
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spiro donor–acceptor TADF emitters: naked TADF free from inhomogeneity caused by donor acceptor bridge bond disorder. Fast rISC and invariant photophysics in solid state hosts†
Larissa Gomes
Franca
 ,
Andrew
Danos
and
Andrew
Monkman
*
,
Andrew
Danos
and
Andrew
Monkman
*
OEM Research group, Department of Physics, Durham University, South Road, Durham, DH1 3LE, UK. E-mail: a.p.monkman@durham.ac.uk
First published on 4th January 2022
Abstract
We have studied the thermally activated delayed fluorescence (TADF) properties of the spiro-bridged donor–acceptor molecule, 10-phenyl-10H,10′H-spiro[acridine-9,9-anthracen]-10′-one, (ACRSA) in guest–host films and used it as a probe to explore the details of host effects on the TADF mechanism in typical OLED host materials. Linked by the rigid spiro C–C bond rather than a flexible C–N bond, we observe no inhomogeneous effects arising from distributions of donor–acceptor bridge dihedral angles. ACRSA displays no time dependent ‘apparent’ red shift of the prompt or delayed charge transfer (CT) emission. Moreover, using a range of different hosts, we show that the ground state dipole moment (dielectric value) of the host has very little effect on the ACRSA CT energy, i.e. there is no so-called ‘solid state solvatochromism’. This leads to weak stabilisation of the CT state in all hosts, but has a very small singlet triplet gap and very fast and efficient monoexponential rISC rates in films (reaching nearly 107 s−1 in zeonex host). We observe no power law decaying DF tail because there is no dispersion of the dihedral angle between donor and acceptor units. The previously much lower reported rISC rates in ACRSA are instead reattributed to intermolecular excimer states. The intermolecular species give rise to additional slow TADF contributions and broaden the overall CT emission band at 10% ACRSA loading and in neat films. Harnessing the rapid and homogenous rISC displayed by isolated ACRSA molecules may unlock higher efficiencies and – crucially – extended operational lifetimes in future TADF OLEDs.
Introduction
We recently revealed the complex solution state molecular photophysics1 of the deceptively simple thermally activated delayed fluorescence (TADF) molecule 10-phenyl-10H,10′H-spiro[acridine-9,9′-anthracen]-10′-one (ACRSA), see Scheme S1 (ESI†).2–4 This is an important TADF molecule for two reasons. First, the acridine donor (D) and anthracenone acceptor (A) units of the ACRSA molecule are bridged by a spiro carbon, not the usual C–N bond found in most D–A TADF molecules. This connecting group, imparts the required orthogonality between the D and A,5 but is also considered to be significantly stronger than bridging C–N bonds. This is one of the suspected main causes of the short operational lifetimes achievable by TADF emitters in devices. Moving away from this architecture to a spiro system for example could greatly enhance emitter lifetime. Second, the spiro bridge is significantly more rigid than C–N bonds. Consequently, there should be far less intrinsic inhomogeneity arising from distortions about this dihedral bond between D and A, which in C–N linked materials results in a distribution of CT state energies. This in turn results in a distribution of singlet–triplet energy gaps, and dispersion in reverse intersystem crossing (rISC) rates ranging up to 4 orders of magnitude in many cases.6 This conformational disorder, originating from the rotation around the C–N bond has also been investigated in other studies.7–10 Furthermore, a molecular design which restricts this torsional motion was reported as an approach to avoid this disorder effect.11–13 However, by using a spiro TADF emitter, the rigidity enforced by the tetrahedral spiro carbon atom gives potential immunity to external conformational distortion of the molecule caused by host interactions. By removing such sources of heterogeneity, the molecular photophysics of ACRSA can be used as an ideal molecule to probe how external factors affect the TADF and rISC mechanisms, including local environment, packing and intermolecular interactions. This should prevent degradation arising through long triplet residency times.The TADF method for harvesting triplet states has attracted great interest for OLEDs providing a means of harvesting triplet excitons without the need for scarce and expensive heavy metals.14,15 To achieve TADF, rISC must be efficient therefore TADF molecules must have a small energy gap between the lowest energy excited singlet (S1) and triplet (T1) states, i.e. small electron exchange energy.15 One way to achieve this is via charge transfer excited states, as found in donor–acceptor (D–A) molecules, that have effective electronic decoupling of ground and excited orbitals, e.g. the D and A units are oriented orthogonally, minimising electron exchange resulting in a very small singlet–triplet energy gap of <50 meV.16 However, when the electron exchange energy is so small, the singlet and triplet CT (charge transfer) orbitals become degenerate and transition between them forbidden because there is no way to change the electron orbital angular moment during the spin flip transition.17 To facilitate TADF a third (triplet) excited state, very close in energy to the 3CT state is required which has a different orbital character which can mix non-adiabatically with 3CT to mediate a spin flip through second order vibronic coupled spin orbit coupling and couple the triplet back to the singlet manifold i.e. rISC.18,19 As we previously demonstrated experimentally and Lyskov and Marian theoretically,20 this mechanism is achieved in ACRSA.
ACRSA was first shown to have both efficient solution state photoluminescence (PL) efficiency of 81%, with a high device EQE of 16.5% by Nasu and Adachi et al.2 However, there are rather few reports of other spiro bridged D A TADF emitters,21–26 as compared to D–A and D–A–D type emitters exploiting the N–C bridging bond motif. Lyskov and Marian have presented a very detailed quantum chemistry analysis of ACRSA highlighting the complex interactions between charge transfer and local states, the role of mixing of high lying excitonic transitions to overcome the forbidden nature of radiative decay from these states, and the potential for rapid nanosecond rISC mediated by n–π* to π–π* ISC transitions, see Scheme S1 (ESI†).20 Following on from this we have made an in-depth study on the molecular photophysics of the ACRSA molecule in solution to experimentally verify many of these theoretical predictions as well as showing unique photophysics. Throughout this new report all comparison to solution state properties of ACRSA refer to our previous paper,1 and the terminology used in describing the energy levels of ACRSA is given Scheme S1 (ESI†), along with a briefly summary of the results from our solution state measurements.
In this new work, we study the molecular photophysics of ACRSA in a range of solid host materials to elucidate the perturbations arising from solid-state interactions. ACRSA's unique spiro derived properties give unprecedented new insight into this realm to answer several major questions of host interactions that form an important part of the overall response of the system. Importantly, we believe these new results offer new understanding on the current short devices working lifetimes suffered by TADF emitters.
Results
Excitation dependent steady state emission spectra of 1% wt ACRSA in zeonex, UGH, DPEPO or neat film and the chemical structures of the hosts are shown in Fig. S1 and S2 (ESI†). In zeonex, the spectra consist of a poorly structured emission, peak energy 2.8 eV (460 nm) and a higher energy knee at 3.05 eV (410 nm). Comparing to previous solution spectra,1 we see that zeonex gives an environment very similar to MCH solution where the 1CT and 1LE states are highly mixed giving strongly overlapped emission. Whereas, in DPEPO and UGH films the 1CT band dominates, peak at 2.4 eV (520 nm), with the appearance of a blue shoulder at ca. 450 nm which is highly dependent on excitation wavelength. We note that in solid-state the 1CT emission is much less red shifted compared to that observed in previously results in toluene solution (a very low polarity solvent). In solution, it is clear that the ACRSA 1CT state causes a large change in dipole moment. As a result of excitation (CT formation) and subsequent strongly red shift, even in low polarity solvents such as toluene, due to rapid re-organisation of the solvent molecules in the solvent shell around the ACRSA molecule which relaxes the Coulomb energy of the excited state.27 Whereas the red shift in all solid films is only around 30 meV, which is far less than toluene (280 meV). As previously discussed by us28,29 and shown by Northey et al.30 a solid host cannot rearrange around the excited guest molecule in the way a solvent molecule can (after excitation) and thus there is little relaxation of the Coulomb energy of the CT state in solid state, i.e. no analogous solid state solvatochromism. The host can organise around the emitter molecule in the ground state if it has a large ground state dipole moment, but no further rearrangement can occur in the excited state. As a consequence, there can be no directly analogous ‘solid state solvatochromic’ effect. Lyskov et al.† calculate that the GS dipole moment of ACRSA is modest, 5.46 D, and so only a small ground state effect should be expected. This is very clear from these ACRSA results.Little emission contribution from the 1LE excited state is observed from measurements made in vacuum (Fig. S3, ESI†), which was also found in solution where degassing results in a very large 1CT DF emission contribution that totally dominated the emission. Film measurements made in air as a function of excitation wavelength do show consistent photophysics with degassed solution measurements, which implies poor oxygen diffusion through the dense films (Fig. S2, ESI†).With 330 nm excitation, we observe emission around 350 nm from the 11B1 exciton state (acridine donor unit fluorescence31), which was very strong in MCH solution for example. Excitation at 350 nm shows well-resolved features on the blue edge of the CT band, which is consistent with the observed 1LE emission in solution. Excitation at 380 nm yields effectively the main CT band (550 nm) and a blue shoulder at 450 nm. From these observations, it is clear that all films behave very similarly to each other and to MCH solution. However, in the films, the overlap between the 1LE and 1CT emission is very large leading to poor spectral resolution. Excitation at 350 nm gives the highest intensity of 1LE emission confirming that the states are present. Moreover, we know from solution (and film results below) that the 1LE state is quenched rapidly by ISC. In film, this leads to an unstructured emission band from the 1LE that is very consistent with emission from a hot, vibrationally unrelaxed state as previously postulated by Greene et al.32 in rather similar acceptor molecules. Thus, it appears to look more Gaussian band shaped than the highly structure band observed in MCH where vibrational cooling to the fluid environment is far more efficient. Such hot emission from local states in the film seems to be the main reason why vibrationally structured emission is lost compared to solution emission bands in CT materials.
As in MCH solution, zeonex films (1% wt ACRSA loading) presented an instantaneous and very broad (half width ca. 125 nm) emission band, centred at 520 nm (2.38 eV) (Fig. 1). This band decays with lifetime of 6.2 ns (Fig. S4, ESI†). We believe that this metastable state arises from a highly conformationally distorted structure, similar to that theoretically described by Fan et al.33 We aim to give a more detailed report about this in a further study. The central energy position is similar to the CT band observed in toluene solution. As this broad band decays, we observe prompt and DF emission centred around 448 nm, having multi-component lifetime of 93.7 ns, 1.2 μs and 8.3 μs. This complex, long lived emission may signify two competing rISC channels from two closely spaced (in energy) excited states, however the dominant channel is very fast, approaching 1 × 107 s−1, Table 1. This we believe strongly reflects the mixed nπ*/ππ* (1LE/1CT) character of the 1CT state and very small energy gap to local triplet states in zeonex. Spectrally it is difficult to resolve these states given the complexity of the overall emission decay, but if we compare to MCH solution we can deduce that the 93.7 ns and 1.21 μs are prompt and DF CT emission, respectively. We believe that zeonex acts very much like the solution state, because the asymmetrically branched polyolefin structure of zeonex which prevents crystallisation and creates a large free volume within the polymer network that the guest molecules occupy with little or no hindrance to conformation motions.
 | ||
| Fig. 1 Time evolution of the emission spectra (area normalised). Laser excitation at 355 nm excitation wavelength, for ACRSA in zeonex (1% loading) film measured at; (a) 300 K, (b) at 80 K. Normalised data shown in Fig. S23 (ESI†). | ||
| ACRSA:HOST | Zeonex 1% | DPEPO 1% | UGH 1% | DPEPO 10% | UGH 10% | NEAT |
|---|---|---|---|---|---|---|
| τ LE (ns) | 6.2 | 7.7 | 4.8 | 6.3 | 5.4 | 4.4 |
| τ PF/1 (ns)/APF/1 (%) | 93.7 | 196 | 287 | 74.8/69 | 69.2/62 | 105/87 |
| τ PF/2 (ns)/APF/2 (%) | — | — | — | 393/31 | 400/38 | 759/13 |
| τ DF/1 (μs)/ADF/1 (%) | 1.2/72 | — | — | — | — | — |
| τ DF/2 (μs)/ADF/2 (%) | 8.3/28 | 7.3 | 5.4 | 8.7 | 7.0 | 6.6 |
| τ AV/PF (ns) | 93.7 | 196 | 287 | 299 | 328 | 437 |
| τ AV/DF (μs) | 6.3 | 7.3 | 5.4 | 8.7 | 7.0 | 6.6 |
| k rad (s−1) × 106 | 1.02 | 0.70 | 0.43 | 1.45 | 1.15 | 3.28 |
| k ISC (s−1) × 106 | 9.66 | 4.41 | 3.06 | 11.9 | 13.3 | 6.23 |
| k rISC (s−1) × 106 | 8.67 | 1.00 | 1.50 | 1.06 | 1.81 | 0.44 |
| 1CT (nm)/(cm−1) | 394/3147 | 419/2959 | 416/2980 | 417/2973 | 417/2973 | 424/2924 |
| 3LE (nm)/(cm−1) | 398/3115 | 403/3076 | 405/3061 | 433/2863 | 426/2910 | 448/2767 |
| |ΔEST| (meV) | 32 | 117 | 81 | 110 | 63 | 157 |
Zeonex films measured at 80 K are more complex. From the kinetic trace (Fig. 2), four distinct decay processes are seen with well-resolved decay times. The initial transient, very broad 520 nm band is not so clearly observed, pointing to this species being strongly thermally activated. A more complex structure is observed at the first few nanoseconds suggesting possible multiple spectral components. This suggests multiple transient emitting states potentially from different metastable molecular configurations.33 We do observe a CT like emission, peak ca. 550 nm decays much more slowly (ca. 250 ns) than at 300 K (Fig. S5, ESI†). After 100 ns, dual emission is observed with a shoulder at 470–500 nm observed, that decays with lifetime of 458 ns. Also, from 100 ns on, a further emission band at 450 nm (peak) is observed. After 25 μs, a highly structure emission band dominates, clearly phosphorescence with lifetime of 1 ms from a local excited triplet state, having a highly developed vibronic progression. This emission is at lower energy than the singlet 1CT (ca. 3.1 eV) having onset energy of 2.97 eV. The energy spacing of the vibronic replicas is 0.19 eV (1530 cm−1), corresponding to the energy of a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch. This identifies the emission as phosphorescence from the anthracenone 3LE π–π* triplet localise around the CO bond.20 This phosphorescence is consistent with the ‘early’ ms phosphorescence observed in UGH and DPEPO films at 80 K (Fig. S6 and S7, ESI†). We also observe a blue shoulder to this emission, onset 3.12 eV. This shoulder does not fit with the 0.19 eV vibronic progression of the main band, and is not truncated by self-absorption. We believe this to be a second underlying higher energy phosphorescence,18,34 potentially from the 3ππ* state of the acridine donor triplet.31 At 300 K in zeonex, we see that this higher energy, unstructured phosphorescence is observed from 20 μs onwards, indicating that thermal equilibrium between these two triplet populations has changed.34 For further comparison, a full data set measured with 337 nm excitation, which excites the 11B1 exciton state as opposed to 355 nm excitation that directly excites the 1LE and 1CT states, giving excess energy to the molecules (Fig. S8 and Table S1, ESI†). A clear result of this excess energy given to the molecule is seen in zeonex films where 337 nm excitation yields higher intensity DF from the 1CT band (510 nm) because of the increased triplet production which 337 nm excitation produces.
O stretch. This identifies the emission as phosphorescence from the anthracenone 3LE π–π* triplet localise around the CO bond.20 This phosphorescence is consistent with the ‘early’ ms phosphorescence observed in UGH and DPEPO films at 80 K (Fig. S6 and S7, ESI†). We also observe a blue shoulder to this emission, onset 3.12 eV. This shoulder does not fit with the 0.19 eV vibronic progression of the main band, and is not truncated by self-absorption. We believe this to be a second underlying higher energy phosphorescence,18,34 potentially from the 3ππ* state of the acridine donor triplet.31 At 300 K in zeonex, we see that this higher energy, unstructured phosphorescence is observed from 20 μs onwards, indicating that thermal equilibrium between these two triplet populations has changed.34 For further comparison, a full data set measured with 337 nm excitation, which excites the 11B1 exciton state as opposed to 355 nm excitation that directly excites the 1LE and 1CT states, giving excess energy to the molecules (Fig. S8 and Table S1, ESI†). A clear result of this excess energy given to the molecule is seen in zeonex films where 337 nm excitation yields higher intensity DF from the 1CT band (510 nm) because of the increased triplet production which 337 nm excitation produces.
 | ||
| Fig. 2 Temperature dependent emission decay kinetics for ACRSA films. Time resolved emission decay for ACRSA in different host matrices ((a) and (b) at 1% loading, (c) and (d) at 10% loading) and in neat film, recorded at 300 K, left, and 80 K right. Decay trace of ACRSA in 20 μM MCH solution is shown in (a) for reference. Efficient thermally activated delayed fluorescence is always observed even at 80 K indicative of small energy splitting and strong vibrational coupling. Laser excitation at 355 nm. | ||
Making measurements at very low ACRSA loadings of 0.01% clearly show that the 550 nm (2.27 eV) band is not present at low ACRSA loadings, (Fig. S9, ESI†) and so we ascribe it to an intermolecular species, as will be seen is readily observed in small molecule hosts in the next section. At 0.01% ACRSA loading in zeonex we find characteristic decay lifetimes of 4 ns and 90 ns prompt emission components and a DF component of 1.3 μs. Because of limited signal to noise ratio at these very low ACRSA concentrations we can observe long lived emission at 430–440 nm but cannot obtain an accurate lifetime.
1% wt ACRSA in UGH and DPEPO films were also investigated with 355 nm and 337 nm excitation. The time resolved emission spectra, Fig. 3, show that the CT emission undergoes minimal time evolution with 337 nm excitation and a very small degree with 355 nm excitation. As we predicted for the rigid spiro-TADF emitter, the CT energy has constant onset at 2.98 eV (UGH) and 2.95 eV (DPEPO), peak at ca 2.5 eV, Table 1 and Fig. S10 and S11 (ESI†). Exciting at 337 nm into the 11B1 exciton state, both UGH and DPEPO films behave in a rather simple fashion (Fig. 3). The spectra remain relatively constant, showing only a minimal temporal shift and no change in band shape. Moreover, photoluminescence decay in both films can be fitted with a simple single exponential expression for prompt and DF. In UGH, we find 30 meV inhomogeneous broadening (from the red shift of emission over 20 μs) and in DPEPO 35 meV. In the first 10 ns, a small blue edge contribution is seen which we ascribe to 1LE emission and a small 11B1 exciton contribution at 350–400 nm. As in MCH solution, a weak red tail which decays very rapidly within the first 10 ns is also observed in UGH and DPEPO films. With 355 nm excitation (into the 1LE/1CT mixed transitions), during the first 100 ns, the emission from UGH films is rather broad, centred at ca 500 nm with a pronounced (but small contribution) red tail in the 550–650 nm region. The major emitting species, especially DF, is a single band, centred at ca. 484 nm, which has some structure, onset 420 nm (2.95 eV). Whereas in DPEPO the earliest emission, is more to the blue, onset 400 nm (3.10 eV), consistent with emission from the 1LE state. However, the major contribution to emission from 100 ns onwards is centred at 490 nm. Overall, we see that the band in UGH has more spectral weight around 450–475 nm whereas in DPEPO the majority spectral weight is from 475 nm to 510 nm. At 80 K, the ACRSA UGH and DPEPO films also show well resolved phosphorescence with lifetime of 0.6 ms, Fig. S6 and S7 (ESI†), having the same spectral shape and onset as observed in zeonex, confirming this to be the anthracenone 3LE π–π* triplet, the lowest monomolecular triplet state of ACRSA independent of host.
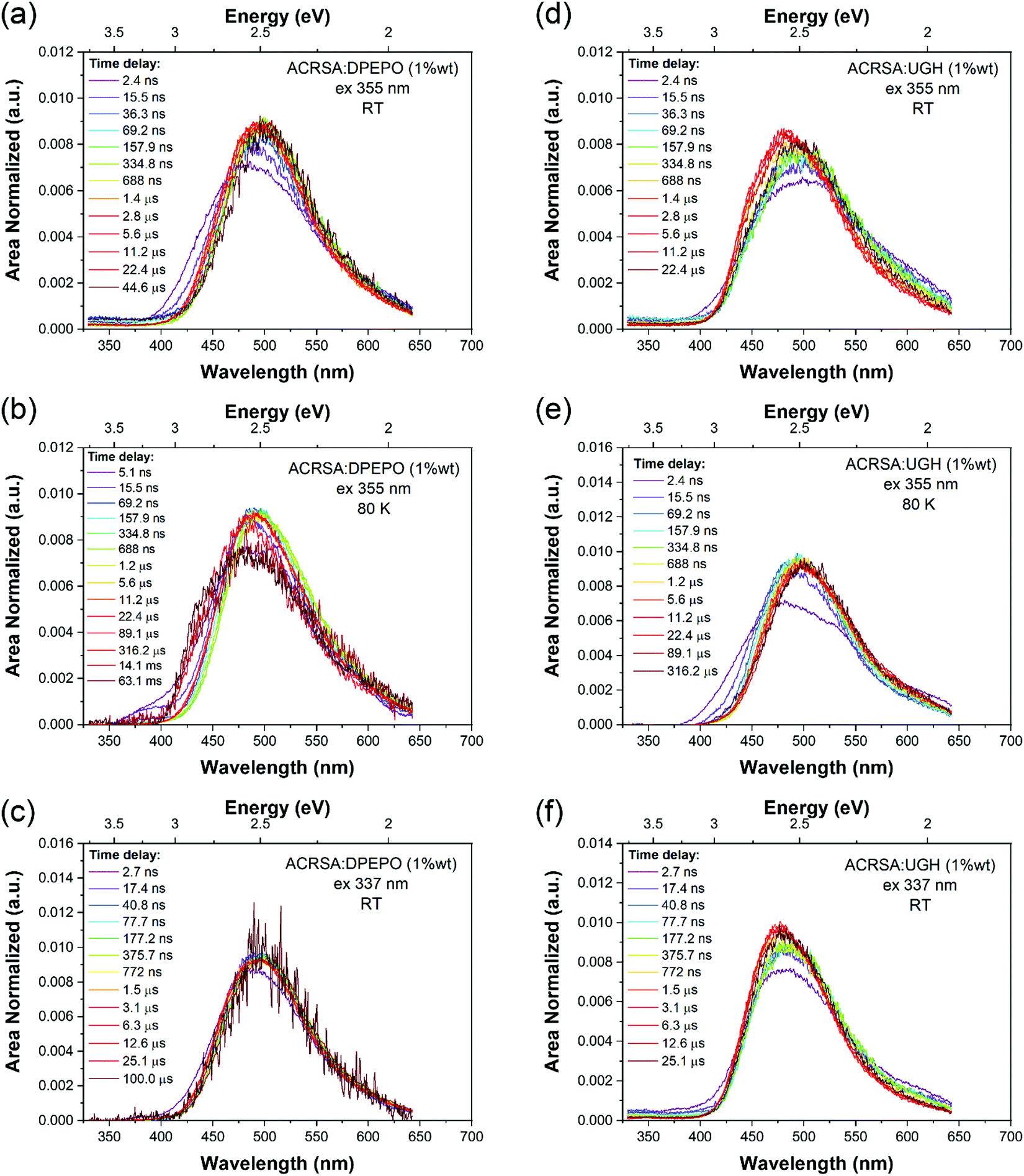 | ||
| Fig. 3 Time evolution of the emission spectra (area normalised). Time resolved emission spectra from ACRSA at 1% loading in UGH and DPEPO host matrices, measured at room temperature and at 80 K, with 355 nm and 337 nm excitation. Normalised data shown in Fig. S24 (ESI†). | ||
Excitation dependent steady state emission spectra of 10% wt ACRSA in UGH, DPEPO and neat film were also studied, Fig. S12 (ESI†). Emission is found to be much less excitation dependent. In all cases the 1CT band dominates, peak at 2.4 eV (520 nm), with the appearance of a blue shoulder at ca. 450 nm, which is more dependent on excitation wavelength. We note that the 1CT emission of ACRSA in UGH, DPEPO and neat film is also much less red shifted compared to that observed in previously observed in toluene (a very low polarity solvent).
At 300 K, in the DPEPO, UGH (10% wt loading) and neat film (Fig. 4), there is little fast 1LE emission observable (similar to degassed solution samples). Moreover, a pair of prompt 1CT decays are observed, lifetimes of 69.2 ns onset at 3 eV and 400 ns onset 2.78 eV, Fig. 4a–c and Fig. S13–S15 (ESI†). This is very different behaviour to that found in the 1% wt loading films. The shorter prompt lifetime species (peak ca. 2.6 eV) is consistent with the solution state 1CT transition and 1% ACRSA zeonex films. Whereas the long lifetime species that grows in very slowly (peak at 2.4 eV) comes from a new excited state species found only at 10% ACRSA loading, which presented a redshifted and broader band as compared to the 1% wt loading films. An isoemissive point is observed in the area normalised time resolve spectra at ca. 505 nm. This implies that there are only two distinct emissive species in the films. Further, we also can clearly see that the blue edge of the emission decays with a concomitant growth in the red edge intensity, Fig. S13–S15 (ESI†). This indicates that the blue species converts to the red species in competition with radiative decay.35
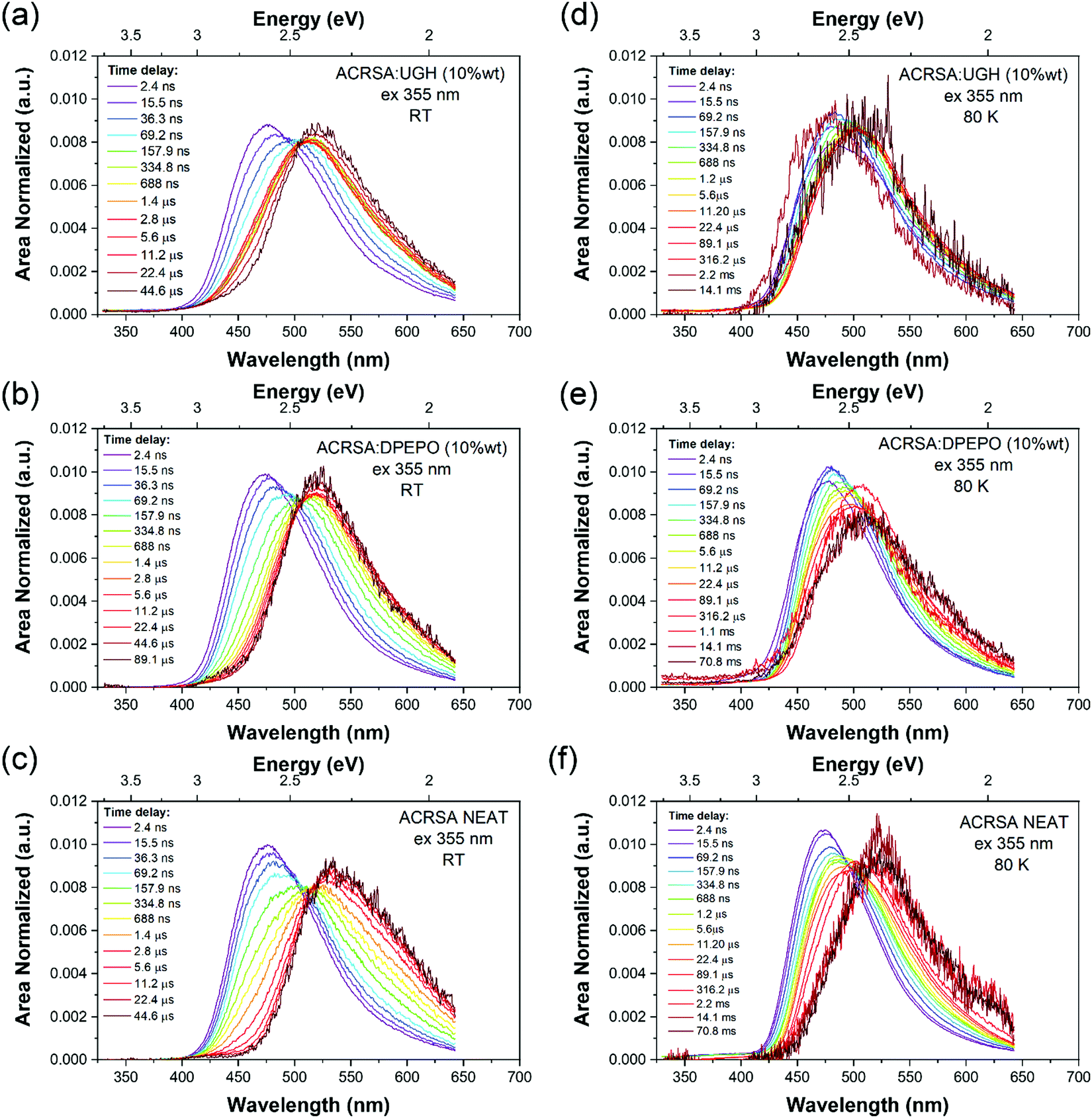 | ||
| Fig. 4 Time evolution of the emission spectra (area normalised). Laser excitation at 355 nm excitation wavelength, for ACRSA in UGH, DPEPO (10% loading) and neat film measured at; (a–c) 300 K, (d–f) at 80 K. Normalised data shown in Fig. S25 (ESI†). | ||
We take this new behaviour in the highly loaded films to indicate formation of some weakly interacting excimeric like state between nearest neighbours. Such a weakly interacting state would be in keeping with intermolecular interactions between two spiro molecules. The very long ‘prompt’ lifetime of this species would also fit with a weakly allowed transition decoupled from its ground state via delocalisation across two molecules. The evolution of the time resolved spectra are totally incompatible with any molecular confirmation change (especially given that we are observing a rigid spiro molecule) or from dispersive rISC rates as found in non-spiro D–A TADF molecules.28 DF in DPEPO, UGH (10% wt loading) and neat film show the same trends and comes from the low energy CT feature, with a single lifetime component of 6–9 μs. UGH films have a slightly faster rISC rate than DPEPO films, but in all cases are krISC is high, around 1 × 106 s−1.
80 K measurements, Fig. 4d–f, we again find very similar kinetics and decay times in prompt and delayed emission in 10% wt loading and neat films, Table S2 (ESI†). Two emission bands are observed, at ca. 470 nm (peak) early times, and at 500–510 nm (peak) later times, with an isoemissive point seen (Fig. S16–S18, ESI†). However, the full spectral evolution takes ca. 2 orders of magnitude longer to achieve at 80 K, indicating that this mechanism is thermally activated. Also, given the redder species is at higher energy compared to at 300 K, we take this as evidence of thermally activated excimer formation. The UGH films show more detailed spectral information (Fig. S16, ESI†). The early time emission, onset ca 420 nm (peak 475 nm) shows some structure indicative of a CT state with a high degree of local character and has strong resemblance to the main emission band observed in zeonex films. We do not observe fast 1LE state emission in solid films because the prompt and delayed CT emission intensity is so much greater.
At very late times, emission has a different band shape and it is centered at 520 nm. From the decay kinetics this is phosphorescence. As can be seen in neat film spectra recorded at 70 ms, Fig. 4f, this distinct new band at the longest times, onset 2.73 eV is ascribed to the phosphorescence of the excimeric species. In UGH there is also evidence of dual phosphorescence from the monomeric species, with a blue component, onset 405 nm (3.06 eV) which decays with lifetime of 0.7 ms to leave a very long lived phosphorescence at 426 nm onset (2.91 eV) that has a lifetime >70 ms,18,34 from the excimeric species. The very long lived monomeric phosphorescence should be from the acridine 3LE state as it was seen in zeonex films at room temperature. Looking at the absolute intensity of DF as a function of temperature, at 80 K it is about an order of magnitude less than at 300 K, indicative of an efficient thermally activated rISC mechanism. The majority of triplet energies quoted here are taken from the 80 K data, see Fig. S5–S7, S16–S18 and Table S2 (ESI†).
Comparing the kinetic decay traces for both 10% wt and 1% wt loaded films, and scaled for film ACRSA absorbance (Fig. S19, ESI†), we see that essentially the kinetics are identical. However, 10% loaded films show quenching of the prompt emission and relatively less DF which implies that in 10% films a fraction of the initial excited states are quenched.
In all cases, independent of ACRSA loading, host and even neat film, we find a linear excitation power dependence for the DF indication that in all cases the DF arises from a monomolecular process, fully in line with rISC, Fig. S20 and S21 (ESI†).
Discussion
We find that ACRSA is a very efficient TADF material with very high rISC rates. Comparing 1% to 10% loaded ACRSA films, has highlighted new photophysical properties for spiro-TADF materials, as well as showing the weak perturbations from host matrices. But most importantly we clearly see that there is no so-called “solid state solvatochromism”. At 1% loading films, the time resolved emission spectra show little or no red shift with time and the energy of the CT state, which differs by only 30 meV in DPEPO compared to UGH. Moreover, the CT emission band is at the same energy as found in MCH solution and in zeonex films, indicating clearly how the static dielectric properties of the host (typically confused as host polarity) has minimal effect on the CT state. This definitively shows that for D–A TADF materials which show large apparent time dependent red shifts of their CT emission, is behaviour that clearly comes from inhomogeneity in the D–A dihedral angle. This different D–A dihedral angle of CT state (with different energies) have different lifetimes, which gives rise to dispersive ISC, rISC and radiative decay rates, leading to an apparent time dependent red shift of emission. 1% ACRSA UGH films behave almost identically to ACRSA dissolved in MCH, both with 355 nm and 337 nm excitation, although we never observe a very fast and highly structured emission from the local 1LE state that we observed in MCH solutions, which may indicate that in solid state this emission comes from a vibrationally unrelaxed states at early times.32 We do though observe weak structure on the emission band ascribed to high LE character of the state. In DPEPO films (1% wt ACRSA loading), we also observe very similar behaviour as UGH films. The main CT emission is seen to be only 30 meV lower in energy than in UGH host, but has more spectral weight in the red which indicates marginally more CT character. However, with 355 nm excitation where the 1LE and 1CT states are excited directly, we see a fast blue component ascribed to 1LE emission. With 337 nm excitation, the 11B1 exciton state is excited and this blue component is much weaker, again as we previously observed in solution, indicating that the 11B1 exciton state couples primarily to the 1CT state as well as decaying by fast intersystem crossing.At 10% loading films we observe rather different photophysics, but again the different hosts, including neat film, show little difference. Two well-resolved bands in the time resolved (area normalised) spectra are observed, with clear isoemissive points. The redder state decays more slowly with long DF lifetimes, whereas the bluer band decays much faster with fast DF, and is the same species as we observe in 1% films (and MCH solution). Clearly, these two bands are not the result of host perturbation through static dielectric/polarizability effects. More importantly, neat film shows near identical behaviour, with the blue band seen at exactly the same energy as in UGH and DPEPO. We also observe that at 80 K there is very little effect on the photophysics apart from the evolution of the spectra and the lifetime of the red state which increases by two orders of magnitude. From this we conclude that the blue emission seen in 1% wt loading films, 10% wt loading films and MCH solution must be a monomolecular state that has mixed LE/CT character, which is totally unaffected by host environment. As we have previously shown, the degree of charge separation and the amount of local wavefunction character dictates the photophysical character of the excited state in such D–A molecules,36,37 we therefore propose that this blue emission band is a mixed 1nπ* (acceptor) 1ππ* CT excited state having a high degree of charge transfer character. This type of highly mixed LE/CT excited state is as predicted in the DFT/MCRI calculations by Lyskov and Marian.20 This state has a high radiative decay rate because of the strong coupling to the ground state (imparted by the high local character) and fast rISC indicating a very small S–T gap. Thus, this vibronically coupled TADF system is very efficient and fast, as seen in the kinetic fit data for UGH films, where >90% of the DF decays with a lifetime less than 5 μs. Gibson and Penfold calculated that the arrangement of 3LE above 3CT but close to 1CT would be the optimum state configuration for fast rISC,19 which we believe is the case for ACRSA.
In highly loaded (10% ACRSA) and neat films, a second red CT band is also observed. Obviously, at higher loading we must have a proportion of ACRSA molecules that interact with nearest neighbours, as well as ‘isolated’ molecules. Such intermolecular interactions must be very similar in all hosts and neat film as the resulting red emission band is observed at very similar energies in all hosts. Most telling is that we observe dual emission from monomeric and this red species simultaneously at high loading, but only from the monomeric species at low loading, again clearly showing that the red shifted species is not a result of ‘solid state solvatochromism’ (oxymoron) but is an excimer state.38 Further, the FWHM of the ACRSA emission band in 1% wt loading film (100 nm) (Fig. S3, ESI†) is much narrower to that of 10% wt loading films (150 nm) (Fig. S12, ESI†) and the FWHM of the steady state emission is also 150 nm. Thus, we can see that the steady state emission spectrum is a weighted average of these two contributions at 10% loading, causing the steady state emission band to red shifted slightly in different hosts. This is in agreement with our previous observations of intermolecular interactions causing emission red shifts in film.12,39 We see from the X-ray structure for ACRSA, given in Fig. S22, that even in this spiro compound, there is face to face overlap of adjacent acceptor groups. We do not believe this to be enough to sustain strongly coupled dimer states, but instead introduces an excimeric type perturbative effect between two acceptors, effectively increasing the delocalisation of the acceptor (a weakly coupled excimer). This increases the effective accepting strength leading to a red shifted intramolecular CT state. In this way the intermolecular interaction mimics polarity but is not affected in anyway by different hosts. It also explains why this red CT is well defined with a single lifetime and energy in all hosts. We note that previously Méhes et al.22 studying a slightly different spiro TADF system also concluded that a bimolecular species was forming in film, but postulated that it was an exciplex between adjacent spiro molecules. We believe that our model of the interacting acceptors, which affect the intramolecular ICT state, has a far greater probability of forming in spiro compounds compared to full exciplex formation. In Fig. 6, we label this state as ‘CTexcimer state’ simply as a way to differentiate it from the isolated molecular 1CT state. To enable rISC from these CT states there must be coupling to an energetically close triplet state. Since in the 10% loaded films, we observe a long-lived emitting species (onset at 2.73 eV) which is not phosphorescence from the 3LE state at 2.9 eV. We assume that this excimeric species has a low energy triplet state giving rise to the 2.73 eV phosphorescence (again noted as an excimer triplet in Fig. 6), yielding a ΔEST of ca. 150 meV and thus the rISC rate will be slower for this low energy CT states as we observe. Cho et al.40 have shown theoretically that dimer states of TADF molecules can have appreciable rISC rates and not greatly reduce overall performance. Thus, we believe that this is the first definitive evidence for TADF from a dimer/excimer like state.
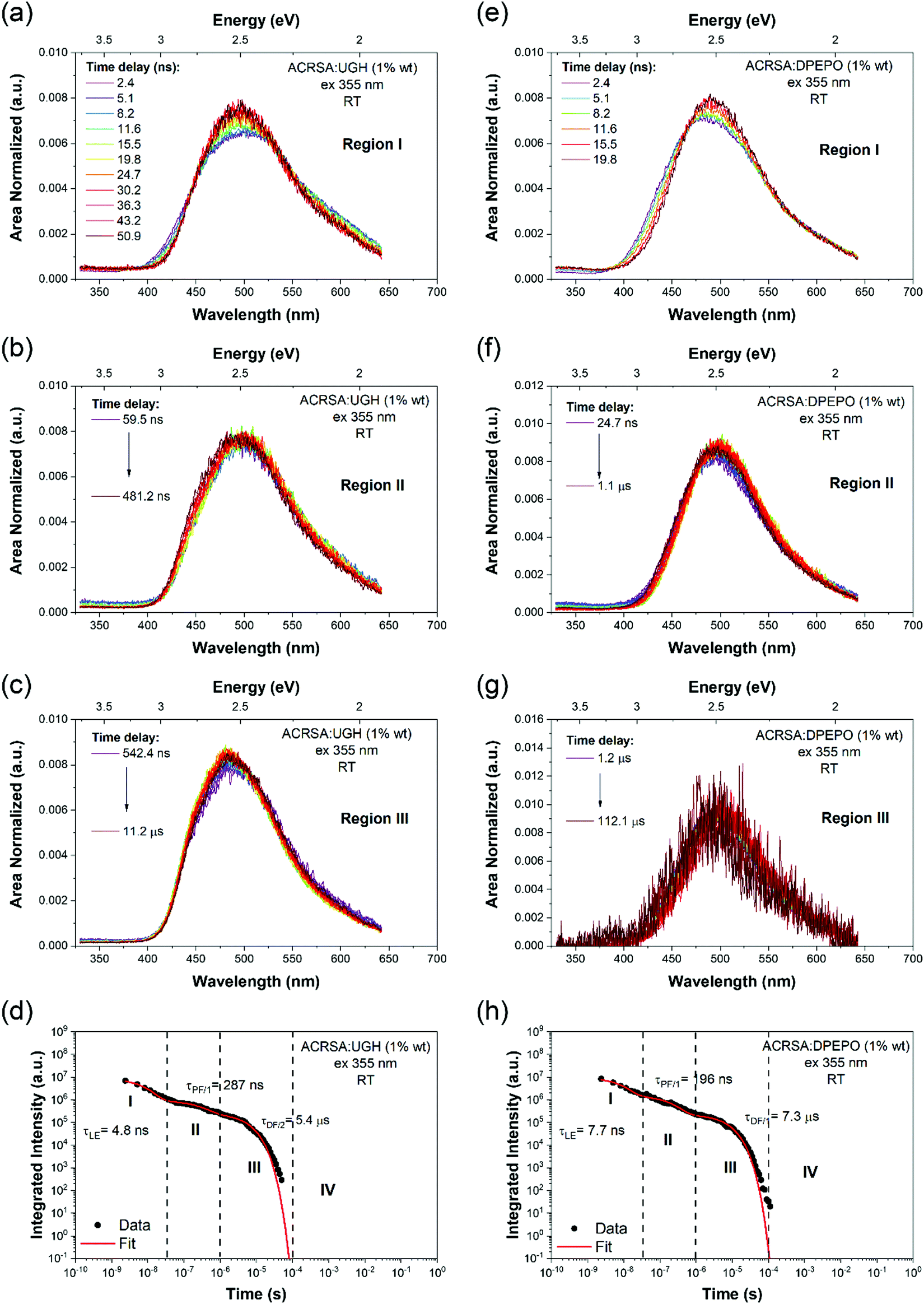 | ||
| Fig. 5 Comparison of long time DF decay kinetics for ACRSA in DPEPO host showing the single exponential decay of the DF with no associated power law decay component. (a)–(c) Area normalised time resolved spectra of ACRSA in UGH (10% wt loading), shown in the three main regimes indicated in (d). (d) Fitting of kinetic decay results at room temperature. (e)–(g) Area normalised time resolved spectra of ACRSA in UGH (1% wt loading), (h) fitting of kinetic decay results at room temperature. Laser excitation at 355 nm. | ||
 | ||
| Fig. 6 Excited state energy diagram for ACRSA in different host matrices; left at 1% ACRSA loading in UGH, DPEPO and Zeonex; right, 10% wt ACRSA loading in UGH and DPEPO and neat film. All quoted energies are taken from the spectral on-set of the associated emission bands at 300 K. Triplet energies measured at 80 K and/or long delay time spectra. We follow nomenclature used by Lyskov and Marian.20 11B1 is the local excited (LE) singlet state associated with the donor unit. 21A2 is the LE singlet state associated with the acceptor unit (transition with nπ* character).1CT is the singlet state with charge transfer character. 23A2 and 13A2 is the LE triplet state associated with the acceptor unit, these transitions have ππ* and nπ* character, respectively. | ||
In zeonex, the presence of intermolecular states described above becomes very clear. The photophysics is very similar to that observed in MCH solution including to observation of a highly red shifted transient emitting species. This we ascribe to the large free volume in this polymer host enabling the phenyl ring bond to the acridine nitrogen to potentially achieve a non-90° dihedral angle in the excited state weakening the donor strength leading to a blue shift of the CT state (higher LE character). This is in line with the theoretical calculations of Fan et al. in ACRSA.33 Whereas, in tightly packed small molecule hosts this is more rigidly fixed in the ground state configuration.
The time resolved emission in zeonex is though dominated by two main, closely spaced (in energy) emitting states. The 1LE state at ca. 430 nm peak (3.14 eV onset) and the mixed (vibrationally coupled) nπ*/ππ* 1CT state at 460 nm.20 The 1LE state has a fast radiative decay rate but is rapidly quenched by allowed SOC to the ππ* 3LEA triplet state, as in solution.1 From the observed local phosphorescence, which is highly structured and very well differentiated spectroscopically from the 1CT state, it is clear that the energy gap between the CT states and the mediating local triplet state is very small, calculated at 32 meV, giving rise to the measured very fast rISC rate approaching 1 × 107 s−1 (1.2 μs decay component) and thus highly efficient TADF. We also observe a slow DF decay (8 μs decay component) having the spectrum of the 1LE state. Given the strong allowed coupling between the nπ* 3LEA and ππ* 3LEA triplet states and allowed SOC between ππ* 3LEA and nπ* 1LE singlet state,1,20 we attribute this slow DF contribution to be from reverse upper triplet state ISC (USrISC) with thermally activated reverse IC driving the initial nπ* 3LEA and ππ* 3LEA step, see Fig. 6. Thus, at long times we only see 1LE delayed emission because all the CT states have been depopulated via fast rISC (TADF). Even at RT, we observe the spectra to blue shift, which we attribute to emergence of phosphorescence from the local 3LE states. At 80 K, this slow USrISC becomes slower because of reduced thermal activation (of the RIC) whereas the CT DF from rISC is still fast indicative of the very small energy gaps and strong vibrational coupling between the CT and local triplet states. We observe very strong local phosphorescence because lees local triplet states are depopulated through the USrISC mechanism. We do not observe USrISC in solution at RT because the triplets are more rapidly quenched by solvent collisions.
With 337 nm excitation both the 1LE and 1CT band are observed with an isoemissive point is observed, whereas with 355 nm only the blue band, onset 3.14 eV, ascribed to 1LE local emission, is observed which is preferentially excited by 355 nm excitation. Again, possible vibrationally hot emission results in the loss of vibrational structure.32 The red 1CT shoulder with 337 nm excitation arises because we excite via the 11B1 exciton state which populates both the 1LE and 1CT states preferentially via IC.1 The red band matches that seen in all other hosts and MCH and is ascribed to the 1CT state which has a long prompt lifetime of 60–90 ns, but as the energy gap to the coupling local triplet states is very small, it has fast rISC which decays with a half-life of 1.2 μs giving a rISC rate of 107 s−1.
At 1% ACRSA loading in zeonex we also see a weak 550 nm emission contribution. Measurements at very low ACRSA loadings (0.01%) show this band not to be present and we conclude that this is from an intermolecular species, as we observe in UGH and DPEPO. We assume that the solubility of ACRSA in zeonex is rather poor in this case.
Thus, the major effect of the solid state host on TADF molecules is to allow stable intermolecular interactions to occur as the guest concentration increases. In ACRSA, we observe this as a long lived low energy CT emission band in 10% wt loading and neat films. The steady state emission spectra then represents a true envelop of both CT bands giving greatly increased spectral width compared to the individual CT emission bands observed in the time resolved data. Potentially a very large emission FWHM of a TADF emitter indicates a high contribution from such intermolecular states39 fully in line with our previous work on the role of dimer/excimer states in TADF emitters. We find, irrespective of host, or ACRSA loading that the DF decays monoexponentially (as always seen in solution) and no long time, non-exponential decay time tail is observed in the decay kinetics, which is usually observed in D–A TADF molecules with a C–N bridge between D and A, Fig. 5. Recently, Serevičius et al. have demonstrated monoexponential decay can be observed in a specifically designed C–N linked D–A molecule.9 From our clear observation, we can also unambiguously ascribe these long lifetime non-exponential DF tails as arising from conformationally distorted (about the D–A dihedral bond) molecules. This gives a broad dispersion of dihedral angles and thus a range of electron exchange energies (i.e. ΔEST gaps) leading to rISC rates ranging anywhere between ca. 106 s−1 and 102 s−1. This then yields the typical power law DF tail. In ACRSA at 1% wt loading films where there is no spectral shift at all, this is abundantly clear. This then will have a major consequence on device lifetime because these slow DF states are much more prone to annihilation with charge carriers leading to degradation. We believe that in ACRSA, lies the template for long lifetime TADF molecules. The immunity to conformational distortion caused by host packing prevents slow rISC rate sites greatly reducing possible degradation from triplet polaron annihilation.
All of our photophysical observations for ACRSA in solid state are summarised in the state energy diagrams shown in Fig. 6.
Conclusions
We have studied the spiro-TADF emitter, ACRSA in different host matrices at 1% wt and 10% wt loading and compared the results to those from neat films. Previously using solution measurements, we identify all of the emissive singlet and triplet states of ACRSA and how they are perturbed by solvent polarity. In the solid state at 1% wt loading, we observe a single CT emission, centred around 485 nm which is both independent of the host matrix and shows no spectral relaxation over more than 20 μs. Clearly this shows that with the rigid spiro bridge between D and A unit there is little inhomogeneity in the dihedral angle between D and A and therefore we see no inhomogeneity in CT state energy or rISC rate, and no power law tail in the DF decay kinetics. As we proposed, this is very different to D–A TADF systems with a C–N bond bridge between D and A which is soft and gives rise to large inhomogeneous rISC rates and CT energies in solid state. This causes a large degree of the unwanted spectral broadening in these D–A TADF systems. Moreover, it also shows unambiguously that there is no extrinsic perturbation from the host due to dielectric constant, i.e. so called “solid state solvatochromism”. At 1% ACRSA loading we see virtually no difference between DPEPO, UGH and neat films. Further, we see that the pure 1CT emission in these films is far less red shifted than it is observed in toluene solution. In zeonex, the energy gap between the CT states and the vibronic coupling mediating local triplet state is very small, 32 meV, giving rise to the measured very fast rISC rate approaching 1 × 107 s−1 and thus highly efficient TADF. We also observe DF from the local singlet state which we propose arises through upper triplet state rISC with thermally activated reverse IC from the strong allowed coupling between the nπ* 3LEA and ππ* 3LEA triplet states and allowed SOC between ππ* 3LEA and nπ* 1LE singlet state.In small molecule hosts at low ACRSA loadings, where the higher dielectric constant of the host materials helps to stabalise the CT state, we observe very efficiently along with mono-exponential DF decay times indicating a homogeneous system. However, surprisingly for a spiro material, at 10% wt loading and in neat film we clearly see strong effects due to intermolecular interaction between neighbouring ACRSA molecules. We believe that these are weakly coupled, not a fully excimeric state, more a co-facial overlap of neighbouring acceptor units which give a more delocalised and thus stronger acceptor. This then gives rise to a lower energy CT state that also contributes to a slow DF because of the large gap between the 3CT and coupling 3LE state which mediates rISC. This state also gives rise to a low energy phosphorescence and the associated triplet state (possibly a 3LE state of the delocalised acridine pair) might be able to mediate rISC independently as well. At low temperatures, in zeonex, we observed a highly structured phosphorescence with 0.19 eV vibronic replicas pointing to strong coupling to the acceptor CO stretching mode, identifying this lowest energy local triplet state as an acceptor 3nπ* state. We also see a long lived, structureless phosphorescence at higher energy which is assigned to the acceptor 3ππ* state. The thermal equilibrium between these two triplet populations is clearly temperature dependent.
ACRSA defines a new blue print for highly efficiency TADF emitters, krISC > 1 × 107 s−1, as it is also a very robust molecule because it removes the weak C–N bridging bond between D and A. This rigidity has great importance, because it is immune to distortions of the D A dihedral bond (driven through host packing) it does not have a long tail of slowly decay DF states. The long residency times of triplet excitation in these distorted states will be far more prone to triplet polaron annihilation preventing a major cause of emitter degradation. This should yield far longer device operating lifetimes. Moreover, because of excimer formation at high loadings, as originally used in devices, we believe EQEs of 16% reflect quenching by excimers and by optimisation of the emitter concentration true state of the art device performance can be achieved. We are actively pursuing this currently.
Author contributions
L. Franca and A. Danos made the presented optical and time resolved spectroscopy measurements. A. Monkman devised the research, undertook the data analysis and supervised the work. A. Monkman wrote the manuscript with L. Franca and A. Danos.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We thank Prof Chihaya Adachi for providing invaluable X-ray data on ACRSA. Funding was gratefully received from the EPSRC (EP/P012167/1) and acknowledge the TADFlife and HyperOLED projects funded by the European Union's Horizon 2020-MCSA-ITN Research and Innovation Programme under grant agreements No. 812872 and No. 732013 under the action ICT-02-2016 for funding the research in Durham.References
- L. G. Franca, Y. Long, C. Li, A. Danos and A. Monkman, The Critical Role of Nπ* States in the Photophysics and Thermally Activated Delayed Fluorescence of Spiro Acridine-Anthracenone, J. Phys. Chem. Lett., 2021, 12(5), 1490–1500, DOI:10.1021/acs.jpclett.0c03314
.
- K. Nasu, T. Nakagawa, H. Nomura, C. J. Lin, C. H. Cheng, M. R. Tseng, T. Yasuda and C. Adachi, A Highly Luminescent Spiro-Anthracenone-Based Organic Light-Emitting Diode Exhibiting Thermally Activated Delayed Fluorescence, Chem. Commun., 2013, 49(88), 10385–10387, 10.1039/c3cc44179b
- H. Nakanotani, T. Higuchi, T. Furukawa, K. Masui, K. Morimoto, M. Numata, H. Tanaka, Y. Sagara, T. Yasuda and C. Adachi, High-Efficiency Organic Light-Emitting Diodes with Fluorescent Emitters, Nat. Commun., 2014, 5(1), 1–7, DOI:10.1038/ncomms5016
- G. Méhes, H. Nomura, Q. Zhang, T. Nakagawa and C. Adachi, Enhanced Electroluminescence Efficiency in a Spiro-Acridine Derivative through Thermally Activated Delayed Fluorescence, Angew. Chem., Int. Ed., 2012, 51(45), 11311–11315, DOI:10.1002/anie.201206289
- F. B. Dias, T. J. Penfold and A. P. Monkman, Photophysics of Thermally Activated Delayed Fluorescence Molecules, Methods Appl. Fluoresc., 2017, 5(1), 012001, DOI:10.1088/2050-6120/aa537e
- K. Stavrou, L. G. Franca and A. P. Monkman, Photophysics of TADF Guest–Host Systems: Introducing the Idea of Hosting Potential, ACS Appl. Electron. Mater., 2020, 2(9), 2868–2881, DOI:10.1021/acsaelm.0c00514
- S. J. Woo, Y. H. Kim and J. J. Kim, Dihedral Angle Distribution of Thermally Activated Delayed Fluorescence Molecules in Solids Induces Dual Phosphorescence from Charge-Transfer and Local Triplet States, Chem. Mater., 2021, 33(14), 5618–5630, DOI:10.1021/acs.chemmater.1c01011
- S. J. Woo and J. J. Kim, TD-DFT and Experimental Methods for Unraveling the Energy Distribution of Charge-Transfer Triplet/Singlet States of a TADF Molecule in a Frozen Matrix, J. Phys. Chem. A, 2021, 125(5), 1234–1242, DOI:10.1021/acs.jpca.0c11322
- T. Serevičius, R. Skaisgiris, I. Fiodorova, G. Kreiza, D. Banevičius, K. Kazlauskas, S. Tumkevičius and S. Juršėnas, Single-Exponential Solid-State Delayed Fluorescence Decay in TADF Compounds with Minimized Conformational Disorder, J. Mater. Chem. C, 2021, 9(3), 836–841, 10.1039/d0tc05503d
- S. Weissenseel, N. A. Drigo, L. G. Kudriashova, M. Schmid, T. Morgenstern, K. H. Lin, A. Prlj, C. Corminboeuf, A. Sperlich, W. Brütting, M. K. Nazeeruddin and V. Dyakonov, Getting the Right Twist: Influence of Donor-Acceptor Dihedral Angle on Exciton Kinetics and Singlet-Triplet Gap in Deep Blue Thermally Activated Delayed Fluorescence Emitter, J. Phys. Chem. C, 2019, 123(45), 27778–27784, DOI:10.1021/acs.jpcc.9b08269
- J. S. Ward, R. S. Nobuyasu, A. S. Batsanov, P. Data, A. P. Monkman, F. B. Dias and M. R. Bryce, The Interplay of Thermally Activated Delayed Fluorescence (TADF) and Room Temperature Organic Phosphorescence in Sterically-Constrained Donor-Acceptor Charge-Transfer Molecules, Chem. Commun., 2016, 52(12), 2612–2615, 10.1039/c5cc09645f
- L. Salah, M. K. Etherington, A. Shuaib, A. Danos, A. A. Nazeer, B. Ghazal, A. Prlj, A. T. Turley, A. Mallick, P. R. McGonigal, B. F. E. Curchod, A. P. Monkman and S. Makhseed, Suppressing Dimer Formation by Increasing Conformational Freedom in Multi-Carbazole Thermally Activated Delayed Fluorescence Emitters, J. Mater. Chem. C, 2021, 9(1), 189–198, 10.1039/D0TC04222F
- M. Hempe, N. A. Kukhta, A. Danos, M. A. Fox, A. S. Batsanov, A. P. Monkman and M. R. Bryce, Vibrational Damping Reveals Vibronic Coupling in Thermally Activated Delayed Fluorescence Materials, Chem. Mater., 2021, 33(9), 3066–3080, DOI:10.1021/acs.chemmater.0c03783
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Highly Efficient Organic Light-Emitting Diodes from Delayed Fluorescence, Nature, 2012, 492(7428), 234–238, DOI:10.1038/nature11687
- F. B. Dias, K. N. Bourdakos, V. Jankus, K. C. Moss, K. T. Kamtekar, V. Bhalla, J. Santos, M. R. Bryce and A. P. Monkman, Triplet Harvesting with 100% Efficiency by Way of Thermally Activated Delayed Fluorescence in Charge Transfer OLED Emitters, Adv. Mater., 2013, 25(27), 3707–3714, DOI:10.1002/adma.201300753
- Y. Im, M. Kim, Y. J. Cho, J.-A. Seo, K. S. Yook and J. Y. Lee, Molecular Design Strategy of Organic Thermally Activated Delayed Fluorescence Emitters, Chem. Mater., 2017, 29(5), 1946–1963, DOI:10.1021/acs.chemmater.6b05324
- B. T. Lim, S. Okajima, A. K. Chandra and E. C. Lim, Radiationless Transitions in Electron Donor-Acceptor Complexes: Selection Rules for S1 → T Intersystem Crossing and Efficiency of S1 → S0 Internal Conversion, Chem. Phys. Lett., 1981, 79(1), 22–27, DOI:10.1016/0009-2614(81)85280-3
- F. B. Dias, J. Santos, D. R. Graves, P. Data, R. S. Nobuyasu, M. A. Fox, A. S. Batsanov, T. Palmeira, M. N. Berberan-Santos, M. R. Bryce and A. P. Monkman, The Role of Local Triplet Excited States and D-A Relative Orientation in Thermally Activated Delayed Fluorescence: Photophysics and Devices, Adv. Sci., 2016, 3(12), 1600080, DOI:10.1002/advs.201600080
- J. Gibson, A. P. Monkman and T. J. Penfold, The Importance of Vibronic Coupling for Efficient Reverse Intersystem Crossing in Thermally Activated Delayed Fluorescence Molecules, ChemPhysChem, 2016, 17(19), 2956–2961, DOI:10.1002/cphc.201600662
- I. Lyskov and C. M. Marian, Climbing up the Ladder: Intermediate Triplet States Promote the Reverse Intersystem Crossing in the Efficient TADF Emitter ACRSA, J. Phys. Chem. C, 2017, 121(39), 21145–21153, DOI:10.1021/acs.jpcc.7b06187
- H. Ohkuma, T. Nakagawa, K. Shizu, T. Yasuda and C. Adachi, Thermally Activated Delayed Fluorescence from a Spiro-Diazafluorene Derivative, Chem. Lett., 2014, 43(7), 1017–1019, DOI:10.1246/cl.140360
- G. Méhes, K. Goushi, W. J. Potscavage and C. Adachi, Influence of Host Matrix on Thermally-Activated Delayed Fluorescence: Effects on Emission Lifetime, Photoluminescence Quantum Yield, and Device Performance, Org. Electron., 2014, 15(9), 2027–2037, DOI:10.1016/j.orgel.2014.05.027
- X. D. Zhu, C. C. Peng, F. C. Kong, S. Y. Yang, H. C. Li, S. Kumar, T. T. Wang, Z. Q. Jiang and L. S. Liao, Acceptor Modulation for Improving a Spiro-Type Thermally Activated Delayed Fluorescence Emitter, J. Mater. Chem. C, 2020, 8(25), 8579–8584, 10.1039/d0tc00743a
- L. Gan, Z. Xu, Z. Wang, B. Li, W. Li, X. Cai, K. Liu, Q. Liang and S. Su, Utilizing a Spiro TADF Moiety as a Functional Electron Donor in TADF Molecular Design toward Efficient “Multichannel” Reverse Intersystem Crossing, Adv. Funct. Mater., 2019, 29(20), 1808088, DOI:10.1002/adfm.201808088
- J. Rao, C. Zhao, Y. Wang, K. Bai, S. Wang, J. Ding and L. Wang, Achieving Deep-Blue Thermally Activated Delayed Fluorescence in Nondoped Organic Light-Emitting Diodes through a Spiro-Blocking Strategy, ACS Omega, 2019, 4(1), 1861–1867, DOI:10.1021/acsomega.8b03296
- X. Tang, L. S. Cui, H. C. Li, A. J. Gillett, F. Auras, Y. K. Qu, C. Zhong, S. T. E. Jones, Z. Q. Jiang, R. H. Friend and L. S. Liao, Highly Efficient Luminescence from Space-Confined Charge-Transfer Emitters, Nat. Mater., 2020, 19(12), 1332–1338, DOI:10.1038/s41563-020-0710-z
-
C. Reichardt and T. Welton, Solvents and Solvent Effects in Organic Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2010, DOI:10.1002/9783527632220
- P. L. dos Santos, M. K. Etherington and A. P. Monkman, Chemical and Conformational Control of the Energy Gaps Involved in the Thermally Activated Delayed Fluorescence Mechanism., J. Mater. Chem. C, 2018, 6(18), 4842–4853, 10.1039/C8TC00991K
- P. L. dos Santos, J. S. Ward, M. R. Bryce and A. P. Monkman, Using Guest–Host Interactions To Optimize the Efficiency of TADF OLEDs, J. Phys. Chem. Lett., 2016, 7(17), 3341–3346, DOI:10.1021/acs.jpclett.6b01542
- T. Northey, J. Stacey and T. J. Penfold, The Role of Solid State Solvation on the Charge Transfer State of a Thermally Activated Delayed Fluorescence Emitter, J. Mater. Chem. C, 2017, 5(42), 11001–11009, 10.1039/C7TC04099G
- P. L. dos Santos, J. S. Ward, M. R. Bryce and A. P. Monkman, Using Guest–Host Interactions To Optimize the Efficiency of TADF OLEDs, J. Phys. Chem. Lett., 2016, 7(17), 3341–3346, DOI:10.1021/acs.jpclett.6b01542
- B. I. Greene, R. M. Hochstrasser and R. B. Weisman, Picosecond Transient Spectroscopy of Molecules in Solution, J. Chem. Phys., 1979, 70(3), 1247–1259, DOI:10.1063/1.437617
- J. Fan, L. Cai, L. Lin and C. Wang, Understanding the Light-Emitting Mechanism of an X-Shape Organic Thermally Activated Delayed Fluorescence Molecule: First-Principles Study, Chem. Phys. Lett., 2016, 664, 33–38, DOI:10.1016/j.cplett.2016.10.009
- R. S. Nobuyasu, Z. Ren, G. C. Griffiths, A. S. Batsanov, P. Data, S. Yan, A. P. Monkman, M. R. Bryce and F. B. Dias, Rational Design of TADF Polymers Using a Donor-Acceptor Monomer with Enhanced TADF Efficiency Induced by the Energy Alignment of Charge Transfer and Local Triplet Excited States, Adv. Opt. Mater., 2016, 4(4), 597–607, DOI:10.1002/adom.201500689
- A. S. R. Koti and N. Periasamy, Time Resolved Area Normalized Emission Spectroscopy (TRANES) of DMABN Confirms Emission from Two States, Res. Chem. Intermed., 2002, 28(7–9), 831–836, DOI:10.1163/15685670260469447
- H. A. Al Attar and A. P. Monkman, Electric Field Induce Blue Shift and Intensity Enhancement in 2D Exciplex Organic Light Emitting Diodes; Controlling Electron-Hole Separation, Adv. Mater., 2016, 28(36), 1–7, DOI:10.1002/adma.201600965
- M. Colella, A. Danos and A. P. Monkman, Less Is More: Dilution Enhances Optical and Electrical Performance of a TADF Exciplex, J. Phys. Chem. Lett., 2019, 10(4), 793–798, DOI:10.1021/acs.jpclett.8b03646
- P. Imbrasas, R. Lygaitis, P. Kleine, R. Scholz, C. Hänisch, S. Buchholtz, K. Ortstein, F. Talnack, S. C. B. Mannsfeld, S. Lenk and S. Reineke, Dimers or Solid-State Solvation? Intermolecular Effects of Multiple Donor–Acceptor Thermally Activated Delayed Fluorescence Emitter Determining Organic Light-Emitting Diode Performance, Adv. Opt. Mater., 2021, 9(14), 2002153, DOI:10.1002/adom.202002153
- M. K. Etherington, N. A. Kukhta, H. F. Higginbotham, A. Danos, A. N. Bismillah, D. R. Graves, P. R. McGonigal, N. Haase, A. Morherr, A. S. Batsanov, C. Pflumm, V. Bhalla, M. R. Bryce and A. P. Monkman, Persistent Dimer Emission in Thermally Activated Delayed Fluorescence Materials, J. Phys. Chem. C, 2019, 123(17), 11109–11117, DOI:10.1021/acs.jpcc.9b01458
- E. Cho, M. Hong, V. Coropceanu and J. Brédas, The Role of Intermolecular Interactions
on the Performance of Organic Thermally Activated Delayed Fluorescence (TADF) Materials, Adv. Opt. Mater., 2021, 9(14), 2002135, DOI:10.1002/adom.202002135.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details; additional photophysical data such as: time resolved emission decays and steady state spectra of ACRSA in different hosts, exciting at different wavelengths, lifetime fittings, X-ray structure. See DOI: 10.1039/d1tc04484b |
| This journal is © The Royal Society of Chemistry 2022 |