
Stimuli-responsive dynamic hydrogels: design, properties and tissue engineering applications
Sivashanmugam
Amirthalingam†
ab,
Arun Kumar
Rajendran†
 b,
Young Gi
Moon
b and
Nathaniel S.
Hwang
*abcd
b,
Young Gi
Moon
b and
Nathaniel S.
Hwang
*abcd
aInstitute of Engineering Research, Seoul National University, Seoul, 08826, Republic of Korea. E-mail: nshwang@snu.ac.kr
bSchool of Chemical and Biological Engineering, Institute of Chemical Processes, Seoul National University, Seoul, 08826, Republic of Korea
cInterdisciplinary Program in Bioengineering, Seoul National University, Seoul, 08826, Republic of Korea
dBio-MAX/N-Bio Institute, Institute of Bio-Engineering, Seoul National University, Seoul, 08826, Republic of Korea
First published on 7th June 2023
Abstract
The field of tissue engineering and regenerative medicine has been evolving at a rapid pace with numerous novel and interesting biomaterials being reported. Hydrogels have come a long way in this regard and have been proven to be an excellent choice for tissue regeneration. This could be due to their innate properties such as water retention, and ability to carry and deliver a multitude of therapeutic and regenerative elements to aid in better outcomes. Over the past few decades, hydrogels have been developed into an active and attractive system that can respond to various stimuli, thereby presenting a wider control over the delivery of the therapeutic agents to the intended site in a spatiotemporal manner. Researchers have developed hydrogels that respond dynamically to a multitude of external as well as internal stimuli such as mechanics, thermal energy, light, electric field, ultrasonics, tissue pH, and enzyme levels, to name a few. This review gives a brief overview of the recent developments in such hydrogel systems which respond dynamically to various stimuli, some of the interesting fabrication strategies, and their application in cardiac, bone, and neural tissue engineering.
 Sivashanmugam Amirthalingam | Sivashanmugam currently works as a post-doctoral researcher at Seoul National University. He has research experience in developing various peptide- and ceramic-loaded hydrogels for bone tissue engineering. His major research interests include chemical modifications of various bio-active polymers and effectively use them in development of scaffolds and hydrogels to promote tissue regeneration. |
 Arun Kumar Rajendran | Arun Kumar, a dental surgeon, holds a master's degree in Nanomedicine and technology. He graduated from Tokyo medical and dental University and currently works as a senior researcher at Seoul National University. His major research interests include designing and synthesis of bioceramics and hydrogels for enhancing bone regeneration and also focus on mechanosignalling. He has worked on the development of various scaffolds and injectable hydrogels for bone tissue engineering. |
 Young Gi Moon | Young Gi is a PhD student at Seoul National University. He has an academic background in materials science and biomedical engineering. His main research area is nanoparticle mediated gene delivery and therapy. |
 Nathaniel S. Hwang | Nathaniel S. Hwang is working as a professor at Seoul National University. He has great experience in development of several biomimicking bone and cartilage regenerative systems and also therapeutic delivery systems. He has keen interest in clinical translation of novel bioceramics and smart delivery systems. He is a member of the scientific program committee of the Korean Tissue Engineering & Regenerative Medicine Society. |
Wider impactHydrogels are being developed at a rapid pace owing to their potential to mimic tissues and deliver a multitude of therapeutic agents. With current advancements, researchers have designed and synthesized various hydrogel systems which dynamically respond to various stimuli, proving beneficial in clinical settings. This review discusses some of the interesting advancements in ‘smart’ hydrogels that respond to various stimuli such as light, heat, electric, magnetic, and acoustic fields, pH, and mechanical forces. The exploitation of benefits from different stimuli by incorporating multiple responsive mechanisms in a single system has also been discussed. Recent clinical trial evaluations of stimuli-responsive hydrogels have been included. This review also provides updates on dynamic hydrogels applied for various tissue engineering applications, which might be of interest to material scientists and clinicians. With the ongoing progress in 3-D printing, new efficient chemistries, minimally invasive treatments, and patient-specific personalized therapeutics, we could expect that, in the future, it will be necessary to utilize such stimuli-responsive dynamic hydrogels to achieve precise spatiotemporal delivery of therapeutic agents as required. Thus, we believe that this review will provide a good understanding of the currently available stimuli-responsive hydrogels and could provide insights for further advancements in this field. |
1. Introduction
Hydrogels are 3-dimensional networks of cross-linked polymeric chains that can absorb and retain water or biological fluids in the matrix, making them promising materials for tissue regeneration.1,2 Additionally, they serve as scaffolds for tissue growth and development, providing a supportive niche for cells to proliferate, migrate, and differentiate.3 A hydrogel can be made of synthetic polymers or natural materials or a combination of both allowing for the utilization of the advantages of both. Progress in the field of hydrogels has been discussed in reviews.4–9 In recent trends, stimuli-responsive dynamic hydrogels have received a lot of attention as they can undergo mechanical and physical changes upon the application of external stimuli or environmental cues.10 Traditional hydrogels have been studied to a large extent as carriers of cells and growth factors. However, they fail to integrate with the defect site, release profile of the growth factors cannot be controlled with respect to the disease condition, and they do not bio-mimic the tissue to a large extent. However, the stimuli-responsive hydrogels are highly adaptable to the dynamics of the defect site in response to external or internal stimuli. Additionally, the release pattern of the growth factors can be controlled on demand and they possess the capability to more closely bio-mimic the physical, mechanical and biological properties of the native tissue.11 Typically, hydrogels can be prepared to respond to various exogenous stimuli, such as temperature, light, electric fields, mechanical forces, and ultrasound, whereas endogenous stimuli, such as pH, enzyme, mechanobiological and metal ions, can cause phase-transition or stiffness change in hydrogels. With careful design of the hydrogel with responsive cross-linkers or chemical modification that responds to the changes in the pathological conditions or injured area in the target, an endogenous activation for intended delivery of cells or growth factors or providing an output signal can be achieved. For example, a hydrogel was designed to change the cross-linking density of the hydrogel in response to vascular morphogenesis.12 In the case of exogeneous stimuli, hydrogels can be designed to modify the properties of the hydrogel remotely or non-invasively. For example, hydrogels were designed to display light-guided regulation of cell attachment.13 The alignment of the gold nanowires was modified with external electric-field stimuli to provide a topographic pattern for better myogenesis.14 In addition, biodegradability is also an important criterion in designing stimuli-responsive hydrogels. Biodegradability of polymers is determined based on the chemical structure. Generally, the polymers that contain ester, amide, anhydride, glyosidic and carbonate bonds can be cleaved by hydrolysis, oxidation or enzymatic action. Furthermore, hydroxyl and carboxyl moieties can increase the hydrophilicity, thereby facilitating biodegradation. Ulery et al. have comprehensively discussed on the biodegradable polymers and their route of degradation.15 Thus, careful design of hydrogels with a combination of endogenous and exogenous stimuli-response characteristics would provide new intelligent materials for tissue regeneration.This review aims to introduce different types of stimuli-responsive dynamic hydrogel systems and provide the discussion on the underlying molecular mechanism that enable these stimuli-responsive reactions. Specifically, this review focuses on dynamic hydrogels capable of responding to various stimuli, including but not limited to light, temperature, electric field, ultrasound, mechanical forces, pH, and enzymes (Fig. 1). Furthermore, a section on dual and multi-stimuli-responsive hydrogels has been included. Additionally, recent updates in the literature on overcoming the previous problems faced by the researchers are discussed. Finally, this review covers recent applications and future perspectives of these stimuli-responsive hydrogels, particularly in the field of tissue regeneration. Thus, this review article offers a comprehensive overview of the latest developments in stimuli-responsive dynamic hydrogels, with an emphasis on their potential use in tissue regeneration.
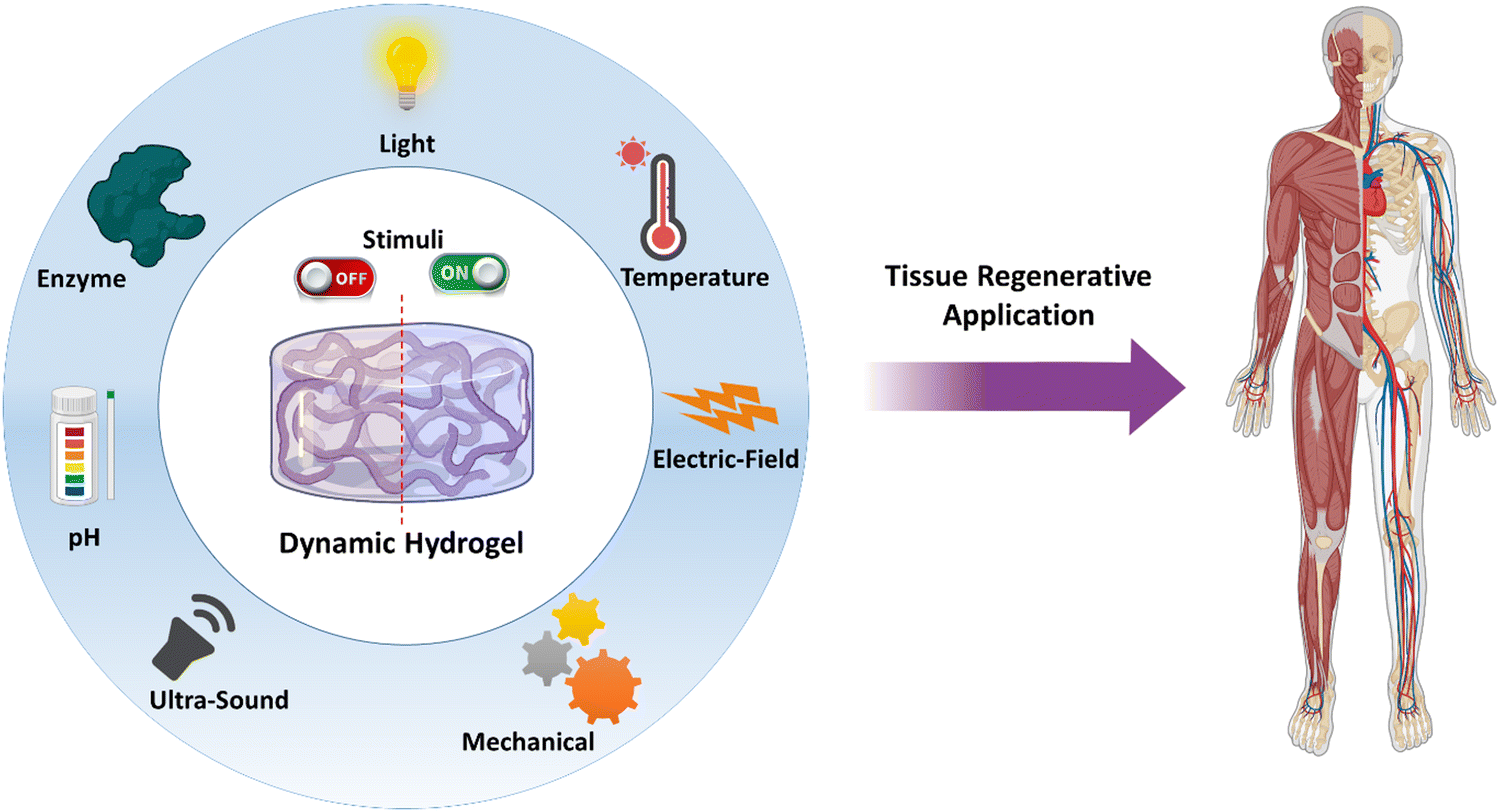 | ||
| Fig. 1 Schematic illustrations of various stimuli that a hydrogel could be designed for tissue regenerative applications. | ||
2. Stimuli-responsive hydrogels
2.1 Photo-responsive hydrogels
Photo-responsive hydrogels have emerged as an attractive option in biomedical applications as they can show spatiotemporal responses. In addition, photo-responsiveness provides many opportunities, as light is a non-invasive stimulus and the remote manipulation of hydrogel could provide fewer by-products.16 Modulating the photo-irradiation parameters, namely, the light intensity and irradiation time provides the precise control of photo-reaction, thus providing spatial control. Temporal control could be easily achieved by turning on or off the light source. Wavelength-selective photochemical reactions can be achieved by tuning the hydrogel properties.17 The interaction of light with photo-responsive moieties in the hydrogel network could lead to degradation, hydrogel contraction or swelling, thermal dissipation, and chemical modifications, leading to variations in material properties such as softening or stiffening. To clarify, hydrogels that are solely cross-linked based on light-stimuli (photo-cross-linked hydrogels) and do not respond to light stimuli later, are not included in the discussion, as they are photo-cross-linked hydrogels, rather than photo-responsive hydrogels.Functional groups play the most important role in defining the hydrogel's physicochemical and biological properties such as bioadhesion, wettability, cell adhesion and spreading, etc. In the case of photo-responsive hydrogels, this can be achieved by introducing functional moieties through photochemical addition reaction or photo-isomerization reaction, or by releasing the photo-caging groups. Conversely, the functionality of the hydrogel can be removed by photo-chemical cleavage reaction or by inactivating the functional group via photo-addition reactions.
With the advent of the two-photon excitation technique and the usage of upconversion nanoparticles, the depth of field can be extended from ∼250 μm (UV/visible light) to 10–100 mm.18 The two-photon approach is a technique that activates photo-responsive moieties using near-infrared (NIR) light, wherein two photons of identical or non-identical energies are absorbed by the molecule, simultaneously, for getting excited. This allows for deeper penetration of light in the tissue, as NIR is less absorbed by the tissue compared to UV and visible light.19 In the case of upconversion nanoparticles, they generally contain trivalent lanthanide dopants with ladder-like energy levels. This permits sequential absorption of multiple photons to attain the excited state, which is in contrast to two-photon approach, wherein simultaneous absorption of photons is required to attain the excited state.20
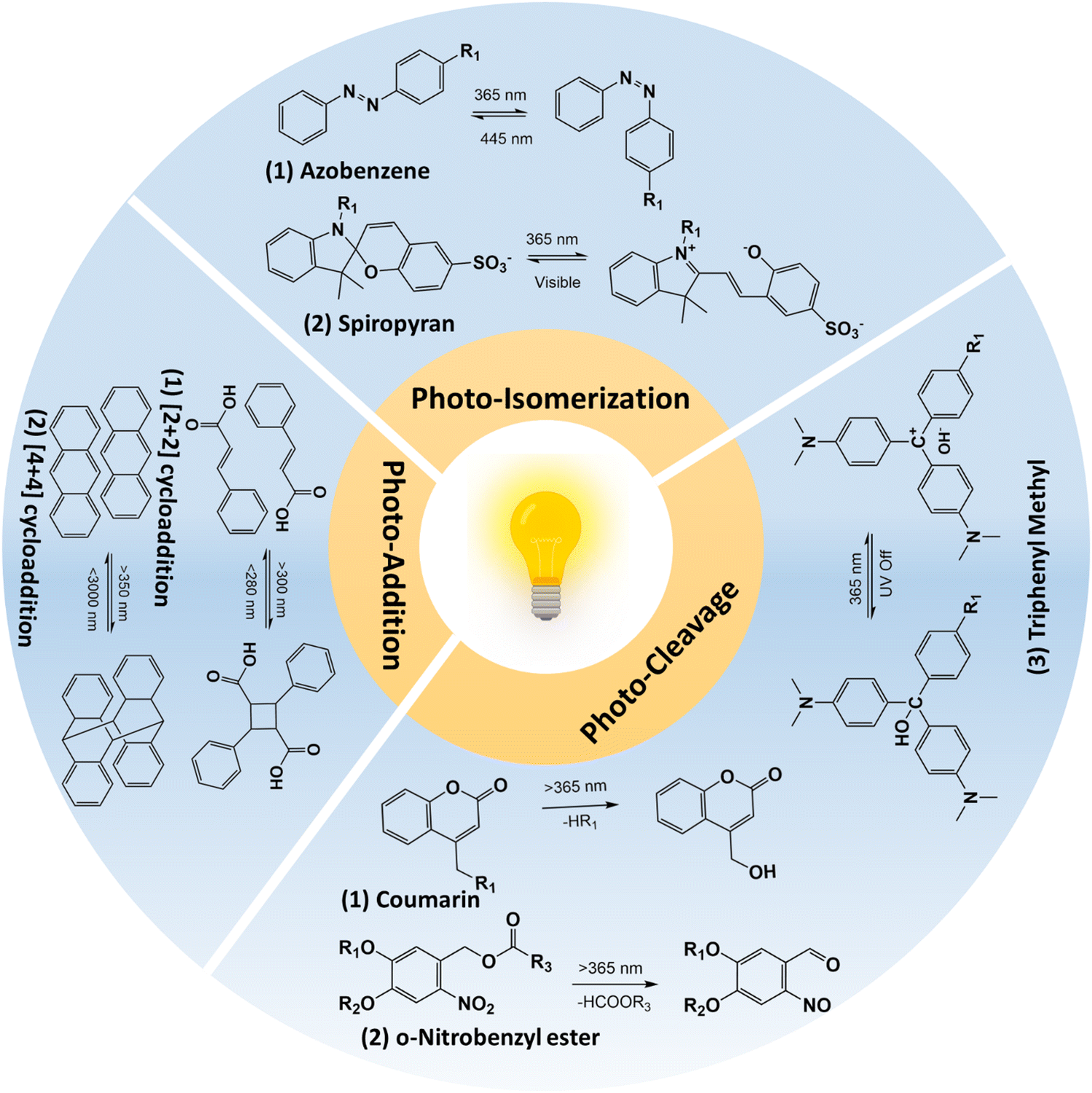 | ||
| Fig. 2 Schematic illustration of photo-reactions that were utilized in designing photo-responsive hydrogels. | ||
2.1.1.1 Photo-cleavage reaction. The photo-cleavage reaction involves the dissociation of chemical bonds triggered by the absorption of photons. The o-nitrobenzyl and o-nitrophenyl ethyl moieties are the most common photolabile group used in biomedical applications because of their biocompatible properties.21,22 The residues that are cleaved by light are biologically inert and do not react with molecules such as proteins or polysaccharides.23,24 The wavelength of light that can cause photo-cleavage depends on the functional group that is attached to the benzene ring, and can be varied from 260 nm to 365 nm. For example, the 4,5-dimethoxy o-nitrobenzyl group undergoes photocleavage reaction at ∼350 nm. However, the inherent phototoxicity and limited penetration depth are associated with UV light18,25 have limited its in vivo application. The use of a two-photon approach and upconversion nanoparticles has been suggested to overcome the above-mentioned problems associated with UV light. For example, Farrukh et al. developed a photo-responsive hydrogel with a o-nitrobiphenyl propyl moiety as the two-photon absorption cross-section is higher (σ2 = 3.2–11 GM; depending upon the substituent in the biphenyl core) compared to the o-nitrobenzyl group (σ2 = 0.0133 GM).13 In this work, authors utilized a p-methoxynitrobiphenyl derivative (PMNB) with σ2 = 3.2 GM at 740 nm that was tethered to a cell adhesive cyclo RGDfC peptide in a PEG hydrogel, thereby achieving light-guided regulation of cell attachment with two-photon excitation at a cytocompatible level (2320 mW cm−2). In another study, Zheng et al. utilized upconversion nanoparticles that can convert the near-infrared light (NIR; λ = 974 nm) to UV light (λ = 340–360 nm) in a PEG hydrogel containing cyclo RGDfC peptides with 3-(4,5-dimethoxy-2-nitrophenyl)-2-butyl ester as the photo-cleavable group.26 In this work, authors could achieve the light-guided regulation of cell attachment with a NIR laser (λ = 974 nm; 10 W cm−2). Further studies should be conducted in comparing use of upconversion nanoparticles and two-photon excitation approaches for finding out efficient photo-cleavage reactions. The coumarin group presents a photolabile alternative to the nitrobenzyl platform, with cleaved residues that exhibit inert properties and expose a less reactive hydroxyl group compared to nitrobenzyl groups.27,28 Unlike other photo-cleavable moieties, coumarin can react to a broad spectrum of wavelengths rather than a specific wavelength. Also, the 6-bromo-7-hydroxy-coumarin-4-yl-methyl linker has shown greater sensitivity compared to o-nitrobenzyl groups29 and Truong et al. showed that coumarin moieties showed a higher degradation rate compared to o-nitrobenzyl derivatives.30 Additionally, these coumarin groups exhibit photodimerization properties that allow them to react reversibly upon exposure to photo-irradiation, resulting in the formation of a cyclo-butyl ring due to cyclization.31 This ring can be easily cleaved by ∼250 nm light, owing to its intense ring strain.32 Meanwhile, triphenylmethane derivatives have been utilized to control swelling properties.33 In contrast to coumarin and o-nitrobenzyl groups, triphenyl methane derivatives dissociate into ionic pairs upon exposure to ultraviolet radiation. Under UV irradiation, triphenyl methane leucohydroxide (TPMLH) derivatives generate triphenylmethyl cations that can form ionic pairs, which are paired once the UV light is removed. As a result, the TPMLH conjugated hydrogel swells under UV light and shrinks in response to the removal of UV irradiation.33,34 A major drawback of using TPMLH derivatives is that it requires several hundred milliwatts of UV light for the reaction to take place. For example, Jiang et al. showed that for the degradation of malachite green leucohydroxide derivatives, 1500 mW cm−2 is required.35 To circumvent this problem, Chen et al. developed a photo-responsive polyacrylamide hydrogel tethered with o-nitrobenzyl and TPMLH moieties.36 The photodissoaction efficiency of the developed system was improved as result of shift in the equilibrium between the TPMLH ion and the hydroxide anion. The hydroxide ion reacts with the proton lost from the o-nitrobenzyl group during photo-cleavage reaction. The authors irradiated the samples using a 365 nm light source at 25 mW cm−2 for 3 min on each side of the hydrogel. In the follow-up study, authors utilized the same hydrogel with a demonstration of bandgap formation and tunability in a vertically suspended photo-responsive hydrogel that is locally activated to form a periodic structure, thereby forming a programmable metamaterial.37
2.1.1.2 Photo-isomerization reaction. Photo-isomerization refers to the transformational change in the chemical structure upon photo-irradiation.38 In contrast to photocleavage reaction, photo-isomerization is a reversible and repeatable process that makes it an attractive method for functionalizing hydrogels in biomedical applications. Azobenzene is one of the most well-known photo-isomerization compounds, composed of two phenyl rings linked by an azo group (–N
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–).39–41 Upon exposure to 365 nm light, this compound undergoes isomerization reaction, specifically, from its trans form to cis form via π–π* transition, and can also be converted to the cis-configuration by irradiating with 445 nm light or by heating. The isomerization of azobenzene in hydrogels results in significant changes in their polarity and hydrophobicity. The cis configuration of hydrogels is more polar than the trans form, which induces steric hindrance for complex structures in the isomerized hydrogel. Through the use of photo-isomerization, the properties of hydrogels such as mechanical strength and swelling ratios can be manipulated.40 Most of the photo-responsive hydrogels with azobenzene moieties are based on azobenzene–cyclodextrin, as a host–guest complex. The host–guest interaction depends on the cis or trans form of azobenzene.42 Liubimtsev et al. developed a double cross-linked hydrogel with changes in the cross-linking density on demand.43 The authors developed a polyacrylamide hydrogel with azobenzene and cyclodextrin (CD) moieties as reversible cross-linkers and N,N′-methylenebis(acrylamide) as an irreversible cross-linker. Cross-linking density of the hydrogel changed with successive irradiation of the hydrogel with UV (λ = 365 nm) and visible (λ = 400 to 500 nm) light. In addition to azobenzene and CD host–guest complexes, recently, Gliozzi et al. utilized azobenzene (such as methyl red) and host polymer matrix made of UV polymerizable diacrylate monomers (such as bisphenol A ethoxylate diacrylate) for developing tunable photo-responsive elastic metamaterials.44 Authors showed that photoisomerization of azobenzene can be utilized to modulate Young's modulus, thereby resulting in a finite waveguide elastic response. Additionally, to overcome the disadvantage of using UV light, azobenzene isomerization was carried out by including upconversion nanoparticles in the hydrogel, utilizing a two-photon approach and by modifying the azobenzene structures to achieve a red-shift in the response.45–47 However, two-photon and upconversion nanoparticles indirectly caused isomerization, where upconversion nanoparticles possessed low efficiency and a high intensity laser targeting a very small area was required for the two-photon approach.45,47 Thus, a visible light response system is required for the isomerization reaction. Recently, Yan et al. developed a tetra-ortho-methoxy-substituted azobenzene dimer (mAzo dimer) and a β-CD dimer as binary gelators.48 Thus, blue light (λ = 450 nm) irradiation caused cis to trans isomerization of mAzo and green light (λ = 550 nm) resulted in the trans to cis isomerization of mAzo. The spiropyran group is a photoisomerization moiety that is widely used in the biomedical field due to its cyclization properties.49–51 Unlike azobenzene, spiropyran undergoes a transformation from an open to a closed form under blue light. In its open form, the compound displays zwitterionic properties, making it hydrophilic. However, upon exposure to photo-irradiation, a ring structure is formed, leading to a switch to hydrophobic properties.16 It is worth noting that the cyclization process is reversible, as the cyclized form is unstable, and the open form exhibits a delicate balance of electrons due to ring strain. As a result, the use of spiropyran in hydrogel functionalization enables the manipulation of hydrogel properties such as mechanical strength and swelling ratios. Recent studies have shown that spiropyran-containing polymers can cause significant changes in both physical and chemical properties upon blue light irradiation.52–54 The change in hydrophilicity of the spiropyran moieties upon photoisomerization could affect the hydrogel swelling properties and this change can be programmed to change the volume of the hydrogel.55 One of the major drawbacks with spiropyran-based materials is that spiropyran molecules discussed above were water insoluble and requires organic solvents for their preparation which limits their biomedical use. To circumvent this, Li et al. developed sulfonate spiropyran-based water soluble photoswitch tethered poly isopropylacrylamide hydrogel.56 Recently, Meeks et al. developed a multiresponsive spiropyran-based hydrogel that is responsive towards pH, temperature and light.57
N–).39–41 Upon exposure to 365 nm light, this compound undergoes isomerization reaction, specifically, from its trans form to cis form via π–π* transition, and can also be converted to the cis-configuration by irradiating with 445 nm light or by heating. The isomerization of azobenzene in hydrogels results in significant changes in their polarity and hydrophobicity. The cis configuration of hydrogels is more polar than the trans form, which induces steric hindrance for complex structures in the isomerized hydrogel. Through the use of photo-isomerization, the properties of hydrogels such as mechanical strength and swelling ratios can be manipulated.40 Most of the photo-responsive hydrogels with azobenzene moieties are based on azobenzene–cyclodextrin, as a host–guest complex. The host–guest interaction depends on the cis or trans form of azobenzene.42 Liubimtsev et al. developed a double cross-linked hydrogel with changes in the cross-linking density on demand.43 The authors developed a polyacrylamide hydrogel with azobenzene and cyclodextrin (CD) moieties as reversible cross-linkers and N,N′-methylenebis(acrylamide) as an irreversible cross-linker. Cross-linking density of the hydrogel changed with successive irradiation of the hydrogel with UV (λ = 365 nm) and visible (λ = 400 to 500 nm) light. In addition to azobenzene and CD host–guest complexes, recently, Gliozzi et al. utilized azobenzene (such as methyl red) and host polymer matrix made of UV polymerizable diacrylate monomers (such as bisphenol A ethoxylate diacrylate) for developing tunable photo-responsive elastic metamaterials.44 Authors showed that photoisomerization of azobenzene can be utilized to modulate Young's modulus, thereby resulting in a finite waveguide elastic response. Additionally, to overcome the disadvantage of using UV light, azobenzene isomerization was carried out by including upconversion nanoparticles in the hydrogel, utilizing a two-photon approach and by modifying the azobenzene structures to achieve a red-shift in the response.45–47 However, two-photon and upconversion nanoparticles indirectly caused isomerization, where upconversion nanoparticles possessed low efficiency and a high intensity laser targeting a very small area was required for the two-photon approach.45,47 Thus, a visible light response system is required for the isomerization reaction. Recently, Yan et al. developed a tetra-ortho-methoxy-substituted azobenzene dimer (mAzo dimer) and a β-CD dimer as binary gelators.48 Thus, blue light (λ = 450 nm) irradiation caused cis to trans isomerization of mAzo and green light (λ = 550 nm) resulted in the trans to cis isomerization of mAzo. The spiropyran group is a photoisomerization moiety that is widely used in the biomedical field due to its cyclization properties.49–51 Unlike azobenzene, spiropyran undergoes a transformation from an open to a closed form under blue light. In its open form, the compound displays zwitterionic properties, making it hydrophilic. However, upon exposure to photo-irradiation, a ring structure is formed, leading to a switch to hydrophobic properties.16 It is worth noting that the cyclization process is reversible, as the cyclized form is unstable, and the open form exhibits a delicate balance of electrons due to ring strain. As a result, the use of spiropyran in hydrogel functionalization enables the manipulation of hydrogel properties such as mechanical strength and swelling ratios. Recent studies have shown that spiropyran-containing polymers can cause significant changes in both physical and chemical properties upon blue light irradiation.52–54 The change in hydrophilicity of the spiropyran moieties upon photoisomerization could affect the hydrogel swelling properties and this change can be programmed to change the volume of the hydrogel.55 One of the major drawbacks with spiropyran-based materials is that spiropyran molecules discussed above were water insoluble and requires organic solvents for their preparation which limits their biomedical use. To circumvent this, Li et al. developed sulfonate spiropyran-based water soluble photoswitch tethered poly isopropylacrylamide hydrogel.56 Recently, Meeks et al. developed a multiresponsive spiropyran-based hydrogel that is responsive towards pH, temperature and light.57
2.1.1.3 Photo-addition/dimerization reaction. The photo-dimerization reaction proceeds to form covalent bonds between the two of the same molecules upon irradiation with light when they are molecularly very close and/or in the right orientation. Some common photo-dimerization reactions are [2+2]- and [4+4]-cycloaddition and these reactions are reversible upon irradiation with a suitable wavelength or upon heating.58 Since the cleavage of the C–C (covalent) bond is a high-energy consumption process, it requires light irradiation with higher energy. For example, Liu et al. developed coumarin-incorporated oligo ethylene glycol bis-urea amphiphile monomers that showed a sol-to-gel transition upon irradiation at 365 nm and the reaction was partially reversible at 254 nm.59 Other examples of [2+2] cycloaddition includes cinnamates,60–63 maleimides,64–66 and stilbenes.67,68 The [4+4] cycloaddition reaction is similar to [2+2]-cycloaddition, wherein exposure to 365 nm causes dimerization and exposure to short UV light (<280 nm) causes dissociation.69–72 Red-shift photodimerization of anthracene was observed when the benzyl imine group was conjugated to anthracene, wherein blue light irradiation causes dimerization.73 Other photo-addition reactions that include allyl sulfides and disulfide addition–recombination reactions, are not of high importance for hydrogels yet.74–76
The clinical translation of photo-responsive hydrogels for tissue engineering applications requires overcoming challenges related to tissue-related light attenuation and phototoxicity. Since the majority of photo-responsive moieties require UV light, direct one-photon activation through tissue is limited to a few hundred micrometers in depth. Red-shifted moieties could moderately increase this depth while adhering to the maximum permissible exposure to skin. Two-photon approaches and upconversion nanoparticles have the potential to increase the depth of the field to a few centimeters, but the use of high-powered pulsed lasers for the two-photon approach may limit their clinical applicability due to associated photodamage, whereas, upconversion nanoparticles are typically doped with lanthanides such as Ho3+, Er3+, and Tm3+, and their biological interactions, excretion, and elimination need to be studied in detail.
2.2 Thermo-responsive hydrogels
Thermo-responsive hydrogels that respond to temperature have been investigated because of the ease of applying external stimuli and their biocompatibility, making them competent candidates for biomedical-related applications: cell therapy, and tissue engineering.77 Thermo-responsive hydrogels are a subclass of supramolecular materials that undergo gelation through hydrophobic interactions. These polymers undergo phase transition because of their amphiphilic nature.78 Thermo-responsive materials, typically undergo sol-to-gel change with an increase in temperature and change back to the solution when the temperature is lowered to a certain value, which is intrinsic to the polymer.79 Gelation does not require any additional cross-linking agents or any other assistance from enzymes; thus, the procedure is considered benign and biocompatible.80 The transition temperature that transforms the state of the polymer (solution to gel transition) is important when considering applications. For example, in many biomedical applications, thermo-responsive polymers have transition temperatures between room temperature and body temperature and they can be injected into the body, where they can undergo in situ gelation.9 Conventionally, they rely on polyacrylates, polyoxyethylene–polyoxypropylene–polyoxyethylene block copolymers (PEO–PPO–PEO), poly(N-isopropyl acrylamide) (PNIPAm), poly(N-vinyl caprolactam) (PNVCL), polysaccharides and polypeptides.2.2.1.1 Lower critical solution temperature. Thermo-responsive hydrogels are classified into two main types. One exhibits a lower critical solution temperature (LCST) and the other exhibits an upper critical solution temperature (UCST). The polymers exhibiting the LCST behavior show the property that polymers undergo phase transition below the critical temperature point or cloud point temperature (TCP). On the other hand, the polymers exhibiting the UCST behavior show the property that polymers undergo phase transition above the TCP (Fig. 3A).81 The UCST characteristic of polymers is enthalpy driven and has very strong supramolecular forces that get weakened upon heating, and thereby they underdo gel-to-sol transition above TCP.82 It is very difficult to achieve UCST features under physiological conditions.83,84 Most of the UCST polymers are zwitterionic and the major drawback of using these polymers is that they are non-biodegradable.83 For example, Sharker et al. developed polyampholytes made of cationic vinyl benzyl trimethylammonium chloride and anionic sodium p-styrene sulfonate that showed UCST behavior above 45 °C.85 Hence, we have focused more on LCST polymers in this review article. The polymers that exhibit an LCST-type behavior in aqueous solution, are soluble below TCP because hydrogen bonding interactions between polymers and the surrounding water molecules are dominant, and intermolecular hydrogen bonding between polymer molecules is restricted. On the other hand, above TCP, hydrogen bonding between water and polymers is disrupted, and intermolecular hydrogen bonding and hydrophobic interactions between polymer molecules become dominant. Hence, the LCST of polymers is entropically driven.86,87 For this reason, above TCP, the polymers become insoluble, forming a hydrogel (Fig. 3B).88 Zhang et al. provided a systematic discussion on the LCST behavior of the polymers and they recommended a standardized set of measurement techniques for TCP.89 Poly(N-alkyl-substituted acrylamides) are the first studied polymers for their thermo-responsive behavior. Among them, PNIPAm is explored widely for biomedical applications not only because of its LCST properties at 32 °C which is close to the body temperature but also because its LCST behavior is not greatly affected by variations in the pH, salt concentration, and other environmental factors.87 Researchers studied the phase transition effect of PNIPAm that undergoes coil-to-globule transition via contraction of the polymeric chain due to the hydrophobic effect and increase in entropy of the system.87,90 The collapse of the PNIPAm chain upon heating is highly dependent on the hydration structure around the monomeric units, wherein upon an increase in temperature, it causes reduced coordination between amide and isopropyl groups with water.90 Even though small variations in salt do not affect the LCST of PNIPAm, molecules with a high charge-to-volume ratio (such as SO42−) can lower the LCST by competing with water molecules for interaction with the polymeric chain, thereby causing dehydration.91,92 In contrast, surfactant addition to the polymeric solution increases the LCST temperature by interacting with the isopropyl group.93 These studies provide us with a deep understanding of the thermogelling behavior of homopolymers. Additionally, LCST of the PNIPAm can be tuned by copolymerizing with other monomeric units, especially adding hydrophilic units such as N-vinyl pyrrolidone causes an increase in LCST,94 whereas the addition of hydrophobic units such as dopamine methacrylamide reduced the LCST.95 However, one of the major drawbacks of the issue is non-biodegradability.96 Biodegradability can be partly addressed by copolymerizing or grafting biodegradable polymers such as gelatin,97 polycaprolactone (PCL),96 and chitosan.98 Calder et al. developed a co-polymer of PNIPAAm, namely, poly(NIPAAm-co-(N-acryloxysuccinimide)-co-(polylactide/-hydroxy methacrylate)-co-(oligo (ethylene glycol) (PNPHO), which showed a promising degradation profile with a tuneable gelation time of 2–10 minutes.99 In their follow-up study, they showed that cross-linking a synthetic peptide, thymosin-β4, brought down the LCST from 24 °C to 21 °C.100 The authors showed the regenerative potential of the developed hydrogel in multiple anatomical defect sites, in vivo. Furthermore, they went on to study the clinical usability of the hydrogel in the mandibular socket after tooth extraction in 10 patients and the hydrogel supported the bone healing cascade at the defect site. Other homopolymers exhibiting a thermo-responsive behavior similar to PNIPAAm include PNVCL (LCST: 32–34 °C)101–103 and poly(2-alkyl-2-oxazoline) (PAOx).104,105 The LCST of PAOx can be fine-tuned based on the alkyl group. For instance, while the addition of a methyl group does not induce a LCST behavior, the incorporation of iso-propyl, cyclo-propyl, and n-propyl moieties in PAOx induces LCST at approximately 35 °C, 30 °C, and 25 °C, respectively.106,107
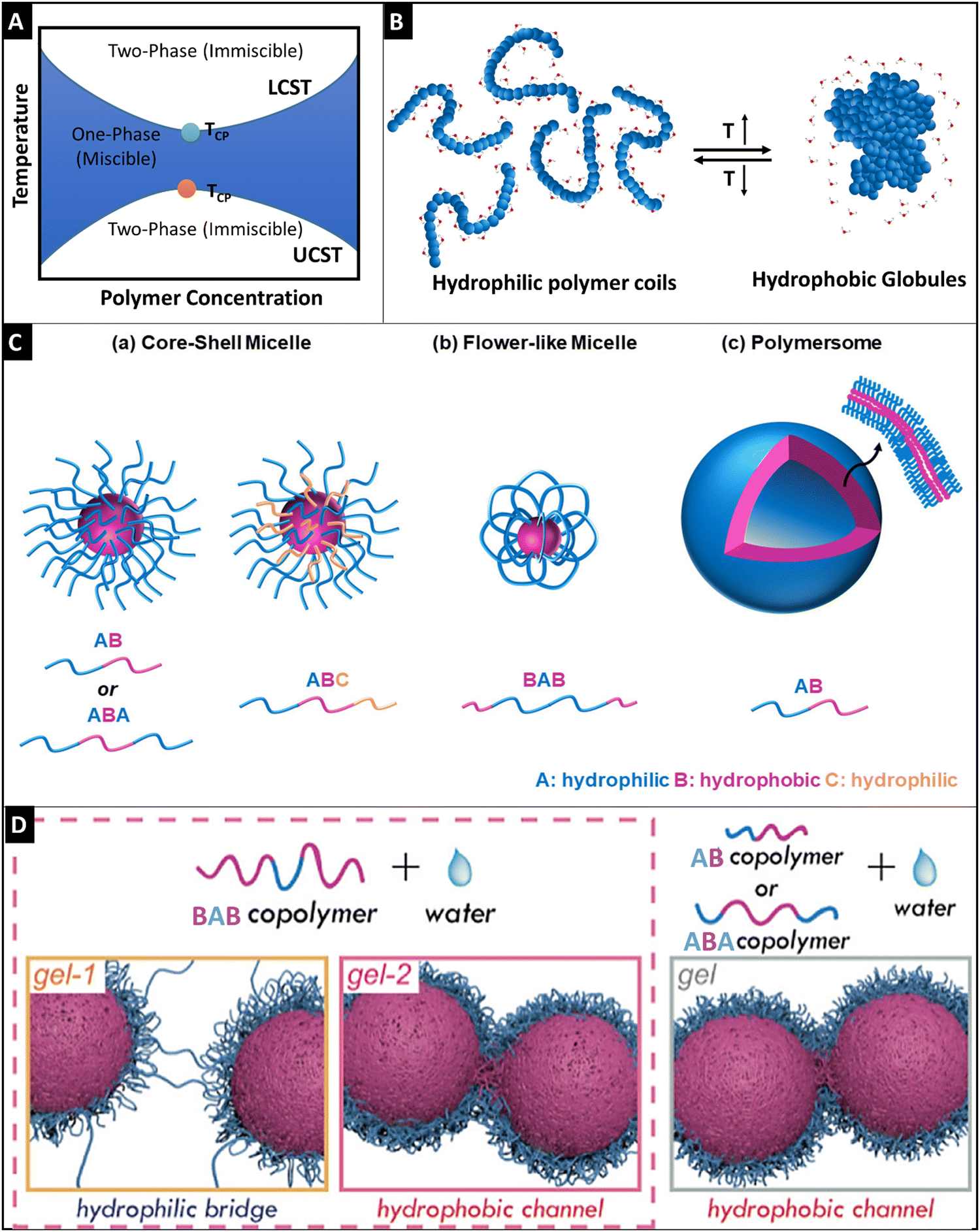 | ||
| Fig. 3 Schematic representation of the phase diagram for the LCST and UCST thermo-responsive polymers (A), schematic diagram showing the phase transition of hydrophilic polymer coils (<LCST) to hydrophobic globule formation (>LCST), reproduced under Creative Commons license,88 (B), a graphical illustration of self-assembly of amphiphilic block polymers (C), wherein core–shell micelle formation was observed for AB and ABA block copolymers (a), flower-like micelle formation was seen for the BAB block polymers and polymersome architecture was observed for AB polymers (c) reproduced under Creative Commons license,88 and schematic diagrams of the gelling behavior of the BAB polymer through a hydrophilic bridge and hydrophobic channel for the BAB polymer (left side) and the gelling behavior of AB and ABA copolymers through hydrophobic channel formation (right side) based on the semi-bald model of micelles (D). Adapted with permission from ref. 114. Copyright © 2019, American Chemical Society. | ||
2.2.1.2 Polymer configuration, micellization, and gelation. Except for PNIPAm, PNVCL, PAOx, and chitosan (wherein, polyol salt such as β-glycerophosphate is added to chitosan solution, the chitosan solution shows thermo-responsive properties);108 thermo-gelling polymers are generally amphiphilic block polymers. Thermo-gelling block polymers could have varied configurations and could be classified as di-block, tri-block, and multi-block polymers (Fig. 3C). In the tri-block polymer, there are multiple configurations such as ABA, BAB, ABC, etc., wherein ‘A’ is a hydrophilic block, ‘B’ is a hydrophobic part and ‘C’ is either hydrophilic or hydrophobic. Water-soluble PEO–PPO–PEO triblock copolymers have a configuration of ABA. PEO–PPO–PEO triblock copolymers have amphiphilic characteristics which are dependent on the molecular weight, block size, and block sequence.109 In this case, PLGA–PEG–PLGA is another studied tri-block polymer having a configuration of BAB.110
Micellization is a unique phenomenon associated with amphiphilic molecules. These molecules undergo micellization after attaining a specific concentration known as critical micellar concentration (CMC) and the temperature at which this property occurs is called critical micellar temperature (CMT). During the thermo-gelation process, these unimers of the polymers undergo self-assembly of micelles, wherein these unimers get inserted into the existing micelles and consequently, the insertion–expulsion and fusion-fragmentation process slowly becomes the dominant process.111 The molecular weight of the polymer and the hydrophobic block length strongly affects the CMC.86 The higher the hydrophobic block length, the denser the accumulation of hydrophobic cores, and thereby the higher stability of micelles is achieved. However, the ratio of hydrophobic block length and hydrophilic part is very important, as exposure of the hydrophobic part in water would destabilize the micelle and gelation would not occur.112 The gelation behavior of these polymers is due to the self-aggregation of these micelles and it is attributed to three different mechanisms, namely, micellar packaging as in the case of di-block and ABA tri-block polymers, inter micellar-bridged packaging is observed for the BAB, BAC tri-block and multi-block polymers, and micellar corona collapse packaging is seen in copolymers containing polymers of low LCST. The cross-linking points of these micelles causing gelation were previously unknown and a series of recent studies from Ding's group has shown that semi-bald micelle formation was observed for ABA and AB block polymers, wherein the physical cross-linking was observed through the hydrophobic channels that were possible through the bald region in the micelles (Fig. 3D).113 On the other hand, gelation was observed for the BAB polymer micelles through two steps: (1) hydrophilic bridging was dominant at lower temperature, whereas, (2) hydrophobic channel cross-linking was predominant at higher temperature (Fig. 3D).114
A crucial aspect in the designing of thermo-responsive hydrogels is the range of temperatures to which the hydrogel will be exposed. This range encompasses the preparation temperature, storage and transportation conditions, and in vivo exposure, including normal body temperature and elevated pathological temperatures such as fever. Given the diverse range of biomaterials and processing techniques available, researchers must carefully evaluate these parameters and adjust the thermo-responsiveness of the hydrogel accordingly to meet the specific requirements. Furthermore, the mechanical strength of thermo-responsive hydrogels may be limited due to their physical cross-linking nature, which can restrict their utilization in tissue engineering applications that demand higher mechanical strength or stability. Moreover, it might be challenging to precisely control the degradation rate of thermo-responsive hydrogels, which can result in degradation that is either too fast or too slow. Additional studies are necessary to investigate the degradation behavior of thermo-responsive polymers under both normal and pathological conditions.
2.3 Electrically-responsive hydrogels
Electric field-responsive hydrogels, in response to external electrical stimuli, can swell or deform. These hydrogels are fast, and reliant and provide spatiotemporal control for their responsiveness. In addition, the inherent property of electrical conductivity can also be utilized to potentiate the intrinsic electrochemical conductivity of the cells, thereby supplementing the biofunctionality of these hydrogels.115,116 The influence of electrical fields on cell processes for osteogenesis, angiogenesis, cardiomyogenesis, myogenesis, and neurogenesis has been recognized well and it can be used to harness microtissue maturation with the help of an electro-active hydrogel.117 Utilizing the programmable and accurate delivery of voltage/current to the desired site can control the spatiotemporal response of the hydrogel. The swelling/deswelling of the hydrogel can be affected by the concentration of the polymer, cross-linking density, and the ionic concentration of the medium.115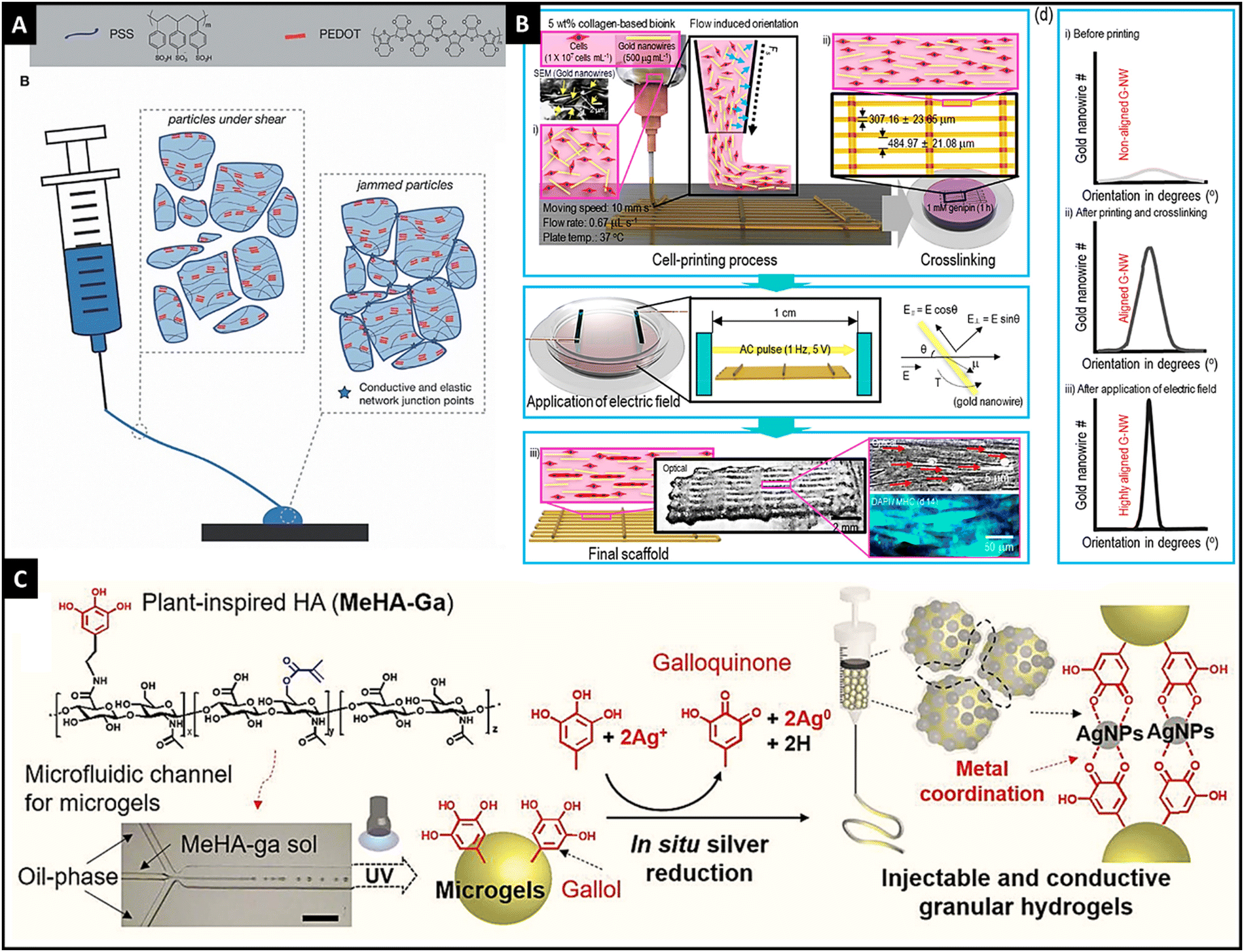 | ||
| Fig. 4 The microgels of PEDOT:PSS showing shear-thinning properties and granular gel return to a jammed configuration resulting in conductivity, reproduced with permission, Copyright © 2021 John Wiley & Sons Ltd125 (A), schematic illustration of obtaining highly aligned Au nanowires through dielectrophoresis and printing in a collagen-based 3-D printed hydrogel, reproduced with permission from ref. 14. Copyright © 2019, American Chemical Society, (B) and schematic graphic showing a granular injectable hydrogel made of gallol-conjugated methacrylated hyaluronic acid that showed in situ Ag ion reduction, thereby forming Ag nanoparticles and hydrogel was jammed using vacuum filtration, reproduced under Creative Commons license,128 (C). | ||
In addition to the use of conductive hydrogels, the inclusion of metallic nanoparticles (such as Ag, Au, and silicon) and carbon-based nanomaterials (such as graphene, carbon nanotubes, and carbon dots) in the polymeric matrix imparts electrical conductivity to the hydrogel.126,127 For instance, Kim et al. utilized Au nanowires for conductivity and geometry of the scaffold in 3-D bio-printed collagen-based hydrogels with C2C12 cells.14 With dielectrophoresis and injection, authors were able to achieve the desired alignment that resulted in a high degree of myoblast alignment and effective myotube formation. Thus, this method provided a new perspective on electro-responsive hydrogels (Fig. 4B). Shin et al. developed an injectable granular microgel with a metal–phenolic coordination network.128 The authors used oxidized gallol (galloquinone) with Ag ions that were reduced to generate Ag nanoparticles. Additionally, gallol acted as chelators of Ag nanoparticles that formed a coordinated network. Vacuum filtration-induced jamming provided electro-conductivity for the developed hydrogel (Fig. 4C). Compared to the embedded nanoparticles in the hydrogel, in situ synthesized nanoparticles have shown higher conductivity and mechanical properties.128,129 Similarly, carbon-based nanomaterials were incorporated into the hydrogel network for imparting electro-responsivity.130,131 Thus, different electro-active materials could be incorporated into the hydrogel network, result in an electro-responsive hydrogel.
Electro-responsive hydrogels have demonstrated potential for use in tissue engineering applications. However, several disadvantages must be taken into account when considering their clinical translation. There is a limited understanding of the long-term behaviour and degradation profile of the electro-responsive polymers in vivo. Care must be taken to prevent potential damage to cells and tissue when applying electric current to stimulate electro-responsiveness. Another drawback of electro-responsive hydrogels is that the biocompatibility of these conductive polymers is not adequate. Researchers have attempted to improve biocompatibility by conjugating or mixing conductive polymers with biocompatible polymers. However, striking a balance between conductivity and biocompatibility is crucial, as biocompatible polymers can act as insulators, thereby reducing the conductivity of the hydrogel. Further studies are required to optimize the balance between biocompatibility and conductivity in electro-responsive hydrogels for tissue engineering applications.
2.4 pH-Responsive hydrogels
The pH-responsive hydrogels are a subset of stimuli-adaptive systems capable of responding to the environmental pH. These hydrogels readily react to certain pH levels and exhibit either swelling or deswelling behavior.132 Such characteristics of pH-adaptive hydrogels are of particular interest for biomedical applications since some parts of the body such as the gastrointestinal tract, vagina, and blood vessels are known to exhibit substantial pH changes.133 Acknowledging that our body parts experience dynamic pH changes, scientists and researchers further shift their interest to the potential therapeutic effects of pH-adaptive hydrogels under specific circumstances. For instance, a lower pH level (between 6.7 and 7.1) than the normal pH level (between 7.3 and 7.4) is a biological parameter characteristic of pathological states.134 Some features of tumors including the oncogene activation, transformation, and hypoxia cause acidosis, a cellular metabolism that generates an excessively high level of acids within tumor microenvironments. To this end, the pH-adaptive hydrogel system is considered to be an ideal option for both delivery and therapy purposes.The swelling behavior of pH-responsive hydrogels upon exposure to water or other physiological fluids depends on the osmotic pressure triggered by multiple factors including the hydrophilicity of polymers, static charges on the polymer, and counter ions within the hydrogel matrix.132 Although all these factors contribute to the swelling behavior to different extents, the general behavior can be divided into three major steps. First is the diffusion of water into the hydrogel network, second is the disintegration of polymer chains, and third is the extension of the hydrogel network followed by the relaxation of polymer chains. In the case of pH-responsive hydrogels, the swelling behavior is affected by two elements – the properties of constituting polymers such as cross-linking density, hydrophilicity, ionic charge, and pKa values and the properties of the surrounding medium such as ionic strength, pH, and counter ions.137,139 Among them, the surrounding pH level is the major factor that controls the swelling behavior of pH-responsive hydrogels. When the surrounding pH value is greater than the pKa value of the acidic pendant group of the polymer chains, the acidic group is ionized (deprotonation) to form fixed negatively charged polymer chains with a positively charged mobile solution.133 This results in an electrostatic repulsion force between the negatively charged chains and thus swelling of hydrogel networks. Similarly, when the surrounding pH value is less than the pKa value of the basic pendant group of the polymer chains, the basic group is ionized (protonation), forming fixed positively charged polymer chains with a negatively charged mobile solution.140 This results in an electrostatic repulsion force between the positively charged chains and thus swelling of hydrogel networks.
Other factors that affect the swelling behavior of pH-responsive hydrogels include the cross-linking density and hydrophobic moieties. The degree of polymer cross-linking controls the overall hydration process in which water or other physiological solutions penetrate through the space between free interstices of polymer chains. A higher degree of cross-linking indicates less room for the solution to stay within the polymer network, allowing for a limited swelling behavior. Hydrophobic moieties also play an important role in the formation of hydrogel networks as they are incorporated to construct amphiphilic hydrogels, consisting of a hydrophilic phase capable of swelling and a hydrophobic phase that aggregates in solution.141,142 As an example, an interpenetrating hydrogel consisting of hydrophilic poly(acrylic acid) [PAA] and hydrophobic poly(butyl acrylate) [PBA], the hydrophobic aggregation of PBA limits swelling and further prevents bursting of the hydrogel at high pH.141 An accurate measurement and monitoring of pH are crucial processes as they play a vital role in controlling the biological functions, microbial activity, and chemical behaviors. pH-Responsive hydrogels are a promising approach for remotely measuring pH levels. Early detection of skin wounds is crucial, as small wounds with minor symptoms may lead to skin necrosis and more severe symptoms if left untreated. Tamayol et al. developed a pH-responsive alginate microfiber system to detect skin damage. Silica dyes were incorporated into the hydrogel, which changed color in response to abnormal pH levels in the range of 7.15–8.93, providing an early warning system for skin damage.143
Although pH-responsive hydrogels have demonstrated numerous applications in various fields ranging from tissue engineering to drug delivery, their relatively low mechanical strength can result in premature dissolution and loss of hydrogel from the intended site. These limitations can be addressed through polymerization, incorporation of reinforcing agents, or through the use of multiple-layer laminates, all of which have the potential to significantly improve the mechanical strength of the hydrogel.
2.5 Enzyme-responsive hydrogels
An enzyme, one of the most vital components in our body, is involved in almost all biological processes. The endogenous enzymes specific to certain tissues and organs serve as biological stimuli with superior selectivity.144 Enzyme-catalyzed reactions are extremely selective for particular substrates, accounting for biological and metabolic processes at the molecular level. Such characteristics that enzymes are ubiquitous in every part of the body and highly specific to the target substrate make them outperform candidate materials for the hydrogel system.Enzyme-responsive hydrogels are prepared either by using intrinsically sensitive natural polymers such as fibrin, collagen, gelatin, and hyaluronic acid or by using biomaterials with enzyme-sensitive linkers.145 By integrating catalyzed reactions of enzymes and responsive polymers, different functional hydrogels can be formed via self-assembly and polymerization reactions. Enzyme-responsive formation and cleavage of chemical bonds are responsible for both construction and degradation of hydrogels used for such different areas as enzyme diagnostic systems, drug delivery systems, and regenerative medicine.146,147
In enzyme-responsive hydrogel systems, a variety of enzymes control the specificity, selectivity, and catalytic efficiency, all of which affect the process of assembly and degradation of hydrogels. Multiple types of enzymes are used in enzyme-responsive hydrogel systems where some of the most frequently used ones include (a) proteases, (b) kinases, (c) phosphatases, and (d) endonucleases.148 Proteases and endonucleases are enzymes that cleave peptides and oligonucleotides, respectively. Abnormally excessive expression of proteases, for instance, is the cause of multiple symptoms including cancer, cardiovascular, inflammatory, neurodegenerative, bacterial, viral, and parasitic diseases. For such reasons, proteases are often considered major targets and stimuli for the selective activation of enzyme-responsive hydrogels. Glycosidases are another type of enzyme that promote the hydrolysis of glycosidic bonds in polysaccharides, allowing them to be ideal targets for carbohydrate-based hydrogels. Among glycosidases, β-mannanase is of particular interest for research as it is secreted by colon microflora and largely localized in the gastrointestinal tract, thereby allowing for colon-targeted drug delivery.149 Not all enzymes cleave and degrade chemical bonds. Transglutaminases, for example, is one of many enzymes that form isopeptide bonds between peptides/proteins.150 By applying such characteristics to the enzyme-responsive hydrogel system, structural strength can be largely enhanced in accordance with the formation of cross-links within the hydrogels.146 Other types of enzymes such as phosphatases and kinases are also frequently used because of their complementary catalytic actions. For instance, kinases are enzymes well known for phosphorylation whereas phosphatases are for dephosphorylation reaction.151 Likewise, different kinds of enzymes as well as enzyme-responsive hydrogels work together in a dynamic manner, forming and deforming hydrogel structures for drug delivery, enzyme sensing, and therapeutic purposes throughout the biomedical research areas.152–154 One promising application of enzyme-responsive hydrogels is in the delivery of growth factors, where the hydrogels are used to sense enzyme at the pathological site and undergo biodegradation, thereby releasing the growth factors, aiding in the tissue regeneration. For instance, Anjum et al. designed a matrix metalloproteinase-responsive hydrogel for delivery of BMP-2 for bone formation.155
Despite the significant advancements in developing novel enzyme-responsive hydrogels with excellent tunable physical and chemical properties, some challenges still remain. These hydrogels exhibit low enzymatic stability and short half-life, which can limit their clinical applications. Further studies are necessary to better understand the enzyme kinetics and swelling behaviors, in order to precisely control drug release and delivery. Furthermore, if the hydrogel relies on enzymes present in the body, detailed studies of enzyme concentration in normal and pathological situations are necessary to optimize their therapeutic efficacy.
2.6 Mechano-responsive hydrogels
Mechanical stimulus is one of the most primitive and easiest stimuli that can be applied to a surface or an object. Mechanical stimuli toward a cell or tissue can be of various types such as compression, tension, shear, shaking, and hydrostatic pressures.156 It has to be noted that, “equal and opposite reaction” is also true in the cellular and tissue microenvironment. The biomaterials such as hydrogels which are implanted in the body experience a variety and combination of mechanical stimuli from the tissues itself.157,158 The hydrogels can be designed to respond to these forces (Fig. 5). Apart from this, the time periods in which the hydrogels could experience mechanical stimuli are when they are being prepared, handled, and implanted. Furthermore, the mechanical stimulus could also be applied to the implanted biomaterial via different strategies, so that the hydrogel could respond and change its intrinsic properties in vivo, to achieve the required outcomes. Altering the intrinsic mechanical or chemical properties of the hydrogel via mechanical stimulation could lead to various outcomes such as strain-stiffening, self-healing, self-growing, shear-thinning, and mechanochromism. Such changes in the properties of the hydrogel can directly affect the mechanosignalling pathways in the cells and tissues, thereby altering the response.159 This enables us to have a spatiotemporal control over biological outcomes such as cell proliferation, differentiation, wound healing, and tissue regeneration. Such hydrogels which could respond to the mechanical stimulus from the body, such as flexion of skin or expansion of lungs, could serve as biosensors to measure various physical activities.160 The mechanosensitive hydrogels are mostly synthesized by a self-assembly process, dynamic cross-linking, and the inclusion of nanostructures.161 The simplest form of mechano-responsive hydrogels changes their behavior or mechanical properties by using a reversible or irreversible conformational change of the polymeric networks or the molecular chains. This can be easily achieved by selecting an appropriate polymer, the concentration of the polymer in the hydrogel, and also the cross-linking densities. When the proper composition is achieved, the polymeric networks act as a spring and can be compressed. By careful choice of cross-linkers and cross-linking density, a reversible shape memory effect can be achieved. Thus these hydrogels can dynamically adjust their mechanical properties and preserve their integrity in accordance with the mechanical stimuli from surroundings.162,163 Such mechanical pressure from the outside can be utilized to trigger a release of drugs or growth factors to achieve a mechano-responsive delivery behavior. The release is achieved when the polymeric networks of the hydrogels are stretched or compressed beyond a limit, which leads to the breakage of a few of the crosslinks, thus releasing the embedded molecules.164 By introducing particles such as LAPONITE® in the polymeric networks, the strength can be greatly increased, sustaining the mechano-responsiveness for multiple cycles.165 One of the major concerns for mechanoresponsive systems is the maintenance of the cross-links or the integrity of the hydrogel network for multiple cycles of triggering. This can be achieved by introducing reversible cross-links by utilizing metal–ligand interactions, dynamic covalent bonds, host–guest interactions, dynamic chemical bonds, boronate linkages, and much more.161,166,167 Another mechanism through which mechano-responsiveness is achieved is shear thinning, wherein, the viscosity of the hydrogel network reduces when sheared but it is regained when shear is removed. This is achieved due to the dynamic cross-links or the non-covalent interactions among the matrix elements.168 Mechanochromism is an interesting property wherein the mechanical stimuli can induce color changes in the mechanophore that is incorporated into the hydrogel network. Some of the molecules that can express mechano-responsive color shifts are spiropyran and rhodamine derivatives.169,170 Such color changes could be easily monitored and quantified to measure the stress–strain experienced by the hydrogels. When mechanochromic hydrogels are employed in skin tissue engineering, they have the potential to alter color based on the contact forces between tissues and hydrogel surfaces. This feature could enable researchers and clinicians to monitor tissue-hydrogel integration and detect hydrogel implant failure. Additionally, as the tissue regenerates and the hydrogel biodegrades, the efficiency of mechanochromism may decrease due to the rupture of internal bonds, thus allowing for effective monitoring of tissue regeneration. Mechanochromic hydrogels could be particularly valuable in cyclically stressed environments such as tendon or cardiac tissue engineering, where they could potentially function as a fracture warning system.171 However, most of the existing mechanochromic hydrogel systems produce color changes within the visible wavelength range, limiting their use for external organs. If researchers devise a means to monitor mechanochromic changes of hydrogels within deeper tissues, it could hold great promise for cardiac and other muscle tissue engineering applications.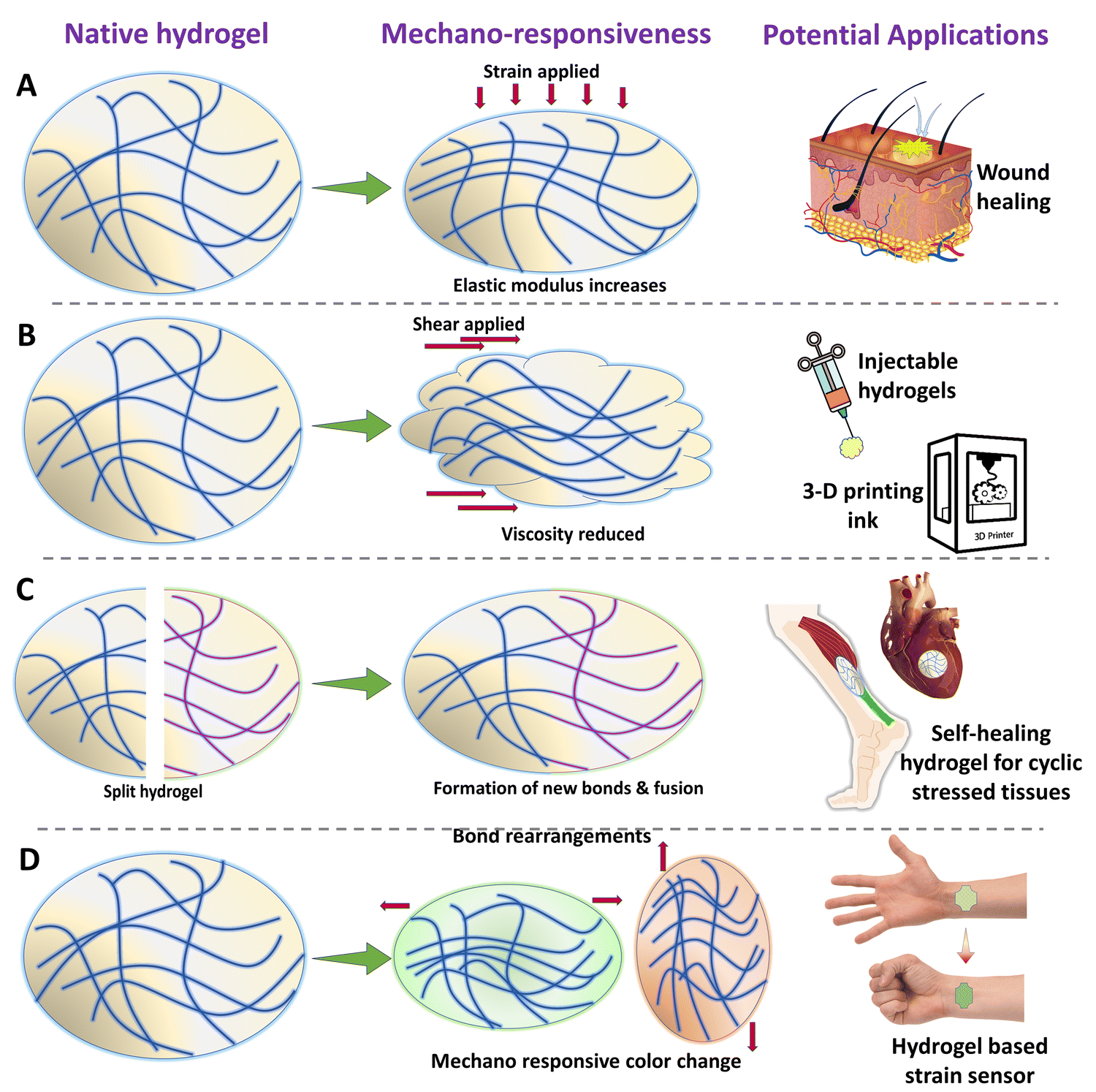 | ||
| Fig. 5 Schematic illustration of various types of mechano-responsive hydrogels and their various bio-medical applications. | ||
2.7 Ultrasound-responsive hydrogels
Ultrasonics have a great advantage in medical applications because they do not need a line of sight environment like the light activation, the ultrasound waves can propagate through the tissues, can be focused, and also the power can be adjusted by changing the frequency and intensity. Ultrasound has already been used widely in clinics for imaging as well as in lithotripsy. Furthermore, it is non-invasive, considered to be safe, and economically a viable option for stimulation.172 Due to these various advantages and a widely available medical appliance, researchers have shown interest in developing materials that could respond to ultrasound waves. By utilizing an ultrasound-responsive hydrogel, technically, researchers can stimulate the hydrogel to undergo phase changes, to break down, to release drugs or molecules upon triggering, and further ultrasonic image the defect filling and tissue regeneration.173,174 Such a diagnostic, therapeutic and monitoring approach proves to be a great advantage of ultrasound-responsive hydrogels.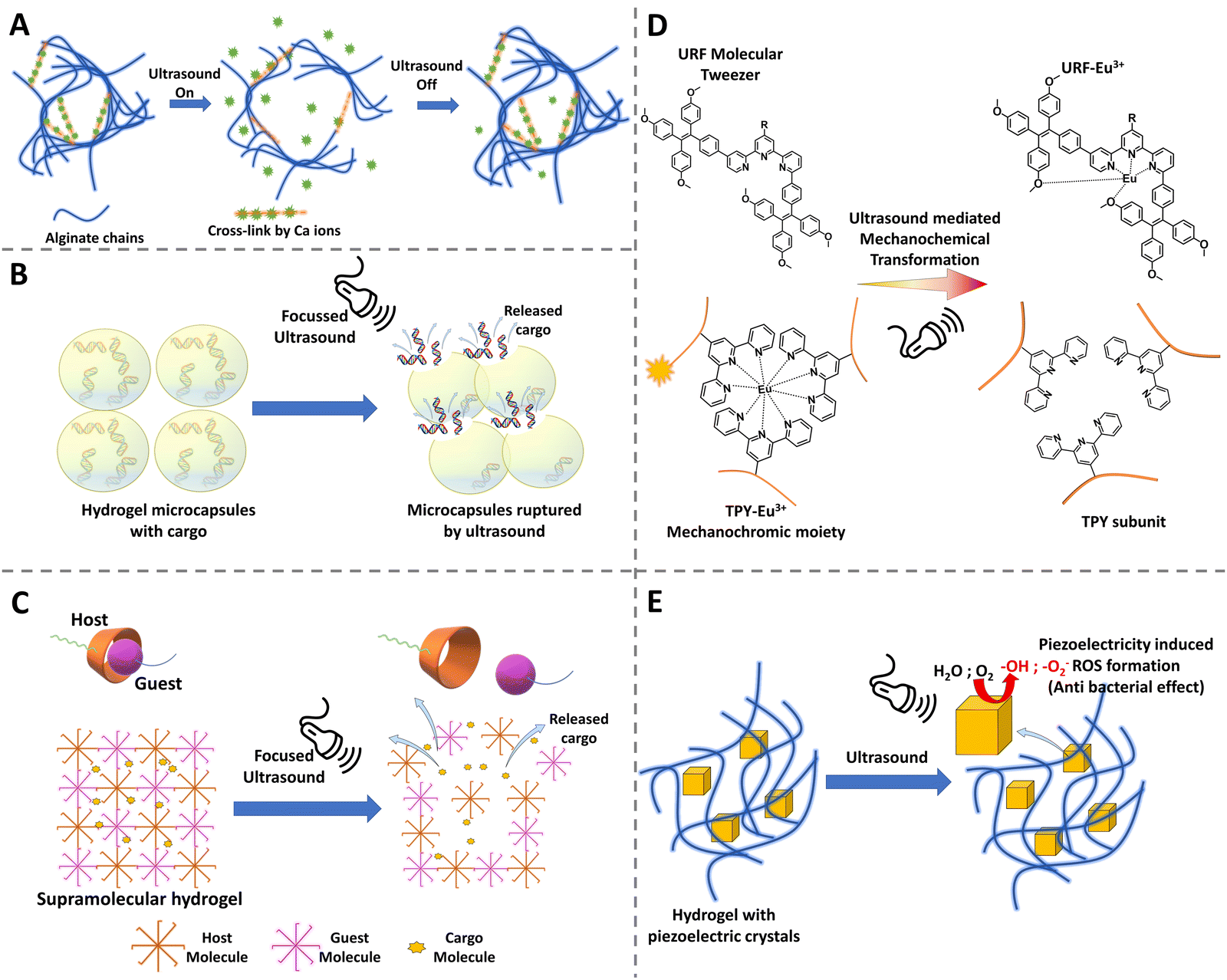 | ||
| Fig. 6 Schematic diagram showing various modalities of ultrasound-triggered responsiveness in hydrogels. (A) Disruption of cross-links to release the cargo; (B) ultrasound cavitation waves rupture the hydrogel microcapsules leading to the release of cargo; (C) disruption of the host–guest interaction in a supramolecular hydrogel thereby delivering the cargo; (D) mechanochromic/chemical transformation by disruption of metal–ligand complexes, leading to change in fluorescence; and (E) induction of piezoelectricity in the piezoelectric crystals incorporated into the hydrogel matrix resulting in ROS production and thus anti-bacterial effect. | ||
2.8 Dual and multiple stimuli-responsive hydrogels
As seen from the above sections, hydrogels can be made responsive to various stimuli according to the need and application. However, each of the stimulatory methods has its advantages and disadvantages. It could be understood that some of the stimulatory methods such as photostimulation could not be used for deeper tissues; however, such chemistries might be much more suitable for deep-tissue applications. To mitigate such circumstances, researchers have opted for dual or multiple stimulatory mechanisms for making the hydrogel dynamic.184,185 For example, a pH-sensitive hydrogel could also respond to thermal or ultrasonic stimulation and so forth. Some of the commonly used triggering systems for hydrogels include light, temperature, ultrasonic waves, magnetic field, electric field, ionic composition, enzymes, reactive oxygen species, matrix metalloproteinases, and glucose and the list keeps ever evolving. Furthermore, by incorporating multiple stimuli triggers in a hydrogel system, it is possible to achieve a great spatiotemporal control throughout the therapy. One of the easiest ways to incorporate multiple stimuli triggers is by carefully combining the already existing single trigger systems into a composite hydrogel system.186 For example, some of the thermo-responsive hydrogels could be easily modified to respond to light stimuli by adding appropriate functional groups. Many hydrogels exhibit shear thinning behaviors, yielding mechano-responsive properties. By the simple addition of thermo-responsive elements such as glycerol phosphate or PNIPAAm, the already existing certain shear-thinning hydrogels could be easily made to respond to thermal stimuli also. Similarly, by incorporating electrically conducting elements such as polymers, and metal nanoparticles, the existing hydrogels can be transformed into electrically responsive hydrogels.187 By incorporating magnetic colloidal particles into the hydrogel matrix, the whole system can be controlled by the external magnetic field to form various structures.188,189 In the recent approaches, most of the stimuli-responsive hydrogels exhibit one or more dynamic responses, which could be seen in the upcoming examples. These multi-triggered hydrogel systems often exhibit the properties of shape-memory, self-healing, and much more complex behaviors.190 Utilizing a single molecule for multiple responsive systems is also an emerging trend in the field. For example, spiropyran is usually used in photo-triggering hydrogels. However, it can also respond to heat or a higher wavelength of light, exhibiting multiple responsiveness.191–193 Similarly, by designing coumarins and hydrazone linkages in a single molecule, the hydrogel system can be made to respond to photo stimulation, pH changes and also to the presence of metal ions.194 By combining the magnetic field and iron oxide nanoparticles, an electric field can be induced in these particles leading to a thermal response leading to the phase change in the polymer matrix. Thus, by carefully choosing the particles and the polymer matrix, magnetic, electric and thermo-responsive hydrogels could be made.195,196 When utilizing magnetic fields and metallic nanoparticles for tissue engineering applications, caution must be exercised due to the potential for adverse effects. Exposure to high and uncontrolled magnetic fields can lead to localized hyperthermia, which can cause tissue damage. To address this issue, nanoparticles can be designed to respond only to specific pulses or a specific combination of pulses, thus limiting inadvertent triggering due to external fields. Furthermore, the presence of metallic nanoparticles is a major drawback of this system, as they may be difficult to clear from the tissue and could pose a risk in long-term implantations. Ultrasonic waves could also be utilized to induce electric fields in a piezoelectric crystal embedded inside the hydrogel, thus making the hydrogel system responsive to both stimuli.197 Thus, various triggering modalities can be carefully combined to produce hydrogels with multiple and interesting triggering mechanisms that could be used for enhancing the diagnostic and therapeutic outcomes.3. Tissue engineering applications of stimuli-responsive hydrogels
3.1 Cardiac tissue
Cardiac tissues are one of the most active and dynamic tissues in the body, which continuously undergo contraction and relaxation, and are subjected to regular electrical stimulations, and further experience the pressure from the blood that is being pumped. It is also well-known that cardiac diseases are one of the major causes of death worldwide. This has prompted researchers to develop biomaterials that could mimic the dynamics of the cardiac tissue, which could lead to better regeneration and 3-D in vitro models. Apart from mimicking the properties of the native tissue, researchers could also dynamically control the hydrogels to express various other properties such as enhanced cell attachment, the release of drugs, growth factors, and even cells. Early works include thermosensitive hydrogels made from PNIPAAm that were utilized to deliver cells to the cardiac tissues. These hydrogels were modified with single-walled carbon nanotubes and the adipose stem cells were embedded within the hydrogel. This hydrogel can respond to the temperature and could become a solid gel at the body temperature of 37 °C.198 This thermo-responsive hydrogel was able to improve cellular adhesion. This system can be further improved to respond to both thermal and pH stimuli by combining with polyacrylamide. The underlying concept is that such pH and thermosensitive hydrogels could sense the slightly acidic pH of an infarcted heart (pH 6–7) and solidify at the required site.199 Pedron et al. were able to construct a bilayer polymeric hydrogel, in which only one layer was thermosensitive and the cells were embedded in the non-sensitive layer. Due to this configuration, the hydrogel could roll into a cylinder at a low temperature, thereby offering protection to the cells during the delivery stage. However, under physiological conditions, upon exposure to sensitive temperature, the bilayer construct can unfurl and release the cells at the required site.200 Instead of organic polymers, a bio-polysaccharide-based thermo-stimulative hydrogel has also been developed using chitosan and glycerol phosphate. Here, an opposite charge-based interaction leads to the formation of thermo-regulated gelation of chitosan chains. Such a system has been utilized to deliver embryonic stem cells to infarcted heart tissue of rats.201 This temperature-responsive hydrogel system improved cell implantation and heart functions in the infarcted region. Researchers have also incorporated gold nanoparticles into this hydrogel system to enhance its electroconductivity, enabling its use in cardiac regenerative applications.202 Chen et al. have utilized a similar system to deliver fibroblast growth factors to the myocardial infarct size in mice models. This delivery of growth factors using a thermo-stimulated delivery system was able to result in improved prognosis.203 Recently, chitosan has been gelled together with glycerol phosphate and vitamin C to reduce the reactive oxygen species, thus proposed as a promising strategy in ischemic infarcts. This hydrogel was able to increase cardiomyocyte survival and adhesion.204 Apart from the thermally stimulated hydrogels, recently there has been a lot of progress in hydrogels that could be exhibit multiple dynamics such as mechanical and electrical coupling, in order to withstand the continuous systolic and diastolic compressions of the heart by self-adaptation and also improved electrical conduction. Such a system was developed using self-doping polyaniline and gelatin mixtures. Furthermore, these were modified with borate side chains and mixed with polyvinyl alcohol to form a mechanical and electrically coupled hydrogel (Fig. 7).205 The dynamic covalent bonds and the reversible interactions among the molecules provided this hydrogel with a self-adaptive resilience which led to the inhibition of ventricular dilation in myocardial infarction and also the maintenance of stroke volume. Light-responsive hydrogel systems have also been developed to aid in cardiac regeneration. Electroconductive polymers such as tetraaniline, PEDOT:PSS, polypyrrole and combinations of these with other biopolymers have been utilized to synthesize electrically responsive hydrogels. These hydrogels help to maintain/improve the rhythm of the heartbeat, thereby enhancing the heart contractions while enhancing the adhesion of cardiomyocytes.206–208 Similarly, hydrogels containing carboxy betaine zwitterions can also respond to electrical impulses and can repair cardiac functions due to their conductivity. Such electro-responsive zwitterionic hydrogels have shown decreased fibrosis areas and recovery of infarcted heart tissues.209 Interestingly, black phosphorous has been used in conjugation with polydopamine and GelMA hydrogels, exploiting the high electrical conductivity and also non-toxic degradation products.210 Apart from these conventional stimulative systems, researchers have come up with an interesting hydrogel incorporating reactive oxygen species-cleavable boronate bonds. These hydrogels can be stimulated by the ROS at the inflammatory infarct site and release nitroxide radicals to counteract the oxidative stress and simultaneously release vascular endothelial growth factors which could enhance the cardiac outcomes.211 Similarly, Cheng et al. have developed a pH/H2O2 and matrix metalloproteinase responsive gel system to deliver the extracellular vesicles for improving cardiac functions.212 Chen et al. developed a supramolecular hydrogel, which is a β-galactosidase (β-gal) enzyme-responsive hydrogel, for delivering curcumin and nitric oxide (NO) donors.213 In the MI model in mice, tail vain injection of β-gal and orthotropic implantation of hydrogel showed restoration of heart functions as shown from the improvement of fraction shortening and ejection fraction. Apart from the conventional light-triggered or light-responsive hydrogel systems, Jing-jing et al. have fabricated a near infrared-II responsive hydrogel system that releases the Mir-196c-3p microRNAs.214 The utilization of NIR-II triggered hydrogel systems allows for the deeper tissue penetration of light and also provides a photothermal effect. This hydrogel has been proposed as a promising on-demand delivery system for treating ischemic reperfusion injury.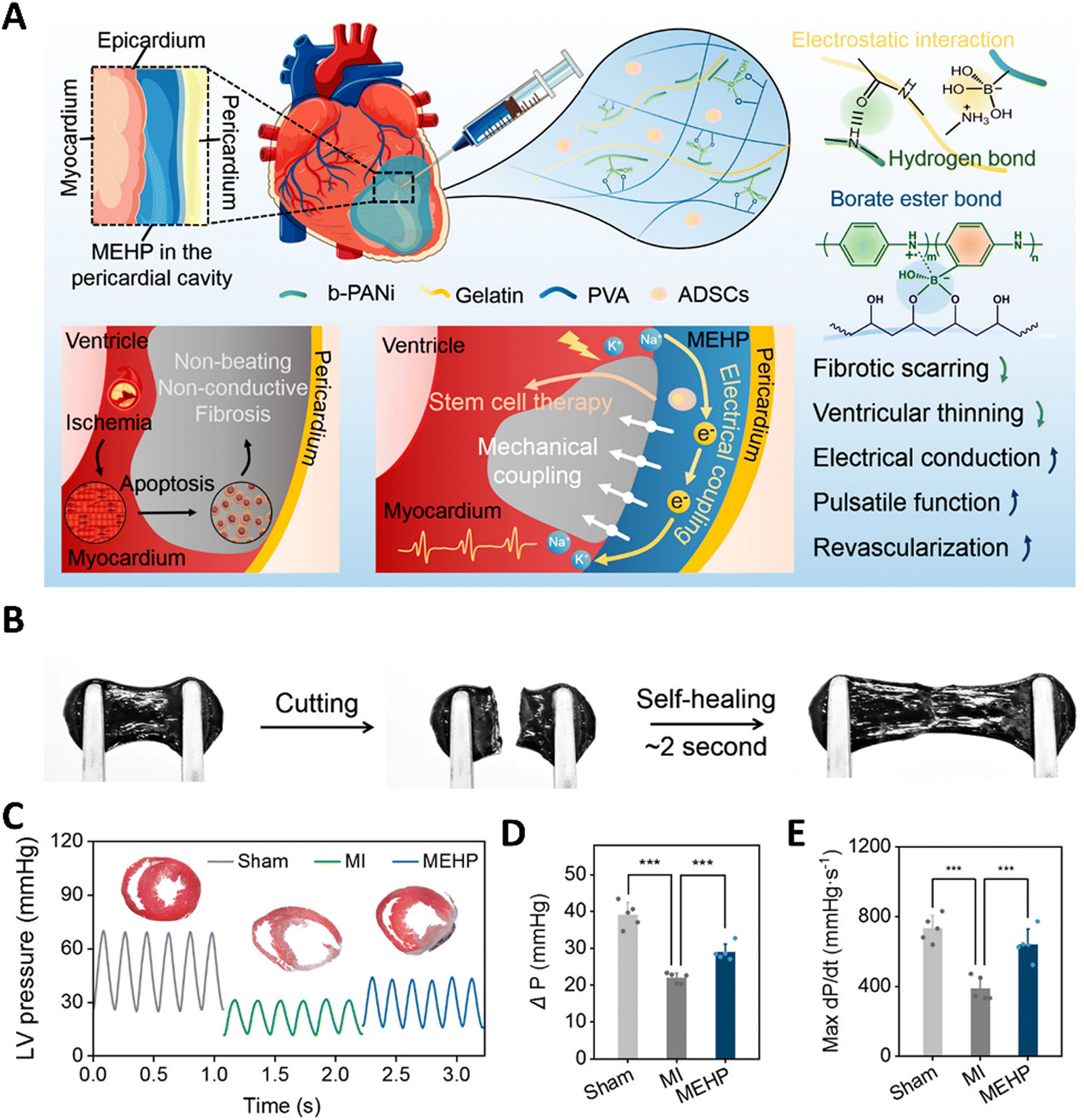 | ||
| Fig. 7 A mechanical–electrical coupled hydrogel patch (MEHP) for treating infarcted myocardium. (A) Schematic diagram of an intrapericardial injectable MEHP. MEHP's cross-linking mechanisms involved reversible noncovalent interactions and dynamic borate ester bonds. When combined with ADSCs, MEHP inhibited fibrotic scarring and ventricular thinning, while enhancing electrical conduction, pulsatile function, and revascularization through mechanical and electrical coupling with the infarcted myocardium. (B) Self-healing property of hydrogel. (C–E) Improvement in heart functions after the application of MEHP. Reproduced with permission from ref. 205. Copyright © 2022, American Chemical Society. | ||
3.2 Bone tissue
Unlike some of the soft tissue like the heart and neurons, bone is mechanically a rigid material, due to its mineral content. However, due to the presence of organic matrix, the bone is also stiff and these organic matrices allow for the cells to reside and remodel the bone as it grows. With these criteria as a priority, researchers try to mimic the mechanical properties of the natural bone through various strategies. When the biomaterial's mechanical, biochemical, and microscopic architectural properties closely resemble those of native bone tissue, a higher bone regeneration could be envisioned due to the activation of various pathways. Such a biomimicking approach could be achieved by mostly incorporating ceramic components, viz., calcium phosphate minerals, into the hydrogel system and delivering it to the bone defect site. However, when incorporating such inorganic mineral components, it becomes difficult to place/fill the biomaterial in the bone defects completely. To rectify these shortcomings, researchers have used various other approaches to utilize soft materials such as scaffolds, powders, or hydrogels to bring out maximum bone regeneration. Hydrogels could also carry various tissue regenerative molecules such as drugs, growth factors, microRNAs, genes, and cells, that could aid in regeneration. However, delivering these through regular scaffolds or gels could lead to rapid delivery and exhaustion of these molecules during various phases of regeneration. Therefore, researchers have resorted to the use of responsive or dynamic hydrogels, which could be made to respond to various external stimuli as discussed in the previous sections.215 Another advantage of dynamic hydrogels is that they can undergo a phase change from a flowable to a solid state under various stimuli, thus enabling the complete filling of bone defects, which might be impossible when high amounts of minerals are implanted via conventional methods such as powders, pellets, or scaffolds. For example, a thermosensitive injectable gel system was developed by Huang et al., for the healing of tendon–bone junctions.216 This hydrogel was synthesized using a combination of methylcellulose, polyvinyl alcohol, and polyvinylpyrrolidone which imparted thermo-sensitive properties. Furthermore, this hydrogel was loaded with bioactive glass. The hydrogel showed self-healing properties by solidifying at 37 °C and also slowly released kartogenin which helped in fibrocartilage and bone regeneration. As seen earlier, a chitosan glycerol phosphate thermosensitive hydrogel system has been used to deliver alendronate in a controlled manner for the prospective treatment of osteoporosis.217 A PEG–PCL–PEG hydrogel system also exhibited a thermosensitive behavior which was used in conjunction with an acellular bone matrix to enhance bone regeneration in rabbits.218 A PNIPAm-based polymeric hydrogel was fabricated along with chitosan and graphene oxide, which was thermosensitive as well as injectable. This hydrogel had improved mechanical properties and enhanced osteogenic differentiation.219 A dipyridamole-loaded PCL and silk fibroin-based thermosensitive hydrogel was synthesized by Taymouri et al., using a chitosan glycerol phosphate system. This system exhibited the highest mechanical strength with the addition of silk fibers and enhanced osteogenic differentiation.220 Zhao et al. synthesized a pH-responsive composite hydrogel composed of carboxymethyl chitosan and amorphous calcium phosphate.221 The hydrogel's pH-responsiveness was achieved using glucono δ-lactone as an acidifier. A combination of BMP-9 overexpressing cells and the hydrogel showed that the hydrogel improves both the effectiveness and maturity of BMP9-induced bone formation in vivo. Motasadizadeh et al. developed a pH-sensitive silk fibroin/sodium alginate hydrogel scaffold loaded with teicoplanin and phenamil-loaded silk fibroin nanoparticles were developed to treat chronic osteomyelitis.222 The developed system exhibited sustained and pH-sensitive drug release with higher rates observed in alkaline pH. In vivo evaluations in methicillin-resistant Staphylococcus aureus-infected rat bones demonstrated promising effectiveness with lower infection and higher regeneration compared to other treatment groups. A hybrid hydrogel system with NIR light stimulation leading to a photothermal effect was synthesized with gelatin, PMMA and polydopamine. This system proved that NIR stimulation enhanced bone repair in the rat skull.223 Another injectable hydrogel system was prepared by compositing calcium phosphate nanoparticles, poly(dimethylaminoethyl methacrylate-co-2-hydroxyethyl methacrylate), indocyanine green along with parathyroid hormones. When triggered with NIR light, this system was able to dynamically release pulses of parathyroid hormones, which proves to be effective in bone repair in ovariectomized rats.224 Hydrogels that could respond to electrical and magnetic fields have been synthesized and successfully demonstrated the effectiveness for bone tissue regeneration (Fig. 8). These hydrogels are typically made of a polymeric matrix and electromagnetic field conducting elements such as graphene oxide and magnetic nanoparticles such as Fe3O4.225–228 It was shown that a pulsed electromagnetic field applied to a responsive hydrogel has a great potential in enhancing the bone regeneration. Aziz et al. developed a hydrogel, containing a matrix metalloproteinase (MMP)-sensitive peptide cross-link and RGD, and demonstrated improved IDG-SW3 osteoblast to osteocyte differentiation and mineralized collagen matrix formation compared to nondegradable hydrogels and standard 2D culture.229 This study showed that the susceptibility of hydrogels to cell-mediated degradation promoted osteocyte differentiation and mineralized collagen matrix formation. Yang et al. developed an injectable MMP1-sensitive hydrogel microsphere hydrogel (LDL-MMP1/GelMA) that could release exosomes in response to MMP1 expression.230In vitro and in vivo experiments demonstrated that the developed hydrogel could promote the migration and osteodifferentiation of BMSCs and recruit CD90+ stem cells via neovascularization. Similarly, the ultrasound triggered release of growth factors and the pH stimulated release of antibiotics, growth factors and cells using the dynamic behavior of hydrogels have also been demonstrated.231–234 Thus, with the advent of the stimuli responsive dynamic hydrogels, bone tissue regeneration has seen vast advancements.235,236 The ability to control the release of specific bioactive molecules at specific time helps the researchers to control the bone regeneration.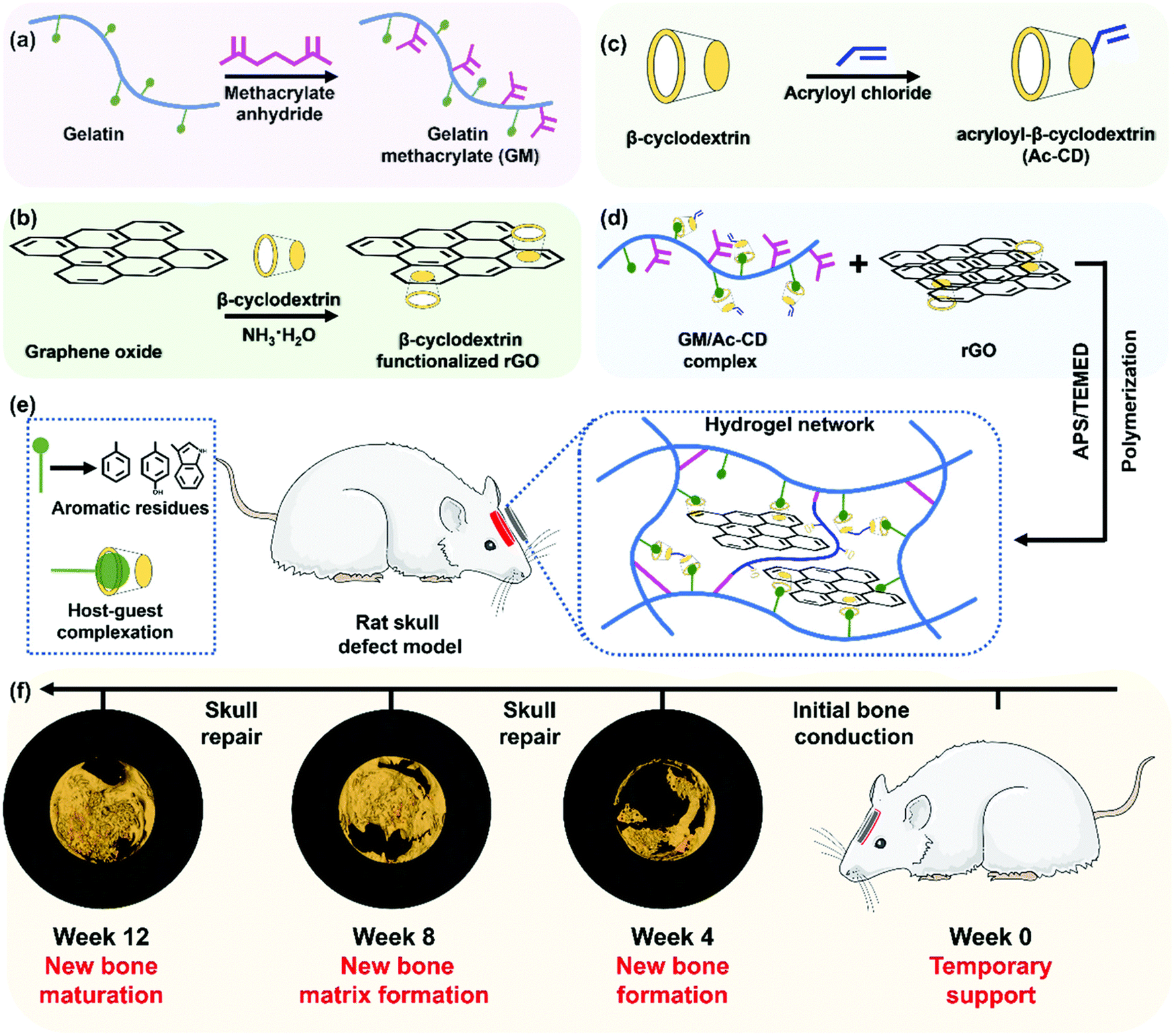 | ||
| Fig. 8 Figure showing the synthesis of an electroconductive and photo-sensitive composite hydrogel for bone defect repair. Reproduced with permission from ref. 225. Copyright © 2022, Royal Society of Chemistry. | ||
3.3 Neural tissues
Neural tissue loss can occur due to various reasons such as trauma, degenerative diseases, spinal cord injury, and traumatic brain injury which can cause a great limitation in the functionality of day-to-day life. Furthermore, the regenerative capacity of neural tissues is limited owing to the scar formation and also the non-recovery of sensory and motor functions. This can be debilitating and lead to poor quality of life. Numerous clinical attempts have been made to improve the functional neural connections between the severed ends of the nerve tissues by close suturing or gluing. Researchers are developing strategies to promote neural regeneration by using relevant scaffolds, which could serve as bridges for the neural tissues to grow. Similar to the cardiac tissues, neuronal tissues, and cells depend on continuous electrical stimulations and conduction. Apart from these, the neuronal tissues also need various growth factors, small molecules, cells and molecules that are necessary for synaptic communications, throughout the regenerative process. Therefore, researchers have worked on various ways to supplement these requirements using hydrogels that could respond to various stimuli and also controllable delivery systems, that could achieve successful and functional neuronal tissue.237 Furthermore, hydrogels could also be stimulated to form guidance conduits in the form of fibers, after the application of certain stimuli, thus making them more functional for neuronal regeneration.238 Similarly, hydrogels can also be stimulated to change their physical properties such as stiffness, to match the relevant stiffness for maximum survival and regeneration of neural populations. For example, a material stiffness of around 1 kPa was found to be suitable for the survival of various types of neural cells.239 The electrical conductivity could also be implemented in the hydrogels, so that the cells which are growing could establish synaptic connections among one another and also with the healthy populations, thereby completing a fully functional neural circuit.As seen in the previous examples of stimuli-responsive hydrogels, a composite hydrogel comprising chitosan, cellulose, hyaluronic acid, and glycerol phosphate was synthesized by Yao et al., which had thermoresponsive properties. This hydrogel could solidify when exposed to a body temperature of 37 °C and remained liquid below 25 °C. It was used to deliver human umbilical cord mesenchymal stem cells in a traumatic brain injury rat model and was found to support the survival of the cells and enhance the recovery of neurological functions.240 A peptide-based hydrogel system was fabricated by Adak et al., with further modifications including a sulfo group for ECM mimicking and a MMP9 cleavable unit. This hydrogel could be injected into the injured brain site and in the presence of MMP9, it could release the neuroprotective peptide, thus promoting the healing of neurons.241 Zheng et al. developed a thermo-responsive hydrogel based on the PLGA–PEG–PLGA system for the delivery of baricitinib, a JAK1/2 inhibitor. This system was able to enhance the microglia formation and also helped in the neurological recovery in the rat spinal cord injury model.242 Wei et al. developed poly(ethylene glycol)-co-polyvaline (mPEG-PLV) and grafted it with tetraniline moieties, resulting in an electroactive and thermosensitive polymer hydrogel matrix. They further loaded nerve growth factors into this matrix and applied it for treating spinal cord injury in rats. Furthermore, transcutaneous electrostimulation was also delivered using electrode needles. This resulted in endogenous neurogenesis and also helped in the recovery of motor function (Fig. 9).243 Similarly, they designed and fabricated a semi-interpenetrating network of hydrogels using poly(N-isopropylacrylamide-co-N-isopropylmethacrylamide) and the conducting polymer polythiophene. The authors showed that this hydrogel could also be used for 3-D printing applications, thus proposing this electroconductive polymer for neural tissue applications.244 Bhattacharyya et al. were able to synthesize a gelatin–genipin hydrogel impregnated with iron oxide nanoparticles. This magnetic field sensitive hydrogel was implanted in a contusion spinal cord injury rat model. With the addition of magnetic field exposure, it was found that there was a significant improvement in spinal reflexes and electrophysiological markers as well as behavioral improvements indicating that magnetic stimulatory hydrogels could enhance neural repair.245 Recently, a magnetic and thermo-responsive hydrogel system was fabricated using chitosan, glycerol phosphate, and iron oxide nanoparticles and was also loaded with dexamethasone. This preparation along with the magnetic field exposure was able to reduce the inflammation of sciatica in rats.246 Tran et al. developed a peptide-based hydrogel system, which could self-assemble. By incorporating magnetic carbonyl iron particles, they were able to control the alignment of the fibrous matrix under the influence of an external magnetic field, and this alignment proved to be helpful in directing the axonal infiltration and also improved axonal growth.189 Thus, various researchers have shown the potential of hydrogels that are sensitive to various stimuli. These stimuli-responsive hydrogels could be exploited by combining various modalities, allowing for the delivery of growth factors on demand.247,248 Systems such as magnetically activated hydrogels could be advantageously applied in clinical settings because of the control and ability to focus the magnetic fields precisely, thus improving the expected outcomes.249
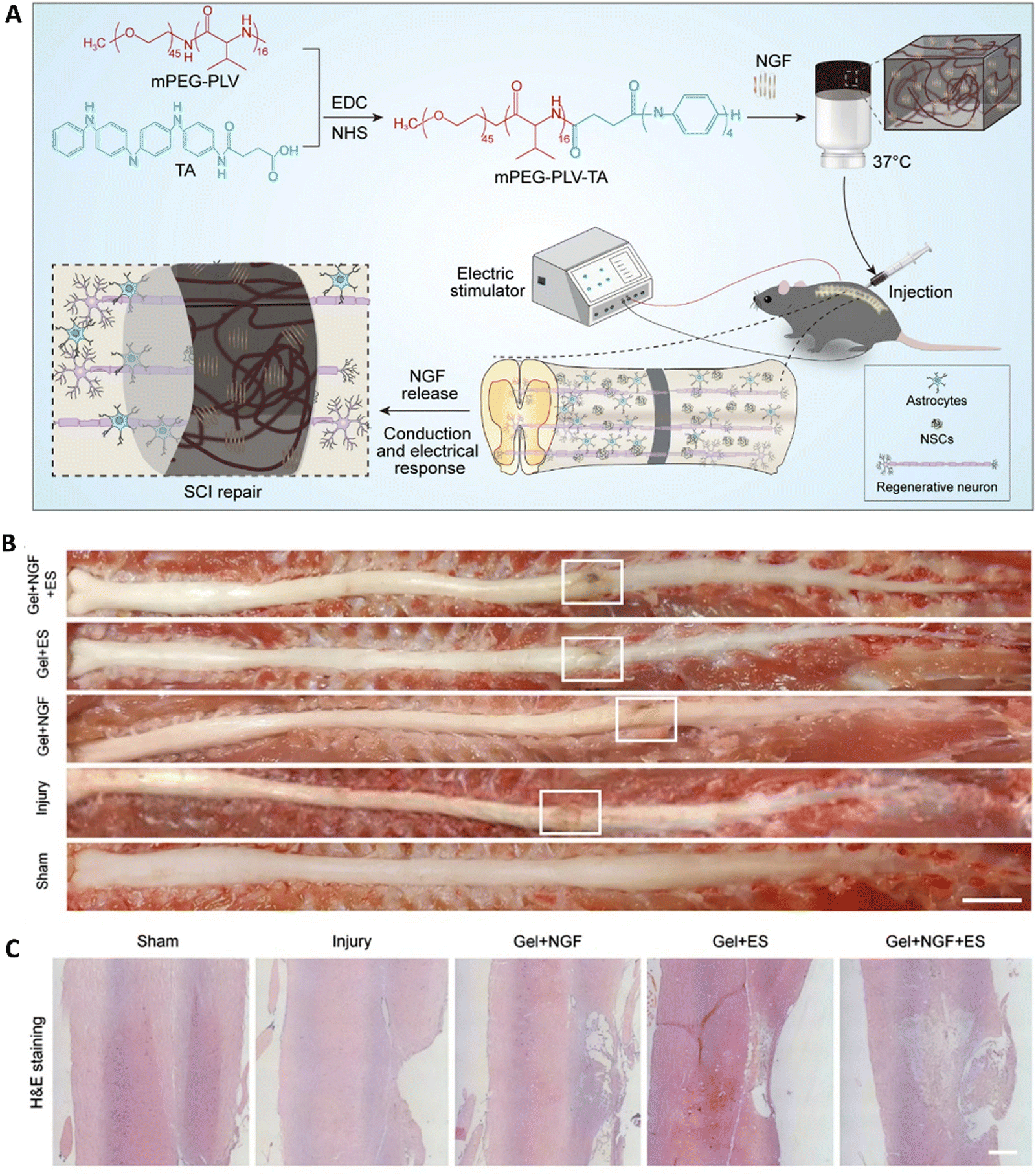 | ||
| Fig. 9 A thermosensitive electroactive hydrogel for spinal cord injury repair. (A) Schematic diagram showing the synthesis and the outline of the in vivo model. (B) Gross morphology of repaired spinal cord injury in various groups and (C) the corresponding H&E sections. Reproduced with permission from ref. 243. Copyright © 2021, Springer Nature. | ||
4. Conclusion and future outlook
The field of stimuli-responsive hydrogel is an emerging field with developments forming new benchmarks in the advanced and bio-functional platforms providing numerous advantages over traditional hydrogel systems. As overviewed here, numerous designs of the hydrogel that respond to various stimuli provides a unique opportunity to carefully design a hydrogel for tissue regenerative application. Additionally, various stimuli-response elements can be rationally added to the hydrogel, providing a multi-functional intelligent hydrogel. Recent updates on tissue regenerative applications, especially in bones, cardiovascular tissue, and nerves using stimuli-responsive hydrogel are provided.Even though many studies have been discussed here, none of the stimuli-responsive hydrogels has received regulatory clearance for use in humans. It is expected that these hydrogels would face severe challenges in translating these platforms to human applications. For example, many hydrogels were designed to respond to endogenous stimuli under pathological conditions. However, there is a lack of robust physiological data during disease progression or in the natural healing cascade of the injured site and there is a lot of patient-to-patient variation in terms of the concentration of the biological molecules and their presence. In this situation, a single stimuli-responsive hydrogel might fail and that is when multi-stimuli-responsive hydrogels can rescue us from the problem by increasing the sensitivity of the hydrogel platform. Another aspect that needs to be considered is that by the introduction of various chemical moieties as stimuli-response elements, the biocompatibility and the degradation profile need to be studied in detail. Furthermore, stimuli-responsive hydrogels implanted in vivo can exhibit variable degradation rates and loss of responsiveness, attributable to material-related and in vivo-related factors. Material-related factors, such as the properties of the precursor materials selected (e.g., hardness, elasticity, hydrophobicity, and degradability), can be adjusted by compositing materials to achieve desired parameters. In vivo factors that influence hydrogel viability in tissue engineering include tissue microenvironmental conditions (e.g., pH, tissue damage, blood and nutrient supply), underlying pathologies, genetic conditions and the individual's immunological response. The type of tissue being regenerated also affects the duration of hydrogel implantation, and matching of the degradation profile of the hydrogel to the regenerative profile of the tissue is essential. For example, skin tissue regenerates faster than bone tissue, requiring hydrogels that last for a few weeks, whereas hydrogels for bone regeneration must last for a few months. By optimizing material properties and controlling in vivo factors with necessary treatment modalities, researchers and clinicians can achieve optimal hydrogel degradation profiles tailored for specific needs. Many researchers are working to overcome these problems for clinical translation. For example, Calder et al. developed a PNIPAm-based hydrogel to study agnostic tissue regeneration and they were able to demonstrate the potential of this hydrogel system through limited clinical studies.100 Moving forward, further developments in such platforms require scientists from chemical, biological, and pharmaceutical backgrounds for an effective clinical translation. With ongoing efforts addressing these challenges, the potential of stimuli-responsive hydrogels for clinical translation in tissue regenerative applications is promising.
Author contributions
Sivashanmugam Amirthalingam: writing – original draft, visualization, and conceptualization. Arun Kumar Rajendran: writing – original draft, visualization, and conceptualization. Young Gi Moon: writing – original draft. Nathaniel S. Hwang: supervision, writing – review and editing, and funding acquisition.Conflicts of interest
The authors declare that they have no conflict of interests.Acknowledgements
This work was supported by the National Research Foundation of Korea [NRF] grant funded by the Korean government (MSIP) (NRF-2021R1A2C2008821). This work was also supported by the Basic Science Research Program through the NRF funded by the Ministry of Education (NRF-2022R1I1A1A01072365). Arun Kumar Rajendran was supported by the Brain Pool program funded by the Ministry of Science and ICT through the National Research Foundation of Korea (2020H1D3A1A04081286).References
- P. Sanchez-Cid, M. Jimenez-Rosado, A. Romero and V. Perez-Puyana, Polymers, 2022, 14, 3022 CrossRef PubMed.
- M. X. Li, J. X. Lv, Y. Yang, G. P. Cheng, S. J. Guo, C. C. Liu and Y. Ding, Gels, 2022, 8, 624 CrossRef CAS PubMed.
- I. M. El-Sherbiny and M. H. Yacoub, Glob. Cardiol. Sci. Pract., 2013, 2013, 316–342 Search PubMed.
- A. Kumar and S. S. Han, Int. J. Polym. Mater. Polym., 2017, 66, 159–182 CrossRef CAS.
- H. B. Zhao, M. Liu, Y. J. Zhang, J. B. Yin and R. J. Pei, Nanoscale, 2020, 12, 14976–14995 RSC.
- R. Kouser, A. Vashist, M. Zafaryab, M. A. Rizvi and S. Ahmad, Mater. Sci. Eng., C, 2018, 84, 168–179 CrossRef CAS PubMed.
- N. S. Kehr, S. Atay and B. Ergun, Macromol. Biosci., 2015, 15, 445–463 CrossRef CAS PubMed.
- S. Yue, H. He, B. Li and T. Hou, Nanomaterials, 2020, 10, 1511 CrossRef CAS PubMed.
- A. Sivashanmugam, R. A. Kumar, M. V. Priya, S. V. Nair and R. Jayakumar, Eur. Polym. J., 2015, 72, 543–565 CrossRef CAS.
- Z. Y. Li, Y. Z. Zhou, T. Y. Li, J. J. Zhang and H. Tian, View, 2022, 3, 20200112 CrossRef CAS.
- H. M. El-Husseiny, E. A. Mady, L. Hamabe, A. Abugomaa, K. Shimada, T. Yoshida, T. Tanaka, A. Yokoi, M. Elbadawy and R. Tanaka, Mater. Today Bio, 2022, 13, 100186 CrossRef CAS PubMed.
- Z. Wei, R. Schnellmann, H. C. Pruitt and S. Gerecht, Cell Stem Cell, 2020, 27, 798–812 CrossRef CAS PubMed.
- A. Farrukh, J. I. Paez and A. del Campo, Adv. Funct. Mater., 2019, 29, 1807734 CrossRef.
- W. Kim, C. H. Jang and G. H. Kim, Nano Lett., 2019, 19, 8612–8620 CrossRef CAS PubMed.
- B. D. Ulery, L. S. Nair and C. T. Laurencin, J. Polym. Sci. Polym. Phys., 2011, 49, 832–864 CrossRef CAS PubMed.
- L. Li, J. M. Scheiger and P. A. Levkin, Adv. Mater., 2019, 31, 1807333 CrossRef PubMed.
- V. X. Truong, F. Y. Li, F. Ercole and J. S. Forsythe, ACS Macro Lett., 2018, 7, 464–469 CrossRef CAS PubMed.
- S. Pearson, J. Feng and A. del Campo, Adv. Funct. Mater., 2021, 31, 2105989 CrossRef CAS.
- S. Ramasundaram, S. Sobha, G. Saravanakumar and T. H. Oh, Polymers, 2022, 14, 5134 CrossRef CAS PubMed.
- G. Lee, H. E. Choi, S. H. Hong, M. Choi, D. W. Han, J. Lee, K. S. Kim and S. K. Hahn, Adv. Drug Delivery Rev., 2022, 188, 114419 CrossRef CAS PubMed.
- A. M. Kloxin, A. M. Kasko, C. N. Salinas and K. S. Anseth, Science, 2009, 324, 59–63 CrossRef CAS PubMed.
- Y. Luo and M. S. Shoichet, Nat. Mater., 2004, 3, 249–253 CrossRef CAS PubMed.
- M. C. Koetting, J. T. Peters, S. D. Steichen and N. A. Peppas, Mat. Sci. Eng. R, 2015, 93, 1–49 CrossRef PubMed.
- C. A. DeForest and K. S. Anseth, Nat. Chem., 2011, 3, 925–931 CrossRef CAS PubMed.
- D. Bennet and S. Kim, Analyst, 2015, 140, 6343–6353 RSC.
- Y. J. Zheng, Z. J. Chen, Q. Y. Jiang, J. Feng, S. Wu and A. del Campo, Nanoscale, 2020, 12, 13654–13661 RSC.
- M. A. Azagarsamy, D. D. McKinnon, D. L. Age and K. S. Anseth, ACS Macro Lett., 2014, 3, 515–519 CrossRef CAS PubMed.
- M. A. Azagarsamy and K. S. Anseth, Angew. Chem., Int. Ed., 2013, 52, 13803–13807 CrossRef CAS PubMed.
- S. Takamori, S. Yamaguchi, N. Ohashi and T. Nagamune, Chem. Commun., 2013, 49, 3013–3015 RSC.
- V. X. Truong, F. Y. Li and J. S. Forsythe, ACS Appl. Mater. Interfaces, 2017, 9, 32441–32445 CrossRef CAS PubMed.
- N. K. Mal, M. Fujiwara and Y. Tanaka, Nature, 2003, 421, 350–353 CrossRef CAS PubMed.
- Q. Zhang, N. R. Ko and J. K. Oh, Chem. Commun., 2012, 48, 7542–7552 RSC.
- Y. Qiu and K. Park, Adv. Drug Delivery Rev., 2001, 53, 321–339 CrossRef CAS PubMed.
- Q. S. Zhang, B. J. Chen, L. Tao, M. Y. Yan, L. Chen and Y. Wei, RSC Adv., 2014, 4, 32475–32481 RSC.
- Y. G. Jiang, P. B. Wan, H. P. Xu, Z. Q. Wang, X. Zhang and M. Smet, Langmuir, 2009, 25, 10134–10138 CrossRef CAS PubMed.
- J. H. Chen, J. H. Huang, H. H. Zhang and Y. H. Hu, ACS Appl. Mater. Interfaces, 2020, 12, 38647–38654 CrossRef CAS PubMed.
- H. Patel, J. H. Chen, Y. H. Hu and A. Erturk, Sci. Rep., 2022, 12, 13033 CrossRef CAS PubMed.
- I. Tomatsu, K. Peng and A. Kros, Adv. Drug Delivery Rev., 2011, 63, 1257–1266 CrossRef CAS PubMed.
- S. Tamesue, Y. Takashima, H. Yamaguchi, S. Shinkai and A. Harada, Angew. Chem., Int. Ed., 2010, 49, 7461–7464 CrossRef CAS PubMed.
- Y. L. Zhao and J. F. Stoddart, Langmuir, 2009, 25, 8442–8446 CrossRef CAS PubMed.
- N. G. Liu, D. R. Dunphy, P. Atanassov, S. D. Bunge, Z. Chen, G. P. Lopez, T. J. Boyle and C. J. Brinker, Nano Lett., 2004, 4, 551–554 CrossRef CAS.
- X. J. Zhang, X. Ma, K. Wang, S. J. Lin, S. T. Zhu, Y. Dai and F. Xia, Macromol. Rapid Commun., 2018, 39, 1800142 CrossRef PubMed.
- N. Liubimtsev, Z. Zagradska-Paromova, D. Appelhans, J. Gaitzsch and B. Voit, Macromol. Chem. Phys., 2023, 224, 2200372 CrossRef CAS.
- A. S. Gliozzi, M. Miniaci, A. Chiappone, A. Bergamini, B. Morin and E. Descrovi, Nat. Commun., 2020, 11, 2576 CrossRef CAS PubMed.
- S. Wu, J. P. Blinco and C. Barner-Kowollik, Chem. – Eur. J., 2017, 23, 8325–8332 CrossRef CAS PubMed.
- T. G. Zhan, M. D. Lin, J. Wei, L. J. Liu, M. Y. Yun, L. Wu, S. T. Zheng, H. H. Yin, L. C. Kong and K. D. Zhang, Polym. Chem., 2017, 8, 7384–7389 RSC.
- D. S. Wang, W. F. Zhao, Q. Wei, C. S. Zhao and Y. H. Zheng, ChemPhotoChem, 2018, 2, 403–415 CrossRef CAS.
- H. C. Yan, Y. Qiu, J. Wang, Q. Jiang, H. Wang, Y. G. Liao and X. L. Xie, Langmuir, 2020, 36, 7408–7417 CrossRef CAS PubMed.
- F. D. Jochum and P. Theato, Chem. Soc. Rev., 2013, 42, 7468–7483 RSC.
- Z. Qiu, H. Yu, J. Li, Y. Wang and Y. Zhang, Chem. Commun., 2009, 3342–3344, 10.1039/b822840j.
- H. Chen, F. Yang, Q. Chen and J. Zheng, Adv. Mater., 2017, 29, 1606900 CrossRef PubMed.
- A. Meeks, R. Mac, S. Chathanat and J. Aizenberg, Chem. Mater., 2020, 32, 10594–10600 CrossRef CAS.
- D. R. Morim, A. Meeks, A. Shastri, A. Tran, A. V. Shneidman, V. V. Yashin, F. Mahmood, A. C. Balazs, J. Aizenberg and K. Saravanamuttu, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 3953–3959 CrossRef CAS PubMed.
- Y. Xiao, S. Zarghami, K. Wagner, P. Wagner, K. C. Gordon, L. Florea, D. Diamond and D. L. Officer, Adv. Mater., 2018, 30, 1801821 CrossRef PubMed.
- J. ter Schiphorst, G. G. Melpignano, H. E. Amirabadi, M. H. J. M. Houben, S. Bakker, J. M. J. den Toonder and A. P. H. J. Schenning, Macromol. Rapid Commun., 2018, 39, 1700086 CrossRef PubMed.
- C. Li, A. Iscen, L. C. Palmer, G. C. Schatz and S. I. Stupp, J. Am. Chem. Soc., 2020, 142, 8447–8453 CrossRef CAS PubMed.
- A. Meeks, M. M. Lerch, T. B. H. Schroeder, A. Shastri and J. Aizenberg, J. Am. Chem. Soc., 2022, 144, 219–227 CrossRef CAS PubMed.
- E. R. Draper and D. J. Adams, Chem. Commun., 2016, 52, 8196–8206 RSC.
- J. Liu, X. W. Lou, M. J. G. Schotman, P. P. M. San Roman and R. P. Sijbesma, Gels, 2022, 8, 615 CrossRef CAS PubMed.
- T. M. Almutairi, H. H. Al-Rasheed, M. Monier, F. S. Alatawi and N. H. Elsayed, Int. J. Biol. Macromol., 2022, 210, 208–217 CrossRef CAS PubMed.
- A. A. H. Bukhari, N. H. Elsayed and M. Monier, Carbohydr. Polym., 2021, 260, 117771 CrossRef CAS PubMed.
- H. Zhang, X. Li, Y. J. Lin, F. Gao, Z. Tang, P. F. Su, W. K. Zhang, Y. Z. Xu, W. G. Weng and R. Boulatov, Nat. Commun., 2017, 8, 1147 CrossRef PubMed.
- T. Peng, A. Y. Dang-i, J. Y. Liu and C. L. Feng, Adv. Fiber Mater., 2019, 1, 241–247 CrossRef.
- M. Aljuaid, H. A. Houck, S. Efstathiou, D. M. Haddleton and P. Wilson, Macromolecules, 2022, 55, 8495–8504 CrossRef CAS PubMed.
- M. Aljuaid, E. Liarou, J. Town, J. R. Baker, D. M. Haddleton and P. Wilson, Chem. Commun., 2020, 56, 9545–9548 RSC.
- T. T. Li, T. J. Ma, J. Li, S. Chen, X. D. Ma, J. Yin and X. S. Jiang, Adv. Mater., 2021, 33, 2007699 CrossRef CAS PubMed.
- S. Ikejiri, Y. Takashima, M. Osaki, H. Yamaguchi and A. Harada, J. Am. Chem. Soc., 2018, 140, 17308–17315 CrossRef CAS PubMed.
- A. M. Castilla, E. R. Draper, M. C. Nolan, C. Brasnett, A. Seddon, L. L. E. Mears, N. Cowieson and D. J. Adams, Sci. Rep., 2017, 7, 8380 CrossRef PubMed.
- Q. B. Chen, L. Cui, X. Y. Zhou, Y. Guan and Y. J. Zhang, J. Mater. Chem. B, 2022, 10, 9914–9922 RSC.
- S. R. Batool, M. A. Nazeer, E. Yildiz, A. Sahin and S. Kizilel, Int. J. Biol. Macromol., 2021, 185, 165–175 CrossRef CAS PubMed.
- Z. Y. Hou, W. M. Nau and R. Hoogenboom, Polym. Chem., 2021, 12, 307–315 RSC.
- K. A. Gunay, T. L. Ceccato, J. S. Silver, K. L. Bannister, O. J. Bednarski, L. A. Leinwand and K. S. Anseth, Angew. Chem., Int. Ed., 2019, 58, 9912–9916 CrossRef PubMed.
- Z. Jiang, M. L. Tan, M. Taheri, Q. Yan, T. Tsuzuki, M. G. Gardiner, B. Diggle and L. A. Connal, Angew. Chem., Int. Ed., 2020, 59, 7049–7056 CrossRef CAS PubMed.
- Z. Y. Wu, P. Cheng, W. G. Zhao, J. L. Fang, T. C. Xu and D. Z. Chen, New J. Chem., 2020, 44, 10902–10910 RSC.
- B. D. Fairbanks, S. P. Singh, C. N. Bowman and K. S. Anseth, Macromolecules, 2011, 44, 2444–2450 CrossRef CAS PubMed.
- N. R. Gandavarapu, M. A. Azagarsamy and K. S. Anseth, Adv. Mater., 2014, 26, 2521–2526 CrossRef CAS PubMed.
- M. R. Matanovic, J. Kristl and P. A. Grabnar, Int. J. Pharm., 2014, 472, 262–275 CrossRef CAS PubMed.
- Y. J. Kim and Y. T. Matsunaga, J. Mater. Chem. B, 2017, 5, 4307–4321 RSC.
- A. A. A. Smith, C. L. Maikawa, H. L. Hernandez and E. A. Appel, Polym. Chem., 2021, 12, 1918–1923 RSC.
- S. Chatterjee, P. C. L. Hui and C. W. Kan, Polymers, 2018, 10, 480 CrossRef PubMed.
- M. A. Ward and T. K. Georgiou, Polymers, 2011, 3, 1215–1242 CrossRef CAS.
- Q. L. Zhang and R. Hoogenboom, Prog. Polym. Sci., 2015, 48, 122–142 CrossRef CAS.
- M. Q. Le, W. Huang, K. F. Chen, C. H. Lin, L. L. Cai, H. T. Zhang and Y. G. Jia, Chem. Eng. J., 2022, 432, 134354 CrossRef CAS.
- J. Seuring and S. Agarwal, ACS Macro Lett., 2013, 2, 597–600 CrossRef CAS PubMed.
- K. K. Sharker, Y. Ohara, Y. Shigeta, S. Ozoe and S. Yusa, Polymers, 2019, 11, 265 CrossRef PubMed.
- M. Sponchioni, U. C. Palmiero and D. Moscatelli, Mat. Sci. Eng. C, 2019, 102, 589–605 CrossRef CAS PubMed.
- A. Halperin, M. Kroger and F. M. Winnik, Angew. Chem., Int. Ed., 2015, 54, 15342–15367 CrossRef CAS PubMed.
- A. P. Constantinou, L. Wang, S. Wang and T. K. Georgiou, Polym. Chem., 2023, 14, 223–247 RSC.
- Q. L. Zhang, C. Weber, U. S. Schubert and R. Hoogenboom, Mater. Horiz., 2017, 4, 109–116 RSC.
- T. E. de Oliveira, D. Mukherji, K. Kremer and P. A. Netz, J. Chem. Phys., 2017, 146, 034904 CrossRef PubMed.
- Y. Yang, J. Maldonado-Valderrama and A. Martin-Molina, J. Mol. Liq., 2020, 303, 112678 CrossRef CAS.
- H. Matsuoka and K. Uda, Langmuir, 2016, 32, 8383–8391 CrossRef CAS PubMed.
- M. M. S. Lencina, E. F. Miconi, M. D. F. Leyes, C. Dominguez, E. Cuenca and H. A. Ritacco, J. Colloid Interface Sci., 2018, 512, 455–465 CrossRef CAS PubMed.
- X. G. Fan, S. Y. Gu, L. Y. Wu and L. Yang, E-Polymers, 2020, 20, 561–570 CrossRef CAS.
- A. Garcia-Penas, C. S. Biswas, W. J. Liang, Y. Wang, P. P. Yang and F. J. Stadler, Polymers, 2019, 11, 991 CrossRef PubMed.
- J. Q. Gan, X. X. Guan, J. Zheng, H. L. Guo, K. Wu, L. Y. Liang and M. G. Lu, RSC Adv., 2016, 6, 32967–32978 RSC.
- F. F. Xin, M. Y. Sui, X. S. Liu, C. Zhao and Y. Q. Yu, Colloid Polym. Sci., 2019, 297, 763–769 CrossRef CAS.
- C. N. Hernandez-Tellez, A. G. Luque-Alcaraz, M. Plascencia-Jatomea, H. J. Higuera-Valenzuela, M. Burgos-Hernandez, N. Garcia-Flores, M. E. Alvarez-Ramos, J. L. Iriqui-Razcon and P. A. Hernandez-Abril, Polymers, 2021, 13, 1704 CrossRef CAS PubMed.
- A. Fathi, S. M. Mithieux, H. Wei, W. Chrzanowski, P. Valtchev, A. S. Weiss and F. Dehghani, Biomaterials, 2014, 35, 5425–5435 CrossRef CAS PubMed.
- D. Calder, A. Fathi, F. Oveissi, S. Maleknia, T. Abrams, Y. W. Wang, J. Maitz, K. H. Y. Tsai, P. Maitz, W. Chrzanowski, I. Canoy, V. A. Menon, K. Lee, B. J. Ahern, N. E. Lean, D. M. Silva, P. M. Young, D. Traini, H. X. Ong, R. S. Mahmoud, H. Montazerian, A. Khademhosseini and F. Dehghani, Adv. Healthcare Mater., 2022, 11, e2201714 CrossRef PubMed.
- N. A. Cortez-Lemus and A. Licea-Claverie, Prog. Polym. Sci., 2016, 53, 1–51 CrossRef CAS.
- J. F. Assis, A. M. Gabriel, L. F. Goncalves, M. R. F. Machado, D. L. Morgado, R. L. Sala, F. H. Cristovan, M. P. Oliveira, T. M. Arantes and E. R. Camargo, Bionanoscience, 2022, 12, 766–773 CrossRef.
- L. Marsili, M. Dal Bo, F. Berti and G. Toffoli, Pharmaceutics, 2021, 13, 1654 CrossRef CAS PubMed.
- Y. Q. You, K. Kobayashi, B. Colak, P. P. Luo, E. Cozens, L. Fields, K. Suzuki and J. Gautrot, Biomaterials, 2021, 269, 120356 CrossRef CAS PubMed.
- O. Sedlacek, A. Van Driessche, A. Uvyn, B. G. De Geest and R. Hoogenboom, J. Controlled Release, 2020, 326, 53–62 CrossRef CAS PubMed.
- A. Lusina, T. Nazim and M. Ceglowski, Polymers, 2022, 14, 4176 CrossRef CAS PubMed.
- V. R. de la Rosa, J. Mater. Sci.: Mater. Med., 2014, 25, 1211–1225 CrossRef CAS PubMed.
- A. Chenite, C. Chaput, D. Wang, C. Combes, M. D. Buschmann, C. D. Hoemann, J. C. Leroux, B. L. Atkinson, F. Binette and A. Selmani, Biomaterials, 2000, 21, 2155–2161 CrossRef CAS PubMed.
- H. Madry, L. Gao, A. Rey-Rico, J. K. Venkatesan, K. Muller-Brandt, X. Y. Cai, L. Goebel, G. Schmitt, S. Speicher-Mentges, D. Zurakowski, M. D. Menger, M. W. Laschke and M. Cucchiarini, Adv. Mater., 2020, 32, 1906508 CrossRef CAS PubMed.
- P. Ghandforoushan, J. Hanaee, Z. Aghazadeh, M. Samiei, A. M. Navali, A. Khatibi and S. Davaran, Int. J. Biol. Macromol., 2022, 201, 270–287 CrossRef CAS PubMed.
- A. Arranja, G. Waton, F. Schosseler and E. Mendes, Soft Matter, 2016, 12, 769–778 RSC.
- T. D. Langridge and R. A. Gemeinhart, J. Controlled Release, 2020, 319, 157–167 CrossRef CAS PubMed.
- S. Q. Cui, L. Yu and J. D. Ding, Macromolecules, 2018, 51, 6405–6420 CrossRef CAS.
- S. Q. Cui, L. Yu and J. D. Ding, Macromolecules, 2019, 52, 3697–3715 CrossRef CAS.
- Z. X. Deng, R. Yu and B. L. Guo, Mater. Chem. Front., 2021, 5, 2092–2123 RSC.
- P. Lavrador, M. R. Esteves, V. M. Gaspar and J. F. Mano, Adv. Funct. Mater., 2021, 31, 2005941 CrossRef CAS.
- V. M. Gaspar, P. Lavrador, J. Borges, M. B. Oliveira and J. F. Mano, Adv. Mater., 2020, 32, 1903975 CrossRef CAS PubMed.
- D. L. Gan, L. Han, M. H. Wang, W. S. Xing, T. Xu, H. P. Zhang, K. F. Wang, L. M. Fang and X. Lu, ACS Appl. Mater. Interfaces, 2018, 10, 36218–36228 CrossRef CAS PubMed.
- S. Wang, J. F. Lei, X. L. Yi, L. Yuan, L. M. Ge, D. F. Li and C. D. Mu, ACS Appl. Polym. Mater., 2020, 2, 3016–3023 CrossRef CAS.
- S. D. Dutta, K. Ganguly, A. Randhawa, T. V. Patil, D. K. Patel and K. T. Lim, Biomaterials, 2023, 294, 121999 CrossRef CAS PubMed.
- A. Mir, A. Kumar and U. Riaz, RSC Adv., 2022, 12, 19122–19132 RSC.
- H. Hussin, S. N. Gan, S. Mohamad and S. W. Phang, Polym. Polym. Compos., 2017, 25, 515–519 CAS.
- B. S. Eftekhari, M. Eskandari, P. A. Janmey, A. Samadikuchaksaraei and M. Gholipourmalekabadi, RSC Adv., 2021, 11, 15795–15807 RSC.
- J. H. Xue, Y. T. Liu, M. A. Darabi, G. H. Tu, L. Huang, L. Ying, B. Xiao, Y. Wu, M. Xing, L. Zhang and L. Zhang, Mater. Sci. Eng., C, 2019, 100, 584–597 CrossRef CAS PubMed.
- V. R. Feig, S. Santhanam, K. W. McConnell, K. Liu, M. Azadian, L. G. Brunel, Z. J. Huang, H. Tran, P. M. George and Z. N. Bao, Adv. Mater. Technol., 2021, 6, 2100162 CrossRef CAS PubMed.
- P. Thoniyot, M. J. Tan, A. A. Karim, D. J. Young and X. J. Loh, Adv. Sci., 2015, 2, 1400010 CrossRef PubMed.
- I. Carayon, A. Gaubert, Y. Mousli and B. Philippe, Biomater. Sci., 2020, 8, 5589–5600 RSC.
- M. Shin, K. H. Song, J. C. Burrell, D. K. Cullen and J. A. Burdick, Adv. Sci., 2019, 6, 1901229 CrossRef CAS PubMed.
- J. H. Min, M. Patel and W. G. Koh, Polymers, 2018, 10, 1078 CrossRef PubMed.
- L. Wang, S. M. Hu, M. W. Ullah, X. H. Li, Z. J. Shi and G. Yang, Carbohydr. Polym., 2020, 249, 116829 CrossRef CAS PubMed.
- M. K. Yazdi and G. H. Motlagh, J. Electron. Mater., 2020, 49, 5326–5334 CrossRef.
- M. Rizwan, R. Yahya, A. Hassan, M. Yar, A. D. Azzahari, V. Selvanathan, F. Sonsudin and C. N. Abouloula, Polymers, 2017, 9, 137 CrossRef PubMed.
- F. Ofridam, M. Tarhini, N. Lebaz, E. Gagniere, D. Mangin and A. Elaissari, Polym. Adv. Technol., 2021, 32, 1455–1484 CrossRef CAS.
- J. C. Berkmann, A. X. H. Martin, A. Ellinghaus, C. Schlundt, H. Schell, E. Lippens, G. N. Duda, S. Tsitsilonis and K. Schmidt-Bleek, Int. J. Mol. Sci., 2020, 21, 2513 CrossRef CAS PubMed.
- Y. Qiu and K. Park, Adv. Drug Delivery Rev., 2001, 53, 321–339 CrossRef CAS PubMed.
- P. Sikdar, M. M. Uddin, T. M. Dip, S. Islam, M. S. Hoque, A. K. Dhar and S. Y. Wu, Mater. Adv., 2021, 2, 4532–4573 RSC.
- G. Kocak, C. Tuncer and V. Butun, Polym. Chem., 2017, 8, 144–176 RSC.
- M. Bustamante-Torres, D. Romero-Fierro, B. Arcentales-Vera, K. Palomino, H. Magana and E. Bucio, Gels, 2021, 7, 182 CrossRef CAS PubMed.
- S. Dai, P. Ravi and K. C. Tam, Soft Matter, 2008, 4, 435–449 RSC.
- R. A. Rather, M. A. Bhat and A. H. Shalla, Carbohydr. Polym. Technol. Appl., 2022, 3, 100202 CAS.
- Y. Y. Liu, X. D. Fan, B. R. Wei, Q. F. Si, W. X. Chen and L. Sun, Int. J. Pharm., 2006, 308, 205–209 CrossRef CAS PubMed.
- G. Frutos, A. Prior-Cabanillas, R. Paris and I. Quijada-Garrido, Acta Biomater., 2010, 6, 4650–4656 CrossRef CAS PubMed.
- A. Tamayol, M. Akbari, Y. Zilberman, M. Comotto, E. Lesha, L. Serex, S. Bagherifard, Y. Chen, G. Q. Fu, S. K. Ameri, W. T. Ruan, E. L. Miller, M. R. Dokmeci, S. Sonkusale and A. Khademhosseini, Adv. Healthcare Mater., 2016, 5, 711–719 CrossRef CAS PubMed.
- P. K. Robinson, Understanding Biochemistry: Enzymes and Membranes, 2015, vol. 59, pp. 75–75 Search PubMed.
- H. M. El-Husseiny, E. A. Mady, L. Hamabe, A. Abugomaa, K. Shimada, T. Yoshida, T. Tanaka, A. Yokoi, M. Elbadawy and R. Tanaka, Mater. Today Bio, 2022, 13, 100186 CrossRef CAS PubMed.
- T. Maki, R. Yoshisaki, S. Akama and M. Yamanaka, Polym. J., 2020, 52, 931–938 CrossRef CAS.
- R. Liu, Z. K. Shi, J. Sun and Z. B. Li, Sci. China: Chem., 2018, 61, 1314–1319 CrossRef CAS.
- M. Sobczak, Int. J. Mol. Sci., 2022, 23, 4421 CrossRef CAS PubMed.
- R. Chandrawati, Exp. Biol. Med., 2016, 241, 972–979 CrossRef CAS PubMed.
- M. P. Savoca, E. Tonoli, A. G. Atobatele and E. A. M. Verderio, Micromachines, 2018, 9, 562 CrossRef PubMed.
- Z. H. Shi, X. Y. Li, M. X. Wei, P. Y. Chen, T. Zhang, X. F. Ling, J. Q. Zhang, C. Zhao, F. Q. Wang and G. L. Liang, Biomaterials, 2021, 269, 120536 CrossRef CAS PubMed.
- O. Sunnapu, R. Khader, M. Dhanka, P. K. Vemula and S. Karuppannan, ChemNanoMat, 2022, 8, e202200334 CrossRef CAS.
- H. Shigemitsu, R. Kubota, K. Nakamura, T. Matsuzaki, S. Minami, T. Aoyama, K. Urayama and I. Hamachi, Nat. Commun., 2020, 11, 3859 CrossRef PubMed.
- G. L. Ma, W. F. Lin, Z. F. Yuan, J. Wu, H. F. Qian, L. B. Xu and S. F. Chen, J. Mater. Chem. B, 2017, 5, 935–943 RSC.
- F. Anjum, P. S. Lienemann, S. Metzger, J. Biernaskie, M. S. Kallos and M. Ehrbar, Biomaterials, 2016, 87, 104–117 CrossRef CAS PubMed.
- S. Ravalli, M. A. Szychlinska, G. Lauretta and G. Musumeci, Appl. Sci., 2020, 10, 2927 CrossRef CAS.
- B. L. Pruitt, A. R. Dunn, W. I. Weis and W. J. Nelson, PLoS Biol., 2014, 12, e1001996 CrossRef PubMed.
- P. Prendergast, R. Huiskes and K. Søballe, J. Biomech., 1997, 30, 539–548 CrossRef CAS PubMed.
- M. S. Ting, J. Travas-Sejdic and J. Malmström, J. Mater. Chem. B, 2021, 9, 7578–7596 RSC.
- N. P. Shetti, A. Mishra, S. Basu, R. J. Mascarenhas, R. R. Kakarla and T. M. Aminabhavi, ACS Biomater. Sci. Eng., 2020, 6, 1823–1835 CrossRef CAS PubMed.
- J. Chen, Q. Peng, X. Peng, L. Han, X. Wang, J. Wang and H. Zeng, ACS Appl. Polym. Mater., 2020, 2, 1092–1107 CrossRef CAS.
- C. Storm, J. J. Pastore, F. C. MacKintosh, T. C. Lubensky and P. A. Janmey, Nature, 2005, 435, 191–194 CrossRef CAS PubMed.
- G. Žagar, P. R. Onck and E. Van Der Giessen, Biophys. J., 2015, 108, 1470–1479 CrossRef PubMed.
- K. Fang, R. Wang, H. Zhang, L. Zhou, T. Xu, Y. Xiao, Y. Zhou, G. Gao, J. Chen and D. Liu, ACS Appl. Mater. Interfaces, 2020, 12, 52307–52318 CrossRef CAS PubMed.
- Z. Chen, J. Tang, N. Zhang, Y. Chen, Y. Chen, H. Li and H. Liu, Colloids Surf., A, 2022, 633, 127867 CrossRef CAS.
- A. Andersen, M. Krogsgaard and H. Birkedal, Biomacromolecules, 2017, 19, 1402–1409 CrossRef PubMed.
- M. E. Smithmyer, C. C. Deng, S. E. Cassel, P. J. LeValley, B. S. Sumerlin and A. M. Kloxin, ACS Macro Lett., 2018, 7, 1105–1110 CrossRef CAS PubMed.
- S. Uman, A. Dhand and J. A. Burdick, J. Appl. Polym. Sci., 2020, 137, 48668 CrossRef CAS.
- M. Li, Q. Zhang, Y.-N. Zhou and S. Zhu, Prog. Polym. Sci., 2018, 79, 26–39 CrossRef CAS.
- L.-J. Wang, K.-X. Yang, Q. Zhou, H.-Y. Yang, J.-Q. He and X.-Y. Zhang, Chin. J. Polym. Sci., 2020, 38, 24–36 CrossRef CAS.
- Y. C. Mao, Y. Kubota, J. Gong, T. Kurose, A. Ishigami, K. Seshimo, T. Watabe, D. Aoki, H. Otsuka and H. Ito, Macromolecules, 2021, 54, 8664–8674 CrossRef CAS.
- R. Chandan, S. Mehta and R. Banerjee, ACS Biomater. Sci. Eng., 2020, 6, 4731–4747 CrossRef CAS PubMed.
- T. J. Yeingst, J. H. Arrizabalaga and D. J. Hayes, Gels, 2022, 8, 554 CrossRef CAS PubMed.
- C.-H. Wu, M.-K. Sun, J. Shieh, C.-S. Chen, C.-W. Huang, C.-A. Dai, S.-W. Chang, W.-S. Chen and T.-H. Young, Ultrasonics, 2018, 83, 157–163 CrossRef CAS PubMed.
- Y. Sun, L.-G. Chen, X.-M. Fan and J.-L. Pang, Int. J. Nanomed., 2022, 5001–5026 CrossRef CAS PubMed.
- H. Jiang, N. M. Carter, A. Zareei, S. Nejati, J. F. Waimin, S. Chittiboyina, E. E. Niedert, T. Soleimani, S. A. Lelièvre and C. J. Goergen, ACS Appl. Bio Mater., 2020, 3, 4012–4024 CrossRef CAS PubMed.
- J. Yi, K.-C. T. Nguyen, W. Wang, W. Yang, M. Pan, E. Lou, P. W. Major, L. H. Le and H. Zeng, ACS Appl. Bio Mater., 2020, 3, 8943–8952 CrossRef CAS PubMed.
- N. Huebsch, C. J. Kearney, X. Zhao, J. Kim, C. A. Cezar, Z. Suo and D. J. Mooney, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 9762–9767 CrossRef CAS PubMed.
- A. Alford, B. Tucker, V. Kozlovskaya, J. Chen, N. Gupta, R. Caviedes, J. Gearhart, D. Graves and E. Kharlampieva, Polymers, 2018, 10, 1342 CrossRef PubMed.
- S. Yamaguchi, K. Higashi, T. Azuma and A. Okamoto, Biotechnol. J., 2019, 14, 1800530 CrossRef PubMed.
- J. H. Arrizabalaga, M. Smallcomb, M. Abu-Laban, Y. Liu, T. J. Yeingst, A. Dhawan, J. C. Simon and D. J. Hayes, ACS Appl. Bio Mater., 2022, 5, 3212–3218 CrossRef CAS PubMed.
- G. Yin, J. Huang, D. Liu, R. Li, S. Wei, M. Si, F. Ni, Y. Zheng, Q. Yang and R. Zhou, Chin. Chem. Lett., 2023, 34, 107290 CrossRef CAS.
- D. Liu, L. Li, B.-L. Shi, B. Shi, M.-D. Li, Y. Qiu, D. Zhao, Q.-D. Shen and Z.-Z. Zhu, Bioact. Mater., 2023, 24, 96–111 CrossRef CAS PubMed.
- M. C. Koetting, J. T. Peters, S. D. Steichen and N. A. Peppas, Mater. Sci. Eng., R, 2015, 93, 1–49 CrossRef PubMed.
- S. Chatterjee and P. C.-L. Hui, in Hydrogels—smart materials for biomedical applications, ed. L. Popa, M. V. Ghica and C. Dinu-Pirvu, Intechopen, 2018 DOI:10.5772/intechopen.80536.
- N. Sood, A. Bhardwaj, S. Mehta and A. Mehta, Drug Delivery, 2016, 23, 748–770 CrossRef CAS PubMed.
- F. Andrade, M. M. Roca-Melendres, E. F. Durán-Lara, D. Rafael and S. Schwartz Jr, Cancers, 2021, 13, 1164 CrossRef CAS PubMed.
- L. Xia, X. Zhao, X. Ma, Y. Hu, Y. Zhang, S. Li, J. Wang, Y. Zhao and R. Chai, Nano Today, 2022, 44, 101507 CrossRef CAS.
- K. A. Tran, Y. Jin, J. Bouyer, B. J. DeOre, Ł. Suprewicz, A. Figel, H. Walens, I. Fischer and P. A. Galie, Biomater. Sci., 2022, 10, 2237–2247 RSC.
- Z. Li, Y. Zhou, T. Li, J. Zhang and H. Tian, View, 2022, 3, 20200112 CrossRef CAS.
- T. Satoh, K. Sumaru, T. Takagi and T. Kanamori, Soft Matter, 2011, 7, 8030–8034 RSC.
- A. Meeks, M. M. Lerch, T. B. Schroeder, A. Shastri and J. Aizenberg, J. Am. Chem. Soc., 2021, 144, 219–227 CrossRef PubMed.
- T. P. Vales, I. W. T. Badon and H.-J. Kim, Macromol. Res., 2018, 26, 950–953 CrossRef CAS.
- J. Hatai, C. Hirschhäuser, J. Niemeyer and C. Schmuck, ACS Appl. Mater. Interfaces, 2019, 12, 2107–2115 CrossRef PubMed.
- A. Najafipour, A. Gharieh, A. Fassihi, H. Sadeghi-Aliabadi and A. R. Mahdavian, Mol. Pharm., 2020, 18, 275–284 CrossRef PubMed.
- V. Nandwana, S.-R. Ryoo, T. Zheng, M. M. You and V. P. Dravid, ACS Biomater. Sci. Eng., 2019, 5, 3049–3059 CrossRef CAS PubMed.
- Y. Du, W. Du, D. Lin, M. Ai, S. Li and L. Zhang, Micromachines, 2023, 14, 167 CrossRef PubMed.
- X. Li, J. Zhou, Z. Liu, J. Chen, S. Lü, H. Sun, J. Li, Q. Lin, B. Yang and C. Duan, Biomaterials, 2014, 35, 5679–5688 CrossRef CAS PubMed.
- Z. Li, Z. Fan, Y. Xu, W. Lo, X. Wang, H. Niu, X. Li, X. Xie, M. Khan and J. Guan, ACS Appl. Mater. Interfaces, 2016, 8, 10752–10760 CrossRef CAS PubMed.
- S. Pedron, S. Van Lierop, P. Horstman, R. Penterman, D. Broer and E. Peeters, Adv. Funct. Mater., 2011, 21, 1624–1630 CrossRef CAS.
- W.-N. Lu, S.-H. Lü, H.-B. Wang, D.-X. Li, C.-M. Duan, Z.-Q. Liu, T. Hao, W.-J. He, B. Xu and Q. Fu, Tissue Eng. A, 2009, 15, 1437–1447 CrossRef CAS PubMed.
- P. Baei, S. Jalili-Firoozinezhad, S. Rajabi-Zeleti, M. Tafazzoli-Shadpour, H. Baharvand and N. Aghdami, Mater. Sci. Eng., C, 2016, 63, 131–141 CrossRef CAS PubMed.
- S. Chen, Y. Li, L. Xie, S. Liu, Y. Fan, C. Fang, X. Zhang, J. Quan and L. Zuo, Chin. J. Tissue Eng. Res., 2021, 25, 2472 Search PubMed.
- Y. Guo, Y. Qu, J. Yu, L. Song, S. Chen, Z. Qin, J. Gong, H. Zhan, Y. Gao and J. Zhang, Int. J. Biol. Macromol., 2022, 202, 102–111 CrossRef CAS PubMed.
- C. Yu, Z. Yue, M. Shi, L. Jiang, S. Chen, M. Yao, Q. Yu, X. Wu, H. Zhang and F. Yao, ACS Nano, 2022, 16, 16234–16248 CrossRef CAS PubMed.
- C. Zhang, M.-H. Hsieh, S.-Y. Wu, S.-H. Li, J. Wu, S.-M. Liu, H.-J. Wei, R. D. Weisel, H.-W. Sung and R.-K. Li, Biomaterials, 2020, 231, 119672 CrossRef CAS PubMed.
- S. Liang, Y. Zhang, H. Wang, Z. Xu, J. Chen, R. Bao, B. Tan, Y. Cui, G. Fan and W. Wang, Adv. Mater., 2018, 30, 1704235 CrossRef PubMed.
- M. Parchehbaf-Kashani, H. Ansari, E. Mahmoudi, M. Barekat, M. Sepantafar, S. Rajabi and S. Pahlavan, ACS Appl. Bio Mater., 2021, 4, 4849–4861 CrossRef CAS PubMed.
- Y. Liu, R. Guo, T. Wu, Y. Lyu, M. Xiao, B. He, G. Fan, J. Yang and W. Liu, Chem. Eng. J., 2021, 418, 129352 CrossRef CAS.
- C. Xu, Y. Xu, M. Yang, Y. Chang, A. Nie, Z. Liu, J. Wang and Z. Luo, Adv. Funct. Mater., 2020, 30, 2000177 CrossRef CAS.
- F. Yang, G. Guo and Y. Wang, Biomaterials, 2022, 289, 121761 CrossRef CAS PubMed.
- P. Cheng, L. Cheng, H. Han, J. Li, C. Ma, H. Huang, J. Zhou, J. Feng, Y. Huang and Y. Lv, Adv. Mater., 2022, 11, 2200971 CAS.
- G. Q. Chen, J. L. Li, M. C. Song, Z. Y. Wu, W. Z. Zhang, Z. Y. Wang, J. Gao, Z. M. Yang and C. W. Ou, Adv. Funct. Mater., 2017, 27, 1701798 CrossRef.
- J.-J. Ji, S.-Y. Chen, Z.-W. Yang, R. Zhang, L.-L. Qian, Y. Jiang, J.-Q. Guo, Y. Wu, Q.-L. Fan and Y.-Y. Yao, Nanomed. Nanotechnol. Biol. Med., 2023, 47, 102618 CrossRef CAS PubMed.
- J. A. Burdick and W. L. Murphy, Nat. Commun., 2012, 3, 1269 CrossRef PubMed.
- K. Huang, J. Du, J. Xu, C. Wu, C. Chen, S. Chen, T. Zhu, J. Jiang and J. Zhao, Mater. Today Chem., 2022, 23, 100720 CrossRef CAS.
- N. Nafee, M. Zewail and N. Boraie, J. Drug Targeting, 2018, 26, 563–575 CrossRef CAS PubMed.
- P. Ni, Q. Ding, M. Fan, J. Liao, Z. Qian, J. Luo, X. Li, F. Luo, Z. Yang and Y. Wei, Biomaterials, 2014, 35, 236–248 CrossRef CAS PubMed.
- N. Amiryaghoubi, N. N. Pesyan, M. Fathi and Y. Omidi, Int. J. Biol. Macromol., 2020, 162, 1338–1357 CrossRef CAS PubMed.
- S. Taymouri, S. Amirkhani and M. Mirian, J. Drug Delivery Sci. Technol., 2021, 64, 102659 CrossRef CAS.
- C. Zhao, N. T. Qazvini, M. Sadati, Z. Y. Zeng, S. F. Huang, A. L. De la Lastra, L. H. Zhang, Y. X. Feng, W. Liu, B. Huang, B. Zhang, Z. Y. Dai, Y. Shen, X. Wang, W. P. Luo, B. Liu, Y. Lei, Z. Y. Ye, L. Zhao, D. G. Cao, L. J. Yang, X. Chen, A. Athiviraham, M. J. Lee, J. M. Wolf, R. R. Reid, M. Tirrell, W. Huang, J. J. de Pablo and T. C. He, ACS Appl. Mater. Interfaces, 2019, 11, 8749–8762 CrossRef CAS PubMed.
- H. Motasadizadeh, M. Tavakoli, S. Damoogh, F. Mottaghitalab, M. Gholami, F. Atyabi, M. Farokhi and R. Dinarvand, Biomater. Adv., 2022, 139, 213032 CrossRef CAS PubMed.
- Y. Wu, X. Zhang, B. Tan, Y. Shan, X. Zhao and J. Liao, Biomater. Res., 2022, 133, 112641 Search PubMed.
- L. Kuang, J. Huang, Y. Liu, X. Li, Y. Yuan and C. Liu, Adv. Funct. Mater., 2021, 31, 2105383 CrossRef CAS.
- Y. Li, J. He, J. Zhou, Z. Li, L. Liu, S. Hu, B. Guo and W. Wang, Biomater. Res., 2022, 10, 1326–1341 RSC.
- X. Ding, J. Shi, J. Wei, Y. Li, X. Wu, Y. Zhang, X. Jiang, X. Zhang and H. Lai, Sci. Adv., 2021, 7, eabj7857 CrossRef CAS PubMed.
- J. Huang, Z. Jia, Y. Liang, Z. Huang, Z. Rong, J. Xiong and D. Wang, RSC Adv., 2020, 10, 541–550 RSC.
- Y. Li, L. Huang, G. Tai, F. Yan, L. Cai, C. Xin and S. Al Islam, Composites, Part A, 2022, 152, 106672 CrossRef CAS.
- A. H. Aziz, R. L. Wilmoth, V. L. Ferguson and S. J. Bryant, ACS Appl. Bio Mater., 2020, 3, 1666–1680 CrossRef CAS PubMed.
- Y. Yang, W. H. Zheng, W. Tan, X. Q. Wu, Z. N. Dai, Z. Y. Li, Z. Yan, Y. L. Ji, Y. L. Wang, W. W. Su, S. Zhong, Y. B. Li, Y. J. Sun, S. Y. Li and W. H. Huang, Acta Biomater., 2023, 157, 321–336 CrossRef CAS PubMed.
- H. Liu, X. Xiang, J. Huang, B. Zhu, L. Wang, Y. Tang, F. Du, L. Li, F. Yan and L. Ma, Chin. Chem. Lett., 2021, 32, 1759–1764 CrossRef CAS.
- T. Levingstone, B. Ali, C. Kearney and N. Dunne, J. Biomed. Mater. Res., Part B, 2021, 109, 1622–1633 CrossRef CAS PubMed.
- C. Zhao, N. T. Qazvini, M. Sadati, Z. Zeng, S. Huang, A. L. De La Lastra, L. Zhang, Y. Feng, W. Liu and B. Huang, ACS Appl. Mater. Interfaces, 2019, 11, 8749–8762 CrossRef CAS PubMed.
- A. Ressler, J. Ródenas-Rochina, M. Ivanković, H. Ivanković, A. Rogina and G. G. Ferrer, Carbohydr. Polym., 2018, 197, 469–477 CrossRef CAS PubMed.
- H. M. El-Husseiny, E. A. Mady, W. A. El-Dakroury, M. B. Zewail, M. Noshy, A. M. Abdelfatah and A. S. Doghish, Appl. Mater. Today, 2022, 29, 101560 CrossRef.
- P. Lavrador, M. R. Esteves, V. M. Gaspar and J. F. Mano, Adv. Funct. Mater., 2021, 31, 2005941 CrossRef CAS.
- M. A. Grimaudo, G. S. Krishnakumar, E. Giusto, F. Furlani, G. Bassi, A. Rossi, F. Molinari, F. Lista, M. Montesi and S. Panseri, Acta Biomater., 2022, 140, 88–101 CrossRef CAS PubMed.
- J. D. Tang, C. Mura and K. J. Lampe, J. Am. Chem. Soc., 2019, 141, 4886–4899 CrossRef CAS PubMed.
- S. Peressotti, G. E. Koehl, J. A. Goding and R. A. Green, ACS Biomater. Sci. Eng., 2021, 7, 4136–4163 CrossRef CAS PubMed.
- M. Yao, Y. Chen, J. Zhang, F. Gao, S. Ma and F. Guan, Mater. Today Chem., 2019, 14, 100192 CrossRef CAS.
- A. Adak, G. Das, J. Khan, N. Mukherjee, V. Gupta, R. Mallesh and S. Ghosh, ACS Biomater. Sci. Eng., 2020, 6, 2287–2296 CrossRef CAS PubMed.
- X.-Q. Zheng, J.-F. Huang, J.-L. Lin, Y.-X. Zhu, M.-Q. Wang, M.-L. Guo, X.-J. Zan and A.-M. Wu, Colloids Surf., B, 2021, 199, 111532 CrossRef CAS PubMed.
- W. Liu, Y. Luo, C. Ning, W. Zhang, Q. Zhang, H. Zou and C. Fu, J. Nanobiotech., 2021, 19, 286 CrossRef CAS PubMed.
- C. Rinoldi, M. Lanzi, R. Fiorelli, P. Nakielski, K. Zembrzycki, T. Kowalewski, O. Urbanek, V. Grippo, K. Jezierska-Woźniak and W. Maksymowicz, Biomacromolecules, 2021, 22, 3084–3098 CrossRef CAS PubMed.
- S. Bhattacharyya, A. Dinda, S. Vishnubhatla, M. F. Anwar and S. Jain, Neurosci. Lett., 2021, 741, 135500 CrossRef CAS PubMed.
- W. Wei, Q. Yang, J. Hu, Y. Yao and H. Yang, Cell Transplant., 2022, 31, 09636897221126088 Search PubMed.
- A. I. Gonçalves and M. E. Gomes, Curr. Opin. Biomed. Eng., 2022, 100431 Search PubMed.
- R. Plen, A. Smith, O. Blum, O. Aloni, U. Locker, Z. Shapira, S. Margel and O. Shefi, Adv. Funct. Mater., 2022, 2204925 CrossRef CAS.
- J. Garcia-Torres, in Hydrogels – From Tradition to Innovative Platforms with Multiple Applications, ed. L. Popa, M. V. Ghica and C. Dinu-Pirvu, Intechopen, 2022 DOI:10.5772/intechopen.102436.
Footnote |
| † Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |