
Progress and perspective for conversion of plastic wastes into valuable chemicals
Kai
Zheng†
a,
Yang
Wu†
a,
Zexun
Hu
a,
Shumin
Wang
a,
Xingchen
Jiao
 *ab,
Juncheng
Zhu
a,
Yongfu
Sun
*a and
Yi
Xie
*a
*ab,
Juncheng
Zhu
a,
Yongfu
Sun
*a and
Yi
Xie
*a
aHefei National Research Center for Physical Sciences at Microscale, University of Science and Technology of China, Hefei, 230026, P. R. China. E-mail: xcjiao@mail.ustc.edu.cn; yfsun@ustc.edu.cn; yxie@ustc.edu.cn
bKey Laboratory of Synthetic and Biological Colloids, Ministry of Education, School of Chemical and Material Engineering, Jiangnan University, Wuxi 214122, China
First published on 5th December 2022
Abstract
Today, discarded plastics in nature have caused serious “white pollution”, however these plastic wastes contain abundant carbon resources that could serve as the feedstock to produce commodities. Because of this, it is requisite to convert these plastic wastes into valuable chemicals. Herein, the state-of-the-art techniques for plastic conversion are divided into two categories, those performed under violent conditions and mild conditions, in which the conversion mechanisms are discussed. The strategies under violent conditions are closer to practical application thanks to their excellent conversion efficiencies, while the strategies under mild conditions are more environmentally friendly, showing enormous development potential in the future. We summarize in detail the pyrolysis, hydropyrolysis, solvolysis and microwave-initiated catalysis for bond cleavage in plastic wastes at temperatures ranging from 448 to 973 K. Also, we overview the photocatalysis, electrocatalysis and biocatalysis for bond cleavage in plastic wastes at near and even normal temperature and pressure. Finally, we present some suggestions and outlooks concerning the improvement of current techniques and in-depth mechanisms of investigation for conversion of plastics into valuable chemicals.
 Kai Zheng | Kai Zheng received his BS degree in the Department of Materials Science and Engineering at Northeast Forestry University, China, in 2018. He is currently pursuing his PhD degree in inorganic chemistry at the University of Science and Technology of China, under the supervision of Prof. Yi Xie. His current interests include the synthesis and characterization of two-dimensional metal oxides and sulfides and their applications in photo/electro-catalysis CO2 conversion. |
 Yang Wu | Wu received his BS degree in the Department of Chemical Engineering at Hefei University of Technology (2020). He is currently pursuing his PhD degree in inorganic chemistry at the University of Science and Technology of China, under the supervision of Prof. Xie Yi and Prof. Sun Yongfu. His current interests include the synthesis and characterization of two-dimensional metal oxides and sulfides and their applications in photo/electro-catalysis CO2 conversion. |
 Zexun Hu | Zexun Hu received his BS degree in the Department of Chemistry at the University of Science and Technology of China (2020). Now, he is pursuing his PhD degree at the Hefei National Laboratory for Physical Sciences at Microscale, University of Science and Technology of China, under the supervision of Prof. Sun Yongfu. His current interests include the synthesis and characterization of nanostructures, especially two-dimensional materials, as well as their applications in energy conversion. |
 Shumin Wang | Shumin Wang received her BS degree in chemical engineering and technology from Anhui University, China, in 2019. She is currently studying for a PhD degree under the supervision of Prof. Yongfu Sun in inorganic chemistry at the University of Science and Technology, China. Her research interest is the conversion of carbon dioxide to high-valued fuels and chemicals, and she currently focuses on materials and electrode design for efficient carbon dioxide conversion and large-scale applications. |
 Xingchen Jiao | Jiao received his BS degree in the Department of Chemistry at Hefei University of Technology (2014) and PhD in Inorganic Chemistry from the University of Science and Technology of China (2019). He currently works as a professor at Jiangnan University. His current interests include the synthesis and characterization of nanostructures, especially two-dimensional materials, as well as their applications in CO2 photo-/electro-conversion and plastic-conversion into valuable chemicals. |
 Juncheng Zhu | JunCheng Zhu received his BS degree in the School of the Gifted Young at the University of Science and Technology of China in 2019. He is currently pursuing his Ph.D. degree in inorganic chemistry at the University of Science and Technology of China, under the supervision of Prof. Yi Xie. His current interests include the synthesis, characterization and theoretical simulation of two-dimensional metallic compounds and their applications in electro/photo/photothermal-catalytic CO2 conversion. |
 Yongfu Sun | Yongfu Sun received his BS degree in the Department of Chemistry at Anhui University (2006) and PhD degree in Inorganic Chemistry from the University of Science and Technology of China (2011). After that, he worked as a postdoctoral fellow at National Synchrotron Radiation Laboratory. In 2014, Dr Sun joined the Hefei National Laboratory for Physical Sciences at Microscale as a professor. His current interests include theoretical computation, controllable synthesis, fine structure characterization of atomically thick two-dimensional materials as well as their photo-/electro-catalytic CO2 reduction into hydrocarbon fuels. |
 Yi Xie | Yi Xie received her BS degree from Xiamen University (1988) and a PhD from the University of Science and Technology of China (USTC, 1996). She is now a principal investigator at Hefei National Laboratory for Physical Sciences at the Microscale and a full professor at the Department of Chemistry, USTC. She was appointed as the Cheung Kong Scholar Professor of inorganic chemistry in 2000 and elected as a member of the Chinese Academy of Sciences in 2013, also a recipient of many awards, including the Chinese National Nature Science Award (2001 and 2012), China Young Scientist Award (2002), China Young Female Scientist Award (2006), IUPAC Distinguished Women in Chemistry/Chemical Engineering Award (2013), TWAS Prize in Chemistry (2014), TWAS member (2015) and L'Oréal-UNESCO for Women in Science Awards (2015). Her research interests are cutting-edge research at four major frontiers: solid-state materials chemistry, nanotechnology, energy science and theoretical physics. |
Key learning points1. Introduction of the hazard of “white pollution” and the traditional disposal methods.2. Systematically overview the mechanism of the state-of-the-art techniques for converting plastic wastes into valuable chemicals under violent conditions. 3. Systematically overview the mechanism of the state-of-the-art techniques for converting plastic wastes into valuable chemicals under mild conditions. 4. Deepen insight into the conversion mechanism to guide the design of high-efficiency catalysts. 5. Perspective for the improvement of the current techniques for practical applications. |
1. Introduction
The word “plastic” refers to the synthetic polymer that has been widely used in all walks of life thanks to its low cost, lightweight, durability, processability and tremendous chemical diversity.1–4 Up to now, a variety of polymers including polyethylene (PE), polypropylene (PP), polyvinyl chloride (PVC), polyethylene terephthalate (PET) and polystyrene (PS) have been used to manufacture commercial plastics. Owing to the enormous demand for plastics in daily life, their global production has ceaselessly increased exponentially in the past several decades. In the 1960s, only 15 million metric tonnes of plastics were consumed, whereas approximately 311 million metric tonnes of plastics were produced in 2014, and it is estimated that more than 500 million metric tonnes of plastics will be required in 2050.5 Accompanied by the rapidly growing production of plastics, a good deal of solid waste is also generated every year. It is predicted that the amount of global plastic waste will increase from 260 to 460 metric tonnes annually from 2016 to 2030.6 Worse still, only roughly 9 percent of this plastic waste is recycled, while 12 percent of it is disposed of by incineration and 79 percent is discarded in landfills, the ocean, or in nature.7 Because of the ultra-stability of these plastic wastes, it is exceedingly difficult for their natural degradation. So, it is estimated that approximately 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 metric tonnes of plastic waste will be accumulated globally by 2050.8 Unfortunately, these discarded plastic wastes will cause huge damage to our lives because they result in the pollution of water sources, the destruction of plowed land, and even the infiltration of food webs.9 More surprisingly, an up-to-date study has reported that multifarious microplastics have been detected in human stools.10 With this in mind, it is urgent to seek more tactics for recycling these plastic wastes.
000 metric tonnes of plastic waste will be accumulated globally by 2050.8 Unfortunately, these discarded plastic wastes will cause huge damage to our lives because they result in the pollution of water sources, the destruction of plowed land, and even the infiltration of food webs.9 More surprisingly, an up-to-date study has reported that multifarious microplastics have been detected in human stools.10 With this in mind, it is urgent to seek more tactics for recycling these plastic wastes.
Today, landfills and incineration are still the most widely used methods for plastic disposal in current life, which are prone to causing serious environmental problems. Apart from these two traditional methods, four main technologies have been developed for plastic disposal, which are referred to as “primary”, “secondary”, “tertiary” and “quaternary” recycling.11 In fact, primary recycling, also called closed-loop recycling, is a process for the direct reuse of unpolluted plastic waste into new products. However, the need for selectional collection and artificial sorting increases the cost of this method,12,13 so this technology is not a popular solution for the general plastic problem today. Secondary recycling is also mechanical recycling that collects plastics after use, which come from various sources, and are often contaminated. A special form of secondary recycling is the dissolution of mixed plastics in an appropriate organic solvent and the subsequent precipitation of various components by adding a non-solvent.14,15 During this process, the mechanical properties of plastics often decrease owing to the reduction of molecular weights or the contamination of the dominating matrix by other mixed plastics. Tertiary recycling, also termed chemical recycling, converts plastics to valuable chemicals by a chemical process such as pyrolysis, hydrocracking, solvolysis, gasification and photoreforming.16,17 In this procedure, the repeating hydrocarbon backbones in plastics are also cleaved to produce oligomers with shorter chains.18 Due to the high economic benefit, this strategy has attracted broad attention. Quaternary recycling is the incineration of plastics for energy recuperation, which is normally used for dealing with seriously polluted plastic waste that is difficult to dispose of by other means.19 In this way, heat energy is created by burning the complex mixtures and some poisonous or greenhouse gases may be synchronously formed. This method is still widely used worldwide thanks to its simple and efficient process. Recently, some gas cleaning technologies have been utilized to purge the gas in a proper incinerator during the incineration procedure.20 In general, all the above methods have some advantages and disadvantages. Because of the abundant carbon resources in plastic wastes that can be used as feedstock for producing value-added fuels, many researchers optimize these four typical technologies or further exploit new techniques for the efficient conversion of plastic waste.
Recently, there have been many reviews concerning the degradation or recycling of plastic waste, most of which mainly enumerate the strategies for their conversion into chemicals.21–24 However, few of them pay much attention to the internal conversion mechanism of various methods. In this regard, we divide the up-to-date techniques into two categories: technologies executed under violent and mild conditions (Scheme 1). Also, we present a timely summary of the mechanism of bond cleavage in plastic waste to produce valuable chemicals (e.g., hydrogen, alkane, olefin, wax gasoline and even carbon). Actually, the strategies under violent conditions involving high temperature and high pressure are closer to practical application thanks to their excellent conversion efficiencies; meanwhile, the strategies under mild conditions such as normal temperature and pressure have enormous development potential in the future due to their environmental friendliness.25 In detail, we outline the pyrolysis of plastics by a consecutive two-stage process at high temperatures. We also review the hydropyrolysis of plastics with a bifunctional metal/acid catalyst at mild temperatures; in addition, we highlight the solvolysis of plastics by a tandem catalytic cross-alkane metathesis process at relatively low temperatures and present the one-step microwave catalysis of plastics at mild temperatures. Subsequently, we elaborate on the photocatalysis of plastics by a two-step procedure under simulated natural environment conditions; also, we overview the electrocatalysis of plastics by a three-step process at ambient temperature and pressure. Furthermore, we summarize the biocatalysis of plastics by a two-enzyme system under normal pressure and temperature (Table 1). Finally, we give a brief conclusion and a perspective on the improvement of current techniques and an in-depth mechanism investigation by in situ studies and theoretical analysis for the conversion of plastics into valuable chemicals. We hope this review enables a deepened comprehension of the conversion mechanism for state-of-the-art techniques and an understanding of the latest developments in plastic disposal.
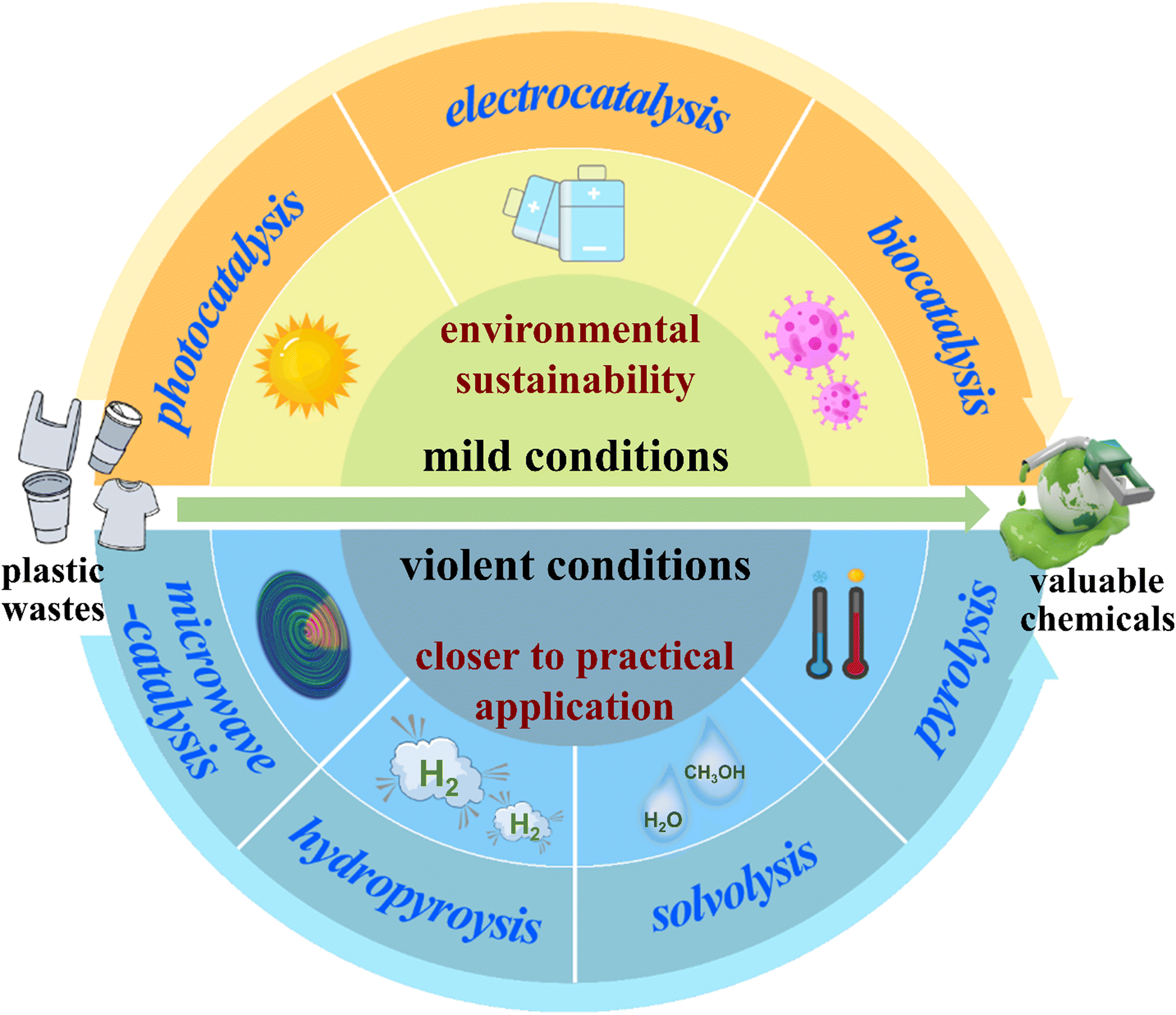 | ||
| Scheme 1 Strategies, divided into two categories, for plastic conversion into valuable chemicals. The strategies executed under violent conditions involve pyrolysis, hydropyrolysis, solvolysis, and microwave catalysis, while the strategies carried out under mild conditions involve photocatalysis, electrocatalysis and biocatalysis. | ||
| Techniques | Temperatures | Advantages | Disadvantages | |
|---|---|---|---|---|
| Strategies under violent conditions | Pyrolysis | 673–973 K | 1. High degradation efficiency | 1. High power consumption |
| 2. Closer to practical application | 2. Poor product selectivity | |||
| 3. Difficulty for the mechanism study | ||||
| Hydropyrolysis | 473–523 K | |||
| Solvolysis | <473 K | |||
| Microwave catalysis | 573–773 K | |||
| Strategies under mild conditions | Photocatalysis | Room temperature | 1. Low power consumption | Low degradation efficiency |
| 2. Environmental protection | ||||
| 3. High product selectivity | ||||
| 4. Explicit degradation mechanism | ||||
| Electrocatalysis | Room temperature | |||
| Biocatalysis | 303 K | |||
2. Conversion of plastic waste into valuable chemicals under violent conditions
Generally speaking, the degradation of plastic waste under violent conditions means the operation is carried out at high temperatures or high pressures, breaking down the stable framework of plastics. That is to say, plastics can be directly converted into valuable chemicals with a high conversion efficiency by means of violent conditions. However, these violent conditions also cause an obstacle to the exploration of conversion mechanisms because many in situ characterization techniques cannot be operated at high temperatures and pressures. To figure out the mechanism for plastic degradation under violent conditions, many researchers have optimized the existing technologies or developed some innovative technologies in recent years.2.1 Pyrolysis for bond cleavage in plastic waste at high temperatures
Today, pyrolysis is a widely used recycling technology conducted in an oxygen-free environment, which commonly uses a high temperature ranging from ca. 300 to 900 °C for the high-efficiency degradation of plastics into all kinds of carbon-based fuels.26–28 In general, pyrolysis usually contains thermal pyrolysis and catalytic pyrolysis. Thermal pyrolysis is simply heating plastics to induce their bond cleavage to produce short-chain molecules, whereas catalytic pyrolysis is often performed with a catalyst in an effort to lower the temperature and decrease the reaction time.21 To date, although much plastic pyrolysis has been reported (Table 2) and even applied in practice,29,30 its degradation mechanism is always indistinct because the pyrolysis process normally undergoes a complicated route.31 During the pyrolysis process, four steps (end-chain depolymerization, random-chain fragmentation, chain-elimination of side chains and cross-linking for the formation of a new chain network) are often presented, so it is quite difficult to summarize a systematic mechanism for plastic pyrolysis.32 In addition, another non-negligible downside of plastic pyrolysis is the uncontrollable product selectivity due to the random bond cleavage in plastics.| Catalysts | Plastics | Advantages | Ref. |
|---|---|---|---|
| Ni/Co pillared montmorillonites | Plastic film waste | The pillared structure possesses abundant Lewis acidity and uniform metal sites for boosting product selectivity. | 87 |
| BaO | Polystyrene based materials | The solid bases play a key role in eliminating hydrogen atoms in polystyrene for the formation of carboanions intermediates. | 88 |
| HZSM-5 zeolite | Low-density polyethylene | The appropriate acidic site density on HZSM-5 catalysts can enhance and tune the product selectivity. | 89 |
| Fe-modified ZSM-5 zeolite | HDPE, LDPE, PP, PS, PVC and PET | High acidity is beneficial to promoting the formation of aromatic and olefins compounds. | 90 |
| Fe(3)-HY zeolite | PVC | Fe sites on HY catalysts not only boost the cracking on the HY surface but also improve the dichlorination of iron oxide. | 91 |
| Fe/Al2O3 zeolite | PP, HDPE, LDPE, HIPS and GPPS | The catalysts make for understanding the relationship between the structure and function. | 92 |
| ZnO zeolite | Polyethylene terephthalate | The strong base of ZnO can promote the generation of benzene-rich aromatic hydrocarbons. | 93 |
| NiO/HY zeolite | Low-density polyethylene | The presence of NiO can improve the formation of aromatics, and the NiO/HY can optimize the ratio between oil yield and oil quality. | 58 |
| SiC foam@ZSM-5 core-shell catalyst | Low-density polyethylene | The pore structure and acidic sties on the catalysts can tune and improve the product selectivity. | 94 |
| Red mud | Plastic mixture (PE, PP, PS, PVC and PVC) | The red mud can boost the aromatization in the liquid over the acidic sites. | 95 |
| Modified mordenite | Plastic mixture (PE, PP, PS, PVC and PVC) | Modified mordenite possesses high surface acidity, which can boost the yield of light liquid products. | 96 |
| Lab-synthesized ZSM-5 zeolite | PET, BOPP, PE and MET | Lab-synthesized ZSM-5 zeolites have strong acidic sites and microporous crystalline structure, which can enhance the cracking and isomerization for improving the performance of plastic conversion. | 97 |
| ZAP USY zeolite | LDPE, HDPE, PP and PS | The catalyst can lower the reaction energy for increasing the production of gases, which enhances the pyrolysis rate of plastic. | 98 |
| Activated carbon | Low-density polyethylene | The porous structure on functional groups modified activated carbon can improve the selectivity and the formation rate of aromatic hydrocarbons. | 99 |
Bearing the above in mind, Joo-Sik Kim et al. have developed a consecutive two-stage process for the pyrolysis of plastics through tandem auger and fluidized bed reactors, wherein the vibrational states of plastic molecules are elevated by the auger reactor, and then injected into the fluidized bed reactor for further bond cleavage to produce various fuels.33 For the degradation of typical PP plastics, the conventional one-step pyrolysis of PP plastic usually undergoes the random cleavage of short C–C and C–H bonds without activation to form multifarious carbonaceous compounds (Fig. 1a). In contrast, the C–C and C–H bonds could be lengthened after activation in the auger reactor, and then these long C–C and C–H bonds could be easily broken to generate abundant ˙CH3, ˙CH2–CH3 and ˙CH2–CH2–CH3 radicals for the eventual formation of short-chain fuels such as hydrogen (H2), methane (CH4) and ethylene (C2H4) in the fluidized bed reactor (Fig. 1b). In other words, upon increasing the temperature in the auger reactor, the weakening of C–C and C–H bonds in PP molecules would promote the production of smaller carbonaceous intermediates and thus result in increasing the yields of short-chain fuels. Additionally, the Flynn–Ozawa–Wall method for the kinetic models in Fig. 1c and d was utilized to evaluate the activation energies for PP molecules with and without activation,34 in which the activator-off and activator-on modes in the thermogravimetric analysis experiments were used to simulate the one-step and two-step pyrolytic PP processes, respectively. The high linear relationship between ln(dx/dt) and 1000 T−1 could be acquired at fractional conversion from 0.1 to 0.9, whereas the average activation energies for the modes of activator-off and activator-on were ca. 188.83 and 170.34 kJ mol−1, hinting at the relatively easy bond cleavage of activated PP molecules for the two-step pyrolytic PP process. That is to say, the formed pyrolysates of PP plastics through the C–C bond cleavage in the first step could be readily evaporated in the second step, thus making for the decrease of activation energy. Profiting from the reduced activation energy, the C–C bond could be cleaved into the shortest carbon-based intermediates as far as possible. In this case, the pyrolysis of PP plastics by this two-stage process could selectively realize product gases containing H2 and CH4 with about 81 wt% at a temperature of 673 K in the auger reactor and a temperature of ca. 973 K in the fluidized bed reactor.33 Compared with the one-step pyrolysis process of plastics, this two-stage pyrolysis plastic demands two kinds of heating equipment, which may be more energy-intensive and high-cost in some respects. Fortunately, the product selectivity of plastics pyrolysis can be regulated via this consecutive two-stage process. Note that relatively unique or uncomplex products can be obtained through these two-stage pyrolysis plastics, which is in favor of the subsequent separation and extraction. In this regard, the whole cost for the recycling of plastic wastes will decrease by several orders of magnitude. It is predictable that both the thermal pyrolysis and the catalytic pyrolysis by a two-stage process will draw more attention in the future. Frankly speaking, the design of the reactor is also an important factor in pyrolysis. Recently, all kinds of reactors have been utilized in plastic pyrolysis (Table 3). High heat transfer rates and reliable temperature control must be taken into consideration upon designing an optimum reactor. In contrast, an inadequate reactor design would result in undesired reaction conditions, thus lowering product quality and forming some by-products.35 As a result, it is necessary to design a typical reactor for high-efficiency pyrolysis of different types of plastics.
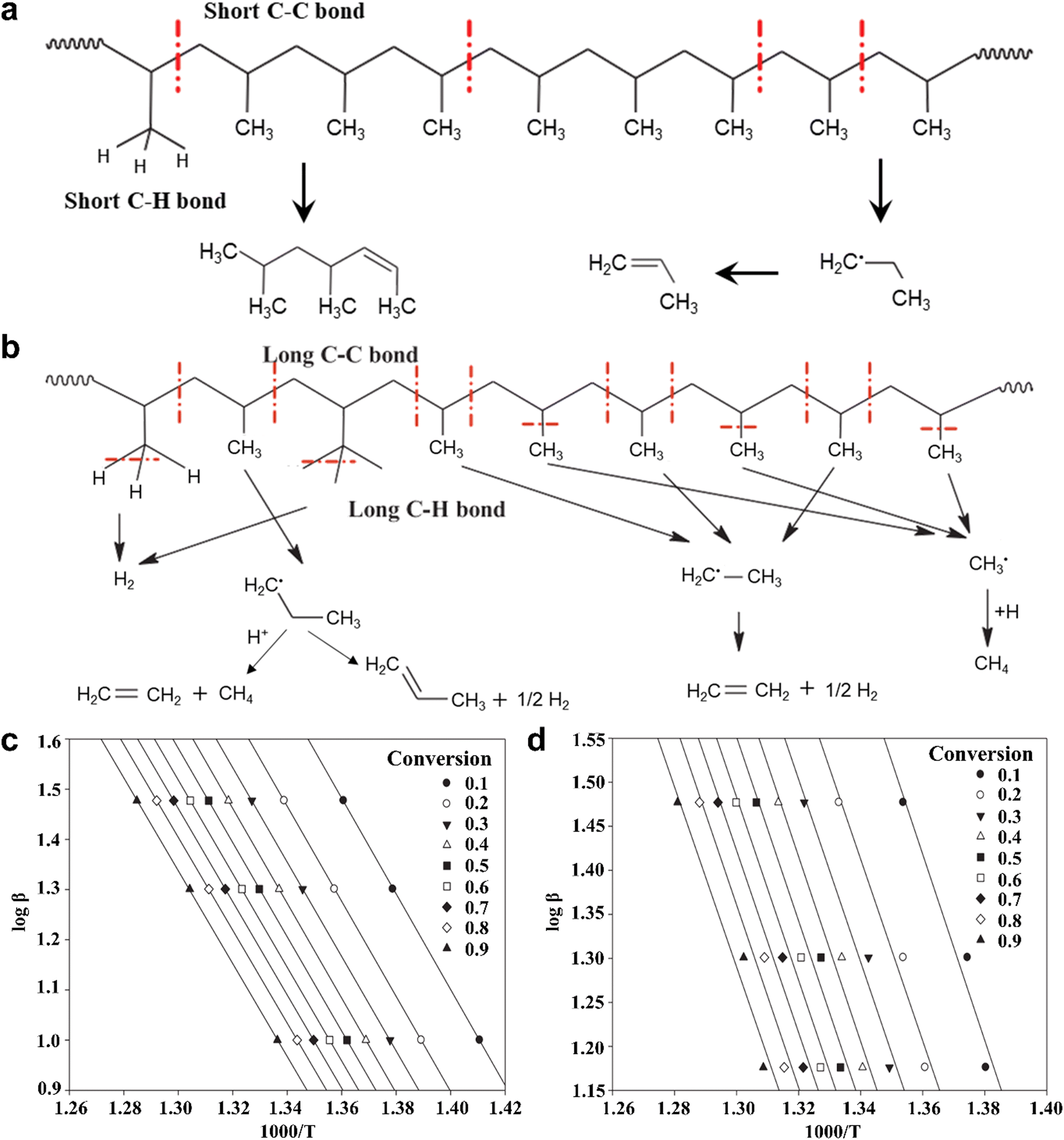 | ||
| Fig. 1 Scheme for the bond cleavage of PP plastics through (a) the conventional one-step pyrolysis and (b) the designed two-step pyrolysis processes. The average activation energies for (c) the conventional one-step pyrolysis and (d) the designed two-step pyrolysis processes by the Flynn–Ozawa–Wall method. Reproduced with permission from ref. 33. Copyright 2019 Elsevier. | ||
| Plastics | Reactors | Temperatures (K) | Ref. |
|---|---|---|---|
| HDPE | Batch | 723 | 100 |
| Fixed bed | 823 | 101 | |
| Fluidised bed | 913 | 102 | |
| LDPE | Batch and fixed bed | 748 | 103 |
| Chromium reactor | 753 | 104 | |
| PP | Batch | 673 | 105 |
| Semi batch | 633 | 106 | |
| Fixed bed | 773–873 | 107 | |
| Semi-batch rotary kiln | 773 | 108 | |
| PS | Stainless steel | 523–703 | 106 |
| Stirred batch | 573–773 | 109 | |
| Semi batch | 923 | 110 | |
| Batch | 753 | 111 | |
| PET | Quartz | 653–1033 | 112 |
| PVC | Stainless steel | 598 | 113 |
2.2 Hydropyrolysis for bond cleavage in plastic waste at mild temperatures
Similar to the pyrolysis of plastics, hydropyrolysis of plastics is a frequently used method for the conversion of long-chain plastics into short-chain molecules, which is usually carried out in a hydrogen atmosphere at high temperatures with the assistance of a catalyst (Table 4). For example, Leshkov et al. realized a variety of PE plastics into liquid alkanes over the catalyst of ruthenium nanoparticles supported on carbon in hydrogen at temperatures ranging from 473 to 523 K.36,37 Compared with the pyrolysis of plastics, the reaction temperature is reduced because of the increased efficiencies of mass and heat transfer in hydrogen.38,39 Some existing strategies for converting plastics into fuels are displayed in Fig. 2a, wherein the thermal or catalytic pyrolysis is usually executed at a temperature exceeding 673 K and the previously reported hydropyrolysis is often carried out at 573 to 723 K.40 Unfortunately, the hydropyrolysis process also follows a random bond cleavage to form complex hydrocarbon with different carbon numbers since the hydrogen atom may attack any carbon atom in plastics (Fig. 2b), and hence, the product selectivity is also difficult to control.| Catalysts | Plastics | Conditions | Ref. |
|---|---|---|---|
| Pt–ZrO2–S | LDPE | 523 K | 101 |
| Pt–ZrO2–SO4 | HDPE, PP and PS | 573–648 K, 5.27–8.38 Mpa | 114 |
| Ni/ZrO2–SO4 | LDPE | 573 K, 1 MPa | 115 |
| Cr/ZrO2–SO4 | LDPE | 523 K, 1 MPa | 116 |
| Pt/WO3/ZrO2 mix HY zeolite | LDPE and PP | 523 K, 1.5 MPa | 40 |
| NiMo/Al2O3 | LDPE, PET and PS | 703 K, 8.3 MPa | 117 |
| CoAl2O4 | PE | 833 K, 0.2 MPa | 118 |
| USY/SBA-16 | LDPE, HDPE, PP and PS | 673 K, 2 MPa | 119 |
| NiW/HY | PE | 743–803 K, 2–5 MPa | 120 |
| Ni/SBA-15 | PE, PP, PET and PS | 923 K, 1 MPa | 121 |
| H-USY | HDPE | 623–663 K | 122 |
| HZSM-5 | HDPE | 673 K, 2 MPa | 123 |
| Pd/trap-HZSM-5 | HDPE | 673 K, 1 MPa | 124 |
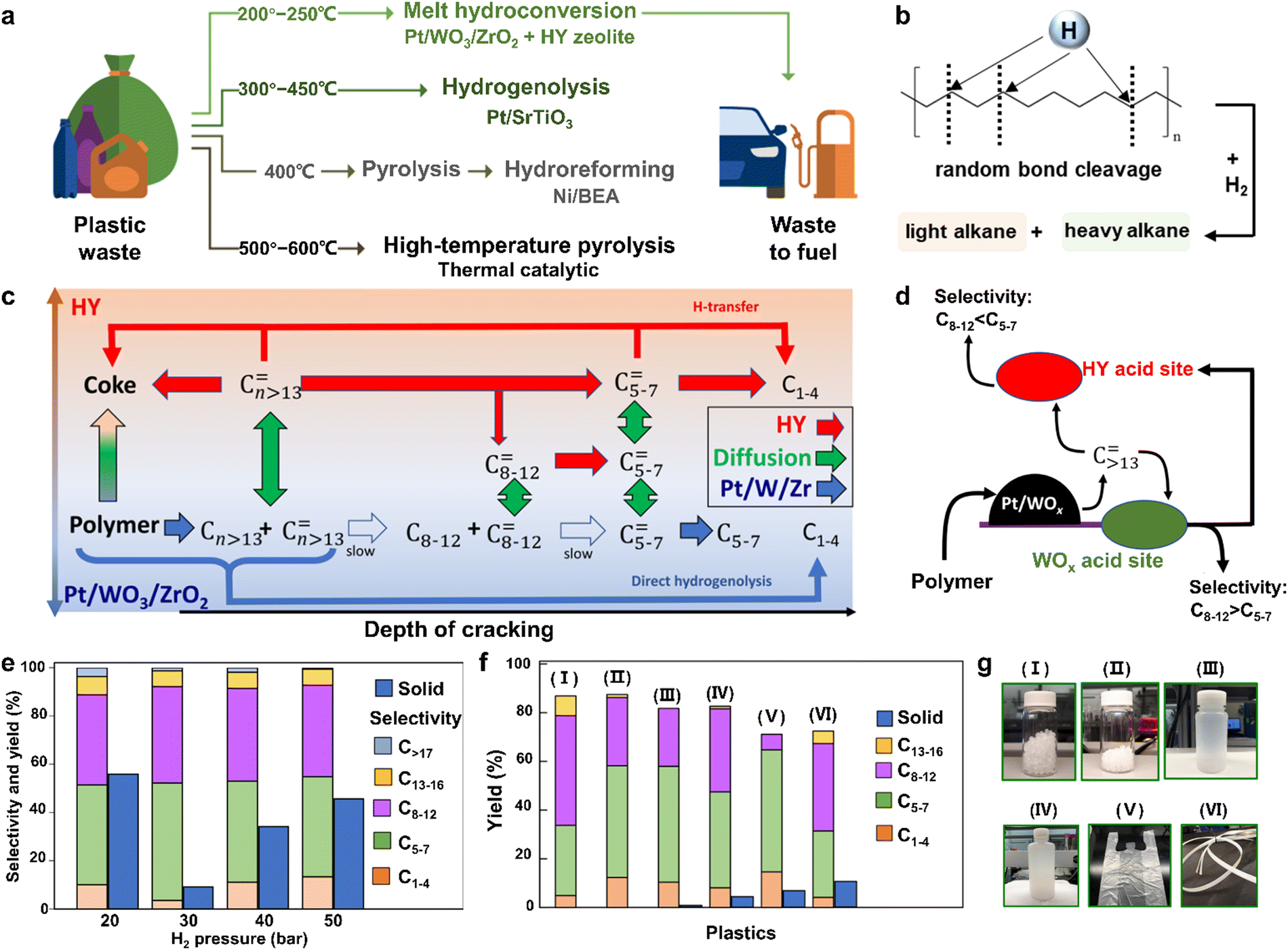 | ||
| Fig. 2 (a) The reported various strategies for the conversion of plastics into fuels at different temperatures. (b) The possible mechanism for random bond cleavage during PE hydropyrolysis. (c) The reaction network emphasizes the acceleration of deep cracking by the HY zeolite in the tandem catalysis process. (d) Scheme for the diffusion of reactive intermediates in the tandem catalysis process, which illustrates the product selectivity tailored by the acid sites of Pt/WO3/ZrO2 and HY zeolite catalysts. (e) LDPE hydropyrolysis by Pt/WO3/ZrO2 and HY zeolite catalysts under different hydrogen pressures at 250 °C. (f) Hydropyrolysis of various plastics by Pt/WO3/ZrO2 and HY zeolite catalysts under 30 bar H2 at 250 °C. (g) I–VI represent PP powders, HDPE powders, LDPE bottle, HDPE bottle, HDPE bag, and composite tape of PP, PE and PS, which correspond to those in f. Reproduced with permission from ref. 40. Copyright 2021 AAAS. | ||
To further reduce the reaction temperature of hydropyrolysis and regulate the product selectivity, many catalysts, especially zeolite catalysts, have been utilized for hydropyrolysis (Table 5). As an example, Vlachos et al. have employed a bifunctional metal/acid catalyst of platinum deposited on tungstate-zirconia (Pt/WO3/ZrO2) and an FAU-type zeolite (HY) for hydropyrolysis of plastics at 473 to 523 K, in which the acid catalyst could break the C–C bonds to form intermediates, while the metal catalyst could enable the succedent hydrogenation of these intermediates to generate various liquid fuels.40 In detail, the plastics were initially hydrocracked into some large olefins (Cn ≥ 13) over Pt/WO3/ZrO2. Then, these large olefins diffused into the acid sites on HY zeolites to quickly be cracked into smaller alkenes (C5–7) thanks to the shape selectivity of their microporous network, whereas the cracking of large olefins into light alkenes over Pt/WO3/ZrO2 was relatively slower owing to their weaker acid sites (Fig. 2c and d). It is worth noting that the product distributions could be manipulated by tailoring the acidity of HY zeolites or the ratio between the zeolite and Pt/WO3/ZrO2 during this tandem catalysis. In addition, the hydrogen pressure was an indispensable factor in affecting the performance of plastic hydropyrolysis, and the optimal hydrogen pressure was 30 bar as shown in Fig. 2e. At lower hydrogen pressure, the speed of hydropyrolysis was decreased, which led to the lower conversion of low-density polyethylene (LDPE). Meanwhile, at higher hydrogen pressure, both the selectivity of C1–4 alkenes and the solid residue increased. Profiting from these advantages, various commercial plastics could be successfully hydrocracked into valuable gasoline-range hydrocarbons through this tandem catalytic procedure at a mild temperature of 523 K (Fig. 2f and g). Unlike the majority of plastic hydropyrolysis, the most eye-catching point of this tandem catalytic process is that the product distribution can be efficaciously manipulated by the acidity and porosity of HY zeolites, which favors obtaining more value-added diesel-range hydrocarbon products. Another admirable merit of this tandem catalytic process lies in its decreased reaction temperature, which conduces to reduced energy consumption. However, using high-purity H2 as a reactant is still potentially hazardous due to its easily explosive feature. Inspired by this tandem catalytic process with a bifunctional metal/acid catalyst, it is necessary to design and prepare a new metal catalyst for capturing the low-concentration H2. In this case, the acid catalyst triggers the C–C bond cleavage to form intermediates that are further hydrogenated into valuable chemicals under low-concentration H2 by the designed metal catalyst. This bifunctional catalyst can both enable the hydropyrolysis at low-concentration of H2 with high efficiency and lower the risks during the hydropyrolysis process.
| Catalysts | Plastics | Conditions | Products | Ref. |
|---|---|---|---|---|
| NiW/HY | PE | 743–803 K, 2–5 MPa | Benzene, toluene and C8 aromatics | 120 |
| Ni/SBA-15 | PE, PP, PET and PS | 923 K, 1 MPa | Paraffins, olefins and aromatics | 121 |
| H-USY | HDPE | 623–663 K | Hydrocarbon (C1–C36) | 122 |
| HZSM-5 | HDPE | 673 K, 2 MPa | Light diesel and heavy diesel | 123 |
| HY zeolite | LDPE and PP | 523 K, 1.5 MPa | Gasoline (C1–C12) and diesel (C9–C22) | 40 |
| Metal/natural zeolite | LDPE | 723 K, 1 MPa | Hydrocarbon (C6–C19) | 125 |
| Pt-beta zeolite | LDPE | 603 K, 2 MPa | Alkanes (C4–C6) | 126 |
| Pt/h-HZSM-5 | PS | 573 K, 18 MPa | Isoparaffins, naphthenes, paraffins, olefins and aromatics | 127 |
| Pt/zeolites | PP, PE and PS | 603 K, 2 MPa | Naphtha (C5–C12) and heavier liquids (C13–C20) | 127 |
2.3 Solvolysis for bond cleavage in plastic waste at relatively low temperatures
Another typical strategy for plastic degradation is solvolysis, which usually involves hydrolysis, methanolysis, glycolysis, etc.22,41,42 The traditional solvolysis, often performed at a temperature from 353 to 553 K, is normally used in the depolymerization of oxygen-containing plastics such as polyesters, polycarbonates, and polyamides into their starting monomers.43 During the solvolysis process, the nucleophilic attack on the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond in these unsaturated polymers cleaves C–O, C–N or hydrophilic groups, resulting in the production of monomers.44,45 The greatest superiority of this method is that the further purification of these acquired monomers allows for the repolymerization to generate brand-new plastics with virgin-grade quality. However, the applied range of conventional solvolysis is relatively narrow because the saturated polymers without oxygen including PE, PP and PVC are rarely reported to be degraded this way, which may be mainly attributed to the extraordinary difficulty of the direct solvolysis of the C–C bond.
O bond in these unsaturated polymers cleaves C–O, C–N or hydrophilic groups, resulting in the production of monomers.44,45 The greatest superiority of this method is that the further purification of these acquired monomers allows for the repolymerization to generate brand-new plastics with virgin-grade quality. However, the applied range of conventional solvolysis is relatively narrow because the saturated polymers without oxygen including PE, PP and PVC are rarely reported to be degraded this way, which may be mainly attributed to the extraordinary difficulty of the direct solvolysis of the C–C bond.
To broaden the application scope of solvolysis, Huang et al. reported a novel solvolysis strategy for commercial PE plastics via a tandem catalytic cross alkane metathesis (CAM) process at a relatively low temperature, wherein a light alkane is employed as the solvent and two catalysts are utilized for the alkane dehydrogenation and the olefin metathesis.46 In this process, the dehydrogenation catalyst initially removes the hydrogen atoms in the PE and light alkane, leading to the production of unsaturated species. Subsequently, the olefin metathesis catalyst induces the C–C bond cleavage and coupling for the inter-conversion of a functional group in these two unsaturated species, thus reducing the chain length of PE. Ultimately, saturated alkanes with short chains are formed by the hydrogenation of newly generated unsaturated species. It is noteworthy that the alkanes with short chains are further capable of reacting with the superfluous light alkanes to become shorter ones through another CAM procedure. In other words, after multiple CAM processes, the solvolysis of PE with the help of a light alkane eventually accounts for the production of short hydrocarbons (Fig. 3a). As for various postconsumer PE plastics, their solvolysis in petroleum ether at 448 K forms oils and waxes by the CAM strategy as shown in Fig. 3b. In addition, a key advantage of this CAM process was that the ratio oil and wax products could be efficaciously tailored by manipulating the reaction time. The wax products would turn into oil products with the extension of reaction time from 2 h to 96 h during the solvolysis of commercial HDPE pellets (Fig. 3c), as verified by the continuously reduced molecular weights (Mw) and polydispersity indexes (PDI) of the wax products shown in Fig. 3d and e. Therefore, this new-concept tandem solvolysis strategy breaks down the C–C bond in the saturated plastics and hence realizes their conversion into smaller carbon-based fuels at relatively low temperatures. In spite of its difference from traditional solvolysis, this tandem solvolysis may contribute to expanding the application into more varieties of plastics such as PP, PVC and PS. The choice of a light alkane is also important in this process because its chain lengths and terminal functional groups have a significant impact on the types of final products. Again, through the targeted selection of an appropriate light alkane, more value-added chemicals may be acquired with an explicit direction by this CAM process. However, there are some unsatisfactory points of this method that need improvement; the noble metal-based catalysts used in the process are expensive and the demanded degradation time is time-consuming. If a cheaper and highly active inorganic semiconductor catalyst for alkane dehydrogenation is discovered and utilized in the CAM process, the reaction time may be greatly shortened at a similar temperature.
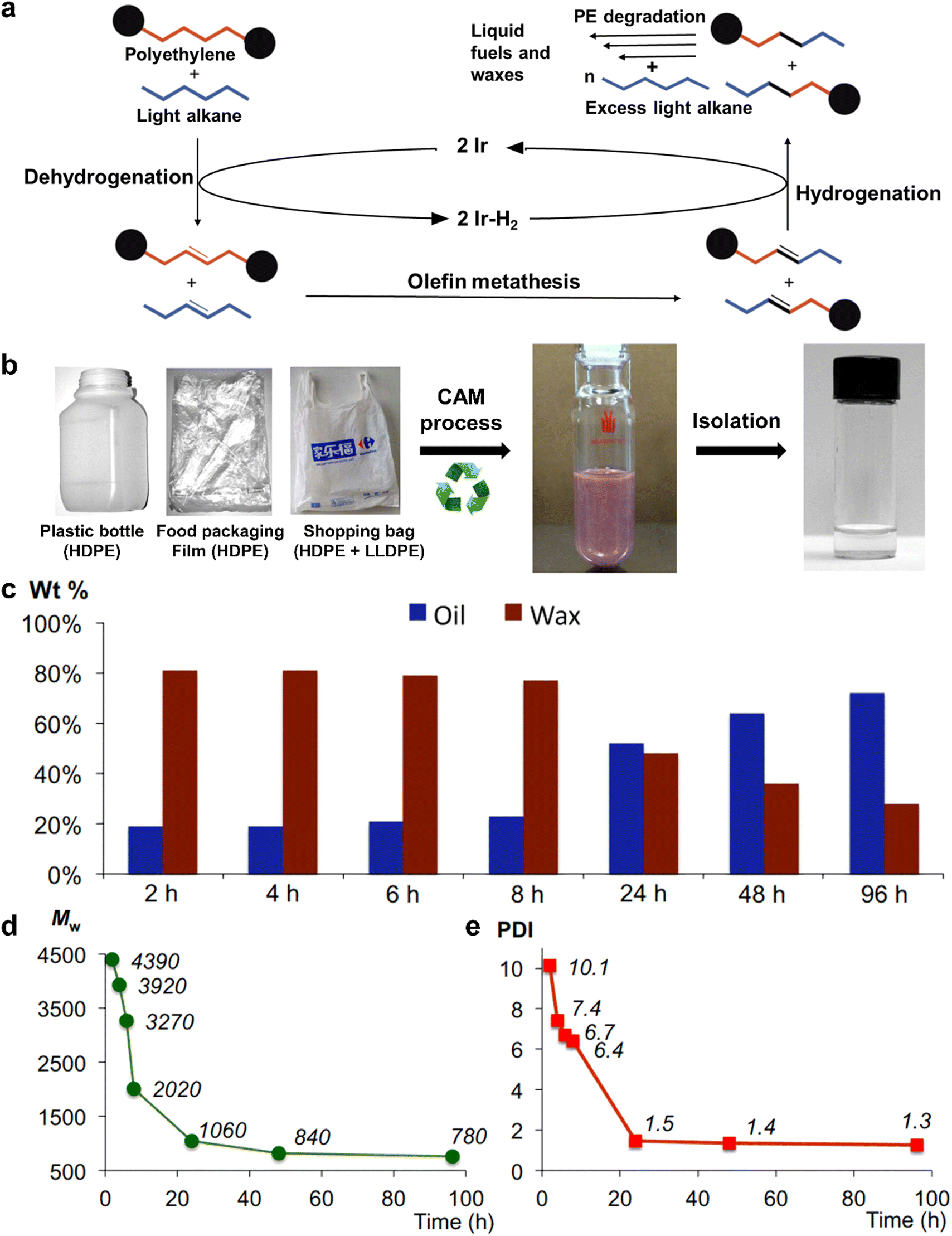 | ||
| Fig. 3 (a) The possible mechanism for the solvolysis of PE with the assistance of light alkanes through the CAM process. (b) Scheme for the solvolysis of various commercial PE plastics through the CAM process. (c) The products for the solvolysis of commercial HDPE pellets. (d) MW and (e) PDI of PE wax products during the solvolysis process. Reproduced with permission from ref. 46. Copyright 2016, AAAS. | ||
2.4 Microwave catalysis for bond cleavage in plastic waste at mild temperatures
Recently, some innovative technologies have been exploited to degrade plastic waste, in which microwave catalysis is regarded as a promising method for realizing efficient bond cleavage to produce value-added fuels.47–49 As is well known, a microwave is an electromagnetic form of energy with wavelengths ranging from 0.001 to 1 m and a frequency band from 0.3 to 300 GHz.50 Compared with the traditional thermo-catalytic plastics degradation, microwave catalysis of plastics causes an entire temperature rise of plastics rather than superficial heating, which provides some advantages such as speedy response rate and fast heating rate.50 Based on these analyses, this technology is beneficial to decreasing energy consumption and improving the overall quality of products from plastics conversion.51,52Bearing the above in mind, Edwards et al. have developed a novel one-step method for microwave catalytic bond cleavage of plastic wastes to produce H2 fuel and multi-walled carbon nanotubes (Fig. 4),53 wherein the microwave-susceptor was the composite of iron oxide and aluminum oxide (FeAlOx). Unlike the traditional pyrolysis-reforming two-step processes of plastics conversion,54,55 this alternative approach is simpler, more efficient and quicker for decomposing plastics into hydrogen-rich gases, as exhibited in Fig. 4a. In this microwave-initiated solid-solid reaction system, the electromagnetic energy derived from microwaving could be directly absorbed by the microwave-absorbing FeAlOx catalyst for generating plentiful heat energy, which was utilized for the ensuing decomposition of high-density polyethylene (HDPE) by the FeAlOx catalyst. That is to say, the FeAlOx catalyst concurrently played two important roles during the procedure. When the microwaves were initiated, the plastic substrate would remain cold at the beginning, whereas the temperature of the catalyst was much higher than that of the substance owing to the interfacial polarization (Fig. 4b), certified by the finite element simulation of electric field strength shown in Fig. 4F. In particular, this catalytic plastic process with the help of a microwave mainly occurred at the interface with a temperature difference between the microwave-activated iron catalyst and plastics (Fig. 4c), in which some intermediates, small alkenes and levels of H2 were formed through a C–H bond cleavage step. Subsequently, the carbon species intruded into the crystal lattice of the iron catalyst to produce Fe3C, which was crucial to the successful generation of multi-walled carbon nanotubes (Fig. 4d and e).56 To this end, the H2 yield of the microwave-initiated HDPE degradation for an FeAlOx catalyst shown in Fig. 4g could come up to 55.6 mmol gplastic−1, roughly thirteen times higher than that of the conventional pyrolysis of HDPE using the same catalyst at a similar temperature of ca. 623 K. It was also worth noting that the dominating products for traditional pyrolytic HDPE were the various oils. In contrast, microwave-initiated catalytic HDPE could mainly account for the production of H2 gas and solid carbon nanotubes. This might be attributed to the microwave electromagnetic energy that was selectively absorbed by the microwave-absorbing catalyst, leading to the preferential escape of hydrogen from HDPE via the microwave-initiated cleavage of C–H chemical bonds and narrowing the product distribution. Consequently, this one-step microwave-initiated catalytic bond cleavage in plastics produces H2 fuels and high-value carbon nanotubes efficiently and selectively. In addition, Hakkarainen et al. achieved the microwave-assisted catalytic conversion of LDPE plastics into platform chemicals and dicarboxylic acids efficiently.57 Also, Ruan et al. used microwave-assisted catalysis to convert LDPE into gasoline-range products with high yield and quality.58 As a burgeoning technique, microwave catalytic plastics have some prominent advantages indeed over the traditional thermo-catalytic plastic degradation. It is predictable that this one-step microwave catalysis may be suitable for the conversion of most plastics into valuable chemicals. However, owing to the extremely drastic reaction procedure, the temperature and pressure cannot be accurately controlled, which may lead to the poor repeatability of experiments, thus impeding their practical application. Hence, a future study may focus on the design of a temperature and pressure controller in the microwave reactor, enabling the precise regulation of temperature and pressure by adjusting the microwave frequency. More importantly, the incorporation of tail gas treatment devices into the microwave reactor helps to collect the produced H2 for other applications. Besides, the continuous extraction of H2 during microwave-induced catalysis can also promote the reaction proceeding on the basis of Le Chatelier's principle.
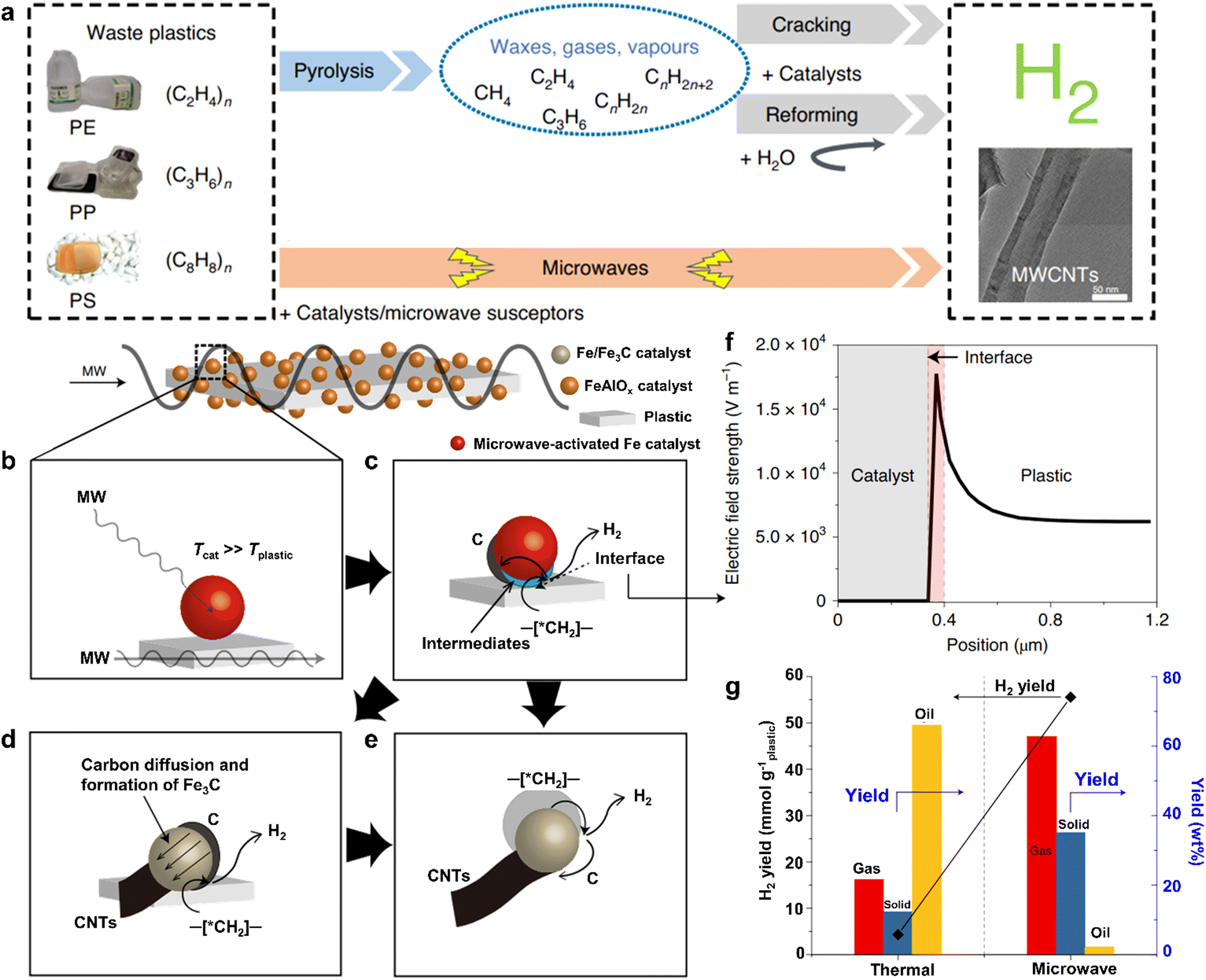 | ||
| Fig. 4 (a) Scheme for the traditional two-step pyrolysis-gasification and the one-step microwave-induced catalysis of plastic conversion. (b–e) A tentative mechanism for the process of microwave-initiated catalytic bond cleavage in plastics to produce H2 and carbon. (f) Finite element simulation of electric field strength on the catalyst and plastics. (g) A comparison of the properties of HDPE conversion though microwave-initiated catalytic and traditional thermal procedures. Reproduced with permission from ref. 53. Copyright 2020 Springer Nature. | ||
2.5 Other violent technologies for the conversion of plastic waste
In addition to the above-mentioned technologies for plastic degradation, some other violent strategies such as high-temperature gasification and plasma-induced catalysis have also been reported for the conversion of plastic waste into valuable chemicals.59,60 For example, Miskolczi et al. have reported the gasification of PE, PP and PET to syngas production with a lanthanum catalyst at 1123 K.61 Also, Williams et al. have developed a two-stage pyrolysis-low-temperature plasma catalysis to convert the PE into H2 at 250 °C efficiently.46 To date, different types of plastics including PE, PP, PS, PVC and PET, etc. have been degraded by these violent technologies (Table 6). Benefiting from the high-energy-induced easy bond cleavage, the degradation of plastic wastes under these violent conditions generally achieves a remarkable efficiency for the conversion of plastics into valuable carbon-based chemicals or gasses. Therefore, these violent methods for plastic degradation may be closer to actual application.| Technologies | Plastics | Conditions | Products | Ref. |
|---|---|---|---|---|
| Thermal pyrolysis | LDPE | 793 K | C5–C15 hydrocarbons and C12–C28 light oil | 128 |
| HDPE | 723 K and 773 K | Paraffin, olefin, naphthene and aromatic | 129 | |
| PP | 793 K | C5–C15 hydrocarbons and C12–C28 light oil | 128 | |
| PS | 703–713 K | Olefins | 130 | |
| Catalytic pyrolysis | LDPE | 825 K | Monoaromatics and C5–C12 alkanes/olefins | 131 |
| HDPE | 723–743 K | C13–C23+ paraffin | 132 | |
| PP | 693–723 K | Paraffin, olefin, naphthene and aromatic | 133 | |
| PS | 673 K | C9–C12 alkylaromatic, methylstyrene (C1-styrene) and isopropylbenzene (C3-benzene) | 134 | |
| PVC | 973 K | Gasoline and kerosene/diesel | 91 | |
| Hydropyrolysis | LDPE | 823 K | Monomers | 135 |
| HDPE | 723 K | C13–C23+ alkane, alkadiene | 136 | |
| PP | 523 K | Gasoline (C1–C12) and diesel (C9–C22) | 40 | |
| PS | 723 K | Monomers | 137 | |
| PET | 923 K | Paraffins, olefins and aromatics | 121 |
3. Conversion of plastic waste into valuable chemicals under mild conditions
Unlike plastic degradation under violent conditions, the conversion of plastic wastes into valuable chemicals under mild conditions generally requires a relatively long time. Also, the conversion efficiency is unable to bear comparison with that under violent conditions. But it is worth noting that degrading plastic wastes under ambient conditions is more energy-saving and environmentally friendly,62 which coincides with the concept of sustainable development for human society. More importantly, the mechanism can be explicitly investigated by in situ characterization techniques. Given this, plastic degradation under mild conditions may attract more and more attention in the coming years.3.1 Photocatalysis for bond cleavage in plastic waste under simulated natural environment conditions
As a clean and green technology, photocatalysis has been widely studied in many fields such as photocatalytic H2 evolution, photocatalytic carbon dioxide (CO2) reduction and photocatalytic nitrogen (N2) fixation.63–65 Many studies concerning plastic photoconversion under ambient conditions have been reported in recent years (Table 7).66,67 During the photocatalytic process, the decomposition of plastics is induced by absorbing the energy from sunlight for the production of active free radicals. When photon energy illuminates on catalysts, the photogenerated electrons (e−) and holes (h+) are produced, which will transfer to the catalyst surface for various reactions. In general, plastics can be directly oxidized by photoexcited holes. Also, plastics can be degraded by some free radicals such as hydroxyl and superoxide radicals, which are derived from the reaction of electrons and holes with O2 or H2O.68 Compared with other techniques, photodegradation of plastics may possess unique virtues in some respects since solar energy is clean and inexhaustible, and usually conducted at normal temperature and pressure. Also, photodegradation of plastics does not require extra energy input.69 However, photodegradation of plastics always suffers from the relatively low conversion efficiency and the superabundant terminal products of CO2.| Plastics | Catalysts | Light types | Conditions | Ref. |
|---|---|---|---|---|
| PE | Goethite | Ultraviolet light | 1 mW cm−2, 300 h | 138 |
| BiOCl | Visible light | 5 h | 139 | |
| TiO2 | Ultraviolet light | 1.4 mW cm−2, 300 h | 140 | |
| TiO2 | Ultraviolet light | 4.95 mW cm−2, 24 h | 62 | |
| TiO2 | Ultraviolet light | 2 mW cm−2, 240 h | 141 | |
| TiO2 | Solar light | 1 mW cm−2, 300 h | 142 | |
| TiO2 | Solar light | 200 h | 143 | |
| PVC | H3BO3 | Visible light | 2 mW cm−2, 250 h | 144 |
| ZnO | Ultraviolet light | 2 h | 145 | |
| CdS | Ultraviolet light | 2 mW cm−2, 250 h | 144 | |
| TiO2 | Ultraviolet light | 1.5 mW cm−2, 300 h | 146 | |
| TiO2 | Ultraviolet light | 1 mW cm−2, 216 h | 147 | |
| PS | TiO2 | Ultraviolet light | 1.75 mW cm−2, 250 h | 143 |
| TiO2/Fe(St)3 | Ultraviolet light | 30 W, 288 h | 148 | |
| TiO2 | Ultraviolet light | 2.5 mW cm−2, 150 h | 149 | |
| TiO2 | Solar light | 600 h | 150 | |
| PET | CN-CNTs-NiMo hybrids | Solar light | 95 mW cm−2, 4 h | 151 |
| CdS/CdOx | Solar light | 100 mW cm−2, 144 h | 152 | |
| CNx/Ni2P | Ultraviolet light | 24 h | 66 | |
| PP | Nb2O5 | Solar light | 100 mW cm−2, 24 h | 70 |
| Co-Ga2O3 | Solar light | 100 mW cm−2, 24 h | 153 | |
| TiO2 | Solar light | 126 h | 154 | |
| TiO2 | Ultraviolet light | 0.35 mW cm−2, 144 h | 155 |
In view of the above disadvantages, our group has presented a two-step procedure of solar-driven C–C bond cleavage and coupling of the plastic photoconversion into carbon-based fuels at normal temperature and pressure.70 Taking PE as an example, PE is initially photooxidized into CO2 through the C–C bond cleavage with the help of O2 and the ˙OH radical, while O2 is photoreduced into H2O through step-by-step reduction into O2˙− and H2O2 in the first step. In the second step, the formed CO2 is further photoreduced to acetic acid (CH3COOH) through the C–C coupling of ˙COOH intermediates. At the same time, H2O is photooxidized to O2 (Fig. 5a). Of note, the ˙OH radicals are generated during the PE photodegradation procedure, while the O2˙− radicals are produced during the oxygen reduction process, as demonstrated by the in situ electron paramagnetic resonance (ESR) spectra in Fig. 5b and c. Also, the ˙COOH intermediates, playing a crucial role in the CO2 photoreduction to CH3COOH, were generated during this process, as confirmed by the in situ Fourier-transform infrared spectroscopy (FTIR) spectra shown in Fig. 5d. All these results and some other labeling experiments certified that the PE underwent the two-step pathway of consecutive photo-induced C–C cleavage and coupling to produce CH3COOH. The photocatalytic experiments revealed that PE, PP, and PVC could be totally photodegraded over the Nb2O5 atomic layers in a short period, where the corresponding CO2 yields were gradually increased at normal temperature and pressure. And the CH3COOH yields also increased gradually, with the CH3COOH formation rates of ca. 47.4, 40.6 and 39.5 μg gcat−1 h−1 for PE, PP, and PVC, respectively (Fig. 5e–g). Accordingly, compared with the conventional one-step photocatalytic plastics, this two-step pathway gives some inspiration to take advantage of the end product of relatively low-value CO2. However, in the above study, the inferior CH3OOH formation yield was limited by the poor activity of CO2 photoreduction over the same catalyst that was also used for plastics degradation. In that aspect, a bifunctional catalyst should be designed, in which one is used for the efficient degradation of plastics into abundant CO2 and the other is employed by the subsequent CO2 photoreduction into target products with high efficiency. Also, a tandem two-chamber reaction cell without mutual interference can be fabricated to separate plastic degradation and CO2 reduction, whereby the generated CO2 in the former cell is transferred into the latter one for CO2 reduction. In this case, some pro-oxidants can be added into the former cell to boost plastic degradation, while some hole sacrificial agents are infused into the latter cell to expedite CO2 reduction. All in all, this two-step pathway may be conducive to guiding the targeted production of valuable chemicals from plastics photodegradation under ambient conditions by designing specific difunctional catalysts and tandem two-chamber reaction cells in the future.
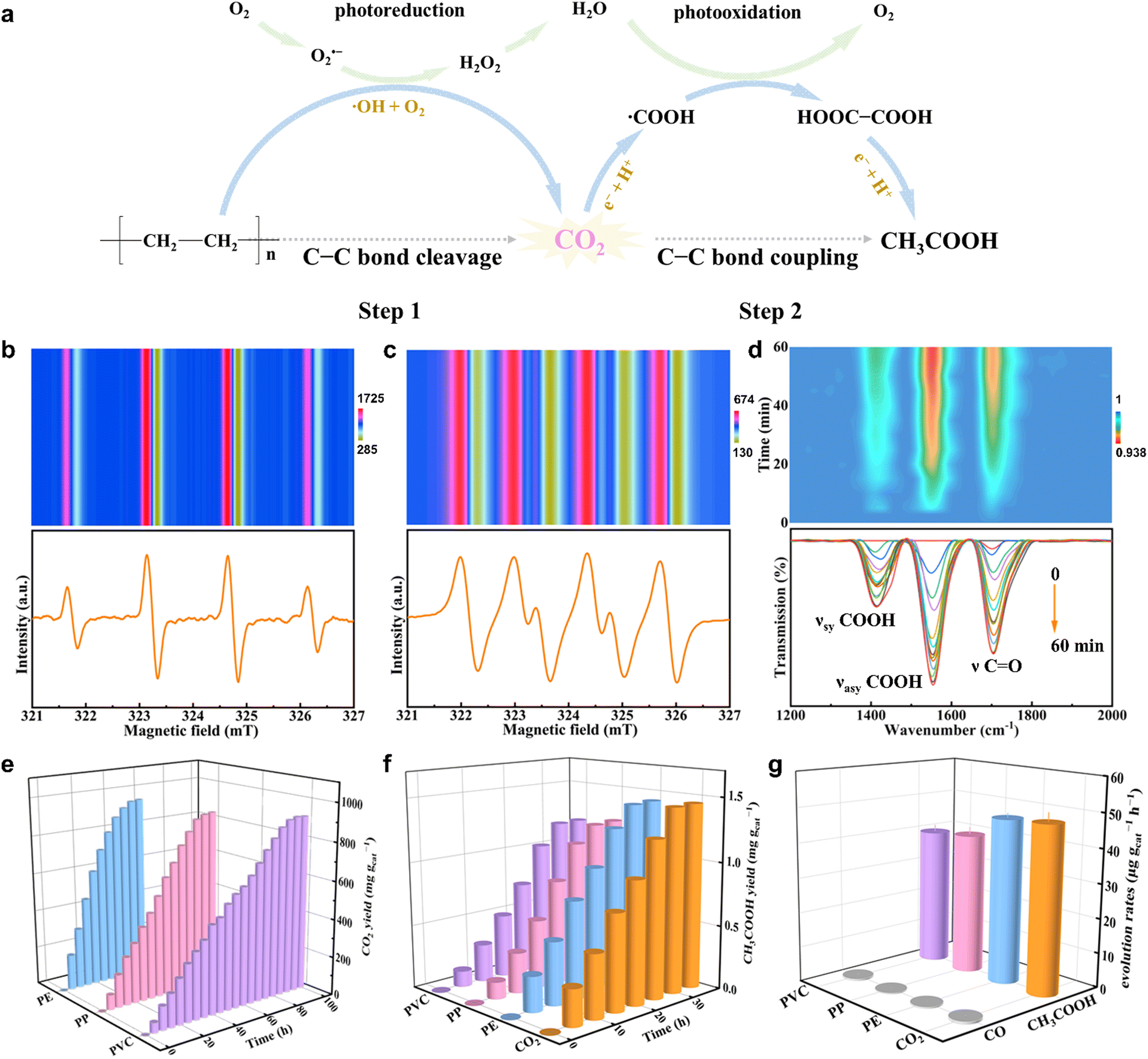 | ||
| Fig. 5 (a) Scheme for photoconverting PE into CH3COOH via the two-step pathway at normal temperature and pressure. In situ ESR spectra for PE photoconversion in (b) pure water and (c) methanol over the Nb2O5 atomic layers. (d) In situ FTIR spectra for PE photoconversion over the Nb2O5 atomic layers. (e) CO2 yield, (f) CH3COOH yield and (g) CH3COOH formation rates of photodegradation for PE, PP, and PVC as well as CO2 photoreduction over the Nb2O5 atomic layers. Reproduced with permission ref. 70. Copyright 2020, Wiley-VCH. | ||
3.2 Electrocatalysis for bond cleavage in plastic waste at ambient temperature and pressure
Similar to photocatalysis technology, electrocatalysis is also a sustainable and attractive method that has been used in widespread fields including electrocatalytic H2 evolution, electrocatalytic O2 evolution, electrocatalytic CO2 reduction and electrocatalytic N2 fixation at ambient temperature and pressure.71–74 During the electrocatalytic process, an externally applied potential is used to trigger the reaction proceeding, enabling access to the reaction pathways and chemical intermediates.75 In detail, a chemical reaction comprises the oxidation reaction and the reduction reaction at two electrodes, which are linked via an exterior electric circuit and the in-between electrolyte. Given this, an electrocatalysis technique possesses some distinct superiorities including the controllable operational process by the electrode potentials, the recyclable supporting electrolytes for lowered overall chemical consumption and the compact, modular and on-demand electrochemical reaction systems for the amplifying application.76 Despite these advantages, there have been few studies about the application of electrocatalysis in plastic degradation to date.Taking this point into account, Duan et al. have reported the electrocatalytic conversion of PET into potassium diformate (KDF) and terephthalic acid (PTA) at ambient temperature and pressure.77 PET was initially decomposed in an alkaline solution to produce their monomers of PTA and ethylene glycol (EG), and then the formed EG was further converted into formate through the electro-driven C–C bond cleavage process (Fig. 6a). The following step was the addition of formic acid as the acidifier into the PET electrolyte to precipitate and regenerate PTA by filtration, thus eventually attaining the solid KDF by concentration and crystallization. In brief, this electrocatalytic PET conversion process comprises three steps of the preliminary alkali-catalyzed PET hydrolysis, the subsequent electroconversion of PET hydrolysate and the ultimate formic acid-assisted separation of products. In this process, the catalysts of nickel foam supported with various Co and Ni phosphides (CoNixP/NF) were fabricated that showed a lower onset potential and higher current density than the Ni2P/NF catalyst and other reported outstanding catalysts for EG electrooxidation at ambient temperature and pressure (Fig. 6b). Particularly, the CoNi0.25P/NF catalyst could realize the EG electrooxidation into the formate (the primary product) with the Faraday efficiency of ca. 90% at the potentials from 1.3 to 1.7 V vs. reversible hydrogen electrode (RHE). The highest formate productivity was 4.1 mmol cm−2 h−1 at 1.7 V vs. RHE in a three-electrode H-cell system, as shown in Fig. 6c. Also, an approximate 90.2% yield of formate was gained with the carbon balance exceeding 90% and with the faradaic efficiency of 82.5% after the 100% conversion of EG at 1.5 V vs. RHE (Fig. 6d). To eliminate the effect of Ohmic resistance in the three-electrode H-cell, membrane-electrode assembly (MEA) flow reactor with a zero gap was utilized to perform the EG electrooxidation, where the current density could reach roughly 500 mA cm−2 at a low cell voltage less than 1.8 V vs. RHE. Additionally, the formate productivity for EG electrooxidation in Fig. 6e could reach 3.6 and 5.1 mmol cm−2 h−1 with excellent faradaic efficiency (>80%) at the constant current densities of 300 and 500 mA cm−2, respectively. More importantly, the Sankey diagram of mass flow analysis showed that 1 kg of PET plastics could obtain 389.2 g of formate and 818.5 g of PTA, after PTA separation (Fig. 6f). KDF powders were acquired by condensation and crystallization processes, affirmed by the XRD patterns shown in Fig. 6g. Consequently, this electrocatalytic strategy realizes profitable and sustainable chemicals from plastic wastes at ambient temperature and pressure. It is really a pathbreaking study concerning electrocatalytic plastics conversion, although there have been some imperfections. For instance, although the high-value KDF is attained by this electrocatalytic process, it is actually a procedure of electrocatalytic EG into formate. The C2 fuel with a higher value is converted into a C1 product with a lower value by this process, which may be a waste of energy. Hence, it is better to choose suitable electrocatalysts for converting the formed EG into valuable ethanol, acetic acid, and even ethanedioic acid. Also, this technique cannot be termed “plastic electrocatalysis” in a real sense because only the EG from PET hydrolysis rather than the pristine PET is electrocatalyzed into chemicals in this process. Future research may emphasize the direct conversion of electrocatalytic plastics into valuable chemicals at ambient pressure and temperature.
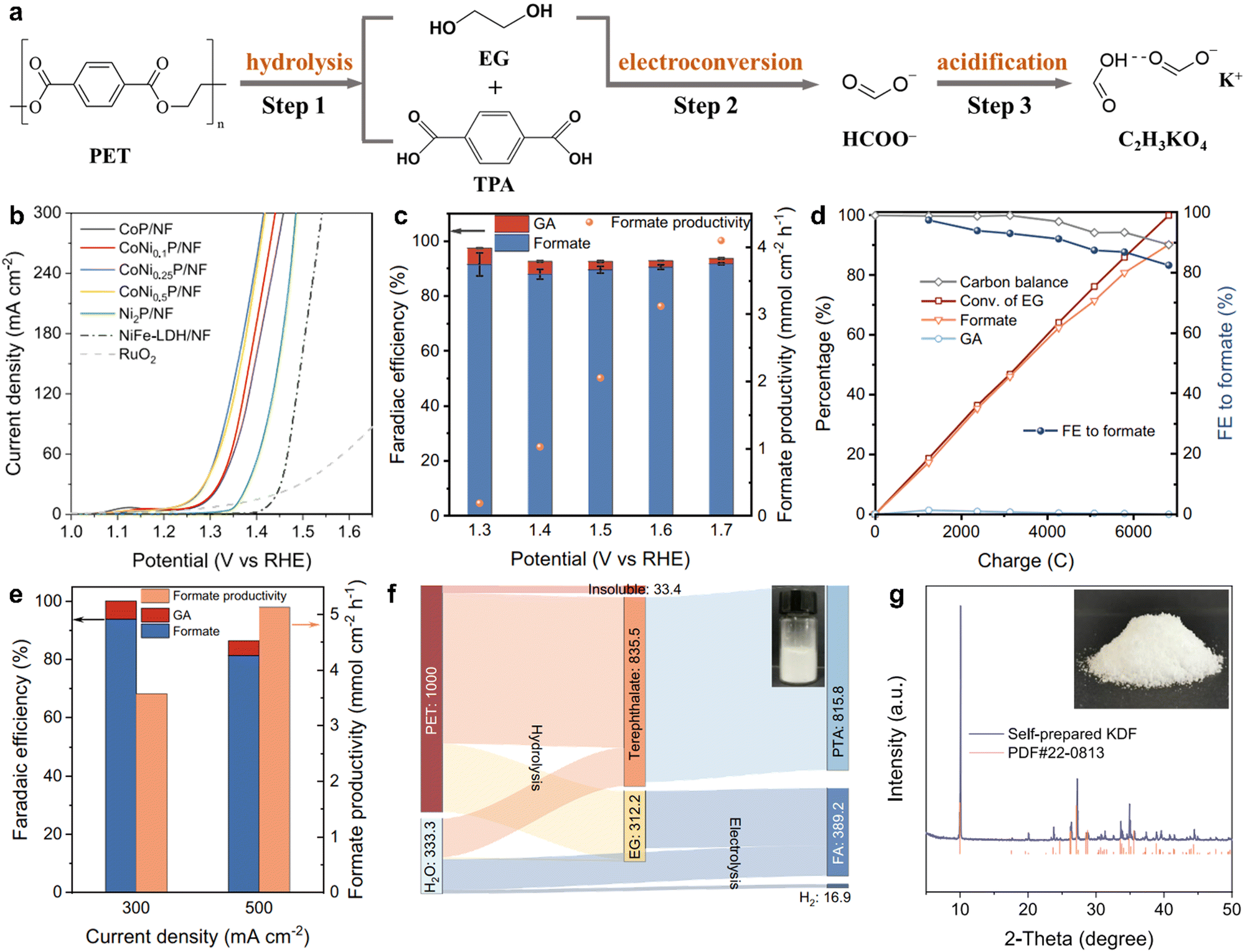 | ||
| Fig. 6 (a) Scheme for electrocatalytic PET conversion into TPA and KDF via the three-step pathway at ambient temperature and pressure. (b) Linear sweep voltammetry curves for EG electrooxidation over various catalysts. (c) Faradaic efficiency and productivity for the formate of EG electrooxidation over CoNi0.25P/NF at different potentials. (d) Kinetic curves of EG electrooxidation over at CoNi0.25P/NF 1.5 V vs. RHE. (e) Faradaic efficiency and productivity for the formate of EG electrooxidation at the current densities of 300 and 500 mA cm−2 by an MEA flow reactor. (f) Sankey diagram showing the mass flow for electrocatalytic PET conversion. (g) XRD pattern for the separated product after acidification. Reproduced with permission from ref. 77. Copyright 2021 Springer Nature. | ||
3.3 Biocatalysis for bond cleavage in plastic waste at normal pressure and temperature
Another eco-friendly strategy for plastic disposal is biodegradation, in which living organisms or microorganisms are able to break down the plastics under normal pressure and temperature.78–80 Generally speaking, the plastic biodegradation technique usually contains two modes. One utilizes the invertebrates to eat plastics for the degradation of plastics in their guts, and the other one uses the microorganisms to cleave the bond in plastics for the conversion of plastics into valuable compounds. Both modes are lucrative and do not produce any detrimental by-products during the plastic biodegradation process.81 Although many significant breakthroughs concerning plastic biodegradation have been achieved recently,82,83 they still encounter low conversion efficiency over a long time period, while the end products often involve low value-added CO2.Taking the disadvantages into consideration, Yoshida et al. have discovered a novel bacterium of Ideonella sakaiensis 201-F6 that could convert PET into the corresponding monomers with two different enzymes, where the PET hydrolase (PETase) cleaved PET into mono(2-hydroxyethyl) terephthalate (MHET), which was further hydrolyzed into TPA and EG by means of the MHET hydrolase (MHETase) (Fig. 7a).84 Inspired by this work, McGeehan et al. also reported a similar two-enzyme system including PETase and MHETase for converting the amorphous PET film into TPA and EG, in which PETase was employed to break the chemical bonds in the amorphous PET film for the production of MHET in the first step, and another one was used to further convert MHET into TPA and EG in the second step.85 Note that the second step contained a sequential acylation and deacylation process that was realized by two active residues of Ser225 and His528 (special enzyme group). During the acylation of MHET, the deprotonation of Ser225 by His528 could trigger their nucleophilic attack of MHET and generate the acyl-enzyme intermediate to liberate EG. Meanwhile, in the deacylation process, His528 also deprotonated the water for proton transfer into the acyl-enzyme intermediate, thus leading to the production of TPA and regeneration of Ser225 for the next catalytic cycle (Fig. 7b). The experiments for the degradation of commercial amorphous PET at 303 K by PETase and MHETase with tailorable concentrations are displayed in Fig. 7c, in which the MHETase alone had inactivity for PET films and the PETase alone could depolymerize PET films with low efficiency. In contrast, the product release for the degradation of PET films was enhanced at a markedly higher level upon adding the mixture of PETase and MHETase to the reaction. In other words, the presence of these two enzymes could synergistically realize the depolymerization of PET films into monomers. In addition, some new chimeric proteins (MP8, MP12 and MP20) were further fabricated via covalently connecting the PETase and the MHETase, wherein the 8, 12, and 20 total glycine and serine residues were used as the glycine-serine linkers (Fig. 7d). It is significant that these chimeric proteins also exhibited excellent performances for degrading the amorphous PET films, further attesting to the synergistic effect of the PETase and the MHETase. As a result, this two-enzyme system for PET degradation can attain the high-value monomers of TPA and EG rather than the low-value CO2. Whether this technique is suitable for saturated plastics? For example, is there a two-enzyme system for PE degradation, where one is used to break the C–C bond in PE to produce the ˙CH2 intermediate, which is further converted into valuable CH4, HCOOH and CH3OH by the other one? In addition, enlightened by this two-enzyme system for PET degradation, designing a multi-enzyme system may be a benefit for depolymerizing plastic wastes with multifold mixtures. On the whole, this two-enzyme system will pave a new way for the biodegradation of plastic wastes at normal pressure and temperature, and future studies should be designed to expand their application into more types of plastic wastes.
 | ||
| Fig. 7 (a) Scheme for the two-enzyme system for PET degradation into TPA and EG through a two-step pathway. (b) The possible mechanism for the conversion of MHET into TPA and EG by MHETase, which contains a sequential acylation and deacylation process. (c) The property for the degradation of amorphous PET films by PETase and MHETase with different concentrations over 96 h at 30 °C. (d) Scheme for the linkage of PETase and MHETase to form new chimeric proteins. Reproduced with permission from ref. 85. Copyright 2020 PNAS. | ||
3.4 Other mild technologies for the conversion of plastic wastes
To the best of our knowledge, apart from the aforesaid mild methods for plastic degradation, there have been no other techniques developed for converting plastic wastes into valuable chemicals under mild conditions until now, which may be because the stable carbon skeleton in plastics is difficult to be broken under this circumstance. An emerging technique of cold plasma has a huge potential for plastic degradation under mild conditions because it produces high-energy electrons that are sensitive to the bond cleavage in plastics. Although the mild avenues for plastic degradation are still far from practical application owing to their relatively low conversion efficiency, it is still worthy pursuing more innovative strategies for plastic degradation under mild conditions in the future because of their low energy consumption and long-term sustainability.4. Conclusions and outlook
In this review, we summarized the multifarious mechanisms of some of the latest strategies for converting plastic wastes into valuable chemicals under violent and mild conditions, respectively. In detail, we elaborated pyrolysis, hydropyrolysis, solvolysis and microwave-initiated catalysis for bond cleavage in plastic wastes under violent conditions. Meanwhile, we highlighted photocatalysis, electrocatalysis and biocatalysis for bond cleavage in plastic wastes under mild conditions. We also discussed the conversion mechanism and future research direction concerning these strategies. In short, thanks to the high conversion efficiencies of the high-energy-driven bond cleavage, the strategies under violent conditions are closer to practical application at present. Owing to environmental protection, the strategies under mild conditions may attract more attention in the future.Although a lot of significant breakthroughs have been unremittingly made in the conversion of plastic wastes into valuable commodities in recent years, there are still many aspects that need to be improved. In this review, we provide some suggestions and outlooks for the future development of plastic recycling as follows:
4.1 Improvement in current techniques for conversion of plastics into valuable chemicals
4.2 In-depth mechanism investigation for conversion of plastics into valuable chemicals
4.3 Replacement of present plastics with recyclable polymeric materials
To date, the best method for plastics disposal is designing and fabricating recyclable polymeric materials to replace modern plastics, wherein self-immolating materials, dynamic covalent substances and vitrimer networks are regarded as the potential candidates for the synthesis of novel reusable plastics. Additionally, the plant-based feedstocks including cellulose, lignin, sugars, vegetable oils and terpenes are also able to fabricate recyclable polymers in spite of the higher cost with respect to traditional plastics at present. Also, chemically recyclable bio-derived polymers are deemed to be competitive candidates relative to petrochemical-derived plastics. That is because the rigid ring structures and stereo-regularity in these bio-renewable resources (e.g., carbohydrates) enable the synthesis of valuable chemicals. Future efforts should focus on designing and preparing recyclable bio-based plastics through a balance between long-term durability and high-efficiency recycling efficiency.Through continual efforts, the future is bright for converting the ubiquitous plastic wastes into valuable chemicals, and much more satisfying progress will be anticipated in the years to come.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Key R&D Program of China (2019YFA0210004), National Natural Science Foundation of China (22125503, 21975242, U2032212, 21890754, 22275178), Strategic Priority Research Program of Chinese Academy of Sciences (XDB36000000), Youth Innovation Promotion Association of CAS (CX2340007003), Major Program of Development Foundation of Hefei Center for Physical Science and Technology (2020HSC-CIP003), Users with Excellence Program of Hefei Science Center (2020HSC-UE001), the University Synergy Innovation Program of Anhui Province (GXXT-2020-001), and the Anhui Provincial Natural Science Foundation of China (2108085QB69).Notes and references
- J. M. García, Chem, 2016, 1, 813–815 Search PubMed.
- D. J. Walsh, M. G. Hyatt, S. A. Miller and D. Guironnet, ACS Catal., 2019, 9, 11153–11188 CrossRef CAS.
- N. Malik, P. Kumar, S. Shrivastava and S. B. Ghosh, Int. J. Plast. Technol., 2017, 21, 1–24 CrossRef CAS.
- M. Day, J. D. Cooney, C. Touchette-Barrette and S. E. Sheehan, J. Anal. Appl. Pyrolysis, 1999, 52, 199–224 CrossRef CAS.
- H. Sardon and P. Dove Andrew, Science, 2018, 360, 380–381 CrossRef CAS PubMed.
- T. Hundertmark, M. Mayer, C. McNally, T. J. Simons, C. Witte, McKinsey Company, 2018, https://www.mckinsey.com/industries/chemicals/our-insights/how-plastics-waste-recycling-could-transform-the-chemical-industry# Search PubMed.
- R. Geyer, R. Jambeck Jenna and L. Law Kara, Sci. Adv., 2017, 3, e1700782 CrossRef.
- A. Chamas, H. Moon, J. Zheng, Y. Qiu, T. Tabassum, J. H. Jang, M. Abu-Omar, S. L. Scott and S. Suh, ACS Sustainable Chem. Eng., 2020, 8, 3494–3511 CrossRef CAS.
- X. Jiao, K. Zheng, Z. Hu, S. Zhu, Y. Sun and Y. Xie, Adv. Mater., 2021, 33, 2005192 CrossRef CAS PubMed.
- P. Schwabl, S. Köppel, P. Königshofer, T. Bucsics, M. Trauner, T. Reiberger and B. Liebmann, Ann. Int. Med., 2019, 171, 453–457 CrossRef PubMed.
- I. A. Ignatyev, W. Thielemans and B. Vander Beke, ChemSusChem, 2014, 7, 1579–1593 CrossRef CAS PubMed.
- S. M. Al-Salem, P. Lettieri and J. Baeyens, Prog. Energy Combust. Sci., 2010, 36, 103–129 CrossRef CAS.
- C. Baillie, D. Matovic, T. Thamae and S. Vaja, Resour., Conserv. Recycl., 2011, 55, 973–978 CrossRef.
- D. S. Achilias, A. Giannoulis and G. Z. Papageorgiou, Polym. Bull., 2009, 63, 449–465 CrossRef CAS.
- A. J. Hadi, G. F. Najmuldeen and K. B. Yusoh, J. Polym. Eng., 2013, 33, 471–481 CAS.
- E. J. Cho, S. Lee, S. J. Oh, M. Han, Y. S. Lee and C. N. Whang, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52, 16443 CrossRef CAS.
- A. L. Andrady, Plastics and the Environment, John Wiley & Sons, New York, 2003 Search PubMed.
- C. Mohanraj, T. Senthilkumar and M. Chandrasekar, Int. J. Energy Res., 2017, 41, 1534–1552 CrossRef CAS.
- E. Butler, G. Devlin and K. McDonnell, Waste Biomass Valorization, 2011, 2, 227–255 CrossRef CAS.
- A. Buekens and K. Cen, J. Mater. Cycles Waste Manage., 2011, 13, 190–197 CrossRef CAS.
- T. Thiounn and R. C. Smith, J. Polym. Sci., 2020, 58, 1347–1364 CrossRef CAS.
- H. Chen, K. Wan, Y. Zhang and Y. Wang, ChemSusChem, 2021, 14, 4123–4136 CrossRef CAS PubMed.
- I. Vollmer, M. J. F. Jenks, M. C. P. Roelands, R. J. White, T. van Harmelen, P. de Wild, G. P. van der Laan, F. Meirer, J. T. F. Keurentjes and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2020, 59, 15402–15423 CrossRef CAS PubMed.
- A. Rahimi and J. M. García, Nat. Rev. Chem., 2017, 1, 0046 CrossRef.
- A. Mohanty, R. K. Borah, A. P. Fatrekar, S. Krishnan and A. A. Vernekar, Chem. Commun., 2021, 57, 10277–10291 RSC.
- D. Chen, L. Yin, H. Wang and P. He, Waste Manage., 2014, 34, 2466–2486 CrossRef CAS.
- T. Maqsood, J. Dai, Y. Zhang, M. Guang and B. Li, J. Anal. Appl. Pyrolysis, 2021, 159, 105295 CrossRef CAS.
- S. Budsaereechai, A. J. Hunt and Y. Ngernyen, RSC Adv., 2019, 9, 5844–5857 RSC.
- I. Velghe, R. Carleer, J. Yperman and S. Schreurs, J. Anal. Appl. Pyrolysis, 2011, 92, 366–375 CrossRef CAS.
- Adnan, J. Shah and M. R. Jan, J. Anal. Appl. Pyrolysis, 2014, 109, 196–204 CrossRef CAS.
- C. D. Clark, H. Kanda, I. Kiflawi and G. Sittas, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 16681 CrossRef CAS.
- A. G. Buekens and H. Huang, Resour., Conserv. Recycl., 1998, 23, 163–181 CrossRef.
- K.-B. Park, Y.-S. Jeong and J.-S. Kim, Appl. Energy, 2019, 253, 113558 CrossRef CAS.
- J. H. Flynn and L. A. Wall, J. Polym. Sci., Part B: Polym. Phys., 1966, 4, 323–328 CrossRef CAS.
- G. Lopez, M. Artetxe, M. Amutio, J. Bilbao and M. Olazar, Renewable Sustainable Energy Rev., 2017, 73, 346–368 CrossRef CAS.
- S. M. Al-Salem, A. Antelava, A. Constantinou, G. Manos and A. Dutta, J. Environ. Manage., 2017, 197, 177–198 CrossRef CAS PubMed.
- J. E. Rorrer, G. T. Beckham and Y. Román-Leshkov, JACS Au, 2020, 1, 8–12 CrossRef PubMed.
- S. L. Gupte and G. Madras, Polym. Degrad. Stab., 2004, 86, 529–533 CrossRef CAS.
- Y. X. Jing, Y. Q. Wang, S. Y. Furukawa, J. Xia, C. Y. Sun, M. J. Hulsey, H. F. Wang, Y. Guo, X. H. Liu and N. Yan, Angew. Chem., Int. Ed., 2021, 60, 5527–5535 CrossRef CAS.
- S. Liu, P. A. Kots, B. C. Vance, A. Danielson and D. G. Vlachos, Sci. Adv., 2021, 7, eabf8283 CrossRef CAS.
- K. Ragaert, L. Delva and K. Van Geem, Waste Manage., 2017, 69, 24–58 CrossRef CAS.
- K. Fukushima, O. Coulembier, J. M. Lecuyer, H. A. Almegren, A. M. Alabdulrahman, F. D. Alsewailem, M. A. McNeil, P. Dubois, R. M. Waymouth and H. W. Horn, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1273–1281 CrossRef CAS.
- J. Payne, P. McKeown and M. D. Jones, Polym. Degrad. Stab., 2019, 165, 170–181 CrossRef CAS.
- S. Dimitris and L. A. Achilias, Material Recycling-Trends and Perspectives, 2012, InTech, 2012, p. 406 Search PubMed.
- D. Achilias, Material Recycling-Trends and Perspectives, BoD–Books on Demand, 2012 Search PubMed.
- X. Jia, C. Qin, T. Friedberger, Z. Guan and Z. Huang, Sci. Adv., 2016, 2, e1501591 CrossRef.
- H. Arshad, S. A. Sulaiman, Z. Hussain, Y. Naz and F. Basrawi, Matec Web Conf., 2017, 131, 02005 CrossRef.
- Y. Zhang, Y. Cui, S. Liu, L. Fan, N. Zhou, P. Peng, Y. Wang, F. Guo, M. Min and Y. Cheng, Bioresour. Technol., 2020, 297, 122480 CrossRef CAS.
- S. Y. Foong, R. K. Liew, Y. Yang, Y. W. Cheng, P. N. Y. Yek, W. A. W. Mahari, X. Y. Lee, C. S. Han, D.-V. N. Vo and Q. Van Le, Chem. Eng. J., 2020, 389, 124401 CrossRef CAS.
- F. Motasemi and M. T. Afzal, Renewable Sustainable Energy Rev., 2013, 28, 317–330 CrossRef CAS.
- J. A. Menéndez, A. Arenillas, B. Fidalgo, Y. Fernández, L. Zubizarreta, E. G. Calvo and J. M. Bermúdez, Fuel Process. Technol., 2010, 91, 1–8 CrossRef.
- E. Grant and B. J. Halstead, Chem. Soc. Rev., 1998, 27, 213–224 RSC.
- X. Jie, W. Li, D. Slocombe, Y. Gao, I. Banerjee, S. Gonzalez-Cortes, B. Yao, H. AlMegren, S. Alshihri and J. Dilworth, Nat. Catal., 2020, 3, 902–912 CrossRef CAS.
- I. Aminu, M. A. Nahil and P. T. Williams, Energy Fuels, 2020, 34, 11679–11689 CrossRef.
- I. Barbarias, G. Lopez, J. Alvarez, M. Artetxe, A. Arregi, J. Bilbao and M. Olazar, Chem. Eng. J., 2016, 296, 191–198 CrossRef CAS.
- L. Reddy Enakonda, L. Zhou, Y. Saih, S. Ould-Chikh, S. Lopatin, D. Gary, P. Del-Gallo and J. M. Basset, ChemSusChem, 2016, 9, 1911–1915 CrossRef CAS PubMed.
- E. Bäckström, K. Odelius and M. Hakkarainen, Ind. Eng. Chem. Res., 2017, 56, 14814–14821 CrossRef.
- K. Ding, S. Liu, Y. Huang, S. Liu, N. Zhou, P. Peng, Y. Wang, P. Chen and R. Ruan, Energy Convers. Manage., 2019, 196, 1316–1325 CrossRef CAS.
- R. Miandad, M. A. Barakat, A. S. Aburiazaiza, M. Rehan and A. S. Nizami, Process Saf. Environ. Prot., 2016, 102, 822–838 CrossRef CAS.
- M. Punčochář, B. Ruj and P. K. Chatterj, Proc. Eng., 2012, 42, 420–430 CrossRef.
- M. Al-asadi, N. Miskolczi and Z. Eller, J. Cleaner Prod., 2020, 271, 122186 CrossRef CAS.
- I. Nabi, K. Li, H. Cheng, T. Wang, Y. Liu, S. Ajmal, Y. Yang, Y. Feng and L. Zhang, iScience, 2020, 23, 101326 CrossRef CAS PubMed.
- X. Zong, H. Yan, G. Wu, G. Ma, F. Wen, L. Wang and C. Li, J. Am. Chem. Soc., 2008, 130, 7176–7177 CrossRef CAS.
- X. Jiao, X. Li, X. Jin, Y. Sun, J. Xu, L. Liang, H. Ju, J. Zhu, Y. Pan and W. Yan, J. Am. Chem. Soc., 2017, 139, 18044–18051 CrossRef CAS.
- J. Li, P. Liu, Y. Tang, H. Huang, H. Cui, D. Mei and C. Zhong, ACS Catal., 2020, 10, 2431–2442 CrossRef CAS.
- T. Uekert, H. Kasap and E. Reisner, J. Am. Chem. Soc., 2019, 141, 15201–15210 CrossRef CAS PubMed.
- S. T. Nguyen, E. A. McLoughlin, J. H. Cox, B. P. Fors and R. R. Knowles, J. Am. Chem. Soc., 2021, 143, 12268–12277 CrossRef CAS.
- Z. Ouyang, Y. Yang, C. Zhang, S. Zhu, L. Qin, W. Wang, D. He, Y. Zhou, H. Luo and F. Qin, J. Mater. Chem. A, 2021, 9, 13402–13441 RSC.
- S. Chatani, C. J. Kloxin and C. N. Bowman, Polym. Chem., 2014, 5, 2187–2201 RSC.
- X. Jiao, K. Zheng, Q. Chen, X. Li, Y. Li, W. Shao, J. Xu, J. Zhu, Y. Pan and Y. Sun, Angew. Chem., Int. Ed., 2020, 59, 15497–15501 CrossRef CAS PubMed.
- W. Cheng, H. Zhang, D. Luan and X. W. D. Lou, Sci. Adv., 2021, 7, eabg2580 CrossRef CAS PubMed.
- H. B. Yang, J. Miao, S.-F. Hung, J. Chen, H. B. Tao, X. Wang, L. Zhang, R. Chen, J. Gao and H. M. Chen, Sci. Adv., 2016, 2, e1501122 CrossRef PubMed.
- S. Gao, Y. Lin, X. Jiao, Y. Sun, Q. Luo, W. Zhang, D. Li, J. Yang and Y. Xie, Nature, 2016, 529, 68–71 CrossRef CAS.
- Y. Luo, G.-F. Chen, L. Ding, X. Chen, L.-X. Ding and H. Wang, Joule, 2019, 3, 279–289 CrossRef CAS.
- L. F. T. Novaes, J. Liu, Y. Shen, L. Lu, J. M. Meinhardt and S. Lin, Chem. Soc. Rev., 2021, 50, 7941–8002 RSC.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- H. Zhou, Y. Ren, Z. Li, M. Xu, Y. Wang, R. Ge, X. Kong, L. Zheng and H. Duan, Nat. Commun., 2021, 12, 4679 CrossRef CAS.
- A. A. Shah, F. Hasan, A. Hameed and S. Ahmed, Biotechnol. Adv., 2008, 26, 246–265 CrossRef CAS PubMed.
- C. DelRe, Y. Jiang, P. Kang, J. Kwon, A. Hall, I. Jayapurna, Z. Ruan, L. Ma, K. Zolkin and T. Li, Nature, 2021, 592, 558–563 CrossRef CAS PubMed.
- V. Tournier, C. M. Topham, A. Gilles, B. David, C. Folgoas, E. Moya-Leclair, E. Kamionka, M. L. Desrousseaux, H. Texier and S. Gavalda, Nature, 2020, 580, 216–219 CrossRef CAS PubMed.
- F. Zhang, Y. Zhao, D. Wang, M. Yan, J. Zhang, P. Zhang, T. Ding, L. Chen and C. Chen, J. Cleaner Prod., 2021, 282, 124523 CrossRef CAS.
- J. Yang and Y. B. Guo, Chin. Chem. Lett., 2018, 29, 252–260 CrossRef CAS.
- K. Janczak, K. Hrynkiewicz, Z. Znajewska and G. Dąbrowska, Int. Biodeterior. Biodegrad., 2018, 130, 65–75 CrossRef CAS.
- S. Yoshida, K. Hiraga, T. Takehana, I. Taniguchi, H. Yamaji, Y. Maeda, K. Toyohara, K. Miyamoto, Y. Kimura and K. Oda, Science, 2016, 351, 1196–1199 CrossRef CAS PubMed.
- B. C. Knott, E. Erickson, M. D. Allen, J. E. Gado, R. Graham, F. L. Kearns, I. Pardo, E. Topuzlu, J. J. Anderson and H. P. Austin, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 25476–25485 CrossRef CAS.
- P. N. Y. Yek, Y. H. Chan, S. Y. Foong, W. A. Wan Mahari, X. Chen, W. Peng, R. K. Liew, L. Ding, Y. F. Tsang and C. Sonne, SSRN, 2022 DOI:10.2139/ssrn.4074339.
- K. Li, Y. Wang, W. Zhou, T. Cui, J. Yang, Z. Sun, Y. Min and J.-M. Lee, Chemosphere, 2022, 299, 134440 CrossRef CAS PubMed.
- H. Ukei, T. Hirose, S. Horikawa, Y. Takai, M. Taka, N. Azuma and A. Ueno, Catal. Today, 2000, 62, 67–75 CrossRef CAS.
- A. Inayat, A. Inayat, W. Schwieger, B. Sokolova and P. Lestinsky, Fuel, 2022, 314, 123071 CrossRef CAS.
- I. Kremer, T. Tomić, Z. Katančić, M. Erceg, S. Papuga, J. P. Vuković and D. R. Schneider, J. Environ. Manage., 2021, 296, 113145 CrossRef CAS PubMed.
- K.-R. Hwang, S.-A. Choi, I.-H. Choi and K.-H. Lee, J. Anal. Appl. Pyrolysis, 2021, 155, 105090 CrossRef CAS.
- N. Cai, X. Li, S. Xia, L. Sun, J. Hu, P. Bartocci, F. Fantozzi, P. T. Williams, H. Yang and H. Chen, Energy Convers. Manage., 2021, 229, 113794 CrossRef CAS.
- S. Kumagai, R. Yamasaki, T. Kameda, Y. Saito, A. Watanabe, C. Watanabe, N. Teramae and T. Yoshioka, Energy Fuels, 2020, 34, 2492–2500 CrossRef CAS.
- Z. Chen, M. Monzavi, M. Latifi, S. Samih and J. Chaouki, Environ. Pollut., 2022, 119573 CrossRef CAS PubMed.
- A. López, I. De Marco, B. M. Caballero, M. F. Laresgoiti, A. Adrados and A. Aranzabal, Appl. Catal., B, 2011, 104, 211–219 CrossRef.
- T. P. Paula, M. d F. V. Marques, M. R. d C. Marques, M. S. Oliveira and S. N. Monteiro, Chemistry, 2022, 4, 297–315 CrossRef CAS.
- K. Sivagami, K. V. Kumar, P. Tamizhdurai, D. Govindarajan, M. Kumar and I. Nambi, RSC Adv., 2022, 12, 7612–7620 RSC.
- A. Palomar-Torres, E. Torres-Jimenez, B. Kegl, G. Bombek, J. Volmajer-Valh and L. Lešnik, Int. J. Environ. Sci. Technol., 2022 DOI:10.1007/s13762-022-04023-z.
- D. Zhang, X. Lin, Q. Zhang, X. Ren, W. Yu and H. Cai, Energy, 2020, 212, 118983 CrossRef CAS.
- S. J. Miller, N. Shah and G. P. Huffman, Energy Fuels, 2005, 19, 1580–1586 CrossRef CAS.
- S. M. Al-Salem, Process Saf. Environ. Prot., 2019, 127, 171–179 CrossRef.
- F. J. Mastral, E. Esperanza, C. Berrueco, M. Juste and J. Ceamanos, J. Anal. Appl. Pyrolysis, 2003, 70, 1–17 CrossRef CAS.
- J. Aguado, D. P. Serrano, G. San Miguel, M. C. Castro and S. Madrid, J. Anal. Appl. Pyrolysis, 2007, 79, 415–423 CrossRef CAS.
- M. Sogancioglu, E. Yel and G. Ahmetli, J. Cleaner Prod., 2017, 165, 369–381 CrossRef CAS.
- W. Kaminsky and I.-J. N. Zorriqueta, J. Anal. Appl. Pyrolysis, 2007, 79, 368–374 CrossRef CAS.
- M. Artetxe, G. Lopez, M. Amutio, G. Elordi, M. Olazar and J. Bilbao, Ind. Eng. Chem. Res., 2010, 49, 2064–2069 CrossRef CAS.
- R. Thahir, A. Altway and S. R. Juliastuti, Energy Rep., 2019, 5, 70–77 CrossRef.
- Y. Zhang, G. Ji, D. Ma, C. Chen, Y. Wang, W. Wang and A. Li, Process Saf. Environ. Prot., 2020, 142, 203–211 CrossRef CAS.
- S.-S. Kim and S. Kim, Chem. Eng. J., 2004, 98, 53–60 CrossRef CAS.
- A. Verma, S. Sharma and H. Pramanik, Waste Manage., 2021, 120, 330–339 CrossRef CAS PubMed.
- R. Prathiba, M. Shruthi and L. R. Miranda, Waste Manage., 2018, 76, 528–536 CrossRef CAS PubMed.
- J. Lee, T. Lee, Y. F. Tsang, J.-I. Oh and E. E. Kwon, Energy Convers. Manage., 2017, 148, 456–460 CrossRef CAS.
- A. Lopez-Urionabarrenechea, I. De Marco, B. M. Caballero, M. F. Laresgoiti and A. Adrados, Fuel Process. Technol., 2015, 137, 229–239 CrossRef CAS.
- K. R. Venkatesh, J. Hu, W. Wang, G. D. Holder, J. W. Tierney and I. Wender, Energy Fuels, 1996, 10, 1163–1170 CrossRef CAS.
- A. K. Amin, K. Wijaya and W. Trisunaryanti, Orient. J. Chem., 2018, 34, 3070–3078 CAS.
- L. Hauli, K. Wijaya and A. Syoufian, Ind. J. Chem., 2020, 20, 422–429 Search PubMed.
- H. K. Joo and C. W. Curtis, Energy Fuels, 1996, 10, 603–611 CrossRef CAS.
- J. Wang, J. Jiang, X. Wang, S. Liu, X. Shen, X. Cao, Y. Sun, L. Dong, X. Meng and A. J. Ragauskas, Chem. Eng. J., 2022, 444, 136360 CrossRef CAS.
- D. Munir and M. R. Usman, J. Anal. Appl. Pyrolysis, 2018, 135, 44–53 CrossRef CAS.
- J. Walendziewski and M. Steininger, Catal. Today, 2001, 65, 323–330 CrossRef CAS.
- P. Narksri, S. Angnanon, J. Guntasub, K. Wijitrattanatri, S. Kingputtapong, S. Phumpradit and N. Hinchiranan, Mater. Today: Proc., 2022, 57, 1040–1047 CAS.
- C. S. Costa, M. Muñoz, M. R. Ribeiro and J. M. Silva, Sustainable Energy Fuels, 2021, 5, 1134–1147 RSC.
- D. Munir and M. R. Usman, J. Porous Mater., 2022, 29, 783–794 CrossRef CAS.
- W. Chen, X. Xu, C. Zhang, Y. Feng, Y. Wang, J. Wang and R. Zhang, Fuel, 2021, 293, 120503 CrossRef CAS.
- W. Sriningsih, M. G. Saerodji, W. Trisunaryanti, R. Armunanto and I. I. Falah, Proc. Environ. Sci., 2014, 20, 215–224 CrossRef CAS.
- A. Bin Jumah, V. Anbumuthu, A. A. Tedstone and A. A. Garforth, Ind. Eng. Chem. Res., 2019, 58, 20601–20609 CrossRef CAS.
- E. G. Fuentes-Ordóñez, J. A. Salbidegoitia, J. L. Ayastuy, M. A. Gutiérrez-Ortiz, M. P. González-Marcos and J. R. González-Velasco, Catal. Today, 2014, 227, 163–170 CrossRef.
- N. Miskolczi, A. Angyal, L. Bartha and I. Valkai, Fuel Process. Technol., 2009, 90, 1032–1040 CrossRef CAS.
- K.-H. Lee and D.-H. Shin, Waste Manage., 2007, 27, 168–176 CrossRef CAS.
- M. N. Siddiqui and H. H. Redhwi, Fuel Process. Technol., 2009, 90, 545–552 CrossRef CAS.
- L. Dai, N. Zhou, H. Li, Y. Wang, Y. Liu, K. Cobb, Y. Cheng, H. Lei, P. Chen and R. Ruan, Sci. Total Environ., 2021, 771, 144995 CrossRef CAS.
- J. Zeaiter, Fuel, 2014, 133, 276–282 CrossRef CAS.
- M. S. Abbas-Abadi, M. N. Haghighi, H. Yeganeh and A. G. McDonald, J. Anal. Appl. Pyrolysis, 2014, 109, 272–277 CrossRef CAS.
- K. H. Lee, Polym. Degrad. Stab., 2008, 93, 1284–1289 CrossRef CAS.
- P. J. Donaj, W. Kaminsky, F. Buzeto and W. Yang, Waste Manage., 2012, 32, 840–846 CrossRef CAS PubMed.
- Y. S. González, C. Costa, M. C. Márquez and P. Ramos, J. Hazard. Mater., 2011, 187, 101–112 CrossRef.
- E. G. Fuentes-Ordóñez, J. A. Salbidegoitia, M. P. González-Marcos and J. R. González-Velasco, Polym. Degrad. Stab., 2016, 124, 51–59 CrossRef.
- G. L. Liu, D. W. Zhu, S. J. Liao, L. Y. Ren, J. Z. Cui and W. B. Zhou, J. Hazard. Mater., 2009, 172, 1424–1429 CrossRef CAS PubMed.
- R. Jiang, G. Lu, Z. Yan, J. Liu, D. Wu and Y. Wang, J. Hazard. Mater., 2021, 405, 124247 CrossRef CAS PubMed.
- W. Asghar, I. Qazi, H. Ilyas, A. Khan, M. Awan and M. Aslam, J. Nanomater., 2011, 2011, 461930 Search PubMed.
- X. U. Zhao, Z. Li, Y. Chen, L. Shi and Y. Zhu, J. Mol. Catal. A: Chem., 2007, 268, 101–106 CrossRef CAS.
- X. Zhao, Z. Li, Y. Chen, L. Shi and Y. Zhu, Appl. Surf. Sci., 2008, 254, 1825–1829 CrossRef CAS.
- L. Zan, W. Fa and S. Wang, Environ. Sci. Technol., 2006, 40, 1681–1685 CrossRef CAS.
- K. Zhang, W. Cao and J. Zhang, Appl. Catal., A, 2004, 276, 67–73 CrossRef CAS.
- S. Chakrabarti, B. Chaudhuri, S. Bhattacharjee, P. Das and B. K. Dutta, J. Hazard. Mater., 2008, 154, 230–236 CrossRef CAS PubMed.
- S. Cho and W. Choi, J. Photochem. Photobiol., A, 2001, 143, 221–228 CrossRef CAS.
- C. Yang, C. Gong, T. Peng, K. Deng and L. Zan, J. Hazard. Mater., 2010, 178, 152–156 CrossRef CAS.
- W. Fa, L. Guo, J. Wang, R. Guo, Z. Zheng and F. Yang, J. Appl. Polym. Sci., 2013, 128, 2618–2622 CrossRef CAS.
- J. Shang, M. Chai and Y. Zhu, J. Solid State Chem., 2003, 174, 104–110 CrossRef CAS.
- Y. Lei, H. Lei and J. Huo, Polym. Degrad. Stab., 2015, 118, 1–9 CrossRef CAS.
- X. Gong, F. Tong, F. Ma, Y. Zhang, P. Zhou, Z. Wang, Y. Liu, P. Wang, H. Cheng and Y. Dai, Appl. Catal., B, 2022, 307, 121143 CrossRef CAS.
- T. Uekert, M. F. Kuehnel, D. W. Wakerley and E. Reisner, Energy Environ. Sci., 2018, 11, 2853–2857 RSC.
- J. Xu, X. Jiao, K. Zheng, W. Shao, S. Zhu, X. Li, J. Zhu, Y. Pan, Y. Sun and Y. Xie, Nat. Sci. Rev., 2022, 9, nwac011 CrossRef PubMed.
- R. Verma, S. Singh, M. K. Dalai, M. Saravanan, V. V. Agrawal and A. K. Srivastava, Mater. Des., 2017, 133, 10–18 CrossRef CAS.
- X. L. García-Montelongo, A. Martínez-De La Cruz, S. Vázquez-Rodríguez and L. M. Torres-Martínez, Mater. Res. Bull., 2014, 51, 56–62 CrossRef.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |