
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photoinduced interfacial electron transfer from perovskite quantum dots to molecular acceptors for solar cells†
Bhagyashree Mahesha
Sachith‡
a,
Zhijing
Zhang‡
a,
Palyam
Subramanyam
*ab,
Challapalli
Subrahmanyam
c,
Akihiro
Furube
 d,
Naoto
Tamai
e,
Takuya
Okamoto
ab,
Hiroaki
Misawa
bf and
Vasudevanpillai
Biju
*abc
d,
Naoto
Tamai
e,
Takuya
Okamoto
ab,
Hiroaki
Misawa
bf and
Vasudevanpillai
Biju
*abc
aGraduate School of Environmental Science, Hokkaido University, N10W5, Sapporo, Hokkaido 060-810, Japan. E-mail: biju@es.hokudai.ac.jp; subbu@es.hokudai.ac.jp
bResearch Institute for Electronic Science, Hokkaido University, N20, W10, Sapporo, Hokkaido 001-0020, Japan
cIndian Institute of Technology Hyderabad, Kandi, Telangana 502285, India
dInstitute of Post-LED Photonics, Tokushima University, 2-1, Minamijosanjima-cho, Tokushima, 770-8506, Japan
eDepartment of Chemistry, School of Science and Technology, Kwansei Gakuin University, 2-1 Gakuen, Sanda, Hyogo 669-1337, Japan
fCenter for emergent Functional Matter Science National Yang Ming Chiao Tung University Hsinchu, 30010, Taiwan
First published on 10th April 2023
Abstract
Bandgap-engineered inorganic and hybrid halide perovskite (HP) films, nanocrystals, and quantum dots (PQDs) are promising for solar cells. Fluctuations of photoinduced electron transfer (PET) rates affect the interfacial charge separation efficiencies of such solar cells. Electron donor- or acceptor-doped perovskite samples help analyze PET and harvest photogenerated charge carriers efficiently. Therefore, PET in perovskite-based donor–acceptor (D–A) systems has received considerable attention. We analyzed the fluctuations of interfacial PET from MAPbBr3 or CsPbBr3 PQDs to classical electron acceptors such as 7,7,8,8-tetracyanoquinodimethane (TCNQ) and 1,2,4,5-tetracyanobenzene (TCNB) at single-particle and ensemble levels. The significantly negative Gibbs free energy changes 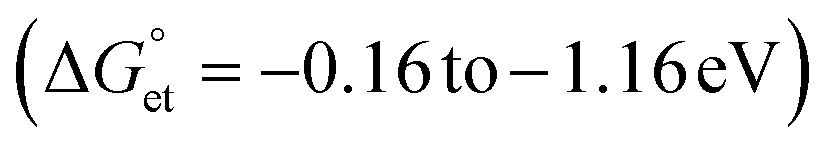 of PET estimated from the donor–acceptor redox potentials, the donor–acceptor sizes, and the solvent dielectric properties help us clarify the PET in the above D–A systems. The dynamic nature of PET is apparent from the decrease in photoluminescence (PL) lifetimes and PL photocounts of PQDs with an increase in the acceptor concentrations. Also, the acceptor radical anion spectrum helps us characterize the charge-separated states. Furthermore, the PL blinking time and PET rate fluctuations (108 to 107 s−1) provide us with single-molecule level information about interfacial PET in perovskites.
of PET estimated from the donor–acceptor redox potentials, the donor–acceptor sizes, and the solvent dielectric properties help us clarify the PET in the above D–A systems. The dynamic nature of PET is apparent from the decrease in photoluminescence (PL) lifetimes and PL photocounts of PQDs with an increase in the acceptor concentrations. Also, the acceptor radical anion spectrum helps us characterize the charge-separated states. Furthermore, the PL blinking time and PET rate fluctuations (108 to 107 s−1) provide us with single-molecule level information about interfacial PET in perovskites.
The large absorption coefficients in the solar spectral range, excellent defect tolerance, wide and tunable photoluminescence (PL) color, high PL quantum yields, and long-range charge carrier diffusion of halide perovskite quantum dots (PQDs) make them attractive for photonic and solar cell technologies.1–15 The efficient extraction of photogenerated charge carriers from PQDs helps optimize perovskite solar cells (PSCs). This is accomplished using different charge transport materials.16 Photoinduced interfacial electron transfer (PET) and subsequent charge separation in perovskite-based donor–acceptor (D–A) systems require further attention to optimize charge separation and improve the power conversion efficiency (PCE). Historically, PET in photovoltaics occurs with various electron acceptors, including TiO2, SnO2, 7,7,8,8-tetracyanoquinodimethane (TCNQ) and 1,2,4,5-tetracyanobenzene (TCNB), and fullerene (C60) derivatives like [6,6]-phenyl C61/71 butyric acid methyl ester (PCBM-C61/71).17–25 Particularly, TCNQ and TCNB are promising and cost-effective strong and multiple electron acceptors,26,27 which are less explored for PSCs. Thermodynamic and kinetic parameters of electron transfer for acceptors on PQDs can be promising during designing next-generation solar cells.
TCNQ- or TCNB-based D–A systems receive considerable attention because of their high electron affinities. For example, Jeong et al. used TCNQ as an electron scavenger in photoelectrochemical studies and reported ultrafast PET from the electrode to TCNQ.28 PET was studied based on in situ spectro-electrochemical and ex situ morphology characterization techniques. Also, Hsu et al. prepared sandwiched multilayer graphene/TCNQ electrodes for organic photovoltaics, where TCNQ molecules were tethered between two graphene layers.29 The strong electron affinity of TCNQ helped achieve high PCE. In a similar study, Nonoguchi et al. successfully measured PET from CdTe nanocrystals to TCNB using time-resolved and steady-state spectroscopic methods.30 For CdTe QDs, they observed PL quenching by static electron transfer to TCNB. The above research subjects explain the importance of TCNQ or TCNB as an electron acceptor in PQD solar cells. However, PET efficiency is known to vary from QD to QD due to different interfacial electronic coupling.31 Therefore, single-particle studies focusing on PQD-TCNB/TCNQ systems can provide further insights into interfacial charge separation in PSCs.
The PL properties of PQD-based D–A interfaces have been studied at the single particle level to correlate the fluctuations of interfacial electron transfer rates with PL blinking. Ultrafast nonradiative relaxation during the PL OFF state inhibits the efficient capturing of photogenerated carriers from ionized PQDs.32 PL blinking denotes the bright (ON-state) and dark (OFF-state) states in a PQD PL intensity trajectory. The current understanding of PL blinking correlates the ON and OFF processes with nonradiative Auger recombination (the charging–discharging model, type-A)33 and trap- or phonon-assisted activation–deactivation (the trapping-de-trapping model, type-B).34 The relaxation of a photo-excited ionized QD is dominated by nonradiative Auger recombination via energy transfer to a successively generated exciton.35 In type-B blinking, shallow defects or multiple recombination centers randomly activated or reactivated in a QD constantly change the nonradiative relaxation rate and cause band-edge blinking. An electron trapped in a surface trap state can be transferred to an acceptor by channeling the PQD neutralization through the electron acceptor, resulting in blinking suppression.36
We investigated the interfacial electron transfer dynamics of molecularly (TCNB or TCNQ)-doped single PQDs. We selected these acceptors based on the feasibility of PET, predicted from the Gibbs free energy changes 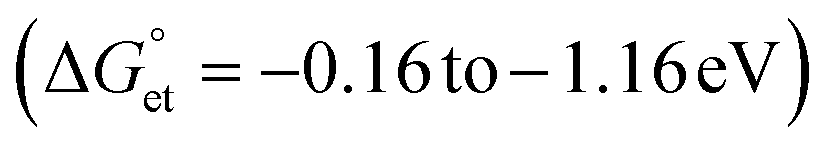 of PET, which were calculated from the ionization potentials of PQDs, the reduction potential of TCNB/TCNQ, the donor–acceptor distances, and the solvent dielectric constants. The negative
of PET, which were calculated from the ionization potentials of PQDs, the reduction potential of TCNB/TCNQ, the donor–acceptor distances, and the solvent dielectric constants. The negative 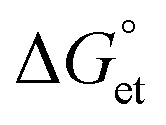 values prompted us to test the PET and charge separation (CS) at the ensemble solution-phase and single-particle levels by steady-state and time-resolved PL (TRPL) measurements. In a PQD sample, PL quenching by PET was dynamic, obviously from the continuous decrease in the PL intensity and lifetime values with the increase in the TCNQ/TCNB concentration. Also, the CS state of a PQD-TCNQ sample was rationalized from the radical anion spectrum of the acceptor, as detected by UV-vis absorption spectroscopy. Next, we examined the PET in single particles by recording the PL ON/OFF kinetics and probability distributions with and without TCNQ/TCNB. We analyzed the ON/OFF-time distributions for ca. 400 single PQDs without or with TCNQ/TCNB. The ON/OFF probability curves showed increased blinking OFF-time, consistent with the PET rates and the stable CS state.
values prompted us to test the PET and charge separation (CS) at the ensemble solution-phase and single-particle levels by steady-state and time-resolved PL (TRPL) measurements. In a PQD sample, PL quenching by PET was dynamic, obviously from the continuous decrease in the PL intensity and lifetime values with the increase in the TCNQ/TCNB concentration. Also, the CS state of a PQD-TCNQ sample was rationalized from the radical anion spectrum of the acceptor, as detected by UV-vis absorption spectroscopy. Next, we examined the PET in single particles by recording the PL ON/OFF kinetics and probability distributions with and without TCNQ/TCNB. We analyzed the ON/OFF-time distributions for ca. 400 single PQDs without or with TCNQ/TCNB. The ON/OFF probability curves showed increased blinking OFF-time, consistent with the PET rates and the stable CS state.
We synthesized colloidal CsPbBr3 PQDs (C-PQDs) by a hot-injection technique and MAPbBr3 PQDs (L-PQDs) by a ligand-assisted reprecipitation (LARP) technique. In contrast, MAPbBr3 PQDs directly synthesized on glass coverslips by a modified spray technique were without any ligands (W-PQDs). The spray method was performed under ambient conditions, for which the details are given in the Experimental section. Sprayed microdroplets of a MAPbBr3 HP precursor solution, without ligands, provided W-PQDs by fast nucleation and growth in the droplet. Moreover, the spray method helped directly deposit well-isolated W-PQDs on glass substrates for single-particle PET studies. The details of PQD preparation, isolation, and characterization are presented in the Experimental section. The PQD structures and schemes of PQDs with/without the quenchers are shown in Fig. 1A. TEM images helped us understand the structural characteristics of the as-synthesized PQDs. The TEM images of both L-PQDs and C-PQDs showed the cubic phase, where the PQD edge lengths were ca. 10 nm for C-PQDs (Fig. 1B) and ca. 10.3 nm for L-PQDs (Fig. 1C). Also, the PQDs showed narrow size distribution. Conversely, the modified spray method gave smaller W-PQDs with an average size of 6.5 nm (Fig. 1D). The particle sizes were estimated by analyzing the TEM images using the ImageJ software. Fig. 1E and F show the UV-vis absorption and PL spectra of PQDs. A scheme of the W-PQD preparation is shown in Fig. 1G. The absorption maxima of the C-PQDs, L-PQDs, and W-PQDs are at 510 nm, 518 nm, and 535 nm, respectively. The PL intensity maxima of C-PQDs, L-PQDs, and W-PQDs are at 517 nm, 523 nm, and 543 nm, respectively. Also, we detected narrow PL spectral full widths at half maxima (fwhm) for C-PQDs (19 nm), L-PQDs (23 nm), and W-PQDs (16 nm), which are consistent with the smaller PQD sizes.37
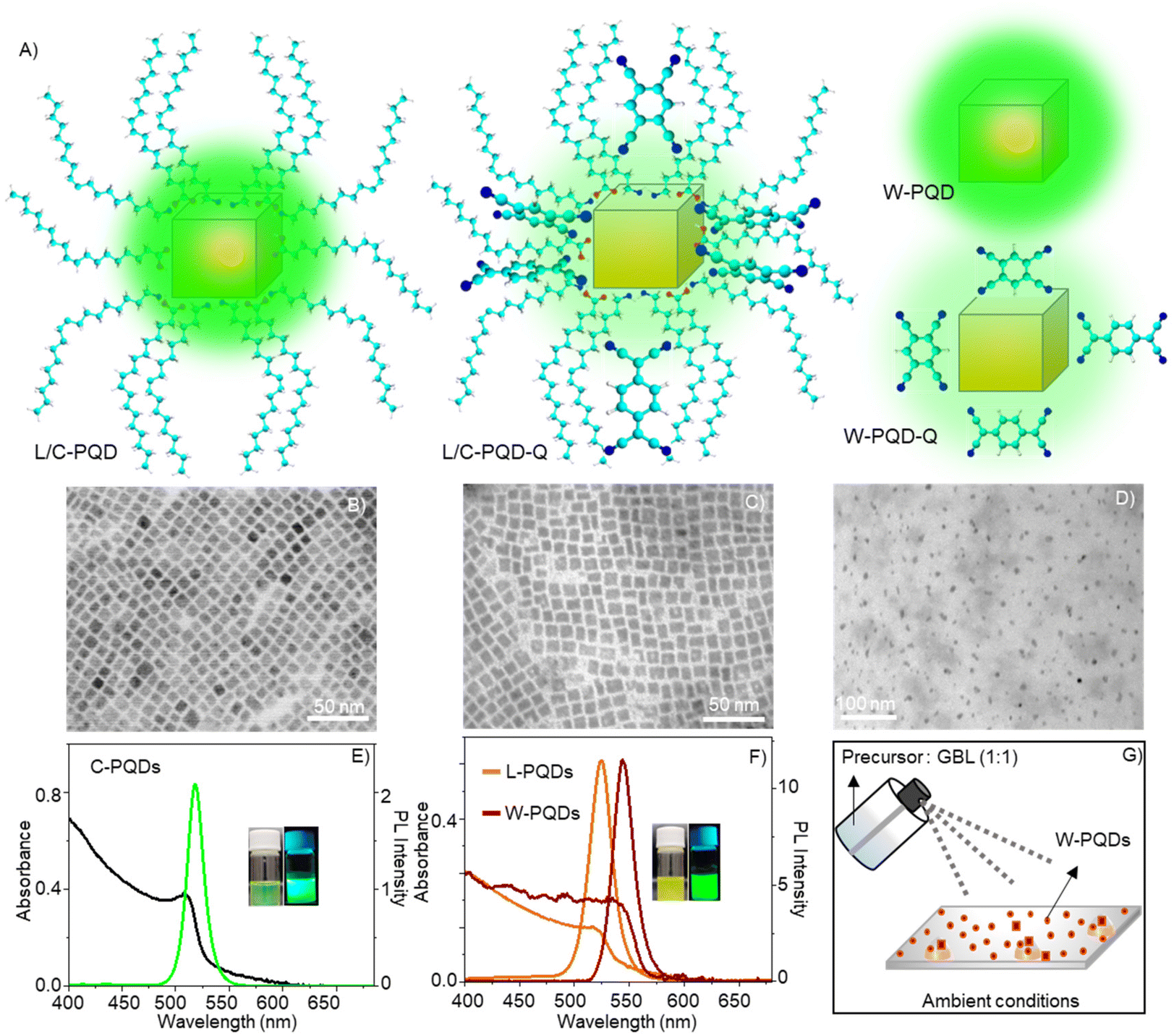 | ||
| Fig. 1 Structures and optical properties of PQDs. (A) Schemes of PQDs with or without electron acceptors (quenchers, Q = TCNQ/TCNB). (B–D) TEM images of (B) C-PQDs, (C) L-PQDs, and (D) W-PQDs. Absorption and PL spectra of (E) C-PQDs and (F) L-PQDs and W-PQDs (insets: photographs of C/L-PQD solutions taken under room or UV light). The PL spectra were recorded under 400 nm excitation. (G) A scheme of the spray synthesis method. | ||
The Gibbs reaction-free energy 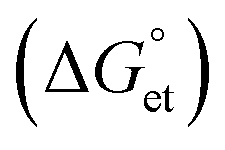 helps predict the electron transfer feasibility.38 We estimated the free energy change
helps predict the electron transfer feasibility.38 We estimated the free energy change 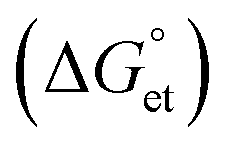 of electron transfer from the Rehm–Weller equation.
of electron transfer from the Rehm–Weller equation.
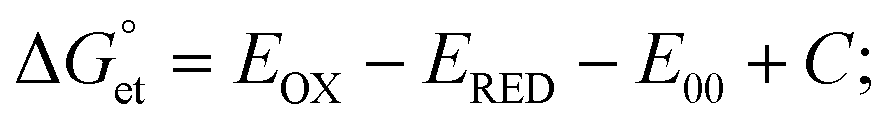 | (1) |
 value can be precisely estimated using the Marcus equation.
value can be precisely estimated using the Marcus equation.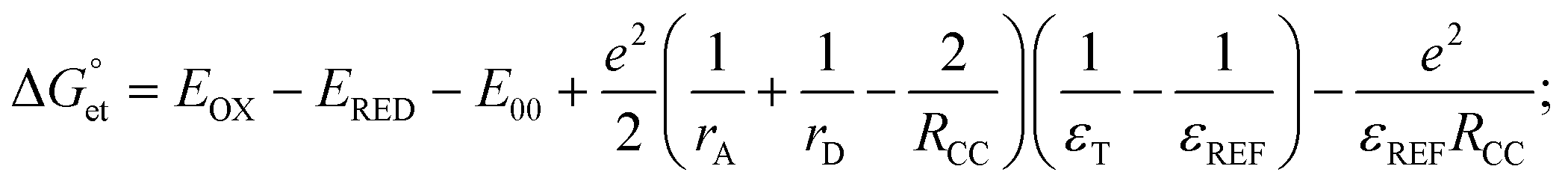 | (2) |
 values are as follows: −1.15 eV for L-PQDs/TCNQ, −0.16 eV for L-PQDs/TCNB, −1.16 eV for C-PQDs/TCNQ, and −0.17 eV for C-PQDs/TCNB. These negative
values are as follows: −1.15 eV for L-PQDs/TCNQ, −0.16 eV for L-PQDs/TCNB, −1.16 eV for C-PQDs/TCNQ, and −0.17 eV for C-PQDs/TCNB. These negative  values predict PET from L-PQDs/C-PQDs to TCNQ/TCNB.
values predict PET from L-PQDs/C-PQDs to TCNQ/TCNB.
Based on the above negative 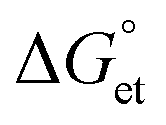 values, we examined PET-induced PL quenching of PQDs by TCNB/TCNQ, by recording the PL spectra and lifetimes of PQD solutions supplemented by different concentrations of TCNQ/TCNB. First, we recorded the PL spectra of L-PQDs or C-PQDs (0.1 mg mL−1 in toluene). Then, we added equal amounts (1 mM solutions in toluene) of TCNQ or TCNB to the PQD solution and systematically increased the quencher concentrations. The PL spectra and decays of these solutions were recorded. As shown in Fig. 2A, C, and S2A, C,† the PL intensities of the PQD solutions were decreased continuously as the TCNQ/TCNB concentration was increased from 0 to 45 μM, showing PET-induced PL quenching. Furthermore, the PL lifetimes of the PQDs were decreased with increases in the TCNQ/TCNB concentration (Table S1,†Fig. 2B, D, and Fig. S2B, D†). The PL decay profiles were fitted using the third-order exponential equation. Table S1† shows the individual amplitudes and the lifetime values. The hydrophobic ligands on L-PQD will likely hinder the interaction between PQDs and the polar quenchers. Conversely, the ligand-free PQDs (W-PQDs) allow for efficient and close interaction between the two. The PL lifetime decreases (Fig. 2B, D, S2B, and S2D†) indicate dynamic interactions between PQD and TCNB/TCNQ. In contrast, during static PL quenching of PQD films by C60,9,13 the number of photons emitted decreased with the increase in the C60 concentration, with the PL lifetime remaining unchanged. Plots of PL intensities/lifetimes vs. TCNQ/TCNB concentration (Stern–Volmer plots) were constructed to understand the PL quenching mechanism further. The fitted Stern–Volmer plots for L-PQDs-TCNB/TCNQ and C-PQDs-TCNB/TCNQ systems are shown in Fig. S3 and S4,† and the fitting parameters are listed in Table 1. From the fitting parameters, we understand similar quenching rate constants (kqs) for TCNB-PQD and TCNQ-PQD systems (Table 1). The Stern–Volmer plot clearly shows the non-linear quenching behavior in both systems (Fig. S3 and S4†), indicating combined dynamic and static interactions that govern the PQD PL quenching by TCNQ/TCNB, even at low quencher concentrations. Furthermore, TCNQ and TCNB show similar Stern–Volmer constants and quenching rate constants, indicating their comparable electron acceptor efficiencies.
values, we examined PET-induced PL quenching of PQDs by TCNB/TCNQ, by recording the PL spectra and lifetimes of PQD solutions supplemented by different concentrations of TCNQ/TCNB. First, we recorded the PL spectra of L-PQDs or C-PQDs (0.1 mg mL−1 in toluene). Then, we added equal amounts (1 mM solutions in toluene) of TCNQ or TCNB to the PQD solution and systematically increased the quencher concentrations. The PL spectra and decays of these solutions were recorded. As shown in Fig. 2A, C, and S2A, C,† the PL intensities of the PQD solutions were decreased continuously as the TCNQ/TCNB concentration was increased from 0 to 45 μM, showing PET-induced PL quenching. Furthermore, the PL lifetimes of the PQDs were decreased with increases in the TCNQ/TCNB concentration (Table S1,†Fig. 2B, D, and Fig. S2B, D†). The PL decay profiles were fitted using the third-order exponential equation. Table S1† shows the individual amplitudes and the lifetime values. The hydrophobic ligands on L-PQD will likely hinder the interaction between PQDs and the polar quenchers. Conversely, the ligand-free PQDs (W-PQDs) allow for efficient and close interaction between the two. The PL lifetime decreases (Fig. 2B, D, S2B, and S2D†) indicate dynamic interactions between PQD and TCNB/TCNQ. In contrast, during static PL quenching of PQD films by C60,9,13 the number of photons emitted decreased with the increase in the C60 concentration, with the PL lifetime remaining unchanged. Plots of PL intensities/lifetimes vs. TCNQ/TCNB concentration (Stern–Volmer plots) were constructed to understand the PL quenching mechanism further. The fitted Stern–Volmer plots for L-PQDs-TCNB/TCNQ and C-PQDs-TCNB/TCNQ systems are shown in Fig. S3 and S4,† and the fitting parameters are listed in Table 1. From the fitting parameters, we understand similar quenching rate constants (kqs) for TCNB-PQD and TCNQ-PQD systems (Table 1). The Stern–Volmer plot clearly shows the non-linear quenching behavior in both systems (Fig. S3 and S4†), indicating combined dynamic and static interactions that govern the PQD PL quenching by TCNQ/TCNB, even at low quencher concentrations. Furthermore, TCNQ and TCNB show similar Stern–Volmer constants and quenching rate constants, indicating their comparable electron acceptor efficiencies.
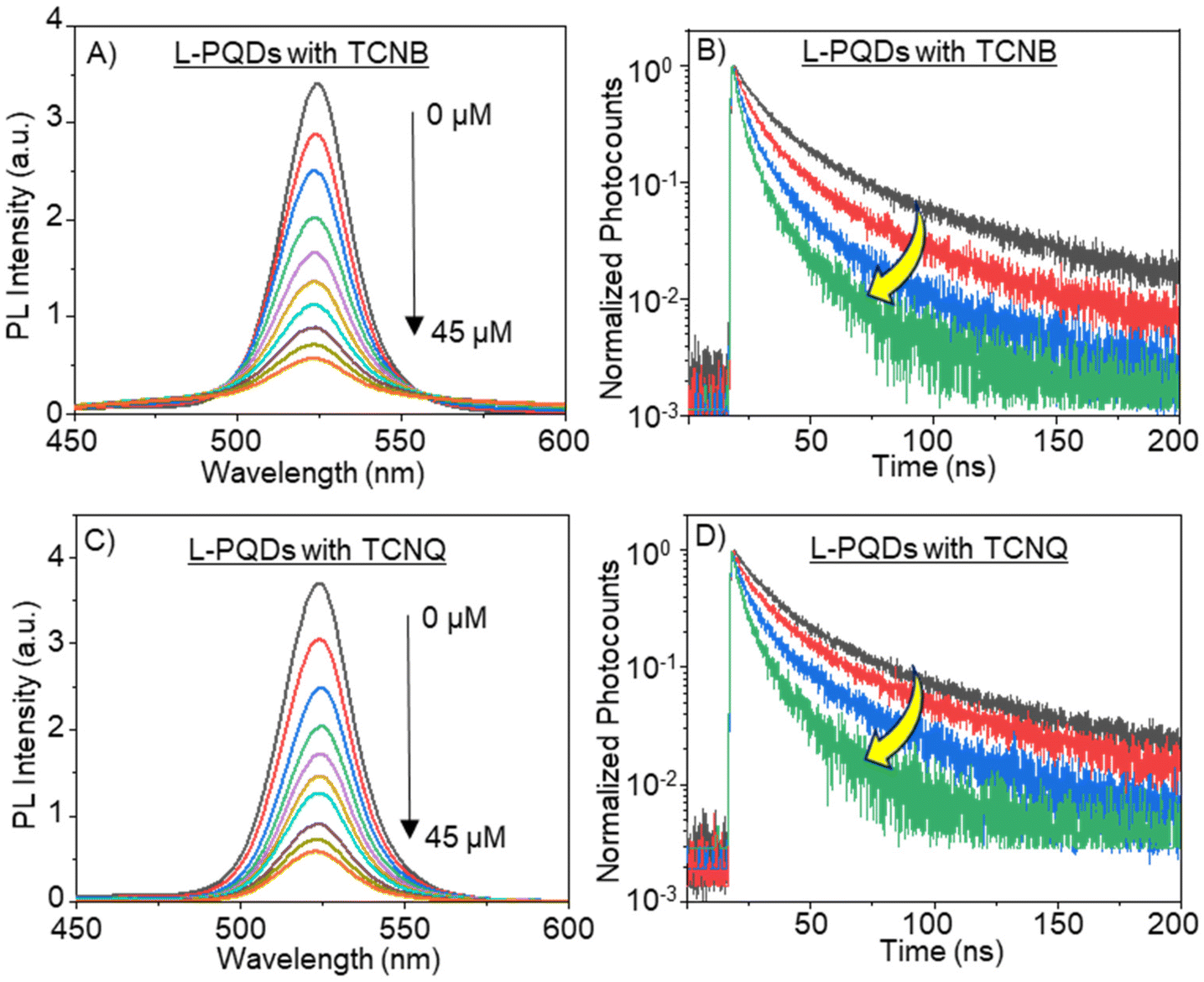 | ||
| Fig. 2 PL spectral and decay profiles of L-PQDs in the presence of electron acceptors. (A and C) PL spectra of L-PQD solutions supplemented with (A) TCNB and (C) TCNQ (λex: 400 nm). (B and D) PL decay profiles of PQD solutions supplemented with (B) TCNB and (D) TCNQ (λex: 400 nm), where the black and green traces indicate the decays with 0 μM and 20 μM TCNB or TCNQ. The spectra from top to bottom in A and C represent the increase in the quencher concentration from 0 to 45 μM at 5 μM intervals. | ||
| System | Stern–Volmer constant KSV (M−1) | Quenching rate parameter kq (M−1 s−1) |
|---|---|---|
| L-PQDs-TCNB | 2.08 × 102 | 0.67 × 1010 |
| L-PQDs-TCNQ | 2.34 × 102 | 0.78 × 1010 |
| C-PQDs-TCNB | 1.46 × 102 | 1.29 × 1010 |
| C-PQDs-TCNQ | 5.30 × 102 | 5.35 × 1010 |
TCNQ and TCNB are well-studied electron acceptors, and these molecules generate radical anions in the presence of electron donors. To understand the formation of TCNQ˙−, we recorded the steady-state absorption spectra (Fig. S5†) for L-PQDs without or with TCNQ. The absorption maximum of the as-synthesized L-PQD was at 518 nm. After adding TCNQ to a PQD solution, we detected three absorption maxima at 393, 755, and 845 nm. The absorption band at 393 nm corresponds to the TCNQ S0 → S1 transition. The unpaired electron in the TCNQ˙− renders it the doublet state electronic structure. TCNQ˙− has the main absorption bands in the 600–900 nm range, with the 755 and 848 nm bands as the characteristic features. These features correspond to the D0 → D1 and D0 → D2 transitions.39,40 However, TCNB˙− was not observed in the steady-state absorption spectra because of its low electron affinity compared to TCNQ, which is also apparent from the lower electron transfer rate for TCNB than that for TCNQ (Table 1). The lack of the characteristic TCNB˙− absorption band in the steady-state spectrum can also be due to the relatively short lifetime of TCNB˙− than that of TCNQ˙−.
To further understand the PQD to TCNQ/TCNB interfacial electron transfer, we examined the roles of TCNQ/TCNB in the PL intensity and ON/OFF times of single PQDs. Generally, PET from a PQD to an acceptor can increase the OFF time by detouring the neutralization of the ionized PQD through a back electron transfer (BET). At first, we investigated the blinking behavior of pristine PQDs using a single-molecule fluorescence imaging system with an EMCCD camera and a spectrometer. The samples were prepared on glass substrates by placing a subnanomolar PQD solution and dragging using a lens-cleaning paper (see the Experimental section). This drop–drag method helped us to control the density of single PQDs at 15–20 per 50 × 50 μm2, as shown in Fig. S6A.† For single W-PQD sample preparation, we followed a spray method,12 where perovskite precursor solutions with controlled concentrations were sprayed on glass coverslips at specific distances between the sprayer and the sample substrate (see the Experimental section). This method helped us to set the single W-PQD density at 20–30 per 50 × 50 μm2 (Fig. 3A). Typical PL blinking plots for the as-prepared W-PQDs and C-PQDs are given in Fig. 3B(i, ii) and S6B(i, ii),† respectively. The PL blinking ON- and OFF-time distributions varied from particle to particle, with some PQDs showing short ON/OFF durations to some showing long-living ON or OFF events. We set the excitation laser intensity far below the biexciton threshold estimated from the particle size and laser intensity. As reported in the literature, pristine MAPbBr3 and CsPbBr3 blink by a combination of charging–discharging and trapping–de-trapping processes.41
 | ||
| Fig. 3 PET-induced blinking of single-PQDs. (A) A PL image of W-PQDs on a cover glass substrate and photoexcited at 404 nm (585 W cm−2). (B) PL intensity trajectories of two pristine W-PQDs. (C) Real-time PL intensity trajectories of single W-PQD immersed in 1-hexadecene (from 0 to 62 s) before and (from 65 to 120 s) after the addition of (i) TCNB (1 mM in toluene) or (ii) TCNQ (1 mM in toluene). The bin size is 33 ms. | ||
To realize the role of electron transfer in PL blinking of PQDs, we recorded the single W-PQD PL intensity trajectories before, during, and after PQD samples were supplemented with TCNQ/TCNB solutions. The intensity trajectory of a pristine W-PQD immersed in 1-hexadecene for 60 s is shown in Fig. 3C(i). A TCNB solution in toluene was introduced to the PQD at 65 s. Generally, PET from the surface trap to an electron acceptor increases the OFF time by redirecting the charge neutralization in a PQD through BET. After treating the PQD with a TCNB solution, the PL trajectory showed long-lived OFF-states, indicating long-lived CS states. Nevertheless, intermittent PL ON events were observable. The PL intensity trajectories represent high-frequency forward and backward electron transfer-mediated nonradiative relaxation processes, with minimal radiative recombination and fluctuations of the PET rates. Conversely, with the addition of the quencher, the short-lived ON–OFF occurrences (Fig. 3C, from 0 to 62 s) of the W-PQDs changed into blinking with prolonged OFF states. In addition to the negative 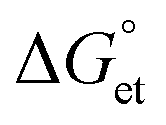 values for both the PQD-TCNQ and PQD-TCNB systems, the oxidative PET is predictable from the MAPbBr3 conduction band and the acceptor LUMO levels.42 Nevertheless, the PET efficiency and rate fluctuations are affected by many factors, such as the PQD–quencher interface and their electronic coupling.43 Therefore, the different blinking ON–OFF times, temporally for a given particle and spatially for different particles, can be attributed to fluctuations of the interfacial electronic coupling element originating from the heterogeneity of the interface, changes to the molecular orientation, and fluctuations in the donor–acceptor distance.44
values for both the PQD-TCNQ and PQD-TCNB systems, the oxidative PET is predictable from the MAPbBr3 conduction band and the acceptor LUMO levels.42 Nevertheless, the PET efficiency and rate fluctuations are affected by many factors, such as the PQD–quencher interface and their electronic coupling.43 Therefore, the different blinking ON–OFF times, temporally for a given particle and spatially for different particles, can be attributed to fluctuations of the interfacial electronic coupling element originating from the heterogeneity of the interface, changes to the molecular orientation, and fluctuations in the donor–acceptor distance.44
To verify the PET-induced blinking change, we collected PL intensity trajectories of more than 400 W-PQDs/C-PQDs before and after treatment with TCNB/TCNQ. We statically analyzed the ON/OFF probability distributions that helped rationalize the blinking when PET is operative. The probability is well-fitted by the truncated power law in eqn (3).
| P(τ) = A0τ−αe−τ/τc | (3) |
 | (4) |
The ON/OFF statistics (Fig. 4A, B, S7A, and S7B†) of pristine W-PQDs (τcON = 1.75 s, and τcOFF = 0.39 s) or C-PQDs (τcON = 0.97 s, and τcOFF = 0.20 s) reveal the blinking probability and the photoionization behavior, which means charging–discharging- or type A blinking. This behavior is evident from the exponential truncation cut-off of the probabilities at short time scales. The ON-τcs and OFF-τcs for W-PQDs and C-PQDs show a higher ionization probability in the latter case. The OFF events in single-particle PL blinking are attributable to nonradiative Auger recombination in charged PQDs. After a hole or an electron localizes in a defect, nonradiative Auger recombination dominates the relaxation of a charged exciton generated by photoexcitation within the carrier de-trapping time. The OFF times of pristine PQDs come from carrier trapping–de-trapping cycles or the stability of the trapped state. However, in the presence of the acceptor, the τcON value for W-PQDs was decreased from 1.75 s to 0.66 s and the τcOFF value was increased from 0.39 s to 0.81 s.
 | ||
| Fig. 4 Blinking time distributions for >400 single PQDs. Probability distributions of (A, C) ON- and (B, D) OFF-times for W-PQDs (A, B) without or (C, D) with TCNB. (E and F) W-PQD's PL decays with or without (E) TCNB and (F) TCNQ. The quencher concentration is 10 μM in both cases. | ||
Similarly, in the presence of the acceptor, the τcON value for C-PQD was decreased from 0.97 s to 0.49 s and the τcOFF value was increased from 0.20 s to 0.31 s. The ON-time (shorter than that of pristine PQDs) and OFF-time (longer than that of pristine PQDs) for W-PQDs or C-PQDs treated with the quencher, as shown in Fig. 4C, D, S7C, and S7D,† suggest a PET-induced additional nonradiative recombination pathway. Conversely, the shorter τcON denotes the probability of charging by PET, and the longer τcOFF suggests repeated PET-BET cycles with a low probability of radiative recombination. Also, a long-living CS state can contribute to the long OFF-time.
Although the dynamic nature of PET is apparent from the ensemble PL intensity and lifetime decrease, we correlate the single-PQD PL ON-/OFF-time with the PL lifetimes to further understand the origin of the dynamic nature. Fig. 4E and F show PL decays of single PQDs with or without TCNQ or TCNB. The PL lifetime was decreased (from 9.7 to 6.3 ns) for a pristine W-PQD after TCNQ treatment, showing the PET rates at 5.56 × 107 s−1 and 1.3 × 108 s−1 for TCNB. These rates are comparable to the ensemble PET rates (Table 1). However, the single PQD PL lifetimes fluctuated occasionally, like single-particle PL intensity fluctuations. The intensity and lifetime fluctuations suggest fluctuations in the interfacial PET efficiency or the dynamic nature of D–A interactions.
In summary, we reveal the PET rate and dynamics for PQD-based D–A systems at the ensemble and single-particle levels. The negative Gibbs free energy changes of PET suggested PET feasibility. Furthermore, PET from PQDs to TCNQ or TCNB was evident from the PL intensity and lifetime decrease. The Stern–Volmer plots helped us identify the dynamic nature of PET even at low acceptor concentrations. The highest PET rate was ca. 8 × 107 s−1 at the ensemble level and ca. 1 × 108 s−1 for single PQDs. The TCNQ radical anion further helped us identify the charge-separated state. Consistent with the PET-induced PL intensity/lifetime decrease at the ensemble and single-particle levels, statistical analysis of the PL intensity trajectories of single PQD-acceptor systems showed an early-time ON-time cut-off and a significant OFF-time increase. This study highlights the fluctuations of interfacial electron transfer rates in PQD-acceptor systems, underscoring the significance of suppressing such fluctuations for high-efficiency perovskite solar cells.
Author contributions
B. M. S. and Z. Z. contributed equally to this work. V. B. conceived the project. B. M. S., Z. Z., P. S., and V. B. carried out the PL experiments. B. M. S., Z. Z., P. S., and C. S. analyzed the electrochemical data and calculated the free energy change values. B. M. S. and T. O. collected the TEM and STEM data. N. T., A. F., H. M. and V. B. analyzed the electron transfer data. All authors contributed to the manuscript writing. V. B. and P. S. finalized the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
V. B. acknowledges the financial support under the MEXT JSPS Grant-in-Aid for Scientific Research B (19H02550 and 23H01781). V. B. and H. M. acknowledge the financial support under the MEXT JSPS Grant-in-Aid for Specially Promoted Research (18H05205). V. B. and C. S. acknowledge a JSPS-DST (Japan-India) bilateral project and the Scheme for Promotion of Academic and Research Collaboration (SPARC) program of the Science and Engineering Research Board of India. We thank the Hokkaido University Photoexcitonix program and the Crossover Alliance to Create the Future with People, Intelligence and Materials. B. M. S. acknowledges a Hokkaido University DX Scholarship for doctoral studies.References
- L. Chouhan, S. Ghimire, C. Subrahmanyam, T. Miyasaka and V. Biju, Chem. Soc. Rev., 2020, 49, 2869–2885 RSC
.
- A. Dey, J. Ye, A. De, E. Debroye, S. K. Ha, E. Bladt, A. S. Kshirsagar, Z. Wang, J. Yin and Y. Wang, ACS Nano, 2021, 15, 10775–10981 CrossRef CAS PubMed
- G. Nedelcu, L. Protesescu, S. Yakunin, M. I. Bodnarchuk, M. J. Grotevent and M. V. Kovalenko, Nano Lett., 2015, 15, 5635–5640 CrossRef CAS PubMed
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed
- J. Shamsi, A. S. Urban, M. Imran, L. De Trizio and L. Manna, Chem. Rev., 2019, 119, 3296–3348 CrossRef CAS PubMed
- Z. Zhang, S. Ghimire, T. Okamoto, B. M. Sachith, J. Sobhanan, C. Subrahmanyam and V. Biju, ACS Nano, 2022, 16, 160–168 CrossRef CAS PubMed
- M. Liu, M. B. Johnston and H. J. Snaith, Nature, 2013, 501, 395–398 CrossRef CAS PubMed
- H. He, S. Mei, Z. Chen, S. Liu, Z. Wen, Z. Cui, D. Yang, W. Zhang, F. Xie, B. Yang, R. Guo and G. Xing, J. Mater. Chem. C, 2021, 9, 11349–11357 RSC
- B. M. Sachith, T. Okamoto, S. Ghimire, T. Umeyama, Y. Takano, H. Imahori and V. Biju, J. Phys. Chem. Lett., 2021, 12, 8644–8651 CrossRef CAS PubMed
- S. Ghimire, L. Chouhan, Y. Takano, K. Takahashi, T. Nakamura, K. I. Yuyama and V. Biju, ACS Energy Lett., 2019, 4, 133–141 CrossRef CAS
- I. Levchuk, A. Osvet, X. Tang, M. Brandl, J. D. Perea, F. Hoegl, G. J. Matt, R. Hock, M. Batentschuk and C. J. Brabec, Nano Lett., 2017, 17, 2765–2770 CrossRef CAS PubMed
- A. P. Pushkarev, V. I. Korolev, D. I. Markina, F. E. Komissarenko, A. Naujokaitis, A. Drabavičius, V. Pakštas, M. Franckevičius, S. A. Khubezhov and D. A. Sannikov, ACS Appl. Mater. Interfaces, 2019, 11, 1040–1048 CrossRef CAS PubMed
- V. C. Nair, C. Muthu, A. L. Rogach, R. Kohara and V. Biju, Angew. Chem., Int. Ed., 2017, 56, 1214–1218 CrossRef CAS PubMed
- S. Rhee, K. An and K. T. Kang, Crystals, 2021, 11, 1–28 Search PubMed
- M. Que, L. Zhu, Y. Guo, W. Que and S. Yun, J. Mater. Chem. C, 2020, 8, 5321–5334 RSC
- K. Wu, G. Liang, Q. Shang, Y. Ren, D. Kong and T. Lian, J. Am. Chem. Soc., 2015, 137, 12792–12795 CrossRef CAS PubMed
- J. Jiménez-López, B. M. D. Puscher, D. M. Guldi and E. Palomares, J. Am. Chem. Soc., 2020, 142, 1236–1246 CrossRef PubMed
- S. Zhou, G. Zhou, Y. Li, X. Xu, Y. J. Hsu, J. Xu, N. Zhao and X. Lu, ACS Energy Lett., 2020, 5, 2614–2623 CrossRef CAS
- I. Jeon, H. Ueno, S. Seo, K. Aitola, R. Nishikubo, A. Saeki, H. Okada, G. Boschloo, S. Maruyama and Y. Matsuo, Angew. Chem., Int. Ed., 2018, 57, 4607–4611 CrossRef CAS PubMed
- P. Schulz, ACS Energy Lett., 2018, 3, 1287–1293 CrossRef CAS
- H. Wang, Y. Rahaq and V. Kumar, Sci. Rep., 2016, 6, 29567 CrossRef CAS PubMed
- T. S. Sherkar, C. Momblona, L. Gil-Escrig, J. Ávila, M. Sessolo, H. J. Bolink and L. J. A. Koster, ACS Energy Lett., 2017, 2, 1214–1222 CrossRef CAS PubMed
- S. H. Wu, M. Y. Lin, S. H. Chang, W. C. Tu, C. W. Chu and Y. C. Chang, J. Phys. Chem. C, 2018, 122, 236–244 CrossRef CAS
- D. Credgington, F. C. Jamieson, B. Walker, T. Q. Nguyen and J. R. Durrant, Adv. Mater., 2012, 24, 2135–2141 CrossRef CAS PubMed
- J. V. Passarelli, C. M. Mauck, S. W. Winslow, C. F. Perkinson, J. C. Bard, H. Sai, K. W. Williams, A. Narayanan, D. J. Fairfield and M. P. Hendricks, Nat. Chem., 2020, 12, 672–682 CrossRef CAS PubMed
- L. Ma, P. Hu, H. Jiang, C. Kloc, H. Sun, C. Soci, A. A. Voityuk, M. E. Michel-Beyerle and G. G. Gurzadyan, Sci. Rep., 2016, 6, 1–7 CrossRef PubMed
- W. T. M. Van Gompel, R. Herckens, K. Van Hecke, B. Ruttens, J. D'Haen, L. Lutsen and D. Vanderzande, Chem. Commun., 2019, 55, 2481–2484 RSC
- H. W. Jeong, T. S. Zsigmond, G. F. Samu and C. Janáky, ACS Energy Lett., 2022, 7, 417–424 CrossRef CAS PubMed
- C. L. Hsu, C. Te. Lin, J. H. Huang, C. W. Chu, K. H. Wei and L. J. Li, ACS Nano, 2012, 6, 5031–5039 CrossRef CAS PubMed
- Y. Nonoguchi, T. Nakashima and T. Kawai, J. Phys. Chem. C, 2009, 113, 11464–11468 CrossRef CAS
- X. Liu, H. Zhao, L. Wei, X. Ren, X. Zhang, F. Li, P. Zeng and M. Liu, Nanophotonics, 2021, 10, 1967–1975 CrossRef CAS
- L. Chouhan, S. Ito, E. M. Thomas, Y. Takano, S. Ghimire, H. Miyasaka and V. Biju, ACS Nano, 2021, 15, 2831–2838 CrossRef CAS PubMed
- G. Yuan, D. E. Gómez, N. Kirkwood, K. Boldt and P. Mulvaney, ACS Nano, 2018, 12, 3397–3405 CrossRef CAS PubMed
- C. T. Trinh, D. N. Minh, K. J. Ahn, Y. Kang and K. G. Lee, ACS Photonics, 2018, 5, 4937–4943 CrossRef CAS
- C. T. Trinh, D. N. Minh, K. J. Ahn, Y. Kang and K. G. Lee, Sci. Rep., 2020, 10, 1–8 CrossRef PubMed
- M. Hamada, S. Nakanishi, T. Itoh, M. Ishikawa and V. Biju, ACS Nano, 2010, 4, 4445–4454 CrossRef CAS PubMed
- M. C. Brennan, A. Forde, M. Zhukovskyi, A. J. Baublis, Y. V. Morozov, S. Zhang, Z. Zhang, D. S. Kilin and M. Kuno, J. Phys. Chem. Lett., 2020, 11, 4937–4944 CrossRef CAS PubMed
- K. Kumar, I. V. Kurnikov, D. N. Beratan, D. H. Waldeck and M. B. Zimmt, J. Phys. Chem. A, 1998, 102, 5529–5541 CrossRef CAS
- L. Ma, P. Hu, C. Kloc, H. Sun, M. E. Michel-Beyerle and G. G. Gurzadyan, Chem. Phys. Lett., 2014, 609, 11–14 CrossRef CAS
- S. Panja, U. Kadhane, J. U. Andersen, A. I. S. Holm, P. Hvelplund, M. B. S. Kirketerp, S. B. Nielsen, K. Støchkel, R. N. Compton and J. S. Forster, J. Chem. Phys., 2007, 127, 124301 CrossRef PubMed
- B. Li, H. Huang, G. Zhang, C. Yang, W. Guo, R. Chen, C. Qin, Y. Gao, V. P. Biju, A. L. Rogach, L. Xiao and S. Jia, J. Phys. Chem. Lett., 2018, 9, 6934–6940 CrossRef CAS PubMed
- T. Golubev, D. Liu, R. Lunt and P. Duxbury, AIP Adv., 2019, 9, 035026 CrossRef
- X. Xian, K. Yan, W. Zhou, L. Jiao, Z. Wu and Z. Liu, Nanotechnology, 2009, 20, 505204 CrossRef PubMed
- J. Wang, A. Li, S. Xu, C. Song, Y. Geng, L. Ye, H. Zhang and W. Xu, ACS Omega, 2019, 4, 10424–10430 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Details of materials, chemicals, synthesis of L-PQDs, synthesis of C-PQDs, and synthesis of W-PQDs; details of absorption and PL measurements, TEM measurements, single-particle PL measurements, PL lifetime measurements, DPV measurements, and PL lifetime calculations; DPV data for donors and acceptors (Fig. S1A–D); ensemble solution-phase PL quenching data (Fig. S2A–D); Stern–Volmer plots (Fig. S3 and S4); TCNQ˙− spectra (Fig. S5); PL images and PL blinking data (Fig. S6); ON-/OFF-time probability plots (Fig. S7); and PL lifetime components and electron transfer rates (Table S1). See DOI: https://doi.org/10.1039/d3nr01032e |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2023 |