
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMagnetocaloric efficiency tuning through solvent-triggered 3D to 2D interconversion in holmium(III)-based dynamic MOFs†
Nadia
El Alouani Dahmouni
a,
Marta
Orts-Arroyo
 b,
Adrián
Sanchis-Perucho
a,
Nicolás
Moliner
a,
Júlia
Mayans
a,
Mario
Pacheco
ac,
Isabel
Castro
*a,
Giovanni
De Munno
b,
Nadia
Marino
*b,
Rafael
Ruiz-García
a and
José
Martínez-Lillo
*a
b,
Adrián
Sanchis-Perucho
a,
Nicolás
Moliner
a,
Júlia
Mayans
a,
Mario
Pacheco
ac,
Isabel
Castro
*a,
Giovanni
De Munno
b,
Nadia
Marino
*b,
Rafael
Ruiz-García
a and
José
Martínez-Lillo
*a
aInstituto de Ciencia Molecular (ICMol), Universitat de València, c/José Beltrán 2, 46980 Paterna (València), Spain. E-mail: isabel.castro@uv.es; f.jose.martinez@uv.es
bCentro di Eccellenza CEMIF.CAL, Dipartimento di Chimica, Università della Calabria, via P. Bucci 14/c, 87030 Arcavacata di Rende (CS), Italy. E-mail: nadia.marino@unical.it
cFacultad de Química, Universidad de la República, Av. Gral. Flores 2124, 11800 Montevideo, Uruguay
First published on 21st June 2024
Abstract
The neutral holmium(III) oxalate octadecahydrate {[Ho2(ox)3(H2O)6]·12H2O}n of mixed hexagonal/decagonal (63·103) 3D net topology shows important changes in the magnetocaloric efficiency upon dehydration/rehydration by heating and water vapor exposition to give the holmium(III) oxalate decahydrate {[Ho2(ox)3(H2O)6]·4H2O}n of hexagonal (63) 2D net topology through the intermediacy of the elusive amorphous anhydrous compound {Ho2(ox)3}n.
Lanthanide-based metal–organic frameworks (Ln MOFs) have attracted considerable attention in the last two decades because of their potential technological applications in cryogenic magnetic refrigeration (CMR).1 Nowadays, the attention in this field is moving toward magnetically anisotropic holmium(III)-based MOFs,2 because they exhibit magnetocaloric effects (MCE) at higher temperatures than their largely investigated magnetically isotropic gadolinium(III) counterparts.3,4 In this respect, metallic holmium itself and the related intermetallic alloys HoB2, HoAl2 and Ho(Co,Ni,Fe)2 are proposed as efficient cryomagnetic coolers operating between the strategically relevant hydrogen and nitrogen liquefaction temperatures,5–8 in place of traditional oxides, such as gadolinium(III)–gallium(III) garnet (GGG) and its dysprosium(III)-substituted derivatives of general formula (DyxGd1−x)3Ga5O12 [x = 0 (GGG), 0.5 (DGGG), and 1 (DGG)], working between the helium and hydrogen liquefaction temperatures.9
Our research focuses on the well-known family of neutral two- and three-dimensional (nD, n = 2 and 3) lanthanide(III) sesquioxalate hydrates (Scheme S1, ESI†).10,11 Herein we report the synthesis, general physico-chemical and structural characterization, thermal stability and water sorption behavior, as well as magnetic and magnetothermal properties of the holmium(III) sesquioxalates hydrates of general formula {[Ho2(ox)3(H2O)6]·xH2O}n [x = 4 (1) and 12 (2)]. Interestingly, a structural interconversion is found within this solvatomorphic pair of neutral oxalato-bridged holmium(III)-based 2D (1) and 3D (2) MOFs upon dehydration/rehydration, which is accompanied by a dramatic magnetocaloric solvatoswitching behavior. This unique feature can be exploited in the proof-of-concept design of a new class of active magnetic regenerators (AMRs) for hydrogen liquefaction based on holmium(III)-based dynamic MOFs.
Slow diffusion of aqueous solutions of Ho(NO3)3·5H2O and H2ox under stoichiometric (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) molar ratios in a H-shaped tube, after several weeks at room temperature, gives a mixture of large plates of 1 and tiny elongated prisms of 2 suitable for single-crystal X-ray diffraction (XRD), whose color varies from yellow to pink depending on the illumination conditions, as it is typical for holmium(III) compounds (Fig. S1, ESI†). Crystals of 1 and 2 were obtained in pure form and good yield [ca. 70 (1)/65% (2)] under the same reaction conditions at 50 and 5 °C, respectively (see Experimental section, ESI†), as revealed by powder XRD (Fig. S2, ESI†). The influence of the reaction temperature on the dimensionality of the resulting lanthanide(III) sesquioxalate hydrates prepared by slow diffusion techniques in a gel medium has been investigated earlier, so that the 2D compounds are favored over the 3D ones at higher temperatures and vice versa.11
:3) molar ratios in a H-shaped tube, after several weeks at room temperature, gives a mixture of large plates of 1 and tiny elongated prisms of 2 suitable for single-crystal X-ray diffraction (XRD), whose color varies from yellow to pink depending on the illumination conditions, as it is typical for holmium(III) compounds (Fig. S1, ESI†). Crystals of 1 and 2 were obtained in pure form and good yield [ca. 70 (1)/65% (2)] under the same reaction conditions at 50 and 5 °C, respectively (see Experimental section, ESI†), as revealed by powder XRD (Fig. S2, ESI†). The influence of the reaction temperature on the dimensionality of the resulting lanthanide(III) sesquioxalate hydrates prepared by slow diffusion techniques in a gel medium has been investigated earlier, so that the 2D compounds are favored over the 3D ones at higher temperatures and vice versa.11
1 and 2 are two different polymorphs (solvatomorphs) that crystallize in the monoclinic and trigonal crystal systems with P21/c and R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) space groups, respectively. The crystal structure is made up of neutral oxalato-bridged holmium(III) hexagonal layers (1) or mixed hexagonal/decagonal nets (2) of (63) and (63·103) topology and overall honeycomb-type architecture, together with both coordinated and a variable amount of disordered hydrogen-bonded crystallization water molecules (Fig. 1), as reported earlier for the related 2D gadolinium(III) and 3D erbium(III) sesquioxalate deca- and octadecahydrates.10,11 Selected crystallographic and structural data for 1 and 2 are given in Tables S1–S4 (ESI†).
space groups, respectively. The crystal structure is made up of neutral oxalato-bridged holmium(III) hexagonal layers (1) or mixed hexagonal/decagonal nets (2) of (63) and (63·103) topology and overall honeycomb-type architecture, together with both coordinated and a variable amount of disordered hydrogen-bonded crystallization water molecules (Fig. 1), as reported earlier for the related 2D gadolinium(III) and 3D erbium(III) sesquioxalate deca- and octadecahydrates.10,11 Selected crystallographic and structural data for 1 and 2 are given in Tables S1–S4 (ESI†).
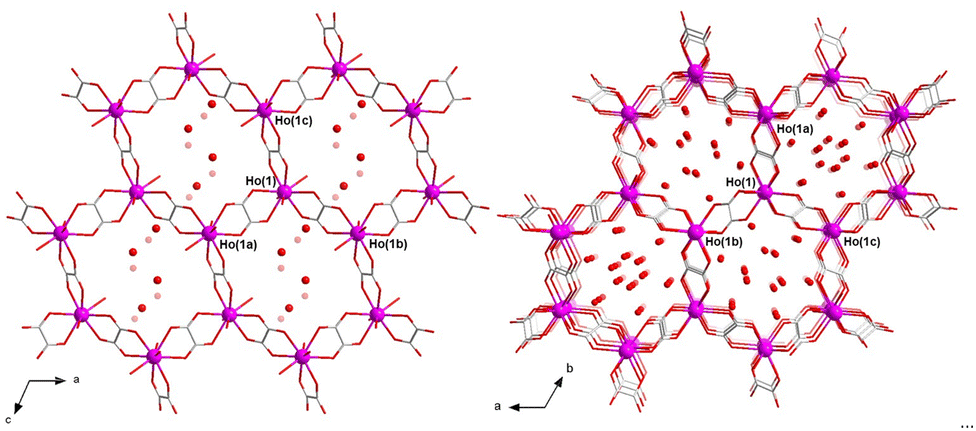 | ||
| Fig. 1 Perspective views of fragments of the neutral oxalato-bridged HoIII hexagonal 2D net of 1 (left) and mixed hexagonal/decagonal 3D net of 2 (right) along the crystallographic b and c axes, respectively [symmetry codes: (a) −x + 1, −y + 1, −z + 1 (1) and x − y + 2/3, x + 1/3, −z + 4/3 (2); (b) −x + 2, −y + 1, −z + 1 (1) and −x + 1, −y + 1, −z + 2 (2); (c) −x + 1, −y + 1, −z (1) and y − 1/3, −x + y + 1/3, −z + 4/3 (2)]. Crystallization water molecules are depicted as red spheres in 2. The crystallization water molecules in 1 are disordered over two positions each; the two alternative sets have both been depicted as dark or light red spheres. H bonds are not shown for clarity. | ||
The HoIII ion of 1 and 2 has a nine-coordinate environment, HoO9, formed by six oxygen atoms from three bis(bidentate) oxalato bridges plus three oxygen atoms from three coordinated water molecules. The metal coordination polyhedra for 1 and 2 are intermediate between tricapped trigonal prism (TCTPR) and monocapped square antiprism (CSAPR) of approximate D3h and C4v molecular symmetries (Tables S2 and S3, ESI†).12 The continuous shape measures (CShM)13 slightly (1) or markedly (2) favor the TCTPR over the CSAPR geometry with a spherical-type distortion of the regular (Johnson-type) polyhedron (Tables S2–S4 and Fig. S3, ESI†).
The asymmetric tris(bidentate) mononuclear [HoIII(ox)3/2(H2O)3] unit of 1 and 2 is chiral, so that the two helical enantiomers are present in the crystal structure (Fig. S4, ESI†).10,11 In both compounds, a regular alternation of (Δ)- and (Λ)-[HoIII(ox)3/2(H2O)3] enantiomers occurs within the resulting achiral oxalato-bridged HoIII layers or nets. In 1, each pair of edge-sharing trinuclear entities around the three oxalate ions per Ho atom adopts a parallel displaced disposition (dihedral angle ϕ = 0°; Table S4 and Fig. S4a, ESI†). Instead, two out of three pairs of edge-sharing trinuclear entities around the three oxalate ions per Ho atom in 2 exhibit a non-planar twisted disposition [ϕ = 49.85(2)°; Table S4, ESI†] (Fig. S4b, ESI†), the remaining one showing a parallel displaced disposition. This feature would be ultimately responsible for achieving a 2D (1) or 3D (2) structure.
The adjacent oxalato-bridged HoIII hexagonal layers in 1, growing in the ac plane and related by a two-fold symmetry axis, are stacked along the crystallographic b axis (Fig. S5a, ESI†). However, they are not eclipsed but displaced by half a unit cell along the crystallographic c axis, giving rise to a densely packed multilayered architecture with an ABABAB sequence. In 2, there are two distinct oxalato-bridged HoIII hexagonal/decagonal nets, related by a simple translation along the c axis, which are mutually interpenetrated in an eclipsed manner (Fig. S5b, ESI†). The intermetallic distances across the oxalato bridges are slightly larger in 2 than in 1 [Ho⋯Ho = 6.245–6.395 (1)/6.312–6.415 Å (2); Table S4, ESI†].
Overall, the parallel-displaced layer arrangement of 1 leads to small hexagonal pores along the [011] direction (Fig. S6a, ESI†), which are filled by the disordered crystallization water molecules that form an hydrogen-bonded 2D reticular net perpendicular to the oxalate-bridged HoIII hexagonal layers (O⋯O = 2.67–3.04 Å) (Fig. S5a, ESI†). In contrast, the doubly interpenetrated (eclipsed) architecture of 2 leads to large hexagonal channels along the [001] direction (Fig. S6b, ESI†), whereby the crystallization water molecules are placed. This gives rise to hydrogen-bonded supramolecular 1D motifs, which ultimately also interact with the oxalate-bridged HoIII 3D net through multiple H-bonds, overall involving both the coordinated and uncoordinated water molecules and the oxalato groups (O⋯O = 1.95–2.94 Å) (Fig. S5b, ESI†). The estimated solvent accessible void (V0) represents about 22 (1) and 35% (2) of the unit cell volume (Table S4, ESI†), thus reflecting the greater porosity of 2 with respect to 1.
The thermal stability of powdered crystalline samples of 1 and 2 has been investigated by thermogravimetric analysis (TGA) under a dry N2 atmosphere. The TGA profiles show an abrupt mass loss from room temperature to around 125 (1) or 100 °C (2), which is followed by a gradual mass loss under further heating just before the decomposition of the HoIII oxalate to the corresponding oxide above 400 °C (Fig. S7, ESI†).11 The percentage values of mass loss (1 − m/m0) are 18.5 (1) and 35% (2) at 300 °C, which would correspond to the release of 8 (1) and 18 H2O (2) molecules per formula unit.
The anhydrous derivative of formula {HoIII2(ox)3}n (3) was then obtained by thermal dehydration of 2 following a slow heating from room temperature to 300 °C under a dry N2 atmosphere (see ESI†). 3 is moisture stable, as revealed by the TGA profile after one day of exposition in the open air at room temperature (Fig. S7, ESI†). Hence, it shows a broad plateau under heating from room temperature to approximately 300 °C, with no mass recovery. The large thermal stability of 3 is remarkable, being a desired choice criterion for practical applications of HoIII sesquioxalate hydrates as cryogenic magnetic coolers when supported on a convenient host device.14 Nevertheless, a partial degree of rehydration occurs in a saturated water vapor atmosphere, as revealed by the TGA profiles (Fig. S7, ESI†). Hence, the percentage values of mass recovery vary from 10 up to 20% after three days or three weeks of exposition of 3 in a closed flask containing water, corresponding to the recovery of 5 and 10 H2O molecules per formula unit, respectively.
The water desorption/adsorption properties of 2 have been further investigated by powder XRD (Fig. 2). The powder XRD pattern of the dehydrated amorphous derivative 3 obtained after heating at 300 °C under a dry N2 atmosphere shows no peaks and remains almost unaltered after one day of exposition in the open air at room temperature (Fig. 2b). After three weeks of exposition to a water vapor saturated atmosphere, however, some peaks appear in the powder XRD pattern. Remarkably, these peaks do not correspond to the original octadecahydrate 3D precursor 2, but they are characteristic of the decahydrate 2D analog 1 (Fig. 2c). This one-way crystalline-to-amorphous-to-crystalline 3D to 2D interconversion upon dehydration/rehydration has been monitored by powder XRD (Fig. S8, ESI†). The thermal amorphization process occurs rapidly upon loss of the first crystallization water molecules, so that it is almost completed at 60 °C with no further change up to 300 °C. On the contrary, the water uptake is a very slow process, being only completed after several weeks (inset of Fig. S7, ESI†).
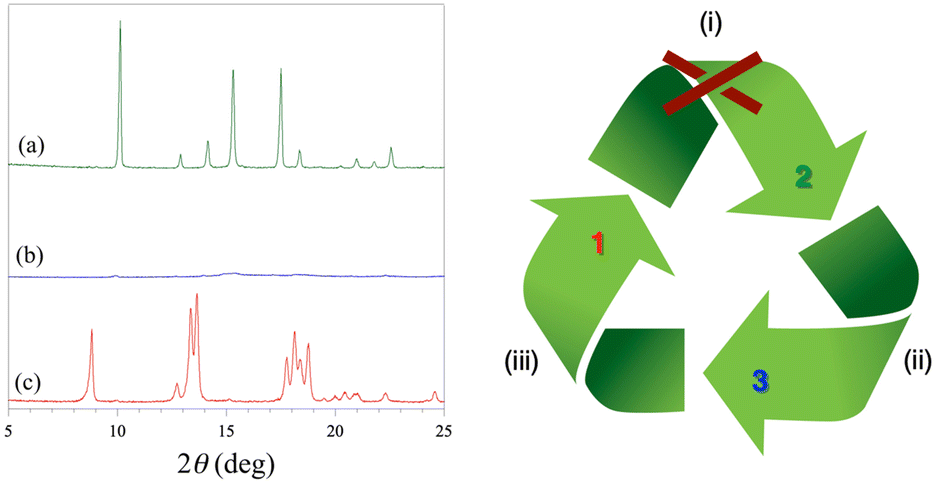 | ||
| Fig. 2 Powder X-ray diffractograms (left) of 2 (a) and the resulting dehydrated 3 (b) and rehydrated 1 (c) derivatives obtained after heating at 300 °C under a dry N2 atmosphere (ii) and subsequent exposition to a saturated water vapor atmosphere for 3 weeks (iii), respectively, as illustrated in the flow diagram (right). Attempts to transform 1 to 2 by prolonged exposition to a saturated water vapor atmosphere or by immersion in water (i) were unsuccessful (red cross in the flow diagram). | ||
The static magnetic properties of 1–3 have been investigated by variable-temperature (T = 2–300 K) direct current (dc) magnetic susceptibility measurements and variable-field (H = 0–8 T) magnetization measurements (see ESI†). The χMT vs. T (χM being the molar dc magnetic susceptibility per formula unit and T the absolute temperature) and M vs. H plots (M being the molar magnetization per formula unit and H the applied dc magnetic field) are qualitatively similar, regardless of the hydration degree along this series (Fig. S9 and S10, ESI†). The magnetic behavior is dominated by the high local magnetic anisotropy of the HoIII ions, possessing a large first-order spin–orbit coupling (SOC). Yet the weak but non-negligible magnetic exchange interactions between the magnetically anisotropic HoIII ions across the oxalato bridge cannot be neglected, as earlier reported for the 2D GdIII sesquioxalate decahydrate of formula {[Gd2(ox)3(H2O)6]·4H2O}n.10
The χMT values at room temperature of 1–3 are close to that calculated for two 4f10 HoIII ions with a 5I8 ground state [χMT = (2Nβ2gJ2/3kB)J(J + 1) = 28.1 cm3 mol−1 K and gJ = 3/2 + [S(S + 1) − L(L + 1)]/2J(J + 1) = 5/4 with S = 2, L = 6, J = 8].15 Upon cooling, χMT continuously decreases down to 6.2 (1)/5.0 (2)/10.2 cm3 mol−1 K (3) at 2 K (Fig. S9, ESI†). However, there is no maximum in the χMvs. T plots (inset of Fig. S9, ESI†), suggesting that the long-range antiferromagnetic order would occur (if any) below 2 K. In this case, the ligand field (LF) effects cause the zero-field splitting (ZFS) of the seventeen (2J + 1) components of the 5I8 ground state into one singlet and eight Kramers doublets (mJ = 0, ±1, ±2, ±3, ±4, ±5, ±6, ±7, and ±8), which is ultimately responsible for the high local magnetic anisotropy of the HoIII ions possessing the highest J value along the lanthanide series. This situation is clearly reflected in the normalized isothermal magnetization curves in the temperature range 2–20 K (Fig. S10, ESI†). Hence, they do not superimpose, reflecting thus the relative importance of the LF effects depending on the particular coordination geometry of the HoIII ions. The maximum M values [11.6 (1)/12.3 (2)/10.8 Nβ (3)] for H = 8 T at T = 2 K are well below the saturation value for the sum of two heptadecyl (2J + 1 = 17) ground states [Ms = 2gJJNβ = 20Nβ with J = 8 and gJ = 5/4]. Anyway, M reaches up to ca. 60 (1)/55 (2)/70% (3) of the maximum value for H = 2 T at T = 2 K, which is sufficiently close to the stronger field that can be achieved with the commercial permanent magnets. These two features (high maximum M values and large slope of the isothermal magnetization curve) are mandatory for the potential applications of HoIII sesquioxalates in CMR.
The magnetothermal properties of 1–3 have been investigated by variable-temperature (T = 2–20 K, with ΔT = 1 K) and variable-field (H = 0–8 T, with ΔH = 0.2 T) magnetization measurements (see ESI†). The temperature and field dependence of the magnetic entropy change (ΔSM), upon connecting the magnetic field (ΔH = H − H0 with H0 = 0), can then be estimated from the M vs. T and H plots (M being the gravimetric magnetization per formula unit) according to the Maxwell equation (Fig. S11 and S12, ESI†).10
The isothermal curves show a rapid saturation of the magnetic entropy change with increasing the magnetic field, so they tend asymptotically to a thermally dependent value, which is reached more quickly at lower temperatures (Fig. S12, right, ESI†). The −ΔSM values in gravimetric units for ΔH = 8 T vary from 2.1 (1)/1.8 (2)/8.2 J kg−1 K−1 (3) (T = 2 K) to 10.6 (1)/8.1 (2)/13.0 J kg−1 K−1 (3) (T = 10 K) (Fig. 3, left), being comparable to those of the commercial cryogenic material GGG [−ΔSM = 6.5 (T = 2 K) and 28.0 J kg−1 K−1 (T = 10 K) for ΔH = 8 T].9a Moreover, the −ΔSM values in molar units (per mol of lanthanide atom) for ΔH = 2 T [1.3 (1), 1.0 (2), and 1.8 J mol−1 K−1 (3) at T = 5 K] are similar to those reported for GGG and its DyIII-substituted derivatives DGGG and DGG [−ΔSM = 3.1–3.3 J mol−1 K−1 for ΔH = 2 T (T = 5 K)] (Fig. S13, ESI†).9d
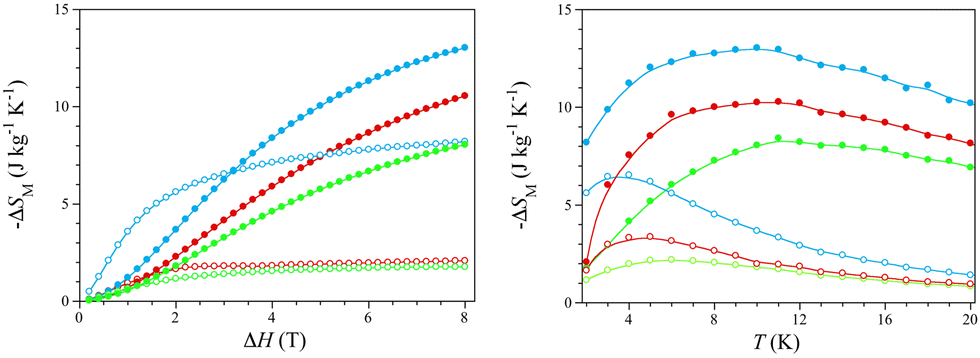 | ||
| Fig. 3 Isothermal (left) and isofield (right) curves of the magnetic entropy change for 1 (red), 2 (green), and 3 (blue) at 2 (○)/10 K (•) and 2 (○)/8 T (•), respectively. The solid lines are only eye-guides. | ||
The overall enhancement of the MCE efficiency in the order 2 < 1 < 3 is as expected because of the molecular weight decrease upon water loss along this series of empirical formula Ho2(ox)3·nH2O [MW = 747 (1), 918 (2), and 594 g mol−1 (3) with n = 10 (1), 18 (2), and 0 (3)]. The maximum −ΔSM values in molar units (per mol of lanthanide atom) for ΔH = 8 T are similar [3.8 (1), 3.7 (2), and 3.9 J mol−1 K−1 (3) at T = 10 K], regardless of the water content, being rather lower than the limiting value for one HoIII ion with no ZFS [−ΔSM = R![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) ln(2J + 1) = 23.6 J mol−1 K−1 with J = 8]. This almost six-fold global reduction of the MCE efficiency is expected because of its high magnetic anisotropy, as discussed above.
ln(2J + 1) = 23.6 J mol−1 K−1 with J = 8]. This almost six-fold global reduction of the MCE efficiency is expected because of its high magnetic anisotropy, as discussed above.
On the other hand, the isofield curves show a maximum of the magnetic entropy change, which dramatically shifts toward higher temperatures with increasing the magnetic field (Fig. S12, left, ESI†). Hence, the −ΔSM maxima cover a wide temperature range, far above the He liquefaction temperature [Tm = 4.8–10.5 (1), 6.0–11.2 (2), and 3.6–9.9 K (3) for ΔH = 2–8 T] (Fig. 3), being even farther than that found for the Ho(III)/Na(I)-based 3D MOF of formula {[Na2HoIII(DTPA)(H2O)]·8H2O}n (DTPA = diethylenetriamine-N,N,N′,N′′,N′′-pentaacetate) [Tm = 3.2–9.5 K for ΔH = 2–8 T].2
The overall enhancement of the optimal working temperature in the order 3 < 1 < 2 is just the opposite to that mentioned above for the −ΔSM values. This feature is also related to the MCE efficiency and, eventually, to the practical applications of HoIII sesquioxalates as cryomagnetic coolers operating near the strategically relevant H2 liquefaction. So, for instance, GGG and its DyIII-substituted derivatives DGGG and DGG have Tm values in a narrower and lower temperature range around the He liquefaction [1.15–1.45 (GGG), 1.25–1.55 (DGGG), and 1.05–2.70 K (DGG) for ΔH = 1–4 T] (Fig. S13, ESI†).9d Yet the Tm values for 1–3 are still far from those found for metallic Ho and the intermetallic alloys HoB2, HoAl2 and Ho(Co,Ni,Fe)2, recently proposed as cryomagnetic coolers between the H2 and N2 liquefaction temperatures.5–7
In summary, the HoIII sesquioxalate deca- and octadecahydrates 1 and 2 constitute a remarkable solvatomorphic pair of 2D and 3D HoIII MOFs. Upon thermal dehydration of 2 and subsequent vapor rehydration, the resulting anhydrous intermediate 3 slowly converts to 1 in a unique crystalline-to-amorphous-to-crystalline transformation. This interconvertible polyreticular series 1–3 exhibits moderate to large MCE with magnetic entropy change maxima at relatively high temperatures, between He and H2 liquefaction. These features allow this novel class of dynamic nD HoIII MOFs (n = 2 and 3) to be proposed as candidates to solvatoswitchable molecular cryomagnetic refrigerant materials instead of traditional oxides, such as GGG and its DyIII derivatives working in the very low-temperature regime, just above the He liquefaction temperature. Current efforts are being devoted to increasing the CMR performance just above the H2 liquefaction temperature before real applications as AMRs can be envisaged.
J. M.-L. and I. C. conceived the project; J. M.-L. and N. Marino designed the experiments; N. E. A. D., M. O.-A., A. S.-P. and M. P. synthesized and characterized the complexes; N. Moliner, N. Marino and G. D. M. carried out the crystallographic study; N. E. A. D., N.Moliner, J. M. and R. R.-G. performed the magnetic and magnetocaloric study; R. R.-G., M. P., N. M. and J. M.-L. wrote and/or reviewed the manuscript. All authors have read and agreed to the published version of the manuscript.
This work was supported by the Spanish MINECO (PID2019-109735GB-I00 and CEX2019-000919-M), the Generalitat Valenciana (AICO/2020/183 and AICO/2021/295) and the Italian MUR through the ElectroLight4Value project (PRIN 2020, 2020927WY3). M. P. acknowledges the PEDECIBA and CSIC for a postdoctoral stage. M. O.-A. thanks the Centro di Eccellenza CEMIF.CAL for a postdoctoral contract.
Data availability
The data provided in this investigation are available from the corresponding authors on reasonable request.Conflicts of interest
There are no conflicts to declare.References
- M. Evangelisti, Molecule-based magnetic coolers: measurement, design and application in Molecular Magnets: Physics and Applications, ed. J. Bartolomé, F. Luis and J. F. Fernández, Springer-Verlag, Heidelberg, 2014, ch. 14, pp. 365–387 Search PubMed.
- B. Rodriguez-Barea, J. Mayans, R. Rabelo, A. Sanchis-Perucho, N. Moliner, J. Martínez-Lillo, M. Julve, F. Lloret, R. Ruiz-García and J. Cano, Inorg. Chem., 2021, 60, 12719–12723 CrossRef CAS PubMed.
- P. Konieczny, W. Sas, D. Czernia, A. Pacanowska, M. Fitta and R. Petka, Dalton Trans., 2022, 51, 12762–12780 RSC.
- X. Zhong, J.-J. Hu, S.-L. Yao, R.-J. Zhang, J.-J. Wang, D.-G. Cai, T.-K. Luo, Y. Peng, S.-J. Liu and H.-R. Wen, CrystEngComm, 2022, 24, 2370–2382 RSC.
- S. Baran, R. Duraj and A. Szytula, Acta Phys. Pol., A, 2015, 127, 815–817 CrossRef.
- B. De Castro, K. Terashima, T. D. Yamamoto, Z. Hou, S. Iwasaki, R. Matsumoto, S. Adachi, Y. Saito, P. Song, H. Takey and Y. Takano, NPG Asia Mater., 2020, 12, 1–7 CrossRef.
- N. Terada and H. Mamiya, Nat. Commun., 2021, 12, 1212 CrossRef CAS PubMed.
- (a) Y. Zhu, K. Asamoto, Y. Nishimura, T. Kouen, S. Abe, K. Matsumoto and T. Numazawa, NPG Asia Mater., 2011, 51, 494–498 CAS; (b) X. Tang, H. Sepehri-Amin, N. Terada, A. Martin-Cid, I. Kurniawan., S. Kobayashi, Y. Kotani, H. Takeya, J. Lai, Y. Matsushita, T. Ohkubo, Y. Miura, T. Nakamura and K. Hono, Nat. Commun., 2022, 13, 1817 CrossRef CAS PubMed.
- (a) B. Daudin, R. Lagnier and B. Salce, J. Magn. Magn. Mater., 1982, 27, 315–322 CrossRef CAS; (b) P. Schiffer, A. P. Ramirez, D. A. Huse and A. J. Valentino, Phys. Rev. Lett., 1994, 73, 2500–2503 CrossRef CAS PubMed; (c) P. Schiffer, A. P. Ramirez, D. A. Huse, P. L. Gammel, U. Yaron, D. J. Bishop and A. J. Valentino, Phys. Rev. Lett., 1995, 74, 2379–2382 CrossRef CAS PubMed; (d) T. Numazawa, K. Kamiya, T. Okano and K. Matsumoto, Physica B, 2003, 329–333, 1656 CrossRef CAS.
- M. Orts-Arroyo, R. Rabelo, A. Carrasco-Berlanga, N. Moliner, J. Cano, M. Julve, F. Lloret, G. De Munno, R. Ruiz-García, J. Mayans, J. Martínez-Lillo and I. Castro, Dalton Trans., 2021, 50, 3801–3805 RSC.
- D. Kustaryono, N. Kerbellec, G. Calvez, S. Freslon, C. Daiguebonne and O. Guillou, Cryst. Growth Des., 2010, 10, 775–781 CrossRef CAS.
- A. Ruiz-Martínez, D. Casanova and S. Alvarez, Chem. – Eur. J., 2008, 14, 1291–1303 CrossRef PubMed.
- 13 SHAPE (Version 2.1), Electronic Structure Group, Universitat de Barcelona, 2013.
- G. Lorusso, E. Natividad, M. Evangelisti and O. Roubeau, Mater. Horiz., 2019, 6, 144–154 RSC.
- O. Kahn, Molecular Magnetism, VCH Publishers, New York, 1993 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional synthetic, structural, physicochemical characterization, magnetic and magnetocaloric data. CCDC 2351940 and 2351941. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc02539c |
| This journal is © The Royal Society of Chemistry 2024 |