
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
α/β-Ni(OH)2 phase control by F-ion incorporation to optimise hybrid supercapacitor performance†
Xuerui
Yi
a,
Veronica
Celorrio
 b,
Haoyu
Zhang
a,
Neil
Robertson
*a and
Caroline
Kirk
*a
b,
Haoyu
Zhang
a,
Neil
Robertson
*a and
Caroline
Kirk
*a
aSchool of Chemistry, EaStCHEM, University of Edinburgh, King's Buildings, David Brewster Road, Edinburgh, Scotland EH9 3FJ, UK. E-mail: neil.robertson@ed.ac.uk; caroline.kirk@ed.ac.uk
bDiamond Light Source Ltd, Harwell Science and Innovation Campus, Oxfordshire, Didcot, OX11 0DE, UK
First published on 4th October 2023
Abstract
We have controlled the formation of flower-like nanostructured pure alpha phase and pure beta phase nickel hydroxide (α-Ni(OH)2, β-Ni(OH)2), as well as mixtures of phases (αβ-Ni(OH)2). Through a range of experiments, we prove that the addition of fluoride ions controls the phase formed and we suggest two possible mechanisms for this effect. Incorporated fluoride ions in the layers disfavour the insertion of other anions and water molecules, resulting in β-Ni(OH)2 which has a smaller interlayer distance; and we suggest that the high electronegativity of F results in less nickel oxidation to Ni3+ also disfavouring extra anions between layers. The nickel hydroxide materials were characterised by a suite of techniques including powder X-ray diffraction, electron microscopy, thermal analysis and in situ X-ray absorption spectroscopy, in combination with electrochemical studies. The redox cycle stability and conductivity were improved through control of phase formation, such that αβ-Ni(OH)2 exhibits 108% capacity retention after 2000 continuous charge–discharge cycles at 20 mA cm−1 while α-Ni(OH)2 displays only 50% capacity retention after 2000 cycles. Finally, the assembled αβ-Ni(OH)2//activated carbon hybrid supercapacitor displays maximum energy density of 32 W h kg−1 at a power density of 900 W kg−1, and retains 86% capacity after 5000 continuous charge–discharge cycles.
Introduction
The increasing demand for portable electronic devices and electric vehicles has highlighted the need for advanced energy storage systems. Hybrid supercapacitors (HBSCs) have emerged as a promising option owing to their ability to provide long-cycle life and fast charge/discharge capabilities. HBSCs consist of two electrodes with different materials; a high-energy density faradaic material and a high-power density electric double layer capacitor material (EDLC). HBSC aims to improve device performance by exploiting the strengths of different electrode materials and also to increase the potential window/range over which the devices perform.1–9Transition metal hydroxides (TMH) are of great interest as cathode materials as their variable valence states enable the ability to reversibly store and release electrons during the charge and discharge cycles. Furthermore, the tuneable morphologies and compositions of TMH materials offer the opportunity to optimize the electrochemical properties, such as charge/discharge rate, cycle stability and capacity retention.10–12 Among TMH materials, nickel hydroxide (Ni(OH)2) has been extensively studied due to its high theoretical capacity and low cost.13,14 There are two phases of Ni(OH)2, denoted α and β, respectively. Well-crystallized β-Ni(OH)2 is commercially available and has an interlayer distance of 4.6 Å along the c-axis (which is equal to the c-axis parameter), with no water molecules or anions inserted between the layers. On the other hand, α-Ni(OH)2 is composed of edge-sharing octahedral (M(OH)6) sheets/layers with anions and water molecules between layers, resulting in relatively weak interactions between the layers and larger interlayer spacing, ≥7.5 Å. Moreover, to maintain electrical neutrality, the average oxidation state of Nin+ in alpha would need to be higher than that of beta, owing to the insertion of additional anions. Exploiting the different phases of nickel hydroxide, research has improved electrochemical performance of nickel hydroxide electrodes by modifying the valence states of nickel and inserting various anions in the interlayer space. For example, to enhance the cycle life of the metal hydroxide electrode, Pan et al. incorporated Al3+ into the structure of alpha-nickel hydroxide to depress changes in the crystal phases effecting the microstructure during long cycling.15 Additionally, Baek et al. improved the wettability of the electrode materials in the aqueous electrolyte by intercalation of the hydrophilic carbonate anion CO32− into the nickel hydroxide structure.16 Li et al. modified the interlayer of nickel hydroxide by incorporating sulfate ions, resulting in the improvement of the conductivity.17
This work develops a strategy for the preparation of different phases of nickel hydroxide through the addition of fluoride ions. We have selectively prepared flower-like nanostructured pure α-Ni(OH)2 and pure β-Ni(OH)2, as well as mixtures of phases (labelled αβ-Ni(OH)2) in a controlled ratio. Electrochemical measurement of different nickel hydroxide electrodes shows αβ-Ni(OH)2 exhibits excellent cycle stability, with 108% capacity retention after 2000 continuous charge–discharge cycles at 20 mA cm−1 in alkaline electrolyte, greater than pure phases of either α-Ni(OH)2 or β-Ni(OH)2. The maximum energy density and power density of αβ-Ni(OH)2//AC (AC = activated carbon) hybrid supercapacitors is 32 W h kg−1 at a power density of 900 W kg−1. The structures and properties of different nickel hydroxides were studied and evaluated to provide perspective for the design and synthesis of TMH applied in energy storage.
Experimental
All chemical reagents were commercially purchased from Sigma-Aldrich and Fisher Scientific UK Ltd and used directly without further purification. Nickel foam was purchased from Liyuan Company. Activated carbon (YEC-8A) was purchased from Fuzhou Yihuan Carbon Co. Ltd.Synthesis of nickel hydroxide
Several Ni-foam pieces (1 cm × 3 cm) were successively treated with acetone, 1 M HCl solution, deionized water (DI water) and absolute ethanol, each for 15 min with the aid of ultrasound to obtain a clean surface. Nickel nitrate (Ni(NO3)2·6H2O, 0.8724 g, 3 mmol), urea (CH4N2O, 0.360 g, 6 mmol) and one of the additives (types and amounts added listed in Table S1†) were dissolved in deionized water (60 mL), followed by ultrasonication for 30 min in order to obtain a pale green solution. After complete dissolution, the mixed solution was transferred into a Teflon-lined stainless-steel autoclave (100 mL) which contained a piece of pre-treated nickel foam (3 × 1 cm2). The autoclave was sealed and heated in an oven at 200 °C for 5 h to allow the growth of Ni(OH)2 on the nickel foam. After the autoclave cooled down naturally to room temperature, a light green precipitate was obtained and separated centrifugally. The precipitate was washed twice with deionized water and then ethanol, and then separated centrifugally again. Finally, the coated nickel foam and the precipitate were dried in a vacuum oven at 60 °C overnight, a mass loading of activated materials around 2 mg cm−2 (Table S1†).Electrochemical measurements
The nickel hydroxide grown in situ on Ni foam could be directly used as a working electrode with a square shape of 1 cm2. Electrochemical performance of the electrodes were tested in a three-electrode configuration with KOH (1 M) aqueous solution as the electrolyte. The as-prepared Ni(OH)2 grown on the nickel foam sample, a platinum foil electrode and a Ag/AgCl electrode (3 M KCl) were used as the working electrode, counter electrode and reference electrode, respectively. Galvanostatic charge–discharge (GCD), cyclic voltammetry (CV) measurements and electrochemical impedance spectroscopy were investigated on an Autolab PGSTAT30 electrochemical workstation at ambient temperature.The capacitance (F g−1) can be estimated by integration of the CV curves showing eqn (1),
 | (1) |
The capacity (C g−1) can be calculated by eqn (2) based on the GCD results,
 | (2) |
Hybrid supercapacitor devices (HSD) were constructed in a two-electrode configuration in 1 M KOH solution using nickel hydroxide as a positive electrode and commercial activated carbon (YEC-8A) as a negative electrode. The negative electrode was prepared with YEC-8A, PTFE binder and Super P with a mass ratio of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:10 ground with a small amount of ethanol. The mixed slurry was pressed on the nickel foam (1 cm × 1 cm) under 10 MPa pressure and then dried at 60 °C under a vacuum overnight. The mass loading of a negative electrode is around 12–16 mg cm−1.
:10:10 ground with a small amount of ethanol. The mixed slurry was pressed on the nickel foam (1 cm × 1 cm) under 10 MPa pressure and then dried at 60 °C under a vacuum overnight. The mass loading of a negative electrode is around 12–16 mg cm−1.
To optimise the supercapacitor performance, the two electrodes are required to be in charge balance, which can be achieved using the two equations below:
| Qelectrode = C × m × E | (3) |
In order to satisfy the charge balance Qpositive electrode = Qnegative electrode, an active material mass balance should be as follows:
 | (4) |
The energy density (E, W h kg−1) and power density (P, W kg−1) for the hybrid supercapacitor device were calculated according to the following equations, respectively:
 | (5) |
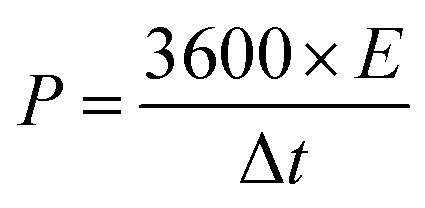 | (6) |
Materials characterization
Powder X-ray diffraction (PXRD) data were collected using a Bruker (D2 PHASER) diffractometer, in reflection geometry, with Cu Kα radiation (λ = 1.5418 Å), a scanning speed of 0.2 steps per second and step size of 0.0365° 2θ over the two-theta range 5 to 60/65° 2θ. Powder samples were prepared as a thin smear on a silicon substrate, or as a piece of active material coated Ni foam located in a deep well sample mount. Fourier transform infrared (FTIR) spectra were collected using a PerkinElmer FTIR spectrometer in attenuated total reflection mode over the range 500 to 4000 cm−1. Raman spectra were recorded on an inVia Renishaw micro-Raman spectrometer with a 514 nm laser as the excitation source from 100 to 3800 cm−1. XPS measurements used a monochromatized (Δε < 300 meV) Scienta 300 X-ray photoelectron spectrometer with X-ray source Al Kα radiation (1486.6 eV). Thermogravimetric analysis (TGA) were carried out using a Netzsch STA 449 F1 thermal analyzer under a N2 flow between room temperature and 600 °C with a heating rate of 5 °C min−1. The morphologies and elemental analysis of the prepared samples were examined by scanning electron microscopy (SEM) with quantitative X-ray spectroscopy (EDX) capabilities with 15 kV accelerating voltage. Nitrogen adsorption and desorption was used to determine Brunauer–Emmett–Teller (BET) surface area and pore size distribution at 77 K using a Micromeritics ASAP 2020 instrument. Transmission Electron Microscopy (TEM) was performed on FEI Titan Themis operated at 200 kV and equipped with a CEOS DCOR probe corrector, a SuperX energy, Gatan Enfinium electron energy loss spectroscopy and a Ceta CMOS camera.X-ray absorption spectroscopy (XAS) measurements were carried out on beamline B18 at the Diamond Light Source (UK), with an electron beam energy of 3.0 GeV and a beam current of 300 mA. The monochromator comprises a Si (111) crystal operating in Quick EXAFS mode. Calibration of the monochromator was achieved by a Ni foil. Combined XANES/EXAFS electrochemical measurements were carried out using in situ custom-designed electrochemical cell18 connected to an OctoStat200 (Ivium). Data analysis was carried out using ATHENA and ARTEMIS software packages.19 To avoid the strong signal from Ni foam, carbon paper was used as the electrode substrate for in situ electrochemical measurements. The preparation of the electrode is the same as the negative electrode of the hybrid supercapacitor device.
Results and discussion
Characterization of materials
As shown in Fig. 1(a), Ni(OH)2 samples were prepared with varying amounts of the additive NH4F. The diffraction peaks of the sample (labelled as “α-Ni(OH)2 no additives”) match with the reference pattern of α-Ni(OH)2 (ICDD-38-715). The diffraction peaks indexed as 003(12.38° 2θ) and 006 (24.83° 2θ) of the α-Ni(OH)2 sample are shifted to a higher 2θ angle, when compared to that of the reference pattern (ICDD_38-715), indicating a contraction in c parameter (Fig. 1). This shift is likely due to differences in the incorporated interlayer species. The two samples labelled αβ_1-Ni(OH)2 and αβ_2-Ni(OH)2 in Fig. 1(a), synthesized using the molar ratios of NH4F to the nickel-salt reagent of 0.2:2 (αβ_1-Ni(OH)2) and 0.5:2 (αβ_2-Ni(OH)2), respectively, show the presence of both alpha and beta Ni(OH)2 type phases. As the molar ratio of the additive NH4F to the nickel-salt reagent increased, the phase(s) of the nickel hydroxide formed contains more β-Ni(OH)2 component. The observed diffraction peaks of the β-Ni(OH)2 sample match well with the reference β-Ni(OH)2 pattern (ICDD-14-117). The PXRD patterns of nickel hydroxide samples in Fig. 1(a) were indexed and refined using the Pawley method20 and the refined unit cell parameters are presented in Table S2.† For the mixed αβ samples in Fig. 1(b), the refined c parameter associated with alpha component is larger than that refined for the pure α-Ni(OH)2 sample. (c = 21.41(2) Å for α-Ni(OH)2; c = 22.051(4) Å for αβ_1-Ni(OH)2; c = 22.065(8) Å for αβ_2-Ni(OH)2).
 | ||
| Fig. 1 (a) PXRD patterns of nickel hydroxide powder prepared with additive NH4F. (b) Expanded 2 theta region of PXRD patterns highlighting the 003 and 006 reflections of the alpha phase for α-Ni(OH)2, αβ_1-Ni(OH)2, αβ_2-Ni(OH)2. | ||
Additionally, the PXRD data collected on samples grown directly on nickel foam are shown in Fig. S1.† Two intense diffraction peaks observed at 44.5° and 51.8° 2θ are due to the nickel foam substrate (ICDD-4-850). As the diffraction peaks from nickel foam are strong and dominate the diffraction pattern, the peaks associated with the nickel hydroxide phase(s) are not as distinct. However, characteristic diffraction peaks for α-Ni(OH)2 (003) and β-Ni(OH)2 (001), (100), (101) can still be observed, which are assigned to the standard ICDD patterns for α-Ni(OH)2 (ICDD-38-715) and β-Ni(OH)2 (ICDD-14-117), respectively, confirming the targeted phases form during the synthesis directly onto the Ni foam. Thus, the Ni(OH)2@Ni foam can be employed directly as a working electrode in supercapacitors without the addition of any extra binders or conductive agents.
The morphology of different phases of nickel hydroxide was characterized by scanning electron microscopy (SEM). As shown in Fig. S2,† the flower-like spheres of nickel hydroxide samples, which are composed of self-assembled nanosheets with an average thickness of 1–2 nm, have open and porous three-dimensional structures. For further insight into the structural morphology of nickel hydroxide, transmission electron microscopy (TEM) was used (Fig. S3†). The TEM images further confirm their nanoflower structures with ultrathin nanosheets. Elemental mapping was carried out and the images are presented in Fig. S3,† revealing the element distribution of different phases of nickel hydroxide, which are consistence with SEM-elemental mapping results in Fig. S2.† It can be clearly observed that Ni (represented by green colour), O (blue colour) and F (orange colour) were detected and uniformly distributed throughout the phases of αβ_1-Ni(OH)2, αβ_2-Ni(OH)2, and β-Ni(OH)2. The F:Ni ratios of αβ_1-Ni(OH)2, αβ_2-Ni(OH)2, and β-Ni(OH)2 were determined to 0.3:2, 0.6:2, and 0.74:2 from EDS, as illustrated in Table S3.† Therefore, these results further confirm the inclusion of F in the materials.
The high-resolution TEM (HRTEM) images and related selected area electron diffraction (SAED) patterns of different nickel hydroxide samples are shown in Fig. 2. The SAED of α-Ni(OH)2 in Fig. 2(a) displays diffuse rings revealing its polycrystalline nature. However, β-Ni(OH)2 in Fig. 2(c) exhibits diffraction spots, highlighting the crystallite analysed is single crystal in nature and has symmetries consistent with trigonal symmetries, as shown by β-Ni(OH)2 (space group P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m1). The adjacent lattice fringes of β-Ni(OH)2 sample in Fig. 2(d) are approximately 0.15 nm and 0.25 nm, corresponding to (110) and (−110) lattice planes of nickel hydroxide, respectively.21 The corresponding fast Fourier transform (FFT) pattern in Fig. S4† further confirms the existence of (100), (010), (110), and (−110) lattice planes. For mixed phases of nickel hydroxide (αβ_1-Ni(OH)2 and αβ_2-Ni(OH)2) in Fig. 2(e) and (g), diffraction rings and spots can both be observed implying that beta and alpha phases are intimately connected in the mixed phases samples. The diffraction rings are more pronounced in the sample of αβ_1-Ni(OH)2, while the diffraction spots are clearer in the sample of αβ_2-Ni(OH)2. The SAED results of mixed phases are in good agreement with their PXRD results, where alpha is the dominant phase of αβ_1-Ni(OH)2 whereas beta is the main phase of αβ_2-Ni(OH)2.
m1). The adjacent lattice fringes of β-Ni(OH)2 sample in Fig. 2(d) are approximately 0.15 nm and 0.25 nm, corresponding to (110) and (−110) lattice planes of nickel hydroxide, respectively.21 The corresponding fast Fourier transform (FFT) pattern in Fig. S4† further confirms the existence of (100), (010), (110), and (−110) lattice planes. For mixed phases of nickel hydroxide (αβ_1-Ni(OH)2 and αβ_2-Ni(OH)2) in Fig. 2(e) and (g), diffraction rings and spots can both be observed implying that beta and alpha phases are intimately connected in the mixed phases samples. The diffraction rings are more pronounced in the sample of αβ_1-Ni(OH)2, while the diffraction spots are clearer in the sample of αβ_2-Ni(OH)2. The SAED results of mixed phases are in good agreement with their PXRD results, where alpha is the dominant phase of αβ_1-Ni(OH)2 whereas beta is the main phase of αβ_2-Ni(OH)2.
 | ||
| Fig. 2 (a and b) HRTEM Images of α-Ni(OH)2 with SAED pattern. (c and d) HRTEM images of β-Ni(OH)2 with SAED pattern. (e and f) HRTEM images of αβ_1-Ni(OH)2 with SAED pattern. (g and h) HRTEM images of αβ_2-Ni(OH)2 with SAED pattern. | ||
The specific surface area of the samples was confirmed by nitrogen adsorption–desorption measurements. The isotherms for all samples (α-Ni(OH)2, αβ_1-Ni(OH)2 and αβ_2-Ni(OH)2 and β-Ni(OH)2) are illustrated in Fig. 3(a) and their BET surface areas are calculated to be 185 m2 g−1, 135 m2 g−1, 112 m2 g−1 and 42 m2 g−1, respectively, based on Brunauer–Emmett–Teller surface analysis.22 The high surface area of the α-Ni(OH)2 sample can provide many active sites for redox reactions during charge–discharge processes which may improve the performance as a supercapacitor.23 A hysteresis loop, over a relative pressure range of ∼0.45–0.9, was observed (Fig. 3(a)), suggesting the presence of pores within nanosheets. It is noted that pore structures can reduce resistance and efficiently facilitate the migration of electrolyte ions during the charging–discharge process.24 The Barrett–Joyner–Halenda (BJH) model was used to calculate the pore size distribution.25 The pore size distribution of the samples is shown in Fig. 3(b) based on the calculation of the BJH method.25 The pore size distribution of α-Ni(OH)2 (red curve in Fig. 3(b)) and β-Ni(OH)2 (green curves) are relatively even with average pore diameters of about 2.1 nm and 1.8 nm respectively. However, the average pore diameters for αβ_1-Ni(OH)2 and αβ_2-Ni(OH)2 samples both are about 4.1 nm in Fig. 3(b).
 | ||
| Fig. 3 (a) Nitrogen adsorption–desorption isotherm of different phases of nickel hydroxide (b) pore size distribution based on the BJH model of different phases of nickel hydroxide. | ||
Fourier transform infrared (FT-IR) spectroscopy was carried out over the range 500 to 4000 cm−1 to confirm the presence of functional groups in the samples. As shown in Fig. 4(a), the broad and weak band centred around 3643 cm−1 from the α-Ni(OH)2 sample can be ascribed to the stretching vibration of the O–H group in nickel hydroxide and intercalated water molecules present in the interlayer space of the turbostratic structure.13 However, the intense and sharp band from the β-Ni(OH)2 sample is situated at 3626 cm−1, which is related to the O–H stretching of the hydroxide anions.21 The adsorption located at 1633 cm−1 and 1600 cm−1 in the α-Ni(OH)2 sample suggests an amount of free H2O attributed to incorporation of water molecules, which is much strong than the β-Ni(OH)2 sample. In addition, the appearance of the signal observed at 2213 cm−1 is assigned to the vibrational mode of the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N triple bond in the OCN− ions, which can be a side product of urea hydrolysis.26 In the α-Ni(OH)2 sample, two types of nitrate symmetries, namely C2v and D3h, have been observed. The presence of peaks at 1053 cm−1, 1280 cm−1 and 1500 cm−1 indicate the existence of nitrate in a C2v symmetry. The peaks at 835 cm−1 and 1370 cm−1 suggest the presence of unbound nitrate anion in a D3h symmetry.27 In the β-Ni(OH)2 sample, peaks associated with unbound nitrate ions are also observed, which may be attributed to a unique feature of layer stacking faulted beta phase, and(or) to the presence of surface nitrate in a small quantities.28 The assignments of the observed bands of FT-IR spectra are listed in Table S4.†
N triple bond in the OCN− ions, which can be a side product of urea hydrolysis.26 In the α-Ni(OH)2 sample, two types of nitrate symmetries, namely C2v and D3h, have been observed. The presence of peaks at 1053 cm−1, 1280 cm−1 and 1500 cm−1 indicate the existence of nitrate in a C2v symmetry. The peaks at 835 cm−1 and 1370 cm−1 suggest the presence of unbound nitrate anion in a D3h symmetry.27 In the β-Ni(OH)2 sample, peaks associated with unbound nitrate ions are also observed, which may be attributed to a unique feature of layer stacking faulted beta phase, and(or) to the presence of surface nitrate in a small quantities.28 The assignments of the observed bands of FT-IR spectra are listed in Table S4.†
 | ||
| Fig. 4 (a) FT-IR spectra of different phases of nickel hydroxide (b) Raman spectra of different phases of nickel hydroxide. | ||
Raman spectroscopy was also carried out to complement the results of the FT-IR analysis and the results are shown in Fig. 4(b). The broad peak due to O–H stretches is observed at 3644 cm−1 of the α-Ni(OH)2 sample and the peak at 464 cm−1 relates to the lattice mode (δ Ni–O). The β-Ni(OH)2 sample is identified by the sharp peak at 3571 cm−1 corresponding to the hydroxide bending mode (v O–H). The mixed-phase samples (blue line and yellow line in Fig. 4(b)) show the main characteristics of both α-Ni(OH)2 and β-Ni(OH)2. The nitrate peak at 1049 cm−1 of the D3h symmetry is observed in both the α-Ni(OH)2 sample and the β-Ni(OH)2 sample but there are also additional bands of the C2v symmetry of nitrate (1291 cm−1 and 1356 cm−1) presented in the α-Ni(OH)2 sample, which supports the findings of the FT-IR results. The assignments of the observed bands of Raman spectroscopy are listed in Table S5.† The results from analysis of the FT-IR and Raman spectra suggest the presence of the interlayer anions and water molecules in the alpha phases, which would require a higher average oxidation state of Nin+ in the alpha phase and mix α/β phases of nickel hydroxide to ensure charge neutrality. On the other hand, beta phases of nickel hydroxide should not have any interlayer species, but they can form disordered structures and result in sorbed water.28
Thermogravimetric analysis (TGA) was carried out for each of the nickel hydroxide samples and a commercial nickel hydroxide (Sigma-Aldrich) to determine decomposition processes during the heat treatment. The weight losses of the nickel hydroxide samples are given in Table S6.† As shown in Fig. 5, the first stage of weight loss between 25 °C and 250 °C is related to the removal of the surface water and structurally bonded water intercalated between the layers of nickel hydroxide. The TGA result for the α-Ni(OH)2 sample shows a relatively sharp weight loss of 12 wt% in the first stage such that intercalated water molecules were removed. Regarding β-Ni(OH)2, the mass change is 2 wt% in the first stage of weight loss. This proves the amount of absorbed or intercalated water molecules and anions contained in the β-Ni(OH)2 samples is much lower than in the α-Ni(OH)2 sample. The weight loss values for the αβ_1-Ni(OH)2 and αβ_2-Ni(OH)2 are in the middle between that of the α-Ni(OH)2 sample and the β-Ni(OH)2 sample (10 wt% and 5 wt%, respectively). The second stage of weight loss occurs above 250 °C, where all samples decompose to form NiO. This was confirmed through analysis of PXRD data collected on the residues of all samples (Fig. S6†) and showed no Ni(OH)2 remained. Furthermore, the NiO residues of α-Ni(OH)2 and β-Ni(OH)2 samples were analyzed by SEM-EDS, as shown in Fig. S7 and S8.† Traces of F are still observed in the NiO residues of β-Ni(OH)2 sample. Table S7† displays the atomic percentage of O, Ni, F in the NiO residue. The decomposition reaction of nickel hydroxide is shown in eqn (7).
| Ni(OH)2 → NiO(s) + H2O↑(g) | (7) |
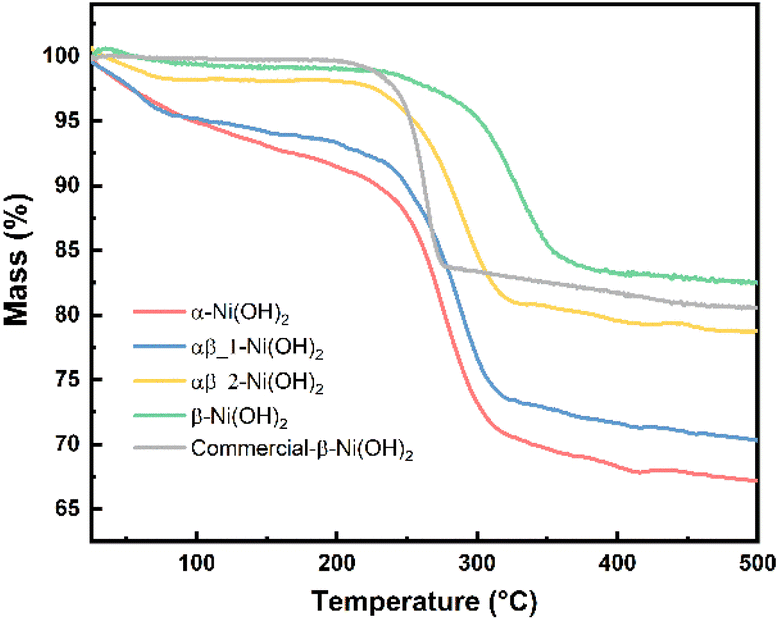 | ||
| Fig. 5 TGA curves of different phases of nickel hydroxide. | ||
Based on eqn (7), the theoretical weight loss after the heating process is 19.42% (the remaining weight-80.52%). However, the remaining weight of α-Ni(OH)2, αβ_1-Ni(OH)2 and αβ_2-Ni(OH)2 samples is 67 wt%, 70 wt% and 78 wt%, respectively. This extra weight loss is likely due to the presence of water molecular and anions intercalated between the layers of the Ni(OH)2-type phases. These results are in agreement with the FT-IR and Raman spectra, which confirmed the presence of anions in various samples. Furthermore, the temperature of phase transformation or decomposition (Ni(OH)2 → NiO) shifts to higher temperature with increased amount of F− added, indicating an enhanced stability of the structure.
All the samples were studied by X-ray photoelectron spectroscopy (XPS) to understand the impact of the fluoride ion addition. The full XPS spectra in Fig. 6(a) show the presence of Ni 2s, Ni 2p, F 1s, and O 1s peaks on the surface of different samples of nickel hydroxide. The Ni 2p3/2 peak of α-Ni(OH)2 in Fig. 6(b) at 856.2 eV is shifted to a higher binding energy region than beta nickel hydroxide (∼855.8 eV) in the ref. 29–33. This is suggested to be due to the average oxidation state of Nin+ in the alpha phase being higher than that of beta-nickel(II) hydroxide in the reference. This is in line with our suggestion that the insertion of anions in the structure of the α-Ni(OH)2 sample, as confirmed by the Raman and FTIR results, requires a higher valence state of nickel to maintain charge neutrality. Fig. S10-1† compares the binding energy peak of Ni 2p3/2 for different nickel hydroxide samples in this work and beta nickel hydroxide found in the literature.29–33 The Ni 2p3/2 peak for our αβ_1-Ni(OH)2, αβ_2-Ni(OH)2 and β-Ni(OH)2 samples (Fig. S10-1†) is positively shifted by 0.3–0.4 eV, when compared to published values, due to the addition of fluorine. Unlike for α-Ni(OH)2, there should be minimal Ni(III) present in the case of β-Ni(OH)2. The observed shift in our sample could be attributed to fluorine replacing some oxygen sites to form Ni–F bonds, in which the Ni ions have higher binding energy than for Ni–O, leading to the positive shift in the spectroscopy.34–36 This is supported by the EDS result in Table S7,† showing traces of F still present in NiO residues of β-Ni(OH)2 sample after TGA. Fig. S9† compares electron energy loss spectroscopy (EELS) of different nickel hydroxide phases at the oxygen K edge, fluorine K edge and Ni L edge to support the XPS results. The fine structure and energy position of the O-K and Ni-L edges give insight into the electronic structure and interactions of the doping element (fluorine) of different phases of nickel hydroxides. Peaks (v) and (vi) of Ni L edge at around ∼853 eV and 872 eV correspond to the energy loss caused by the transitions of electrons of nickel from p1/2(L2) and 2p3/2(L3) states to empty 3d states.37 As illustrated in Fig. S9,† different fine structures of the O-K edge are observed between different phases of nickel hydroxides. The first pre-edge feature of oxygen at around 527 eV is not observed in the spectrum of the α-Ni(OH)2 sample suggesting a different electronic environment of oxygen for α-Ni(OH)2 than the nickel hydroxides with some fluorine present. The broad bump (indicated with the dashed line, (ii)) at the energy loss of 532 eV can be attributed to the electronic transition from the 1s core state to a hybridized state of O 2p bond with Ni 3d in the LDH cationic layer.38 The decrease in intensity of the bump (ii) from the alpha phase to the beta phase reveals fewer holes in 2p–3d orbitals suggesting the lower valence state of nickel in beta samples.
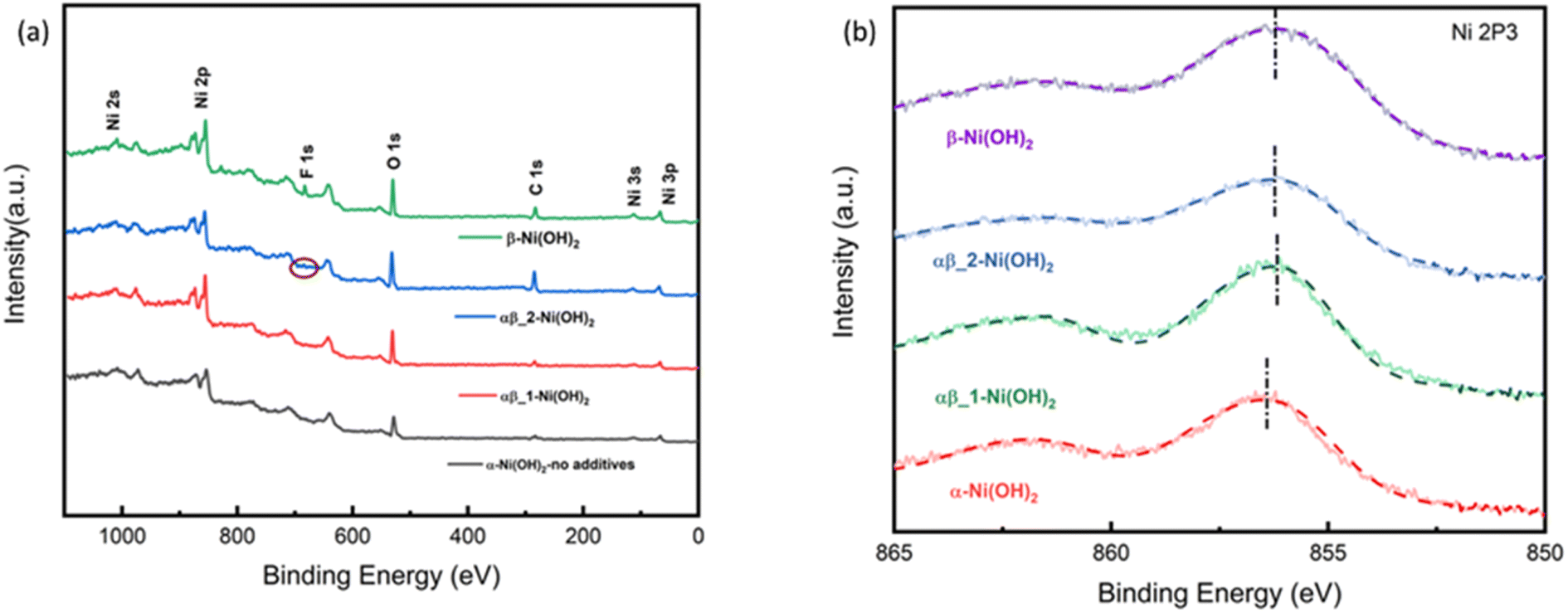 | ||
| Fig. 6 (a) XPS survey spectrum of nickel hydroxide (b) Ni 2p XPS spectra of nickel hydroxide of different phases. | ||
The impact on nickel hydroxide of the addition of fluoride
To investigate the key factor in synthetic phase formation, a series of experiments were conducted using different additions of NaF and NH4Cl to determine the role of NH4F in terms of cations or anions. Ni(OH)2 samples were prepared with varying amounts of an additive, either NH4F, NaF or NH4Cl (Table S1†). All samples were characterized by PXRD to identify the phase(s) of nickel hydroxide present. The PXRD data presented in Fig. S11† show the phases identified as present for samples prepared using NaF as the fluoride source, which also shows that as the F− amount increases, the phases formed are more beta-like in nature. However, with the addition of NH4Cl, the phases identified as present in these samples were all found to be α-Ni(OH)2 (Fig. S11†), regardless of the NH4Cl–Ni salt reagent ratios. Thus, F− ion is determined as the phase-controlling agent in these experiments for the synthesis of different nickel hydroxide-type phases.Table S2† displays the refined unit cell parameters to investigate the impact of fluoride addition on as-prepared nickel hydroxides. As mentioned in Fig. 1(b), the mixed αβ samples exhibit a larger refined c parameter associated with the alpha component compared to the pure α-Ni(OH)2 sample. Furthermore, the results from FTIR and Raman reveal the nitrate species as present in the interlayer of alpha-nickel hydroxide, caused by the higher average oxidation state of nickel in alpha-nickel hydroxide, which gives the charge neutrality. Based on these results, the formation of nickel hydroxide phases may occur via two mechanisms: the inclusion of fluoride may disfavor incorporation of other anions and water molecules, leading to β-Ni(OH)2. Additionally, the high electronegativity of fluorine may discourage nickel oxidation to Ni3+ and therefore the requirement of extra anions to incorporate to achieve charge balance. Additionally, TGA results exhibit that the decomposition temperature for the different samples increases with the increased amount of F present in the nickel hydroxide, suggesting improved stability of the structure with the addition of fluorine.
Electrochemical performance of nickel hydroxide electrodes
Electrodes of different nickel hydroxide samples were measured in a three-electrode configuration in 1 M KOH as the electrolyte. Fig. 7(a) exhibits CV curves of the α-Ni(OH)2, αβ_1-Ni(OH)2, αβ_2-Ni(OH)2, β-Ni(OH)2 with the scan rate of 5 mV s−1 in the potential range of 0–0.55 V vs. Ag/AgCl. The mechanism for the charge–discharge process is related to the diffusion of protons, corresponding to a reversible reaction in eqn (8):| Nin+OOH + H+ + (n − 2)e− = Ni(OH)2(3 < n < 4) | (8) |
 | ||
| Fig. 7 (a) CV curves at 5 mV s−1. (b) GCD curves at 3 A g−1. (c) Cycling performance for different nickel hydroxide samples in the three-electrode configuration. | ||
As shown in Fig. 7(a), a pair of faradaic redox peaks is observed for all nickel hydroxide samples, showing a separation of around 0.2–0.3 V between oxidation and reduction peaks. It can be observed that the oxidation peak in the β-Ni(OH)2 sample shifts to a higher potential than the α-Ni(OH)2 sample. This suggests that Ni in the beta phase is harder to oxidize than that in the alpha phase, which is consistent with our suggestion that the nickel is less electron-rich with additional fluorine. The capacitance of different nickel hydroxide electrodes was calculated by eqn (1) in Experimental and can be found in Table S9.†Fig. 7(b) shows galvanostatic charge–discharge (GCD) curves of different nickel hydroxide samples at 3 A g−1. The GCD profiles of α-Ni(OH)2 display remarkable specific capacities of 2223 C g−1, 1970 C g−1, 1710 C g−1, 1531 C g−1, 1400 C g−1 and 936 C g−1 with increasing current densities from 3 A g−1 to 20 A g−1. The excellent capacity of the α-Ni(OH)2 electrode might be caused by its large surface area, the largest of the four materials, providing more active sites for the electrochemical reaction. At the same time, the larger interlayer spacing of the α-Ni(OH)2 sample will include water molecules and anions that facilitate the penetration of the electrolyte and provide faster reaction kinetics, as well as the fast replenishment of OH− for electrochemical reactions. Fig. 7(c) shows the cycling performance for different nickel hydroxide samples in the three-electrode configuration. The α-Ni(OH)2 electrode only keeps 50% capacitance retention after 2000 cycles. The poor cycle stability may be caused by the inserted foreign ions and water molecules, which will lead to structural collapse in the alkaline electrolyte during continuous charge–discharge processes. However, the αβ_1-Ni(OH)2 electrode offers excellent cycle stability with 108% after 2000 cycles. This could be attributed to the presence of beta phase facilitating the overall stability of the αβ_1-Ni(OH)2 electrode.39–41 Therefore, the moderate amount of fluorine addition in the structure of nickel hydroxide can modify the interlayer spacing of nickel hydroxide to benefit the OH– insertion/extraction and improve the stability of the electrode. Furthermore, Fig. S1† present the PXRD data for all nickel hydroxide electrodes before and after undergoing cycling test.
Electrochemical impedance spectroscopy (EIS) in the three-electrode configuration was used to characterize the electrochemical performance of nickel hydroxide electrodes in the frequency range of 10 kHz to 0.01 Hz. The results were fitted to an equivalent circuit as shown in an inset of Fig. 8(a). In an equivalent circuit, R1 is the internal resistance, R2 is the charge-transfer resistance, CPE1 is a constant phase element to approximate the double-layer capacitance, CPE2 models the low-frequency mass capacitance, Wo is the Warburg element and R leak is the low-frequency leakage resistance.42,43 The sum of R1 and R2 is equivalent series resistance, ESR, which is one of the key factors in determining the power density of a supercapacitor. R1 of β-Ni(OH)2 at 0.87 Ω is the smallest of all samples, suggesting its low intrinsic electronic resistance and excellent conductivity. This suggests that the addition of fluorine in nickel hydroxide can benefit its conductivity. In contrast, R2 of β-Ni(OH)2 is the largest (1.73 Ω) while that of α-Ni(OH)2 exhibits 0.76 Ω indicating anions and water molecules in the structures improve the charge transfer during the electrochemical cycling process. It is noted that the ESR of αβ_1-Ni(OH)2 is 1.93 Ω presenting the synergistic effect, i.e. this mixed phase is better than pure phases (α-Ni(OH)2-2.06 Ω and β-Ni(OH)2-2.60 Ω). Electrochemical parameters associated with the equivalent circuit are summarized in Table S8.†
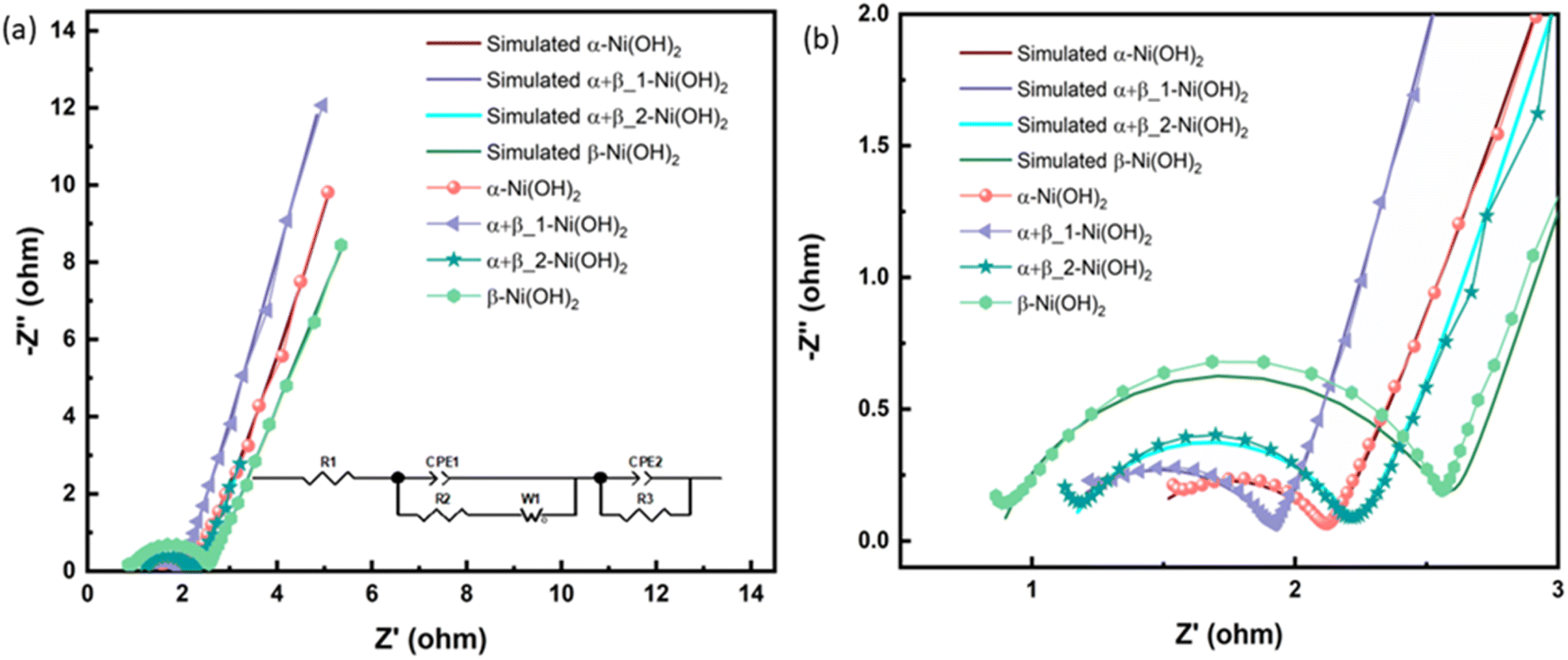 | ||
| Fig. 8 (a) Nyquist plots, inset show the corresponding equivalent circuit of different electrodes of nickel hydroxide. (b) The enlarged Nyquist plots. | ||
In situ X-ray absorption spectroscopy (XAS)
In situ XAS measurements were employed to study the chemical reduction and oxidation process of nickel hydroxide electrodes, using a purpose-made electrochemical cell based on the design published.18Fig. 9 presents the Ni K-edge X-ray absorption near-edge structure (XANES) for the four different samples, α-Ni(OH)2, αβ_1-Ni(OH)2, αβ_2-Ni(OH)2 and β-Ni(OH)2, during both oxidation and reduction scans. It can be observed that with increasing potential, the absorption edge position of the four samples undergoes a positive edge shift, indicating the oxidation of the Ni atoms. Based on previous studies,13,44 the divalent polymorphs of α-Ni(OH)2 and β-Ni(OH)2 are expected to oxidize to their respective forms with different average oxidation states, γ-NiOOH and β-NiOOH. Therefore, the positive shifts to different extents for the four samples may be attributed to the presence of two reversible couples (α-Ni(OH)2/γ-NiOOH and β-Ni(OH)2/β-NiOOH). To provide the comparison of the difference of structural changes during electrochemical processes undergone by Ni in all samples, XANES data for four samples are shown in Fig. S15† under consistent conditions of the open-circuit potential (OCP) and 0.6 V vs. Ag/AgCl (oxidation scan directions). The adsorption edge in the four samples remains relatively the same position under the OCP in Fig. S15(a),† which is in line with our XPS results. However, when the potential is increased to 0.6 V, the absorption edge position in α-Ni(OH)2 and αβ_1-Ni(OH)2 samples moves to higher energy in comparison to αβ_2-Ni(OH)2 and β-Ni(OH)2 samples. This suggests more electron transfer occurs in the oxidation process from the α-Ni(OH)2 and αβ_1-Ni(OH)2 electrodes, resulting in higher capacities provided by α-Ni(OH)2 and αβ_1-Ni(OH)2 than the other two electrodes. | ||
| Fig. 9 In situ Ni K-edge XANES spectra of (a) α-Ni(OH)2, (b) αβ_1-Ni(OH)2, (c) αβ_2-Ni(OH)2 and (d) β-Ni(OH)2 under a oxidation and reduction scans. | ||
Furthermore, Fig. 9(d) exhibits negligible energy edge shifts in the β-Ni(OH)2 electrode throughout oxidation and reduction scans, whereas notable changes are observed in the α-Ni(OH)2 electrode (Fig. 9(a)). This observation suggests that the electrochemical reaction of the β-Ni(OH)2 electrode may involve a smaller proportion of the material being oxidized, for example only at the surface of the electrode. In comparison, that of the α-Ni(OH)2 electrode takes place within the volumetric bulk phase. These different shifts of the energy edges observed between the β-Ni(OH)2 and α-Ni(OH)2 electrodes might be attributed to their different crystal structures. The α-Ni(OH)2 sample displays a more open three-dimensional structure with the insertion/presence of anions and water molecules in its layers. This benefits the mobility and replenishment of protons in the redox reaction (eqn (8)) throughout the bulk of the electrode. In contrast, the compact structure of the β-Ni(OH)2 electrode hampers the availability of sufficient paths for proton hopping, blocking the migrations of protons/electrons between the surface and the bulk of the electrode. This may result in only the surface charges participating in the electrochemical reactions while hindering the involvement of the bulk phase. As revealed by the Ni K-edge XANES spectra in Fig. 9(b) and (c) for the mix phases, αβ_1-Ni(OH)2 shows a notable energy edge shift, rendering it more like α-Ni(OH)2. Conversely, αβ_2-Ni(OH)2 exhibits a lack of obvious shift and displays a greater resemblance to β-Ni(OH)2. The PXRD characterization also supports that alpha is the dominant phase in αβ_1-Ni(OH)2 whereas beta is the dominant phase in αβ_2-Ni(OH)2, consistent with the XANES results.
The Fourier transforms of the extended X-ray absorption fine structure (FT-EXAFS) at the Ni K-edge of four different samples are presented in Fig. S15(c) and (d),† corresponding to the OCP and 0.6 V vs. Ag/AgCl, respectively. The FT-EXAFS spectrum in Fig. S15(c)† shows scattering peaks at ∼1.5 Å and 2.6 Å, corresponding to the Ni–O distance and Ni–Ni distance, respectively. Upon an increase in the potential to 0.6 V, significant structural changes are observed in α-Ni(OH)2 and αβ_1-Ni(OH)2 samples in Fig. 10(a) and (b). This change can be attributed to the transformation of α-Ni(OH)2 to γ-NiOOH, accompanied by a reduction in the distance between the first Ni–O shell and the second Ni–Ni shell, as evidenced by the corresponding grey line that aligns with the NiOOH reference data (Fig. S15(d)†).45 However, the structure of the β-Ni(OH)2 electrode in FT-EXAFS exhibits slight changes throughout the whole redox scans (Fig. 10(d)). This suggests that negligible phase changes happen in the bulk of the β-Ni(OH)2 electrode during oxidation and reduction scans, which also supports the suggestion that protons/electrons only release from the surface of the β-Ni(OH)2 electrode based on XANES results (Fig. 9(d)). To further study the reversibility of nickel hydroxide electrodes, Fig. 10(a)–(d) of FT-EXANES analysis compared their structural changes of electrodes before the oxidation process and after a whole redox process. The result confirms that the variations in the bond lengths can be reversible upon subsequent reduction process in the β-Ni(OH)2 electrode. Conversely, the bond lengths of the α-Ni(OH)2 electrode are partly recovered to their original values. These findings also suggest that the β-Ni(OH)2 electrode exhibits better electrochemical cycle stability, while the α-Ni(OH)2 electrode suffers irreversible structural effect after the redox reaction.
 | ||
| Fig. 10 The FT signal of the κ2-weighted EXAFS (a) α-Ni(OH)2, (b) αβ_1-Ni(OH)2, (c) αβ_2-Ni(OH)2 and (d) β-Ni(OH)2 under a oxidation and reduction scans. | ||
In light of the above characterization results in this work, the addition of fluorine plays an important role in controlling phases formed, from the open-structural α-Ni(OH)2 phase to compact β-Ni(OH)2, leading to their different electrochemical performance. Combined with the in situ XAS results, it is suggested that electrochemical redox reactions occur at the surface of the β-Ni(OH)2 electrode with compact structures, exhibiting pseudocapacitors-like performance. This surface-controlled mechanism contributes to improved cycling stability. In contrast, the α-Ni(OH)2 electrode with a more open structure predominately relies on the faradaic reaction (eqn (8)) taking place within the bulk of the electrode, via more channels and active sites for electron/proton transfer, contributing to the high capacity of the electrode, and presenting battery-like performance characteristics. Considering the individual performance of the pure phases of alpha and beta, it suggests that the αβ_1-Ni(OH)2 sample, comprising a mixture of α and β, exhibits a synergistic effect on its electrochemical performance, as shown in Fig. 7. The αβ_1-Ni(OH)2 sample presents the merits of the combination of the excellent cycle stability inherited from the beta phase (Fig. 7(c)), along with the high capacity retained from the alpha phase (Fig. 7(b)).
Hybrid supercapacitor performance
To further evaluate the practical application of nickel hydroxide, we used α-Ni(OH)2, αβ_1-Ni(OH)2 and β-Ni(OH)2 as the positive electrodes, respectively and activated carbon as the negative electrode to assemble hybrid supercapacitors. Fig. S17† displays the CV curves of nickel hydroxide//AC in the range of 0–1.8 V at scan rates of 10–100 mV s−1. According to the typical GCD curves at different current densities, the specific capacitance was calculated in Table S9.† The CV curves of αβ_1-Ni(OH)2//AC at different scan rates present similar shapes in Fig. S13(c).† A high capacitance of 71 F g−1 can be achieved at energy density of 1 A g−1. Fig. 11(b) shows that αβ_1-Ni(OH)2//AC retained 86% of its initial specific capacitance after 5000 cycles. In the initial 1000 continue charge and discharge process, the capacitance improves because of the increased exposed active sites caused by the activation of electrodes. As shown in Fig. S19,† Nyquist plots compare the different performance before and after 5000 cycles of devices in the frequency range of 100 kHz–0.01 Hz. Nearly identical Nyquist plots are observed after 5000 cycles for β-Ni(OH)2 while large changes occur in the devices of α-Ni(OH)2 and αβ_1-Ni(OH)2, respectively. Additionally, a straight and nearly vertical line at the low-frequency range of the β-Ni(OH)2 device demonstrates excellent capacitive behaviour without diffusion limitation. The Ragone plot in Fig. 12 exhibits the relationship between the power density and energy density of electrode materials. The power densities and energy densities of different nickel hydroxide hybrid supercapacitors in Table S9† are calculated using eqn (5) and (6) in Experimental section. The maximum energy density of the αβ_1-Ni(OH)2//AC device is 32 W h kg−1 at a power density of 900 W kg−1. The assembled αβ_1-Ni(OH)2//AC hybrid supercapacitors show a high power density and good cycle stability. Thus, αβ_1-Ni(OH)2 is a promising material for energy storage.46–51 | ||
| Fig. 11 (a) The specific capacitance evolution under various current densities of αβ_1-Ni(OH)2//AC. (b) Cycling performance at 100 mA cm−1 for 5000 cycles. | ||
 | ||
| Fig. 12 Ragone plot of Ni(OH)2//AC hybrid supercapacitors.46–51 | ||
Conclusions
In summary, we present a novel, one-step method to synthesize flower-like nanostructured α-Ni(OH)2, β-Ni(OH)2, as well as mixtures of phases (αβ-Ni(OH)2). The PXRD results show that fluoride, in the addition of NH4F is a phase directing agent, controlling which phase(s) form. The high electronegativity of fluorine could disfavor the oxidation to Ni3+, the presence of which favors the formation of the β-Ni(OH)2 phase, avoiding the introduction of extra anions to interlayers. Moreover, the addition of fluoride in the structure may give a stronger interaction between octahedral layers enhancing the stability of nickel hydroxide. Comparison of electrochemical performances and morphologies of the different nickel hydroxides, shows that the αβ_1-Ni(OH)2 electrode is an excellent electrode candidate for hybrid-supercapacitors applications. The cycle stability of αβ_1-Ni(OH)2 electrode exhibits remarkable capacity retention of 108% after 2000 continuous charge–discharge cycles at 20 mA cm−1 in alkaline electrolyte. This may be attributed to the beta component in the mixed phase improving its cycle stability. The maximum energy density of αβ-Ni(OH)2//AC hydride supercapacitors is 32 W h kg−1 at a power density of 900 W kg−1. Thus, the moderate amount of additional fluoride in the nickel hydroxide, that modifies the interlayer spacing and the valence state of the nickel element, leads to the αβ-Ni(OH)2 sample inheriting the higher capacity of alpha nickel hydroxide and also keeping the excellent cycling stability due to the more stable framework of β-Ni(OH)2.Author contributions
XY designed, carried out and analysed most of the practical work and drafted the manuscript. VC carried out X-ray Absorption Spectroscopy (XAS) and its data analysis. HZ carried out a range of preliminary experiments and electrochemical performance of the work. NR and CK directly supervised this work. All authors inputted into manuscript preparation and results interpretation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to acknowledge the Diamond Light Source for the provision of beamtime (SP28636). We thank Dr Bingyu Lei and Matilda Rhodes for help with measurement of in situ XAS. We thank Dr Nicola Cayzer for help with the SEM images acquisition. We thank Dr Andrey Gromov for helping to perform Raman measurements. We thank Dr Rebecca Rae for help with TGA and PXRD measurements. We thank Dr John Tobin and Amin Md Khairul for help with nitrogen adsorption and desorption measurements. We thank Dr Aaron Naden for performing the TEM measurements in the University of St. Andrews. We thank Dr Stephen Francis in the University of St. Andrews for help in performing XPS measurements.Notes and references
- A. Borenstein, O. Hanna, R. Attias, S. Luski, T. Brousse and D. Aurbach, J. Mater. Chem. A, 2017, 5, 12653–12672 RSC.
- Y. Shao, M. F. El-Kady, J. Sun, Y. Li, Q. Zhang, M. Zhu, H. Wang, B. Dunn and R. B. Kaner, Chem. Rev., 2018, 118, 9233–9280 CrossRef CAS PubMed.
- C. Ye, A. Wang, C. Breakwell, R. Tan, C. Grazia Bezzu, E. Hunter-Sellars, D. R. Williams, N. P. Brandon, P. A. A. Klusener, A. R. Kucernak, K. E. Jelfs, N. B. McKeown and Q. Song, Nat. Commun., 2022, 13, 3184 CrossRef CAS PubMed.
- H. Lei, Y. Pan, R.-G. Guo, J. Zhang, X. Han, L.-M. Yu and W.-Q. Jiang, Rare Met., 2022, 41, 1977–1982 CrossRef CAS.
- D. Shao, Q. Wang, X. Yao, Y. Zhou and X.-Y. Yu, J. Mater. Chem. A, 2022, 10, 21848–21855 RSC.
- B. Jia, B. Zhang, Z. Cai, X. Yang, L. Li and L. Guo, eScience, 2023, 3, 100112 CrossRef.
- M. Dixit, Electrochem. Solid-State Lett., 1999, 2, 170 CrossRef CAS.
- G. Fu, X. Kang, Y. Zhang, X. Yang, L. Wang, X.-Z. Fu, J. Zhang, J.-L. Luo and J. Liu, Nano-Micro Lett., 2022, 14, 200 CrossRef CAS PubMed.
- T. Wang, Y. Wang, J. Lei, K.-J. Chen and H. Wang, Exploration, 2021, 1, 20210178 CrossRef PubMed.
- H. Liu, X. Liu, S. Wang, H.-K. Liu and L. Li, Energy Storage Mater., 2020, 28, 122–145 CrossRef.
- Z. Pu, T. Liu, I. S. Amiinu, R. Cheng, P. Wang, C. Zhang, P. Ji, W. Hu, J. Liu and S. Mu, Adv. Funct. Mater., 2020, 30, 2004009 CrossRef CAS.
- Y. Zhang, C. Kirk and N. Robertson, ChemPhysChem, 2020, 21, 2643–2650 CrossRef CAS PubMed.
- D. S. Hall, D. J. Lockwood, C. Bock and B. R. MacDougall, Proc. R. Soc. A, 2015, 471, 20140792 CrossRef PubMed.
- K. Lawson, PhD thesis, Loughborough University, 2021.
- T. Pan, J. M. Wang, Y. L. Zhao, H. Chen, H. M. Xiao and J. Q. Zhang, Mater. Chem. Phys., 2003, 78, 711–718 CrossRef CAS.
- S.-H. Baek, Y.-M. Jeong, D. Y. Kim and I.-K. Park, Chem. Eng. J., 2020, 393, 124713 CrossRef CAS.
- T. Li, S. Yang, Y. Zuo, W. Li, H. Yue, Š. Kment and Y. Chai, Inorg. Chem. Front., 2023, 10, 1001–1010 RSC.
- A. M. Wise, P. W. Richardson, S. W. T. Price, G. Chouchelamane, L. Calvillo, P. J. Hendra, M. F. Toney and A. E. Russell, Electrochim. Acta, 2018, 262, 27–38 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- G. S. Pawley, J. Appl. Crystallogr., 1981, 14, 357–361 CrossRef CAS.
- H. Li, S. Liu, C. Huang, Z. Zhou, Y. Li and D. Fang, Electrochim. Acta, 2011, 58, 89–94 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- E. Y. L. Teo, L. Muniandy, E.-P. Ng, F. Adam, A. R. Mohamed, R. Jose and K. F. Chong, Electrochim. Acta, 2016, 192, 110–119 CrossRef CAS.
- Y. Xin, X. Dai, G. Lv, X. Wei, S. Li, Z. Li, T. Xue, M. Shi, K. Zou, Y. Chen and Y. Liu, ACS Appl. Mater. Interfaces, 2021, 13, 28118–28128 CrossRef CAS PubMed.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373–380 CrossRef CAS.
- X. Yi, H. Sun, N. Robertson and C. Kirk, Sustainable Energy Fuels, 2021, 5, 5236–5246 RSC.
- D. S. Hall, D. J. Lockwood, S. Poirier, C. Bock and B. R. MacDougall, J. Phys. Chem. A, 2012, 116, 6771–6784 CrossRef CAS PubMed.
- K. Lawson, S. P. Wallbridge, A. E. Catling, C. A. Kirk and S. E. Dann, J. Mater. Chem. A, 2023, 11, 789–799 RSC.
- X. Yu, J. Zhao, L.-R. Zheng, Y. Tong, M. Zhang, G. Xu, C. Li, J. Ma and G. Shi, ACS Energy Lett., 2018, 3, 237–244 CrossRef CAS.
- D. Xiong, W. Li and L. Liu, Chem.–Asian J., 2017, 12, 543–551 CrossRef CAS PubMed.
- W. He, X. Li, S. An, T. Li, Y. Zhang and J. Cui, Sci. Rep., 2019, 9, 10838 CrossRef PubMed.
- D. Zhu, J. Liu, L. Wang, Y. Du, Y. Zheng, K. Davey and S.-Z. Qiao, Nanoscale, 2019, 11, 3599–3605 RSC.
- A. N. Mansour, Surf. Sci. Spectra, 1994, 3, 239–246 CrossRef CAS.
- B. Lei, L. Warren, C. Morrison, G. Kerherve, W. S. J. Skinner, D. J. Payne and N. Robertson, Phys. Chem. Chem. Phys., 2023, 25, 4701–4709 RSC.
- S. J. Patil, N. R. Chodankar, S.-K. Hwang, G. S. Rama Raju, Y.-S. Huh and Y.-K. Han, Small, 2022, 2103326 CrossRef CAS PubMed.
- D. H. Lee, K. J. Carroll, S. Calvin, S. Jin and Y. S. Meng, Electrochim. Acta, 2012, 59, 213–221 CrossRef CAS.
- H. K. Schmid and W. Mader, Micron, 2006, 37, 426–432 CrossRef CAS PubMed.
- C. Hobbs, C. Downing, S. Jaskaniec and V. Nicolosi, npj 2D Mater. Appl., 2021, 5, 1–9 CrossRef.
- H. Chen, Z. Wu, Y. Zhong, T. Chen, X. Liu, J. Qu, W. Xiang, J. Li, X. Chen, X. Guo and B. Zhong, Electrochim. Acta, 2019, 308, 64–73 CrossRef CAS.
- X. Li, Y. Tang, J. Zhu, H. Lv, L. Zhao, W. Wang, C. Zhi and H. Li, Small, 2020, 16, 2001935 CrossRef CAS PubMed.
- B. Guo, J. Zhao, X. Fan, W. Zhang, S. Li, Z. Yang, Z. Chen and W. Zhang, Electrochim. Acta, 2017, 236, 171–179 CrossRef CAS.
- M. Adamič, S. D. Talian, A. R. Sinigoj, I. Humar, J. Moškon and M. Gaberšček, J. Electrochem. Soc., 2019, 166, A5045–A5053 CrossRef.
- Q. A. Huang, Y. Li, K. C. Tsay, C. Sun, C. Yang, L. Zhang and J. Zhang, J. Power Sources, 2018, 400, 69–86 CrossRef CAS.
- S. Klaus, Y. Cai, M. W. Louie, L. Trotochaud and A. T. Bell, J. Phys. Chem. C, 2015, 119, 7243–7254 CrossRef CAS.
- R. Lin, L. Kang, T. Zhao, J. Feng, V. Celorrio, G. Zhang, G. Cibin, A. Kucernak, D. J. L. Brett, F. Corà, I. P. Parkin and G. He, Energy Environ. Sci., 2022, 15, 2386–2396 RSC.
- W. He, G. Zhao, P. Sun, P. Hou, L. Zhu, T. Wang, L. Li, X. Xu and T. Zhai, Nano Energy, 2019, 56, 207–215 CrossRef CAS.
- Y. Li, Q. Li, S. Zhao, C. Chen, J. Zhou, K. Tao and L. Han, ChemistrySelect, 2018, 3, 13596–13602 CrossRef CAS.
- Y. Tang, Y. Liu, S. Yu, Y. Zhao, S. Mu and F. Gao, Electrochim. Acta, 2014, 123, 158–166 CrossRef CAS.
- Z. Tang, C. Tang and H. Gong, Adv. Funct. Mater., 2012, 22, 1272–1278 CrossRef CAS.
- H. B. Li, M. H. Yu, F. X. Wang, P. Liu, Y. Liang, J. Xiao, C. X. Wang, Y. X. Tong and G. W. Yang, Nat. Commun., 2013, 4, 1894 CrossRef CAS PubMed.
- J. Huang, P. Xu, D. Cao, X. Zhou, S. Yang, Y. Li and G. Wang, J. Power Sources, 2014, 246, 371–376 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of experimental conditions, PXRD pattern for nickel hydroxide with three different additions, refined cell parameters of nickel hydroxides, PXRD pattern of nickel hydroxide grown on nickel foam, SEM images, TEM images, PXRD pattern of NiO residue after TGA, tables of band assignments FT-IR and Raman, EELS pattern, patterns of electrochemical performance of different nickel hydroxide electrodes. See DOI: https://doi.org/10.1039/d3ta04731h |
| This journal is © The Royal Society of Chemistry 2023 |