
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLarge scale recyclable monolithic methyltrimethoxysilane aerogels formed by self-reinforcement†
Gylen
Odling
 *a,
Hannah
Logan
b,
Aaron
Chan
b,
Andrew J.
Bissel
a,
Colin R.
Pulham
b and
David E.
Oliver
a
*a,
Hannah
Logan
b,
Aaron
Chan
b,
Andrew J.
Bissel
a,
Colin R.
Pulham
b and
David E.
Oliver
a
aSunamp Ltd., Unit 1 Satellite Park, Macmerry, East Lothian, Scotland EH33 1RY, UK. E-mail: gylen.odling@sunamp.com
bEaStChem School of Chemistry, Joseph Black Building, David Brewster Road, Edinburgh, EH9 3FJ, UK
First published on 27th July 2023
Abstract
A method by which large scale monoliths of methyltrimethoxysilane (MTMS) aerogels may be prepared using ambient pressure conditions is discussed. It has been found that the addition of powdered dry MTMS aerogel into the precursor mixture substantially increases the size of the aerogel monolith which may be formed without cracking, decreases shrinkage and improves the mechanical strength of the resulting monoliths, which are termed self-reinforced aerogels (SRAs). A discussion of the mechanism by which this enhanced strength arises is presented, with observations of the microscopic differences to the gelation process and final dried product when a self-reinforcement strategy is used.
Introduction
Due to their high surface area, porosity, low densities and highly insulative nature, aerogels have garnered considerable interest in recent years. Aerogels appear in work in the fields of environmental remediation,1 catalysis,2 energy storage,3–5 gas separation and storage6–8 and thermal insulation9,10 amongst others, however they are yet to become ubiquitous in modern life. An oft cited reason for this is the inherent fragility of the nanoporous network,11 which leads most examples to be powdered, granular or relatively small monoliths in nature and supported or protected by a matrix.12 Indeed, current commercially available aerogel thermal insulation involves aerogel particles integrated into a supporting blanket or panel. As these such products are becoming more widely available and their production scaled up, thought should be given to their end of lifecycle fate. Development of processes which allow facile routes for material re-use or recycling, and production processes with zero or near-zero waste streams are therefore key at this stage of the commercialisation of aerogel materials and products.Typically, aerogels are prepared by sol–gel processes to produce an alco/hydrogel which are subsequently dried to give the aerogel. To avoid pore collapse, supercritical drying13 or freeze drying14 have been applied, however more recently ambient pressure drying methods have gained success.15 To dry an aerogel under ambient pressure, organic moieties are needed in the pore network, allowing the polar solvent to be removed without pore collapse resulting in densification of the material. Such an organic moiety may be added to the network particle surfaces,16 or present in the silicon precursor material. For this purpose, trialkoxyorganosilanes being used frequently to produce aerogels via ambient pressure drying, and of these, methyltrimethoxysilane (MTMS) is one of the most thoroughly investigated.17 Hydrolysable methoxy groups allowing the wet gel to form via acidic hydrolysis followed by basic condensation, and the methyl moiety allows ambient pressure drying to proceed. Ideally, on drying an aerogel undergoes initial densification and release back to its original form in a process known as spring-back as the solvent leaves the pore network. The hydrophobic nature of the network surface imparted by the methyl groups reduces the capillary force between the solvent and network pore surface and thus allows drying to proceed without pore collapse and the resulting densification. While aerogels may be successfully formed in this manner, the forces applied to the network during drying can be significant and fundamentally inhomogeneous (e.g. they may proceed from the outside inward), and therefore a typically fragile network is highly susceptible to damage if it is not able to withstand said spring-back. Thus cracking of ambient pressure dried aerogels during the drying process remains a challenge, requiring very slow drying processes, careful tuning of the drying conditions, or the use of stabilising additives18,19 or support matrices20 to give a monolithic product. Even with these considerations it is often not possible to produce litre scale monoliths. Many factors contribute to the mechanical strength of an aerogel network, and therefore its susceptibility to crack. Key parameters such as the particle nucleation and growth characteristics, their aggregation and the size and distribution of pores are known to contribute to the macroscopic strength of the material. Tuning such characteristics during the sol–gel process may consist of control over phase separation (i.e. the growing hydrophobic network separating from the hydrophilic solvent), the number and distribution of particle nucleation sites, and the rate of hydrolysis and local reagent availability. Each of these may play a significant role in the production of a large scale macroscopic aerogel monolith piece.
Herein is reported a method of producing an MTMS aerogel monoliths where cracking is avoided by introduction of powdered MTMS aerogel into the preparation process as an additive. Inclusion of an MTMS aerogel additive into the preparation process is found to impart increased mechanical strength and flexibility to the final aerogel, allowing large scale MTMS aerogel monoliths to be prepared. The samples produced in this manner are referred to as self-reinforced aerogels (SRAs). The formation process is observed microscopically, and thereby the potential mechanism by which improved mechanical strength is imparted is discussed. In addition, an investigation into the SRA thermal insulation performance and thermal stability is presented. It is furthermore demonstrated herein that the preparation process may re-use MTMS aerogel produced by this same process, and thereby recycle the material. Furthermore, it is shown that the material produced by this process may be iteratively recycled up to at least 3 times without significant change in the thermally insulative properties of the resulting aerogel.
Experimental
Materials
Methyltrimethoxysilane (98%) was purchased from Fisher Scientific. Glacial acetic acid was purchased from (SOURCE). NH3OH solution (35%) was purchased from Alfa Aesar. Isopropyl alcohol (IPA) (>99%) was purchased from ReAgent. Tap water was used throughout. Materials were used as received without further purification.Aerogel preparation
Water (10 g) and IPA (11.775 g) are mixed with 0.1 M acetic acid solution (1 g). To this is added MTMS (4.775 g) and the whole mixture stirred at ca. 45 °C for 1 hour. To this is then added 1 M NH3OH solution (1.5 g) with vigorous stirring. The mixture is then poured into a mould and held at 65 °C while covered for 3 hours. The cover is then removed, and the aerogel allowed to dry for around 16 hours at 65 °C. This gives a material referred to as 0×SRA, or unmodified MTMS aerogel. This process is then repeated, with the dried aerogel being crudely ground and then added into the mixture after the 1 M NH3OH solution in amounts corresponding to 10 wt% of the total wet mixture. This gives a material referred to as 1×SRA. This process was repeated at various scales up to around 7 L of starting sol.Samples of dried 1×SRA were recycled back into the same process in the place of the original dried aerogel to give 2×SRA. 3×SRA and 4×SRA were produced in the same way, using 2×SRA and 3×SRA as additive aerogel precursors respectively.
Samples of dried aerogel were calcined in a covered dish in a furnace at 450 °C for 1 hour to give hydrophilic insulation panels.
Analytical methods
Thermal conductivity testing was carried out using a Thermtest HFM-100 with samples of 21.23 mm thickness. The thermal conductivity measurements were taken at an average temperature of 20 °C. Three repeat measurements were used to gauge reproducibility. Thermogravimetric measurements were made on a Netzsch STA 449F1 instrument in air (20 ml min−1), with a ramp rate of 5 °C min−1. SEM images were captured on a Zeiss Crossbeam 550 instrument, and the samples were platinum coated before imaging.Results and discussion
Preparation of large scale MTMS aerogel monoliths
While small (i.e. <cm3/<dm3) pieces of MTMS aerogel may be easily prepared without cracks, attempts to create large scale monolithic panels, blocks or discs of MTMS aerogel directly were unsuccessful. Instead of a monolith, severely cracked and oft-times deformed aerogel pieces were obtained (Fig. 1). | ||
| Fig. 1 Example of cracked MTMS aerogel (0×SRA) formed without any stabilising agent in a ca. 12 × 12 cm metal container. | ||
During drying, the aerogel network is known to be subject to strain,21 undergoing a shrink-spring back process as the solvent is removed from the pore structure. This phenomenon, and the resulting cracking, has severely limited the size of aerogel monoliths which may be prepared, leading most large-scale applications to favour the use of reinforcing additives.22,23 Further images of cracked MTMS aerogels prepared as part of this work can be found in the ESI† (Fig. S1–S5).
However, it was found that large scale monoliths of MTMS aerogels can be formed when reinforced by the material itself. Recycling of the cracked material back into the aerogel preparation process in this manner allows large SRA pieces, including some moulded shapes, to be prepared without any supporting network material. A schematic representation of the process is given in Fig. 2.
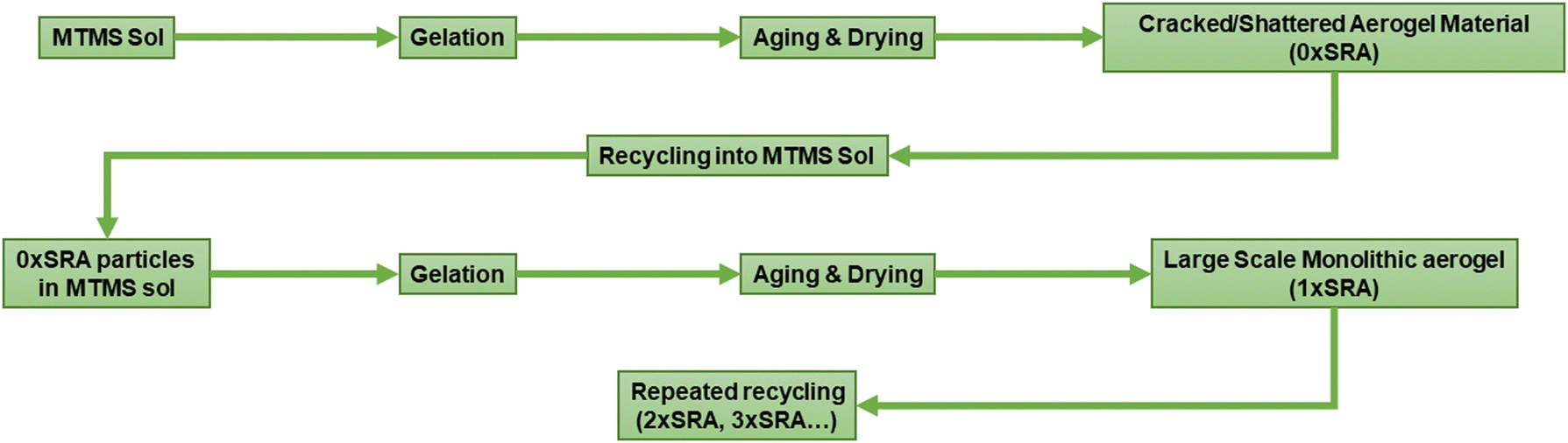 | ||
| Fig. 2 Process used to generate MTMS aerogel and SRAs. | ||
Addition of 10 wt% dried MTMS aerogel into a subsequent aerogel preparation to give an SRA was found to be sufficient to avoid cracking up to at least 45 × 60 × 5 cm pieces (Fig. 3). At lower loadings (i.e. 5 wt%) larger pieces of SRA could be produced, however in scaling up beyond ca. 10 × 10 × 3 cm pieces, cracking was found to occur.
 | ||
| Fig. 3 (A) A 45 × 30 × 5 cm panel of an SRA (B) a 45 × 60 × 5 panel of the 1×SRA. | ||
A possible factor for the effectiveness of using self-reinforcement to produce large scale aerogel monoliths is that of the dispersibility of the reinforcing additive in the precursor sol. MTMS aerogels are typically hydrophobic due to the presence of surface methyl groups,24 however were found to still be able to be dispersed in the precursor sol. The dispersibility of the additive materials was therefore investigated for comparison. A variety of commercially available hydrophobic and hydrophilic silicas (Cab-O-Sil®) and commercial aerogel products (Enersens Kwark®) were substituted for MTMS aerogel powder in the reinforcement process. These comparison additives were chosen according to their compatibility (i.e. hydrophilic/phobic) and their densities relative to the sol. This was found to be unsuccessful, producing cracked and powdery aerogel samples, with no observed monolith stabilising effects gained by their use. Generally hydrophilic silicas were found to wet and segregate, forming a dense layer at the bottom of the container above which the MTMS aerogel would form with no obvious changes to its mechanical strength. Hydrophobic silicas and the commercial hydrophobic aerogel would float on the MTMS sol surface rather than disperse, and therefore be effectively non-interacting with the sol and giving no strengthening effect. Conversely, use of the MTMS aerogel itself gives a dispersed network of reinforcing particles, which stay dispersed in the sol until gelation has occurred. Thus, the wettability and dispersibility of an additive used in this manner is key to gain any monolith strengthening effect. The aerogel additive clearly has some surfactant like role in the gelation process, however a surfactant effect appears to be not sufficient alone to produce large scale monolithic aerogels.
It is proposed that the highly dispersed MTMS aerogel particles act to reduce phase separation and provide nucleation sites for siloxane particle formation, templating the growth of the siloxane network due to the chemical similarity of the MTMS aerogel particle surface to the wet siloxane network. To investigate this, micrographs and recordings were taken of the sol precursor during the gelation process, both with and without the presence of the reinforcing aerogel additive (Fig. 4, 5 and Supporting media 1–12, ESI†). Initially in the blank aerogel preparation the sol appears homogeneous, however over time phase separation occurs (Fig. 4 and Supporting media 1–6, ESI†) as hydrophobic oligomers separating from the polar solvent system. Over time this heterogeneity becomes severe, and by the time full gelation occurs these hydrophobic zones have grown to be significant in size (Fig. 4B–D). Furthermore, it can be observed in the microscope video recordings that during the transition from sol to gel that significant flow occurs in the solution despite the lack of agitation (Supporting media 1–6, ESI†). Thus, the likelihood of hydrophobic oligomeric zones coalescing and becoming larger is increased as the sol flow promotes collision between said zones, giving the opportunity for them to combined and thereby reduce their surface energy. By comparison, where the aerogel additive is present in an SRA preparation, the sol is far more stationary during gelation (Supporting media 7–12, ESI†) and the incidence of hetereogeneous zones is greatly reduced (Fig. 5). The causes of this can be considered to be twofold; the aforementioned reduction in the sol flow during gelation, but also the disruption of surface tension of hydrophobic zones by the hydrophobic aerogel particles.
 | ||
| Fig. 4 Micrographs of the unmodified MTMS aerogel sol during network formation and growth, gelation time increasing (A–D). | ||
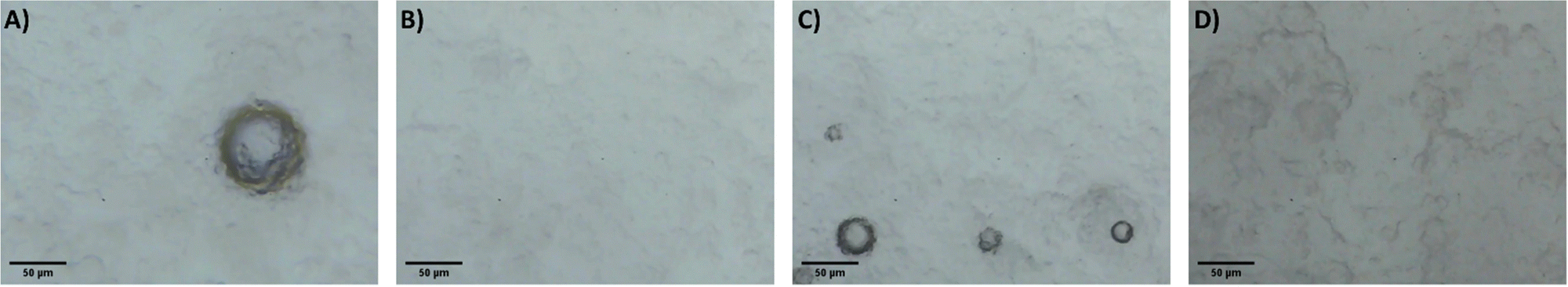 | ||
| Fig. 5 Micrographs of the 1×SRA during gelation, gelation time increasing (A–D). Note higher magnification used in these images compared to blank. | ||
It has been found previously that limiting the extent of the phase separation using surfactants may improve the aerogel mechanical properties.25–27 However, when repeating the un-reinforced aerogel preparation with the addition of a range of concentrations of the surfactants Cetrimmonium bromide (hexadecyltrimethylammonium bromide; CTAB) or sodium dodecylsulfate (SDS), no improvement to the cracking was observed (Fig. S6 and associated text, ESI†). Conversely, an increase in cracking was found on higher surfactant content, while lower amounts allowed only moderately sized pieces to be prepared. Thus, it is proposed that the stabilising effect of the self-reinforcing additive may not be due to a reduction in phase separation alone.
Prior work has explored adding surfactants such as SDS and CTAB into aerogel preparations to reduce phase separation and thereby produce a stronger, more homogeneous preparation. For example, Hayase et al.25 demonstrated the use of cetrimmonium chloride for suppression of aggregate phase separation during gelation, and noted its effect on optimising the resulting materials strength. Finding an optimum surfactant concentration, the authors suggested a combination of suppression of phase segregation and hindrance of particle aggregation at high concentrations gives an optimum surfactant loading. Similarly, Cheng et al.26 found that CTAB may increase the strength of the network formation, assigning this to a uniform homogeneous phase on gelation allowing increased interparticle connectivity. He et al.27 also discuss the introduction of surfactants into an MTMS aerogel preparation with an aim to increase the homogeneity of the mix and note again a resulting strengthening of the network.
Here however, the lack of any significant increase in the size of aerogel monolith which may be prepared by inclusion of a surfactant additive suggests that the mechanism by which the reintroduction of powdered aerogel as an additive into the formulation provides improved strength may not be only one of improved homogeneity during gelation. While the reinforcing powdered aerogel additive does reduce phase separation, as can be observed when comparing Fig. 4 and 5, and in the supporting media, a similar effect may be found in the literature when using surfactants. However, as can be observed in Fig. S6 (ESI†), including a surfactant to the 0×SRA preparation, thereby reducing phase separation, does nothing to allow a larger monolith to be prepared. Thus, while the reduction in phase separation provided by the self-reinforcement may be a contributing factor to the monoliths formation, it is likely that other factors are also contributory. For instance, the work of both Cheng and He suggests that the surfactant property allows the formation of smaller particles and pores within the structure by enabling the use of higher water contents. Smaller pores are known to encourage densification of the gel on drying, due to the inverse relationship between pore size and capillary force, and thus a denser, but stronger, gel may result.
One explanation which has been somewhat overlooked in the past work is a concurrent alteration to the gelation nucleation and growth regime in the sol. A templating MTMS aerogel additive, which can be dispersed throughout the sol bulk, provides a large surface area from which nucleation and growth of the siloxane network may proceed. Work by Du et al.28 studied the effect of polymeric substances on the gelation of metal oxide networks, their conclusions including the modulation of nucleation and growth characteristics of metal oxide gel networks by the templating, coordinating nature of the polymer additive. It is suggested herein that the self-reinforcement by the aerogel additive achieves a similar effect. Nucleation from such a large number of points in the bulk may aide with the homogeneity of the network formation and avoid macroscopic failure points (i.e. points of non-connection). Further to this, a recent review by Karamkimar et al.29 discussed the binodal nature of aerogel gelation and raise a point which is highly relevant to the SRA system, that a high nucleation rate coupled with a low growth rate gives many small siloxane clusters. As suggested earlier, in an SRA there exist a very large number of nucleation sites, and the feed sol is inherently mass transfer limited due to the heterogeneous, static nature of the precursor-aerogel suspension. As gelation occurs, the consumption of reagents to grow the network chains can cause large local changes in concentration which alter the growth characteristics of the network as the monomeric precursors are consumed.30–32 Where the growth is occurring on and between pre-formed heterogeneous surfaces, this feed can easily become depleted, limiting network growth and favouring smaller particles.
Hence, the theory of SRA formation is thus; nucleation induced from a large number of points, mass transfer limitation during gelation and simultaneous reduction of phase separation and heterogeneous zone growth promotes formation of an aerogel with enhanced physical strength and thus enables the production of large scale monoliths.
Electron microscopy
To gain an insight into the network structure and the effect of the SRA modification, and to test the hypothesis of modifications to the nucleation and growth regime of the aerogel, scanning electron micrographs were recorded of un-reinforced aerogel samples (0×SRA) and 1×SRA. Fig. 6 shows a comparison of the outer surfaces of the aerogel monoliths. Generally, the aerogels form a highly porous network of particles with voids characteristic of reported aerogel networks. It can be observed in Fig. 6B and D, that the SRAs exhibit slightly smaller particle sizes, and a more tightly packed network. | ||
| Fig. 6 Scanning electron micrographs of the outer surface of the un-reinforced MTMS aerogel (A and C) and SRA-1x (B and D). | ||
However, when observing the cross-section of the prepared materials were imaged (Fig. 7), a greater incidence of very small (approx. 1–4 μm diameter) secondary particles grown from the main network forming particles can be observed in the 1×SRA sample only (Fig. 7B–D). It is suggested that the growth of these secondary particles is a result of the inclusion of the aerogel additive into the sol during preparation, due to the aforementioned changes to siloxane chain nucleation and growth, and hindrance of large heterogeneous zone formation during gelation. Indeed, it is intuitive that an additive which both provides nucleation sites and a reduction in mass transport would result in many particles which have been limited in size. Differences between the core and outer layers of the aerogel monoliths may be understood by the differences in mass transport in these zones on gelation, with the sol being more readily able to flow around the outside of the gelled monolith and feed the growing particles, allowing these particles to grow to a greater extent than those in the core. Reinforcement of aerogel materials around the “neck” region between the main network forming particles, which the self-reinforcement provides, has been reported as a means to improve aerogel strength,22,33 but typically requires introduction of additives or post-gelation processing which is detrimental to the aerogel thermal properties.
 | ||
| Fig. 7 Scanning electron micrographs of the cross-section surface of the un-reinforced MTMS aerogel (A) and SRA-1x (B–D). | ||
Recycling of MTMS aerogel monoliths
Iterative recycling of the SRA was investigated to gauge whether the SRA material could be simply re-used in the same process at the end of its use. A series of SRA pieces named n×SRA were prepared, where n is the number of recycles carried out according to Fig. 2. It was found that monolithic SRA pieces could be generated using materials which had been reformed by the preparation process at least 3 times (Fig. S8, ESI†). Thus, the preparation process described herein can be used to capture a valuable waste stream by reforming old, used, off cut or broken pieces of SRA into new monoliths. Combined with an easily recapturable alcohol/water solvent system and low concentrations of acids and bases to induce hydrolysis and condensation, the process of producing the SRA materials lends itself to minimal waste production.Thermal properties
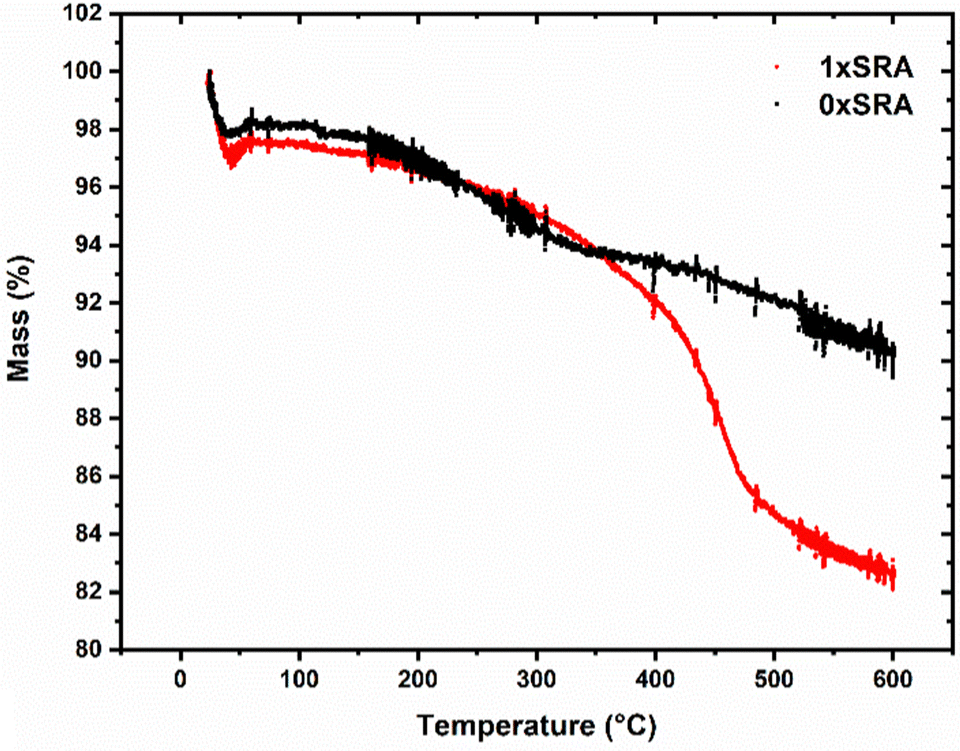 | ||
| Fig. 8 TGA scans for the unmodified (SRA-0x) and SRA-1x samples in air. | ||
The presence of the organic methyl groups in the MTMS aerogel structure are necessary to allow ambient pressure drying to proceed and leaves the resulting aerogel structure hydrophobic. Removal of such using high temperatures therefore leaves the aerogels, including the SRAs, completely inorganic in nature and hydrophilic. Such a calcination process renders the materials non-combustible, allowing their use at high temperatures. On a larger scale, applying a 450 °C thermal treatment to the SRA monoliths for 1 hour results in a fully hydrophilic monolith which does not burn when subjected to a propane blowtorch (Fig. S9, ESI†). It was observed that this calcination process results in an embrittled monolith, which when applied to an unmodified MTMS aerogel piece rather than an SRA resulted in severe further cracking and granularization. In contrast, the SRAs modified in this manner remained reasonably robust. Self-reinforcement may allow monolithic aerogels with a variety of different properties to be produced, which would otherwise give powdery or delicate results.
Thermal conductivity
Low thermal conductivities were observed for all samples, including the non-reinforced aerogel, in line with reported literature values for MTMS based ambient pressure dried aerogels34–36 (Table 1).| Sample | Thermal conductivity (mW mK−1) | Density (g cm−3) |
|---|---|---|
| Unmodified MTMS aerogel (0×SRA) | 38.4 | 0.18 |
| 1×SRA | 40.6 | 0.20 |
| 2×SRA | 40.9 | 0.20 |
| 3×SRA | 41.4 | 0.20 |
It was found that the thermal conductivity of the control MTMS aerogel is largely unaffected by forming an SRA, with a slight increase in thermal conductivities measured for samples containing recycled aerogel, concurrent with a slight increase in density. However, the increases are relatively small and thus it is shown that that this material may be recycled at least 3× without significant loss of thermal performance. A slight increase in density is in line with the SEM observations, where the presence of strengthening small secondary particles fill in some of the free space present in the pore network. A concurrent rise in the thermal conductivity of the monoliths upon self-reinforcement (i.e. going from the unmodified 0×SRA to 1×SRA) is similar to previous reports where the material density increases, and has been explained by a loss of porosity causing increased thermal transport through the structure compared to the alternative trapped gas.37 The work of Wong et al.38 demonstrated that higher densities typically result in a more brittle monolith with higher thermal conductivity, with a rapid increase in both properties above a density of about 0.2 g cm−3. The self-reinforcement strategy applied here therefore appears to give a good increase in strength, without risking increasing the brittle nature of the materials overmuch.
Conclusions
A preparation method of large scale monolithic MTMS aerogels is reported herein. It has been found that reintroduction of particulate MTMS aerogel into the production process allows cracking to be avoided and thereby allows large panels and shaped pieces of SRA to be prepared. This material has been found to have improved mechanical strength and stability over the unmodified MTMS aerogel, superhydrophobicity and low thermal conductivity, making it a good candidate for thermal insulation applications. It has been postulated that the addition of recycled MTMS aerogel powders leads to a reduction in hydrophobic zone growth and coalescence during gelation and therefore detrimental phase separation is avoided without the use of surfactants. A further effect of aerogel recycling is an increase in the nucleation site density and reduction in sol feed rate in the gelation step, leading to a greater incidence of small aerogel particles in the aerogel monolith bulk. This combination of increased homogeneity during preparation and gel phase nucleation improvements are thought to increase the strength of the resulting aerogel and thereby avoid cracking during drying. This processing technique allows the aerogel to be recycled back into the production process, reducing waste, and may be recycled in this manner at least 3 times without loss of thermal performance.Author contributions
The enclosed work was devised and carried out in part by G. O. A. C. was responsible for unmodified MTMS aerogel formulation determination. H. L. recorded the TGA scans. A. J. B., C. R. P. and D. E. O. provided supervision and guidance throughout. G. O. authored the manuscript which has been approved by all contributors.Conflicts of interest
This work pertains to a patent application filed with the UK IPO (GB202112550D0).Acknowledgements
The authors thank Dr Fraser Laidlaw (The University of Edinburgh) for assistance in recording the SEM micrographs.Notes and references
- A. S. Jatoi, Z. Hashmi, S. A. Mazari, R. Abro and N. Sabzoi, J. Porous Mater., 2021, 28, 1919–1933 CrossRef CAS.
- H. Maleki and N. Hüsing, Appl. Catal., B, 2018, 221, 530–555 CrossRef CAS.
- Y. B. Pottathara, H. R. Tiyyagura, Z. Ahmad and K. K. Sadasivuni, J. Electrochem. Energy Convers. Storage, 2020, 30, 101549 CrossRef.
- H. Liu, H. Du, T. Zheng, K. Liu, X. Ji, T. Xu, X. Zhang and C. Si, Chem. Eng. J., 2021, 426, 130817 CrossRef CAS.
- J. Mao, J. Iocozzia, J. Huang, K. Meng, Y. Lai and Z. Lin, Energy Environ. Sci., 2018, 11, 772–799 RSC.
- S. Yu, S. Song, R. Li and B. Fang, Nanoscale, 2020, 12, 19536–19556 RSC.
- J. E. Amonette and J. Matyáš, Microporous Mesoporous Mater., 2017, 250, 100–119 CrossRef CAS.
- L. Keshavarz, M. R. Ghaani, J. M. D. MacElroy and N. J. English, Chem. Eng. J., 2021, 412, 128604 CrossRef CAS.
- S. Ahankari, P. Paliwal, A. Subhedar and H. Kargarzadeh, ACS Nano, 2021, 15, 3849–3874 CrossRef CAS PubMed.
- J. Wang, D. Petit and S. Ren, Nanoscale Adv., 2020, 2, 5504–5515 RSC.
- X. Hou, R. Zhang and D. Fang, ACS Sustainable Chem. Eng., 2021, 9, 7638–7648 CrossRef CAS.
- T. Linhares, M. T. Pessoa De Amorim and L. Durães, J. Mater. Chem. A, 2019, 7, 22768–22802 RSC.
- F. Mißfeldt, P. Gurikov, W. Lölsberg, D. Weinrich, F. Lied, M. Fricke and I. Smirnova, Ind. Eng. Chem. Res., 2020, 59, 11284–11295 CrossRef.
- C. Ruan, Y. Ma, G. Shi, C. He, C. Du, X. Jin, X. Liu, S. He and Y. Huang, Appl. Surf. Sci., 2022, 592, 153280 CrossRef CAS.
- R. Ganesamoorthy, V. K. Vadivel, R. Kumar, O. S. Kushwaha and H. Mamane, J. Cleaner Prod., 2021, 329, 129713 CrossRef CAS.
- M. V. Khedkar, S. B. Somvanshi, A. V. Humbe and K. M. Jadhav, J. Non-Cryst. Solids, 2019, 511, 140–146 CrossRef CAS.
- K. Kanamori and K. Nakanishi, Chem. Soc. Rev., 2011, 40, 754–770 RSC.
- L. Wang, J. Feng, Y. Jiang, L. Li and J. Feng, RSC Adv., 2019, 9, 10948–10957 RSC.
- B. N. Nguyen, M. A. B. Meador, A. Medoro, V. Arendt, J. Randall, L. McCorkle and B. Shonkwiler, ACS Appl. Mater. Interfaces, 2010, 2, 1430–1443 CrossRef CAS PubMed.
- E. Ul Haq, S. F. A. Zaidi, M. Zubair, M. R. Abdul Karim, S. K. Padmanabhan and A. Licciulli, Energy Build., 2017, 151, 494–500 CrossRef.
- S. Iswar, W. J. Malfait, S. Balog, F. Winnefeld, M. Lattuada and M. M. Koebel, Microporous Mesoporous Mater., 2017, 241, 293–302 CrossRef CAS.
- H. Maleki, L. Durães and A. Portugal, J. Non-Cryst. Solids, 2014, 385, 55–74 CrossRef CAS.
- J. Lin, G. Li, W. Liu, R. Qiu, H. Wei, K. Zong and X. Cai, J. Mater. Sci., 2021, 56, 10812–10833 CrossRef CAS.
- Z. Yang, H. Yu, X. Li, H. Ding and H. Ji, J. Non-Cryst. Solids, 2019, 525, 119677 CrossRef CAS.
- G. Hayase, K. Kanamori and K. Nakanishi, Microporous Mesoporous Mater., 2012, 158, 247–252 CrossRef CAS.
- X. Cheng, C. Li, X. Shi, Z. Li, L. Gong and H. Zhang, Mater. Lett., 2017, 204, 157–160 CrossRef CAS.
- S. He and X. Chen, J. Non-Cryst. Solids, 2017, 463, 6–11 CrossRef CAS.
- A. Du, B. Zhou, J. Shen, J. Gui, Y. Zhong, C. Liu, Z. Zhang and G. Wu, New J. Chem., 2011, 35, 1096–1102 RSC.
- S. Karamikamkar, H. E. Naguib and C. B. Park, Adv. Colloid Interface Sci., 2020, 276, 102101 CrossRef CAS PubMed.
- K. Mawhinney and S. C. Jana, ACS Appl. Polym. Mater., 2019, 1, 3115–3129 CrossRef CAS.
- L. D. Gelb, J. Phys. Chem. C, 2007, 111, 15792–15802 CrossRef CAS.
- R. W. Pekala and D. W. Schaefer, Macromolecules, 1993, 26, 5487–5493 CrossRef CAS.
- H. Maleki, L. Durães and A. Portugal, J. Mater. Chem. A, 2015, 3, 1594–1600 RSC.
- L. Durães, A. Maia and A. Portugal, J. Supercrit. Fluids, 2015, 106, 85–92 CrossRef.
- H. Gao, L. Bo, P. Liu, D. Chen, A. Li, Y. Ou, C. Dong, J. Wang, X. Chen, C. Hou, W. Dong and G. Wang, Sol. Energy Mater. Sol. Cells, 2019, 201, 110122 CrossRef CAS.
- D. Du, Y. Jiang, J. Feng, L. Li and J. Feng, Vacuum, 2020, 173, 109117 CrossRef CAS.
- X. Lu, R. Caps, J. Fricke, C. T. Alviso and R. W. Pekala, J. Non-Cryst. Solids, 1995, 188, 226 CrossRef CAS.
- J. C. H. Wong, H. Kaymak, S. Brunner and M. H. Koebel, Microporous Mesoporous Mater., 2014, 183, 23 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00085k |
| This journal is © The Royal Society of Chemistry 2023 |