
Hybrid core–shell nanoparticles for cell-specific magnetic separation and photothermal heating†
Cristina
de la Encarnación‡
ab,
Felix
Jungwirth‡
 ac,
David
Vila-Liarte
abd,
Carlos
Renero-Lecuna
ad,
Safiyye
Kavak
e,
Iñaki
Orue
f,
Claire
Wilhelm
g,
Sara
Bals
e,
Malou
Henriksen-Lacey
ad,
Dorleta
Jimenez de Aberasturi
*adh and
Luis M.
Liz-Marzán
*adh
ac,
David
Vila-Liarte
abd,
Carlos
Renero-Lecuna
ad,
Safiyye
Kavak
e,
Iñaki
Orue
f,
Claire
Wilhelm
g,
Sara
Bals
e,
Malou
Henriksen-Lacey
ad,
Dorleta
Jimenez de Aberasturi
*adh and
Luis M.
Liz-Marzán
*adh
aCIC biomaGUNE, Basque Research and Technology Alliance (BRTA), 20014 Donostia-San Sebastián, Spain. E-mail: djimenezdeaberasturi@cicbiomagune.es; llizmarzan@cicbiomagune.es
bDepartment of Applied Chemistry, University of the Basque Country, 20018, Donostia-San Sebastián, Spain
cGoethe-Universität Frankfurt, Physikalisches Institut, 60438 Frankfurt am Main, Germany
dCentro de Investigación Biomédica en Red de Bioingeniería, Biomateriales y Nanomedicina (CIBER-BBN), 20014 Donostia-San Sebastián, Spain
eEMAT and NANOlab Center of Excellence, University of Antwerp, Antwerp, Belgium
fSGIKER, Servicios Generales de Investigación, University of the Basque Country, 48940 Leioa, Spain
gLaboratoire Physico Chimie Curie, PCC, CNRS UMR168, Institut Curie, Sorbonne University, PSL University, Paris, 75005, France
hIkerbasque, Basque Foundation for Science, 48009 Bilbao, Spain
First published on 5th April 2023
Abstract
Hyperthermia, as the process of heating a malignant site above 42 °C to trigger cell death, has emerged as an effective and selective cancer therapy strategy. Various modalities of hyperthermia have been proposed, among which magnetic and photothermal hyperthermia are known to benefit from the use of nanomaterials. In this context, we introduce herein a hybrid colloidal nanostructure comprising plasmonic gold nanorods (AuNRs) covered by a silica shell, onto which iron oxide nanoparticles (IONPs) are subsequently grown. The resulting hybrid nanostructures are responsive to both external magnetic fields and near-infrared irradiation. As a result, they can be applied for the targeted magnetic separation of selected cell populations – upon targeting by antibody functionalization – as well as for photothermal heating. Through this combined functionality, the therapeutic effect of photothermal heating can be enhanced. We demonstrate both the fabrication of the hybrid system and its application for targeted photothermal hyperthermia of human glioblastoma cells.
10th Anniversary statementLuis Liz-Marzán has been an author in The Journal of Materials Chemistry since 2000, he was also a guest editor of a special issue in 2006, soon after entering the Editorial Advisory Board. After the journal splitting in 2013, Liz-Marzán continued being in the EAB of The Journal of Materials Chemistry B. We have naturally continued publishing our work in the journal and are happy to be part of the 10th anniversary celebration. HAPPY BIRTHDAY! |
Introduction
The development of minimally invasive cancer treatments is of great interest toward reducing the worldwide burden of cancer-associated morbidity and mortality.1 Tumour ablation via hyperthermal processes is one of the methods being explored, aiming to cause local and intense heating of cancerous tissue to temperatures above 42 °C, thereby resulting in the initiation of cell death associated mechanisms, such as apoptosis and necrosis.2 Among the various methods available to induce hyperthermia, the use of noble metal plasmonic nanoparticles (NPs) as sensitizers for photothermal heating has raised significant interest. When irradiated with a laser wavelength matching a localized surface plasmon resonance (LSPR) mode in the NPs, part of the absorbed photon energy is converted into heat and released to the nearby environment. As a result, rapid heating can be achieved, radiating from the NPs location. Another interesting feature of plasmonic NPs is the possibility of tuning the LSPR wavelength through geometrical parameters (size and shape). For example, in gold nanorods (AuNRs), the longitudinal LSPR absorbance band can be tuned into the near infrared (NIR) region, where human tissue is most transparent, allowing for selective heating of the nanostructures with negligible harm to healthy tissue when a NIR laser is used.3 The efficacy of such photothermal heating (PTH) is thus dependent on properties associated with the laser (intensity, duration, etc.), the tissue (refractive index, heat-sink effects, etc.), and the morphological properties of the gold NPs (AuNPs), including their LSPR wavelength, concentration, and stability in biofluids.In addition to AuNPs, magnetic iron oxide NPs (IONPs) based on either magnetite (Fe3O4) or maghemite (γ-Fe2O3), have also shown promise as nanoheaters, by magnetic hyperthermia.4 In contrast to plasmonic NPs, the method of heat induction is fundamentally different in this case, involving the dissipation of magnetic energy by hysteresis losses and/or Néel or Brown relaxation, upon exposure to high frequency alternating magnetic fields.5 Besides thermal therapeutic applications, both AuNPs and IONPs can be used in a wide variety of diagnostic imaging applications, for example AuNPs are useful in computed tomography (CT), photoacoustic imaging (PAI), or surface-enhanced Raman scattering (SERS), whereas IONPs are excellent contrast agents for magnetic resonance imaging (MRI). Each technique offers differing levels of resolution, contrast, penetration depth, and ultimately feasibility for their use in clinical settings. Therefore, the combination of both materials in single NP entities, thus forming hybrid NPs, is of great interest to maximise the multimodal and correlative nature of such systems.4 Indeed, not only can multiple imaging modalities be combined (i.e. MRI and SERS),6 but also diagnostic and therapeutic applications, resulting in the commonly used term of theranostics.7 Finally, the elemental components of such hybrid NPs offer a wide range of supplementary applications, such as magnetically controlled cell separation8,9 or heat-induced drug delivery,3 both of which could be included to achieve a truly multimodal hybrid NP system.
Whereas most of the literature relating to hybrid magnetic-plasmonic NPs in biomedical applications has been based of using IONPs covered by a gold shell,10,11 we chose to explore a reverse NP construction, in which a gold core is surrounded by IONPs. By working with pre-synthesized AuNPs, a better control can be achieved over the LSPR, which is essential for PTH. AuNR synthesis in which the longitudinal LSPR maximum can be tailored to the wavelength of interest is well documented.12 Subsequent growth of IONPs on the outer surface of the hybrid NPs provides accurate control over their coverage, via sequential thermal decomposition steps, which could not be realized were the IONP located in the hybrid NP interior. We thus propose a core–shell structure comprising an AuNR core – sensitive to NIR irradiation – coated with a silica shell, on which small IONPs are firmly adsorbed to allow for magnetic separation. Both components could additionally be used in various other bio-applications related to theranostics, such as MRI, CT, or photothermal drug release. NPs can also be targeted through surface functionalization, and herein we further demonstrate the possibility for targeted PTH by functionalizing the hybrid NPs with antibodies against the cell membrane CD44 protein, a key receptor for hyaluronic acid that is involved in biological processes such as cell–cell interactions, cell adhesion, migration, and, crucially, tumour metastasis.13 As a model cell type we employed human glioblastoma cells, but the technique could be used for any other CD44-expressing cells, and equally other receptors could be targeted by using the corresponding antibodies.
Results and discussion
Synthesis and characterization of hybrid nanoparticles
The fabrication of multifunctional hybrid NPs involves multiple synthetic steps. AuNRs were synthesized by means of a standard seeded-growth method12 and subsequently coated with mesoporous silica shells,14 which provided stability as well as surface silanol groups that could be used for further functionalization. Then, magnetic IONPs were directly grown onto the silica shells, through a stepwise thermal decomposition of organometallic iron precursors.15The synthesis of IONPs on top of SiO2 NPs required selection of the appropriate IONP density on the silica surface, through the number of thermal decomposition steps. Therefore, we first carried out an optimization process, using mesoporous SiO2 NPs of 150 nm and 240 nm diameter, synthesized using the same modified Stöber method,14 but in the absence of the AuNR cores. The growth of IONPs on the SiO2 NPs surface involved the thermal decomposition of iron oleate, as reported by Nistler et al.16 Briefly, mesoporous silica particles in ethanol were mixed with iron(III) oleate dissolved in 1-octadecene. Oleic acid, which acts as a stabilizing agent, was then added, the mixture was heated at a controlled rate of 200 °C h−1, and then maintained at 317 °C for 30 min, to promote iron oleate decomposition and formation of IONPs. To obtain the final product, the reaction mixture was centrifuged to separate IONP-decorated silica (SiO2@IONPs) from free IONPs in the reaction medium. The IONP coverage on SiO2 NPs was gradually increased for an increasing number of thermal decomposition steps (up to five). A schematic representation of the synthesis is shown in Fig. S1A (ESI†). The resulting SiO2@IONPs were imaged by TEM, observing that the density and size of IONPs could be gradually increased by increasing the number of thermal decomposition steps (Fig. S1B, ESI†).
Although the synthesis was successful for both 150 and 240 nm SiO2 particles, we selected the smaller ones for magnetic characterization because of their higher specific surface area. Hysteresis loops and zero field cooled/field cooled (ZFC/FC) curves were recorded, and the results are shown in Fig. S2 (ESI†). We first measured the magnetic properties of the SiO2@IONPs with the lowest IONP density, i.e., those obtained from a single thermal decomposition step (SiO2@1IONPs). The hysteresis loop (M(H)) for this sample (Fig. S2A, ESI†) shows a high saturation magnetization of 75 emu g−1, in agreement with values reported in the literature.17 These results indicate a superparamagnetic behaviour for SiO2@1IONPs, with low remanence and coercivity, which was found to lower further when increasing the number of thermal decomposition steps (Fig. S2B, ESI†). This behaviour was confirmed by the ZFC/FC curves for both samples. The blocking temperature, estimated as the point where the ZFC curve exhibits an apex, was determined to be TB = 100 K, but shifted to TB = 225 K for a higher number of decomposition steps (Fig. S2C and D, ESI†). Analysis of the normalized magnetization M/Ms (H) at 300 K according to the Langevin model18 (see Experimental Section) and fitting of ZFC/FC measurements, yielded a size (diameter) distribution centred around 10 nm in SiO2@1IONPs (one growth step). Fitting of ZFC/FC measurements for SiO2@4IONPs was performed using the same parameters (magnetization, thermal dependence etc.) as those for SiO2@1IONPs, for meaningful comparison. The estimated average size in this case was 15 nm, thereby confirming NP growth. The 3D structure of the SiO2@IONPs was characterized by electron tomography (Fig. S3, ESI†). Additionally, the ZFC curve indicates that IONPs are made of maghemite (γ-Fe2O3), due to the absence of a shoulder near 110 K, which is known to correspond to a peculiar metal–insulator transition in Fe3O4.19 The SiO2@IONPs could be efficiently separated by placing a hand-held magnet near the solution, resulting in NP accumulation into a pellet. We observed that SiO2@IONPs with a lower density of IONPs were difficult to separate magnetically, whereas those synthesized with 3–5 thermal decomposition steps were readily separated. However, whereas SiO2@3IONPs and SiO2@4IONPs were easily redispersed upon removal of the magnet, SiO2@5IONPs remained as a hard pellet.
Once the growth of IONPs on the surface of silica particles was optimized, the method was implemented for hybrid NPs including AuNRs as plasmonic cores. A schematic representation of the complete process is shown in Fig. 1A. AuNRs were synthesized by a three-step seed-mediated method,9 involving the controlled growth of pre-made AuNR seeds (7.5 nm × 21.0 nm) into larger AuNRs (16.4 ± 2.7 nm × 66.5 ± 5.7 nm) with a longitudinal LSPR at 790 nm (Fig. 1B and Fig. S4, ESI†). AuNRs were then coated with a mesoporous silica shell using a modified Stöber method.14 Briefly, a colloidal dispersion containing AuNRs, CTAB, ethanol, and water was prepared. After adjusting the pH to 9 by addition of aqueous ammonia, a selected amount of tetraethyl orthosilicate (TEOS) was added dropwise under strong stirring, to trigger TEOS hydrolysis and SiO2 condensation. After TEOS addition, the temperature was increased to 60 °C and the mixture was stirred for three days. Then, the particles were recovered by centrifugation and thoroughly washed with ethanol and dilute HCl, to remove the surfactant (CTAB). Normalized UV-vis spectra and TEM images were recorded for initial characterization (Fig. 1C and Fig. S4, ESI†). The TEM images showed uniform encapsulation of every AuNR by a silica shell, with a thickness of 70.8 ± 8.2 nm, while the UV-vis spectrum showed a small redshift of the longitudinal LSPR, as expected for an increased local refractive index upon silica deposition.
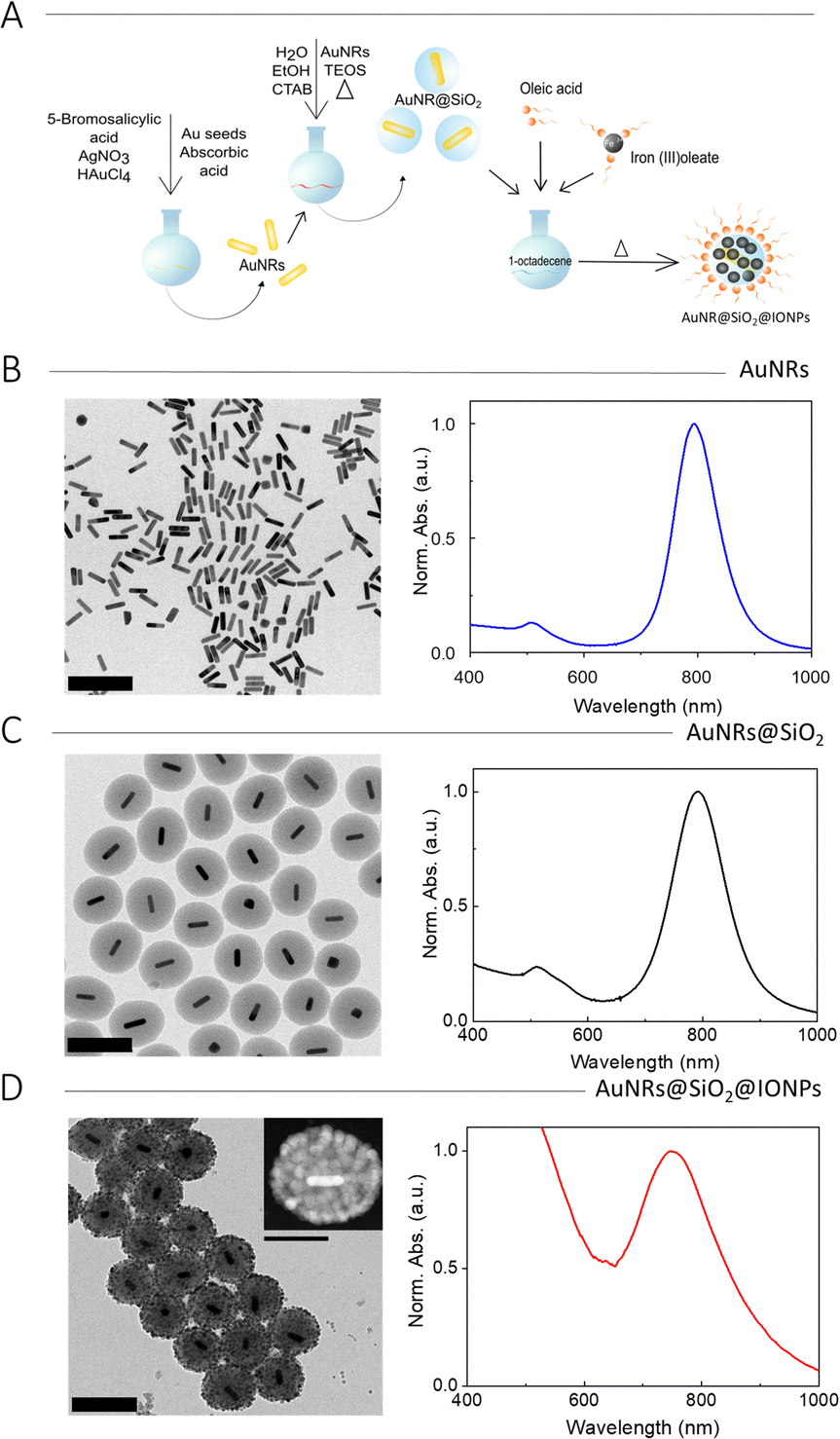 | ||
| Fig. 1 (A) Schematic representation of the synthesis of AuNR@SiO2@IONPs. (B–D) TEM images and normalized (at LSPR maximum) UV-visible spectra for AuNRs (B), AuNR@SiO2 (C), and AuNR@SiO2@IONPs (D). The inset in (D) shows a single particle imaged using high-angle annular dark field (HAADF) STEM. Scale bars correspond to 200 nm (100 nm in the inset). | ||
Finally, IONPs were grown on the silica surface by using the procedure optimized for SiO2@IONPs. In short, AuNR@SiO2 NPs were added to a solution of oleic acid and iron(III) oleate in 1-octadecene, followed by controlled temperature increase, leading to the decomposition of the iron complex and formation of AuNR@SiO2@IONPs, which were recovered and washed by centrifugation. The thermal decomposition step was repeated up to three times to increase IONP surface coverage, yielding densely covered hybrid NPs (Fig. S5A1–3, ESI†). It should be noted that, as found in an initial optimization process using AuNRs with an LSPR at 760 nm, the thermal treatment induced partial shortening of AuNRs, with a corresponding LSPR blueshift to 730 nm (Fig. S5B, ESI†). Therefore, we chose to compensate for AuNR reshaping by initially using longer AuNRs (16.4 ± 2.7 nm × 66.5 ± 5.7 nm) with a longitudinal LSPR at 790 nm. By using an optimized number of three decomposition steps, dense and homogenous IONP coverage was achieved. The 3D structure was confirmed by electron tomography (Fig. S6, ESI†). Representative results of TEM and UV-vis spectroscopy characterization are shown in Fig. 1D. It should be noted that the colloidal stability of these NPs decreased for higher IONP loadings. When comparing the UV-vis spectra of AuNRs and AuNR@SiO2 (Fig. 1B and C) with that of AuNR@SiO2@IONPs (Fig. 1D) a significant LSPR blue shift from 790 to 760 nm was observed, in agreement with partial thermal AuNR reshaping at the high temperature employed for IONP formation.20,21 This AuNR shortening effect also became apparent in TEM images, the initial AuNRs appeared longer (66.5 ± 5.7 nm length, 16.4 ± 2.7 nm width, 4.1 aspect ratio) than the final AuNRs (58.3 ± 5.1 nm length, 15.1 ± 2.4 nm width, 3.8 aspect ratio). In the final sample, the AuNR LSPR bands show an apparent damping caused by the high background signal produced due the presence of IONPs, which shows as a broad shoulder rising into the UV region.22
Upon completion of the synthetic process, the external layer of oleate-capped IONPs renders the hybrid AuNR@SiO2@IONPs hydrophobic. However, for their use in biological environments, stability in aqueous media is required. Therefore, an additional phase transfer step was carried out, through encapsulation with an amphiphilic polymer.23,24 Dodecylamine-modified polyisobutylene-alt-maleic anhydride (PMA) was employed for the encapsulation of both AuNR@SiO2@IONPs and SiO2@IONPs, the latter to be used as controls in subsequent experiments. No phase transfer was required for AuNRs@SiO2 (also used as a control in some experiments) because these particles were readily stable in polar solvents after synthesis. However, AuNR@SiO2 were functionalized with a polyethylene glycol (PEG)-silane of a similar size as PMA, to enhance their stability and to mimic the coating of AuNR@SiO2@IONPs. These coatings allowed us to study the hybrid NPs and their controls (AuNR@SiO2 and SiO2@IONPs) under similar conditions. The detailed procedure is described in the Experimental section.
The spatial elemental distribution of Au, Si, and Fe in the hybrid NPs was characterized using energy dispersion X-ray spectroscopy (EDX) analysis in electron microscopy. As shown in Fig. 2A, all three components were observed at their expected locations (AuNRs inside the silica shell and IONPs on the outer silica surface). We also characterized the magnetic properties of AuNR@SiO2@IONPs, and the corresponding hysteresis loop and ZFC/FC curves are plotted in Fig. 2B. Again, the hysteresis loop revealed a superparamagnetic behaviour at room temperature, with no remanence and low coercivity. On the other hand, the ZFC/FC curves showed a blocking temperature of 230 K, similar to that obtained for SiO2@4IONPs (Fig. S2D, ESI†). Finally, the response of AuNR@SiO2@IONPs to an external magnetic field was evaluated toward their use for rapid magnetic separation. To this end, an aqueous dispersion of AuNRs@SiO2@IONPs was placed in an Eppendorf tube and a magnet placed at the side, resulting in fast magnetic separation and accumulation into a pellet at the tube wall within 20 s (Fig. S7, ESI†). Upon removal of the magnet, the NPs could be redispersed again in water and remained colloidally stable. The described separation process could be repeated for any desired number of times.
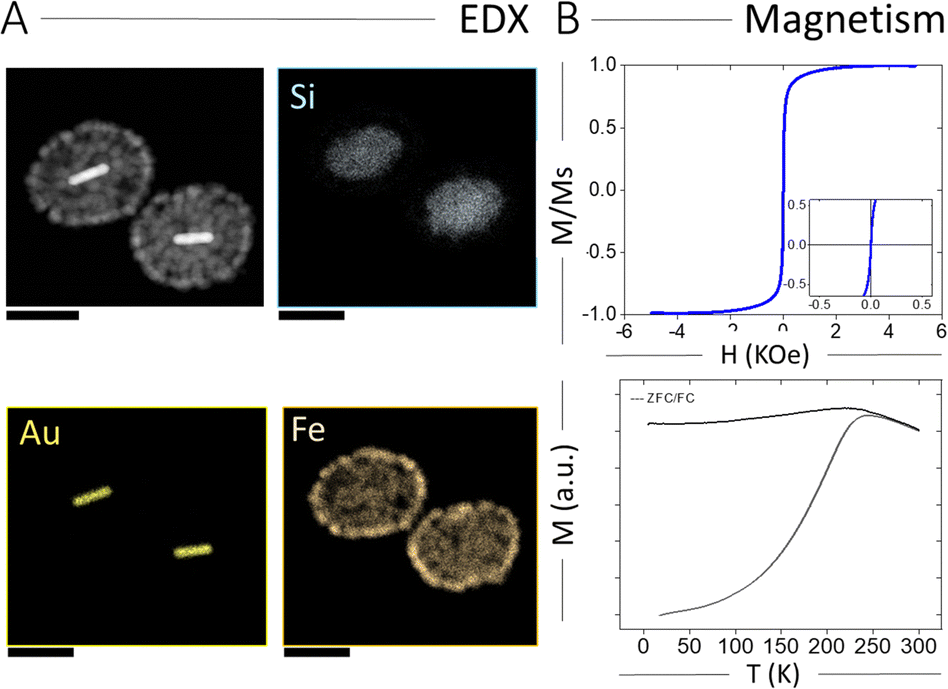 | ||
| Fig. 2 (A) EDX analysis of AuNR@SiO2@IONPs, showing the elemental distribution of Au, Fe and Si. Scale bars correspond to 100 nm. (B) Hysteresis loop and ZFC/FC curves for AuNR@SiO2@IONPs. The inset in (B) shows a magnification of the central area of the curve, in which coercivity and remanence are appreciated to be negligible. | ||
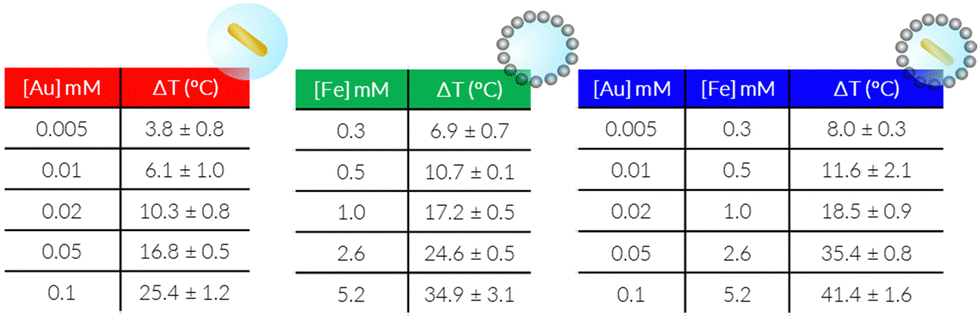
We next measured the PTH performance of the hybrid NPs using an 808 nm laser, which closely matches the LSPR maximum of the AuNRs. We chose to use a laser irradiation setup in which the spot size was matched to the sample diameter, thus allowing us to measure the true power density in the exposed sample area. Dispersions with different NP concentrations (measured using inductively coupled plasma mass spectrometry, ICP-MS) were irradiated for 5 min and temperature increments were registered using a NIR thermal camera, collecting data from a region of interest (ROI), corresponding to the NP dispersion. The absolute temperature was measured at different times and temperature increments were calculated by subtracting the initial temperature (time = 0) from the final temperature (time = 5 min) (Fig. 3, left column, Table 1). The specific absorption rates (SAR), which represent the power absorbed per mass of material,25 were also calculated (Fig. 3, right column). Specific measurement details can be found in the Experimental Section. As expected, for each system, an increase in temperature was observed with increasing NP concentration (as expressed in [Fe], [Au], or OD808), although it was found not be linear because of light attenuation at higher concentrations.
 | ||
| Fig. 3 Average PTH temperature increase (ΔT, °C) and estimated SAR, using conditions of 5 min irradiation (808 nm) at 2.5 W cm−2 in solution, at different concentrations of AuNR@SiO2@IONPs (A), AuNR@SiO2 (B), and SiO2@IONPs (C). Results show mean ± SD of triplicate measurements (error bars for SAR are within the symbols). | ||
|
Through comparison across the different NP systems, we observed that, for a certain concentration of Au and or Fe in AuNR@SiO2@IONPs, the equivalent control samples resulted in weaker heating, and that the AuNR@SiO2 control consistently heated less than the SiO2@IONPs. In other words, at comparable concentrations, the presence of both metals always resulted in a cumulative heating. Indeed, it has been recently shown that iron oxide nanomaterials can be efficient nano-heaters in the NIR, where an absorption band can be assigned to an intervalence charge transfer between Fe3+ and Fe2+ cations.26 Besides, under the conditions studied, the contribution of iron oxide is found to be higher than that of gold due to the higher proportion of IONPs versus AuNRs per hybrid particle (see the difference in global Fe versus Au concentration). Upon a recalculation taking the relative concentrations into account, it is clear that AuNRs remain much more efficient heaters, as expected from the presence of an LSPR mode in close resonance with the laser wavelength (808 nm vs. LSPR@760 nm). From these calculations, we estimate that 25 times more Fe in SiO2@IONPs compared to Au in AuNRs@SiO2 was needed to reach the same temperature increment. We attempted to relate these heating properties based on the optical density (OD808) due to the different NPs. However, taking into consideration the significant differences in metal concentration, that the maximum OD of Fe is not at 808 nm, and that the sum of both metals does not result in a sum of OD, such a comparison proved difficult (Table S1, ESI†).
Biological application of hybrid nanoparticles
Biological assays were carried out using a human glioblastoma cell line, U87. We first screened cell viability upon exposure to AuNR@SiO2@IONPs for 24 h and 48 h (Fig. S8, ESI†). At concentrations ranging between 0.16 and 1.3 mM [Fe] (corresponding to 3–25 μM [Au]), no cytotoxicity was observed after 24 h. However, after a further 24 h (total of 48 h) significant cytotoxicity at the highest concentrations was observed. We thus chose to work with 0.33 and 0.65 mM [Fe] (6.3 and 12.5 μM [Au]) for further cell studies, i.e., incubating U87 cells with AuNR@SiO2@IONPs for 24 h followed by removal of any non-endocytosed fraction. Brightfield imaging showed high levels of uptake (Fig. S8C, ESI†), corroborated by TEM analysis showing endosomal NP uptake after 24 h (Fig. 4A). Considering literature reports that claim increased photothermal effects when NPs have been internalized by cells,25,27,28 we studied the heating efficiency of AuNR@SiO2@IONPs upon cell internalization (Fig. 4B). We subsequently proceeded to irradiate the samples (see Experimental section for full details), evaluating biocompatibility by using the Alamar Blue cell viability assay, immediately post irradiation and after a further 24 h, and by using live/dead fluorescence labelling up to 5 days post irradiation (Fig. 4C, D and Fig. S9, ESI†). The results show that PTH induces cytotoxicity in a time-dependent manner, suggestive of apoptosis or ferroptosis cell death mechanisms, as previously documented in other works.29–31 Furthermore, considering that the photothermal component of AuNR@SiO2@IONPs (i.e., AuNR) is in relatively low concentration compared to the Fe component (52-fold less Au than Fe), high laser power densities were required to observe efficient PTH-induced cell death.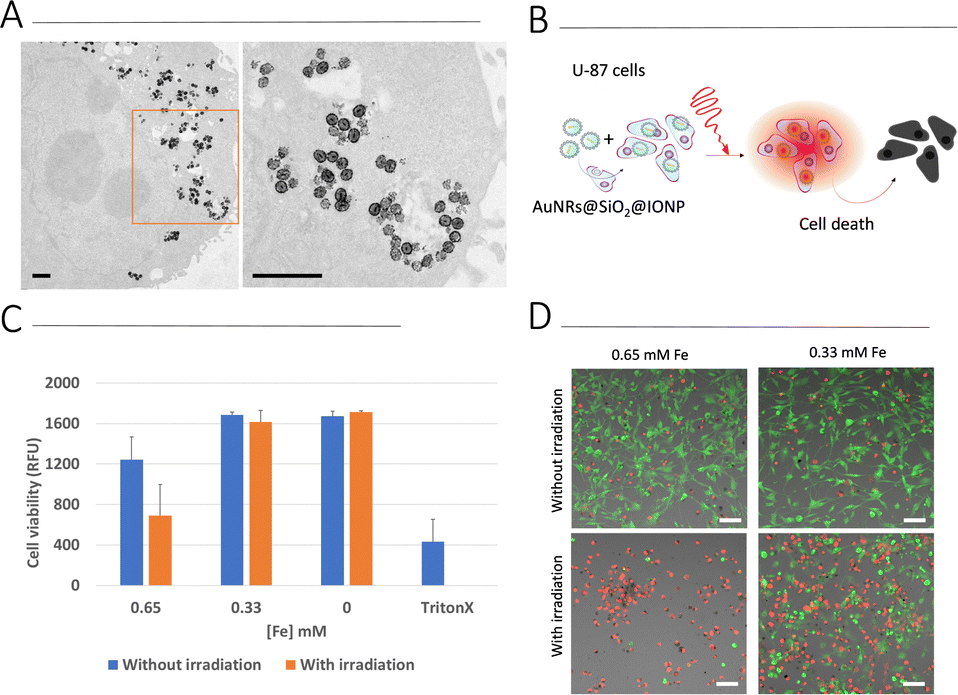 | ||
| Fig. 4 (A) TEM images of U87 cells exposed to AuNR@SiO2@IONPs for 24 h. The right-hand image corresponds to the area highlighted by an orange box in the left-hand image. Scale bars: 1 μm. (B) Schematic illustration of biological PTH. (C) U87 cell viability after exposure to AuNR@SiO2@IONPs for 24 h, followed by irradiation with an 808 nm laser. The relative fluorescence intensity (RFU), a measure of cell viability is shown after additional 24 h, determined using the Alamar Blue assay. Mean ± SD of triplicate measurements. (D) Live/dead fluorescence imaging of cells 5 days post irradiation, showing dead cells in red (propidium iodide) and live cells in green (Calcein). Scale bars: 100 μm. | ||
As it is well known that 2D cell cultures have severe limitations to reproduce the real biological environment, 3D models have become a more reliable testbed to evaluate cell behaviour under different conditions.32 Therefore, we used a 3D model to further evaluate the PTH properties of our NPs in a more realistic scenario. Under certain growth conditions, U87 cells spontaneously form cell spheroids, which can be used as miniaturized 3D models for high-throughput testing. U87 cells were incubated for 24 h with AuNR@SiO2@IONPs (0.33 and 0.65 mM [Fe], equivalent to 6.3 and 12.5 μM [Au], respectively) in 2D cell growth substrates, and subsequently transferred to spheroid microplates to obtain an array of spheroids placed at the same imaging plane. After 72 h, spheroids were collected into an Eppendorf tube and irradiated for 5 min. After irradiation, spheroids were transferred back to spheroid microplates for subsequent imaging using live/dead fluorophores, to evaluate the extent of cell death (Fig. 5 and Fig. S10, ESI†). Controls were used to confirm the non-toxic nature of the NPs in spheroids, in addition to the lack of induced cell death upon irradiation of spheroids without NPs. Regarding NP-exposed spheroids, we again found a time-dependent increase in cell death upon irradiation, studied up to 48 h post irradiation.
 | ||
| Fig. 5 Live/dead images of spheroids in which cells were pre-incubated with AuNR@SiO2@IONPs (6.3 and 12.5 μM [Au], equivalent to 0.33 and 0.65 mM [Fe], respectively) prior to forming spheroids. Irradiations were for 5 min using an 808 nm laser, with cell viability determined 24 h and 48 h post irradiation. Scale bar: 100 μm. Additional examples of spheroids are shown in the ESI† (Fig. S10). | ||
As observed here and widely reported in the literature, NPs are readily uptaken by cells, even if they do not carry any specific surface-targeting ligands.24 If specific cancer cell targeting is to be achieved, NPs should be conjugated with antibodies (or other biorecognition elements), so that intracellular uptake would be limited by specific antibody-antigen receptor interactions at the cell membrane.33 For this study we chose to target the transmembrane glycoprotein CD44, a commonly expressed receptor in various cancer types and especially upregulated in metastatic tumour cells.34 U87 can be effectively targeted using anti-CD44 molecules, thanks to its ubiquitous membrane expression.35 Functionalization of AuNR@SiO2@IONPs with antibodies was performed using a protocol based on the activation of free carboxylic groups in the PMA structure, via 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide/N-hydroxysuccinimide (EDC/NHS) chemistry,36 and subsequent binding of an anti-human anti-CD44 antibody via protein G (see Experimental section). The use of protein G assisted the correct orientation of the antibody, which maintained free its fragment antigen-binding region (Fab region), to interact with CD44 on the cell surface.
Immunostaining was first used to verify that U87 cells were positive for the expression of CD44. We explored the use of non-labelled, and Alexa Fluor (AF) labelled, antibodies both for this step and for conjugation to AuNR@SiO2@IONPs. The experiments presented herein were conducted with AF488-labelled CD44 (DF1485), but other examples and alternatives are shown in the ESI† (Fig. S11). Cells were grown as an adherent culture and fixed prior to normal direct immunostaining protocols. As can be observed in Fig. 6A, U87 cells were positive for CD44 expression. We thus added AuNR@SiO2@IONP-AB to cells and incubated for 2 h, followed by washing to remove non-associated NPs and carry out live cell imaging. We examined the presence of NPs using both fluorescence (detecting AF488) and multiphoton (MP) confocal microscopy, the latter being used to detect the NPs directly. As shown in Fig. 6A, both the fluorescence and MP signals correlated perfectly, confirming that MP confocal microscopy can be used to image NPs in live cells, thereby avoiding the requirement to fix and locate the NPs using immunostaining protocols when non-labelled antibodies are used. Similar surface expression of CD44 was observed by incubating the breast cancer cell line MDA.MB.231 with AuNR@SiO2@IONP-AB, indicating that this system can work with multiple cell types (Fig. S12, ESI†). In all cases, adjusting NP concentration was crucial to avoid overloading: a concentration at 0.33 mM Fe (6.3 μM Au) gave good results. ICP-MS data suggests that ca. 70% of added AuNR@SiO2@IONP-AB NPs were associated with cells, giving sufficient signal in both fluorescence and MP imaging to detect in situ the spatial location of the NPs (Fig. S13, ESI†). In contrast, AuNR@SiO2@IONP added at the same concentration for also 2 h resulted in ca. 10% association. Furthermore, we employed TEM imaging to verify the membrane location of AuNR@SiO2@IONP-ABs, which was further corroborated by fluorescence confocal imaging (Fig. 6B).
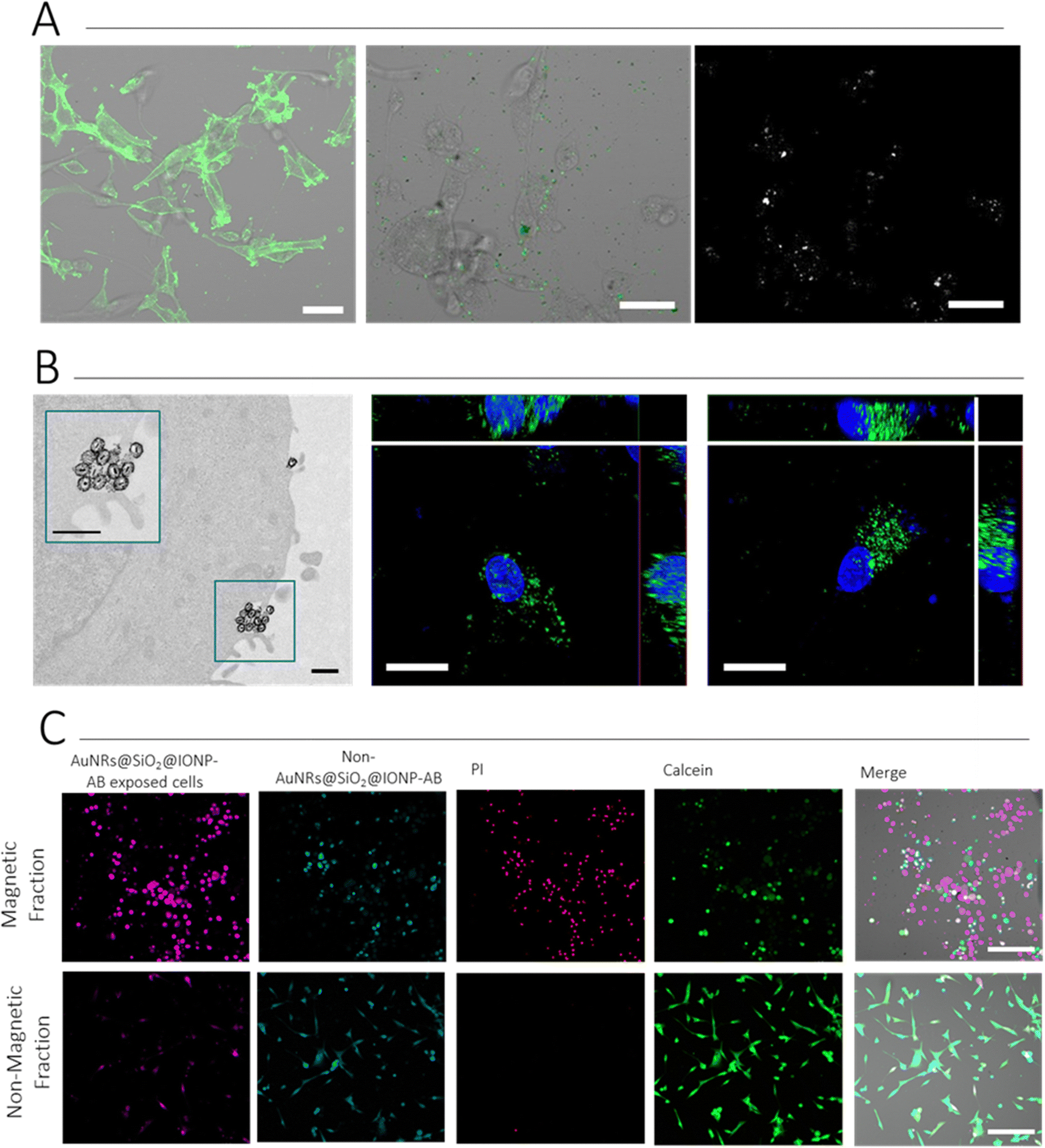 | ||
| Fig. 6 (A) Verification of CD44 expression on U87 cells (left), and subsequent AuNR@SiO2@IONP-AB targeting of cells, viewed using brightfield and fluorescence detection of AF488 (centre), as well as multiphoton microscopy (right). Scale bar: 50 μm (B) AuNR@SiO2@IONP-AB incubation with U87 cells for 2 h results in surface binding, as observed via TEM (left) and fluorescence confocal microscopy (centre). In comparison with AuNR@SiO2@IONPs (right), surface association is clearly observed. Scale bars: 0.5 μm (TEM), 20 μm (confocal). (C) Fluorescence confocal microscopy showing magnetic separation of AuNR@SiO2@IONP-AB associated with CD44 expressing cells, and subsequent PTH-induced cytotoxicity. Scale bars: 200 μm. | ||
We subsequently proceeded to determine the suitability of CD44-targeted NPs for magnetic cell selection, followed by inducing cell death via PTH. U87 cells were divided into two groups, each labelled with a different fluorophore (“Cell Tracker” fluorophores). Only one group was incubated with AuNR@SiO2@IONP-AB. Subsequently, both populations were mixed, and a magnet was placed in contact with the side of the Eppendorf tube. After 5 min, ca. 75% of the solution was carefully removed (herein termed “Non-Magnetic Fraction”, taking special care to avoid the side in contact with the magnet. Both the non-magnetic fraction and the remaining “Magnetic Fraction” were analysed via fluorescence microscopy to determine the proportion of each cell population, and via ICP-MS to determine the proportion of AuNR@SiO2@IONP-AB in each fraction. Upon confirmation of successful separation of the cells exposed to AuNR@SiO2@IONP-AB in the Magnetic Fraction, we proceeded to irradiate both fractions with an 808 nm laser, followed by live/dead staining the following day, to determine PTH-induced cytotoxicity. As can be seen in Fig. 6C, few AuNR@SiO2@IONP-AB expressing cells (those in pink) were imaged in the non-magnetic fraction in agreement with the high level of magnetic cell separation. Indeed, ICP-MS analyses of both the Magnetic and Non-Magnetic fractions suggest that the efficiency of magnetic separation is close to 75%. Regarding the ability to cause PTH-induced cytotoxicity, PI-expressing cells (dead cells) were only observed in the Magnetic Fraction (pink fraction), as expected because this was the cell population containing hybrid NPs. We additionally noted that the cell morphology was drastically different in both fractions, indicating that the irradiated Magnetic Fraction was indeed non-viable whereas the irradiated Non-Magnetic fraction was composed of viable cells (due to the lack of hybrid NPs in this fraction which can cause PTH). Taken together, these results suggest that PTH induces the specific death only on those cells containing AuNR@SiO2@IONP-AB, thereby confirming the suitability of the AB-labelled hybrid NPs for CD44 targeting, cell separation, and subsequent PTH-induced cytotoxicity protocols.
Conclusions
A multistep method was developed for the synthesis of hybrid, multifunctional NPs. Starting from AuNRs and silica coating, IONPs were grown using thermal decomposition of iron oleate. The density of IONPs on the silica surface can be increased by performing multiple thermal decomposition steps, which number was optimized using core-free SiO2 NPs. Electron microscopy confirmed that the number of decomposition steps could be used not only to tailor surface coverage, but also the size of IONPs, which reached 19 nm after 3 thermal decomposition steps. We hypothesize that, IONPs are formed in the first step and then act as nucleation and preferential growth sites for subsequent steps.The obtained AuNR@SiO2@IONPs were characterized by TEM, HAADF and EDX, thereby confirming the structure and composition of the hybrid NPs. UV-vis spectra were also recorded, showing partial reshaping of AuNRs due to the high temperature used during IONP synthesis. Additionally, magnetic characterization showed superparamagnetic behavior, characterized by low remanence and coercivity. The photothermal characterization of the NPs displayed promising properties as nanoheaters, achieving high temperature increments, even for low Au concentrations. By comparing with suitable controls and considering the molar concentration of each element, the heating capacity of AuNRs is ca. 25 times higher. The hybrid NPs were shown to induce PTH-based cytotoxicity in both 2D and 3D cell cultures, and their magnetic properties allowed the magnetic selection of cells. Specific targeting of CD44-expressing cancer cells was achieved by conjugating the corresponding antibodies to the AuNR@SiO2@IONPs surface. This strategy, in combination with magnetic selection and PTH, proves the suitability of these hybrid NPs as a potential cancer therapeutic method. A future interesting direction might take advantage of the hydrophobic drug loading ability of mesoporous silica, thus creating a heat-sensitive drug-release sponge.37
Experimental
Materials
Ferric chloride hexahydrate (FeCl3·6H2O, ≥98%), sodium oleate (≥99%), ammonia (≥99.5%,), absolute ethanol (CH3CH2OH,), tetraethyl orthosilicate (TEOS), hexane (≥95%), 1-octadecene (≥90%), hexadecyltrimethylammonium bromide (CTAB, ≥99%), 1-decanol (98%), hydrogen tetrachloroaurate trihydrate (HAuCl4·3H2O, ≥99.9%,), sodium citrate tribasic dihydrate (≥98%), silver nitrate (AgNO3, ≥99%), L-ascorbic acid (AA, ≥99%), O-[2-(3-mercaptopropionylamino)ethyl]-O′-methylpolyethylene glycol (SH-PEH,), 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC, ≥98%), N-hydroxysuccinimide (NHS, ≥98%), phosphate buffered saline (PBS), phosphate buffer (PB), 2-(N-morpholino)ethanesulfonic acid (MES buffer) were purchased from Sigma-Aldrich and COOH-PEG-silane (Mw 5000 Da) was purchased from Nanocs. All the reactants were prepared and used without further treatments. Milli-Q water was used for all the experiments.Nanoparticle synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–15000 rpm for 45–60 min in 2 mL tubes. The precipitates were collected and diluted with 20 mL of a 10 mM CTAB solution. The AuNRs dispersion was centrifuged twice under the same conditions. The final volume of small AuNRs dispersion was fixed to [Au] = 4.6 mM.
000–15000 rpm for 45–60 min in 2 mL tubes. The precipitates were collected and diluted with 20 mL of a 10 mM CTAB solution. The AuNRs dispersion was centrifuged twice under the same conditions. The final volume of small AuNRs dispersion was fixed to [Au] = 4.6 mM.
To perform the thermal decomposition, 3 g of iron(III) oleate was dissolved in 21 mL of 1-octadecene in a 3 neck 150 mL round bottom flask equipped with a magnetic stirring bar. The reaction vessel was connected to a vacuum line and the mixture was degassed at 70 °C in a bath for 30 min. Afterwards, the water bath was removed, and the flask was equipped with a reflux condenser, a thermocouple and a septum while applying a N2-flow. 130 mg of mesoporous silica was dispersed in 3 mL of ethanol using a sonication bath and then added to the reaction mixture together with 535 μL of oleic acid. The flask was equipped with a septum and a thermocouple, and the reaction mixture was heated to 317 °C with a heating ramp of 200 °C h−1 and a holding time at 317 °C of 30 min. During the whole heating process, a gentle N2-flow was applied, furthermore the water cooling for the reflux condenser was only switched on when the reaction reached a temperature of 200 °C, ensuring that all ethanol was removed from the reaction, which otherwise could drip back into the hot solvent causing splashing of the reaction mixture due to spontaneous evaporation. After cooling to room temperature, hexane was added and the product was recovered by centrifugation (9000 rpm, 10 min, 20 °C). The product was washed several times with hexane to remove unbound IONPs. For overgrowth, the same process was employed. First 130 mg of the prepared NPs was dissolved in 7 mL of 1-octadecene and then added to 3 g iron(III) oleate dissolved in 14 mL 1-octadecene. The mixture was degassed for 30 min, the flask was equipped with a reflux condenser and 535 μL of oleic acid was added. Then the same heating ramp was used, and the product was collected via centrifugation (9000 rpm, 10 min, 20 °C) and washed several times with hexane. This process was repeated until the desired coverage was obtained.
Functionalization
Nanoparticles characterization
Photothermal measurements
Since the mass of Fe and Au are significantly lower than those of water (Cw = 4.18 J g−1 K−1, Ccell = 4.125 J g−1 K−1), the initial equation can be written as:
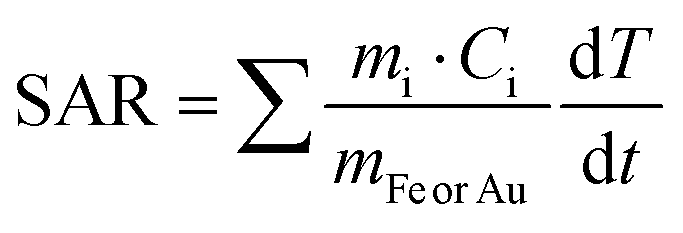
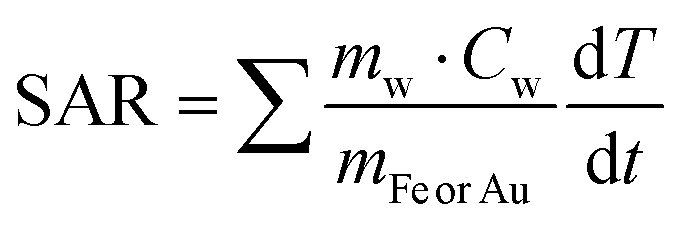
Cell culture
Author contributions
C. de la Encarnación performed essential experimental work and wrote the first draft of the manuscript; F. Jungwirth performed essential experimental work; D. Vila-Liarte performed auxiliary experimental work; C. Renero-Lecuna performed auxiliary experimental work; S. Kavak performed electron tomography analysis; I. Orue performed magnetic characterisation; C. Wilhelm supported hyperthermia experiments; S. Bals performed electron tomography analysis; M. Henriksen-Lacey performed cell experiments; D. Jimenez de Aberasturi and L. M. Liz-Marzán conceived, coordinated and supervised the project. All authors revised and approved the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the European Research Council (ERC AdG 787510, 4DBIOSERS and ERC CoG 815128, REALNANO) and MCIN/AEI/10.13039/501100011033 through grant PID2019-108854RA-I00. S.K acknowledges the Flemish Fund for Scientific Research (FWO Vlaanderen) through a PhD research grant (1181122N). We thank Itziar Galarreta for assistance with magnetic measurements and Marco Möller for support with EDX measurements.Notes and references
- G. M. Cooper, The Development and Causes of Cancer, in The Cell: A Molecular Approach, Sinauer Associates, Sunderland (MA), 2nd edn, 2000. Available from: https://www.ncbi.nlm.nih.gov/books/NBK9963/ Search PubMed.
- K. Richter, M. Haslbeck and J. Buchner, Mol. Cell, 2010, 40, 253–266 CrossRef CAS PubMed.
- N. R. Datta, S. G. Ordóñez, U. S. Gaipl, M. M. Paulides, H. Crezee, J. Gellermann, D. Marder, E. Puric and S. Bodis, Cancer Treat. Rev., 2015, 41, 742–753 CrossRef CAS PubMed.
- C. de la Encarnación, D. Jimenez de Aberasturi and L. M. Liz-Marzán, Adv. Drug Deliv. Rev., 2022, 189, 114484 CrossRef PubMed.
- X. Liu, Y. Zhang, Y. Wang, W. Zhu, G. Li, X. Ma, Y. Zhang, S. Chen, S. Tiwari, K. Sji, S. Zhang, H. M. Pan, Y. X. Zhao and X.-J. Liang, Theranostics, 2020, 10, 3793–3815 CrossRef CAS PubMed.
- C. de la Encarnación, E. Lenzi, M. Heriksen-Lacey, B. Molina, K. Jenkinson, A. Herrero, L. Colás, P. Ramos Cabrer, J. Toro-Mendoza, J. Langer, S. Bals, D. Jimenez de Aberasturi and L. M. Liz-Marzán, J. Phys. Chem. C, 2022, 126, 19519–19531 CrossRef.
- S. M. Dadfar, K. Roemhild, N. I. Drude, S. von Stillfried, R. Knüchel, F. Kiessling and T. Lammers, Adv. Drug Deliv. Rev., 2019, 138, 302–332 CrossRef CAS.
- J. Chen, L. Chen, S. Du, J. Wu, M. Quan, H. Yin, Y. Wu, X. Ye, X. Liang and H. Jiang, J. Nanobiotechnology, 2019, 17, 116 CrossRef PubMed.
- C. Liu, B. Yang, X. Chen, Z. Hu, Z. Dai, D. Yang, X. Zheng and X. She, Nanotechnology, 2019, 30, 285706 CrossRef CAS PubMed.
- D. L. Miyasato, A. W. Mohamed and C. A. Zavaleta, Rev. Nanomed. Nanobiotechnol., 2021, 13, e1721 CAS.
- Z. Abed, J. Beik, S. Laurent, N. Eslahi, T. Khani, E. S. Davani, H. Ghaznavi and A. Shakeri-Zadeh, J. Cancer Res. Clin. Oncol., 2019, 145, 1213–1219 CrossRef CAS.
- G. González-Rubio, V. Kumar, P. Llombart, P. Díaz-Núñez, E. Bladt, T. Altantzis, S. Bals, O. Peña-Rodríguez, E. G. Noya, L. G. Macdowell, A. Guerrero-Martínez and L. M. Liz-Marzán, ACS Nano, 2019, 13, 4424–4435 CrossRef PubMed.
- K. Endo and T. Terada, J. Hepatol., 2000, 32, 78–84 CrossRef CAS PubMed.
- M. Sanz-ortiz, K. Sentosun, S. Bals and L. M. Liz-Marzán, ACS Nano, 2015, 9, 10489–10497 CrossRef CAS PubMed.
- J. Park, K. An, Y. Hwang, J. E. G. Park, H. J. Noh, J. Y. Kim, J. H. Park, N. M. Hwang and T. Hyeon, Nat. Mater., 2004, 3, 891–895 CrossRef CAS PubMed.
- A. Nistler, C. Hartmann, C. Rümenapp, M. Opel, B. Gleich, N. P. Ivleva, R. Niessner and M. Seidel, J. Magn. Magn. Mater., 2017, 442, 497–503 CrossRef CAS.
- N. Lee and T. Hyeon, Chem. Soc. Rev., 2012, 41, 2575–2589 RSC.
- M. Knobel, W. C. Nunes, L. M. Socolovsky, E. De Biasi, J. M. Vargas and J. C. Denardin, J. Nanosci. Nanotechnol., 2008, 8, 2836–2857 CrossRef CAS PubMed.
- B. S. Yadav, R. Singh, A. K. Vishwakarma and N. Kumar, J. Supercond. Nov. Magn., 2020, 33, 2199–2208 CrossRef CAS.
- M. B. Mohamed, K. Z. Ismail, S. Link and M. A. El-Sayed, J. Phys. Chem. B, 1998, 102, 9370–9374 CrossRef CAS.
- W. J. Kennedy, S. Izor, B. D. Anderson, G. Frank, V. Varshney and G. J. Ehlert, ACS Appl. Mater. Interfaces, 2018, 10, 43865–43873 CrossRef CAS PubMed.
- J. Tang, M. Myers, K. A. Bosnick and L. E. Brus, J. Phys. Chem. B, 2003, 107, 7501–7506 CrossRef CAS.
- A. B. Serrano-Montes, D. Jimenez de Aberasturi, J. Langer, J. J. Giner-Casares, L. Scarabelli, A. Herrero and L. M. Liz-Marzán, Langmuir, 2015, 31, 9205–9213 CrossRef CAS PubMed.
- D. Jimenez de Aberasturi, A. B. Serrano-Montes, J. Langer, M. Henriksen-Lacey, W. J. Parak and L. M. Liz-Marzán, Chem. Mater., 2016, 28, 6779–6790 CrossRef CAS.
- A. Espinosa, J. Kolosnjaj-Tabi, A. Abou-Hassan, A. Plan Sangnier, A. Curcio, A. K. A. Silva, R. Di Corato, S. Neveu, T. Pellegrino, L. M. Liz-Marzán and C. Wilhelm, Adv. Funct. Mater., 2018, 28, 1803660 CrossRef.
- E. Bertuit, E. Benassai, G. Meriguet, J. M. Greneche, B. Baptiste, S. Neveu, C. Wilhelm and A. Abou-Hassan, ACS Nano, 2022, 16, 271–284 CrossRef CAS.
- A. Espinosa, A. K. A. Silva, A. Sánchez-Iglesias, M. Grzelczak, C. Péchoux, L. Desboeufs, L. M. Liz-Marzán and C. Wilhelm, Adv. Healthc. Mater., 2016, 5, 1040–1048 CrossRef CAS PubMed.
- A. Espinosa, J. Reguera, A. Curcio, A. Muñoz-Noval, C. Kuttner, A. Van de Walle, L. M. Liz-Marzán and C. Wilhelm, Small, 2020, 16, 1904960 CrossRef CAS.
- Q. Chen, X. Ma, L. Xie, W. Chen, Z. Xu, E. Song, X. Zhu and Y. Song, Nanoscale, 2021, 13, 4855–4870 RSC.
- C. Blanco-Andujar, D. Ortega, P. Southern, S. A. Nesbitt, N. T. K. Thanh and Q. A. Pankhurst, Nanomedicine, 2016, 11, 121–136 CrossRef CAS PubMed.
- Q. Feng, Y. Liu, J. Huang, K. Chen, J. Huang and K. Xiao, Sci. Rep., 2018, 8, 2082 CrossRef PubMed.
- J. C. Fontoura, C. Viezzer, G. Fabiana, R. A. Ligabue, R. Weinlich, R. D. Puga, D. Antonow, P. Severino and C. Bonorino, Mater. Sci. Eng., C, 2020, 107, 110264 CrossRef CAS PubMed.
- J. Majumder, O. Taratula and T. Minko, Adv. Drug Deliv. Rev., 2019, 144, 57–77 CrossRef CAS PubMed.
- C. Chen, S. Zhao, A. Karnad and J. W. Freeman, J. Hematol. Oncol., 2018, 11, 1–23 CrossRef PubMed.
- M. Kesik, B. Demir, F. B. Barlas, C. Geyik, S. C. Cevher, D. O. Demirkol, S. Timur, A. Cirpan and L. Toppare, RSC Adv., 2015, 5, 83361–83367 RSC.
- A. C. Marques, P. J. Costa, S. Velho and M. H. Amaral, J. Controlled Release, 2020, 320, 180–200 CrossRef CAS PubMed.
- R. P. Castillo, M. Colilla and M. Vallet-Regí, Expert Opin. Drug Deliv., 2017, 14, 229–243 CrossRef CAS PubMed.
- W. van Aarle, W. J. Palenstijn, J. de Beenhouwer, T. Altantzis, S. Bals, K. J. Batenburg and J. Sijbers, Ultramicroscopy, 2015, 157, 35–47 CrossRef CAS PubMed.
- K. Sentosun, M. N. Sanz Ortiz, K. J. Batenburg, L. M. Liz-Marzán and S. Bals, Part. Part. Syst. Charact., 2015, 32, 1063–1067 CrossRef CAS.
- J. Reguera, D. Jiménez de Aberasturi, M. Henriksen-Lacey, J. Langer, A. Espinosa, B. Szczupak, C. Wilhelm and L. M. Liz-Marzán, Nanoscale, 2017, 9, 9467–9480 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Further characterisation data and cell assays. See DOI: https://doi.org/10.1039/d3tb00397c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |