
Selective placement of modifiers on hematite thin films for solar water splitting†
Fabio A.
Pires
 ab,
Gabriel T.
dos Santos
ac,
Jefferson
Bettini
a,
Carlos A. R.
Costa
a,
Renato V.
Gonçalves
d,
Ricardo H. R.
Castro
e and
Flavio L.
Souza
*abf
ab,
Gabriel T.
dos Santos
ac,
Jefferson
Bettini
a,
Carlos A. R.
Costa
a,
Renato V.
Gonçalves
d,
Ricardo H. R.
Castro
e and
Flavio L.
Souza
*abf
aBrazilian Nanotechnology National Laboratory (LNNano), Brazilian Center for Research in Energy and Materials (CNPEM), Campinas, SP, Brazil. E-mail: flavio.souza@lnnano.cnpem.br
bInstitute of Chemistry, State University of Campinas (UNICAMP), Campinas, SP, Brazil
cEngineering School, Federal University of Rio Grande do Sul (UFRGS), Porto Alegre, RS, Brazil
dSão Carlos Institute of Physics, University of São Paulo (USP), São Carlos, SP, Brazil
eDepartment of Materials Science and Engineering, Lehigh University, Bethlehem, PA 18015, USA
fCenter for Natural and Human Sciences, Federal University of ABC (UFABC), Santo André, SP, Brazil
First published on 7th September 2023
Abstract
The design of nanostructured materials for photoelectrochemical water splitting relies on a detailed understanding of the reactional bottlenecks. For hematite, a model system for photoanodes, the challenges concern poor charge transfer and separation, carrier recombination rate, and sluggish water oxidation kinetics. Several methods have been proposed to address each individually, with complex multi-step processes offered as solutions to improve overall performance. Here, we introduce a single polymeric precursor solution that enables the design of hematite (α-Fe2O3) with synergistic bulk and interfacial engineering using Ga3+, Hf4+ and NiFeOx. The solution causes Ga3+ to dope hematite lattice to reduce polaronic effects, while simultaneously induces Hf4+ enrichment at both surface and grain boundaries, improving charge separation and reducing recombination. Hf4+ also led to a refined microstructure derived from interface stabilization, which associated with Ga3+ bulk doping and NiFeOx electrodeposition resulted in a thin film with 65% of overall photoelectrode efficiency. As a consequence, the modified hematite photoanode (176 nm-thick) delivered a water oxidation photocurrent of 2.30 mA cm−2 in contrast to 0.37 mA cm−2 for the pristine system measured at 1.23 V against hydrogen reversible electrode (RHE). The results suggest the simplicity of this new polymeric solution may offer a cost-effective, scalable and versatile alternative for multiple chemical modifications in oxides beyond hematite.
Introduction
The urgency for renewable energy production in the past years has drawn significant attention for the development of photoelectrodes for solar water splitting. As the limiting step of water splitting is water oxidation reaction, designing efficient photoanodes is a key challenge that requires a clear understanding at the atomic and microstructural levels. Along with TiO2,1,2 hematite (α-Fe2O3) is the most studied photoanode for photoelectrochemical water splitting because of its abundance, stability, ability to absorb a significant fraction of solar radiation, and band edges suitable for water oxidation.3 These characteristics also make hematite an interesting material for proof-of-concept studies, enabling testing for potential solutions to process bottlenecks.Over the years, record-breaking performances of hematite as photoanodes for solar water splitting have been associated with columnar morphology obtained by hydrothermal/solvothermal methods.4,5 The columnar structure mitigates common hematite deficiencies, such as poor hole (h+) diffusion length and electron–hole pair short lifetime.6,7 This contrasts with typical interface-rich microstructures obtained by sol–gel methods, which show poor efficiency due to a high concentration of recombination sites and low electronic conduction.8,9 However, the benefits of simple solution-based processes continued to inspire strategies to solve those intrinsic microstructural drawbacks to enable effective photoanode design.
More recently, homogeneous hematite nanostructured thin films were produced by a low-cost spin-coating deposition of a polymeric precursor solution.10,11 The produced films still presented limited photoelectrochemical performance,11 but the optimization of thermal treatments led to improved wettability and roughness for an incremental enhancement of photocurrent.12 Subsequently, a better understanding of the impact of annealing processes on electron trapping,13 the establishment of the performance dependence on dopant positioning (i.e., interface versus crystal lattice doping),13 and the practical benefits of solvent exchange to improve wettability,12 resulted in a paradigm-shifting on photoanode design, scilicet, from the morphology-centered to one focused on interfacial properties and dopant engineering to enable cost-effective production routes.14
The concept of dual (or multi) chemical modifications for mitigating efficiency-hindering features in hematite photoanodes has brought further performance improvement.15,16 For instance, tetravalent ions, such as Ti4+,17 Zr4+,18 Si4+,19 and Sn4+,20,21 commonly used in hematite as single modifiers have been employed in multi modification investigations22 combined with Be2+,23 Zn2+,24 Al3+,25 In3+,26 Nb5+,27 N3−,28 F−,29,30 among others, in the search for higher water oxidation photocurrents. However, the modifications increased the synthesis and device production complexity by demanding multi-step protocols to deliver each chemical change.29 Moreover, dopants are not necessarily synergistic or additive, and it is not uncommon to observe dopants weakening each other in multi-doping systems,31 requiring further study to augment the understanding of doping engineering.
Fundamentally, modification by dopants can be grouped based on their placement in the system. When the dopant is within the crystal lattice, i.e., the ions form solid solution with hematite in its corundum structure, the dopant affects donor density in response to charge balancing32 and may also minimize polaronic effects by structural stabilization.33 On the other hand, if ions preferentially segregate to interfaces, or in other words form interfacial excesses,34 the changes in the local chemistry may impact energy barriers between grains and grains-substrate for improved charge transport and, when located at the surface, impact the response of the system to gas phase chemistry.35,36 From a microstructural perspective, interface excess is also associated with the stability of nanostructures,37 as the excesses lead to local energy reductions, as described by the Gibbs adsorption isotherm.38 This interfacial energy reduction results in coarsening inhibition, for smaller grain sizes and greater thermal stability.34
In this work, we hypothesize that the design of a specific dopant placement can potentially mitigate dopant competitive behaviors to enable unprecedented photoanode efficiencies. Therefore, we introduce a solution process for the fabrication of synergistically engineered bulk (crystal lattice) and interface on hematite thin films. The process entails a hematite polymeric precursor containing trivalent modifying cations mixed with tetravalent ions added at different synthesis stages to form a single liquid precursor to be either spin coated, dip coated, or sprayed onto a conductive surface to build the components of a photoelectrochemical cell.
The proper dopant selection combined with the synthesis strategy allows for a predictive allocation of the dopants in the nanostructure. For demonstration, Ga3+ dopant was selected for hematite (crystal) because of its similarity in terms of valence, ionic radius size (Ga3+ = 62 pm; Fe3+ = 64) pm, and coordination number to Fe3+,39 theoretically benefiting solid solution formation.40 Targeting the interfacial chemical manipulation, Hf4+ was selected following segregation design strategies,34i.e., it shows ionic radius mismatch with both Ga3+ and Fe3+ (Hf4+ = 72 pm39), suggesting unfavorable formation of solid solution, while being unlikely to form a second phase in the concentrations and thermal treatment temperatures adopted.41
Targeting an enhancement on the surface charge transfer kinetics, NiFeOx oxygen evolution reaction (OER) catalyst was electrodeposited onto Ga–Hf-co-doped photoanode to reduce the overpotential for water oxidation.42 Nickel-iron alloys, such as hydroxides, oxyhydroxides and oxides are well-known non-noble metal alternatives with high stability and faradaic efficiency43 to modify hematite surface. The introduced modifications resulted in a remarkable performance enhancement for the hematite-based photoanodes, with an overall photoelectrode efficiency of 65% in the conversion of absorbed light into photocurrent density, which by itself demonstrates the success of the design strategies and open great perspectives for future technological developments.
Experimental section
Synthesis of hematite-based photoanodes by the polymeric precursor (PPS) method
For the synthesis of the polymeric precursor, 2.6 × 10−2 mol of citric acid (C6H8O7, Sigma-Aldrich, 99%) was dissolved in 10 mL of milli-Q ultra-pure water (18.2 MΩ cm) at room temperature (25 °C). After dissolution, a quantified aliquot of 0.45 mol L−1 gallium nitrate (Ga(NO3)3·xH2O, Alfa Aesar, 98%) aqueous solution was added to obtain a precursor with 0.5% Ga3+ proportionally to the stoichiometric quantity of Fe3+ to be added. Next, 8.7 × 10−3 mol of iron(III) nitrate (Fe(NO3)3·9H2O, Sigma-Aldrich, 98%) was added and the system was heated at 60–70 °C under stirring. After complete homogenization, 5.4 × 10−2 mol of ethylene glycol (C2H6O2, Synth, 99%) was added. The system was kept under heat and stirring for 30 min, then left to cool down to 25 °C, when its volume was measured. The polymeric precursor was concentrated at 60–70 °C until it reached 50% of its initial volume. With the solution still hot, stoichiometric quantity of a previously prepared 0.2 mol L−1 hafnium chloride (HfCl4, Sigma-Aldrich, 98%) ethanolic solution was added under stirring. This last step resulted in a precursor solution with 3.0% Hf4+ stoichiometric to the Fe3+ concentration. The solution was then cooled down to 25 °C. An aliquot of 2.5 mL was diluted in 750 μL of anhydrous ethanol (Merck, 99.9%) and 500 μL of isopropyl alcohol (Synth, 99.5%), both slowly added to the solution under stirring for 5 min. The as-prepared solutions were stored in a refrigerator (T ∼7.0 °C) for 24 h, and then spin-coated at 25 °C onto the substrate surface.Control solutions were previously prepared following the same steps to obtain (i) pristine hematite (H), (ii) Ga3+-modified (Ga0.1H, Ga0.5H and Ga1.0H) samples, and (iii) Hf4+-modified (HHf1.0, HHf2.0, HHf3.0, HHf4.0, HHf5.0, HHf6.0 and HHf7.0) samples. The optimal concentrations for the co-doped solution (0.5% Ga3+ and 3.0% Hf4+) were selected based on the responses of those control products.
All thin films were prepared on aluminium borosilicate glass (1.0 mm thick) substrates coated with fluorine-doped tin oxide (FTO, Solaronix, RΩ 8–10 Ω cm−1, >80% transmission from 500 to 800 nm, 2 cm × 1 cm). First, the substrates were cleaned by successive rinsing in acetone and ethanol for 30 min each at ∼70 °C and in Milli-Q water (18.2 MΩ cm) under ∼100 °C heat for 30 min. After, the substrates were thermally treated at 550 °C for 60 min (Lindberg/Blue M Mini-Mite horizontal tube furnace, model TF55035A). Then, 100.0 μL of polymeric solution was deposited onto the FTO surface with a spin-coating equipment (Laurell Tech. Corp., model WS-650MZ-23NPP) using 500 rpm for 5 s and 7000 rpm for 30 s. After coating, the thin films were thermally treated at 550 °C in air atmosphere for 30 min (Lindberg/Blue M Mini-Mite horizontal tube furnace, model TF55035A) and then at 750 °C in N2 flux (18 mL min−1) for 30 min (OTF-1200X-50-SL automatic sliding quartz tube furnace, MTI Corporation). Annealing under N2 flux was carried out by sealing the furnace tube to a coupled vacuum pump and pressure gauge. The quartz tube where the samples were placed underwent three consecutive steps of evacuation/pressurization with N2 gas, which flux was kept throughout the thermal treatment. After this step, the furnace was slide to the sample position and the thermal treatment started. The furnaces were preheated at employed temperatures and the region where the samples were placed had their temperatures verified by an external thermocouple. Temperatures and duration of thermal treatments adopted in this work were optimized as reported elsewhere44,45 to preserve the substrate integrity and conductivity. Fig. S1† shows a schematic visualization of all processes involved in the fabrication of the photoelectrodes.
NiFeOx alloy (photo)electrodeposition
NiFeOx alloy was electrodeposited under illumination onto the Ga0.5HHf3.0 sample by immersing the film in a pH 5.3 aqueous solution consisting of 16.0 mM NiSO4·6H2O (Sigma Aldrich, 99.9%), 5.0 mM Fe2(SO4)3·3H2O (Sigma Aldrich, 97%), and 0.1 M NaCH3COO·3H2O (Sigma Aldrich, 99%). The procedure was carried out in a three-electrode electrochemical cell containing a platinum counter electrode, a commercial (Metrohm Autolab) Ag/AgCl(sat) reference electrode, and the as-prepared solution in which the working electrode was immersed. Using a potentiostat/galvanostat (Autolab PGSTAT 129N), a linear sweep voltammetry was performed from 0.5 to 0.9 V vs. Ag/AgCl, at scan rate of 10 mV s−1, under 100 mW cm−2 simulated sunlight illumination. No additional thermal treatments were employed after NiFeOx (photo)electrodeposition.Structural characterization
The X-ray diffraction (XRD) pattern of the as-prepared photoelectrodes were identified with the aid of a Bruker diffractometer D8 Advance ECO-AXS, with Bragg–Brentano standard configuration and Kα-Cu incident radiation (λ = 1.54 Å), measuring an angular range from 15° ≤ 2θ ≤ 110°, with a 0.02° step and 18.0 s per step. Surface chemical composition of the photoelectrodes was investigated by X-ray photoelectron spectroscopy (XPS) employing Scienta-Omicron ESCA+ spectrometer equipped with a hemisphere analyzer (EAC-2000 sphere), a monochromatic Kα-Al source (hν = 1486.6 eV) and a low-energy electron flood gun (charge neutralizer). The XPS spectra were recorded at a pass energy of 30 eV with 0.05 eV per step. Adventitious carbon (C–C/C–H) at 284.8 eV was used to calibrate the XPS spectra. XPS data analysis was performed using Gaussian–Lorentzian function GL(30) and a Shirley-type background subtraction (CasaXPS software). Ultraviolet photoelectron spectroscopy (UPS) measurements were carried out in a Specs XPS/UPS system with a Phoibos 150 analyzer and a CMOS 2D detector using He I line (21.22 eV) from a UHV gas discharge lamp. The acceleration potential of −4 V was applied to the samples.Morphological characterization
Focused-ion beam scanning electron microscopy (FIB-SEM) cross-section images were acquired with the microscope Quanta FEG 650, FEI Company, with an T2 detector (mode A + B), accelerating voltage of 5 kV and 50 pA current. Working distance was 7.0 mm and the horizontal field width was kept at 2.07 μm. Scanning transmission electron microscopy (STEM) analysis was performed in a JEOL JEM 2100F microscope equipped with energy dispersive X-ray spectroscopy (EDS) module Oxford SDD X-Max 80 mm2. EDS data was acquired simultaneously to STEM using a 0.7 nm probe size and a 0.3 s time acquisition. The elemental and atomic concentration maps were obtained using the Gatan GMS plugin.Electrical characterization
Conductive atomic force microscopy (c-AFM) measurements were performed in a NX-10 Park Systems microscope, with NanoSensorsTM Pt/Ir-coated silicon probe, PPP-EFM model, resonance frequency (nominal) 75 kHz, force constant (nominal) 2.8 N m−1. A 5 V bias was applied to the probe. The experiments were carried out in an environment-controlled chamber with 0.5% relative humidity and 25 °C. Scanning image areas of 1 μm × 1 μm with a resolution of 512 × 512 pixels were recorded.Optical characterization
Optical measurements were performed in a Shimadzu UV-VIS-NIR spectrophotometer UV-3600 Plus equipped with an integrating sphere. The photoelectrodes were positioned in a sample holder for thin films, and absorption spectra were recorded from 240 to 800 nm. The maximum current generated due to the light absorption of the nanoporous structures (Jabs) was calculated from the optical measurements as presented in eqn (1),46 where q is the elementary electron charge, ø is the photon flux in the AM 1.5 G filter, and λ is in the visible range for the absorption spectra of all samples analyzed.| Jabs = −qø(1 − e(−∫Absdy)) | (1) |
The optical band gap energies of the photoelectrodes were estimated from UV-vis absorption spectra according to the Tauc relation (eqn (2)), where α is the absorption coefficient, h is the Planck constant, ν is the frequency, n is ½ for allowed indirect electronic transitions and 2 for allowed direct transitions, A is a constant, and Eg is the estimated optical band gap energy.47
| (αhν)n = A(hν − Eg) | (2) |
Photoelectrochemical characterization
The photoelectrodes were assembled as photoanodes for water oxidation in a three-electrode electrochemical cell containing a platinum counter electrode, a commercial (Metrohm Autolab) Ag/AgCl(sat) reference electrode, and 1.0 mol L−1 NaOH (Sigma-Aldrich, 98%) electrolyte solution (pH = 13.6). The area of the photoanodes measured in this configuration was 0.283 cm2. Using a potentiostat/galvanostat (Autolab PGSTAT 129N), linear sweep voltammetry (LSV) measurements were conducted under sunlight illumination (100 mW cm−2) simulated by a 450 W Xe lamp (Osram, ozone free) equipped with an AM 1.5 global filter at a scan rate of 50 mV s−1. The power (1 sun) was adjusted and calibrated to the position of the photoanodes with the aid of an optical-meter (Newport 843-R-USB). Chopped illumination measurements were carried out with 20 mV s−1 scan rate and chopping frequency of 0.8 Hz. Stability chrono amperometry experiments at 1.23 V vs. reversible hydrogen electrode (RHE) were carried out for 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s and 0.5 s interval time under 100 mW cm−2 illumination. Intensity modulated photocurrent spectroscopy (IMPS) analysis was performed using a potentiostat (PGSTAT302N) with a FRA32M module coupled with a LED driver. A monochromatic blue LED (470 nm) was used to generate a sinusoidal oscillating light intensity in a frequency range from 20 kHz to 0.1 Hz under calibrated 30 mW cm−2 illumination. The amplitude of the modulation was set at 10% of the base light intensity. All potentials were converted to RHE using the Nernst eqn (3):
000 s and 0.5 s interval time under 100 mW cm−2 illumination. Intensity modulated photocurrent spectroscopy (IMPS) analysis was performed using a potentiostat (PGSTAT302N) with a FRA32M module coupled with a LED driver. A monochromatic blue LED (470 nm) was used to generate a sinusoidal oscillating light intensity in a frequency range from 20 kHz to 0.1 Hz under calibrated 30 mW cm−2 illumination. The amplitude of the modulation was set at 10% of the base light intensity. All potentials were converted to RHE using the Nernst eqn (3):| ERHE = EAg/AgCl + E0Ag/AgCl + 0.059 pH | (3) |
The overall photoelectrode efficiencies (ηoverall) representing the total yield of current generated in relation to the maximum current permitted by the absorption properties of the photoanodes were calculated from eqn (4), where Jph is the photocurrent at water oxidation potential (1.23 V vs. RHE) and Jabs was calculated from eqn (1).
| ηoverall (%) = (Jph/Jabs) × 100 | (4) |
Gas chromatography
The amount of H2 and O2 produced in the photoelectrochemical cell with the optimized photoanode was monitored by gas chromatography (GC System 7890B Agilent Technologies equipped with a thermal conductivity detector – TCD) in a three-electrode PEC cell with an Ag/AgCl reference electrode, Pt foil counter electrode, and 1.0 mol L−1 NaOH as electrolyte. A constant potential of 1.23 V vs. RHE under 100 mW cm−2 illumination was applied during the experiment. Argonium (Ar) was used as carrier gas. The photoelectrochemical cell was evacuated three times to exchange atmospheric air with the Ar flux. The GC data was extracted from the software Agilent OpenLab CDS coupled to the chromatograph. The faradaic efficiency was calculated from the ratio of gas charge (from the evolved gas amount data) and the overall water splitting charge, which was measured by chrono amperometry during the experiment.Results and discussion
The polymeric precursors for hematite thin films were synthesized by the poly-condensation of citric acid containing Fe3+ ions followed with the addition of ethylene glycol. For Ga3+ doping, gallium nitrate was added along with Fe3+ ions to maximize mixing before polymerization. In contrast, for Hf4+ doping, an ethanol solution containing Hf4+ ions was introduced after the Fe3+ based solution was polymerized and homogenized. The latter addition targeted interface enrichment on the crystallized film by creating a separate chemical environment surrounding the polymeric network.13 For both doped and undoped precursors, the solutions were concentrated under heating and spin coated onto FTO glass sheets, followed by annealing. Furthermore, the electrodeposition of NiFeOx alloy was carried out in the Ga-Hf-co-doped photoanode, as detailed in the experimental section.Fig. 1a–d shows the cross-section images of the pristine hematite (H), Ga-doped (Ga0.5H), Hf-doped (HHf3.0) and Ga–Hf-co-doped hematite (Ga0.5HHf3.0) thin films. The images show homogenous mesoporous morphologies for all samples, revealing commonly reported hematite worm-like nanograins.12,48 When comparing the films, it is apparent Hf4+ caused grain size reduction during the thin film production,49 a phenomenon commonly associated with ion segregation to interfaces.50 Ga3+ insertion does not cause crystal size reduction but promotes measurable increase in porosity. This suggests Ga3+ is predominantly doping hematite crystal lattice and could favor the active surface area for catalysis. The co-doped film showed a microstructure combining the individual effects of the dopants, for a relatively porous fine-grained structure.
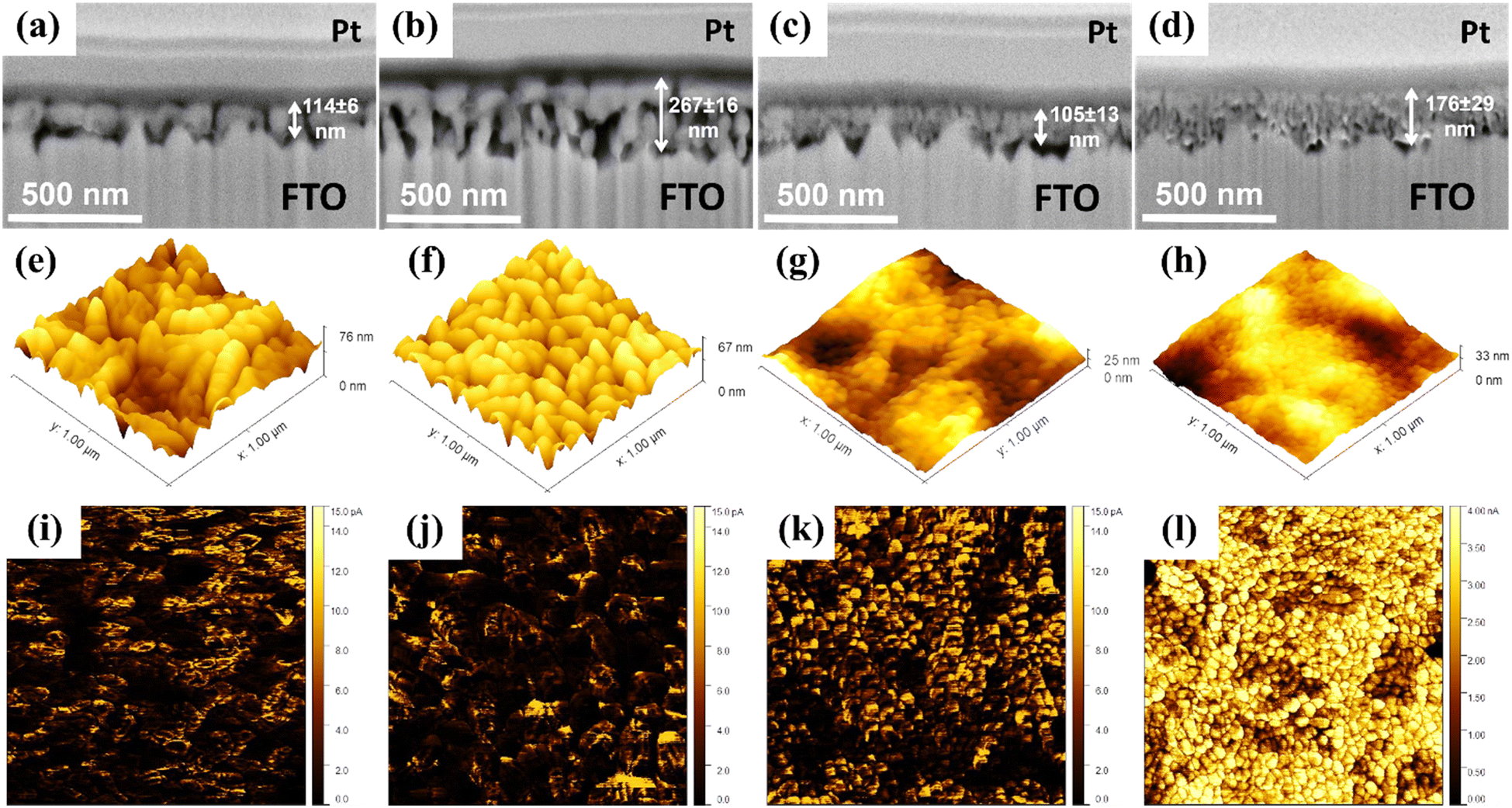 | ||
| Fig. 1 Cross-section images of (a) pristine hematite, (b) Ga0.5H, (c) HHf3.0, and (d) Ga0.5HHf3.0 acquired by focused-ion beam scanning electron microscopy (FIB-SEM). Atomic force microscopy (AFM) topological maps of (e) pristine hematite, (f) Ga0.5H, (g) HHf3.0, and (h) Ga0.5HHf3.0. Electrical conductivity profiles in the surface of (i) pristine hematite, (j) Ga0.5H, (k) HHf3.0, and (l) Ga0.5HHf3.0 obtained in the conductive mode of atomic force microscopy (c-AFM). | ||
Topological maps in Fig. 1e–h show the modification with Hf4+ reduced surface roughness in comparison to both undoped and Ga0.5H photoanodes (note the vertical z-direction scale bars). A similar result has been reported for Zr4+ in photoanodes produced by a polymeric precursor solution,12 with the smoother surface being attributed to better substrate wetting (smaller nanograins) and possibly correlated with more efficient electron collection at the back-contact. Current maps displayed in Fig. 1i–l highlight the synergistic effect of Ga3+ and Hf4+. While the single-doped hematite photoanodes showed limited electric conductivity enhancement, the Ga–Hf-co-doped photoanode exhibited uniform and constant conductivity pattern over the surface. This is likely an attribution of the spatial distribution of the dopants and their intrinsic electronic effects.
Fig. 2 shows the chemical composition mapping of Ga0.5HHf3.0_NiFeOx photoanode determined by energy dispersive X-ray spectroscopy (EDS) performed during scanning transmission electron microscopy (STEM) image acquisitions. The representative spectrum region where the chemical composition was evaluated shows Sn from the substrate (light blue) does not significantly diffuse into the hematite layer despite the thermal treatment, implying that any performance indicator cannot be attributed to Sn doping.51 The image also displays the oxygen (green) distributed over FTO and α-Fe2O3, iron (red) from hematite, and electrodeposited nickel (pink) spread over the photoanode surface. Gallium (yellow) is homogeneously distributed in the hematite grains as an indication of crystal lattice doping. Hafnium, though, besides segregating at hematite grain boundaries, is also found at the substrate–hematite interface, which would likely improve electrical contact and prevent electron loss due to FTO exposition to the electrolyte.52
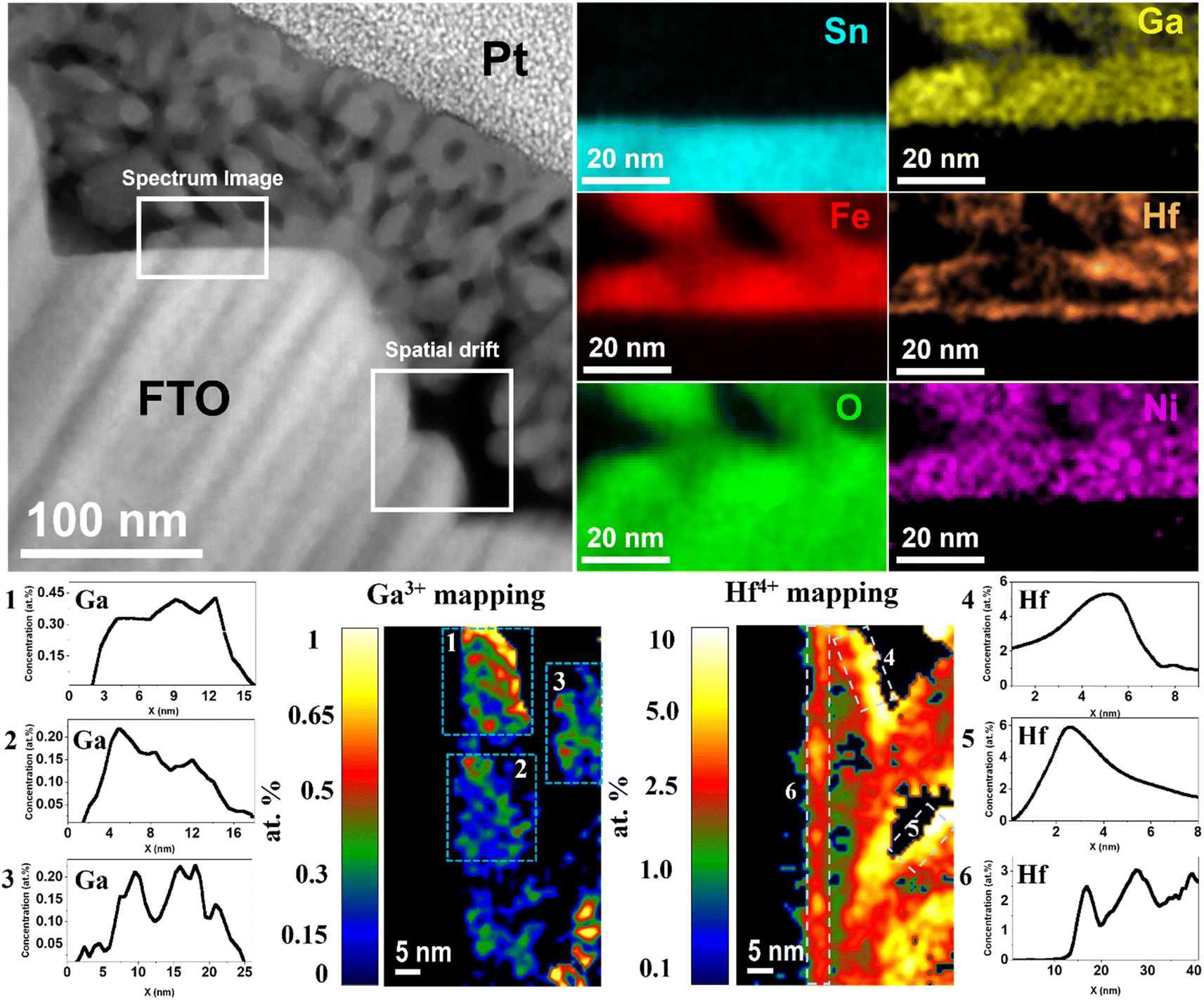 | ||
| Fig. 2 Scanning transmission electron microscopy (STEM) micrograph (left) and chemical maps obtained from energy dispersive X-ray spectroscopy (EDS) (right) for Ga0.5HHf3.0_NiFeOx photoanode. Below is an analysis of the atomic concentration of Ga3+ doping in the hematite grains (left) and the segregation of Hf4+ at the grain–grain interfaces and surface (right). | ||
Atomic concentration maps from Fig. 2 show gallium concentration between 0.2 and 0.45 at%. The average atomic concentration profile of three regions was calculated and identified by dashed windows (1–3) represented by their respective numbered graphs. Besides the concentrations in good agreement with the precursor added, it is remarkable the absence of Ga3+ at the surface. Hafnium atomic concentration maps identified by dashed windows 4–6 show that the average maximum concentration of Hf4+ segregated at the interfaces was approximately 6 at%, while at the FTO/hematite interface the average maximum concentration was 2.5 at%. As observed, the heterogeneous distribution of Hf4+ due segregation at polycrystalline grain boundaries and accumulation at the surface delivered atomic concentration distribution tending to zero towards the bulk (see x-axis of graphs 4–6). These results are particularly interesting, considering the stoichiometric control stablished by the synthesis method and its selectivity to deliver bulk and interfacial doping.
Furthermore, the observed dopant distribution has a cause-effect relationship with the conductivity maps presented in Fig. 1. As Ga3+ is dissolved in the lattice substituting Fe3+, it causes local negative strain due to it is relatively smaller ionic radius. Moreover, gallium–oxygen bonds are expected to be more ionic because of the difference in electronegativity, causing minor changes in the local energy levels and affecting polaronic migration for an improved conductivity.33 Since Ga3+ is not observed at the grain boundary regions, those remain similar to undoped hematite, showing high resistivity and annihilating benefits from the lattice doping across the film. Consequently, the conductivity profile shown in Fig. 1j for Ga0.5H is still within the picoampère (pA) range and similar to that for undoped hematite. Conversely, the large ionic radius of Hf4+ combined with its valence state are responsible for its segregation to grain boundaries, where it is expected to satisfy broken chemical bonds and ionic coordination.53 This grain boundary ‘healing’ effect decreases charge trapping across the grains by reducing the number of charge recombination sites, enhancing electron transport as observed in Fig. 1k. The Hf4+ impact becomes prominent when the synergistic effects with Ga3+ in the lattice are combined (Fig. 1l).
More in-depth understanding of the role of the dopants on the properties of the photoanodes was obtained from structural characterizations. X-ray diffraction (XRD) confirmed the crystalline phase α-Fe2O3 diffraction pattern indexed to the JCPDS card No. 33-0664 (rhombohedral crystalline system, space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c),54,55 with no detectable second phases associated with Ga3+ or Hf4+ (Fig. S2†). The diffraction patterns also remark the slight preferential growth in the (110) direction, due to its vertical orientation to the substrate.56
c),54,55 with no detectable second phases associated with Ga3+ or Hf4+ (Fig. S2†). The diffraction patterns also remark the slight preferential growth in the (110) direction, due to its vertical orientation to the substrate.56
X-ray photoelectron spectroscopy (XPS) identified gallium and hafnium and the respective oxidation states in the photoanodes (Fig. 3). The doublet spin–orbit component corresponding to Fe 2p3/2 (between 710.6 and 711.2 eV) and 2p1/2 (between 724.0 and 724.6 eV) with respective satellite peaks at approximately 719 and 733 eV are typical of α-Fe2O3.57 The Fe 2p3/2 energy ∼14 eV lower than Fe 2p1/2 is characteristic of Fe3+ state, corroborating XRD data and consistent with hematite polycrystalline phase. XPS analysis was not able to reliably detect gallium 2p3/2 and 2p1/2 (ref. 58) in the high-resolution spectra, corroborating Ga3+ species are distributed in the interior of hematite grains rather than on the surface. Binding energies for Hf 4f7/2 (16.5 eV) and Hf 4f5/2 (18.1 eV) are somewhat lower than found for HfO2 in the literature,59 consistent with the existence of HfOx species (suboxides) in the surface of the photoanodes.
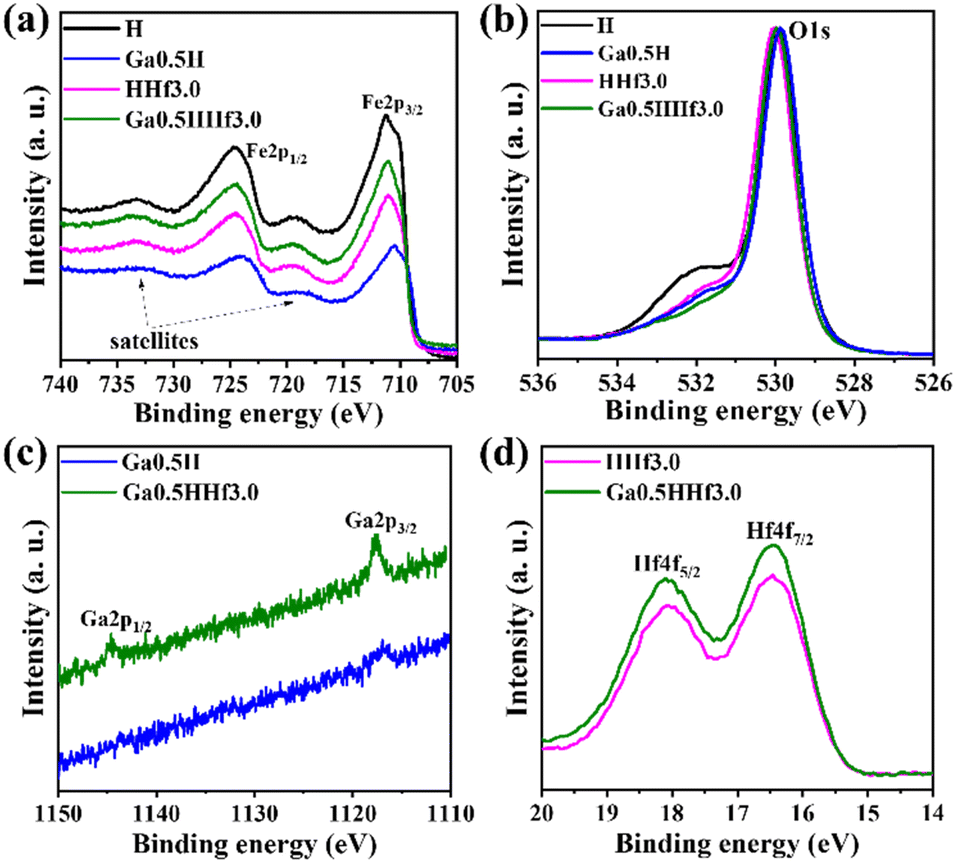 | ||
| Fig. 3 X-ray photoelectron spectroscopy (XPS) high-resolution spectra of (a) Fe 2p, (b) O 1s (c) Ga 2p, and (d) Hf 4f for the single-modified (Ga0.5H and HHf3.0) and Ga–Hf-co-doped (Ga0.5HHf3.0) photoanodes. | ||
Moreover, a shift to lower binding energies is observed for Fe 2p3/2 and Fe 2p1/2 peaks of Ga0.5H, indicating Ga3+ is interfering significantly on Fe3+ chemical environment, i.e., doping the crystal lattice. A minor shift for Fe 2p3/2 and Fe 2p1/2 peaks of HHf3.0 can be attributed to the mentioned presence of hafnium suboxides at the surface, as indicated by Hf 4f high-resolution spectra. Hafnium atoms must dispute oxygen atoms in the surface for charge balancing, as the thermal treatment in inert atmosphere (750 °C, N2) for surface photoelectrochemical activation produces superficial oxygen vacancies, possibly distorting Fe–O bonds locally and slightly interfering in Fe3+ chemical environment.60 This result reinforces that hafnium is most likely to form excess at the interfaces rather than doping.
The Ga–Hf-co-doped photoanode has binding energies for the Hf4f peaks similar to the single-modified Hf sample, consistent with the hypothesis of non-stoichiometric hafnium oxides here as well (Fig. S3†). Moreover, the addition of both modifiers did not shift Ga 2p3/2 and Ga 2p1/2 or Hf 4f7/2 and Hf 4f5/2 binding energies when compared to single-modified photoanodes. This result supports the synthesis process can drive both modifiers to their specific role without creating conflicting drawbacks.
The designed dopant distribution was expected to affect the efficiency of the photoanodes. Interestingly, the absorption properties were not significantly affected by the modifications, as observed in the absorption profiles (Fig. S4†) and estimated optical band gap values (Fig. S5 and Table S1†). However, Fig. 4 shows the current density profile curves and photocurrent measured at 1.23 V vs. RHE (Jph) with clear improvements for the chemically modified photoanodes with optimized Ga3+ and Hf4+ content (see Fig. S6† for Ga3+ and Hf4+ optimization).
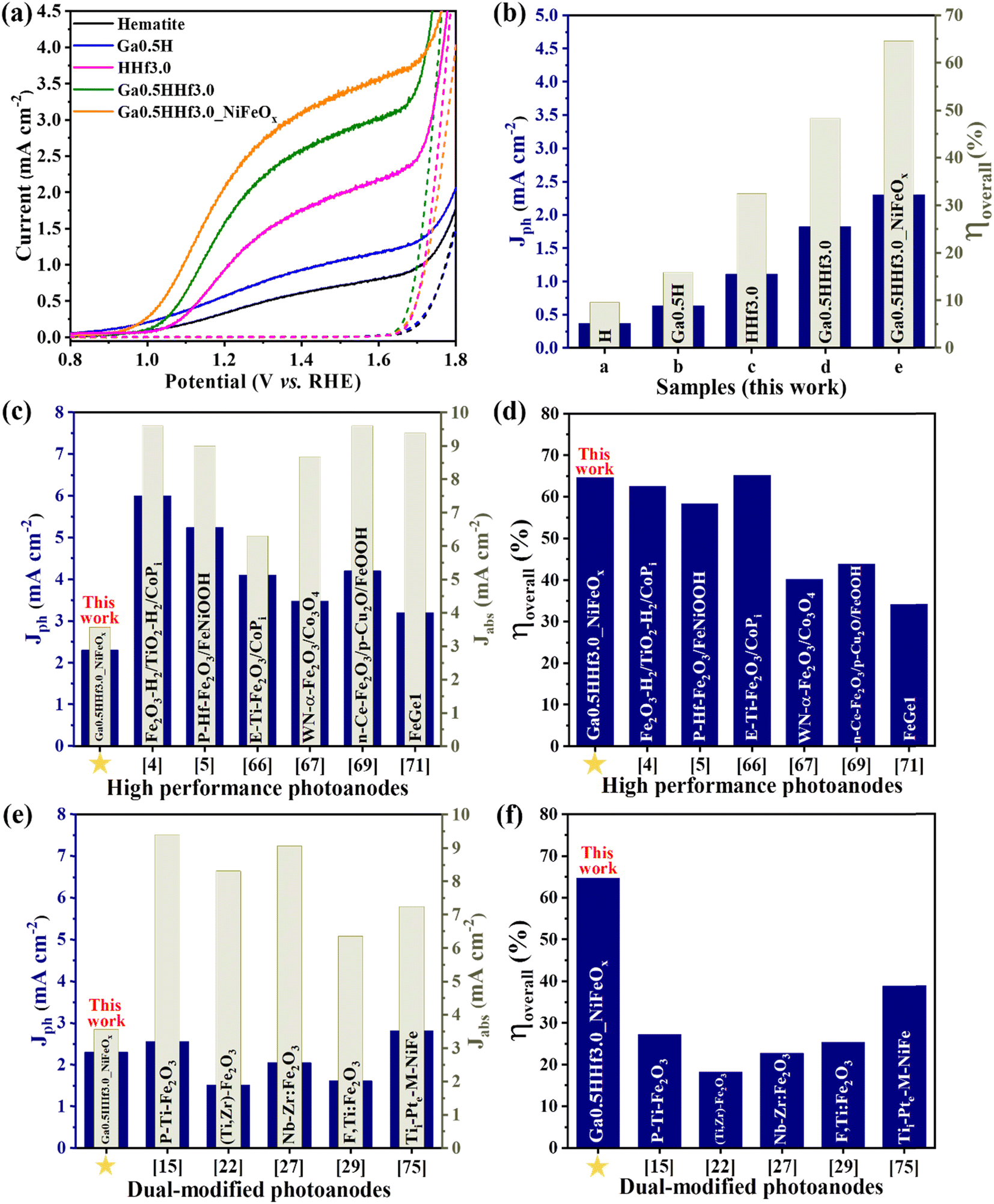 | ||
| Fig. 4 (a) Photocurrent curves of pristine hematite, Ga-modified hematite, Hf-modified hematite, and Ga–Hf-co-doped photoanodes under (straight lines) and without (dashed lines) simulated sunlight illumination. (b) Photocurrent values measured at 1.23 V vs. RHE (Jph) and overall photoelectrode efficiency (ηoverall) values for a) pristine hematite (H); (b) Ga0.5H; (c) HHf3.0; (d) Ga0.5HHf3.0; and (e) Ga0.5HHf3.0_NiFeOx. (c) Photocurrent values at 1.23 V vs. RHE under 100 mW cm−2 illumination (Jph) and calculated current values due to light absorption (Jabs) for some hematite-based photoanodes with highest photocurrents and available UV-vis optical absorption data reported in the literature compared to this work [4,5,66,67,69,71]. (d) Calculated overall photoelectrode efficiencies of the photoanodes shown in item c compared to the ηoverall of the optimized Ga0.5HHf3.0_NiFeOx photoanode. (e) Photocurrent values at 1.23 V vs. RHE under 100 mW cm−2 illumination (Jph) and calculated current values due to light absorption (Jabs) for hematite-based photoanodes produced by dual modification strategies with available UV-vis optical absorption data reported in the literature [15,22,27,29,75]. (f) Overall photoelectrode efficiency comparison of hematite-based photoanodes produced by dual modification strategies recently reported in the literature with Ga0.5HHf3.0_NiFeOx photoanode. | ||
The photocurrent curves (Fig. 4a) show Ga3+ doping promotes a cathodic shift in the water oxidation onset potential of the photoanodes (∼100 mV), but the synergistic effect of Ga3+ and Hf4+ exhibit approximately the same onset as pristine hematite. This indicates Hf4+ features predominate over Ga3+ at the surface because it does not change the overpotential necessary for water oxidation. On the other hand, Ga3+ and Hf4+ addition considerably improved the photocurrent density throughout the water oxidation potential range (from 0.37 mA cm−2 for pristine hematite to 1.82 mA cm−2 for Ga0.5HHf3.0 at 1.23 V vs. RHE), indicating better electron injection to the external circuit. NiFeOx (photo)electrodeposition can enable the collection of minority charge carriers at lower potentials, and the passivation of surface-trapping states created by the addition of modifiers. It is apparent that NiFeOx improved the overall J–V curve by showing a shift in the onset to a lower potential and increasing the photocurrent response, suggesting that passivation of surface states occur prior to charge transport and transfer enhancement in the photoanode.61–64
The synergistic effect of the chemical modifications led to a photocurrent improvement up to 2.30 mA cm−2. While this number is still relatively modest when contrasted with the literature,4,5,65–73 the film thickness is only 176 nm, as shown in Fig. 1, representing a mass of material of less than 1 mg. This is a consequence of the high efficiency achieved by Ga–Hf-co-doping as illustrated in Fig. 4b by the overall photoelectrode efficiency (ηoverall). The overall photoelectrode efficiency values calculated from the ratio between Jph and the current values from light absorption (Jabs) does not correspond to the overall water splitting efficiency, since only the anodic photocurrent associated with the water oxidation processes taking place on the photoanode is considered. However, these values are extremely useful for comparing the total yield of current generated in relation to the maximum current permitted by the absorption properties of the photoanodes studied. In this work, for instance, while an incremental increase is provided when each dopant is individually introduced, the photoanode containing both Ga3+ and Hf4+ added by the polymeric precursor solution method showed 48% of overall photoelectrode efficiency, with NiFeOx bringing it up to 65%.
Fig. 4c and d show the overall photoelectrode efficiency of the optimized Ga0.5HHf3.0_NiFeOx photoanode compared to state-of-the-art hematite-based materials from the literature (Table S2†). Despite its modest photocurrent at 1.23 V vs. RHE and lower current generated due to light absorption (Jabs), the ηoverall of Ga0.5HHf3.0_NiFeOx is only rivaled by E-Ti-Fe2O3 photoanode,66 which was prepared by a multi-step process including hydrothermal synthesis followed by electrodeposition of CoPi and a subsequent KOH treatment. When compared with recent dual modification strategies reported in the literature (Table S3†),15,22,24,25,27–30,74–76 it becomes clear the new method leads to synergistic roles for Ga3+ and Hf4+, resulting in superior overall photoelectrode efficiency, as shown in Fig. 4e and f.
The Ga0.5HHf3.0_NiFeOx photoanode optimized by the selective doping was further tested to assess performance and structural stability over time using chopped illumination measurements (Fig. 5a) and operation tests for 3 h under illumination (Fig. 5b). Its reproducibility was also validated (Fig. S7†). Fig. 5c displays the amount of gas produced by this photoanode during 5 h operation, detected by gas chromatography.
 | ||
| Fig. 5 (a) Chopped illumination voltammetry of pristine hematite, Ga-modified hematite, Hf-modified hematite, and GaHf-modified photoanodes. (b) Chrono amperometry measurements at 1.23 V vs. RHE under 100 mW cm−2 illumination (3 h) for pristine hematite and the optimized Ga0.5HHf3.0_NiFeOx photoanode. (c) Amount of gas produced by Ga0.5HHf3.0_NiFeOx monitored by gas chromatography for 5 h and faradaic efficiencies of H2 and O2 evolution. (d) Linear sweep voltammetry (LSV) of Ga0.5HHf3.0_NiFeOx photoanode measured before and after stability test. (e–g) High-resolution XPS spectra of (e) Ga 2p, (f) Hf 4f, and (g) Ni 2p for Ga0.5HHf3.0_NiFeOx photoanode measured before and after stability test shown in (b). | ||
As observed in the chopped illumination curves (Fig. 5a) and the chrono-amperometry measurements (Fig. 5b), the current generated by the engineered photoanodes is preserved in strongly alkaline media. These results validate the doping method in terms of incorporating modifications without damaging hematite well-known stability.12,64Fig. 5c further demonstrates the optimized photoanode can produce H2 and O2 with high faradaic efficiency, indicating almost all charge photogenerated is used for overall water splitting reaction. These results suggest structural and chemical stability of the photoanodes. To test this, the Ga–Hf-co-doped photoanode performance and surface composition were characterized before and after 3 h operation in the photoelectrochemical cell. Voltammetry measurement after stability test (Fig. 5d) corroborated the sustained performance indicated by the chrono amperometry, revealing identical photocurrent value at 1.23 V vs. RHE and same current density profile.
High resolution XPS spectra of Ga3+, Hf4+ and Ni2+ after the stability test (Fig. 5e–g) show that the surface composition remains unaltered during the 3 h of PEC operation. This result is summarized in Table 1. It is important to note that Ga3+ and Ni2+ percentages lie in the limit of detection of the XPS equipment, therefore determining their quantities is not trivial and the results in the table must be analyzed only for figure of merit and relative comparison. The presence of nickel was monitored only in the component Ni 2p3/2 because the doublet Ni 2p1/2 is not distinguishable from signal noise. The only measurable effect after the 3 h operation was a slight increase in –OH species adsorbed on the surface of the photoanode (Fig. S8 and Table S4†), as shown in the assigned peak centered at 532.7 eV,77,78 but no significant impact on the overall photoelectrochemical performance was observed. It is also interesting to note that the atomic percentages of Ga3+ and Hf4+ obtained by XPS analysis are in good agreement with the EDS average percentages shown in Fig. 2.
| Sample | O (%) | Fe (%) | Gaa (%) | Hf (%) | Nia (%) |
|---|---|---|---|---|---|
| a Gallium and nickel percentages lie in the limit of detection of the equipment, therefore are not reliably detected. | |||||
| Ga0.5HHf3.0_NiFeOx before 3 h operation | 68.16 ± 0.30 | 27.59 ± 0.30 | 0.16 ± 0.10 | 3.96 ± 0.10 | 0.13 ± 0.10 |
| Ga0.5HHf3.0_NiFeOx after 3 h operation | 67.64 ± 0.30 | 28.06 ± 0.30 | 0.17 ± 0.10 | 4.02 ± 0.10 | 0.11 ± 0.10 |
Complementarily, a mechanistical overview on the role of the dopants was investigated by intensity modulated photocurrent spectroscopy (IMPS) performed under 30 mW cm−2 monochromatic illumination of a blue LED (λ = 470 nm). From the obtained data (see IMPS plot of pristine hematite in Fig. S10†), important parameters such as charge separation and transfer efficiency values were calculated using the IMPS general theory,79 accordingly described in Fig. S9† and presented in Fig. 6. As displayed in Ga0.5H photocurrent density profile (Fig. 6a), the onset potential cathodic shift when compared to pristine hematite is a signature of the reduction of polaronic effects, indicating the local symmetry break favored the electron mobility. Hence, charge separation (Fig. 6g) and transfer (Fig. 6j) efficiencies are mildly improved with a constant profile throughout the potential range of IMPS analysis, suggesting the local chemistry environment changes caused by Ga3+ addition can improve only the bulk conductivity. Analyzing the effect of Hf4+ addition, observed in both onset potential anodic shift in the HHf3.0 photocurrent density profile (Fig. 6b) and in the shift of charge transfer efficiency (ηtransf.) toward more positive potentials (Fig. 6k), there are indicatives that one of hafnium major roles is to form excess at the surface, thus creating surface states. Charge separation efficiency (CSExLHE) values for HHf3.0 (Fig. 6h) also confirm the other major role of Hf4+ which is its segregation at the hematite grain/grain interfaces. The reduction of the energy barrier heights at the grain boundaries introduced effectively enhanced electronic conduction and, therefore, charge separation efficiency.
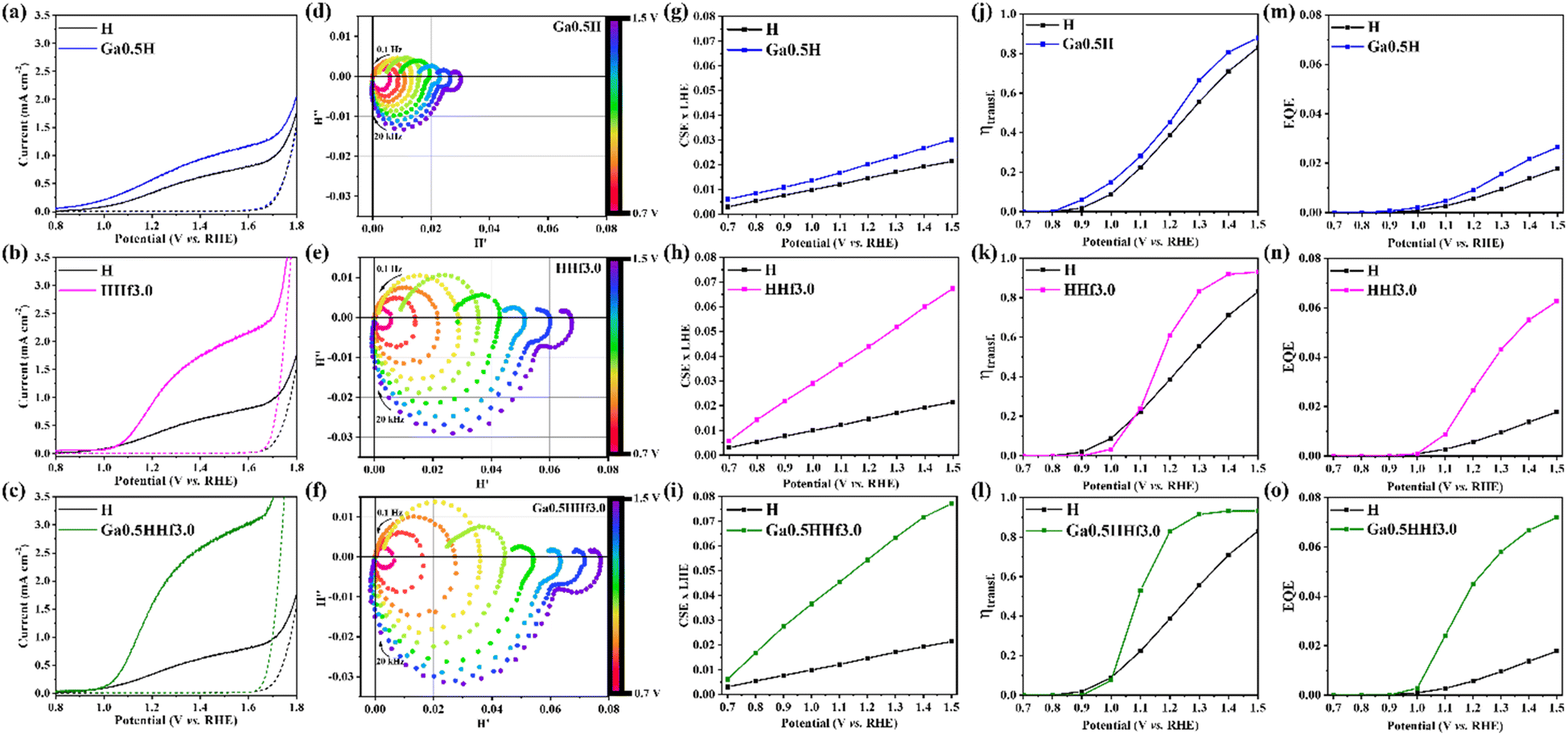 | ||
| Fig. 6 (a–c) Photocurrent density profiles of Ga0.5H, HHf3.0, and Ga0.5HHf3.0, respectively, compared to pristine hematite (measured under 100 mW cm−2 illumination). (d–f) Intensity modulated photocurrent spectroscopy (IMPS) plots of Ga0.5H, HHf3.0, and Ga0.5HHf3.0, respectively, measured from 0.7 V to 1.5 V, with a step of 0.1 V, under 30 mW cm−2 monochromatic illumination (λ = 470 nm). (g–i) Charge separation efficiency (CSE) versus light harvesting efficiency (LHE) values for Ga0.5H, HHf3.0, and Ga0.5HHf3.0, respectively, extracted from IMPS analysis. (j–l) Charge transfer efficiency (ηtransf.) values for Ga0.5H, HHf3.0, and Ga0.5HHf3.0, respectively, extracted from IMPS analysis. (m–o) External quantum efficiency (EQE) values for Ga0.5H, HHf3.0, and Ga0.5HHf3.0, respectively, extracted from IMPS analysis (30 mW cm−2 monochromatic illumination at 470 nm from a blue LED). | ||
From external quantum efficiency (EQE) values obtained under 30 mW cm−2 illumination at 470 nm for Ga0.5H (Fig. 6m) and HHf3.0 (Fig. 6n), it is evident the greater efficacy of segregation to overcome interfacial drawbacks over bulk doping since the electronic conduction is mainly limited by high energy barriers at the grain boundaries. Furthermore, the synergistic combination of both dopants resulted in a highly efficient photoanode with enhanced charge separation efficiency (Fig. 6i) and improved charge transfer efficiency (Fig. 6l).
The band edge positions (valence band maximum and work function) of the photoanodes (Fig. 7) were determined from ultraviolet photoelectron spectroscopy (UPS) spectra (Fig. S10†) with the input of bandgap energies (Eg) from Tauc plots. Gallium doping did not significantly displace the band edge positions but shifted the Fermi energy (EF) towards the conduction band (CB), justifying the increase in charge separation efficiency observed by IMPS analysis.32 This result is consistent with the assumption that gallium doping is reducing structural polaronic effects since small polarons are mainly responsible for hematite Fermi level pinning. For hafnium doping, there is an observable shift of valence band edge to a more negative value (VB = −7.34 eV) compared to pristine hematite (VB = −6.9 eV), which explains its superior PEC performance. On the other hand, the energy difference between the conduction band (CB) and the water reduction potential (−4.5 eV in Evacuum) for HHf3.0, which is 0.74 eV compared to 0.3 eV of pristine hematite, shows its requirement of a higher overpotential to drive the water oxidation reaction, corroborating the charge transfer efficiency profiles observed in Fig. 6k. Contrasting these findings with the profiles of J–V curves, it is possible to conclude the entangled relation between the band positioning of the semiconducting absorber materials and their photoelectrochemical behavior.
 | ||
| Fig. 7 Band diagrams obtained from UPS spectra of pristine hematite, Ga0.5H, HHf3.0, and Ga0.5HHf3.0. Valence band maximum and work function were extracted from the expansion of the region near the Fermi level and the determination of the secondary electron cutoff (Fig. S10†). Gap energies were extracted from Tauc plots. | ||
Conclusions
A new method for dual modification of hematite nanostructures which can deliver selectively allocation of dopants, i.e., interface versus crystal lattice, was introduced and demonstrated in the manufacturing of hematite ultrathin films doped with Ga3+ and Hf4+. A combination of characterization techniques, including FIB-SEM, STEM of the deposited films, and XPS demonstrated Ga3+ was indeed selectively doping the crystal lattice while Hf4+ was preferentially located at the interfaces, resulting in a synergistic impact reducing polaronic effects and improving charge transport between grains, respectively. The photoanode modified with Ga3+/Hf4+/NiFeOx showed a seven-fold increase in the overall photoelectrode efficiency compared to pristine hematite, demonstrating the synthetic strategy can be employed to produce highly efficient photoelectrodes for photoelectrochemical (PEC) water splitting. The PEC operation study showed the robustness of the method on maintaining the modifications incorporated in the hematite structure without damaging α-Fe2O3 stability. Moreover, charge carrier dynamic analysis and experimental band edge positions determination corroborated the hypothesis of this work by displaying bulk and interfacial doping signatures attributed to gallium and hafnium, respectively.While the results are encouraging in this particular field, the nature of the proposed new polymeric precursor solution method possibly offers a powerful and overreaching synthetic alternative for the design of nanostructured materials with dopants purposefully positioned in strategic regions. This refined dopant addition could enable the simultaneous control of multiple challenges found in the nanostructures applied for device implementation, including improvement of electric contacts, increase in surface area and stability, manipulation of charge carrier densities, sintering of grains and passivation of surface states.
Author contributions
Fabio A. Pires: investigation, data curation, formal analysis, writing – original draft, writing – review & editing, visualization. Gabriel T. dos Santos: formal STEM-EDS analysis. Jefferson Bettini: formal STEM-EDS analysis. Carlos Alberto Rodrigo Costa: formal AFM analysis. Renato V. Gonçalves: formal XPS analysis, writing – review & editing. Ricardo H. R. Castro: writing – review & editing, visualization. Flavio L. Souza: conceptualization, writing – review & editing, visualization, supervision, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge CNPq, CAPES and FAPESP (Grants 2017/02317-2, 2021/04967-0 and 2022/04150-6). F. L. S. acknowledges FAPESP (Grant 2017/11986-5), Shell and the strategic importance of the support given by ANP (Brazil's National Oil, Natural Gas and Biofuels Agency) through the R&D levy regulation. The authors are thankful to J. M. da Silva and F. E. Montoro for STEM sample preparation. R. H. R. C. thanks NSF DMR Ceramics 2015650.Notes and references
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef PubMed.
- M. H. Richter, W.-H. Cheng, E. J. Crumlin, W. S. Drisdell, H. A. Atwater, D. Schmeißer, N. S. Lewis and B. S. Brunschwig, Chem. Mater., 2021, 33, 1265–1275 CrossRef CAS.
- B. Scherrer, T. Li, A. Tsyganok, M. Döbeli, B. Gupta, K. D. Malviya, O. Kasian, N. Maman, B. Gault, D. A. Grave, A. Mehlman, I. Visoly-Fisher, D. Raabe and A. Rothschild, Chem. Mater., 2020, 32, 1031–1040 CrossRef CAS.
- T. H. Jeon, G. Moon, H. Park and W. Choi, Nano Energy, 2017, 39, 211–218 CrossRef CAS.
- C. Lu, D. Zhang, Z. Wu, X. Zhao, K. Feng, G. Zhang, S. Wang, Z. Kang and J. Zhong, Appl. Catal., B, 2023, 331, 122695 CrossRef CAS.
- S. R. Pendlebury, M. Barroso, A. J. Cowan, K. Sivula, J. Tang, M. Grätzel, D. Klug and J. R. Durrant, Chem. Commun., 2011, 47, 716–718 RSC.
- C. Y. Cummings, F. Marken, L. M. Peter, A. A. Tahir and K. G. U. Wijayantha, Chem. Commun., 2012, 48, 2027–2029 RSC.
- X. Lian, X. Yang, S. Liu, Y. Xu, C. Jiang, J. Chen and R. Wang, Appl. Surf. Sci., 2012, 258, 2307–2311 CrossRef CAS.
- W. Hamd, S. Cobo, J. Fize, G. Baldinozzi, W. Schwartz, M. Reymermier, A. Pereira, M. Fontecave, V. Artero, C. Laberty-Robert and C. Sanchez, Phys. Chem. Chem. Phys., 2012, 14, 13224–13232 RSC.
- D. N. F. Muche, T. M. G. dos Santos, G. P. Leite, M. A. Melo Jr., R. V. Gonçalves and F. L. Souza, Mater. Lett., 2019, 254, 218–221 CrossRef CAS.
- D. N. F. Muche, S. A. Carminati, A. F. Nogueira and F. L. Souza, Sol. Energy Mater. Sol. Cells, 2020, 208, 110377 CrossRef CAS.
- K. C. Bedin, B. Mouriño, I. Rodríguez-Gutiérrez, J. B. Souza Jr, G. T. dos Santos, J. Bettini, C. A. R. Costa, L. Vayssieres and F. L. Souza, Chin. J. Catal., 2022, 43, 1247–1257 CrossRef CAS.
- K. C. Bedin, I. Rodríguez-Gutiérrez, L. R. P. Peregrino, L. Vayssieres and F. L. Souza, J. Am. Ceram. Soc., 2023, 106, 79–92 CrossRef CAS.
- J. B. Souza Jr, F. L. Souza, L. Vayssieres and O. K. Varghese, Appl. Phys. Lett., 2021, 119, 200501 CrossRef.
- T. K. Sahu, A. K. Shah, A. Banik and M. Qureshi, ACS Appl. Energy Mater., 2019, 2, 4325–4334 CrossRef CAS.
- T. Wang, L. Gao, P. Wang, X. Long, H. Chai, F. Li and J. Jin, J. Colloid Interface Sci., 2022, 624, 60–69 CrossRef CAS PubMed.
- D. Wang, H. Chen, G. Chang, X. Lin, Y. Zhang, A. Aldalbahi, C. Peng, J. Wang and C. Fan, ACS Appl. Mater. Interfaces, 2015, 7, 14072–14078 CrossRef CAS PubMed.
- I. K. Jeong, M. A. Mahadik, S. Kim, H. M. Pathan, W.-S. Chae, H. S. Chung, G. W. Kong, S. H. Choi and J. S. Jang, Chem. Eng. J., 2020, 390, 124504 CrossRef.
- M. Allieta, M. Marelli, F. Malara, C. L. Bianchi, S. Santangelo, C. Triolo, S. Patane, A. M. Ferretti, Š. Kment, A. Ponti and A. Naldoni, Catal. Today, 2019, 328, 43–49 CrossRef CAS.
- Y. Ling, G. Wang, D. A. Wheeler, J. Z. Zhang and Y. Li, Nano Lett., 2011, 11, 2119–2125 CrossRef CAS PubMed.
- M. Li, Y. Yang, Y. Ling, W. Qiu, F. Wang, T. Liu, Y. Song, X. Liu, P. Fang, Y. Tong and Y. Li, Nano Lett., 2017, 17, 2490–2495 CrossRef CAS PubMed.
- Z. Sun, G. Fang, J. Li, J. Mo, X. He, X. Wang and Z. Yu, Chem. Phys. Lett., 2020, 754, 137736 CrossRef CAS.
- A. Annamalai, H. H. Lee, S. H. Choi, S. Y. Lee, E. Gracia-Espino, A. Subramanian, J. Park, K. Kong and J. S. Jang, Sci. Rep., 2016, 6, 23183 CrossRef CAS PubMed.
- Q. Zhu, C. Yu and X. Zhang, J. Energy Chem., 2019, 35, 30–36 CrossRef.
- I. K. Jeong, M. A. Mahadik, J. B. Hwang, W.-S. Chae, S. H. Choi and J. S. Jang, J. Colloid Interface Sci., 2021, 581, 751–763 CrossRef CAS PubMed.
- A. P. Singh, C. Tossi, I. Tittonen, A. Hellman and B. Wickman, RSC Adv., 2020, 10, 33307–33316 RSC.
- L. K. Dhandole, T. S. Koh, P. Anushkkaran, H.-S. Chung, W.-S. Chae, H. H. Lee, S. H. Choi, M. Cho and J. S. Jang, Appl. Catal., B, 2022, 315, 121538 CrossRef CAS.
- T. Jiao, C. Lu, K. Feng, J. Deng, D. Long and J. Zhong, J. Colloid Interface Sci., 2021, 585, 660–667 CrossRef CAS PubMed.
- K. Kang, H. Zhang, J. H. Kim, W. J. Byun and J. S. Lee, Nanoscale Adv., 2022, 4, 1659–1667 RSC.
- L. K. Dhandole, P. Anushkkaran, W.-S. Chae, H.-S. Chung, H.-H. Lee, S. H. Choi, M. Cho and J. S. Jang, Chem. Eng. J., 2022, 446, 136957 CrossRef.
- K.-Y. Yoon, J. Park, M. Jung, S.-G. Ji, H. Lee, J. H. Seo, M.-J. Kwak, S. I. Seok, J. H. Lee and J.-H. Jang, Nat. Commun., 2021, 12, 4309 CrossRef CAS PubMed.
- Gurudayal, S. Y. Chiam, M. H. Kumar, P. S. Bassi, H. L. Seng, J. Barber and L. H. Wong, ACS Appl. Mater. Interfaces, 2014, 6, 5852–5859 CrossRef CAS PubMed.
- T. J. Smart, V. U. Baltazar, M. Chen, B. Yao, K. Mayford, F. Bridges, Y. Li and Y. Ping, Chem. Mater., 2021, 33, 4390–4398 CrossRef CAS.
- R. H. R. Castro, Curr. Opin. Solid State Mater. Sci., 2021, 25, 100911 CrossRef CAS.
- D. R. Clark, H. Zhu, D. R. Diercks, S. Ricote, R. J. Kee, A. Almansoori, B. P. Gorman and R. P. O'Hayre, Nano Lett., 2016, 16, 6924–6930 CrossRef CAS PubMed.
- X. Xu, Y. Liu, J. Wang, D. Isheim, V. P. Dravid, C. Phatak and S. M. Haile, Nat. Mater., 2020, 19, 887–893 CrossRef CAS PubMed.
- K. Nakajima, F. L. Souza, A. L. M. Freitas, A. Thron and R. H. R. Castro, Chem. Mater., 2021, 33, 3915–3925 CrossRef CAS.
- H. Peng, Z. Jian and F. Liu, Int. J. Ceram. Eng. Sci., 2020, 2, 49–65 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A, 1976, 32, 751–767 CrossRef.
- H. J. Van Hook, J. Am. Ceram. Soc., 1965, 48, 470–472 CrossRef.
- V. Raghavan, Trans. Indian Inst. Met., 1992, 819–822 Search PubMed.
- G. Dong, L. Yan and Y. Bi, J. Mater. Chem. A, 2023, 11, 3888–3903 RSC.
- H. Hajibabaei, A. R. Schon and T. W. Hamann, Chem. Mater., 2017, 29, 6674–6683 CrossRef CAS.
- N. M. Ito, W. M. Carvalho Jr, D. N. F. Muche, R. H. R. Castro, G. M. Dalpian and F. L. Souza, Phys. Chem. Chem. Phys., 2017, 19, 25025–25032 RSC.
- I. Rodríguez-Gutiérrez, B. Mouriño, A. L. M. Freitas, C. A. R. Costa, E. L. Pires, R. V. Gonçalves, L. Vayssieres and F. L. Souza, ECS J. Solid State Sci. Technol., 2022, 11, 043001 CrossRef.
- A. E. Nogueira, M. R. S. Soares, J. B. Souza Jr., C. A. O. Ramirez, F. L. Souza and E. R. Leite, J. Mater. Chem. A, 2019, 7, 16992–16998 RSC.
- P. Makuła, M. Pacia and W. Macyk, J. Phys. Chem. Lett., 2018, 9, 6814–6817 CrossRef PubMed.
- K. Sivula, R. Zboril, F. Le Formal, R. Robert, A. Weidenkaff, J. Tucek, J. Frydrych and M. Grätzel, J. Am. Chem. Soc., 2010, 132, 7436–7444 CrossRef CAS PubMed.
- F. C. De Lima, G. R. Schleder, J. B. Souza Jr., F. L. Souza, F. B. Destro, R. H. Miwa, E. R. Leite and A. Fazzio, Appl. Phys. Lett., 2021, 118, 201602 CrossRef CAS.
- R. H. R. Castro and D. Gouvêa, J. Am. Ceram. Soc., 2016, 99, 1105–1121 CrossRef CAS.
- K. C. Bedin, A. L. M. Freitas, A. Tofanello, I. Rodríguez-Gutiérrez and F. L. Souza, Ceram. Eng. Sci., 2020, 2, 204–227 CAS.
- P. Shadabipour and T. W. Hamann, Chem. Commun., 2020, 56, 2570–2573 RSC.
- J. P. Buban, K. Matsunaga, J. Chen, N. Shibata, W. Y. Ching, T. Yamamoto and Y. Ikuhara, Science, 2006, 311, 212–215 CrossRef CAS PubMed.
- L. Pauling and S. B. Hendricks, J. Am. Chem. Soc., 1925, 47, 781–790 CrossRef CAS.
- R. L. Blake, R. E. Hessevick, T. Zoltai and L. W. Finger, Am. Mineral., 1966, 51, 123–129 CAS.
- S. Kment, P. Schmuki, Z. Hubicka, L. Machala, R. Kirchgeorg, N. Liu, L. Wang, K. Lee, J. Olejnicek, M. Cada, I. Gregora and R. Zboril, ACS Nano, 2015, 9, 7113–7123 CrossRef CAS PubMed.
- T. Yamashita and P. Hayes, Appl. Surf. Sci., 2008, 254, 2441–2449 CrossRef CAS.
- J. L. Bourque, M. C. Biesinger and K. M. Baines, Dalton Trans., 2016, 45, 7678–7696 RSC.
- P. C. Silva Neto, D. A. Ramirez, A. R. Terto, J. Y. E. Santos, J. C. V. Dos Santos, F. M. T. Mendes, F. L. Serafini, M. C. M. Farias and E. K. Tentardini, Surf. Coat. Technol., 2021, 415, 127097 CrossRef CAS.
- N. Kaiser, T. Vogel, A. Zintler, S. Petzold, A. Arzumanov, E. Piros, R. Eilhardt, L. Molina-Luna and L. Alff, ACS Appl. Mater. Interfaces, 2022, 14, 1290–1303 CrossRef CAS PubMed.
- K. C. Bedin, D. N. F. Muche, M. A. Melo Jr., A. L. M. Freitas, R. V. Gonçalves and F. L. Souza, ChemCatChem, 2020, 12, 3156–3169 CrossRef CAS.
- J. Qiu, H. Hajibabaei, M. R. Nellist, F. A. L. Laskowski, S. Z. Oener, T. W. Hamman and S. W. Boettcher, ACS Energy Lett., 2018, 3, 961–969 CrossRef CAS.
- W. Wang, M. Heggen, W. Cui, B. Probst, R. Alberto and C. Cui, Chem. Commun., 2020, 56, 10179–10182 RSC.
- A. L. M. Freitas, D. N. F. Muche, E. R. Leite and F. L. Souza, J. Am. Ceram. Soc., 2020, 103, 6833–6846 CrossRef CAS.
- Gurudayal, R. A. John, P. P. Boix, C. Yi, C. Shi, M. C. Scott, S. A. Veldhuis, A. M. Minor, S. M. Zakeeruddin, L. H. Wong, M. Grätzel and N. Mathews, ChemSusChem, 2017, 10, 2449–2456 CrossRef CAS PubMed.
- Q. Wu, K. Zhang, D. Wang, Y. Lin and T. Xie, Electrochim. Acta, 2023, 449, 142241 CrossRef CAS.
- C. Li, Z. Chen, W. Yuan, Q.-H. Xu and C. M. Li, Nanoscale, 2019, 11, 1111–1122 RSC.
- J. Wu, M. Qi, G. Wang, B. Yu, C. Liu, W. Hou and W. Liu, ACS Sustain. Chem. Eng., 2020, 8, 5200–5208 CrossRef CAS.
- J. Wu, P. Huang, H. Fan, G. Wang and W. Liu, ACS Appl. Mater. Interfaces, 2020, 12, 30304–30312 CrossRef CAS PubMed.
- D. Chen, Z. Liu, Z. Guo, M. Ruan and W. Yan, ChemSusChem, 2019, 12, 3286–3295 CrossRef CAS PubMed.
- M. H. M. Rodrigues, I. Rodriguez-Gutierrez, C. A. O. Ramirez, C. A. R. Costa, C. A. Biffe, J. B. Souza Jr, F. L. Souza and E. R. Leite, J. Mater. Chem. A, 2022, 10, 13456–13466 RSC.
- Z. Zhang, I. Karimata, H. Nagashima, S. Muto, K. Ohara, K. Sugimoto and T. Tachikawa, Nat. Commun., 2019, 10, 4832 CrossRef PubMed.
- Z. Zhang, H. Nagashima and T. Tachikawa, Angew. Chem., 2020, 59, 9047–9054 CrossRef CAS PubMed.
- A. Subramanian, M. A. Mahadik, J.-W. Park, I. K. Jeong, H.-S. Chung, H. H. Lee, S. H. Choi, W.-S. Chae and J. S. Jang, Electrochim. Acta, 2019, 319, 444–455 CrossRef CAS.
- Z. Zhong, G. Zhan, B. Du, X. Lu, Z. Qin and J. Xiao, J. Colloid Interface Sci., 2023, 641, 91–104 CrossRef CAS PubMed.
- P. Anushkkaran, M. A. Mahadik, W.-S. Chae, H. H. Lee, S. H. Choi and J. S. Jang, Appl. Surf. Sci., 2023, 623, 157035 CrossRef CAS.
- J. Xiao, F. Zhao, J. Zhong, Z. Huang, L. Fan, L. Peng, S.-F. Zhou and G. Zhan, Chem. Eng. J., 2020, 402, 126163 CrossRef CAS.
- S.-Y. Yuan, L.-W. Jiang, J.-S. Hu, H. Liu and J.-J. Wang, Nano Lett., 2023, 23, 2354–2361 CrossRef CAS PubMed.
- E. A. Ponomarev and L. M. Peter, J. Electroanal. Chem., 1995, 396, 219–226 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis procedure; structural, optical, and photoelectrochemical characterizations; literature review; Tables S1–S4, and Fig. S1–S11† cited in the main text. See DOI: https://doi.org/10.1039/d3se00998j |
| This journal is © The Royal Society of Chemistry 2023 |