771 CrossRef CAS
- K. Ding, T. Shan, J. Xu, M. Li, Y. Wang, Y. Zhang, Z. Xie, Z. Ma, F. Liu and H. Zhong, Chem. Commun., 2020, 56, 11433–11436 RSC
- M. Hecht and F. Würthner, Acc. Chem. Res., 2021, 54, 642–653 CrossRef CAS PubMed
- D. M. Stoltzfus, J. E. Donaghey, A. Armin, P. E. Shaw, P. L. Burn and P. Meredith, Chem. Rev., 2016, 116(21), 12920–12955 CrossRef CAS PubMed
- J. C. Blakesley, F. A. Castro, W. Kylberg, G. F. A. Dibb, C. Arantes, R. Valaski, M. Cremona and J.-S. Kim, Org. Electron., 2014, 15, 1263–1272 CrossRef CAS
- E. Sebastian, A. M. Philip, A. Benny and M. Hariharan, Angew. Chem., Int. Ed., 2018, 57, 15696–15701 CrossRef CAS
- E. Castiglioni, S. Abbate, G. Longhi and R. Gangemi, Chirality, 2007, 19, 491–496 CrossRef CAS PubMed
- Y. Yang, R. C. d. Costa, M. Fuchter and A. J. Campbell, Nat. Photonics, 2013, 7, 634–638 CrossRef CAS
Footnotes |
| † Dedicated to Prof. Daoben Zhu on the occasion of his 80th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2106433–2106437. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1tc04116a |
| This journal is © The Royal Society of Chemistry 2022 |