
Organic synthesis in the study of terpene-derived oxidation products in the atmosphere
Mary Alice
Upshur
,
Ariana Gray
Bé
,
Jingyi
Luo
 ,
Jonathan G.
Varelas
,
Franz M.
Geiger
and
Regan J.
Thomson
*
,
Jonathan G.
Varelas
,
Franz M.
Geiger
and
Regan J.
Thomson
*
Department of Chemistry, Northwestern University, 2145 Sheridan Rd, Evanston, IL 60208, USA. E-mail: r-thomson@northwestern.edu
First published on 20th March 2023
Abstract
Covering: 1997 up to 2022
Volatile biogenic terpenes involved in the formation of secondary organic aerosol (SOA) particles participate in rich atmospheric chemistry that impacts numerous aspects of the earth's complex climate system. Despite the importance of these species, understanding their fate in the atmosphere and determining their atmospherically-relevant properties has been limited by the availability of authentic standards and probe molecules. Advances in synthetic organic chemistry directly aimed at answering these questions have, however, led to exciting discoveries at the interface of chemistry and atmospheric science. Herein we provide a review of the literature regarding the synthesis of commercially unavailable authentic standards used to analyze the composition, properties, and mechanisms of SOA particles in the atmosphere.
 Mary Alice Upshur | Mary Alice Upshur is currently a research scientist working in the paper coating materials group at the Dow Chemical Company in Collegeville, PA. Prior to this, she specialized in FTIR and Raman spectroscopy in the analytical sciences group as well as completed brief research assignments in the Performance Silicones and Home and Personal Care businesses at Dow Chemical. She received her PhD in chemistry from Northwestern University in 2017 studying the synthesis and spectroscopic analysis of secondary organic aerosol constituents under the supervision of Prof. Regan Thomson and Prof. Franz Geiger. |
 Ariana Gray Bé | Ariana Gray Bé was born in Cleveland, Ohio. She received her B.S. in Chemistry from Denison University in 2015, where she worked under the direction of Prof. Joseph Reczek. She received her PhD in 2020 from Northwestern University and was the recipient of an NSF Graduate Research Fellowship from 2006 to 2009. Jointly advised by Prof. Regan Thomson and Prof. Franz Geiger, her doctoral studies focused on the synthesis and surface chemical analysis of terpene-derived oxidation products in biogenic secondary organic aerosol particles. She then spent two years in the Innovation Department at Beyond Meat, and in 2022 she joined the Research and Development team at Ambercycle, a materials company working to create an infinite textile ecosystem to eliminate waste in fashion. |
 Jingyi Luo | Jingyi (Jenny) Luo received her B.S. in chemistry from the University of Michigan in 2018, where she worked with Prof. John P. Wolfe on the palladium-catalyzed synthesis of nitrogen-containing heterocycles. She is currently a PhD candidate in the lab of Prof. Regan Thomson, conducting research in the synthesis of deuterated analogues of monoterpenes for the investigation of their oxidation mechanisms in the atmosphere. |
 Jonathan G. Varelas | Jonathan Varelas was born in Madison, CT. He completed his BS in chemistry from Providence College in 2015 under the supervision of Prof. Seann Mulcahy. He then received his doctorate in chemistry from Northwestern University in 2021 as a joint student in the laboratories of Prof. Regan Thomson and Prof. Franz Geiger, where he synthesized and studied isoprene-derived organosulfates. After completing his doctorate, Jonathan moved into the realm of science communications, operating as a freelance consultant and multimedia producer for various pharmaceutical companies. He now serves as the director of multimedia engagement and eLearning at LectureLinx, a pharmaceutical marketing and compliance firm. |
 Franz M. Geiger | Franz Geiger is currently the Charles E. and Emma H. Morrison Professor of Chemistry at Northwestern University, where he leads research projects that involve experiments and computations to study the special role that surfaces and interfaces play in the world. He is a native of Berlin, Germany, where he received his Vordiplom in chemistry at the Technische Universitaet in 1993. He earned his PhD in 1998 at Georgetown University working with Janice Hicks as a NASA Fellow in Earth Systems Science, and was a NOAA Postdoctoral Fellow in Climate and Global Change with Mario Molina at MIT. |
 Regan J. Thomson | Regan J. Thomson was born in New Zealand in 1976 and received his PhD in 2003 at The Australian National University working with Prof. Lewis N. Mander. Following postdoctoral studies with Prof. David A. Evans at Harvard University, he joined the faculty at Northwestern University in 2006 where he is currently a Professor of Chemistry. Regan's research interests include reaction development, total synthesis, natural product discovery and biosynthesis, and atmospheric chemistry. He is the recipient of an NSF CAREER Award (2009), an Amgen Young Investigator Award (2010), an Illinois Division American Cancer Society Research Scholar Award (2012), and a Novartis Chemistry Lectureship (2015 and 2016). |
1. Introduction
Atmospheric fine particulate matter (PM2.5) has been shown to scatter and/or absorb radiation, influence cloud formation and climate processes,1–4 as well as have detrimental effects on human health.5–7 Organic species constitute the majority of PM2.5 in both pristine and urban environments.2,8 Secondary organic aerosol (SOA) particles (SOA), in particular, make up a larger fraction of atmospheric organic aerosol than primary organic aerosol (POA) particles and are formed as a result of the chemical transformation of volatile organic compounds (VOCs) via atmospheric oxidation. Reactions of both biogenic and anthropogenic VOCs with atmospheric oxidants generate compounds of lower volatility, and often higher O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :C ratios, which can then partition into the condensed aerosol phase, continue to react heterogeneously on existing particles, or nucleate to form new particles.9–14
:C ratios, which can then partition into the condensed aerosol phase, continue to react heterogeneously on existing particles, or nucleate to form new particles.9–14
Although great progress has been made toward identifying key molecular precursors that form biogenic and anthropogenic SOA, the current level of mechanistic understanding regarding the formation of SOA particles remains low. These gaps in our knowledge of the chemistry of SOA particles have resulted in difficulties in the prediction of SOA particle effects on the climate system by atmospheric chemistry and transport models,9,15–17 and have thus contributed greatly to the large uncertainties in the effects of aerosols on radiative forcing and global warming.18 Furthermore, the large number as well as variability of chemical constituents, sources and chemical reactions creates numerous challenges in determining the chemistry, composition, and properties of SOA particles. For instance, it has been estimated from mass spectrometric studies that 10000–100000 different organic compounds have been detected in the atmosphere.19,20
Over the past decade, organic synthesis has begun to emerge in the field of atmospheric chemistry as a powerful tool in the study of SOA material. While several SOA precursors and surrogate compounds, along with select oxidation products, are available from commercial sources, the majority of SOA constituents are not. Synthetic organic chemistry addresses this challenge by enabling access to authentic standards, which, when combined with physical measurements, laboratory chamber studies, and field studies, can be used to provide a molecular characterization of SOA particles.19 Indeed, pure and homogenous synthetic reference standards of compounds that are not available from commercial sources have not only proven critical for benchmarking and chemical identification in mass spectrometric studies of SOA particles, but have also provided insight into important physical properties of aerosol particles.
This manuscript seeks to provide a comprehensive review of the current state of the literature regarding the synthesis of commercially unavailable authentic standards used to analyze the composition, properties, and mechanisms of SOA particles in the atmosphere. We discuss studies reporting the synthesis of molecular standards derived from biogenic SOA precursors, namely isoprene (Section 2), monoterpenes (Section 3), and sesquiterpenes (Section 4). We also discuss the synthesis of standards relevant to SOA material influenced by anthropogenic pollutants, namely organosulfates and organonitrates (Section 5), and probe compounds (Section 6) to help facilitate mechanistic investigations. Where possible, and to the best of our ability, yields are provided and structures are depicted showing relative or absolute stereochemical relationships if reported in the cited work. If the original work did not provide yields or indicate stereochemistry, none are shown.
2. Isoprene-derived species
Isoprene (2-methyl-1,3-butadiene, C5H8, 1) is the most abundant non-methane hydrocarbon emitted into the Earth's atmosphere with emissions estimated to exceed 500 Tg per year.21–23 Isoprene dominates emissions from many tropical forest ecosystems as well as deciduous plants. It was originally proposed that atmospheric oxidation of isoprene yielded only highly volatile gaseous products that did not contribute to SOA formation.24,25 In 2004, however, Claeys and coworkers reported the synthesis of a mixture of the two diastereomeric compounds, 2-methylthreitol (8) and 2-methylerythritol (9), which were then used in combination with mass spectrometric studies to identify both compounds in field aerosol samples collected in a pristine environment in the Amazon rainforest (Fig. 1).26 While detailed synthesis procedures were not provided, this groundbreaking work tied the formation of low volatility oxidation products formed in the aerosol phase under low nitrogen oxide (NOx = NO + NO2) concentrations (generally considered to be pristine conditions) to the photooxidation of isoprene, resulting in a rapid growth of studies aimed at understanding the role of isoprene-derived aerosol particles in the climate system.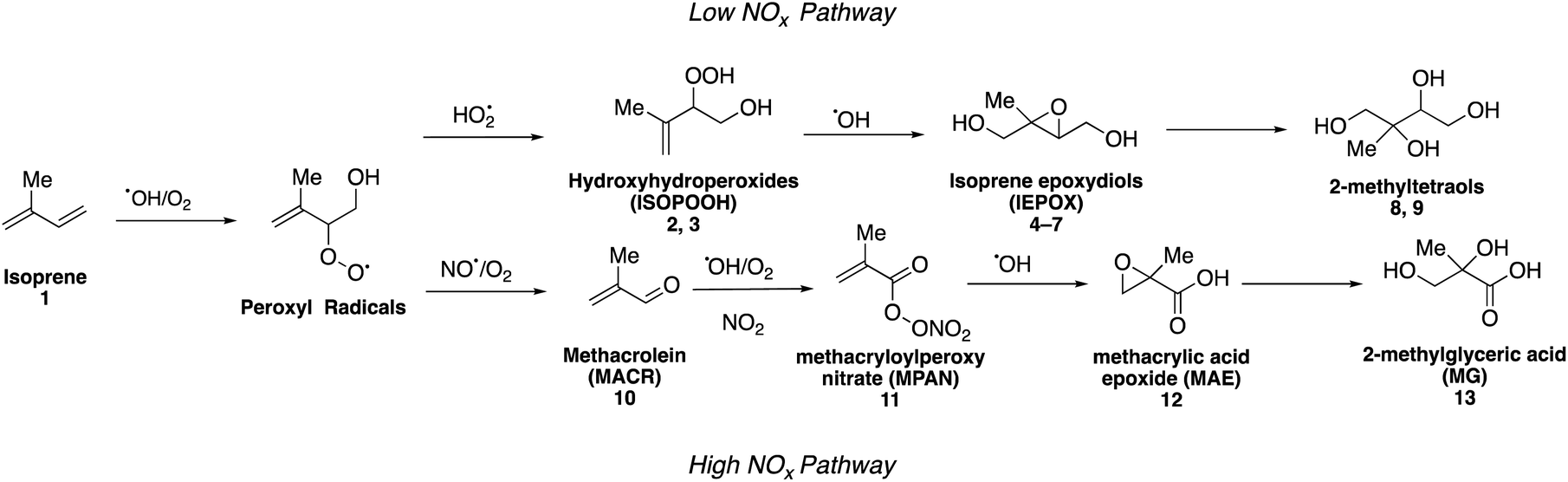 | ||
| Fig. 1 Atmospheric oxidation pathways for isoprene. | ||
Isoprene (1) predominantly reacts with hydroxyl (OH) radicals in the atmosphere, which, along with slower oxidation by nitrate (NO3) radicals and ozone (O3), accounts for up to 50% of the global SOA budget.27–30 Oxidation of isoprene by OH radicals results in the formation of peroxyl radicals (RO2), which can react with other atmospheric oxidants to form organic compounds that contribute to SOA formation (Fig. 1). In the presence of higher concentrations of NOx (a marker for anthropogenic pollution due to combustion processes), these radicals can decompose to form smaller volatile products, such as formaldehyde and methyl vinyl ketone, which results in minimal amounts of SOA particles.10,30–32 Other studies using high NOx conditions have demonstrated that isoprene SOA particles can also form from the oxidation of a variety of isoprene-derived intermediates, including methacrolein (MACR, 10) and methacryloylperoxynitrate (MPAN, 11).31,33–36 In the absence of NOx, these radicals can react with hydroperoxy radicals (HO2) to form various isomers of isoprene hydroxyhydroperoxides (ISOPOOH, 2 and 3),37 which then go on to form isomeric isoprene epoxydiols (IEPOX) compounds (4–7). These epoxides have been proposed as key intermediates in the formation of biogenic SOA particles.33,38–41 Acid-catalyzed uptake and multiphase chemical reactions of epoxide compounds as well as aqueous reactions of MACR (10) and methyl vinyl ketone with sulfate radical anions have been shown to enhance isoprene-derived SOA formation.33,35,42–44
Recent studies have led to the development of a variety of syntheses of authentic standards of intermediates in the atmospheric isoprene oxidation pathway. These standards have been essential in the characterization of isoprene-derived SOA particles and have yielded valuable insight into SOA formation and growth, as outlined in the sections below.
2.1. Isoprene hydroxyhydroperoxides (ISOPOOH)
As previously mentioned, ISOPOOH isomers (2, 3) are considered to be some of the first closed shell reaction products formed from the reaction of isoprene with HO2. These hydroperoxides have very different properties compared to their urban counterparts formed under high concentrations of NOx.45,46 Atmospheric models estimate that only 30% of global emissions of isoprene react with NOx, emphasizing the abundance of ISOPOOH and other intermediates formed under low NOx conditions.38,45,47 The use of synthetic ISOPOOH has only appeared in the literature in the past few years, with the first report in 2014, although this report and others do not include specific synthesis details.48 Various organic synthetic studies have, however, used photooxygenation (via an ene reaction), as well as more recent synthetic methodology involving oxidation by H2O2 catalyzed by phosphomolybdic acid (PMA)49,50 to access similar peroxides.51,52 Chamber experiments involving ISOPOOH isomers, 2-hydroperoxy-3-methyl-but-3en-1-ol (4,3-ISOPOOH, 2) and 2-hydroperoxy-2-methylbut-3-en-1-ol (1,2-ISOPOOH, 3),38,53,54 revealed that reactions with OH radicals can lead to the production of IEPOX at yields greater than 75%.53 ISOPOOH standards were also used to demonstrate that products directly derived from ISOPOOH have the potential to form SOA compared to highly oxidized compounds detected in monoterpene oxidation.54 Access to a synthetic standard also allowed for direct studies of its uptake under atmospheric conditions, which revealed that uptake is slow and reduced by the presence of surface-active organic species on the surfaces of aerosols.41,55,56 Chamber experiments using authentic ISOPOOH standards (2, 3) also revealed an additional route to SOA formation that does not proceed through the formation of the IEPOX intermediate.56,57 These standards continue to be used in both field and chamber studies to better quantify ISOPOOH in the atmosphere and characterize SOA formation pathways from isoprene.48,58–622.2. Isoprene-derived epoxy diols (IEPOX)
Gas-phase IEPOX isomers (4–7) form from the oxidation of ISOPOOH in the atmosphere and laboratory studies indicate that these compounds are likely key intermediates in the production of low volatility products in SOA material derived from isoprene under low NOx conditions.33,38,39,63,64 IEPOX was first detected in 2009 through the synthesis of a surrogate standard (2,3-epoxy-1,4-butanediol, BEPOX) lacking the methyl group present in IEPOX, since authentic standards of IEPOX had not yet been accessed. By quantifying IEPOX using the synthetic BEPOX standard, this important study established that OH-initiated oxidation of isoprene gave high yields (∼50%) of IEPOX and suggested that IEPOX may serve as a precursor to the formation of 2-methyltetraols, C5-alkene triols, dimers, and organosulfates.38While syntheses of two isomers of IEPOX were previously reported in synthetic organic literature,65–67 the first authentic isomers of IEPOX were synthesized and used in atmospheric studies in 2010 (Fig. 2).67
 | ||
| Fig. 2 Synthesis of δ-IEPOX (4). | ||
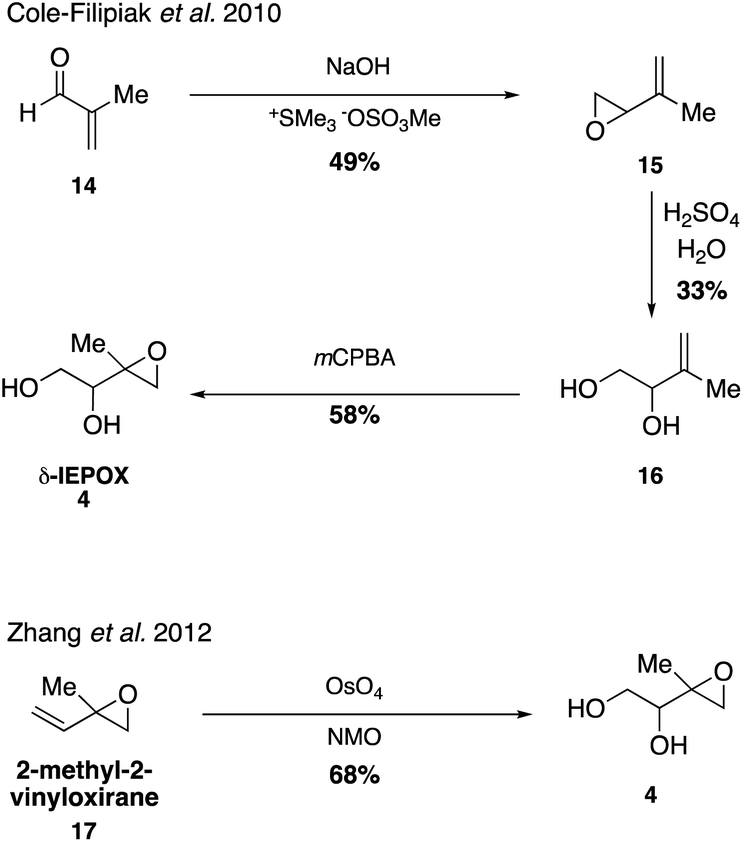
Synthesis of three different isomers of IEPOX confirmed that hydrolysis lifetimes of epoxides in the atmosphere are on the order of SOA lifetimes at atmospherically relevant pH values,67 improving upon similar studies using surrogate compounds.63 While the first syntheses resulted in a mixture of IEPOX isomers, optimization of synthetic routes towards the various isomers of IEPOX in recent years has resulted in a rapid growth of studies using synthetic standards of IEPOX in order to quantify its abundance in the environment as well as analyze effects of atmospheric conditions on its role in SOA formation through both chamber and field studies.32,39–41,68–73 Of the four isomers, δ-IEPOX (4) is the most easily accessed (Fig. 2).
While it was originally synthesized in three separate steps from methacrolein (14) in 2010,67 subsequent syntheses accessed δ-IEPOX (4) in one step through the dihydroxylation of commercially available 2-methyl-2-vinyloxirane (17).74 α-IEPOX (5) can be accessed in two steps either from 2-methyl-2-vinyloxirane (17) via epoxide opening under aqueous conditions followed by epoxidation, or from hydroxyacetone (18) via a vinyl Grignard reaction followed by epoxidation (Fig. 3).74–76cis-β-IEPOX (6) can also be accessed in two steps through the reduction of 3-methyl furan-2(5H)-one (20) or citraconic anhydride (21), followed by epoxidation (Fig. 4).74,75,77 Synthetic routes were also established to access isomeric tetrahydrofurans (Fig. 5, 23 and 24), which were detected as rearrangement products of IEPOX isomers in acidic aerosols during chamber experiments.39,74,78 A mixture of cis- and trans-methyltetrahydrofurans (22, 23) can also be accessed in four steps from dihydroxytetrahydrofuran (25) and then separated to access the pure isomers by column chromatography.74
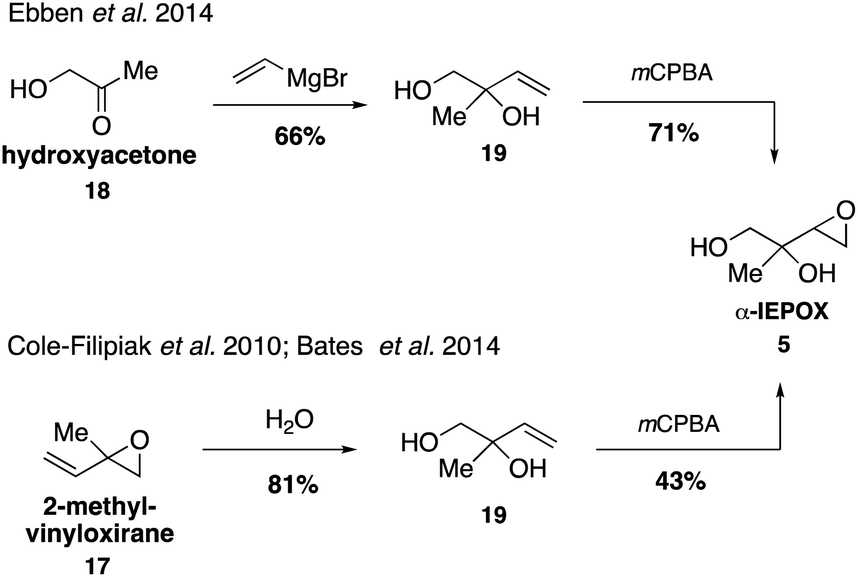 | ||
| Fig. 3 Synthesis of α-IEPOX (5). | ||
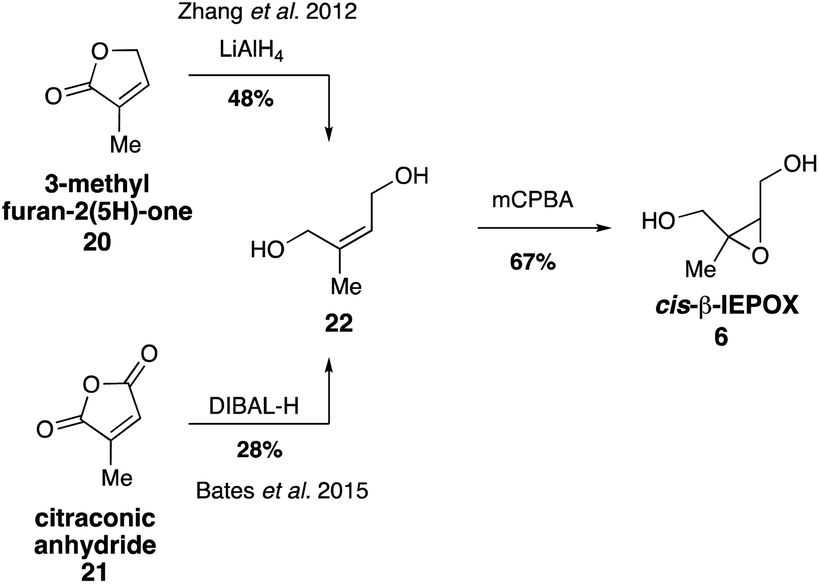 | ||
| Fig. 4 Synthesis of cis-β-IEPOX (6). | ||
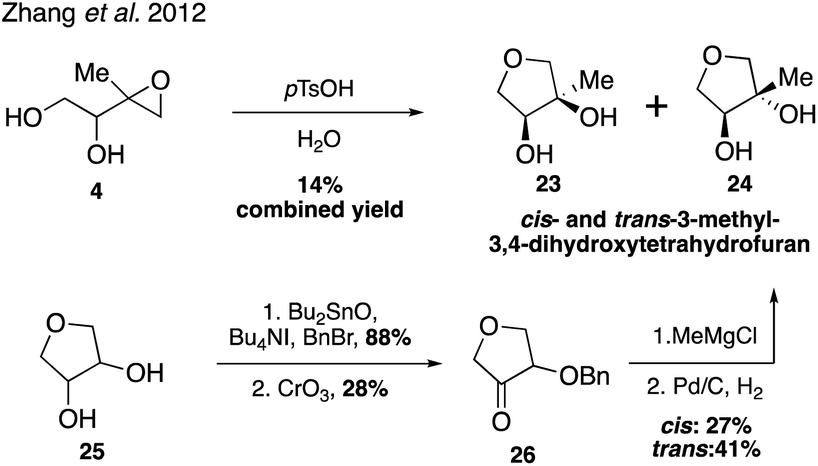 | ||
| Fig. 5 Synthesis of tetrahydrofurans 23 and 24. | ||
Trans-β-IEPOX (7) requires the greatest number of synthetic steps out of all the epoxides, but access to standards of all four isomers has determined that it is the longest-lived and most abundant isomer in the atmosphere (Fig. 6).76,79Trans-β-IEPOX (7) can be accessed in four steps from isoprene (1) by a procedure involving initial bromination, hydrolysis and epoxidation,67 or in five steps from 3-methyl-2-buten-1-ol (prenol, 30)74,75 by a route involving allylic oxidation followed by epoxidation. Synthetic trans-β-IEPOX (7) was observed to have enhanced surface activity and was tentatively identified on the surfaces of isoprene-derived laboratory aerosols using a surface-specific vibrational spectroscopy technique, sum frequency generation (SFG) spectroscopy.75,80 These results indicate that higher concentrations of trans-β-IEPOX at the surfaces of aerosols may significantly decrease supersaturation ratios and increase the propensity of SOA to form cloud condensation nuclei (CCN). Additional studies have shown that uptake of trans-β-IEPOX (7) at the surface of aerosols accounts for half of the particle-phase material produced from isoprene photooxidation in chamber studies.79
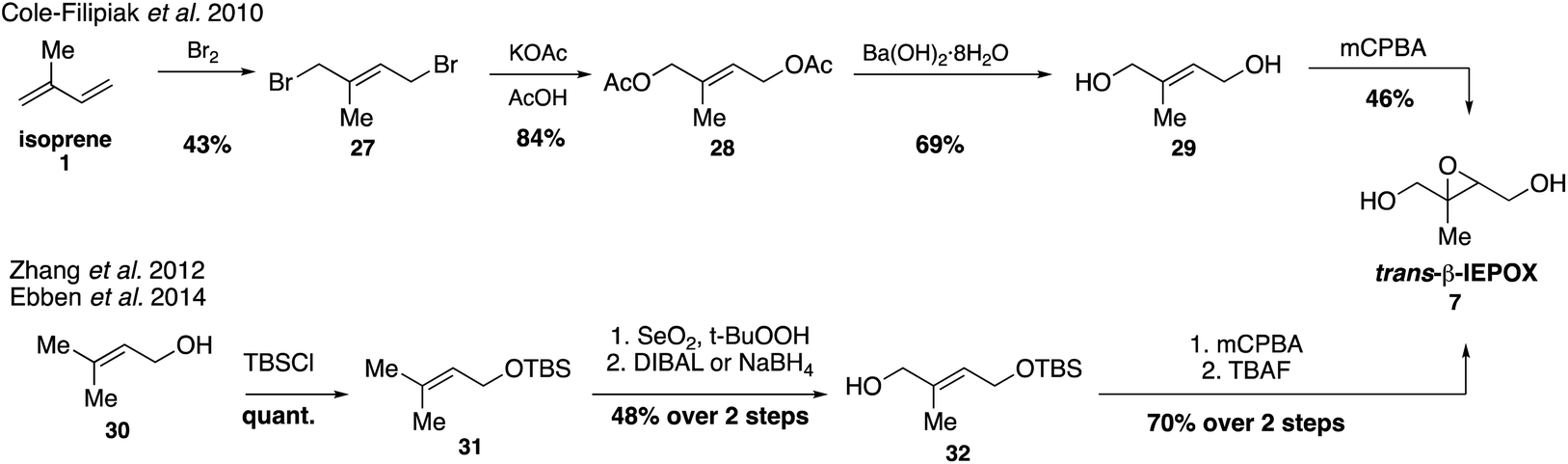 | ||
| Fig. 6 Syntheses of trans-β-IEPOX (7) from either isoprene (1) or prenol (30). | ||
Access to greater quantities of each of the IEPOX isomers has led to a more detailed understanding of their physical properties, their rates of chemical reactions with other atmospheric constituents, and has also allowed for further identification of higher order oxidation products in the atmosphere.55,60,67,68,75,76,79–82
2.3. 2-Methyltetraol diastereomers
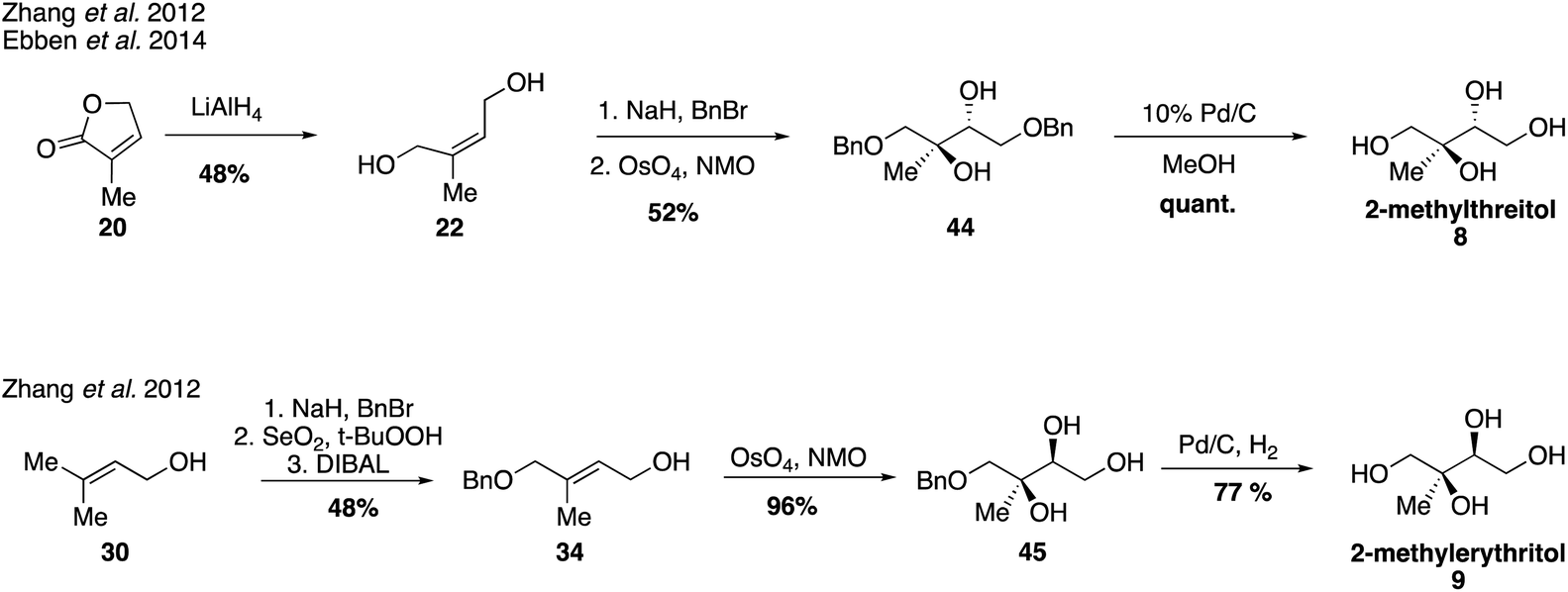
Since their first synthesis in the atmospheric community in 2004, 2-methyltetraol standards have been used ubiquitously in atmospheric studies and have been recognized as definitive markers of aerosols derived from isoprene detected in a variety of regions.26,31,64,83–89 While the first synthetic route towards these standards yielded a mixture of the two diastereomers 2-methylthreitol (8) and 2-methylerythritol (9), additional routes have since been developed to access pure standards of each possible stereoisomer.Prior to studies involving the atmospheric chemistry of 2-methyltetraols, an enantioselective synthesis of 2-methylthreitol (8) and 2-methylerythritol (9) was published in 2000 in order to investigate their biosynthesis in plant terpenoids (Fig. 7).90 2-Methylthreitol (8) and 2-methylerythritol (9) were synthesized in six and seven steps, respectively, from dimethylfumarate (33).90 For the case of compound 8, control of absolute stereochemistry was achieved using a Sharpless epoxidation of allylic alcohol 34 to deliver epoxide 35 with high enantioselectivity (93:7 er). Epoxide opening and debenzylation delivered the optically enriched tetraol (2S,3R)-2-methylthreitol (8). Enantioselective access to the diastereomeric tetraol 9 was accomplished through a Sharpless dihydroxylation of enoate 36, which produced diol 37 in a 92:8 ratio of enantiomers. Reduction of the ester and cleavage of the benzyl ether yielded optically enriched (2S,3R)-2-methylerythritol (9). Several years later, in 2007, Moen and coworkers established a chemoenzymatic route that provided access to all four stereoisomers of the methyltetraol compounds through an enantioselective resolution (Fig. 8).91 Access to all four of these isomers was highly valuable and enabled subsequent studies determining enantiomeric compositions in atmospheric aerosols.92,93
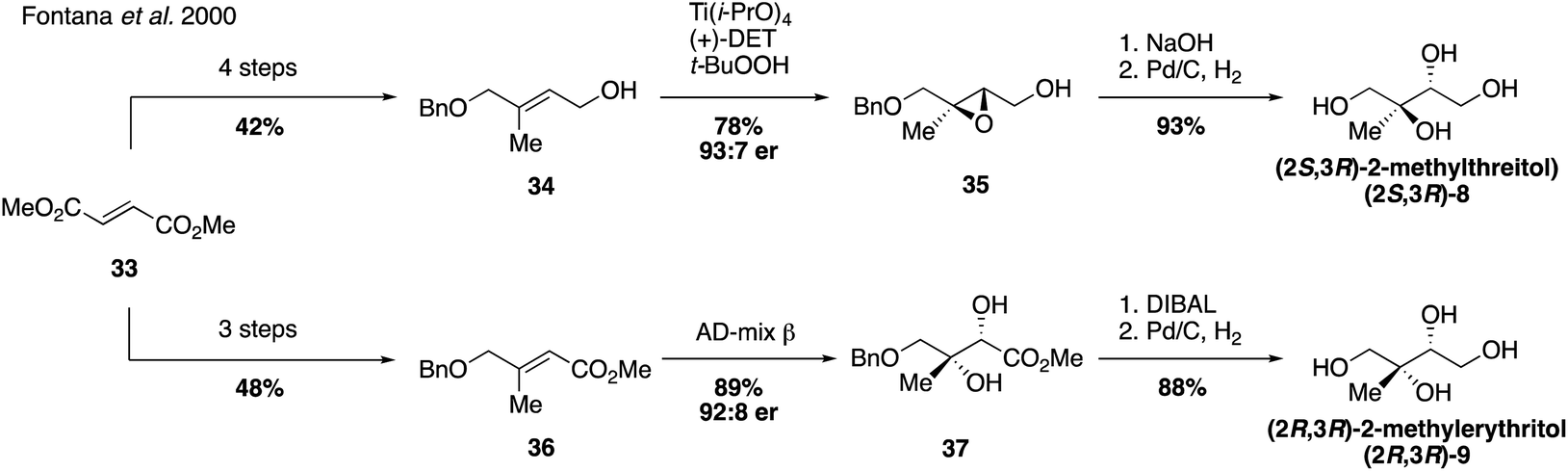 | ||
| Fig. 7 Enantioselective synthesis of the diastereomeric 2-methyltetraols. | ||
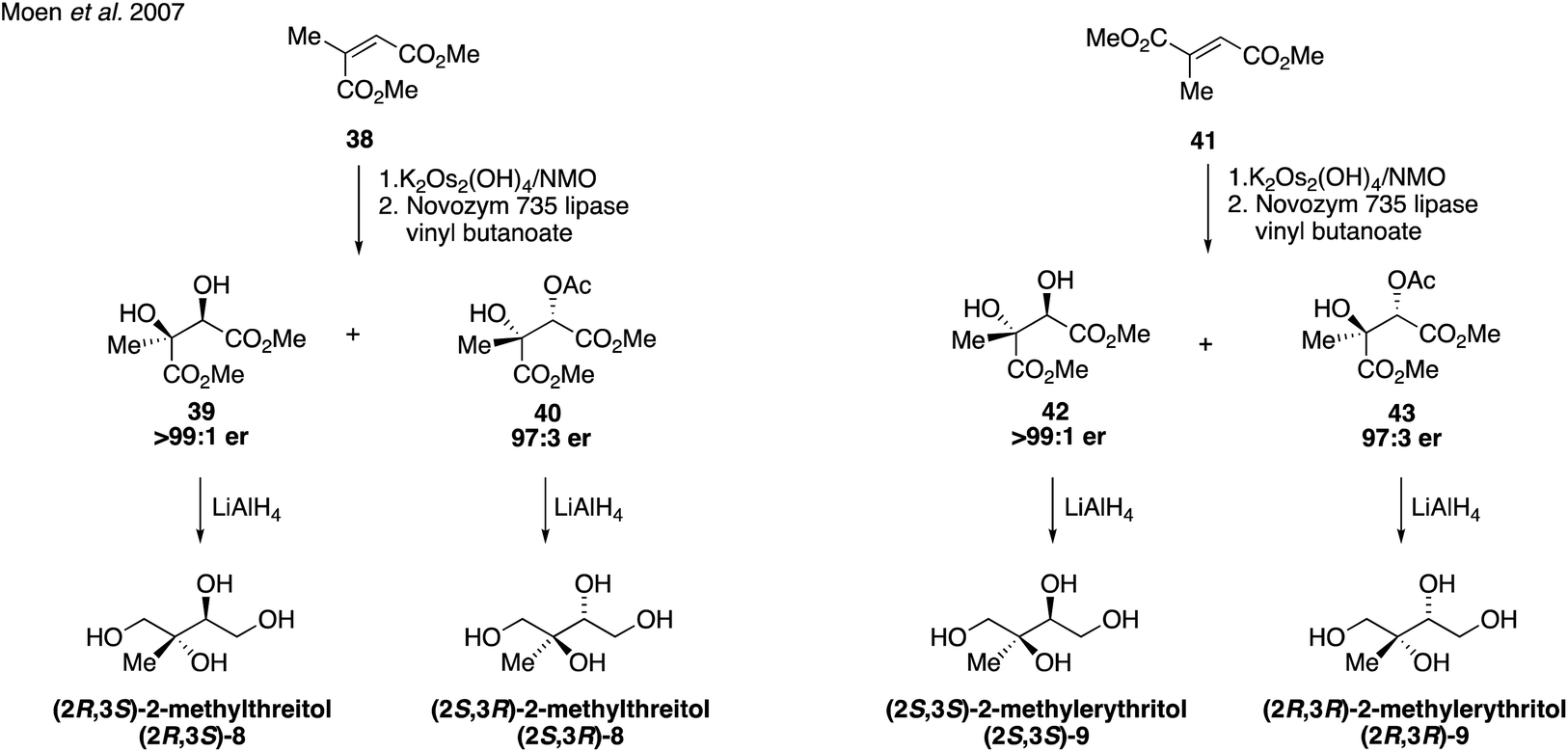 | ||
| Fig. 8 A chemoenzymatic approach to accessing all stereoisomers of the 2-methyltetraols. | ||
Additional synthetic work fueling further atmospherically-relevant research focused on efficient routes to racemic material (Fig. 9), generating 2-methylthreitol (8) in four steps from 3-methyl furan-2(5H)-one (20) and 2-methylerythritol (9) in five steps from 3-methyl-2-buten-1-ol (30).75 Access to these standards continues to aid in the study of isoprene oxidation products formed under low NOx conditions and their quantification in field and chamber studies.33,36,39,63,67
 | ||
| Fig. 9 Efficient access to racemic 2-methyltetraols that fueled atmospheric chemistry studies. | ||
2.4. Isoprene-derived C5H10O3 Products
Reactive uptake products possessing the molecular formula C5H10O3 have served as tracers for the study of isoprene-derived SOA, but the structural identity of these species has remained elusive until recently. In order to provide authentic standards of two possible C5H10O3 candidates the Gold lab established synthetic routes to access unsaturated triol 46 and hemiacetal 51 (Fig. 10).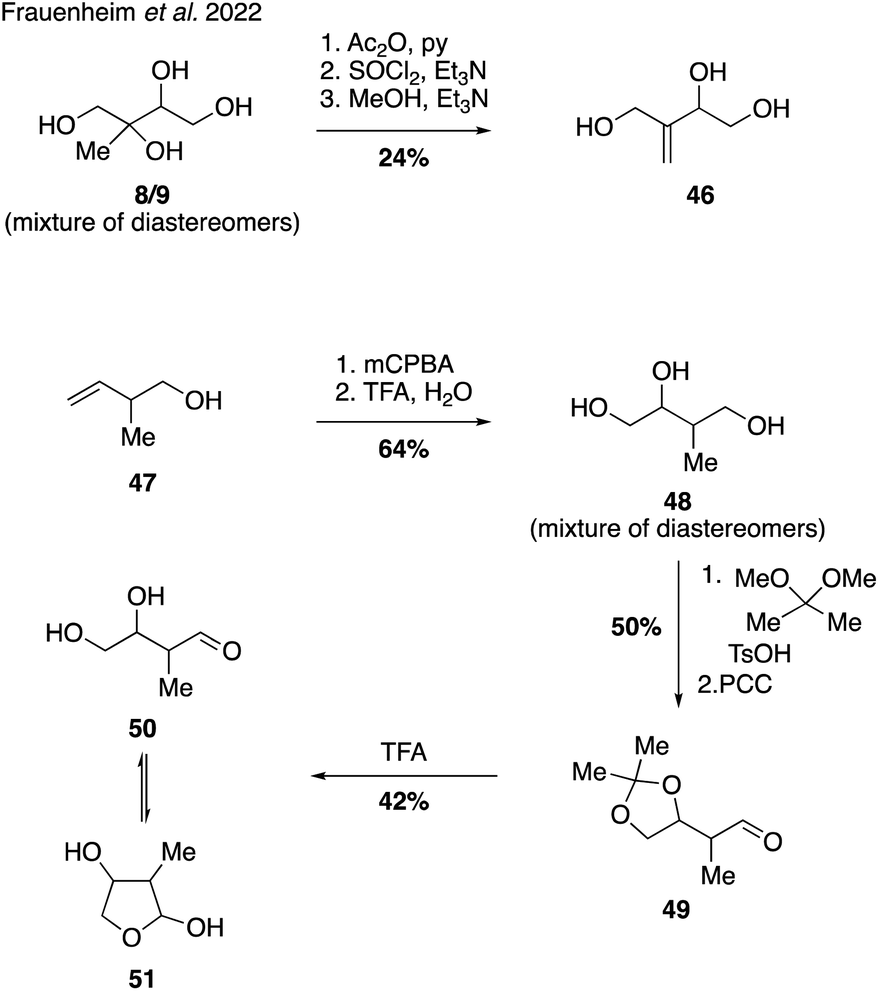 | ||
| Fig. 10 Synthesis of “C5H10O3” products. | ||
The synthesis of triol 46 was conducted using a mixture of 2-methyltetraol diastereomers (i.e., 8/9) as the starting material, which was first treated with acetic anhydride to protect the less hindered primary and secondary alcohols. Dehydration of the free tertiary alcohol using thionyl chloride followed by removal of the acetate protecting groups produced the desired triol 46 in good overall yield, albeit as a mixture of diastereomers. The synthesis of hemiacetal 51 commenced from alkene 47, which underwent epoxidation and hydrolysis to yield triol 48. Preferential formation of the 5-membered cyclic acetonide followed by oxidation using PCC delivered aldehyde 49, which was treated with trifluoroacetic acid to cleave the protecting group. Analysis of the NMR spectral data of the product showed no diagnostic aldehyde resonances associated with the open-tautomer 50, indicating an equilibrium strongly favoring the cyclic hemiacetal 51. Access to these two authentic standards allowed confirmation of their presence in laboratory-generated IEPOX SOA and in atmospheric SOA samples. Quantitation of chamber-derived yields led the authors to tentatively estimate that these C5H10O3 isomers could contribute 8.7 Tg C per year to the atmosphere.
2.5. Methacrolein-derived oxidation products
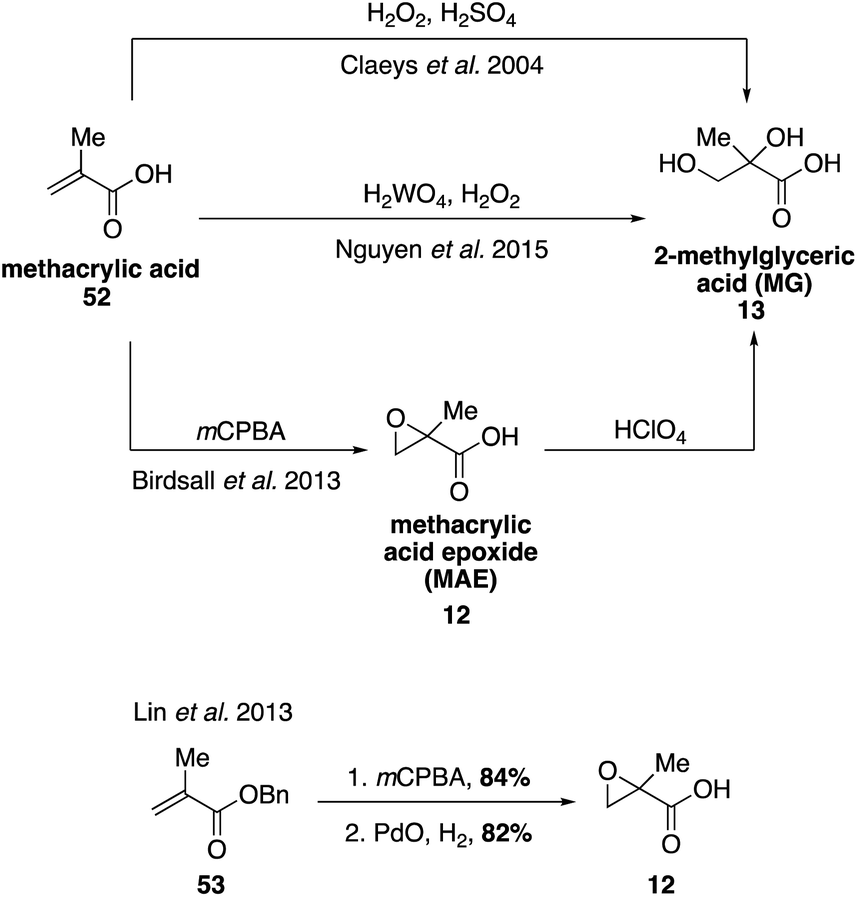
Emission of NOx from anthropogenic sources can alter the oxidation pathways of biogenic VOCs in the atmosphere. Laboratory experiments have confirmed that under high NOx conditions, essentially all SOA material derived from isoprene originates from photochemistry of its first generation oxidation product, methacrolein (MACR, 10).33,39,94–96 Oxidation of MACR (10) forms methylglyceric acid (MG, 13) and methacrylic acid (52), which have both been detected in both laboratory and field SOA samples.31,36,97–99 Methacrylic acid epoxide (MAE, 12) was also identified as a precursor to the formation of MG (13).100 Access to synthetic standards of many of these oxidation products has allowed for detailed studies on the mechanisms leading to formation of 13 as well as other dimers, oligomers, and organosulfates or nitrates that are structurally related to MG and detected in aerosol samples.39,101–106The commercial availability of methacrylic acid (52) has facilitated the synthesis of higher order oxidation products derived from methacrolein (Fig. 11). MG (13) was first prepared from methacrylic acid (45) using H2O2 and sulfuric acid,107 and later using H2O2 and H2WO4.108 Epoxidation of methacrylic acid (52) resulted in the isolation of MAE (12), from which MG was also accessed via acid-catalyzed hydrolysis of 12.109 MAE (12) has also been synthesized in two steps from benzyl methacrylate (53) via epoxidation followed by hydrogenation.100
 | ||
| Fig. 11 Synthesis of methacrolein-derived oxidation products. | ||
2.6. 2-Methyl-3-buten-2-ol (MBO)
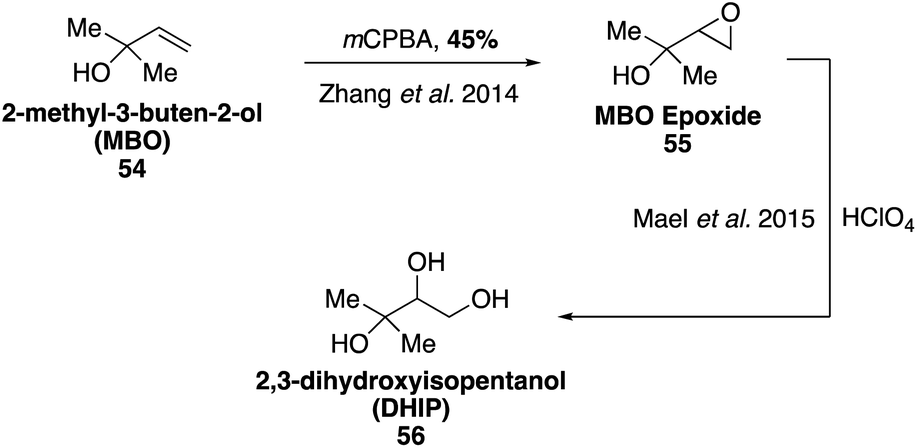
2-Methyl-3-buten-2-ol (MBO, 54) is a structurally similar BVOC to isoprene that is emitted by a few specific species of pine tree and has been demonstrated to contribute to photooxidation chemistry in the atmosphere.110,111 Estimates of global MBO emissions are only around 1.2–2.2 Tg per year,23,112 however MBO has been shown to affect local production of ozone and hydroxyl radicals in some forested areas.113 Major oxidation products detected in SOA constituents formed from the photooxidation of MBO (54) include 2-hydroxy-2-methylproponal, glycoaldehyde, 2,3-dihydroxyisopentanol (DHIP, 56), and MBO-derived organosulfates.114–117 Glycoaldehyde can undergo further oxidation to form glyoxal products, which have been shown to contribute to SOA formation.118–121 SOA formation from MBO photooxidation is also enhanced at higher aerosol acidities and inhibited in the presence of nitric oxide.39,116,117 Ozonolysis of MBO has been shown to form only very slight amounts of aerosols (with yields less than 1%).122MBO (54) is commercially available and has been used to synthesize MBO epoxide ((3,3-dimethyloxiran-2-yl)methanol, 55) through epoxidation using mCPBA (Fig. 12).115 Access to MBO epoxide 55 allowed for the study internal hydrogen transfer (H-shift), which may lead to the formation of epoxides.115,123 Synthesis of MBO-derived epoxide 55 also enabled studies for the reactive uptake of MBO epoxide resulting in SOA formation as well as detailed studies on the formation of organosulfates and organonitrates from MBO.115,124 DHIP (56) was also later synthesized from MBO epoxide using HClO4.124
 | ||
| Fig. 12 Synthesis of MBO-derived oxidation products. | ||
3. Monoterpene-derived SOA material
Monoterpenes represent an important fraction of VOCs emitted from biogenic sources in the atmosphere, with global emission rates estimated to be over 150 Tg per year.23 The lifetime of monoterpenes is quite short due to rapid reactions with hydroxyl radicals (OH), nitrate radicals (NO3), and ozone (O3), generating a complex mixture of products including carbonyls, alcohols, and carboxylic acids.9,125,126The most abundant monoterpene emitted in the troposphere is α-pinene (57, 66.1 Tg per year), with dominant emissions from coniferous trees in Boreal forests.23,127 Ozonolysis of α-pinene is an important reaction pathway,126 with ozone accounting for approximately 40% conversion of α-pinene emitted in the atmosphere.128 The oxidation of α-pinene by ozone has become one of the most well studied SOA systems in the atmospheric community.129–135 A variety of multifunctional gas phase and particle phase products have been identified, however pathways to higher order dimers and oligomers remain elusive.136–140 Recently, a class of extremely low-volatility VOCs (ELVOCs) was identified as an important component of aerosols derived from α-pinene ozonolysis.141–143 However, structural identification of ELVOCs and their formation are still uncertain and syntheses of authentic standards for these proposed products have not yet been reported.
Other abundant monoterpenes in the atmosphere whose oxidation products also contribute to SOA formation include β-pinene, limonene and 3-carene with emissions of 18.9, 11.4, and 7.1 Tg per year, respectively.23 A variety of authentic standards of oxidation products derived from α-pinene and β-pinene have been synthesized and aided in the identification and quantification of SOA constituents in both laboratory and field studies. While there are several studies focused on the characterization of SOA derived from limonene and 3-carene, only a few have used authentic synthetic standards to identify oxidation products.125,144–150 Here we focus on the synthetic chemistry of pinene-derived oxidation products, which is much more developed.
3.1. α-Pinene- and β-pinene-derived oxidation products
Pinonaldehyde (59) was identified early on as a major first generation oxidation product of α-pinene ozonolysis and was first synthesized as a standard for atmospheric studies in 1997 via cleavage of commercially available pinanediol (58) using periodic acid (Fig. 13A).151 A shorter synthetic route involves ozonolysis of α-pinene with a reductive workup.152 Pinonaldehyde (59) was identified as the most abundant compound in SOA particles collected over Boreal forests and has been used ubiquitously in field and chamber studies.153–159 Yields of pinonaldehyde (59) from oxidation of α-pinene (57) are proposed to be as high as 87%,160,161 and access to pinonaldehyde (59) has allowed for detailed investigations of its reactivity and physical properties.162–166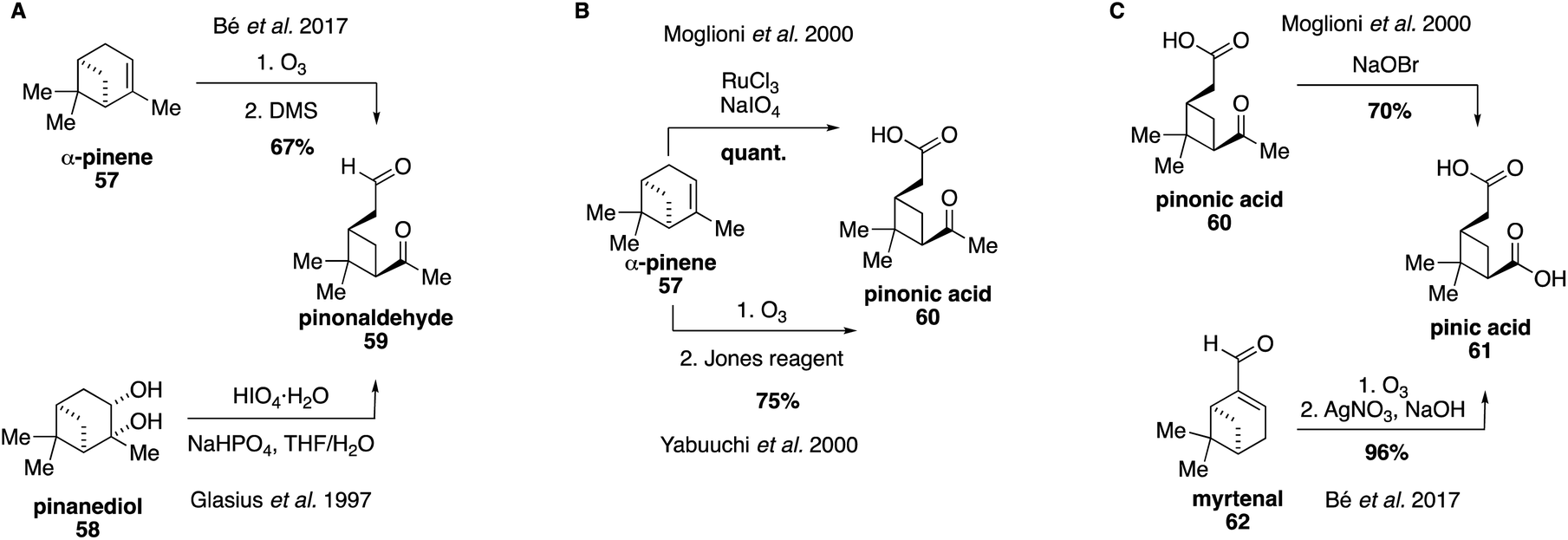 | ||
| Fig. 13 Synthetic access to pinonaldehyde, pinonic acid, and pinic acid. | ||
Both pinonic acid (60) and pinic acid (61) have been identified as atmospheric tracers for α-pinene oxidation and both are commercially available.167 However, synthetic routes to access each compound have also been developed, which can provide access to more pure samples (Fig. 13B and C).146,168,169 Pinonic acid (60) can be accessed through ruthenium-catalyzed oxidative cleavage168 or ozonolysis of α-pinene (57) followed by an oxidative workup.170 Pinic acid (61) has been previously synthesized through degradation of methyl ketone 60 using aqueous sodium hypobromite,168 as well as via ozonolysis of myrtenal (62).152 These compounds have also been used as surrogate standards to identify organic acids and dimers derived from α-pinene since authentic standards have not yet been synthesized.131,171 Recent work has expanded on these efforts by focusing on the synthesis of α-pinene-derived norpinic and norpinonic acids,172 as well as peroxypinic acids as proxies for highly oxygenated molecules in biogenic SOA.173 For instance, in 2018, a mixture of diperoxypinic acid and two different monoperoxypinic acids was synthesized from pinic acid (55), however, no separation of the peroxy acids was performed, likely due to the instability of such compounds.173 Moreover, recently, α-pinene-derived α-acyloxyalkyl hydroperoxides have been accessed via ozonolysis of α-pinene (57) in the presence of an excess amount of various organic acids.14,174
Hydroxy-pinonaldehyde (65) and hydroxy-pinonic acid (66) are also oxidation products that are proposed to form from more highly substituted Criegee radicals during ozonolysis of α-pinene.130 Both compounds have been detected in numerous field and chamber aerosol studies131,175–180 and were first synthesized in 2002 from the oxidative cleavage of myrtenol (63), exploiting aldehyde 64 as a common intermediate (Fig. 14).181
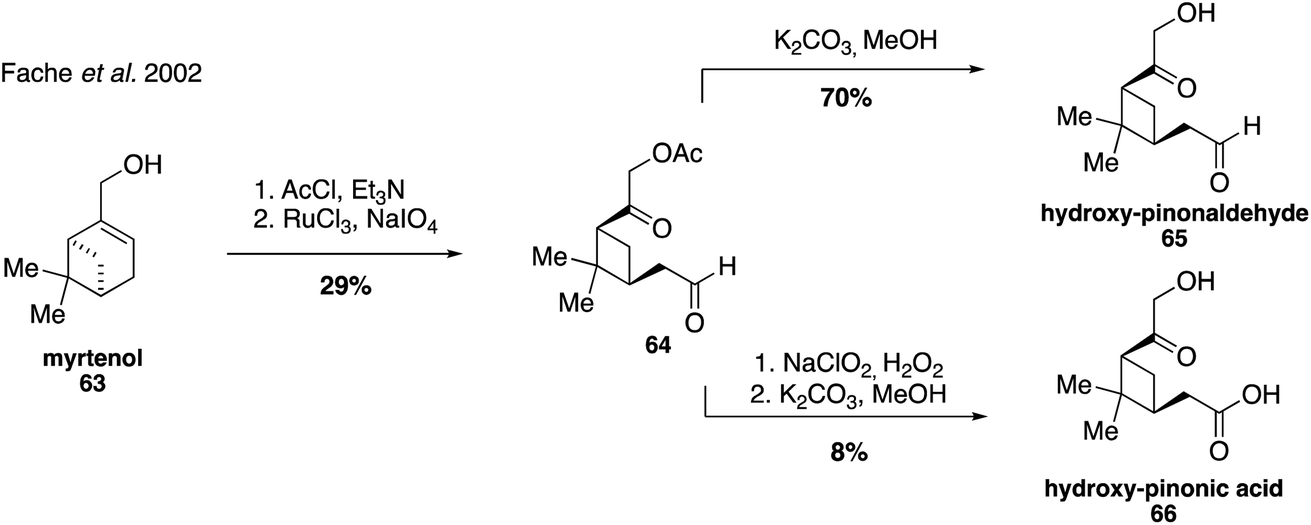 | ||
| Fig. 14 Synthetic access to hydroxyl-pinonaldehyde and hydroxy-pinonic acid. | ||
Lactone-containing terpenoic acids derived from α-pinene and β-pinene have recently attracted a great deal of attention due to their potential relevance in dimer formation as well as other high-MW SOA products.131,134,135,182,183 They were first detected in 2009 through the use of authentic synthetic standards.129 Terpenylic acid (68) can be prepared starting from pinonic acid (58) by treatment with sulfuric acid, which results in the rearrangement product, homoterpenyl methyl ketone (67), which can then be oxidized to form terpenylic acid (68) using KMnO4 (Fig. 15A).129 Recently, an alternative procedure was reported for the direct synthesis of terpenylic acid (68) by oxidative cleavage of cyclopentene 69.184 Campholenic aldehyde (71) has been synthesized in the literature through a ZnBr2-catalyzed rearrangement of α-pinene oxide (70) in refluxing toluene (Fig. 15B).185,186 These synthetic standards were used to determine that terpenylic acid (68) forms from the atmospheric oxidation of α-pinene (57) and campholenic aldehyde (71). Oxidation of α-pinene (57) leads to the formation of α-pinene oxide (70), which undergoes rearrangement to generate campholenic aldehyde (71).185 Once formed, campholenic aldehyde (71) can further react with ozone to form terpenylic aldehyde, which forms the corresponding acid upon additional oxidation.187,188
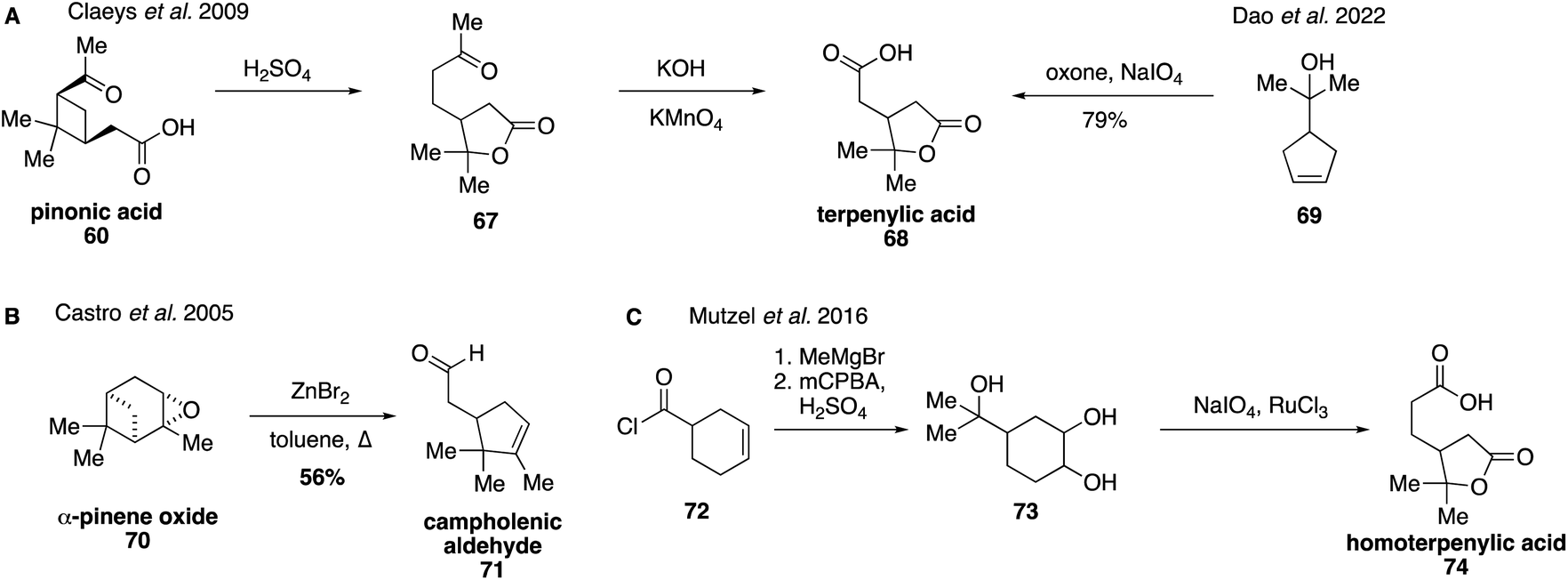 | ||
| Fig. 15 Synthetic access to terpenylic acid, campholenic aldehyde, and homoterpenylic acid. | ||
Homoterpenylic acid (74) was accessed in three steps from 3-cyclohexene-1-carbonyl chloride (72) through a series of oxidations (Fig. 15C).147 In addition to these species, diaterpenylic acid acetate (78) was also accessed synthetically for atmospheric studies and identified as products formed from both photooxidation and ozonolysis of α-pinene.129,134,189 Diaterpenylic acid acetate (78) was synthesized in eight steps starting from dimethyl malonate (75) (Fig. 16).183 A modification on this route was reported recently, which included more detailed experimental conditions.190 Access to these synthetic standards revealed that an abundant MW 358 dimer compound detected in field and chamber studies140,191–194 may be a dimeric compound formed from pinic acid and terpenylic acid (or related terpenoic acids).129,134
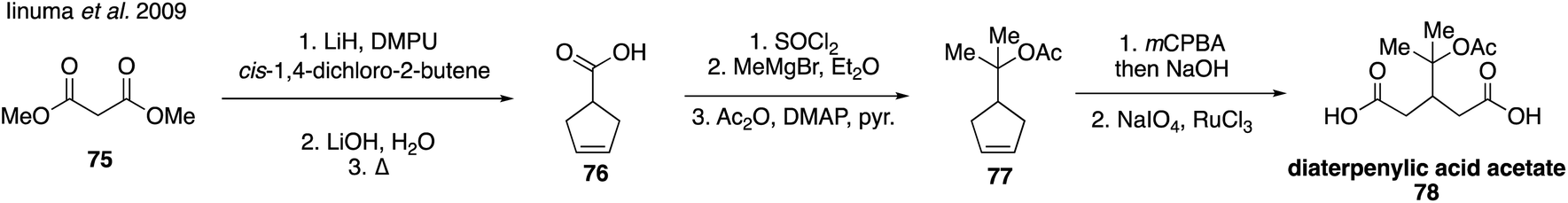 | ||
| Fig. 16 Synthesis of diaterpenylic acetate. | ||
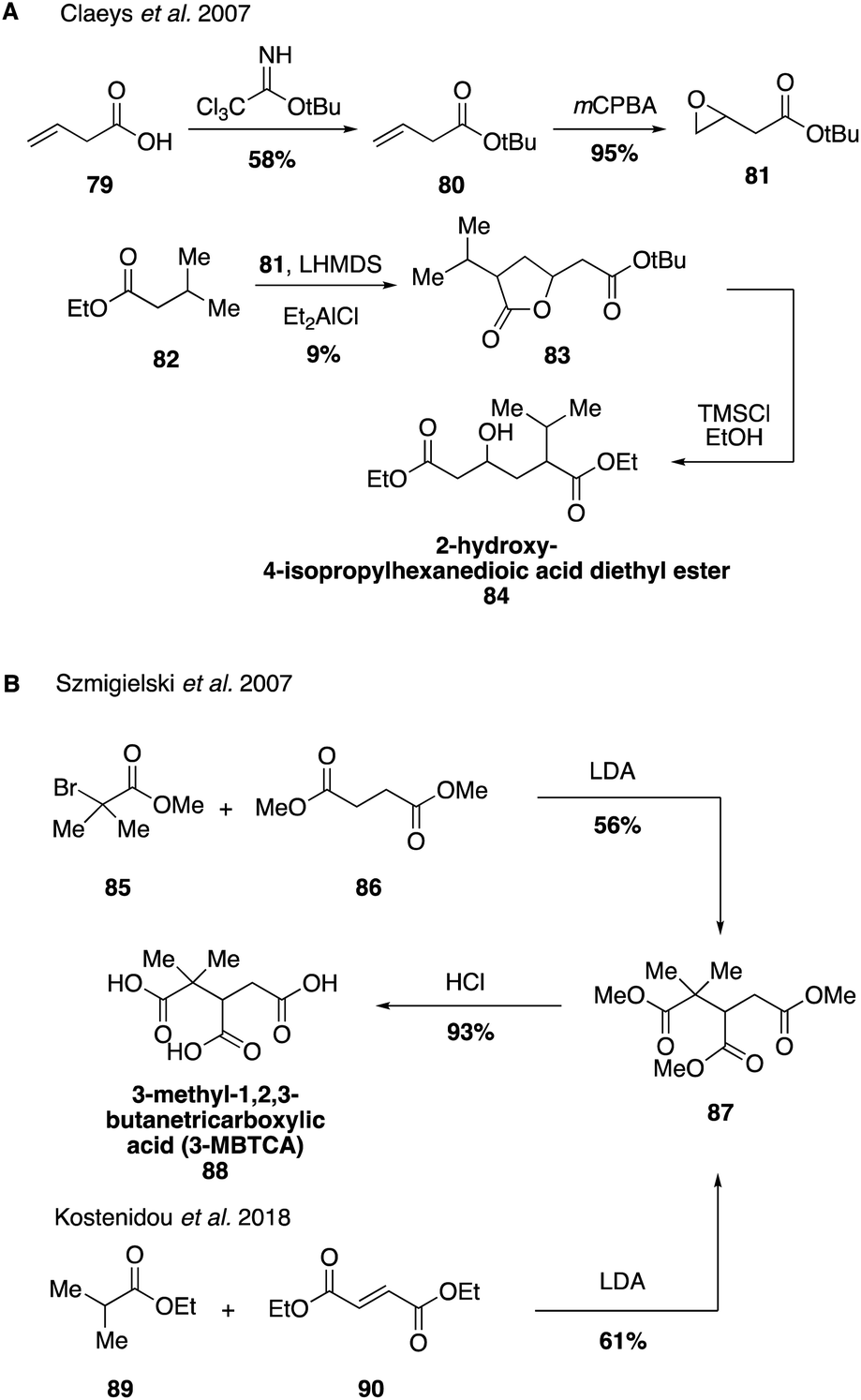
Routes to other highly oxidized and polar species derived from pinene have also been reported (Fig. 17). Hydroxycarboxylic acids have been identified as tracers for the photooxidation of α-pinene.84,195,196 2-Hydroxy-4-isopropylhexanedioic acid was also identified in atmospheric aerosols as a higher order reaction product derived from pinic acid (61). Its structural elucidation was achieved through the synthesis of the ethyl di-ester derivative 84, which can be formed in four steps from acid 74 (Fig. 17A).195 3-MBTCA (3-methyl-1,2,3-butanetricarboxylic acid, 88) is formed by photooxidation of cis-pinonic acid (58) and its structure was unambiguously identified through the synthesis of an authentic standard in 2007.195 However, it was first detected from aerosol samples from Amazonia and Belgium in 2000 and 2002.197,198 3-MBTCA (88) can be generated in two synthetic steps through the nucleophilic reaction between methyl-2-bromoisobutyrate (85) with dimethylsuccinate (86) to form the trimethyl ester 87, which can undergo acid hydrolysis to yield the authentic standard of 3-MBTCA (Fig. 17B).199 Alternatively, triester 87 may be accessed by the conjugate addition of ethyl isobutyrate (89) to diethyl fumarate (90) in a more efficient manner.190,200 Since its synthesis, 3-MBTCA (88) has been used in a variety of field and chamber studies as a tracer for the chemical aging of biogenic SOA particles by OH radicals and its formation mechanism from cis-pinonic acid has been studied in detail.201–203
 | ||
| Fig. 17 Synthesis of highly oxidized species. | ||
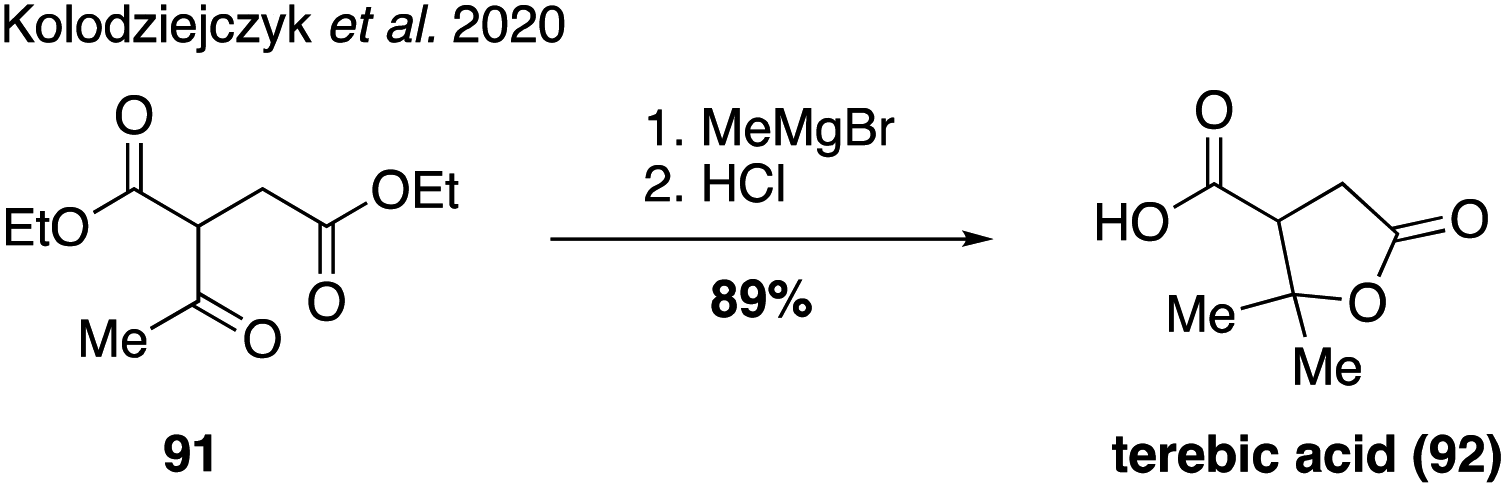
In a recent report investigating the physiochemical properties of several pinene-derived oxidation products, Szmigielski and coworkers reported a concise synthesis of terebic acid (92) from diethyl acetylsuccinate (91, Fig. 18).190 Access to this compound allowed a detailed investigation into its thermal properties, water solubility and acidity.
 | ||
| Fig. 18 Synthesis of terebic acid. | ||
3.2. α-Pinene-derived dimers
An increasing number of field and laboratory studies have reported that α-pinene-derived dimers and higher order oxidation products formed from accretion reactions may make up a significant portion of the total organic mass present in atmospheric aerosol.171,204,205 However, the diversity of monomer precursors and the chemical transformations that couple them together presents a great challenge for structural elucidation and analysis of their relevant properties.194,206–208 As such, the vast majority of dimers and oligomers derived from α-pinene, such as those proposed to form via aldol, ester, or anhydride pathways, have yet to be synthesized. Two recent studies, however, have provided initial examples of synthetic pathways leading to pinene-derived dimers to fuel investigations in to their physical properties. The first of these reports established access to three ester dimers (i.e., 96, 97 and 98) using (+)-cis-pinonic acid (60) as a starting material (Fig. 19).209 Diacid 93 was prepared by oxidation of the methyl ketone within 60, while a Steglich esterification of 60 followed by reduction delivered benzyl ester 94. Secondary alcohol 95 was accessed from 60 by reduction with sodium borohydride. Both 94 and 95 were formed as mixtures of epimeric alcohols of which the major isomer was used for subsequent reactions, but whose stereochemistry was not assigned. Benzyl ester 94 was coupled with either 93, 60 or 95 to deliver ester dimers 96, 97 and 98, respectively. While these three pinene-derived esters are not thought to be naturally occurring in the atmosphere they served as useful model compounds to investigate the effect of different functional groups and molecular size on the efficiency of electrospray ionization mass spectrometry (ESI-MS) applied to monoterpene SOA material. The authors found that the ionization efficiencies of the dimer species were 19–36 times higher than cis-pinonic acid itself, providing evidence suggesting that the mass contribution of pinene dimers to the fraction of SOA material has been significantly overestimated in prior studies.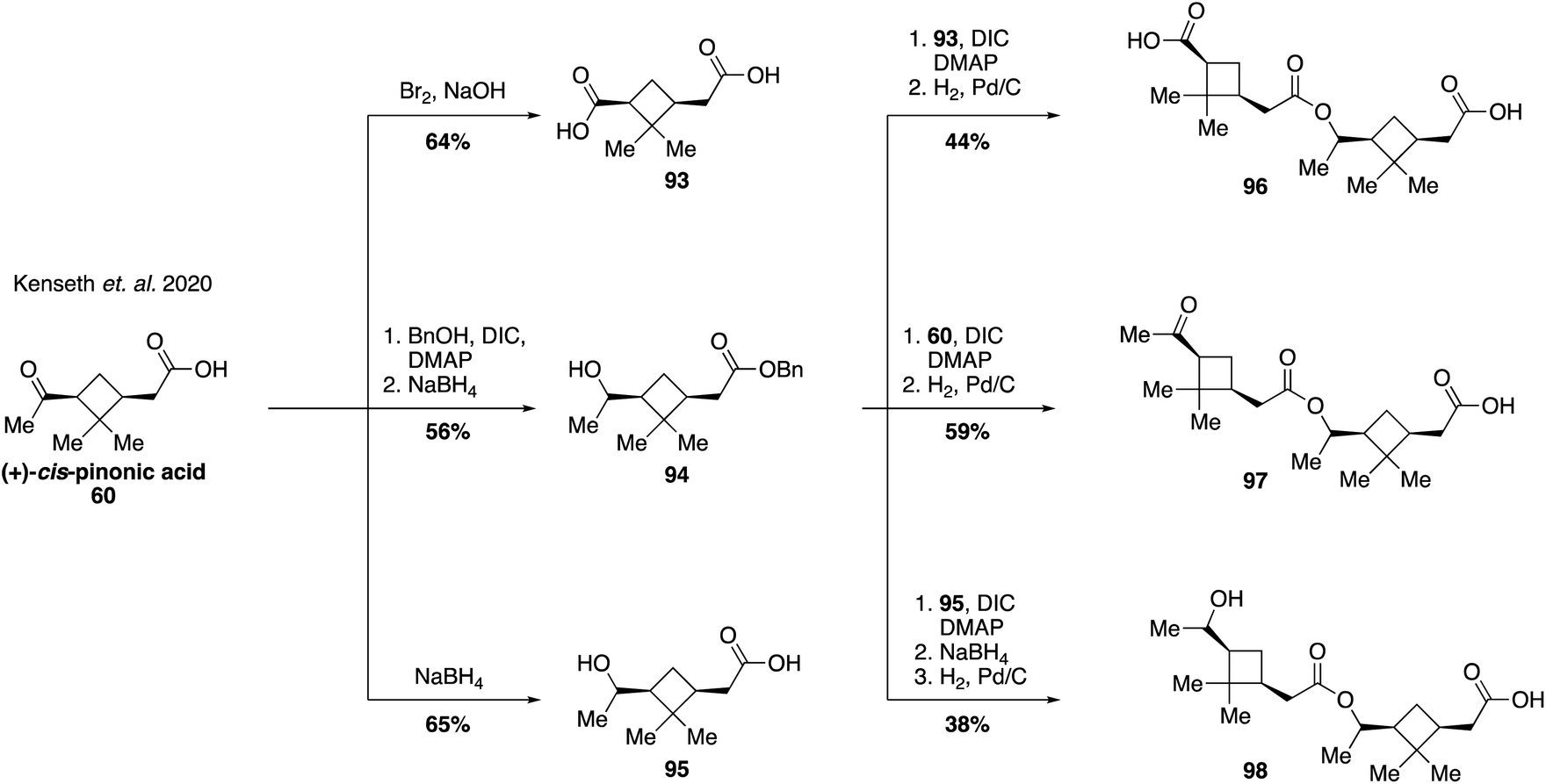 | ||
| Fig. 19 Synthesis of pinene-derived ester dimers as model compounds for mass spectroscopy studies. | ||
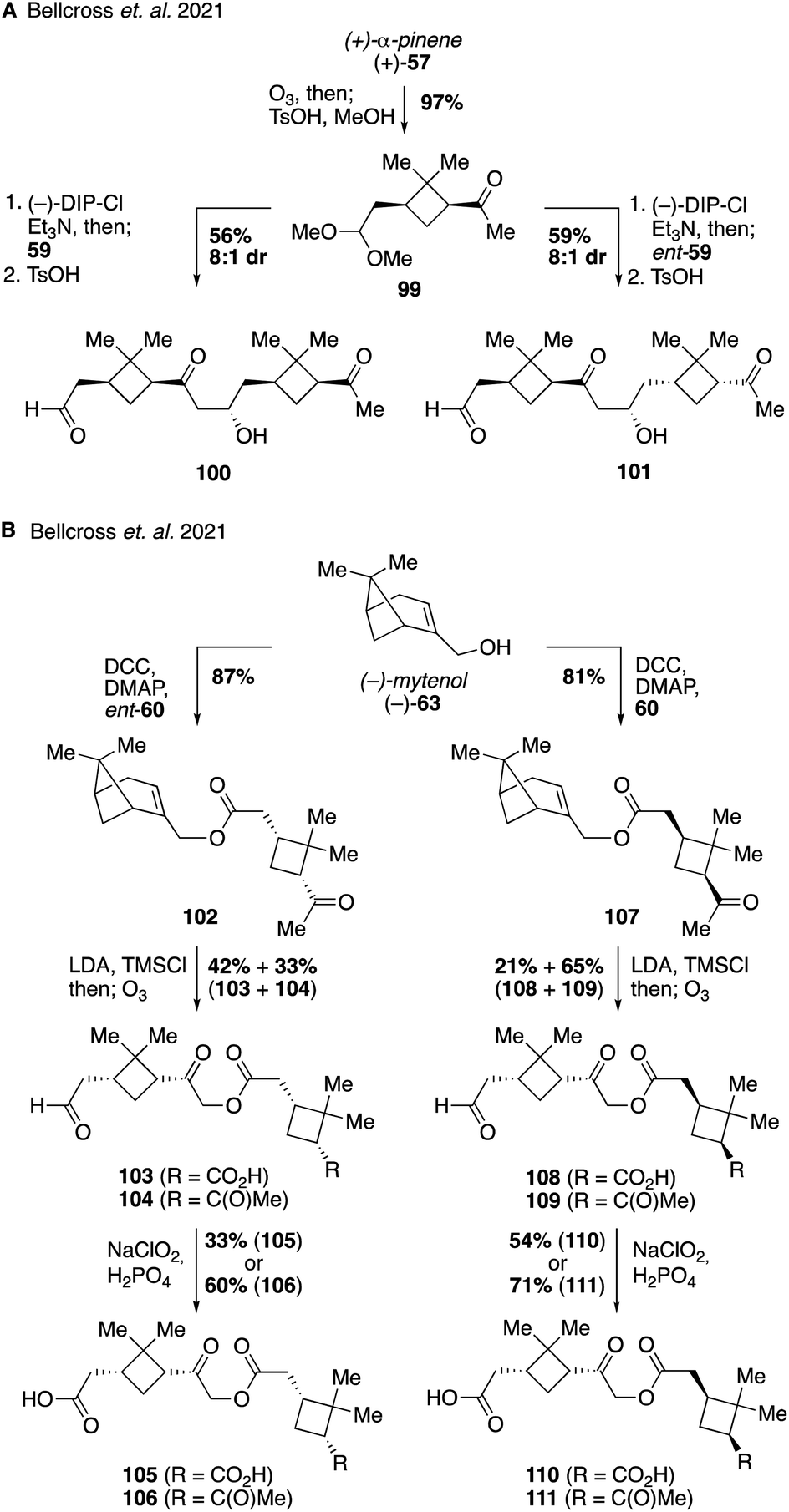
The second study on pinene-derived dimers targeted both aldol and ester-based dimers with the specific goal of investigating the influence that backbone stereochemistry might have on the physical properties of the compounds (Fig. 20). Access to each of the aldol diastereomers 100 and 101 was accomplished from protected pinonaldehyde derivative 99 through the use of Paterson's procedure for stereocontrolled aldol reactions via (−)-B-chlorodiisopinocampheylborane [(−)-DIP-Cl] derived boron enolates.210,211 Thus, aldol reaction of 99 with either one of the enantiomers of pinonaldehyde provided efficient access to either 100 or 101 in a stereodivergent manner (Fig. 20A). In a similar fashion, stereodivergent synthesis of the ester-linked dimers commenced from (−)-myrtenol (63) as a common starting material (Fig. 20B). An initial Steglich esterification of 63 with ent-pinonic acid (ent-60) gave ester 102 in 87% yield. The methyl ketone within 102 was converted to its enol silane, and the resultant unpurified compound was exposed to ozonolysis conditions to generate a separable mixture of acid-aldehyde 103 and keto-aldehyde 104 in 42% and 33% yield, respectively. Subsequent oxidation provided the corresponding carboxylic acids, 105 and 106 (from 103 and 104, respectively). Access to the diastereomeric suite of compounds (i.e., 107–111) was accomplished using the same sequence of reactions but using the opposite enantiomer of pinonic acid (60) in the initial Steglich esterification. With these compounds in hand the authors investigated their ability to depress the surface tension of water droplets, a property linked to cloud formation in the atmosphere. Dynamic surface tension measurements revealed significant difference between diastereomeric pairs of compounds, indicating that backbone stereochemistry may play an important role in various atmospheric processes and that chirality should be considered more frequently in future models of the climate system.212,213
 | ||
| Fig. 20 Synthesis of pinene-derived aldol and ester dimers. | ||
4. Sesquiterpene-derived SOA material
While isoprene and monoterpenes boast the largest emission rates of biogenic VOCs, sesquiterpenes (C15H24) emitted from various coniferous and deciduous plants have been found to have higher reactivity with atmospheric oxidants leading to a potential to form large amounts of aerosols.9,214 The total global sesquiterpene emission is estimated to be ∼14.8 Tg per year, and ∼9–16% of total terpene emissions.215 In particular, their high reactivity towards O3,216,217 coupled with the very low volatilities of their oxidation products may indicate that SOA formation from sesquiterpenes is currently underestimated.218–221 Therefore, aerosol yields from the ozonolysis of sesquiterpenes may be one of the major determinants in the SOA budget in the atmosphere.160,161,222–224A variety of sesquiterpenes have been detected in the atmosphere due to commercially available standards, including β-caryophyllene, α-humulene, longifolene, α-copaene, α-farnesene and α-cedrene.215,225–228 β-Caryophyllene (112) is the most abundant sesquiterpene emitted in the atmosphere and is proposed to play an important role in the formation of SOA.225,229–231 Previous studies indicate that low molecular weight compounds form the majority of SOA from the ozonolysis of β-caryophyllene (112).218,232–235 While few oxidation products derived from β-caryophyllene (112) have been identified, it is the only sesquiterpene for which authentic standards of oxidation products have been synthesized. Due to the availability of these standards there has been accelerated progress in the study of first and second generation oxidation products derived from ozonolysis of β-caryophyllene (112) as outlined below.
4.1. Oxidation products derived from β-caryophyllene
Aldehydes derived from β-caryophyllene (112) were identified early on in large amounts in both the gas and aerosol phases.161,236 In 2008, authentic standards were synthesized through the ozonolysis of β-caryophyllene with a reductive workup, leading to the formation of β-nocaryophyllene aldehyde (113) and β-caryophyllene aldehyde (114) (Fig. 21A).229 These standards were used to unambiguously identify and quantify the presence of β-nocaryophyllene aldehyde in field samples from Boreal forests as well as determine atmospherically relevant properties.229,237 Additional oxidation products derived from β-caryophyllene (112) were accessed in 2013, including β-caryophyllonic acid (115), β-caryophyllinic acid (116) and β-nocaryophyllonic acid (117).238,239 While β-caryophyllinic acid (116) has been tentatively proposed as an abundant tracer for β-caryophyllene derived SOA,222,240 synthesis of these standards unambiguously identified constituents of β-caryophyllene-derived SOA in both chamber and field experiments and indicated that β-nocaryophyllonic acid (117) and β-caryophyllonic acid (115) may be important tracers in the particle phase for β-caryophyllene ozonolysis pathways.238 More recently, β-caryophyllene aldehyde (113) and β-caryophyllonic acid (115) were identified as highly surface active and therefore may play an important role at the surfaces of β-caryophyllene-derived material, where they would significantly enhance CCN activity (Fig. 21B).152 This study also developed an alternative route to access β-caryophyllinic acid (116) and β-nocaryophyllinic acid (117) via oxidation of an iodolactone intermediate (119) accessed from β-caryophyllonic acid (115).152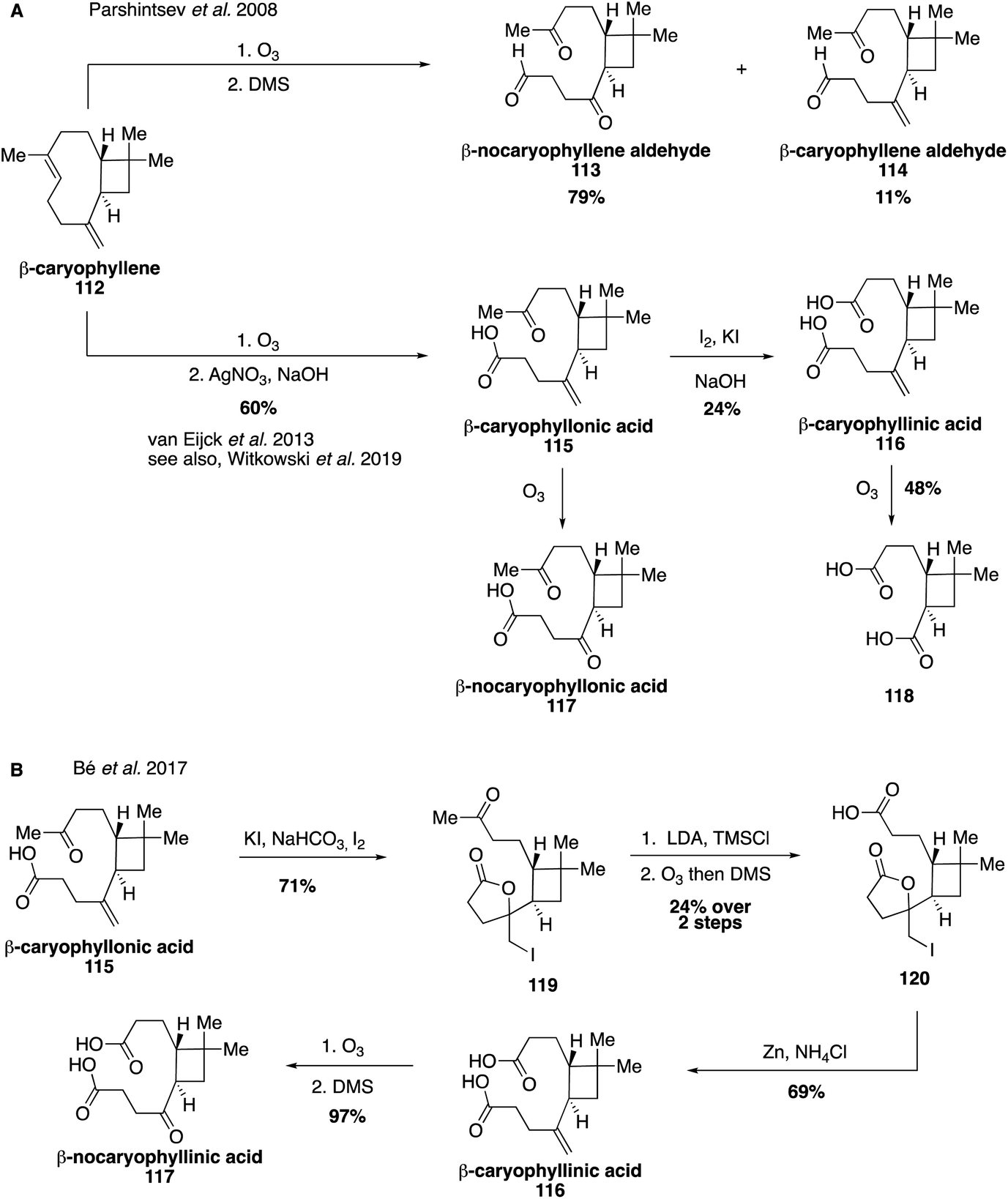 | ||
| Fig. 21 Synthetic routes accessing a number of caryophyllene oxidation products. | ||
Many recent studies have proposed a variety of additional tracers for oxidation products derived from β-caryophyllene (104) as well as other sesquiterpenes.218,232,235,241,242 Synthesis of additional oxidation products derived from β-caryophyllene and expansion to studies of other sesquiterpenes will be essential for analyzing the impact of sesquiterpenes on SOA formation and accurately quantifying their abundance in the atmosphere.
5. Biogenic SOA material influenced by atmospheric pollutants
The large numbers of anthropogenic sources around the world result in abundant emissions of primary organic aerosols (POAs) and also influence SOA formation. Processes such as biomass burning and combustion all yield VOCs that can influence the chemistry and composition of SOA particles. Despite the estimate that biogenic sources for VOCs heavily outweigh anthropogenic sources,2 it has been established that SOA formation is greatly enhanced by anthropogenic emissions composed primarily of nitrogen oxides and sulfur dioxide,243 which can serve as a source of acidic aerosol in the atmosphere.9,10,244 Many proposed SOA constituents influenced by NOx or SOx are highly polar and synthetically challenging to access. As a result, very few synthetic routes have been published to access authentic standards of organosulfate and organonitrate aerosol components. Development of additional synthetic procedures to access these standards would significantly enhance understanding of their chemical composition, atmospheric abundance, as well as chemical and physical properties.5.1. Organosulfate species
Organosulfate compounds have recently gained recognition in the atmospheric community as potential markers for SOA particles influenced by acidic sulfate.244–247 The main source of sulfate in the atmosphere is oxidation of SO2 emitted from the combustion of fossil fuels, indicating that detection of organosulfates may also serve as a marker for anthropogenic pollution. Monosulfate esters present in atmospheric aerosols can be produced from acid-catalyzed particle-phase reactions and are generally highly water-soluble, nonvolatile, and negatively charged at acidic pH, such that they remain in the particle phase of aerosols. Organosulfate compounds have been detected in a variety of both laboratory and field studies as a significant component of SOA particles246,248–252 and were observed to form from a variety of precursors such as isoprene, monoterpenes, sesquiterpenes, MBO as well as other aromatic and aliphatic compounds.102,114,185,235Despite their abundance, until recently only a few organosulfate species have been identified and quantified in the atmosphere since authentic standards of organosulfate products are not commercially available. Several studies have used surrogate standards such as camphorsulfonic acid and galactose sulfate to quantify organosulfates and aerosol material.246,253,254 Sulfate esters present in organosulfates often have high aqueous solubilities and a propensity to undergo hydrolysis, which can make their isolation and purification challenging. Despite these challenges many of the putative atmospheric organosulfates have been prepared in recent years.
The most abundant and ubiquitous organosulfates are those derived from isoprene.35,39,102,246,255 The organosulfate derived from IEPOX and 2-methyltetraol oxidation products (m/z 215) was identified as one of the most abundant single organic compounds in atmospheric aerosols.32,33,256 An NMR spectrum for a mixture of IEPOX-derived organosulfate isomers as tetrabutylammonium salts has been published59 and this mixture has been used as a standard in several field studies.59,70,257 More recently, one of the four possible IEPOX-derived organosulfate regioisomers (121) was synthesized under Bu4NHSO4 conditions from epoxide 5 (Fig. 22),258 yet the development of synthetic routes to access all regio- and stereoisomers of the IEPOX-derived organosulfates has been challenging.
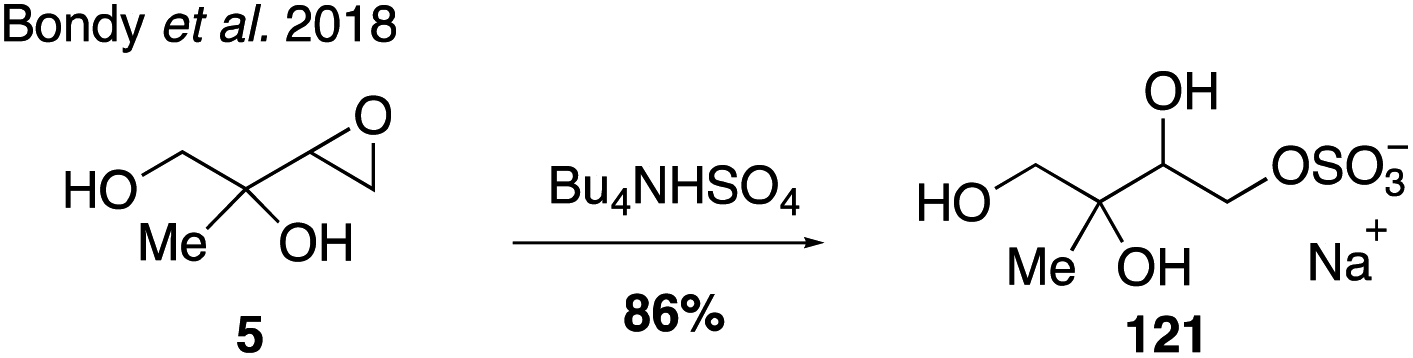 | ||
| Fig. 22 Sulfation through epoxide opening. | ||
Since primary, secondary and tertiary isomers (as well as syn and anti stereoisomers) may have different rates of hydrolysis and therefore atmospheric lifetimes and properties,81,259 access to atmospherically relevant salts of each IEPOX-derived sulfate isomer is desirable.257 Isolation of pure isomers will also aid in quantifying the relative amounts of these isomers in the atmosphere and provide insight into the aging and fate of SOA influenced by anthropogenic emissions. Recent work from our own labs has established detailed synthetic routes that have enabled access to all eight possible regio- and stereoisomers of the IEPOX-derived organosulfate isomers as racemates. Access to the anti-configured sulfate regioisomers (corresponding to threitol stereochemistry) was accomplished using prenol 30 as a common starting material (Fig. 23A). The tertiary and secondary sulfates anti-125 and anti-127, respectively, were prepared from protected tetraol 123 as a point of divergence, with the desired stereochemical configuration arising from the stereoselective basic hydrolysis of epoxide 122. Direct sulfation of tertiary alcohol 123 was accomplished using sulfur trioxide pyridine complex to afford sulfate ester 124, which was further processed to the desired tertiary sulfate anti-125 by counter ion exchange and global protecting group removal by hydrogenolysis. Alternatively, the secondary organosulfate anti-127 could be synthesized from protected tetraol 123via benzyl ether 126. Sulfation of the free secondary alcohol within 126 followed by sulfation and protecting group removal delivered the desired compound in an efficient fashion. The two primary organosulfate isomers, anti-131 and anti-135, were also generated using prenol 30 as the starting material (Fig. 23B). Differentiation of the two primary hydroxyl groups was accomplished early in each route through the selective introduction of benzyl or para-methoxybenzyl ether groups on either of the terminal alcohols, allowing the isomeric epoxides, 128 and 132 to be prepared. Epoxide opening followed by a series of protecting group manipulations and sulfate installation analogous to the secondary and tertiary sulfate synthesis, then provided the two primary organosulfates, anti-131 and anti-135.
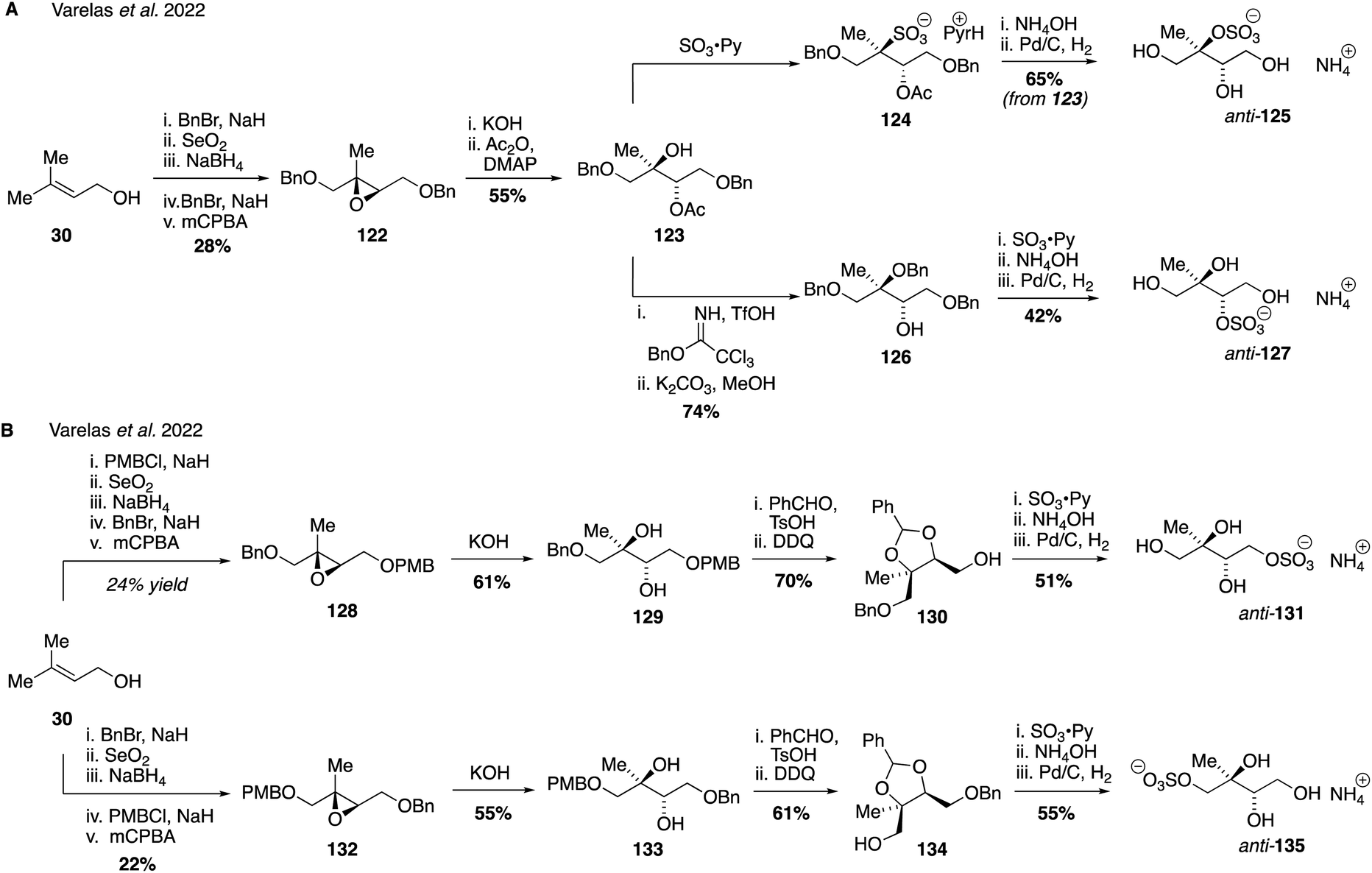 | ||
| Fig. 23 Diastereoselective synthesis anti-configured IEPOX-derived organosulfate isomers. | ||
In an analogous series of reactions that also used prenol 30 as the starting material, the series of syn-configured sulfate regioisomers (corresponding to the erthyritol stereochemistry) was also prepared (Fig. 24). The key difference between the approach used to access these diastereomeric compounds was the method employed to introduce the secondary and tertiary hydroxyl groups: while the anti-stereochemistry was generated through epoxidation followed by hydrolytic ring-opening, the syn-stereochemistry was obtained by stereospecific osmium-catalyzed dihydroxylation. In this way, diols 136, 141 and 144 were forged and then manipulated to produce each of the requisite syn-sulfates using a series of protecting groups similar to those employed in the anti-sulfate series. Access to this complete set of all eight isomers in diastereomeric form allowed for a series of investigations into their stability and acidity properties. The tertiary sulfates were shown to be the least stable in aqueous solution with half-lives of 4.5 days, while the secondary and primary sulfates were stable for greater that 60 and 180 days, respectively. Interestingly, the pKa of the tertiary sulfates were reported to be lower than the secondary and primary isomers (ca. 3 for 125, 4 for 127 and 5 for 131/135), although at this stage no rational explanation for these differences has been provided. Ultimately, while the synthetic chemistry required to access each of these isomers was somewhat laborious this careful approach led to some important new insights and lays the foundation for future studies on pure samples, such as investigating their role in gas particle uptake and hygroscopic growth.260
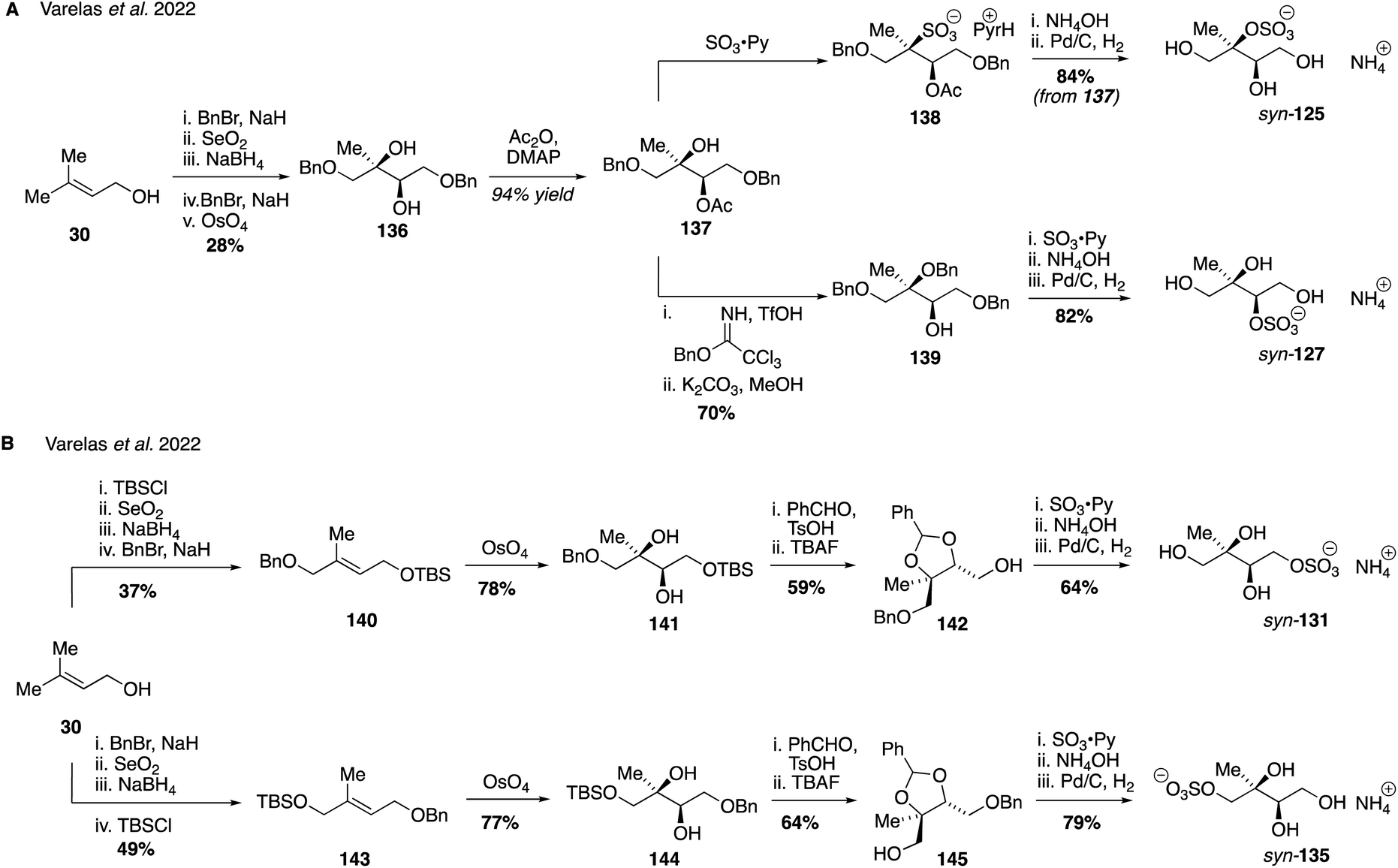 | ||
| Fig. 24 Diastereoselective synthesis syn-configured IEPOX-derived organosulfate isomers. | ||
Glycolic acid sulfate (147) was first synthesized and identified in 2009, which confirmed its previous misidentification in field and chamber studies as glyoxal sulfate.261 Glycolic acid sulfate (147) and its structurally related lactic acid sulfate (149) were synthesized from glycolic acid (146) and lactic acid (148), respectively, using diisopropylethylamine (DIEA) and chlorosulfonic acid (Fig. 25).261 Access to these standards allowed for studies to determine that they are ubiquitously detected in a variety of locations but only account for less than 0.2% of organic aerosols in those areas.262,263 Glycolic acid sulfate (147) was later synthesized as the potassium salt from 146 using a pyridine sulfur trioxide complex. A hydroxyacetone sulfate (151) and benzyl sulfate were accessed using the same procedure from hydroxyacetone (150) and benzyl alcohol.263 Their syntheses allowed for their quantification in aerosol samples and the advancement of hydrophilic interaction liquid chromatography coupled with mass spectrometry, which provides excellent separation of isoprene-derived carboxy-organosulfates.263–265
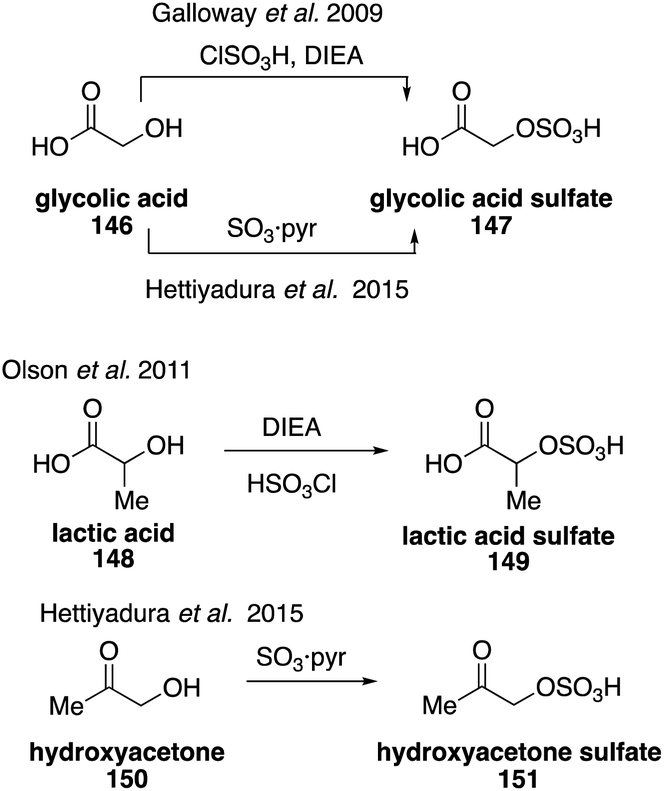 | ||
| Fig. 25 Synthesis of sulfates derived from glycolic acid, lactic acid and hydroxyacetone. | ||
Synthesis of additional dihydroxybutanone and dihydroxybutanal organosulfate isomers (154, 155, 158, 159) also led to identification of the sulfate isomers of 3,4-dihydroxybutan-2-one (158 and 159) in atmospheric aerosols (Fig. 26).44
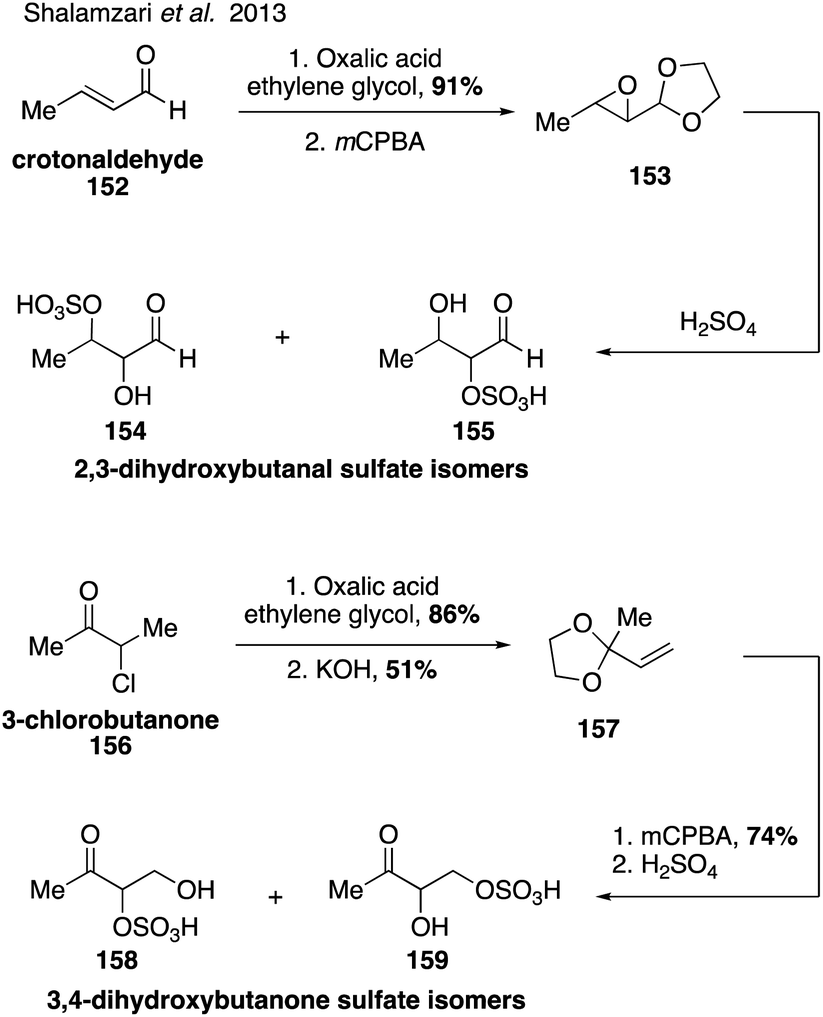 | ||
| Fig. 26 Synthesis of sulfated butanal and butanone species. | ||
Organosulfates derived from monoterpenes, such as α-pinene, β-pinene, and limonene, have also been detected in field and chamber aerosol studies around the world.103,156,245,254,266–268 Epoxides containing tertiary carbons have been shown to react quickly with acidic sulfate sources,269 indicating that compounds such as β-pinene oxide (160) may react readily to form organosulfates.270 In order to investigate formation of organosulfates from monoterpenes, a mixture of β-pinene organosulfates (2-pinanol-10-hydrogensulfate and 10-pinanol-2-hydrogensulfate, 161 and 162) were synthesized via the addition of sulfuric acid to 160 (Fig. 27).185 These standards were used to demonstrate that β-pinene organosulfates may be present in concentrations up to 23 ng m−3 ambient SOA, however it is possible that the structural assignments of 161 and 162 are incorrect given the propensity for of β-pinene oxide to undergo acid catalyzed rearrangement. Detailed structural assignment using NMR spectroscopy was not reported.
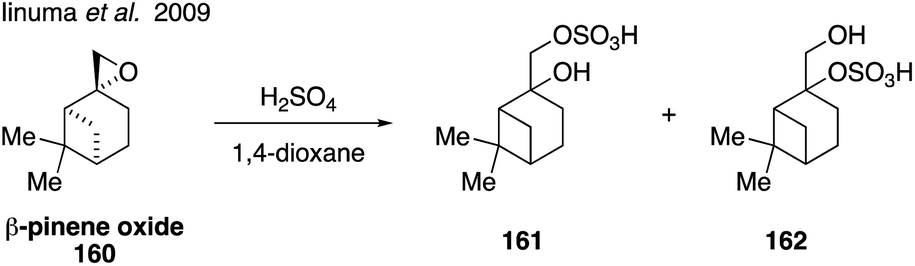 | ||
| Fig. 27 Synthesis of pinene-derived organosulfates. | ||
Limonene-derived organosulfates were recently synthesized271 by treating a mixture of limonene oxides (163 and 164) with sodium bisulfate following synthetic protocols developed by Cavdar and coworkers (Fig. 28).272 While the limonene-derived organosulfates (165–168) were unable to be separated and contained other organosulfate impurities, the surface activity and hygroscopicity of the resulting mixture were analyzed in order to further understand the atmospheric impact of organosulfates derived from monoterpenes.271
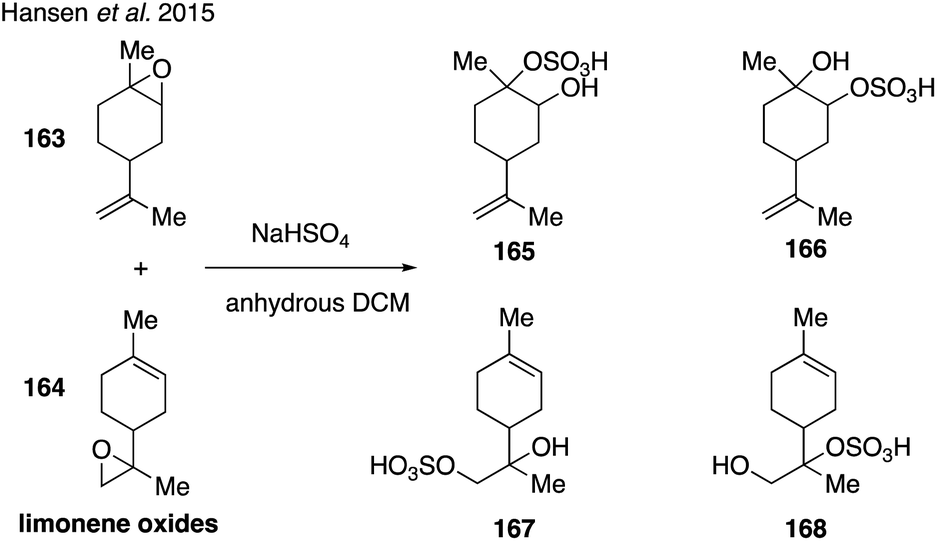 | ||
| Fig. 28 Synthesis of limonene-derived organosulfates. | ||
In 2017, Wang and coworkers reported a strategy for the synthesis of monoterpene-derived organosulfates via diastereoselective dihydroxylation followed by monosulfation with sulfur trioxide-pyridine complex (Fig. 29).273 Upjohn dihydroxylation of α-pinene and β-pinene with catalytic osmium tetraoxide and stoichiometric NMO provided the corresponding diols 58 and 171 as single diastereomers due to steric hindrance from the bridged gem-dimethyl groups. However, when limonene (173) was subjected to the same conditions, a mixture of diastereomers was obtained due to lack of steric differentiation. This problem was addressed by employing Sharpless asymmetric dihydroxylation using AD-mix-β, which provided major diastereomer 174 in 20:1 dr. The diols were subsequently treated with sulfur trioxide-pyridine complex to provide the corresponding organosulfate standards (i.e., 169, 172 and 175), with the sulfate group installed at the less sterically hindered position. With authentic standards in hand, the authors discovered that β-pinene derived organosulfate 172 degrades to a dehydrogenated organosulfate compound and a hydroperoxylorganosulfate compound over prolonged storage. In addition, the authors were the first to report identification of limonene and limonaketone organosulfates in ambient samples collected in the Pearl River Delta, China.273
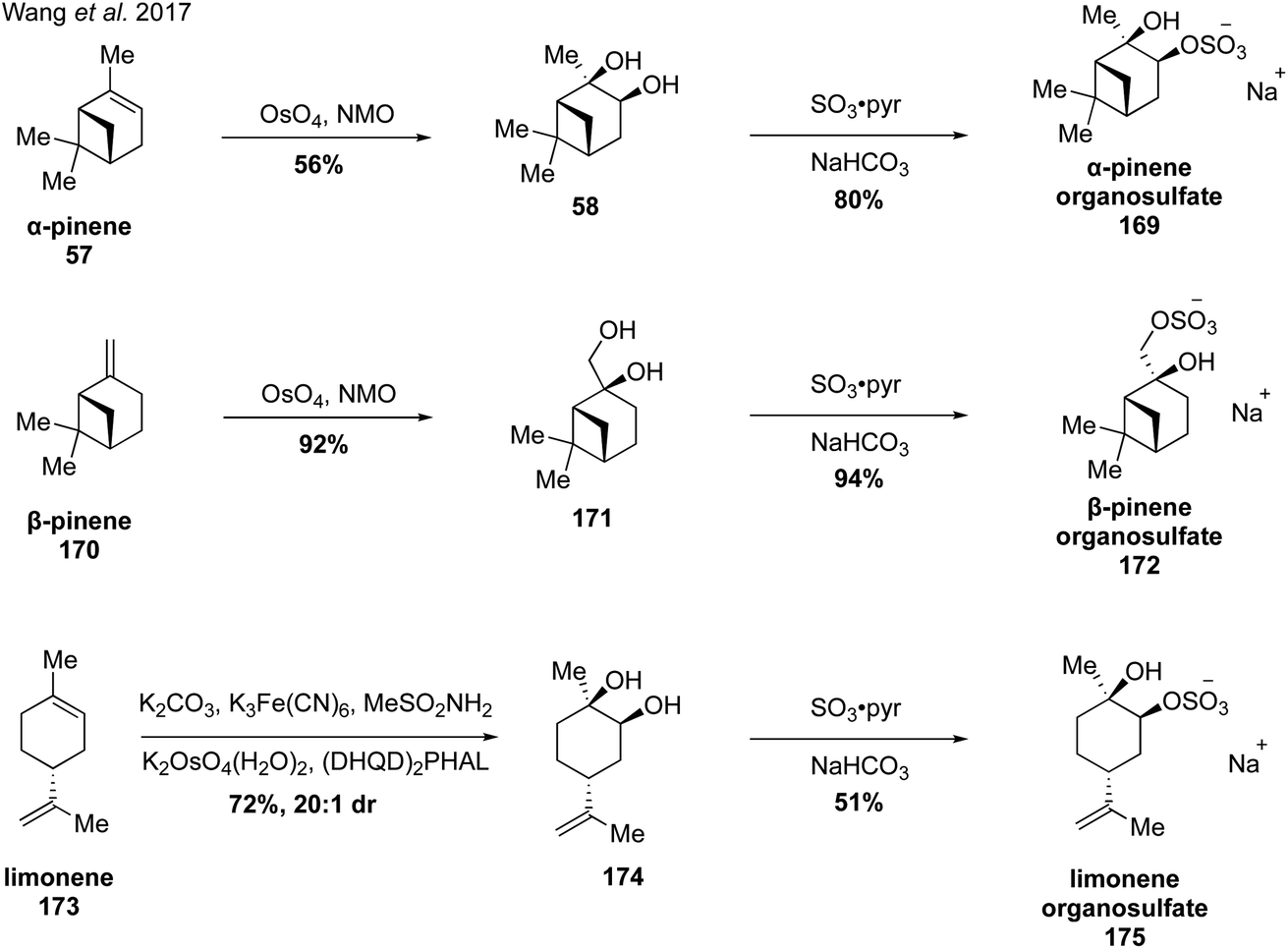 | ||
| Fig. 29 Access to complex organosulfate species through diastereoselective dihydroxylation followed by regioselective sulfation. | ||
Wang and coworkers expanded on this work in their groundbreaking 2019 study, generalizing their two-step dihydroxylation/sulfation route to eight monoterpenes (i.e., α- and β-pinene, limonene, sabinene, Δ3-carene, terpinolene, and α- and γ-terpinene) and two sesquiterpenes (i.e., α-humulene and β-caryophyllene) to synthesize a total of 26 α-hydroxy organosulfate standards, of which major isomers were isolated (Fig. 30) and the minor isomers were analyzed via LC/MS and MS/MS.274 Retention times and MS/MS characteristics of the synthesized standards subsequently enabled the detection of α-hydroxy organosulfates derived from Δ3-carene, sabinene, terpinolene, and β-caryophyllene in ambient samples for the first time.274 An additional outcome of this work were new potential insights into the formation mechanisms of organosulfates in the atmosphere and the importance of whether sulfation occurs from acidic opening of an epoxide or by direct sulfation of an alcohol. Epoxide precursors typically yield rearranged organosulfate products, while diol precursors allow the formation of products with the cyclobutane intact. Further work in this important area will likely rely on extensive use of synthetic standards.
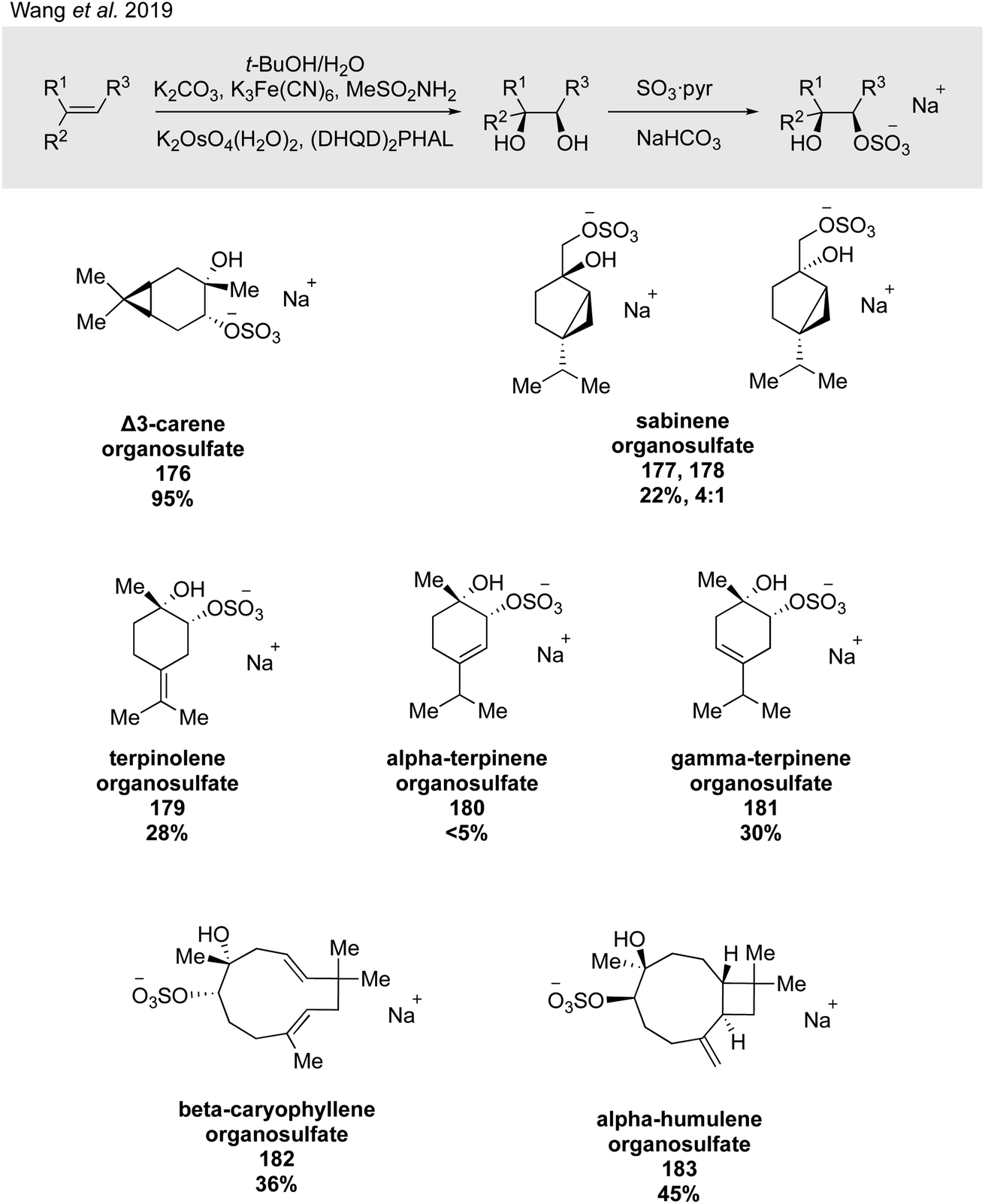 | ||
| Fig. 30 A general protocol for the diastereo- and regioselective installation of sulfate esters across alkenes. | ||
5.2. Organonitrate species
Nitrogen oxides (NOx) are predominantly emitted from combustion of fossil fuels and change the oxidation pathways of VOCs through the formation of organonitrates.275–277 In high NOx environments, nitric oxide can add to peroxy radicals in the atmosphere to form organonitrate compounds. The nitrate radical (NO3) serves has a strong oxidant in the nighttime atmosphere (since it undergoes rapid photolysis in sunlight) and has been shown to react quickly with unsaturated BVOCs.278–280 Organonitrates are often stable but can also hydrolyze readily in the particle phase, especially at tertiary positions and at high humidities, to form nitric acid.259,281–284 While this reaction is likely acid-catalyzed, its mechanism remains uncertain.81,285 The effect of organonitrates on atmospheric nitrogen levels and secondary organic aerosol has therefore been the topic of a variety of studies.30,286–288 However, due to a current lack of authentic standards, elucidation of information related for mechanisms of oxidation at high NOx concentrations and identification of organonitrate species remains challenging, making their synthesis a priority for future studies.289The synthesis of organonitrates has been achieved through nitration of alkyl halides with silver nitrate, nitration of alcohols or alkanes using nitric acid,290–293 or treatment of alcohols with dinitrogen pentoxide.294 More recently hydroxy nitrate esters have been accessed through epoxide opening in the presence of Bi(NO3)3·5H2O.295–297 Multifunctional nitrates have also been accessed298–301 and carbonyl containing nitrate compounds have been synthesized through the nitration of hydroxy ketones with dinitrogen pentoxide.294,302 Many of these syntheses have resulted in the isolation of a mixture of products, indicating that additional work is needed to isolate pure authentic standards. These initial studies have established several physical properties of small molecule organonitrates and may prove useful in future attempts to access atmospheric standards, since multifunctional organonitrates (possessing four or more oxygen atoms) dominate nitrate mass in submicron particles.303
Organonitrates formed from isoprene under high NOx conditions have been detected in the gas phase and can partition to the particle phase due to their low volatilities, while under low NOx conditions in pristine environments organonitrates are expected to have negligible yields.304–307 Isoprene-derived hydroxynitrates are some of the few standards that have been synthesized and analyzed in atmospheric studies.298,300,301 Four of the eight possible isoprene hydroxyl nitrate isomers 2-methyl-1-nitrooxybut-3-ene-2-ol (β-2,1 184), 2-methyl-2-nitrooxybut-3-ene-1-ol (β-1,2 185), 2-methyl-4-nitrooxybut-2-ene-1-ol (δ-1,4 186 and 187), Fig. 31 were synthesized in 2010 via the addition of nitric acid to 2-methyl-2-vinyl oxirane (17).308 While the original synthesis did not allow for the separation of the cis and trans isomers of the δ-1,4 nitrate (186, 187), this was later achieved in the synthesis of both the cis and trans δ-1,4 nitrate (186, 187) as well as the additional synthesis of another isomer, 3-methyl-2-nitrooxybut-3-ene-1-ol (β-4,3 188) via the ring-opening reaction of the corresponding epoxide precursor with Bi(NO3)3·5H2O (Fig. 32).309
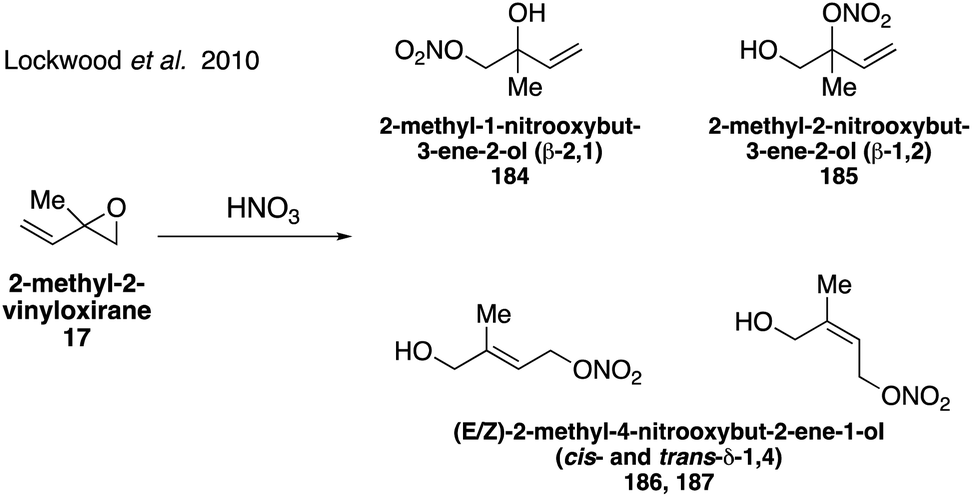 | ||
| Fig. 31 Unselective synthesis of isoprene-derived organonitrates using nitric acid. | ||
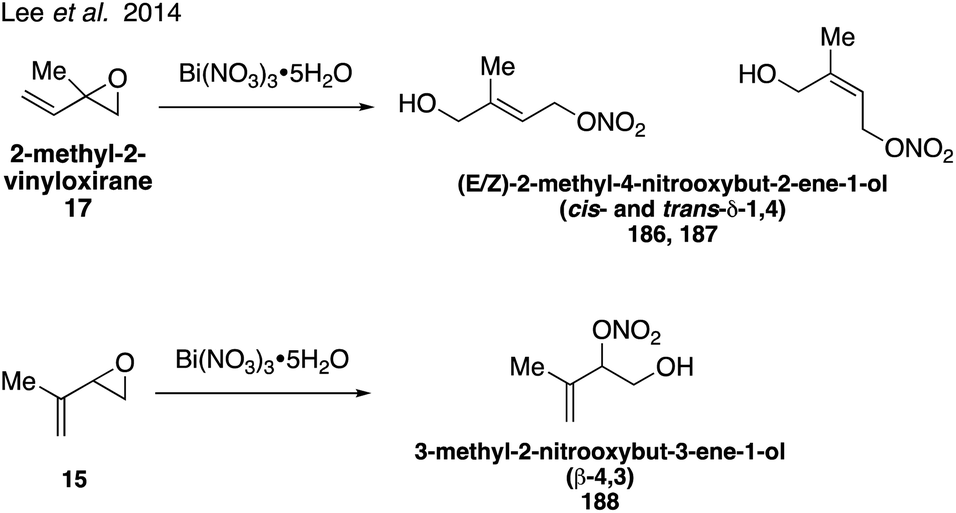 | ||
| Fig. 32 Improved selectivity for organonitrate synthesis using bismuth nitrate. | ||
Synthesis of these authentic standards demonstrated the relative importance of the individual isoprene nitrate isomers as well as their different reactivities with atmospheric oxidants.308,309 Since these isoprene nitrates retain a degree of unsaturation, they are highly reactive and can produce higher order organonitrate products proposed in the literature.289,309 For instance, chamber studies have identified methacryloylperoxynitrate (MPAN, 190) as a second-generation product.32,33 The synthesis of MPAN from the peroxidation of methacrylic anhydride (189) followed by nitration310 confirmed that acyl peroxynitrates play an important role in SOA formation under high NOx conditions (Fig. 33).33,108
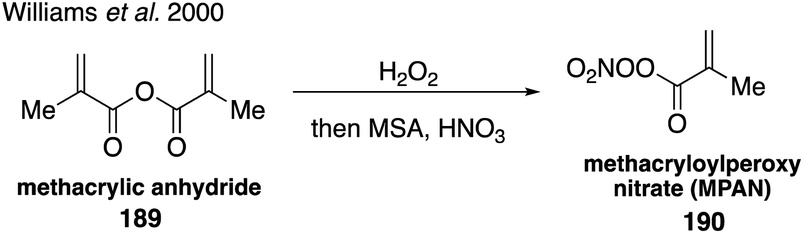 | ||
| Fig. 33 Synthesis of MPAN. | ||
Recently, Nguyen and coworkers expanded the current library of isoprene-derived hydroxynitrate standards with the synthesis of 2-methyl-3-nitrooxybutane-1,2,4-triol (193) and 2-methyl-1,4-dinitrooxybutane-2,3-diol (198) from isoprene (1) (Fig. 34).311 The synthesis of both 193 and 198 began with the direct bromination of isoprene (1) to yield dibromoalkene 191. Direct nitration of bromide 191 with AgNO3 followed by dihydroxylation with KMnO4 gave 193. To access compound 198 dibromide 191 was treated with potassium acetate followed by hydrolysis and bromination to obtain dihydroxy dibromide compound 196. Subsequent nitration with AgNO3 afforded the unstable dinitrate 197 which spontaneously hydrolyzed to 198. These new organonitrate standards were used along with previously synthesized organosulfate standards to study their OH-initiated oxidation kinetic rate coefficients, leading the authors to conclude that aqueous photooxidation of isoprene-derived SOA compounds may affect the SOA budget when incorporated into global models.311
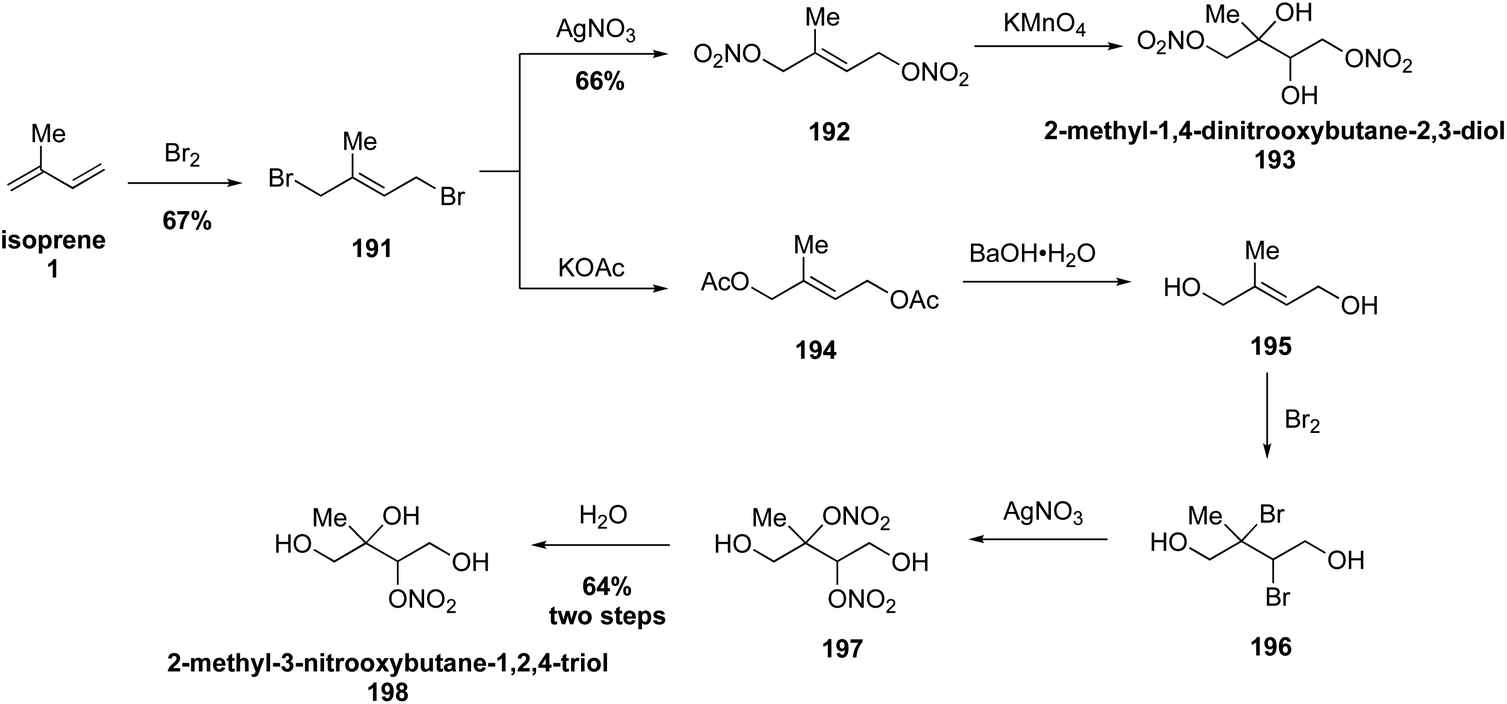 | ||
| Fig. 34 Synthetic route for accessing complex organonitrates derived from isoprene. | ||
NOx initiated oxidation of monoterpenes can compete with ozonolysis in the atmosphere to result in SOA formation.312–314 Monoterpene-derived organonitrates may represent a significant fraction of SOA.315,316 Organonitrate 199 derived from α-pinene was synthesized starting from α-pinene oxide (70) in the presence of Bi(NO3)3·5H2O (Fig. 35).317 Along with rate constant data, product identification confirmed that a unimolecular specific acid-catalyzed mechanism is responsible for hydrolysis of the α-pinene derived organonitrate under acidic conditions.317
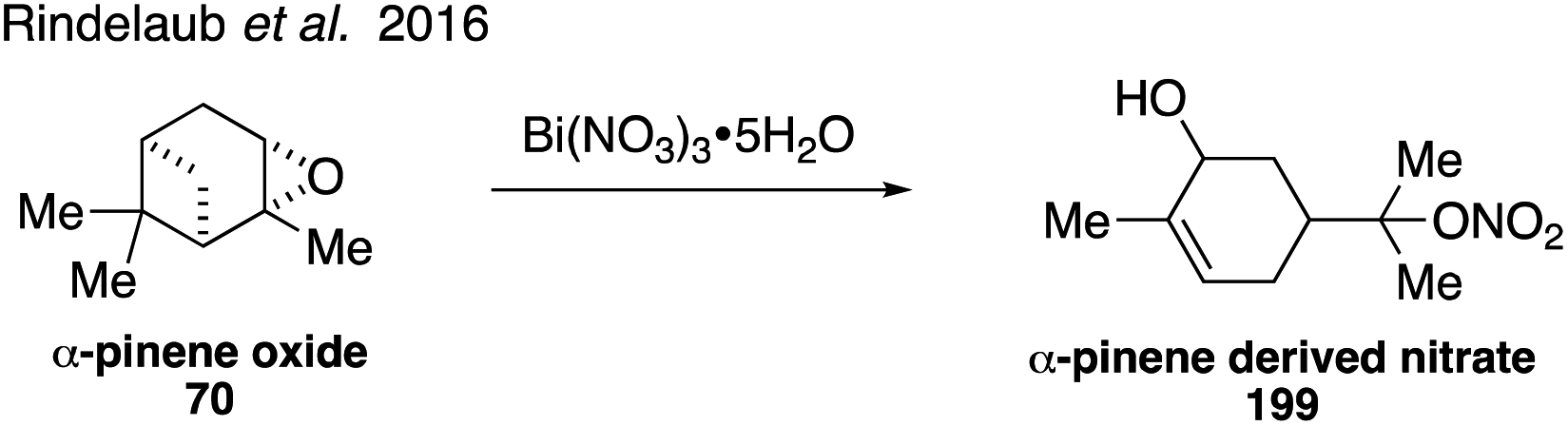 | ||
| Fig. 35 Synthesis of a pinene-derived organonitrate. | ||
In 2020, McKnight and coworkers expanded this method to synthesize nine organonitrates derived from monoterpenes (Fig. 36) including Δ3-carene, limonene, α-pinene, β-pinene and perillic alcohol.318 Exposing α-pinene oxide and β-pinene oxide to Bi(NO3)3·5H2O both resulted in multiple carbocation rearrangement products.318 A 2021 study by Wang and coworkers reported the regioselective synthesis of several terpene-derived organonitrates.319 Access to these species was accomplished via two routes: epoxidation of the terpene to deliver an epoxide that was opened with concentrated nitric acid (Fig. 37) or regioselective bromohydration of the terpene with NBS followed by nitration with AgNO3 (Fig. 38). Two of the synthesized organonitrates, β-pinene derived organonitrate 221 and β-caryophyllene derived organonitrate 219, had not been previously reported. The same synthesis methods were used in a later study to further investigate the hydrolysis mechanism of terpene-derived organonitrates. Consistent with Rindelaub's findings in 2016,317 tertiary and secondary organonitrates were found to hydrolyze via a unimolecular acid-catalyzed mechanism.320 Primary organonitrates were found to hydrolyze via competing unimolecular and bimolecular mechanisms.320
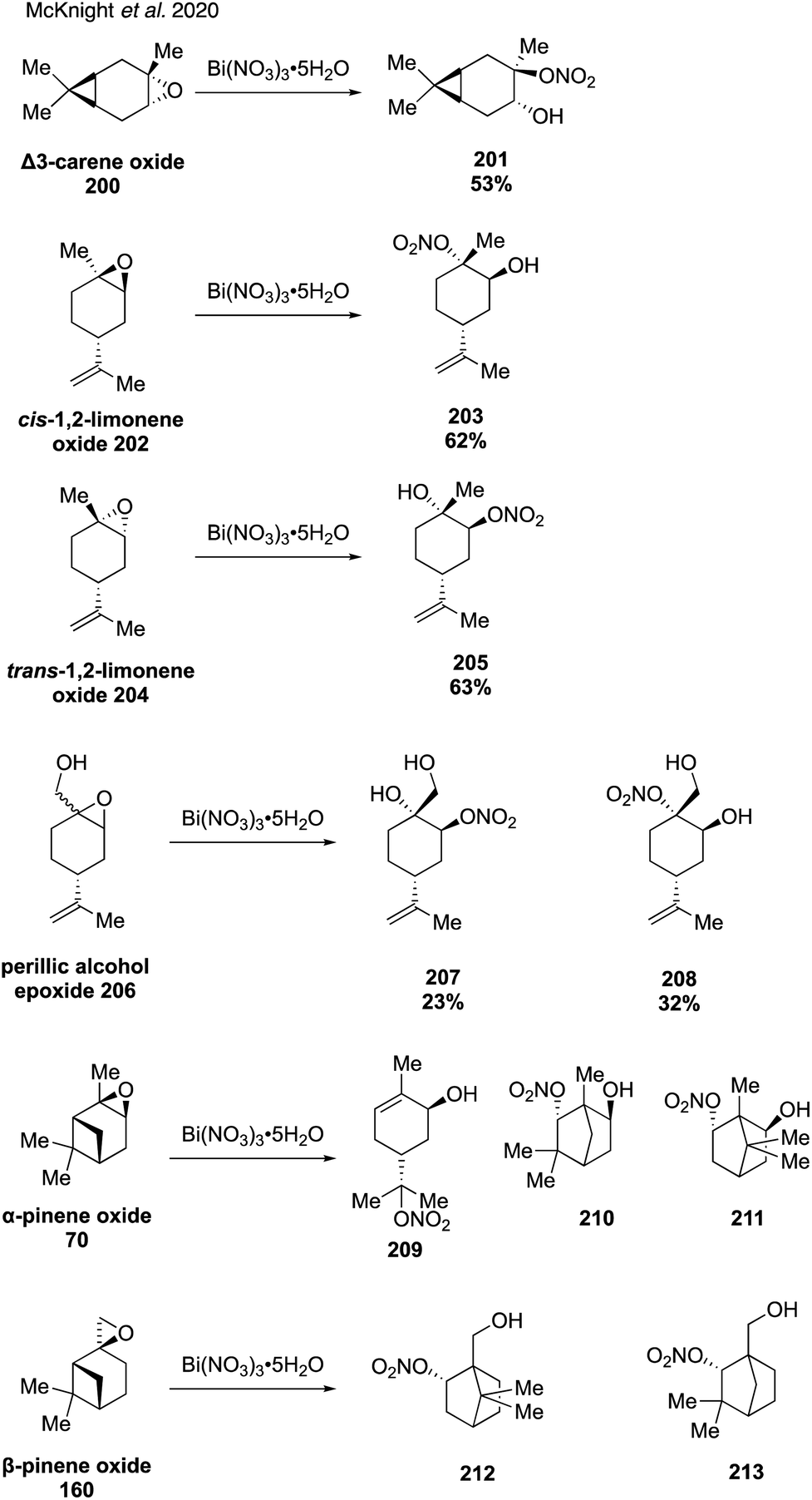 | ||
| Fig. 36 Diverse organonitrates accessed using bismuth nitrate. | ||
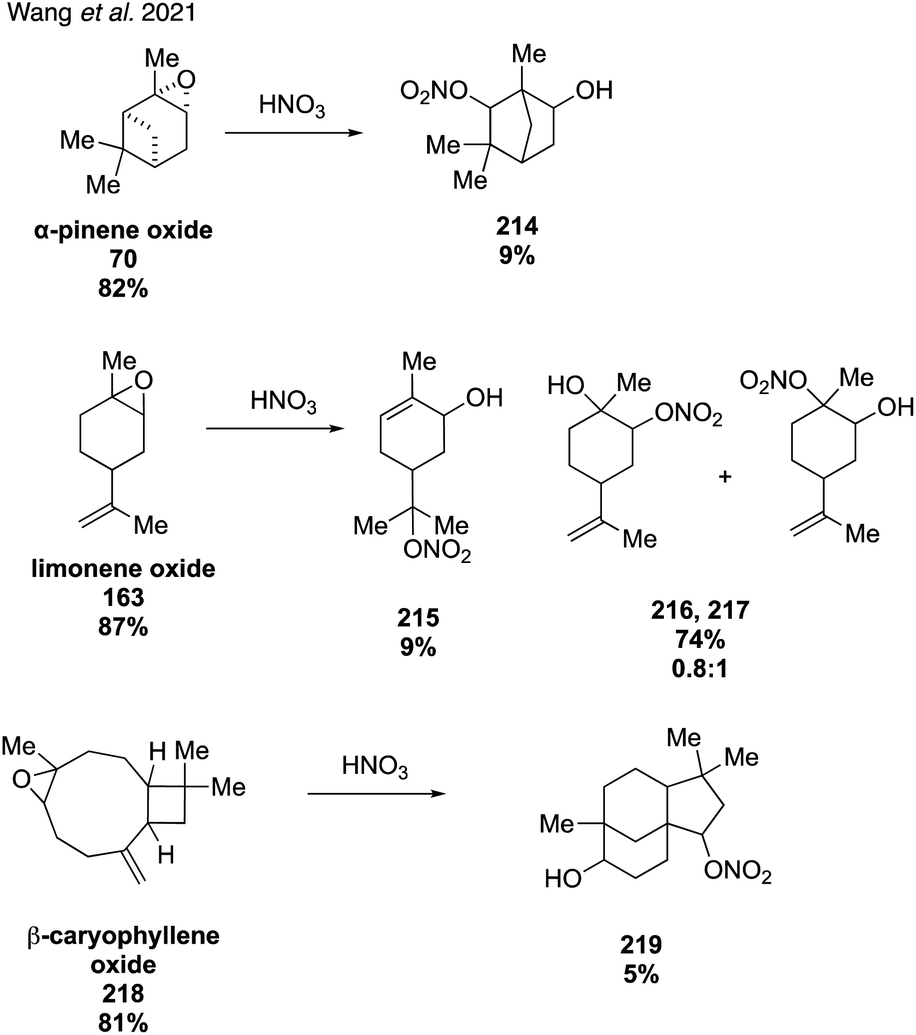 | ||
| Fig. 37 Organonitrates derived from epoxide opening with nitric acid. | ||
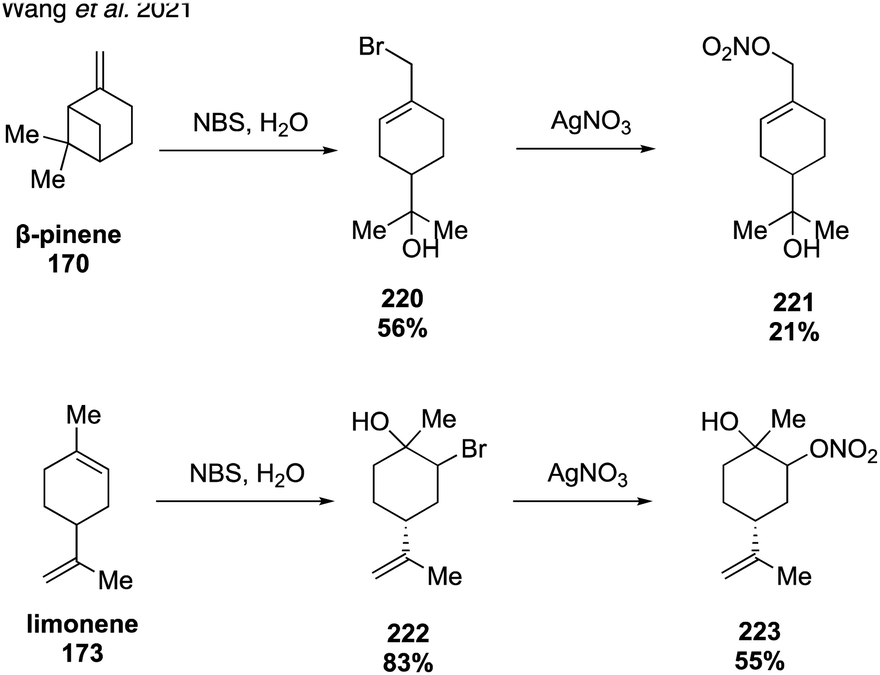 | ||
| Fig. 38 Organonitrate from halides by displacement with silver nitrate. | ||
In addition, Wang and coworkers demonstrated the feasibility of sulfating complex organonitrate species using sulfur trioxide (Fig. 39).319 Several of the resulting nitrooxy organosulfates were then identified in ambient aerosol samples for the first time, correcting previous misidentifications and confirming the existence of previously unrecognized rearrangement pathways in the atmosphere.320
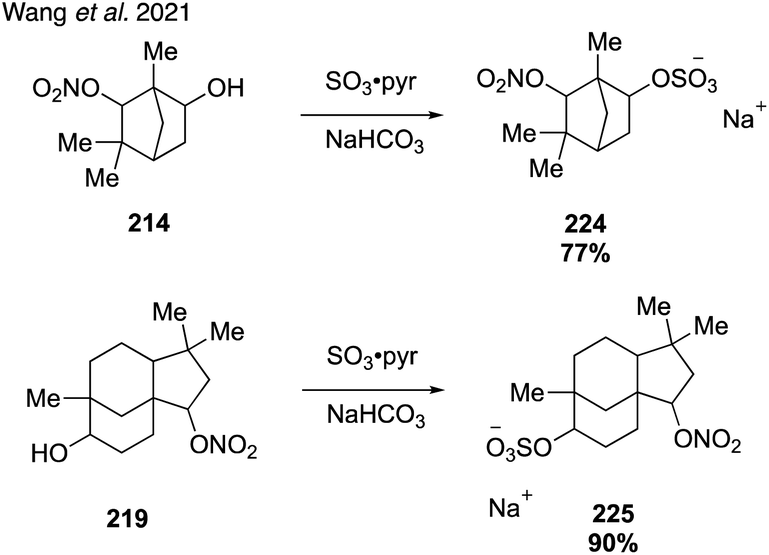 | ||
| Fig. 39 Complex nitroxy organosulfates. | ||
6. Synthesis of mechanistic probes
6.1. Synthesis of isotopically-labelled species
The synthesis and use of isotopologues derived from BVOCs holds the promise of enabling detailed molecular studies of biogenic SOA particle chemistry and physics. Recently, synthetic strategies to access isotopically-labeled oxidation products of BVOCs, namely α-pinene, have been developed to aid in studying the composition and formation chemistry of SOA particles as well as chemical exchange between particles.321,322 Synthetic access to the isotopologues of α-pinene coupled with the ability to synthesize laboratory-derived SOA material has also opened up new approaches for studying SOA material using vibrational spectroscopy323,324 and mass spectrometry.325In 2016, Upshur and coworkers developed protocols for accessing pinene isotopologues containing deuterium at the C10 vinyl methyl group and the C9 endo disposed methyl group (Fig. 40).323 The routes rely on nopinone (226) as a common intermediate, which can be converted to the vinyl isotopologue 227 by a straightforward addition of the CD3-cuprate to the corresponding enol triflate in 57% yield. On the other hand, addition of methyllithium to nopinone (226) allowed for the generation of caged ether 228 by means of a Suarez functionalization. Opening of the ether bond led to the C9 alcohol 230 which was oxidized to the corresponding carboxylic acid 234 with PDC. Reduction of 234 with LiAlD4 produced deuterated alcohol 236, which was further transformed to the desired C9 dueterated methyl analog 240 by LiAlD4 displacement of the corresponding tosylate 238. The route was sufficiently flexible to enable access to other isotopolgues with varying degrees of deuterium incorporation at C9 and C10 (i.e., 233, 241 and 242).
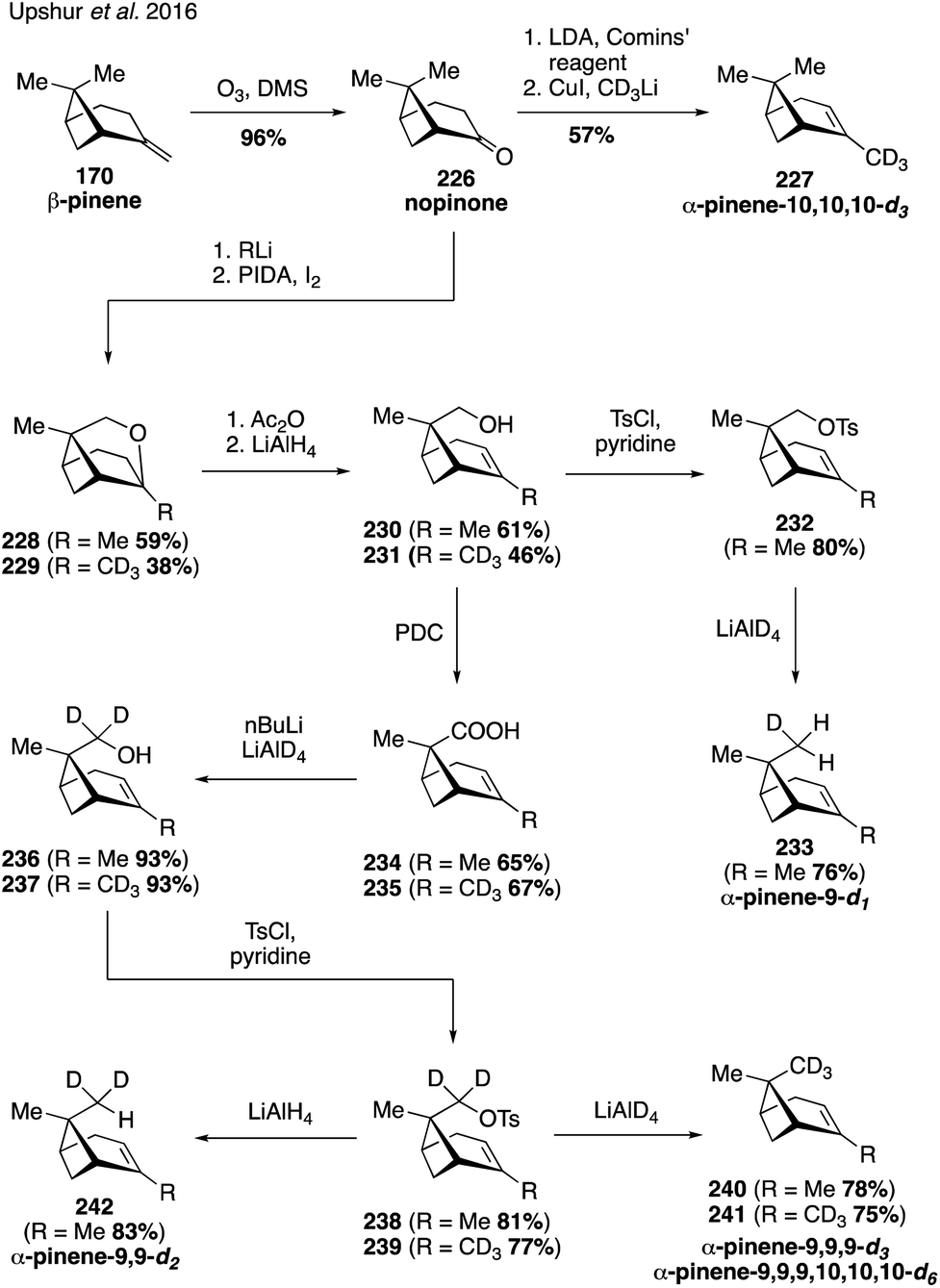 | ||
| Fig. 40 Access to deuterated isotopologues using nopinone as a starting material. | ||
To gain further access to a wider range of pinene isotopologues, such as those with methylene bridge, bridgehead methine, allylic, and vinyl deuteration the same group devised a strategy for the de novo synthesis of α-pinene isotopologues adapted from a previous total synthesis completed by Thomas and Fallis (Fig. 41).326,327 The syntheses of α-pinene-6,6-d2 (Fig. 41A) and α-pinene-1,3,4,4,5,-d5 and α-pinene-1,3,4,4,5,6,6-d7 (Fig. 41B) all utilize a bottom-up approach starting from Hagemann's ester (243), allowing for the generation of multiple site-specifically labeled analogues in a unified fashion.324 For the case of α-pinene-d2 (251), introduction of the desired deuterium atoms took place in the first step of the synthesis with the LiAlD4 reduction of Hagemann's ester (243) to deliver diol 244 (Fig. 41A). Oxidation with DDQ then gave enone 245 which was converted through a series of steps to tosylate 247, which underwent intramolecular cyclization to afford the key cyclobutene ring as was shown by Thomas and Fallis in their pioneering pinene synthesis. Cleavage of the benzylidene group then allowed for the final formation of the alkene using the cuprate procedure devised in the earlier synthesis of isotopologues (see Fig. 40). By modifying this route to include an additional deuteration of Hagemann's ester (243) to form bis-deuterated species 257, the authors were able to access several other highly deuterated compounds(i.e., 265 and 256) in an effective fashion highlighting the generality of the approach (Fig. 41B).
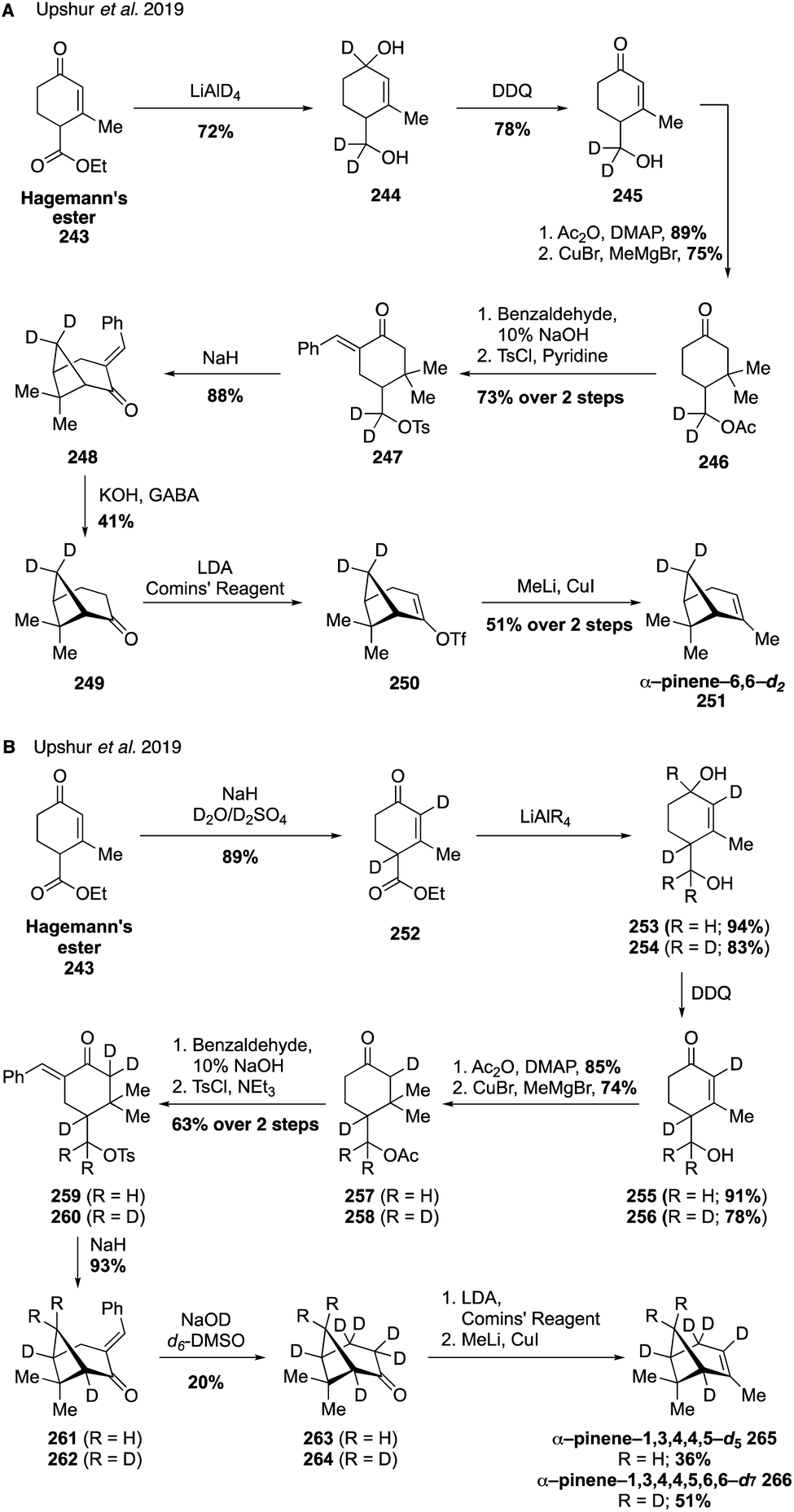 | ||
| Fig. 41 Total synthesis of pinene isotopologues from Hagemann's ester. | ||
By subsequently preparing and collecting surface vibrational spectra of SOA material derived from the α-pinene isotopologues, initial attempts were made toward identifying the surface oscillators in α-pinene-derived SOA particles.324 These α-pinene isotopologues have also been used in mechanistic chamber-based studies, including the demonstration of particle–particle chemical exchange using single-particle mass spectrometry.321 Specifically, SOA mixing experiments using these isotopologues revealed that there is little limitation to diffusion among SOA particles derived from unlabeled and deuterated α-pinene SOA, however, diffusion and uptake of semi-volatile compounds is limited when deuterated α-pinene SOA is mixed with SOA derived from other BVOCs such as limonene and β-caryophyllene.321 Additionally, work aiming to identify intramolecular hydrogen migration in autoxidation leading to highly oxygenated multifunctional organic compounds has utilized isotopologue 227.325 The distribution patterns of SOA products generated from ozonolysis and OH-oxidation of unlabeled α-pinene and isotopologue 227 revealed a systematic shift in the mass of monomers in the deuterated system that was explained by the decomposition of isomeric vinylhydroperoxides to release vinoxy radical isotopologues, which serve as precursors to a sequence of autooxidation reactions that ultimately yield ELVOCs contributing to SOA formation and growth.325 These studies would not have been possible without access to isotopically labeled analogues, given that mass spectra of SOA populations generated from different terpenes cannot be differentiated otherwise.
While current synthetic efforts are primarily dedicated to deuterium labeling of α-pinene, a limited number of studies have focused on isotopic labeling of other SOA precursors and oxidation products. Trans-β-IEPOX-d2 (270) was synthesized from aldehyde 267 in a four-step route involving a Corey–Gilman–Ganem oxidation, deuterium-installation via reduction, epoxidation, and TBS deprotection (Fig. 42).328,329 Deuterated analogues 275 and 277 derived from β-caryophyllene, along with other sesquiterpenes (i.e. aromadendrene and isoledene), were prepared in 2016 via synthetic routes hinging on the use of Wittig olefination and α-hydrogen-deuterium exchange (Fig. 43).330 Beyond deuterium incorporation, 13C-labeled β-pinene (278) was recently synthesized from 13C-iodomethane and nopinone (226) using Wittig olefination chemistry to aid in investigating O3- and OH-initiated oxidation of β-pinene (Fig. 44).322
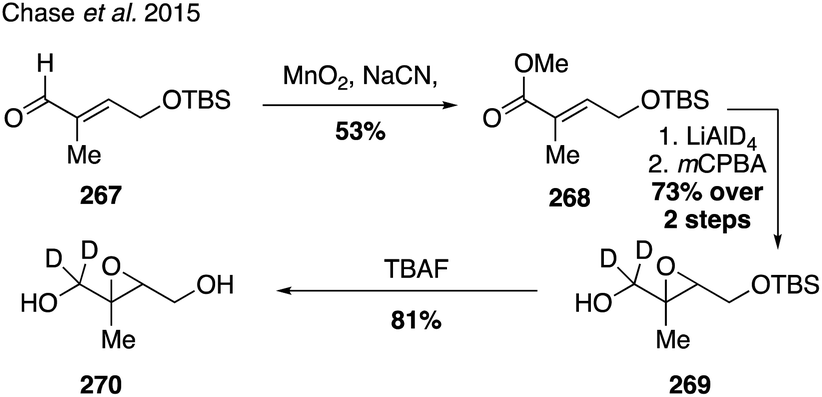 | ||
| Fig. 42 Synthesis of deuterium labelled IEPOX. | ||
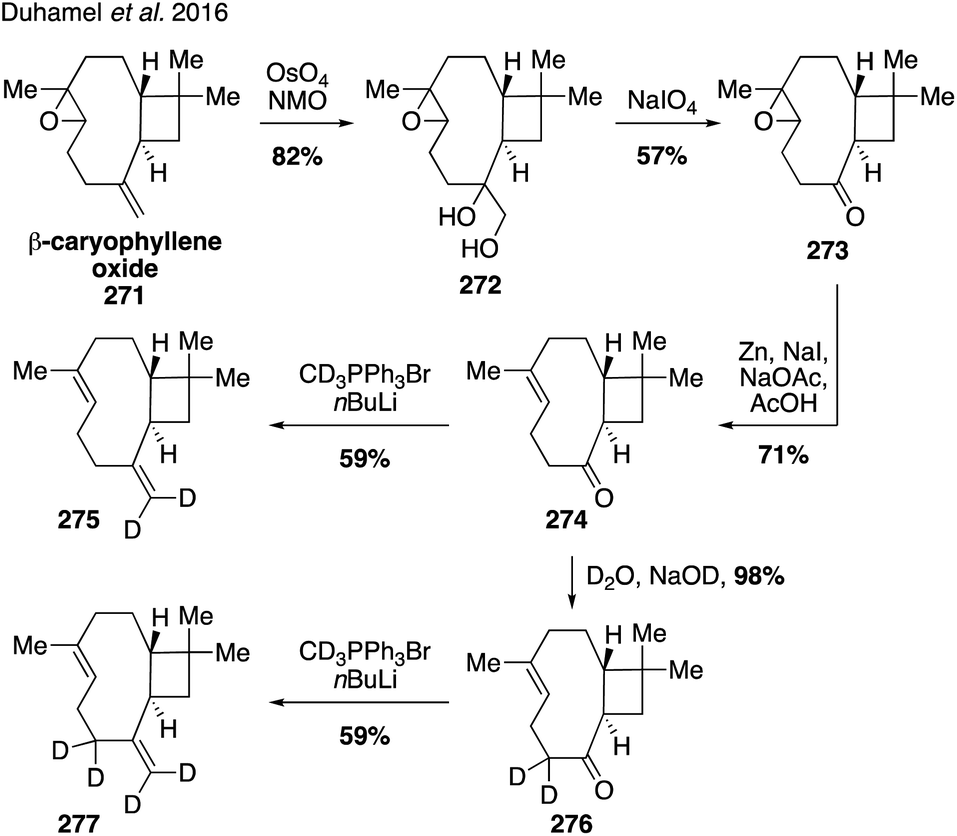 | ||
| Fig. 43 Preparation of deuterated caryophyllene species. | ||
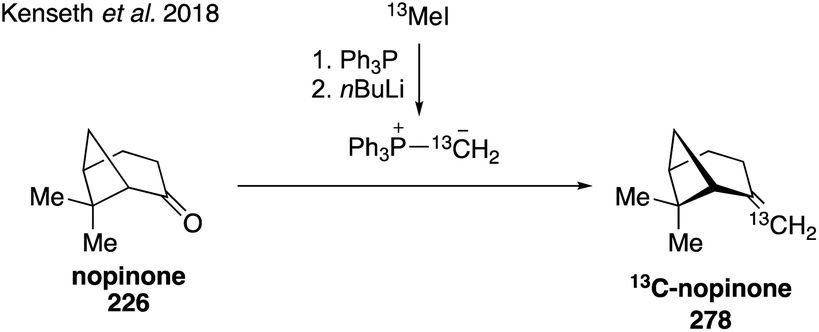 | ||
| Fig. 44 Direct access to 13C-labeled pinene through a Wittig reaction. | ||
Further studies focused on the synthesis of BVOC and oxidation product isotopologues, along with the generation of particles derived from such molecules, will bring the field steps closer to elucidating properties and formation mechanisms of biogenic SOA particles.
6.2. Miscellaneous probe compounds
A recent publication from the Zhang lab reported the synthesis of two unsaturated analogs of pinonaldehyde (i.e., 279 and 281) in order to gain new insights into the breakdown of Criegee intermediates formed during the ozonolysis of α-pinene in the atmosphere (Fig. 45).331 A selective methylenation of the more reactive aldehyde group within pinonaldehyde (59) allowed for a direct synthesis of analog 279, while a slightly more circuitous route involving a protecting group was required to deliver analog 281. Ozonolysis of each of these species then allowed for detailed studies into the different pathways for SOA formation from the two regioisomeric Criegee intermediates. Future synthetic work along these lines and in concert with computational modeling could revel important new levels of understanding of these complex processes.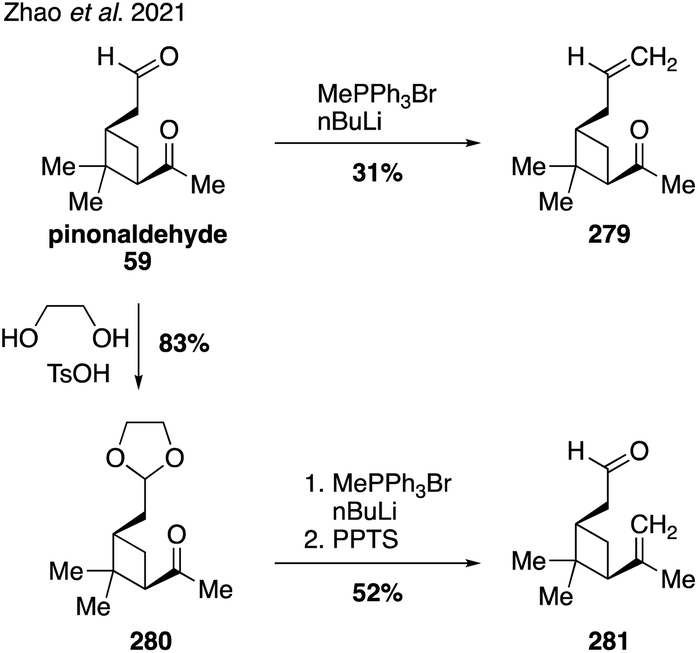 | ||
| Fig. 45 Synthesis of unsaturated species as ozonolysis probes. | ||
7. Conclusions
The complex products produced by the atmospheric oxidation of volatile terpenes has provided significant stimulation for the development of many creative synthetic pathways to enable their access. Access to these products has enabled verification of tentative assignments made based on initial mass spectrometry data, and has provided important material to investigate the atmospherically-relevant properties of these important constituents of the climate system. More recent work has focused on providing robust access to organosulfate and organonitrate species formed in the atmosphere by the interplay of biogenic terpene species with the products of anthropogenic pollution, while advances in the preparation of isotopically-labelled compounds has enabled a greater understanding of the complex mechanistic pathways involved in the formation of secondary organic aerosol material. Much of the advances made in this area are due to a rich interplay between synthetic organic chemistry, physical chemistry, atmospheric science, and environmental engineering. Given the importance of understanding our climate, continued interdisciplinary efforts supported by synthetic chemistry will likely remain of high value.8. Conflicts of interest
There are no conflicts to declare.9. Acknowledgements
We gratefully acknowledge support from the National Science Foundation (CHE-1607640 and CHE-2003359). MAU gratefully acknowledges support from a National Aeronautics and Space Administration Earth and Space (NASA ESS) Fellowship, a National Science Foundation (NSF) Graduate Research Fellowship (NSF-GRFP), and a P.E.O. Scholar Award. AGB gratefully acknowledges support from a National Science Foundation (NSF) Graduate Research Fellowship (NSF-GRFP).10. Notes and references
- J. H. Seinfeld and S. N. Pandis, Atmospheric Chemistry and Physics: From Air Pollution to Climate Change, John Wiley & Sons, Hoboken, NJ, 2nd edn, 2006 Search PubMed.
- M. Kanakidou, J. H. Seinfeld, S. N. Pandis, I. Barnes, F. J. Dentener, M. C. Facchini, R. Van Dingenen, B. Ervens, A. Nenes, C. J. Nielsen, E. Swietlicki, J. P. Putaud, Y. Balkanski, S. Fuzzi, J. Horth, G. K. Moortgat, R. Winterhalter, C. E. L. Myhre, K. Tsigaridis, E. Vignati, E. G. Stephanou and J. Wilson, Atmos. Chem. Phys., 2005, 5, 1053–1123 CrossRef CAS.
- U. Poschl, Y. Rudich and M. Ammann, Atmos. Chem. Phys., 2007, 7, 5989–6023 CrossRef.
- U. Lohmann and J. Feichter, Atmos. Chem. Phys., 2005, 5, 715–737 CrossRef CAS.
- J. L. Mauderly and J. C. Chow, Inhalation Toxicol., 2008, 20, 257–288 CrossRef CAS PubMed.
- J. O. Anderson, J. G. Thundiyil and A. Stolbach, J. Med. Toxicol., 2011, 8, 166–175 CrossRef PubMed.
- K. H. Kim, E. Kabir and S. Kabir, Environ. Int., 2015, 74, 136–143 CrossRef CAS PubMed.
- J. L. Jimenez, M. R. Canagaratna, N. M. Donahue, A. S. H. Prevot, Q. Zhang, J. H. Kroll, P. F. DeCarlo, J. D. Allan, H. Coe, N. L. Ng, A. C. Aiken, K. S. Docherty, I. M. Ulbrich, A. P. Grieshop, A. L. Robinson, J. Duplissy, J. D. Smith, K. R. Wilson, V. A. Lanz, C. Hueglin, Y. L. Sun, J. Tian, A. Laaksonen, T. Raatikainen, J. Rautiainen, P. Vaattovaara, M. Ehn, M. Kulmala, J. M. Tomlinson, D. R. Collins, M. J. Cubison, J. Dunlea, J. A. Huffman, T. B. Onasch, M. R. Alfarra, P. I. Williams, K. Bower, Y. Kondo, J. Schneider, F. Drewnick, S. Borrmann, S. Weimer, K. Demerjian, D. Salcedo, L. Cottrell, R. Griffin, A. Takami, T. Miyoshi, S. Hatakeyama, A. Shimono, J. Y. Sun, Y. M. Zhang, K. Dzepina, J. R. Kimmel, D. Sueper, J. T. Jayne, S. C. Herndon, A. M. Trimborn, L. R. Williams, E. C. Wood, A. M. Middlebrook, C. E. Kolb, U. Baltensperger and D. R. Worsnop, Science, 2009, 326, 1525–1529 CrossRef CAS PubMed.
- M. Hallquist, J. C. Wenger, U. Baltensperger, Y. Rudich, D. Simpson, M. Claeys, J. Dommen, N. M. Donahue, C. George, A. H. Goldstein, J. F. Hamilton, F. Herrmann, T. Hoffmann, Y. Iinuma, M. Jang, M. E. Jenkin, J. L. Jimenez, A. Kiendler-Scharr, W. Maenhaut, G. McFiggans, T. F. Mentel, A. Monod, A. S. H. Prevot, J. H. Seinfeld, J. D. Surratt, R. Szmigielski and J. Wildt, Atmos. Chem. Phys., 2009, 9, 5155–5236 CrossRef CAS.
- J. H. Kroll and J. H. Seinfeld, Atmos. Environ., 2008, 42, 3593–3624 CrossRef CAS.
- I. Riipinen, J. Pierce, T. Yli-Juuti, T. Nieminen, S. Hakkinen, M. Ehn, H. Junninen, K. Lehtipalo, T. Petaja and J. Slowik, Atmos. Chem. Phys., 2011, 11, 3865 CrossRef CAS.
- N. M. Donahue, S. A. Epstein, S. N. Pandis and A. L. Robinson, Atmos. Chem. Phys., 2011, 11, 3303–3318 CrossRef CAS.
- U. Baltensperger, M. Kalberer, J. Dommen, D. Paulsen, M. R. Alfarra, H. Coe, R. Fisseha, A. Gascho, M. Gysel, S. Nyeki, M. Sax, M. Steinbacher, A. S. H. Prevot, S. Sjogren, E. Weingartner and R. Zenobi, Faraday Discuss., 2005, 130, 265–278 RSC.
- R. Zhao, C. M. Kenseth, Y. Huang, N. F. Dalleska and J. H. Seinfeld, Environ. Sci. Technol., 2018, 52, 2108–2117 CrossRef CAS PubMed.
- C. L. Heald, H. Coe, J. L. Jimenez, R. J. Weber, R. Bahreini, A. M. Middlebrook, L. M. Russell, M. Jolleys, T. M. Fu, J. D. Allan, K. N. Bower, G. Capes, J. Crosier, W. T. Morgan, N. H. Robinson, P. I. Williams, M. J. Cubison, P. F. DeCarlo and E. J. Dunlea, Atmos. Chem. Phys., 2011, 11, 12673–12696 CrossRef CAS.
- A. Karambelas, H. O. T. Pye, S. H. Budisulistiorini, J. D. Surratt and R. W. Pinder, Environ. Sci. Technol. Lett., 2014, 1, 278–283 CrossRef CAS.
- H. O. T. Pye, R. W. Pinder, I. R. Piletic, Y. Xie, S. L. Capps, Y.-H. Lin, J. D. Surratt, Z. Zhang, A. Gold, D. J. Luecken, W. T. Hutzell, M. Jaoui, J. H. Offenberg, T. E. Kleindienst, M. Lewandowski and E. O. Edney, Environ. Sci. Technol., 2013, 47, 11056–11064 CrossRef CAS PubMed.
- IPCC, 2013: Summary for Policymakers, in Climate Change 2013: The Physical Science Basis, Cambridge University Press, Cambridge, UK, 2013 Search PubMed.
- B. Noziere, M. Kaberer, M. Claeys, J. Allan, B. D'Anna, S. Decesari, E. Finessi, M. Glasius, I. Grgic, J. F. Hamilton, T. Hoffmann, Y. Iinuma, M. Jaoui, A. Kahno, C. J. Kampf, I. Kourtchev, W. Maenhaut, N. Marsden, S. Saarikoski, J. Schnelle-Kreis, J. D. Surratt, S. Szidat, R. Szmigielski and A. Wisthaler, Chem. Rev., 2015, 115, 3919–3983 CrossRef CAS PubMed.
- A. H. Goldstein and I. E. Galbally, Environ. Sci. Technol., 2007, 41, 1514–1521 CrossRef CAS PubMed.
- P. O. Wennberg, K. H. Bates, J. D. Crounse, L. G. Dodson, R. C. McVay, L. A. Mertens, T. B. Nguyen, E. Praske, R. H. Schwantes, M. D. Smarte, J. M. St Clair, A. P. Teng, X. Zhang and J. H. Seinfeld, Chem. Rev., 2018, 118, 3337–3390 CrossRef CAS PubMed.
- A. Guenther, T. Karl, P. Harley, C. Wiedinmyer, P. I. Palmer and C. Geron, Atmos. Chem. Phys., 2006, 6, 3181–3210 CrossRef CAS.
- A. B. Guenther, X. Jiang, C. L. Heald, T. Sakulyanontvittaya, T. Duhl, L. K. Emmons and X. Wang, Geosci. Model Dev., 2012, 5, 1471–1492 CrossRef.
- R. M. Kamens, M. W. Gery, H. E. Jeffries, M. Jackson and E. I. Cole, Int. J. Chem. Kinet., 1982, 14, 955–975 CrossRef CAS.
- S. N. Pandis, S. E. Paulson, J. H. Seinfeld and R. C. Flagan, Atmos. Environ., 1991, 25, 997–1008 CrossRef.
- M. Claeys, B. Graham, G. Vas, W. Wang, R. Vermeylen, V. Pashynska, J. Cafmeyer, P. Guyon, M. O. Andreae, P. Artaxo and W. Maenhaut, Science, 2004, 303, 1173–1176 CrossRef CAS PubMed.
- A. G. Carlton, C. Wiedinmyer and J. H. Kroll, Atmos. Chem. Phys., 2009, 9, 4987–5005 CrossRef CAS.
- D. K. Henze and J. H. Seinfeld, Geophys. Res. Lett., 2006, 33, L09812 CrossRef.
- R. Atkinson, J. Phys. Chem. Ref. Data, 1997, 26, 215–290 CrossRef CAS.
- J. H. Kroll, N. L. Ng, S. M. Murphy, R. C. Flagan and J. H. Seinfeld, Environ. Sci. Technol., 2006, 40, 1869–1877 CrossRef CAS PubMed.
- J. D. Surratt, S. M. Murphy, J. H. Kroll, N. L. Ng, L. Hildebrandt, A. Sorooshian, R. Szmigielski, R. Vermeylen, W. Maenhaut, M. Claeys, R. C. Flagan and J. H. Seinfeld, J. Phys. Chem. A, 2006, 110, 9665–9690 CrossRef CAS PubMed.
- M. N. Chan, J. D. Surratt, M. Claeys, E. S. Edgerton, R. L. Tanner, S. L. Shaw, M. Zheng, E. M. Knipping, N. C. Eddingsaas, P. O. Wennberg and J. H. Seinfeld, Environ. Sci. Technol., 2010, 44, 4590–4596 CrossRef CAS PubMed.
- J. D. Surratt, A. W. Chan, N. C. Eddingsaas, M. Chan, C. L. Loza, A. J. Kwan, S. P. Hersey, R. C. Flagan, P. O. Wennberg and J. H. Seinfeld, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6640–6645 CrossRef CAS PubMed.
- V. S. Nguyen and J. Peeters, J. Phys. Chem. A, 2015, 119, 7270–7276 CrossRef CAS PubMed.
- Y. H. Lin, E. M. Knipping, E. S. Edgerton, S. L. Shaw and J. D. Surratt, Atmos. Chem. Phys., 2013, 13, 8457–8470 CrossRef.
- E. O. Edney, T. E. Kleindienst, M. Jaoui, M. Lewandowski, J. H. Offenberg, W. Wang and M. Claeys, Atmos. Environ., 2005, 39, 5281–5289 CrossRef CAS.
- J. D. Crounse, H. C. Knap, K. B. Ornso, S. Jorgensen, F. Paulot, H. G. Kjaergaard and P. O. Wennberg, J. Phys. Chem. A, 2012, 116, 5756–5762 CrossRef CAS PubMed.
- F. Paulot, J. D. Crounse, H. G. Kjaergaard, A. Kurten, J. M. St Clair, J. H. Seinfeld and P. O. Wennberg, Science, 2009, 325, 730–733 CrossRef CAS PubMed.
- H. F. Zhang, Y. H. Lin, Z. F. Zhang, X. L. Zhang, S. L. Shaw, E. M. Knipping, R. J. Weber, A. Gold, R. M. Kamens and J. D. Surratt, Environ. Chem., 2012, 9, 247–262 CrossRef CAS.
- T. B. Nguyen, M. M. Coggon, K. H. Bates, X. Zhang, R. H. Schwantes, K. A. Schilling, C. L. Loza, R. C. Flagan, P. O. Wennberg and J. H. Seinfeld, Atmos. Chem. Phys., 2014, 14, 3497–3510 CrossRef.
- C. J. Gaston, T. P. Riedel, Z. F. Zhang, A. Gold, J. D. Surratt and J. A. Thornton, Environ. Sci. Technol., 2014, 48, 11178–11186 CrossRef CAS PubMed.
- J. D. Surratt, M. Lewandowski, J. H. Offenberg, M. Jaoui, T. E. Kleindienst, E. O. Edney and J. H. Seinfeld, Environ. Sci. Technol., 2007, 41, 5363–5369 CrossRef CAS PubMed.
- J. Schindelka, Y. Iinuma, D. Hoffmann and H. Herrmann, Faraday Discuss., 2013, 165, 237–259 RSC.
- M. S. Shalamzari, O. Ryabtsova, A. Kahnt, R. Vermeylen, M. F. Herent, J. Quetin-Leclercq, P. Van der Veken, W. Maenhaut and M. Claeys, Rapid Commun. Mass Spectrom., 2013, 27, 784–794 CrossRef PubMed.
- J. D. Crounse, F. Paulot, H. G. Kjaergaard and P. O. Wennberg, Phys. Chem. Chem. Phys., 2011, 13, 13607–13613 RSC.
- J. D. Crounse, L. B. Nielsen, S. Jorgensen, H. G. Kjaergaard and P. O. Wennberg, J. Phys. Chem. Lett., 2013, 4, 3513–3520 CrossRef CAS.
- D. R. Worton, J. D. Surratt, B. W. Lafranchi, A. W. Chan, Y. Zhao, R. J. Weber, J. H. Park, J. B. Gilman, J. de Gouw, C. Park, G. Schade, M. Beaver, J. M. Clair, J. Crounse, P. Wennberg, G. M. Wolfe, S. Harrold, J. A. Thornton, D. K. Farmer, K. S. Docherty, M. J. Cubison, J. L. Jimenez, A. A. Frossard, L. M. Russell, K. Kristensen, M. Glasius, J. Mao, X. Ren, W. Brune, E. C. Browne, S. E. Pusede, R. C. Cohen, J. H. Seinfeld and A. H. Goldstein, Environ. Sci. Technol., 2013, 47, 11403–11413 CrossRef CAS PubMed.
- J. C. Rivera-Rios, T. B. Nguyen, J. D. Crounse, W. Jud, J. M. St Clair, T. Mikoviny, J. B. Gilman, B. M. Lerner, J. B. Kaiser, J. de Gouw, A. Wisthaler, A. Hansel, P. O. Wennberg, J. H. Seinfeld and F. N. Keutsch, Geophys. Res. Lett., 2014, 41, 8645–8651 CrossRef CAS.
- Y. Li, H. D. Hao and Y. K. Wu, Org. Lett., 2009, 11, 2691–2694 CrossRef CAS PubMed.
- Y. Li, H. D. Hao, Q. Zhang and Y. K. Wu, Org. Lett., 2009, 11, 1615–1618 CrossRef CAS PubMed.
- P. H. Dussault and K. R. Woller, J. Org. Chem., 1997, 62, 1556–1559 CrossRef CAS.
- W. Adam, N. Bottke, O. Krebs, I. Lykakis, M. Orfanopoulos and M. Stratakis, J. Am. Chem. Soc., 2002, 124, 14403–14409 CrossRef CAS PubMed.
- J. M. St. Clair, J. C. Rivera-Rios, J. D. Crounse, H. C. Knap, K. H. Bates, A. P. Teng, S. Jorgensen, H. G. Kjaergaard, F. N. Keutsch and P. O. Wennberg, J. Phys. Chem. A, 2015, 120, 1441–1451 CrossRef PubMed.
- J. E. Krechmer, M. M. Coggon, P. Massoli, T. B. Nguyen, J. D. Crounse, W. W. Hu, D. A. Day, G. S. Tyndall, D. K. Henze, J. C. Rivera-Rios, J. B. Nowak, J. R. Kimmel, R. L. Mauldin, H. Stark, J. T. Jayne, M. Sipila, H. Junninen, J. M. S. Clair, X. Zhang, P. A. Feiner, L. Zhang, D. O. Miller, W. H. Brune, F. N. Keutsch, P. O. Wennberg, J. H. Seinfeld, D. R. Worsnop, J. L. Jimenez and M. R. Canagaratna, Environ. Sci. Technol., 2015, 49, 10330–10339 CrossRef CAS PubMed.
- T. P. Riedel, Y. H. Lin, H. Budisulistiorini, C. J. Gaston, J. A. Thornton, Z. F. Zhang, W. Vizuete, A. Gold and J. D. Surrattt, Environ. Sci. Technol. Lett., 2015, 2, 38–42 CrossRef CAS.
- M. Riva, S. H. Budisulistiorini, Y. Z. Chen, Z. F. Zhang, E. L. D'Ambro, X. Zhang, A. Gold, B. J. Turpin, J. A. Thornton, M. R. Canagaratna and J. D. Surratt, Environ. Sci. Technol., 2016, 50, 9889–9899 CrossRef CAS PubMed.
- J. M. Liu, E. L. D'Ambro, B. H. Lee, F. D. Lopez-Hilfiker, R. A. Zaveri, J. C. Rivera-Rios, F. N. Keutsch, S. Iyer, T. Kurten, Z. F. Zhang, A. Gold, J. D. Surratt, J. E. Shilling and J. A. Thornton, Environ. Sci. Technol., 2016, 50, 9872–9880 CrossRef CAS PubMed.
- T. B. Nguyen, J. D. Crounse, R. H. Schwantes, A. P. Teng, K. H. Bates, X. Zhang, J. M. St Clair, W. H. Brune, G. S. Tyndall, F. N. Keutsch, J. H. Seinfeld and P. O. Wennberg, Atmos. Chem. Phys., 2014, 14, 13531–13549 CrossRef.
- S. H. Budisulistiorini, X. Li, S. T. Bairai, J. Renfro, Y. Liu, Y. J. Liu, K. A. McKinney, S. T. Martin, V. F. McNeill, H. O. T. Pye, A. Nenes, M. E. Neff, E. A. Stone, S. Mueller, C. Knote, S. L. Shaw, Z. Zhang, A. Gold and J. D. Surratt, Atmos. Chem. Phys., 2015, 15, 8871–8888 CrossRef CAS.
- A. J. Kramer, W. Rattanavaraha, Z. F. Zhang, A. Gold, J. D. Surratt and Y. H. Lin, Atmos. Environ., 2016, 130, 211–218 CrossRef CAS.
- L. Xu, A. M. Middlebrook, J. Liao, J. A. de Gouw, H. Y. Guo, R. J. Weber, A. Nenes, F. D. Lopez-Hilfiker, B. H. Lee, J. A. Thornton, C. A. Brock, J. A. Neuman, J. B. Nowak, I. B. Pollack, A. Welti, M. Graus, C. Warneke and N. L. Ng, J. Geophys. Res.: Atmos., 2016, 121, 11137–11153 CrossRef CAS.
- L. P. Su, E. G. Patton, J. V. G. de Arellano, A. B. Guenther, L. Kaser, B. Yuan, F. L. Z. Xiong, P. B. Shepson, L. Zhang, D. O. Miller, W. H. Brune, K. Baumann, E. Edgerton, A. Weinheimer, P. K. Misztal, J. H. Park, A. H. Goldstein, K. M. Skog, F. N. Keutsch and J. E. Mak, Atmos. Chem. Phys., 2016, 16, 7725–7741 CrossRef CAS.
- N. C. Eddingsaas, D. G. VanderVelde and P. O. Wennberg, J. Phys. Chem. A, 2010, 114, 8106–8113 CrossRef CAS PubMed.
- W. Wang, I. Kourtchev, B. Graham, J. Cafmeyer, W. Maenhaut and M. Claeys, Rapid Commun. Mass Spectrom., 2005, 19, 1343–1351 CrossRef CAS PubMed.
- W. Adam, K. Peters and M. Renz, J. Org. Chem., 1997, 62, 3183–3189 CrossRef CAS PubMed.
- C. Chiappe, A. De Rubertis, V. Tinagli, G. Amato and P. G. Gervasi, Chem. Res. Toxicol., 2000, 13, 831–838 Search PubMed.
- N. C. Cole-Filipiak, A. E. O'Connor and M. J. Elrod, Environ. Sci. Technol., 2010, 44, 6718–6723 CrossRef CAS PubMed.
- Y.-H. Lin, S. H. Budisulistiorini, K. Chu, R. A. Siejack, H. Zhang, M. Riva, Z. Zhang, A. Gold, K. E. Kautzman and J. D. Surratt, Environ. Sci. Technol., 2014, 48, 12012–12021 CrossRef CAS PubMed.
- S. H. Budisulistiorini, M. R. Canagaratna, P. L. Croteau, W. J. Marth, K. Baumann, E. S. Edgerton, S. L. Shaw, E. M. Knipping, D. R. Worsnop, J. T. Jayne, A. Gold and J. D. Surratt, Environ. Sci. Technol., 2013, 47, 5686–5694 CrossRef CAS PubMed.
- W. Rattanavaraha, K. Chu, H. Budisulistiorini, M. Riva, Y. H. Lin, E. S. Edgerton, K. Baumann, S. L. Shaw, H. Y. Guo, L. King, R. J. Weber, M. E. Neff, E. A. Stone, J. H. Offenberg, Z. F. Zhang, A. Gold and J. D. Surratt, Atmos. Chem. Phys., 2016, 16, 4897–4914 CrossRef CAS PubMed.
- Y. J. Zhang, L. L. Tang, Y. L. Sun, O. Favez, F. Canonaco, A. Albinet, F. Couvidat, D. T. Liu, J. T. Jayne, Z. Wang, P. L. Croteau, M. R. Canagaratna, H. C. Zhou, A. S. H. Prevot and D. R. Worsnop, Geophys. Res. Lett., 2017, 44, 2035–2043 CAS.
- Y.-H. Lin, Z. Zhang, K. S. Docherty, H. Zhang, S. H. Budisulistiorini, C. L. Rubitschun, S. L. Shaw, E. M. Knipping, E. S. Edgerton, T. E. Kleindienst, A. Gold and J. D. Surratt, Environ. Sci. Technol., 2012, 46, 250–258 CrossRef CAS PubMed.
- S. J. Stropoli, C. R. Miner, D. R. Hill and M. J. Elrod, Environ. Sci. Technol., 2019, 53, 176–184 CrossRef CAS PubMed.
- Z. Zhang, Y. H. Lin, H. Zhang, J. D. Surratt, L. M. Ball and A. Gold, Atmos. Chem. Phys., 2012, 12, 8529–8535 CrossRef CAS.
- C. J. Ebben, B. F. Strick, M. A. Upshur, H. M. Chase, J. L. Achtyl, R. J. Thomson and F. M. Geiger, Atmos. Chem. Phys., 2014, 14, 2303–2314 CrossRef.
- K. H. Bates, J. D. Crounse, J. M. St Clair, N. B. Bennett, T. B. Nguyen, J. H. Seinfeld, B. M. Stoltz and P. O. Wennberg, J. Phys. Chem. A, 2014, 118, 1237–1246 CrossRef CAS PubMed.
- K. H. Bates, T. B. Nguyen, A. P. Teng, J. D. Crounse, H. G. Kjaergaard, B. M. Stoltz, J. H. Seinfeld and P. O. Wennberg, J. Phys. Chem. A, 2016, 120, 106–117 CrossRef CAS PubMed.
- T. V. Robinson, D. S. Pedersen, D. K. Taylor and E. R. T. Tiekink, J. Org. Chem., 2009, 74, 5093–5096 CrossRef CAS PubMed.
- Y. J. Liu, M. Kuwata, B. F. Strick, F. M. Geiger, R. J. Thomson, K. A. McKinney and S. T. Martin, Environ. Sci. Technol., 2015, 49, 250–258 CrossRef CAS PubMed.
- M. A. Upshur, B. F. Strick, V. F. McNeill, R. J. Thomson and F. M. Geiger, Atmos. Chem. Phys., 2014, 14, 10731–10740 CrossRef.
- A. I. Darer, N. C. Cole-Filipiak, A. E. O'Connor and M. J. Elrod, Environ. Sci. Technol., 2011, 45, 1895–1902 CrossRef CAS PubMed.
- T. Cui, Z. Zeng, E. O. dos Santos, Z. Zhang, Y. Chen, Y. Zhang, C. A. Rose, S. H. Budisulistiorini, L. B. Collins, W. M. Bodnar, R. A. F. de Souza, S. T. Martin, C. M. D. Machado, B. J. Turpin, A. Gold, A. P. Ault and J. D. Surratt, Environ. Sci.: Processes Impacts, 2018, 20, 1524–1536 RSC.
- I. Kourtchev, T. Ruuskanen, W. Maenhaut, M. Kulmala and M. Claeys, Atmos. Chem. Phys., 2005, 5, 2761–2770 CrossRef CAS.
- I. Kourtchev, T. M. Ruuskanen, P. Keronen, L. Sogacheva, M. Dal Maso, A. Reissell, X. Chi, R. Vermeylen, M. Kulmala, W. Maenhaut and M. Claeys, Plant Biology, 2008, 10, 138–149 CrossRef CAS PubMed.
- S. Decesari, S. Fuzzi, M. C. Facchini, M. Mircea, L. Emblico, F. Cavalli, W. Maenhaut, X. Chi, G. Schkolnik, A. Falkovich, Y. Rudich, M. Claeys, V. Pashynska, G. Vas, I. Kourtchev, R. Vermeylen, A. Hoffer, M. O. Andreae, E. Tagliavini, F. Moretti and P. Artaxo, Atmos. Chem. Phys., 2006, 6, 375–402 CrossRef CAS.
- A. L. Clements and J. H. Seinfeld, Atmos. Environ., 2007, 41, 1825–1830 CrossRef CAS.
- X. Xia and P. K. Hopke, Environ. Sci. Technol., 2006, 40, 6934–6937 CrossRef CAS PubMed.
- T. M. Cahill, V. Y. Seaman, M. J. Charles, R. Holzinger and A. H. Goldstein, J. Geophys. Res.: Atmos., 2006, 111, D16312 CrossRef.
- J. Dommen, A. Metzger, J. Duplissy, M. Kalberer, M. R. Alfarra, A. Gascho, E. Weingartner, A. S. H. Prevot, B. Verheggen and U. Baltensperger, Geophys. Res. Lett., 2006, 33, L13805 CrossRef.
- A. Fontana, R. Messina, A. Spinella and G. Cimino, Tetrahedron Lett., 2000, 41, 7559–7562 CrossRef CAS.
- A. R. Moen, K. Ruud and T. Anthonsen, Eur. J. Org. Chem., 2007, 1262–1266 CrossRef CAS.
- N. J. D. Gonzalez, A. K. Borg-Karlson, J. P. Redeby, B. Noziere, R. Krejci, Y. X. Pei, J. Dommen and A. S. H. Prevot, J. Chromatogr. A, 2011, 1218, 9288–9294 CrossRef CAS PubMed.
- J. M. Cash, M. R. Heal, B. Langford and J. Drewer, Environ. Sci.: Processes Impacts, 2016, 18, 1369–1380 RSC.
- J. J. Orlando, G. S. Tyndall and S. E. Paulson, Geophys. Res. Lett., 1999, 26, 2191–2194 CrossRef CAS.
- T. Gierczak, J. B. Burkholder, R. K. Talukdar, A. Mellouki, S. B. Barone and A. R. Ravishankara, J. Photochem. Photobiol., A, 1997, 110, 1–10 CrossRef CAS.
- M. M. Galloway, A. J. Huisman, L. D. Yee, A. W. H. Chan, C. L. Loza, J. H. Seinfeld and F. N. Keutsch, Atmos. Chem. Phys., 2011, 11, 10779–10790 CrossRef CAS.
- M. Jaoui, E. O. Edney, T. E. Kleindienst, M. Lewandowski, J. H. Offenberg, J. D. Surratt and J. H. Seinfeld, J. Geophys. Res.: Atmos., 2008, 113, D09303 CrossRef.
- R. Szmigielski, J. D. Surratt, R. Vermeylen, K. Szmigielska, J. H. Kroll, N. L. Ng, S. M. Murphy, A. Sorooshian, J. H. Seinfeld and M. Claeys, J. Mass Spectrom., 2007, 42, 101–116 CrossRef CAS PubMed.
- H. Zhang, J. D. Surratt, Y. H. Lin, J. Bapat and R. M. Kamens, Atmos. Chem. Phys., 2011, 11, 6411–6424 CrossRef CAS.
- Y. H. Lin, H. F. Zhang, H. O. T. Pye, Z. F. Zhang, W. J. Marth, S. Park, M. Arashiro, T. Q. Cui, H. Budisulistiorini, K. G. Sexton, W. Vizuete, Y. Xie, D. J. Luecken, I. R. Piletic, E. O. Edney, L. J. Bartolotti, A. Gold and J. D. Surratt, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6718–6723 CrossRef CAS PubMed.
- A. W. H. Chan, M. N. Chan, J. D. Surratt, P. S. Chhabra, C. L. Loza, J. D. Crounse, L. D. Yee, R. C. Flagan, P. O. Wennberg and J. H. Seinfeld, Atmos. Chem. Phys., 2010, 10, 7169–7188 CrossRef CAS.
- J. D. Surratt, J. H. Kroll, T. E. Kleindienst, E. O. Edney, M. Claeys, A. Sorooshian, N. L. Ng, J. H. Offenberg, M. Lewandowski, M. Jaoui, R. C. Flagan and J. H. Seinfeld, Environ. Sci. Technol., 2007, 41, 517–527 CrossRef CAS PubMed.
- Y. Gómez-González, J. D. Surratt, F. Cuyckens, R. Szmigielski, R. Vermeylen, M. Jaoui, M. Lewandowski, J. H. Offenberg, T. E. Kleindienst, E. O. Edney, F. Blockhuys, C. Van Alsenoy, W. Maenhaut and M. Claeys, J. Mass Spectrom., 2008, 43, 371–382 CrossRef PubMed.
- L. E. Hatch, J. M. Creamean, A. P. Ault, J. D. Surratt, M. N. Chan, J. H. Seinfeld, E. S. Edgerton, Y. X. Su and K. A. Prather, Environ. Sci. Technol., 2011, 45, 5105–5111 CrossRef CAS PubMed.
- L. E. Hatch, J. M. Creamean, A. P. Ault, J. D. Surratt, M. N. Chan, J. H. Seinfeld, E. S. Edgerton, Y. X. Su and K. A. Prather, Environ. Sci. Technol., 2011, 45, 8648–8655 CrossRef CAS PubMed.
- A. W. Birdsall, C. R. Miner, L. E. Mael and M. J. Elrod, Atmos. Chem. Phys., 2014, 14, 12951–12964 CrossRef.
- M. Claeys, W. Wang, A. C. Ion, I. Kourtchev, A. Gelencser and W. Maenhaut, Atmos. Environ., 2004, 38, 4093–4098 CrossRef CAS.
- T. B. Nguyen, K. H. Bates, J. D. Crounse, R. H. Schwantes, X. Zhang, H. G. Kjaergaard, J. D. Surratt, P. Lin, A. Laskin, J. H. Seinfeld and P. O. Wennberg, Phys. Chem. Chem. Phys., 2015, 17, 17914–17926 RSC.
- A. W. Birdsall, C. A. Zentner and M. J. Elrod, Atmos. Chem. Phys., 2013, 13, 3097–3109 CrossRef.
- C. D. Geron, R. W. Daly, R. R. Arnts, A. B. Guenther and F. L. Mowry, Sci. Total Environ., 2016, 565, 730–741 CrossRef CAS PubMed.
- P. Harley, V. Fridd-Stroud, J. Greenberg, A. Guenther and P. Vasconcellos, J. Geophys. Res.: Atmos., 1998, 103, 25479–25486 CrossRef CAS.
- K. Sindelarova, C. Granier, I. Bouarar, A. Guenther, S. Tilmes, T. Stavrakou, J. F. Muller, U. Kuhn, P. Stefani and W. Knorr, Atmos. Chem. Phys., 2014, 14, 9317–9341 CrossRef.
- P. D. Goldan, W. C. Kuster, F. C. Fehsenfeld and S. A. Montzka, Geophys. Res. Lett., 1993, 20, 1039–1042 CrossRef CAS.
- H. F. Zhang, D. R. Worton, M. Lewandowski, J. Ortega, C. L. Rubitschun, J. H. Park, K. Kristensen, P. Campuzano-Jost, D. A. Day, J. L. Jimenez, M. Jaoui, J. H. Offenberg, T. E. Kleindienst, J. Gilman, W. C. Kuster, J. de Gouw, C. Park, G. W. Schade, A. A. Frossard, L. Russell, L. Kaser, W. Jud, A. Hansel, L. Cappellin, T. Karl, M. Glasius, A. Guenther, A. H. Goldstein, J. H. Seinfeld, A. Gold, R. M. Kamens and J. D. Surratt, Environ. Sci. Technol., 2012, 46, 9437–9446 CrossRef CAS PubMed.
- H. F. Zhang, Z. F. Zhang, T. Q. Cui, Y. H. Lin, N. A. Bhathela, J. Ortega, D. R. Worton, A. H. Goldstein, A. Guenther, J. L. Jimenez, A. Gold and J. D. Surratt, Environ. Sci. Technol. Lett., 2014, 1, 242–247 CrossRef CAS PubMed.
- A. W. H. Chan, M. M. Galloway, A. J. Kwan, P. S. Chhabra, F. N. Keutsch, P. O. Wennberg, R. C. Flagan and J. H. Seinfeld, Environ. Sci. Technol., 2009, 43, 4647–4652 CrossRef CAS PubMed.
- M. Jaoui, T. E. Kleindienst, J. H. Offenberg, M. Lewandowski and W. A. Lonneman, Atmos. Chem. Phys., 2012, 12, 2173–2188 CrossRef CAS.
- J. H. Kroll, N. L. Ng, S. M. Murphy, V. Varutbangkul, R. C. Flagan and J. H. Seinfeld, J. Geophys. Res.: Atmos., 2005, 110, D23207 CrossRef.
- J. Liggio, S. M. Li and R. McLaren, J. Geophys. Res.: Atmos., 2005, 110, D10304 CrossRef.
- I. Magneron, A. Mellouki, G. Le Bras, G. K. Moortgat, A. Horowitz and K. Wirtz, J. Phys. Chem. A, 2005, 109, 4552–4561 CrossRef CAS PubMed.
- N. Sareen, A. G. Carlton, J. D. Surratt, A. Gold, B. Lee, F. D. Lopez-Hilfiker, C. Mohr, J. A. Thornton, Z. F. Zhang, Y. B. Lim and B. J. Turpin, Atmos. Chem. Phys., 2016, 16, 14409–14420 CrossRef CAS.
- N. Carrasco, J. F. Doussin, M. O'Connor, J. C. Wenger, B. Picquet-Varrault, R. Durand-Jolibois and P. Carlier, J. Atmos. Chem., 2007, 56, 33–55 CrossRef CAS.
- H. C. Knap, J. A. Schmidt and S. Jorgensen, Atmos. Environ., 2016, 147, 79–87 CrossRef CAS.
- L. E. Mael, M. I. Jacobs and M. J. Elrod, J. Phys. Chem. A, 2015, 119, 4464–4472 CrossRef CAS PubMed.
- A. Calogirou, B. R. Larsen and D. Kotzias, Atmos. Environ., 1999, 33, 1423–1439 CrossRef CAS.
- R. Atkinson and J. Arey, Acc. Chem. Res., 1998, 31, 574–583 CrossRef CAS.
- A. Guenther, C. N. Hewitt, D. Erickson, R. Fall, C. Geron, T. Graedel, P. Harley, L. Klinger, M. Lerdau, W. A. Mckay, T. Pierce, B. Scholes, R. Steinbrecher, R. Tallamraju, J. Taylor and P. Zimmerman, J. Geophys. Res.: Atmos., 1995, 100, 8873–8892 CrossRef CAS.
- R. J. Griffin, D. R. Cocker, J. H. Seinfeld and D. Dabdub, Geophys. Res. Lett., 1999, 26, 2721–2724 CrossRef CAS.
- M. Claeys, Y. Iinuma, R. Szmigielski, J. D. Surratt, F. Blockhuys, C. Van Alsenoy, O. Boge, B. Sierau, Y. Gomez-Gonzalez, R. Vermeylen, P. Van der Veken, M. Shahgholi, A. W. H. Chan, H. Herrmann, J. H. Seinfeld and W. Maenhaut, Environ. Sci. Technol., 2009, 43, 6976–6982 CrossRef CAS PubMed.
- M. E. Jenkin, D. E. Shallcross and J. N. Harvey, Atmos. Environ., 2000, 34, 2837–2850 CrossRef CAS.
- K. Kristensen, T. Cui, H. Zhang, A. Gold, M. Glasius and J. D. Surratt, Atmos. Chem. Phys., 2014, 14, 4201–4218 CrossRef.
- R. K. Pathak, A. A. Presto, T. E. Lane, C. O. Stanier, N. M. Donahue and S. N. Pandis, Atmos. Chem. Phys., 2007, 7, 3811–3821 CrossRef CAS.
- A. A. Presto and N. M. Donahue, Environ. Sci. Technol., 2006, 40, 3536–3543 CrossRef CAS PubMed.
- F. Yasmeen, R. Vermeylen, R. Szmigielski, Y. Iinuma, O. Boge, H. Herrmann, W. Maenhaut and M. Claeys, Atmos. Chem. Phys., 2010, 10, 9383–9392 CrossRef CAS.
- X. Zhang, R. C. Mcvay, D. D. Huang, N. F. Dalleska, B. Aumont, R. C. Flagan and J. H. Seinfeld, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 14168–14173 CrossRef CAS PubMed.
- W. A. Hall and M. V. Johnston, Aerosol Sci. Technol., 2011, 45, 37–45 CrossRef CAS.
- W. I. V. Hall and M. Johnston, J. Am. Soc. Mass Spectrom., 2012, 23, 1097–1108 CrossRef CAS PubMed.
- L. Müller, M. C. Reinnig, H. Hayen and T. Hoffmann, Rapid Commun. Mass Spectrom., 2009, 23, 971–979 CrossRef PubMed.
- L. Muller, M. C. Reinnig, J. Warnke and T. Hoffmann, Atmos. Chem. Phys., 2008, 8, 1423–1433 CrossRef.
- S. Gao, N. L. Ng, M. Keywood, V. Varutbangkul, R. Bahreini, A. Nenes, J. He, K. Y. Yoo, J. L. Beauchamp, R. P. Hodyss, R. C. Flagan and J. H. Seinfeld, Environ. Sci. Technol., 2004, 38, 6582–6589 CrossRef CAS PubMed.
- M. Ehn, J. A. Thornton, E. Kleist, M. Sipila, H. Junninen, I. Pullinen, M. Springer, F. Rubach, R. Tillmann, B. Lee, F. Lopez-Hilfiker, S. Andres, I. H. Acir, M. Rissanen, T. Jokinen, S. Schobesberger, J. Kangasluoma, J. Kontkanen, T. Nieminen, T. Kurten, L. B. Nielsen, S. Jorgensen, H. G. Kjaergaard, M. Canagaratna, M. D. Maso, T. Berndt, T. Petaja, A. Wahner, V. M. Kerminen, M. Kulmala, D. R. Worsnop, J. Wildt and T. F. Mentel, Nature, 2014, 506, 476–479 CrossRef CAS PubMed.
- A. Mutzel, L. Poulain, T. Berndt, Y. Iinuma, M. Rodigast, O. Boge, S. Richters, G. Spindler, M. Sipila, T. Jokinen, M. Kulmala and H. Herrmann, Environ. Sci. Technol., 2015, 49, 7754–7761 CrossRef CAS PubMed.
- T. Jokinen, T. Berndt, R. Makkonen, V. M. Kerminen, H. Junninen, P. Paasonen, F. Stratmann, H. Herrmann, A. B. Guenther, D. R. Worsnop, M. Kulmala, M. Ehn and M. Sipila, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 7123–7128 CrossRef CAS PubMed.
- F. Yasmeen, R. Szmigielski, R. Vermeylen, Y. Gomez-Gonzalez, J. D. Surratt, A. W. H. Chan, J. H. Seinfeld, W. Maenhaut and M. Claeys, J. Mass Spectrom., 2011, 46, 425–442 CrossRef CAS PubMed.
- N. M. Donahue, J. E. Tischuk, B. J. Marquis and K. E. H. Hartz, Phys. Chem. Chem. Phys., 2007, 9, 2991–2998 RSC.
- M. Glasius, M. Lahaniati, A. Calogirou, D. Di Bella, N. R. Jensen, J. Hjorth, D. Kotzias and B. R. Larsen, Environ. Sci. Technol., 2000, 34, 1001–1010 CrossRef CAS.
- A. Mutzel, M. Rodigast, Y. Iinuma, O. Boge and H. Herrmann, Atmos. Environ., 2016, 130, 136–144 CrossRef CAS.
- M. Hallquist, I. Wangberg and E. Ljungstrom, Environ. Sci. Technol., 1997, 31, 3166–3172 CrossRef CAS.
- B. Witkowski, S. Jurdana and T. Gierczak, Environ. Sci. Technol., 2018, 52, 3402–3411 CrossRef CAS PubMed.
- J. V. Amorim, S. Wu, K. Klimchuk, C. Lau, F. J. Williams, Y. Huang and R. Zhao, Environ. Sci. Technol., 2020, 54, 12484–12492 CrossRef CAS PubMed.
- M. Glasius, A. Calogirou, N. R. Jensen, J. Hjorth and C. J. Nielsen, Int. J. Chem. Kinet., 1997, 29, 527–533 CrossRef CAS.
- A. G. Bé, M. A. Upshur, P. Liu, S. T. Martin, F. M. Geiger and R. J. Thomson, ACS Cent. Sci., 2017, 3, 715–725 CrossRef PubMed.
- M. Boy, T. Petaja, M. Dal Maso, U. Rannik, J. Rinne, P. Aalto, A. Laaksonen, P. Vaattovaara, J. Joutsensaari, T. Hoffmann, J. Warnke, M. Apostolaki, E. G. Stephanou, M. Tsapakis, A. Kouvarakis, C. Pio, A. Carvalho, A. Rompp, G. Moortgat, C. Spirig, A. Guenther, J. Greenberg, P. Ciccioli and M. Kulmala, Atmos. Chem. Phys., 2004, 4, 657–678 CrossRef CAS.
- A. Laaksonen, M. Kulmala, C. D. O'Dowd, J. Joutsensaari, P. Vaattovaara, S. Mikkonen, K. E. J. Lehtinen, L. Sogacheva, M. Dal Maso, P. Aalto, T. Petaja, A. Sogachev, Y. J. Yoon, H. Lihavainen, D. Nilsson, M. C. Facchini, F. Cavalli, S. Fuzzi, T. Hoffmann, F. Arnold, M. Hanke, K. Sellegri, B. Umann, W. Junkermann, H. Coe, J. D. Allan, M. R. Alfarra, D. R. Worsnop, M. L. Riekkola, T. Hyotylainen and Y. Viisanen, Atmos. Chem. Phys., 2008, 8, 2657–2665 CrossRef CAS.
- B. Noziere, I. Barnes and K. H. Becker, J. Geophys. Res.: Atmos., 1999, 104, 23645–23656 CrossRef CAS.
- N. C. Eddingsaas, C. L. Loza, L. D. Yee, M. Chan, K. A. Schilling, P. S. Chhabra, J. H. Seinfeld and P. O. Wennberg, Atmos. Chem. Phys., 2012, 12, 7413–7427 CrossRef CAS.
- N. C. Eddingsaas, C. L. Loza, L. D. Yee, J. H. Seinfeld and P. O. Wennberg, Atmos. Chem. Phys., 2012, 12, 6489–6504 CrossRef CAS.
- M. Jaoui and R. M. Kamens, Atmos. Environ., 2003, 37, 1835–1851 CrossRef CAS.
- I. G. Kavouras, N. Mihalopoulos and E. G. Stephanou, Nature, 1998, 395, 683–686 CrossRef CAS.
- A. Lee, A. H. Goldstein, M. D. Keywood, S. Gao, V. Varutbangkul, R. Bahreini, N. L. Ng, R. C. Flagan and J. H. Seinfeld, J. Geophys. Res.: Atmos., 2006, 111, D07302 Search PubMed.
- A. Lee, A. H. Goldstein, J. H. Kroll, N. L. Ng, V. Varutbangkul, R. C. Flagan and J. H. Seinfeld, J. Geophys. Res.: Atmos., 2006, 111, D17305 CrossRef.
- B. Noziere, M. Spittler, L. Ruppert, I. Barnes, K. H. Becker, M. Pons and K. Wirtz, Int. J. Chem. Kinet., 1999, 31, 291–301 CrossRef CAS.
- H. J. Chacon-Madrid and N. M. Donahue, Atmos. Chem. Phys., 2011, 11, 10553–10563 CrossRef CAS.
- H. J. Chacon-Madrid, K. M. Henry and N. M. Donahue, Atmos. Chem. Phys., 2013, 13, 3227–3236 CrossRef.
- G. Duporte, J. Parshintsev, L. M. F. Barreira, K. Hartonen, M. Kulmala and M. L. Riekkola, Environ. Sci. Technol., 2016, 50, 4693–4700 CrossRef CAS PubMed.
- S. R. Jackson, J. E. Ham, J. C. Harrison and J. R. Wells, J. Atmos. Chem., 2016, 1–14, DOI:10.1007/s10874-016-9344-6.
- T. S. Christoffersen, J. Hjorth, O. Horie, N. R. Jensen, D. Kotzias, L. L. Molander, P. Neeb, L. Ruppert, R. Winterhalter, A. Virkkula, K. Wirtz and B. R. Larsen, Atmos. Environ., 1998, 32, 1657–1661 CrossRef CAS.
- A. G. Moglioni, E. Garcia-Exposito, G. P. Aguado, T. Parella, V. Branchadell, G. Y. Moltrasio and R. M. Ortuno, J. Org. Chem., 2000, 65, 3934–3940 CrossRef CAS PubMed.
- Y. Ma, T. R. Willcox, A. T. Russell and G. Marston, Chem. Commun., 2007, 1328–1330 RSC.
- T. Yabuuchi and T. Kusumi, J. Org. Chem., 2000, 65, 397–404 CrossRef CAS PubMed.
- K. Kristensen, K. L. Enggrob, S. M. King, D. R. Worton, S. M. Platt, R. Mortensen, T. Rosenoern, J. D. Surratt, M. Bilde, A. H. Goldstein and M. Glasius, Atmos. Chem. Phys., 2013, 13, 3763–3776 CrossRef.
- A. Kołodziejczyk, P. Pyrcz, A. Pobudkowska, K. Błaziak and R. Szmigielski, J. Phys. Chem. B, 2019, 123, 8261–8267 CrossRef PubMed.
- S. S. Steimer, A. Delvaux, S. J. Campbell, P. J. Gallimore, P. Grice, D. J. Howe, D. Pitton, M. Claeys, T. Hoffmann and M. Kalberer, Atmos. Chem. Phys., 2018, 18, 10973–10983 CrossRef CAS.
- R. Zhao, C. M. Kenseth, Y. Huang, N. F. Dalleska, X. M. Kuang, J. Chen, S. E. Paulson and J. H. Seinfeld, J. Phys. Chem. A, 2018, 122, 5190–5201 CrossRef CAS PubMed.
- R. Kamens, M. Jang, C. J. Chien and K. Leach, Environ. Sci. Technol., 1999, 33, 1430–1438 CrossRef CAS.
- B. R. Larsen, D. Di Bella, M. Glasius, R. Winterhalter, N. R. Jensen and J. Hjorth, J. Atmos. Chem., 2001, 38, 231–276 CrossRef CAS.
- J. Z. Yu, R. C. Flagan and J. H. Seinfeld, Environ. Sci. Technol., 1998, 32, 2357–2370 CrossRef CAS.
- J. Z. Yu, R. J. Griffin, D. R. Cocker, R. C. Flagan, J. H. Seinfeld and P. Blanchard, Geophys. Res. Lett., 1999, 26, 1145–1148 CrossRef CAS.
- A. A. Presto, K. E. H. Hartz and N. M. Donahue, Environ. Sci. Technol., 2005, 39, 7036–7045 CrossRef CAS PubMed.
- J. D. Rindelaub, J. S. Wiley, B. R. Cooper and P. B. Shepson, Rapid Commun. Mass Spectrom., 2016, 30, 1627–1638 CrossRef CAS PubMed.
- F. Fache, O. Piva and P. Mirabel, Tetrahedron Lett., 2002, 43, 2511–2513 CrossRef CAS.
- A. Kahnt, Y. Iinuma, F. Blockhuys, A. Mutzel, R. Vermeylen, T. E. Kleindienst, M. Jaoui, J. H. Offenberg, M. Lewandowski, O. Böge, H. Herrmann, W. Maenhaut and M. Claeys, Environ. Sci. Technol., 2014, 48, 4901–4908 CrossRef CAS PubMed.
- Y. Iinuma, O. Boge, M. Keywood, T. Gnauk and H. Herrmann, Environ. Sci. Technol., 2009, 43, 280–285 CrossRef CAS PubMed.
- P. D. Q. Dao, J.-H. Park, H.-J. Lim and C. S. Cho, Chem. Pap., 2022, 76, 1885–1889 CrossRef CAS.
- Y. Iinuma, O. Boge, A. Kahnt and H. Herrmann, Phys. Chem. Chem. Phys., 2009, 11, 7985–7997 RSC.
- J. M. Castro, P. J. Linares-Palomino, S. Salido, J. Altarejos, M. Nogueras and A. Sanchez, Tetrahedron, 2005, 61, 11192–11203 CrossRef CAS.
- Y. Iinuma, A. Kahnt, A. Mutzel, O. Boge and H. Herrmann, Environ. Sci. Technol., 2013, 47, 3639–3647 CrossRef CAS PubMed.
- A. Kahnt, Y. Iinuma, A. Mutzel, O. Boge, M. Claeys and H. Herrmann, Atmos. Chem. Phys., 2014, 14, 719–736 CrossRef.
- Y. Gomez-Gonzalez, W. Wang, R. Vermeylen, X. Chi, J. Neirynck, I. A. Janssens, W. Maenhaut and M. Claeys, Atmos. Chem. Phys., 2012, 12, 125–138 CrossRef CAS.
- A. Kołodziejczyk, P. Pyrcz, K. Błaziak, A. Pobudkowska, K. Sarang and R. Szmigielski, ACS Omega, 2020, 5, 7919–7927 CrossRef PubMed.
- T. Hoffmann, R. Bandur, U. Marggraf and M. Linscheid, J. Geophys. Res.: Atmos., 1998, 103, 25569–25578 CrossRef CAS.
- Y. Gao, W. A. Hall and M. V. Johnston, Environ. Sci. Technol., 2010, 44, 7897–7902 CrossRef CAS PubMed.
- K. S. Docherty, W. Wu, Y. B. Lim and P. J. Ziemann, Environ. Sci. Technol., 2005, 39, 4049–4059 CrossRef CAS PubMed.
- L. Müller, M. C. Reinnig, J. Warnke and T. Hoffmann, Atmos. Chem. Phys., 2008, 8, 1423–1433 CrossRef.
- M. Claeys, R. Szmigielski, I. Kourtchev, P. Van der Veken, R. Vermeylen, W. Maenhaut, M. Jaoui, T. E. Kleindienst, M. Lewandowski, J. H. Offenberg and E. O. Edney, Environ. Sci. Technol., 2007, 41, 1628–1634 CrossRef CAS PubMed.
- M. Lewandowski, M. Jaoui, T. E. Kleindienst, J. H. Offenberg and E. O. Edney, Atmos. Environ., 2007, 41, 4073–4083 CrossRef CAS.
- A. Kubatova, R. Vermeylen, M. Claeys, J. Cafmeyer and W. Maenhaut, J. Geophys. Res.: Atmos., 2002, 107, 8343 CrossRef.
- A. Kubatova, R. Vermeylen, M. Claeys, J. Cafmeyer, W. Maenhaut, G. Roberts and P. Artaxo, Atmos. Environ., 2000, 34, 5037–5051 CrossRef CAS.
- R. Szmigielski, J. D. Surratt, Y. Gómez-González, P. Van der Veken, I. Kourtchev, R. Vermeylen, F. Blockhuys, M. Jaoui, T. E. Kleindienst, M. Lewandowski, J. H. Offenberg, E. O. Edney, J. H. Seinfeld, W. Maenhaut and M. Claeys, Geophys. Res. Lett., 2007, 34, L24811 CrossRef.
- E. Kostenidou, E. Karnezi, A. Kolodziejczyk, R. Szmigielski and S. N. Pandis, Environ. Sci. Technol., 2018, 52, 1150–1155 CrossRef CAS PubMed.
- Y. Y. Zhang, L. Muller, R. Winterhalter, G. K. Moortgat, T. Hoffmann and U. Poschl, Atmos. Chem. Phys., 2010, 10, 7859–7873 CrossRef CAS.
- L. Muller, M. C. Reinnig, K. H. Naumann, H. Saathoff, T. F. Mentel, N. M. Donahue and T. Hoffmann, Atmos. Chem. Phys., 2012, 12, 1483–1496 CrossRef.
- M. Claeys, R. Szmigielski, R. Vermeylen, W. Wang, M. S. Shalamzari and W. Maenhaut, in Disposal of Dangerous Chemicals in Urban Areas and Mega Cities: Role of Oxides and Acids of Nitrogen in Atmospheric Chemistry, ed. I. Barnes and K. J. Rudziński, Springer Netherlands, Dordrecht, 2013, pp. 227–238, DOI:10.1007/978-94-007-5034-0_18.
- J. W. DePalma, A. J. Horan, W. A. Hall IV and M. V. Johnston, Phys. Chem. Chem. Phys., 2013, 15, 6935–6944 RSC.
- W. A. Hall and M. V. Johnston, Aerosol Sci. Technol., 2011, 45, 37–45 CrossRef CAS.
- K. Kristensen, Å. K. Watne, J. Hammes, A. Lutz, T. Petäjä, M. Hallquist, M. Bilde and M. Glasius, Environ. Sci. Technol. Lett., 2016, 3, 280–285 CrossRef CAS.
- J. Liggio and S. M. Li, Atmos. Chem. Phys., 2013, 13, 2989–3002 CrossRef.
- M. P. Tolocka, M. Jang, J. M. Ginter, F. J. Cox, R. M. Kamens and M. V. Johnston, Environ. Sci. Technol., 2004, 38, 1428–1434 CrossRef CAS PubMed.
- C. M. Kenseth, N. J. Hafeman, Y. Huang, N. F. Dalleska, B. M. Stoltz and J. H. Seinfeld, Environ. Sci. Technol., 2020, 54, 12829–12839 CrossRef CAS PubMed.
- I. Paterson and J. M. Goodman, Tetrahedron Lett., 1989, 30, 997–1000 CrossRef CAS.
- I. Paterson, J. M. Goodman, M. Anne Lister, R. C. Schumann, C. K. McClure and R. D. Norcross, Tetrahedron, 1990, 46, 4663–4684 CrossRef CAS.
- J. Byron, J. Kreuzwieser, G. Purser, J. van Haren, S. N. Ladd, L. K. Meredith, C. Werner and J. Williams, Nature, 2022, 609, 307–312 CrossRef CAS PubMed.
- D. Leppla, N. Zannoni, L. Kremper, J. Williams, C. Pöhlker, M. Sá, M. C. Solci and T. Hoffmann, Atmos. Chem. Phys., 2023, 23, 809–820 CrossRef CAS.
- R. J. Griffin, D. R. Cocker, R. C. Flagan and J. H. Seinfeld, J. Geophys. Res.: Atmos., 1999, 104, 3555–3567 CrossRef CAS.
- T. R. Duhl, D. Helmig and A. Guenther, Biogeosciences, 2008, 5, 761–777 CrossRef CAS.
- Y. H. Shu and R. Atkinson, J. Geophys. Res.: Atmos., 1995, 100, 7275–7281 CrossRef CAS.
- J. Kesselmeier and M. Staudt, J. Atmos. Chem., 1999, 33, 23–88 CrossRef CAS.
- Y. J. Li, Q. Chen, M. I. Guzman, C. K. Chan and S. T. Martin, Atmos. Chem. Phys., 2011, 11, 121–132 CrossRef.
- M. Frosch, M. Bilde, A. Nenes, A. P. Praplan, Z. Juranyi, J. Dommen, M. Gysel, E. Weingartner and U. Baltensperger, Atmos. Chem. Phys., 2013, 13, 2283–2297 CrossRef.
- Q. Chen, Y. L. Li, K. A. McKinney, M. Kuwata and S. T. Martin, Atmos. Chem. Phys., 2012, 12, 3165–3179 CrossRef CAS.
- B. Bonn and G. K. Moortgat, Geophys. Res. Lett., 2003, 30, 1585 CrossRef.
- M. Jaoui, T. E. Kleindienst, K. S. Docherty, M. Lewandowski and J. H. Offenberg, Environ. Chem., 2013, 10, 178–193 CrossRef CAS.
- T. Hoffmann, J. R. Odum, F. Bowman, D. Collins, D. Klockow, R. C. Flagan and J. H. Seinfeld, J. Atmos. Chem., 1997, 26, 189–222 CrossRef CAS.
- A. Tasoglou and S. N. Pandis, Atmos. Chem. Phys., 2015, 15, 6035–6046 CrossRef CAS.
- D. Helmig, J. Ortega, T. Duhl, D. Tanner, A. Guenther, P. Harley, C. Wiedinmyer, J. Milford and T. Sakulyanontvittaya, Environ. Sci. Technol., 2007, 41, 1545–1553 CrossRef CAS PubMed.
- T. Sakulyanontvittaya, T. Duhl, C. Wiedinmyer, D. Helmig, S. Matsunaga, M. Potosnak, J. Milford and A. Guenther, Environ. Sci. Technol., 2008, 42, 1623–1629 CrossRef CAS PubMed.
- N. C. Bouvier-Brown, A. H. Goldstein, J. B. Gilman, W. C. Kuster and J. A. de Gouw, Atmos. Chem. Phys., 2009, 9, 5505–5518 CrossRef CAS.
- M. Jaoui and R. M. Kamens, J. Atmos. Chem., 2003, 46, 29–54 CrossRef CAS.
- J. Parshintsev, J. Nurmi, I. Kilpelainen, K. Hartonen, M. Kulmala and M. L. Riekkola, Anal. Bioanal. Chem., 2008, 390, 913–919 CrossRef CAS PubMed.
- T. E. Kleindienst, M. Jaoui, M. Lewandowski, J. H. Offenberg, C. W. Lewis, P. V. Bhave and E. O. Edney, Atmos. Environ., 2007, 41, 8288–8300 CrossRef CAS.
- M. Lewandowski, M. Jaoui, J. H. Offenberg, T. E. Kleindienst, E. O. Edney, R. J. Sheesley and J. J. Schauer, Environ. Sci. Technol., 2008, 42, 3303–3309 CrossRef CAS PubMed.
- B. Kanawati, F. Herrmann, S. Joniec, R. Winterhalter and G. K. Moortgat, Rapid Commun. Mass Spectrom., 2008, 22, 165–186 CrossRef CAS PubMed.
- T. L. Nguyen, R. Winterhalter, G. Moortgat, B. Kanawati, J. Peeters and L. Vereecken, Phys. Chem. Chem. Phys., 2009, 11, 4173–4183 RSC.
- R. Winterhalter, F. Herrmann, B. Kanawati, T. L. Nguyen, J. Peeters, L. Vereecken and G. K. Moortgat, Phys. Chem. Chem. Phys., 2009, 11, 4152–4172 RSC.
- M. N. Chan, J. D. Surratt, A. W. H. Chan, K. Schilling, J. H. Offenberg, M. Lewandowski, E. O. Edney, T. E. Kleindienst, M. Jaoui, E. S. Edgerton, R. L. Tanner, S. L. Shaw, M. Zheng, E. M. Knipping and J. H. Seinfeld, Atmos. Chem. Phys., 2011, 11, 1735–1751 CrossRef CAS.
- M. Jaoui, S. Leungsakul and R. M. Kamens, J. Atmos. Chem., 2003, 45, 261–287 CrossRef CAS.
- K. Hartonen, J. Parshintsev, V. P. Vilja, H. Tiala, S. Knuuti, C. K. Lai and M. L. Riekkola, Atmos. Environ., 2013, 81, 330–338 CrossRef CAS.
- A. van Eijck, T. Opatz, D. Taraborrelli, R. Sander and T. Hoffmann, Atmos. Environ., 2013, 80, 122–130 CrossRef CAS.
- B. Witkowski, M. Al-sharafi and T. Gierczak, Atmos. Environ., 2019, 213, 231–238 CrossRef CAS.
- M. Jaoui, M. Lewandowski, T. E. Kleindienst, J. H. Offenberg and E. O. Edney, Geophys. Res. Lett., 2007, 34, L05816 CrossRef.
- Y. Zhao, L. M. Wingen, V. Perraud and B. J. Finlayson-Pitts, Atmos. Chem. Phys., 2016, 16, 3245–3264 CrossRef CAS.
- S. Kundu, R. Fisseha, A. L. Putman, T. A. Rahn and L. R. Mazzoleni, Atmos. Environ., 2017, 154, 70–81 CrossRef CAS.
- R. Volkamer, J. L. Jimenez, F. San Martini, K. Dzepina, Q. Zhang, D. Salcedo, L. T. Molina, D. R. Worsnop and M. J. Molina, Geophys. Res. Lett., 2006, 33, L17811 CrossRef.
- J. D. Surratt, Y. Gómez-González, A. W. H. Chan, R. Vermeylen, M. Shahgholi, T. E. Kleindienst, E. O. Edney, J. H. Offenberg, M. Lewandowski, M. Jaoui, W. Maenhaut, M. Claeys, R. C. Flagan and J. H. Seinfeld, J. Phys. Chem. A, 2008, 112, 8345–8378 CrossRef CAS PubMed.
- Y. Iinuma, C. Muller, T. Berndt, O. Boge, M. Claeys and H. Herrmann, Environ. Sci. Technol., 2007, 41, 6678–6683 CrossRef CAS PubMed.
- E. A. Stone, L. M. Yang, L. Y. E. Yu and M. Rupakheti, Atmos. Environ., 2012, 47, 323–329 CrossRef CAS.
- G. Spolnik, P. Wach, K. J. Rudzinski, K. Skotak, W. Danikiewicz and R. Szmigielski, Anal. Chem., 2018, 90, 3416–3423 CrossRef CAS PubMed.
- L. N. Hawkins, L. M. Russell, D. S. Covert, P. K. Quinn and T. S. Bates, J. Geophys. Res.: Atmos., 2010, 115, D13201 CrossRef.
- A. A. Frossard, P. M. Shaw, L. M. Russell, J. H. Kroll, M. R. Canagaratna, D. R. Worsnop, P. K. Quinn and T. S. Bates, J. Geophys. Res.: Atmos., 2011, 116, D05205 CrossRef.
- M. P. Tolocka and B. Turpin, Environ. Sci. Technol., 2012, 46, 7978–7983 CrossRef CAS PubMed.
- K. M. Shakya and R. E. Peltier, Environ. Sci. Technol., 2013, 47, 9332–9338 CrossRef CAS PubMed.
- S. L. Blair, A. C. MacMillan, G. T. Drozd, A. H. Goldstein, R. K. Chu, L. Pasa-Tolic, J. B. Shaw, N. Tolic, P. Lin, J. Laskin, A. Laskin and S. A. Nizkorodov, Environ. Sci. Technol., 2017, 51, 119–127 CrossRef CAS PubMed.
- Q. T. Nguyen, M. K. Christensen, F. Cozzi, A. Zare, A. M. K. Hansen, K. Kristensen, T. E. Tulinius, H. H. Madsen, J. H. Christensen, J. Brandt, A. Massling, J. K. Nojgaard and M. Glasius, Atmos. Chem. Phys., 2014, 14, 8961–8981 CrossRef.
- G. Duporte, P. M. Flaud, E. Geneste, S. Augagneur, E. Pangui, H. Lamkaddam, A. Gratien, J. F. Doussin, H. Budzinski, E. Villenave and E. Perraudin, J. Phys. Chem. A, 2016, 120, 7909–7923 CrossRef CAS PubMed.
- K. Kristensen and M. Glasius, Atmos. Environ., 2011, 45, 4546–4556 CrossRef CAS.
- K. D. Froyd, S. M. Murphy, D. M. Murphy, J. A. de Gouw, N. C. Eddingsaas and P. O. Wennberg, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 21360–21365 CrossRef CAS PubMed.
- A. P. S. Hettiyadura, T. Jayarathne, K. Baumann, A. H. Goldstein, J. A. de Gouw, A. Koss, F. N. Keutsch, K. Skog and E. A. Stone, Atmos. Chem. Phys., 2017, 17, 1343–1359 CrossRef CAS.
- A. L. Bondy, R. L. Craig, Z. Zhang, A. Gold, J. D. Surratt and A. P. Ault, J. Phys. Chem. A, 2018, 122, 303–315 CrossRef CAS PubMed.
- K. S. Hu, A. I. Darer and M. J. Elrod, Atmos. Chem. Phys., 2011, 11, 8307–8320 CrossRef CAS.
- P. E. Ohno, J. Wang, F. Mahrt, J. G. Varelas, E. Aruffo, J. Ye, Y. Qin, K. J. Kiland, A. K. Bertram, R. J. Thomson and S. T. Martin, ACS Earth Space Chem., 2022, 6, 2481–2490 CrossRef CAS.
- M. M. Galloway, P. S. Chhabra, A. W. H. Chan, J. D. Surratt, R. C. Flagan, J. H. Seinfeld and F. N. Keutsch, Atmos. Chem. Phys., 2009, 9, 3331–3345 CrossRef CAS.
- C. N. Olson, M. M. Galloway, G. Yu, C. J. Hedman, M. R. Lockett, T. Yoon, E. A. Stone, L. M. Smith and F. N. Keutsch, Environ. Sci. Technol., 2011, 45, 6468–6474 CrossRef CAS PubMed.
- A. P. S. Hettiyadura, E. A. Stone, S. Kundu, Z. Baker, E. Geddes, K. Richards and T. Humphry, Atmos. Meas. Tech., 2015, 8, 2347–2358 CrossRef CAS.
- M. Riva, T. D. Barbosa, Y. H. Lin, E. A. Stone, A. Gold and J. D. Surratt, Atmos. Chem. Phys., 2016, 16, 11001–11018 CrossRef CAS.
- M. S. Shalamzari, R. Vermeylen, F. Blockhuys, T. E. Kleindienst, M. Lewandowski, R. Szmigielski, K. J. Rudzinski, G. Spolnik, W. Danikiewicz, W. Maenhaut and M. Claeys, Atmos. Chem. Phys., 2016, 16, 7135–7148 CrossRef CAS.
- L. E. Meade, M. Riva, M. Z. Blomberg, A. K. Brock, E. M. Qualters, R. A. Siejack, K. Ramakrishnan, J. D. Surratt and K. E. Kautzman, Atmos. Environ., 2016, 145, 405–414 CrossRef CAS.
- J. Liggio and S.-M. Li, J. Geophys. Res.: Atmos., 2006, 111, D24303 CrossRef.
- K. Kristensen, M. Bilde, P. P. Aalto, T. Petaja and M. Glasius, Atmos. Environ., 2016, 130, 36–53 CrossRef CAS.
- E. C. Minerath and M. J. Elrod, Environ. Sci. Technol., 2009, 43, 1386–1392 CrossRef CAS PubMed.
- D. A. Cortés and M. J. Elrod, J. Phys. Chem. A, 2017, 121, 9297–9305 CrossRef PubMed.
- A. M. K. Hansen, J. Hong, T. Raatikainen, K. Kristensen, A. Ylisirnio, A. Virtanen, T. Petaja, M. Glasius and N. L. Prisle, Atmos. Chem. Phys., 2015, 15, 14071–14089 CrossRef CAS.
- H. Cavdar and N. Saracoglu, Tetrahedron, 2009, 65, 985–989 CrossRef CAS.
- Y. Wang, J. Ren, X. H. H. Huang, R. Tong and J. Z. Yu, Environ. Sci. Technol., 2017, 51, 6791–6801 CrossRef CAS PubMed.
- Y. Wang, Y. Ma, X. Li, B. Y. Kuang, C. Huang, R. Tong and J. Z. Yu, Environ. Sci. Technol., 2019, 53, 12278–12290 CrossRef CAS PubMed.
- P. J. Ziemann and R. Atkinson, Chem. Soc. Rev., 2012, 41, 6582–6605 RSC.
- L. H. Renbaum and G. D. Smith, Phys. Chem. Chem. Phys., 2009, 11, 8040–8047 RSC.
- R. Szmigielski, in Disposal of Dangerous Chemicals in Urban Areas and Mega Cities, Springer, 2013, pp. 211–226 Search PubMed.
- R. Atkinson and J. Arey, Atmos. Environ., 2003, 37, S197–S219 CrossRef CAS.
- R. P. Wayne, I. Barnes, P. Biggs, J. P. Burrows, C. E. Canosamas, J. Hjorth, G. Lebras, G. K. Moortgat, D. Perner, G. Poulet, G. Restelli and H. Sidebottom, Atmos. Environ., 1991, 25, 1–203 CrossRef.
- S. S. Brown and J. Stutz, Chem. Soc. Rev., 2012, 41, 6405–6447 RSC.
- S. Liu, J. E. Shilling, C. Song, N. Hiranuma, R. A. Zaveri and L. M. Russell, Aerosol Sci. Technol., 2012, 46, 1359–1369 CrossRef CAS.
- J. D. Rindelaub, K. M. McAvey and P. B. Shepson, Atmos. Environ., 2015, 100, 193–201 CrossRef CAS.
- C. M. Boyd, J. Sanchez, L. Xu, A. J. Eugene, T. Nah, W. Y. Tuet, M. I. Guzman and N. L. Ng, Atmos. Chem. Phys., 2015, 15, 7497–7522 CrossRef CAS.
- J. K. Bean and L. Hildebrandt Ruiz, Atmos. Chem. Phys., 2016, 16, 2175–2184 CrossRef CAS.
- M. I. Jacobs, W. J. Burke and M. J. Elrod, Atmos. Chem. Phys., 2014, 14, 8933–8946 CrossRef.
- H. O. T. Pye, D. J. Luecken, L. Xu, C. M. Boyd, N. L. Ng, K. R. Baker, B. R. Ayres, J. O. Bash, K. Baumann, W. P. L. Carter, E. Edgerton, J. L. Fry, W. T. Hutzell, D. B. Schwede and P. B. Shepson, Environ. Sci. Technol., 2015, 49, 14195–14203 CrossRef CAS PubMed.
- N. L. Ng, P. S. Chhabra, A. W. H. Chan, J. D. Surratt, J. H. Kroll, A. J. Kwan, D. C. McCabe, P. O. Wennberg, A. Sorooshian, S. M. Murphy, N. F. Dalleska, R. C. Flagan and J. H. Seinfeld, Atmos. Chem. Phys., 2007, 7, 5159–5174 CrossRef CAS.
- B. R. Ayres, H. M. Allen, D. C. Draper, S. S. Brown, R. J. Wild, J. L. Jimenez, D. A. Day, P. Campuzano-Jost, W. Hu, J. de Gouw, A. Koss, R. C. Cohen, K. C. Duffey, P. Romer, K. Baumann, E. Edgerton, S. Takahama, J. A. Thornton, B. H. Lee, F. D. Lopez-Hilfiker, C. Mohr, P. O. Wennberg, T. B. Nguyen, A. Teng, A. H. Goldstein, K. Olson and J. L. Fry, Atmos. Chem. Phys., 2015, 15, 13377–13392 CrossRef CAS.
- N. L. Ng, S. S. Brown, A. T. Archibald, E. Atlas, R. C. Cohen, J. N. Crowley, D. A. Day, N. M. Donahue, J. L. Fry, H. Fuchs, R. J. Griffin, M. I. Guzman, H. Herrmann, A. Hodzic, Y. Iinuma, J. L. Jimenez, A. Kiendler-Scharr, B. H. Lee, D. J. Luecken, J. Mao, R. McLaren, A. Mutzel, H. D. Osthoff, B. Ouyang, B. Picquet-Varrault, U. Platt, H. O. T. Pye, Y. Rudich, R. H. Schwantes, M. Shiraiwa, J. Stutz, J. A. Thornton, A. Tilgner, B. J. Williams and R. A. Zaveri, Atmos. Chem. Phys., 2017, 17, 2103–2162 CrossRef CAS PubMed.
- P. L. Nichols Jr, A. B. Magnusson and J. D. Ingham, J. Am. Chem. Soc., 1953, 75, 4255–4258 CrossRef.
- O. Luxenhofer, M. Schneider, M. Dambach and K. Ballschmiter, Chemosphere, 1996, 33, 393–404 CrossRef CAS.
- S. Woidich, O. Froescheis, O. Luxenhofer and K. Ballschmiter, Fresenius' J. Anal. Chem., 1999, 364, 91–99 CrossRef CAS.
- X. Yang, F. Luo, J. Li, D. Chen, E. Ye, W. Lin and J. Jin, J. Am. Soc. Mass Spectrom., 2019, 30, 2762–2770 CrossRef CAS PubMed.
- J. Kames, U. Schurath, F. Flocke and A. Volzthomas, J. Atmos. Chem., 1993, 16, 349–359 CrossRef CAS.
- H. Cavdar and N. Saracoglu, Eur. J. Org. Chem., 2008, 4615–4621 CrossRef CAS.
- R. M. A. Pinto, J. A. R. Salvador and C. Le Roux, Tetrahedron, 2007, 63, 9221–9228 CrossRef CAS.
- X. Shi, X. Qiu, Z. Cheng, Q. Chen, Y. Rudich and T. Zhu, Environ. Sci. Technol., 2020, 54, 707–713 CrossRef CAS PubMed.
- K. Muthuramu, P. B. Shepson and J. M. Obrien, Environ. Sci. Technol., 1993, 27, 1117–1124 CrossRef CAS.
- J. Kastler and K. Ballschmiter, Fresenius' J. Anal. Chem., 1998, 360, 812–816 CrossRef CAS.
- G. Werner, J. Kastler, R. Looser and K. Ballschmiter, Angew. Chem., Int. Ed., 1999, 38, 1634–1637 CrossRef CAS PubMed.
- K. Treves, L. Shragina and Y. Rudich, Environ. Sci. Technol., 2000, 34, 1197–1203 CrossRef CAS.
- R. Suarez-Bertoa, B. Picquet-Varrault, W. Tamas, E. Pangui and J. F. Doussin, Environ. Sci. Technol., 2012, 46, 12502–12509 CrossRef CAS PubMed.
- B. H. Lee, C. Mohr, F. D. Lopez-Hilfiker, A. Lutz, M. Hallquist, L. Lee, P. Romer, R. C. Cohen, S. Iyer, T. Kurten, W. W. Hu, D. A. Day, P. Campuzano-Jost, J. L. Jimenez, L. Xu, N. L. Ng, H. Y. Guo, R. J. Weber, R. J. Wild, S. S. Brown, A. Koss, J. de Gouw, K. Olson, A. H. Goldstein, R. Seco, S. Kim, K. McAvey, P. B. Shepson, T. Starn, K. Baumann, E. S. Edgerton, J. M. Liu, J. E. Shilling, D. O. Miller, W. Brune, S. Schobesberger, E. L. D'Ambro and J. A. Thornton, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 1516–1521 CrossRef CAS PubMed.
- E. C. Tuazon and R. Atkinson, Int. J. Chem. Kinet., 1990, 22, 1221–1236 CrossRef CAS.
- M. Sprengnether, K. L. Demerjian, N. M. Donahue and J. G. Anderson, J. Geophys. Res.: Atmos., 2002, 107, 4268 CrossRef.
- F. Paulot, J. D. Crounse, H. G. Kjaergaard, J. H. Kroll, J. H. Seinfeld and P. O. Wennberg, Atmos. Chem. Phys., 2009, 9, 1479–1501 CrossRef CAS.
- T. B. Nguyen, J. Laskin, A. Laskin and S. A. Nizkorodov, Environ. Sci. Technol., 2011, 45, 6908–6918 CrossRef CAS PubMed.
- A. L. Lockwood, P. B. Shepson, M. N. Fiddler and M. Alaghmand, Atmos. Chem. Phys., 2010, 10, 6169–6178 CrossRef CAS.
- L. Lee, A. P. Teng, P. O. Wennberg, J. D. Crounse and R. C. Cohen, J. Phys. Chem. A, 2014, 118, 1622–1637 CrossRef CAS PubMed.
- J. Williams, J. M. Roberts, S. B. Bertman, C. A. Stroud, F. C. Fehsenfeld, K. Baumann, M. P. Buhr, K. Knapp, P. C. Murphy, M. Nowick and E. J. Williams, J. Geophys. Res.: Atmos., 2000, 105, 28943–28960 CrossRef CAS.
- K. A. Abellar, J. D. Cope and T. B. Nguyen, Environ. Sci. Technol., 2021, 55, 13728–13736 CrossRef CAS PubMed.
- T. Berkemeier, M. Ammann, T. F. Mentel, U. Poschl and M. Shiraiwa, Environ. Sci. Technol., 2016, 50, 6334–6342 CrossRef CAS PubMed.
- M. Spittler, I. Barnes, I. Bejan, K. J. Brockmann, T. Benter and K. Wirtz, Atmos. Environ., 2006, 40, S116–S127 CrossRef CAS.
- J. L. Fry, D. C. Draper, K. J. Zarzana, P. Campuzano-Jost, D. A. Day, J. L. Jimenez, S. S. Brown, R. C. Cohen, L. Kaser, A. Hansel, L. Cappellin, T. Karl, A. H. Roux, A. Turnipseed, C. Cantrell, B. L. Lefer and N. Grossberg, Atmos. Chem. Phys., 2013, 13, 8585–8605 CrossRef.
- L. Xu, H. Y. Guo, C. M. Boyd, M. Klein, A. Bougiatioti, K. M. Cerully, J. R. Hite, G. Isaacman-VanWertz, N. M. Kreisberg, C. Knote, K. Olson, A. Koss, A. H. Goldstein, S. V. Hering, J. de Gouw, K. Baumann, S. H. Lee, A. Nenes, R. J. Weber and N. L. Ng, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 37–42 CrossRef CAS PubMed.
- A. W. Rollins, J. D. Smith, K. R. Wilson and R. C. Cohen, Environ. Sci. Technol., 2010, 44, 5540–5545 CrossRef CAS PubMed.
- J. D. Rindelaub, C. H. Borca, M. A. Hostetler, J. H. Slade, M. A. Lipton, L. V. Slipchenko and P. B. Shepson, Atmos. Chem. Phys., 2016, 16, 15425–15432 CrossRef CAS.
- E. A. McKnight, N. P. Kretekos, D. Owusu and R. L. LaLonde, Atmos. Chem. Phys., 2020, 20, 4241–4254 CrossRef CAS.
- Y. Wang, R. Tong and J. Z. Yu, Environ. Sci. Technol., 2021, 55, 8573–8582 CrossRef CAS PubMed.
- Y. Wang, I. R. Piletic, M. Takeuchi, T. Xu, S. France and N. L. Ng, Environ. Sci. Technol., 2021, 55, 14595–14606 CrossRef CAS PubMed.
- Q. Ye, M. A. Upshur, E. S. Robinson, F. M. Geiger, R. C. Sullivan, R. J. Thomson and N. M. Donahue, Chem, 2018, 4, 318–333 CAS.
- C. M. Kenseth, Y. Huang, R. Zhao, N. F. Dalleska, J. C. Hethcox, B. M. Stoltz and J. H. Seinfeld, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 8301–8306 CrossRef CAS PubMed.
- M. A. Upshur, H. M. Chase, B. F. Strick, C. J. Ebben, L. Fu, H. Wang, R. J. Thomson and F. M. Geiger, J. Phys. Chem. A, 2016, 120, 2684–2690 CrossRef CAS PubMed.
- M. A. Upshur, M. M. Vega, A. G. Bé, H. M. Chase, Y. Zhang, A. Tuladhar, Z. A. Chase, L. Fu, C. J. Ebben, Z. Wang, S. T. Martin, F. M. Geiger and R. J. Thomson, Chem. Sci., 2019, 10, 8390–8398 RSC.
- X. Zhang, A. T. Lambe, M. A. Upshur, W. A. Brooks, A. Gray Bé, R. J. Thomson, F. M. Geiger, J. D. Surratt, Z. Zhang, A. Gold, S. Graf, M. J. Cubison, M. Groessl, J. T. Jayne, D. R. Worsnop and M. R. Canagaratna, Environ. Sci. Technol., 2017, 51, 5932–5940 CrossRef CAS PubMed.
- M. T. Thomas and A. G. Fallis, J. Am. Chem. Soc., 1976, 98, 1227–1231 CrossRef CAS.
- M. T. Thomas and A. G. Fallis, Tetrahedron Lett., 1973, 14, 4687–4690 CrossRef.
- H. M. Chase, B. T. Psciuk, B. L. Strick, R. J. Thomson, V. S. Batista and F. M. Geiger, J. Phys. Chem. A, 2015, 119, 3407–3414 CrossRef CAS PubMed.
- H. M. Chase, B. Rudshteyn, B. T. Psciuk, M. A. Upshur, B. F. Strick, R. J. Thomson, V. S. Batista and F. M. Geiger, J. Phys. Chem. B, 2016, 120, 1919–1927 CrossRef CAS PubMed.
- N. Duhamel, D. Martin, R. Larcher, B. Fedrizzi and D. Barker, Tetrahedron Lett., 2016, 57, 4496–4499 CrossRef CAS.
- Z. Zhao, W. Zhang, T. Alexander, X. Zhang, D. B. C. Martin and H. Zhang, Environ. Sci. Technol., 2021, 55, 6700–6709 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |