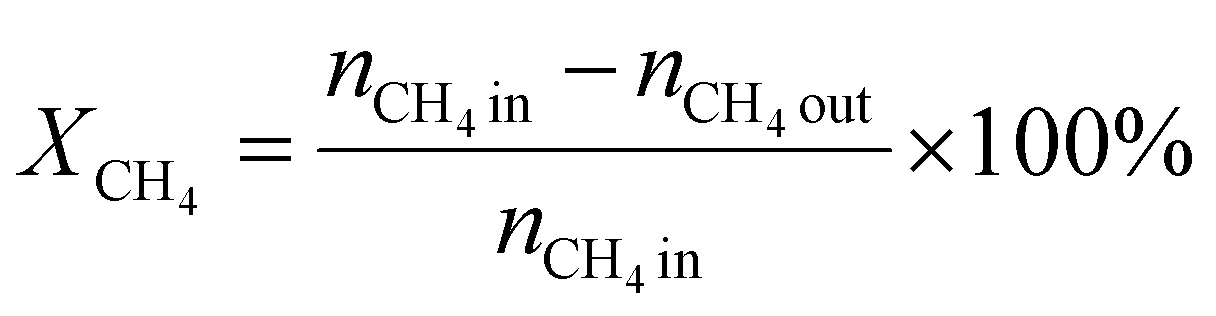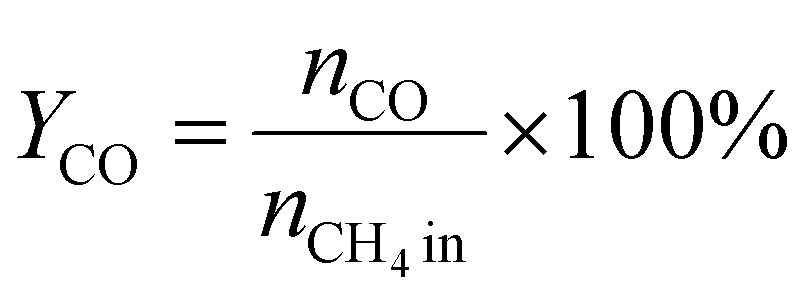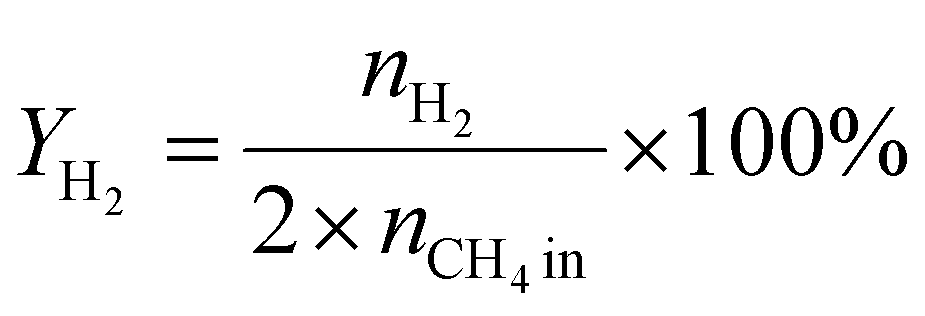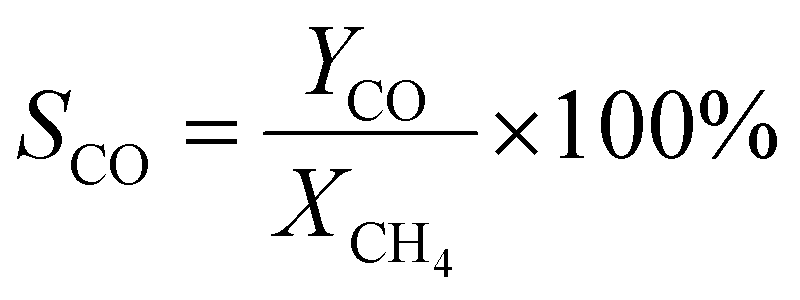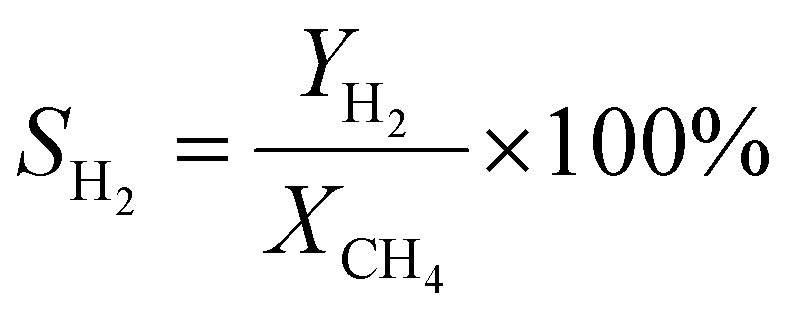DOI:
10.1039/D4DT02421D
(Paper)
Dalton Trans., 2024,
53, 18021-18026
Catalytic partial oxidation of methane over oxide-ion-conductive lanthanum silicate apatites†
Received
26th August 2024
, Accepted 18th October 2024
First published on 21st October 2024
Abstract
A lanthanum silicate La9.33Si6O26 (LSO) crystallizes in an apatite-type structure and has been known as a promising oxide-ion conductor. Here, we report the activity of LSO for catalytic partial oxidation of methane (CPOX) to synthesis gas. The LSO catalyst demonstrated relatively high catalytic activity from 500 to 700 °C, with CH4 conversion reaching 22.1% at 700 °C while retaining moderate CO and H2 selectivities of 20–60%. Notably, LSO exhibited higher CPOX activity than non-apatite-type La2SiO5 despite their similar specific surface areas. The higher CPOX activity of LSO is likely attributed to its structural superiority involving mobile oxide ions in the crystal structure. The reaction kinetic study showed that the reaction orders for methane and oxygen in the CPOX reaction over the LSO catalyst were 0.69–0.73 and 0.08–0.21, respectively. Furthermore, the small contribution of adsorbed O species generated from gas-phase O2 molecules indicated that the lattice oxygen may be involved in the reaction mechanism. The kinetic isotope effect (KIE) study using a CD4 suggested that C–H bond breaking is the rate-determining step of CPOX over LSO.
Introduction
Methane is the primary component of natural gas and a vital carbon feedstock due to its abundance. However, converting methane into value-added chemicals, such as methanol and ethylene, is challenging because of its high stability and the complexity of multistep reactions. This challenge has driven research on selective methane oxidation. One approach to selective methane conversion is a direct route that yields products from methane in a single step. Alternatively, an indirect process converts methane into syngas, an intermediate raw material, which offers several advantages. Syngas, composed of CO and H2, is industrially significant as a precursor for synthesizing valuable chemicals such as acetic acid and methanol.1 Furthermore, syngas production is more energy-efficient than direct routes, which typically suffer from low yield problems.2
Various processes to produce syngas include dry reforming of methane (DRM) (1), steam reforming of methane (SRM) (2), catalytic partial oxidation of methane (CPOX) (3), and autothermal reforming of methane (ATR) (4), which combines SRM and CPOX. Among these, CPOX potentially offers notable benefits. It requires less energy input owing to its exothermic nature, in contrast to the endothermic processes of DRM and SRM. Furthermore, this reaction produces syngas with an H2 to CO ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, which is optimal for methanol synthesis.
:1, which is optimal for methanol synthesis.
| | | CH4 + x/2O2 + (1 − x)H2O → CO + (3 − x)H2 | (4) |
Catalytic oxidation reactions generally involve an oxygen transfer step, where the oxygen species migrates from the catalyst surface to the reactant. To ensure the smooth process of oxygen transfer, oxygen mobility on catalysts is important due to its impact on reactivity. High oxygen mobility allows for fast surface diffusion and absorption into the subsurface, thereby affecting the reaction rate.3 Moreover, catalysts with oxide-ion conductive capability can supply active oxygen species to active sites, further enhancing catalytic activity.4
Previous research has shown that high oxide ion mobility in the catalyst correlates with faster oxygen transfer, thereby influencing the reaction rate. For example, Zhu et al. demonstrated that Y-doped ZrO2 facilitates a faster CPOX reaction than pure ZrO2 because of the enhanced oxygen diffusion.5 In the study on n-butene oxidative dehydrogenation over γ-Bi2MoO6, Jung et al. observed that catalytic performance is closely related to the oxygen mobility of the catalyst.6 Yan et al. investigated the oxidative dehydrogenation of 1-butene with CO2 over Fe2O3/γAl2O3 catalyst and found that the catalytic activity increases linearly with oxygen mobility.7 Borchert et al. reported that increased oxygen conductivity improves the C2 selectivity during the oxidative coupling of methane (OCM) over metal oxide-doped La2O3.8 Whereas OCM does not involve the oxygen species migration to the reactant (CH4), the conductivity would still benefit OCM by enhancing oxygen adsorption and its transformation into lattice oxygen. Furthermore, it was found that oxygen-conducting catalysts (Pt/(Ce0.71Gd0.29)O2−x) suppress carbon deposit formation more effectively compared to non-oxygen conducting catalysts (Pt/γAl2O3).9
Motivated by these findings, we conducted screening tests for CPOX activity on various compounds exhibiting oxide ion conductivity capabilities. Notably, high CPOX activity was discovered in a lanthanum silicate La9.33Si6O26, hereafter denoted as “LSO”, which possesses an apatite-type structure. Many research groups have previously focused on the ionic conductive capability of LSO.10–14 Nevertheless, to our knowledge, no studies have dealt with the CPOX catalytic activity of LSO. Here, we report the CPOX activity of the LSO catalyst, demonstrating its high selectivity for syngas and superior catalytic activity compared to a non-apatite-type lanthanum silicate.
Experimental
Catalyst preparation
The compounds investigated in this study were synthesized using a solid-state reaction method. The target chemical compositions, starting reagents used, and synthesis conditions for these compounds are summarized in Table S1 of the ESI.† Polycrystalline samples of LSO and its substituted derivatives were also prepared. The phase purity of the resultant samples was verified using X-ray powder diffraction (XRD; Rigaku Ultima IV) with Cu-Kα radiation. Nitrogen gas adsorption/desorption isotherms were measured at 77 K (Microtrac BEL, BELSORP-mini II), and the specific surface area was calculated using the Bruauer–Emmett–Teller (BET) method.
Catalytic activity tests
A 50 mg catalyst was loaded into a quartz tube (inner diameter ≈ 0.5 cm) and supported by quartz wool on both sides. The tube, with both ends connected to a gas line, was positioned inside a tubular furnace. A gas mixture containing CH4 (2.5 mL min−1), O2 (1.25 mL min−1), and N2 (6.25 mL min−1) was passed through the catalyst before the temperatures were raised. The effluent gas was analyzed using a gas chromatograph (GC-TCD; INFICON Micro GC Fusion) connected to the gas line outlet. The conversion, yield, and selectivity were calculated using the following formulas, where X, Y, and S represent conversion, yield, and selectivity, respectively.| | 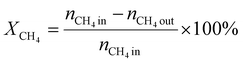 | (5) |
| | 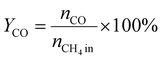 | (6) |
| | 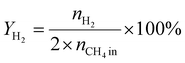 | (7) |
| | 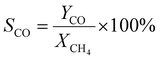 | (8) |
| | 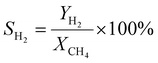 | (9) |
The kinetic isotope effect (KIE) was investigated using equipment similar to that employed for the conventional catalytic activity tests. The catalytic activity of a 100 mg catalyst was measured with a gas mixture containing either normal methane or deuterated methane (CH4 or CD4, 5 mL min−1), O2 (2.5 mL min−1), and N2 (12.5 mL min−1). The KIE value was calculated from the ratio of the CH4 oxidation rate to the CD4 oxidation rate.
Results and discussion
Screening tests for CPOX activity
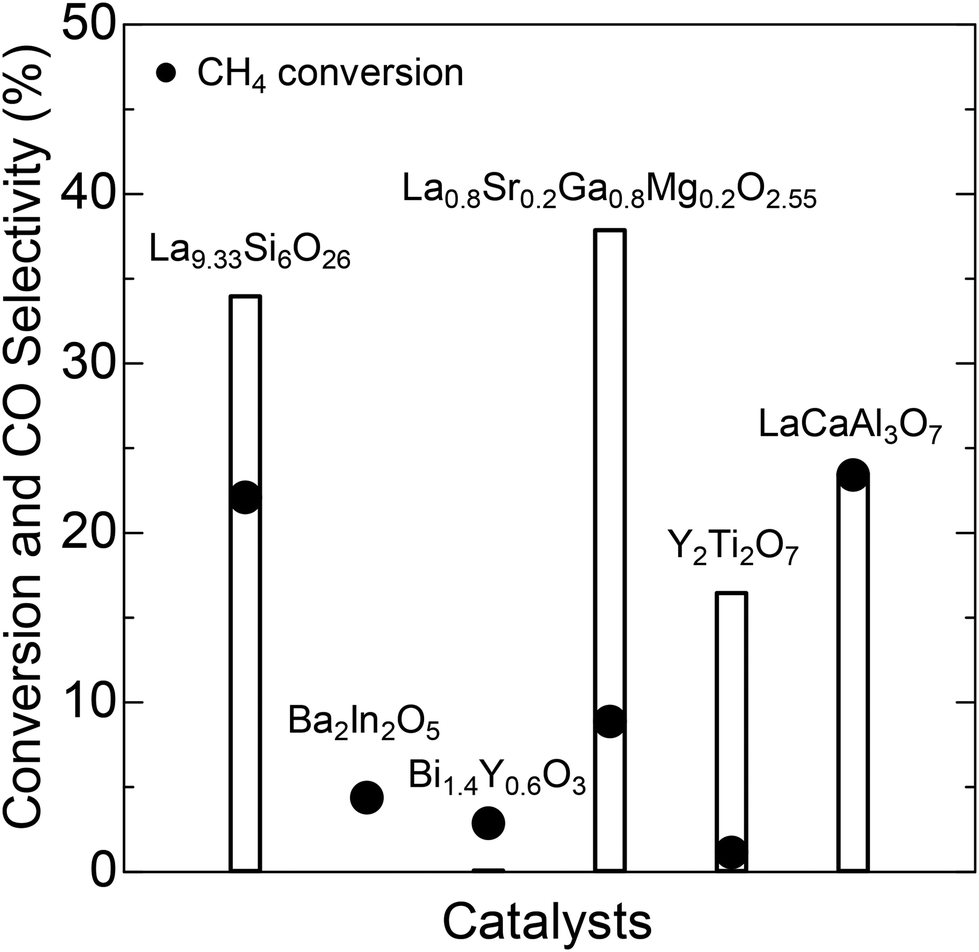
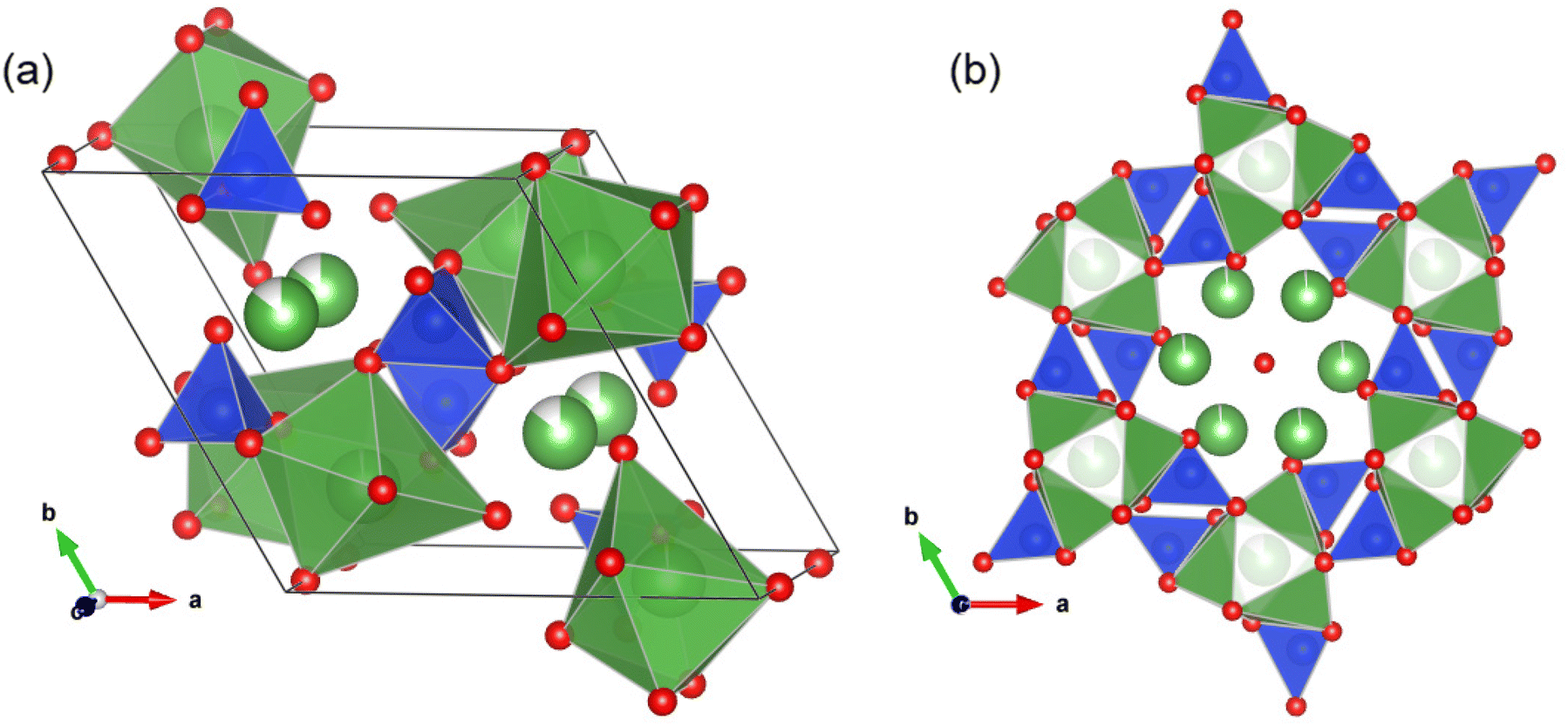
The compounds tested as CPOX catalysts include perovskite-type La0.8Sr0.2Ga0.8Mg0.2O2.55 (LSGM), brownmillerite-type Ba2In2O5 (BIO), fluorite-type Bi1.4Y0.6O3 (BYO), pyrochlore-type Y2Ti2O7 (YTO), and melilite-type LaCaAl3O7 (LCA). As demonstrated in Fig. 1, the CPOX activity appears to depend strongly on the chemical composition and/or crystal structure of the catalysts. LSGM and LCA exhibit relatively high conversion and CO selectivity. Similarly, YTO displays high conversion but low selectivity. We also discovered high CPOX activity in the apatite-type La9.33Si6O26 (LSO). Fig. 2a presents a schematic illustration of the LSO crystal structure.15,16 Apatites with the general formula of A10−x(BO4)6Xy typically belong to the P63/m (176) space group within a hexagonal system. In this formula, “A” represents cations such as Na, Ca, La, etc.; the BO4 tetrahedral group includes SiO44−, PO43−, etc.; and “X” denotes anions, including O2−, OH−, F−, etc.17 The anions “X” occupy channels along the c-axis and exhibit high mobility, which is responsible for the unique ionic conductivity of apatites.
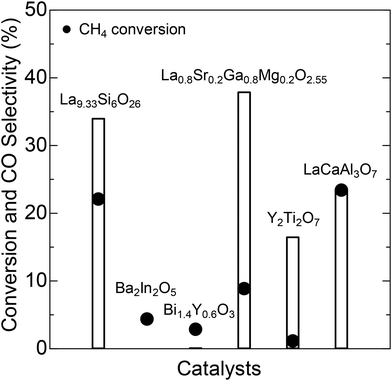 |
| | Fig. 1 CH4 conversion and CO selectivity (at 700 °C) over various catalysts exhibiting oxide-ion-conductive capabilities. | |
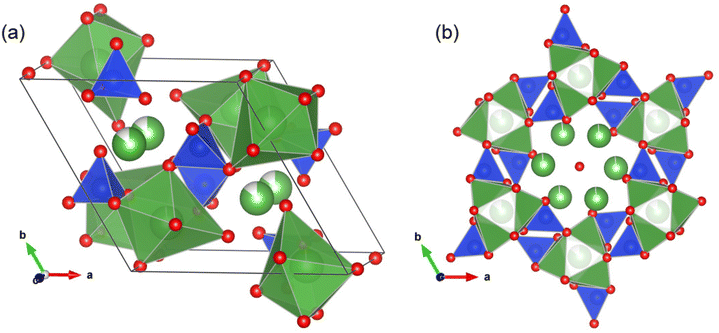 |
| | Fig. 2 (a) Schematic representation of the La9.33Si6O26 apatite structure drawn with VESTA software.15 (b) La and Si polyhedra enclose a one-dimensional tunnel containing mobile oxide ions (a red sphere in the middle). Color coding: green = La, blue = Si, red = O. Crystallographic data were extracted from ref. 16 | |
Catalyst characterization of LSO
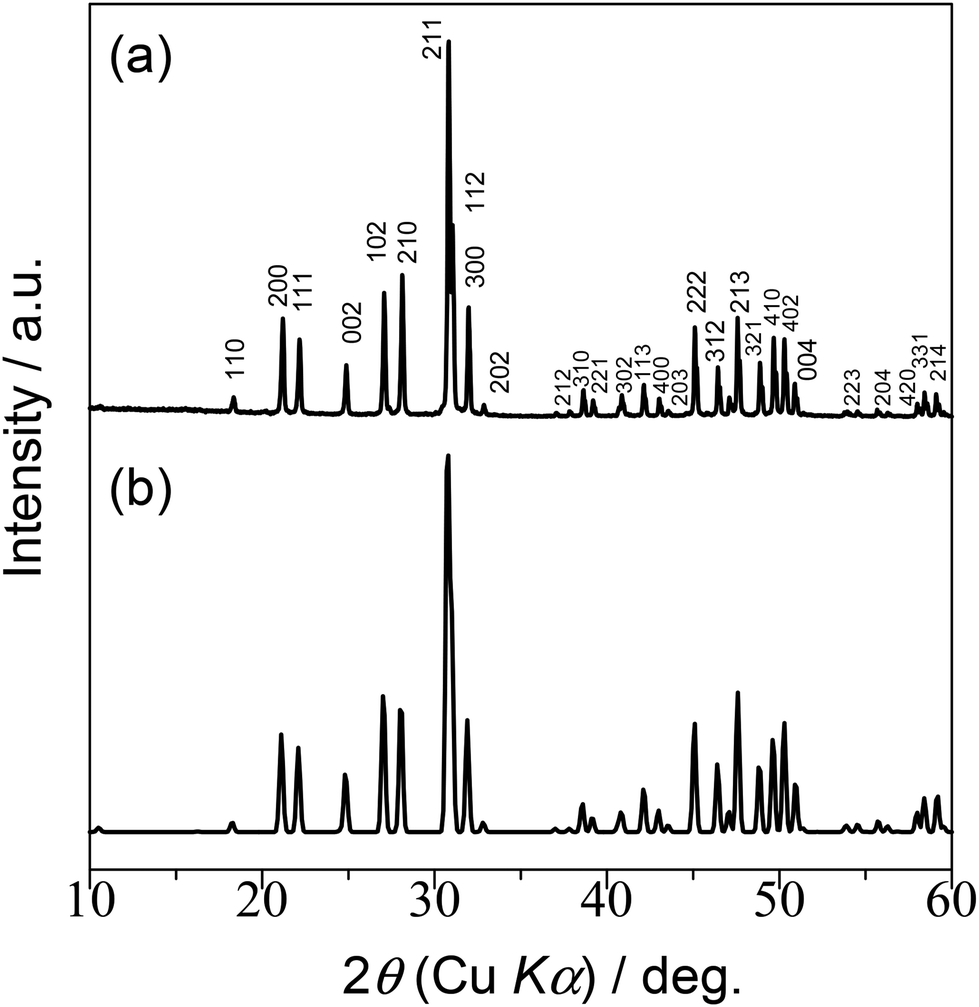
XRD analysis indicates that the diffraction pattern for the LSO sample is essentially identical to that previously reported for apatite-type La9.33Si6O2618 (Fig. 3). The lattice parameter values agree with those for the reference (Table S2†). No diffraction peaks from secondary phases are visible, demonstrating the high quality of the sample. The specific surface area of the sample is estimated to be 0.32 m2 g−1. The low value of the surface area is indicative of the presence of coarse grains with high crystallinity.
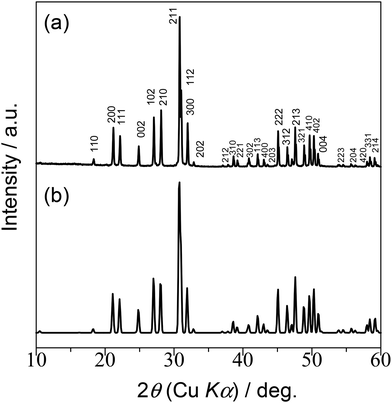 |
| | Fig. 3 XRD pattern of LSO from (a) this work and (b) ref. 18 | |
Catalytic activity of LSO
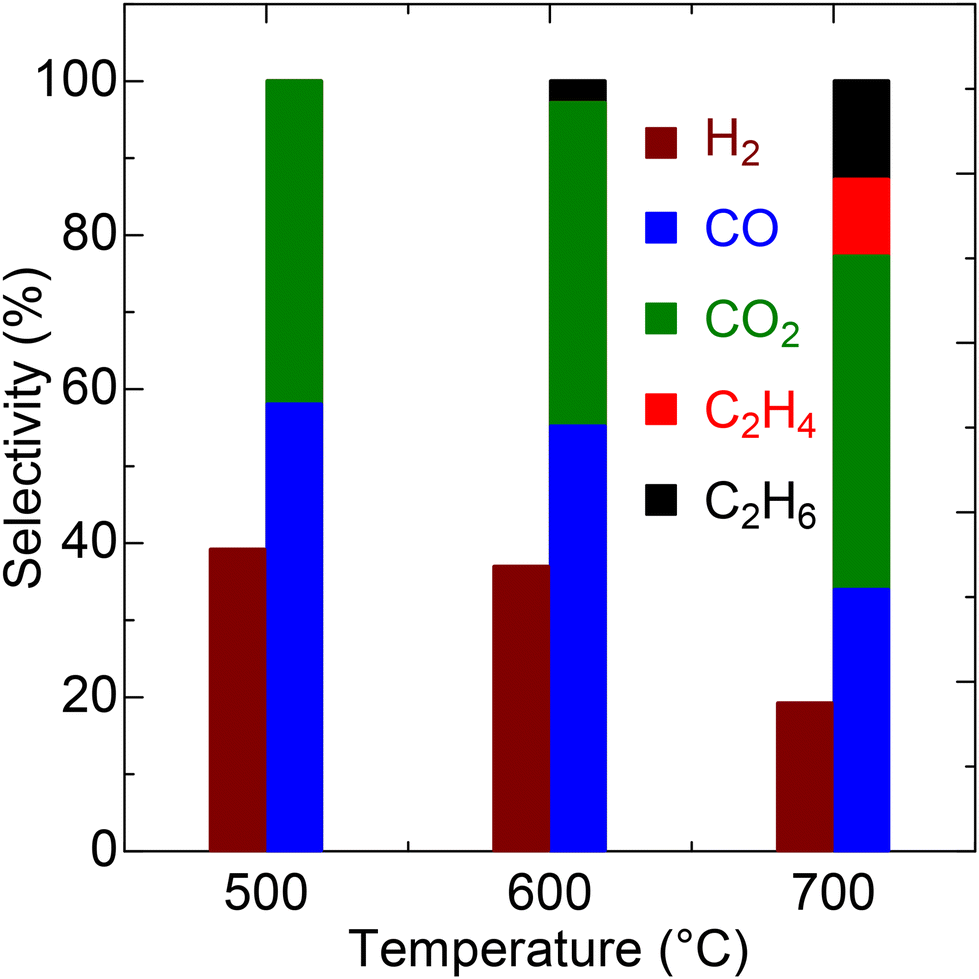
The LSO catalyst predominantly produces CO as the main carbon product at 500 and 600 °C (Fig. 4 and Table S3†). The CO selectivity exceeds 50% at these temperatures, with a downward trend at higher temperatures. Conversely, the CO2 selectivity remains relatively constant at approximately 40% from 500 to 700 °C. Additionally, a small amount of C2 hydrocarbons, likely resulting from OCM, is observed, with C2 selectivity surpassing 20% at 700 °C. Hydrogen evolution exhibits a similar trend to that of CO, with the highest H2 selectivity recorded at 39.2% at 500 °C, decreasing progressively to 19.2% at 700 °C due to the successive oxidation to water at elevated temperatures. The long-term catalytic test was conducted on LSO at 700 °C for 72 h. As demonstrated in Fig. S1,† LSO exhibits stable conversion and syngas selectivity throughout the reaction period. This result indicates that the catalyst maintains high stability during extended catalytic testing.
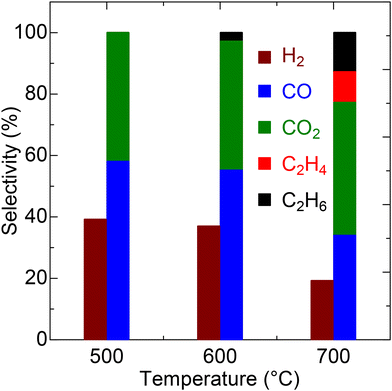 |
| | Fig. 4 Selectivity values for LSO in CPOX at different temperatures. | |
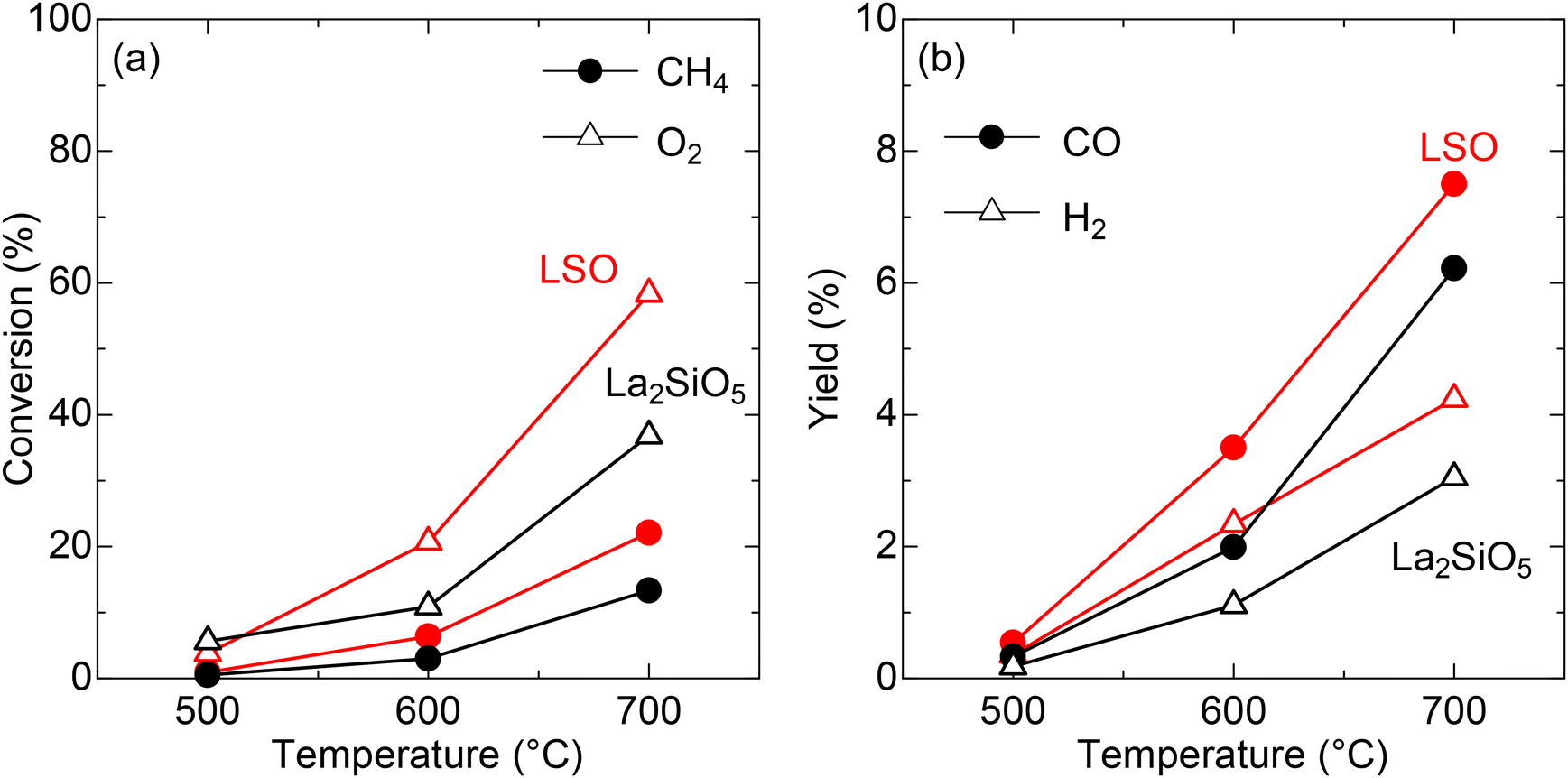
To discuss the crystallographic superiority of LSO on the CPOX performance, the catalytic activity was compared to that of La2SiO5, a non-apatite-type lanthanum silicate (Fig. S2 and Table S4†). As discussed in the previous section, LSO has mobile oxide ions in its structure (Fig. 2b). These anions are prone to oxide ion conduction and can promote catalytic oxidation.19 Moreover, oxide ion conductivity facilitates oxygen supply and, consequently, enhances the reactivity.20,21 Unlike LSO, La2SiO5 possesses a monoclinic structure22 and does not contain mobile oxide ions. As presented in Fig. 5a, b and Table S3,† methane conversion over LSO is 22.1% at 700 °C; the CO and H2 yields at this temperature are 7.5% and 4.2%, respectively. These values are higher than XCH4 = 13.3%, YCO = 6.2, and YH2 = 3.1 for La2SiO5, as summarized in Table S5.† Although the specific surface area of LSO is slightly larger than that of the La2SiO5 sample (0.24 m2 g−1), the higher CH4/O2 conversions by a factor of ∼2 cannot be solely attributed to differences in surface area. Further analysis reveals that the specific surface area of the catalyst is not a decisive factor for catalytic performance (see “Reaction kinetic study” section), suggesting that the enhanced activity of LSO is likely originating from its oxide ion conductivity.
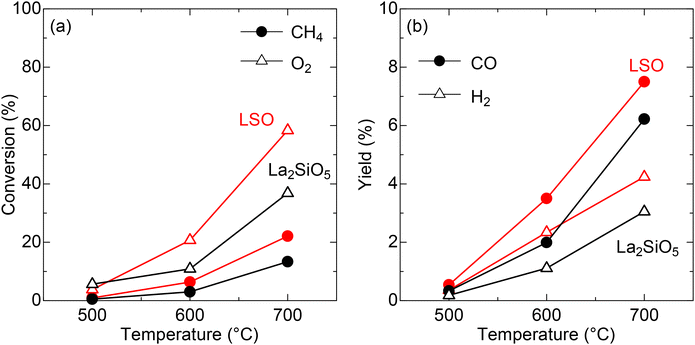 |
| | Fig. 5 (a) Catalytic conversions over LSO (red) and La2SiO5 (black). Circles and triangles denote values for CH4 and O2, respectively. (b) Catalytic yields over LSO (red) and La2SiO5 (black). Circles and triangles denote values for CO and H2, respectively. | |
The results of CPOX activity tests for chemically substituted LSO (Fig. S3†) further support our hypothesis. As demonstrated in Fig. S4,† sodium-substituted LSO (La9NaSi6O26, LNSO) and calcium-substituted LSO (La8Ca2Si6O26, LCSO) exhibit significantly lower methane conversion than pristine LSO. Previous studies on oxide-ion conductive properties of chemically substituted LSO have reported that substituting Na and Ca at the La site reduces oxide-ion conductivity due to the decreased concentration of cation vacancies.23,24 Considering the similar grain microstructures in LSO, LNSO, and LCSO, the substantially lower methane conversion activity of LNSO and LCSO can be attributed to their reduced oxide-ion mobility within the crystal structures. Notably, a favorable impact of oxide-ion conductivity (and thereby oxide-ion mobility) on oxidative catalytic activity has been suggested.25,26 Jung et al. investigated the oxidative dehydrogenation reaction of n-butene to 1,3-butadiene over Ni9Fe3Bi1Mo12O51 catalysts prepared under various pH conditions. They found that the catalyst prepared at pH 8 exhibited the highest oxygen mobility and catalytic activity.27 Additionally, another study on the same reaction revealed superior catalytic activity of multicomponent bismuth molybdate (Co9Fe3Bi1Mo12O51) over pristine bismuth molybdate (γ-Bi2MoO6) due to the enhanced oxygen mobility of the former.28
Reaction kinetic study
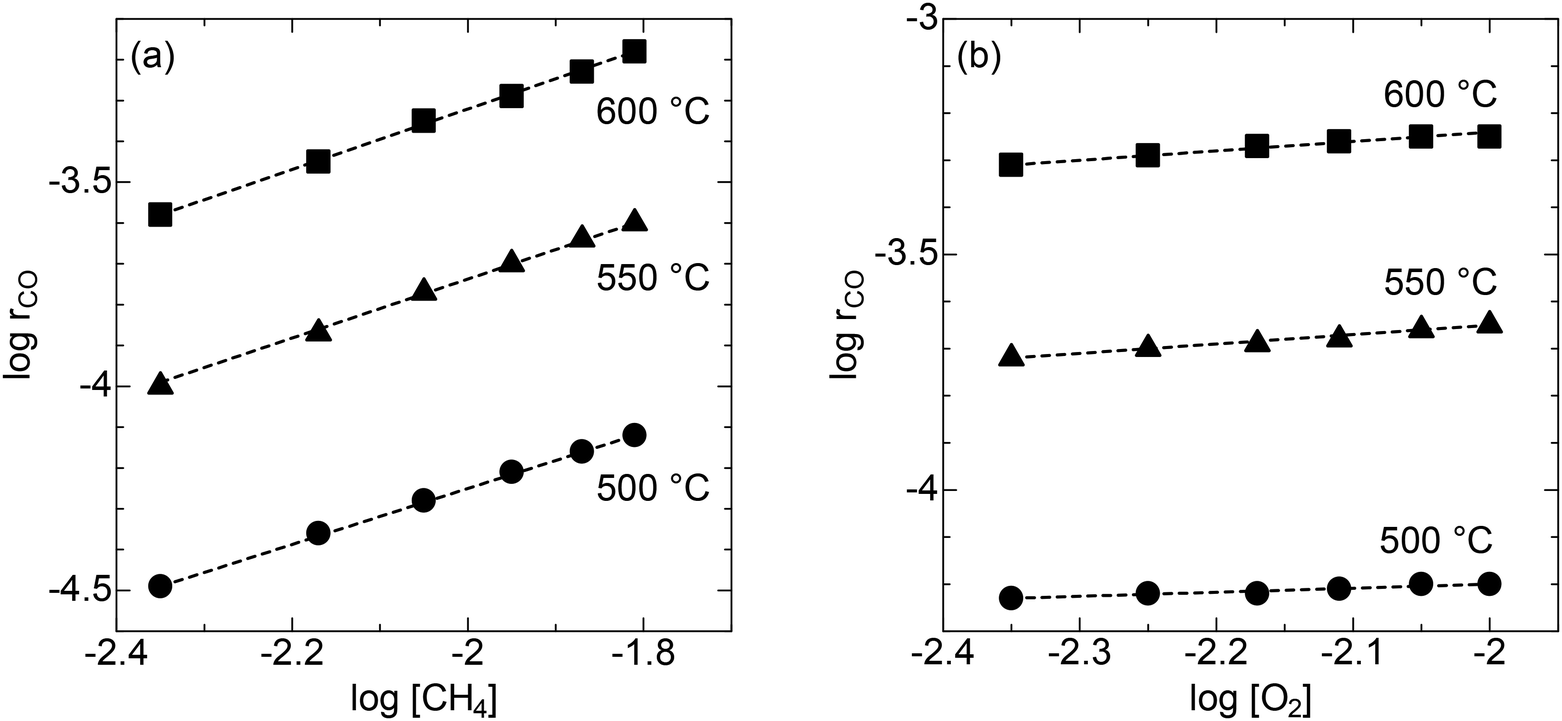
The reaction kinetic study was performed to gain mechanistic insights into CPOX over the LSO catalyst. For CPOX reactions over oxide catalysts, the methane oxidation rate as a function of methane and oxygen partial pressures depends on the different oxygen species that activate methane.29–33 The analysis focused on methane conversions of less than 10% to avoid heat and mass transfer issues.34 Three different temperatures were chosen for this experiment: 500 °C, 550 °C, and 600 °C. The methane oxidation rate over LSO increases with increasing methane concentration (Fig. 6a). In contrast, the methane oxidation rate is nearly independent of oxygen concentration (Fig. 6b). From the reaction rate equation rCH4 = k[CH4]α [O2]β, the estimated reaction orders for methane and oxygen in the CPOX reaction are 0.69–0.73 and 0.08–0.21, respectively (Table 1). The reaction order of O2 indicates that the contribution of adsorbed oxygen species on the surface, which are generated from gaseous O2 molecules, is small in the CPOX reaction on LSO.29,35 Considering the significant oxide-ion conductive capability of LSO, the results suggest that CPOX proceeds through the Mars–Van Krevelen mechanism, in which methane is oxidized by lattice oxygen. Additionally, the high reaction order for methane suggests that methane activation is the rate-determining step for methane oxidation on LSO.
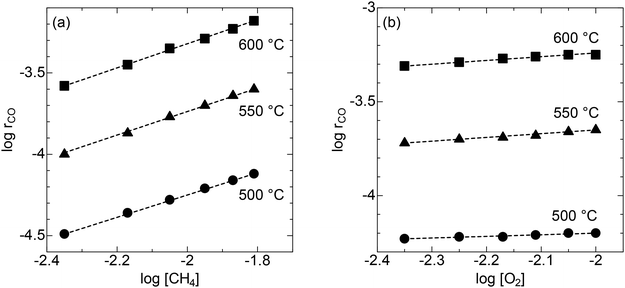 |
| | Fig. 6 Linear correlations between logrCO and (a) log [CH4] (O2 concentration: 12.5%) and (b) log [O2] (CH4 concentration: 25%) at 500, 550, and 600 °C. The total flow rate is set at 10 mL min−1. | |
Table 1 Summary of α, and β values at 500, 550, 600 °C
|
T/°C |
α
|
β
|
| 500 |
0.69 |
0.08 |
| 550 |
0.73 |
0.21 |
| 600 |
0.73 |
0.19 |
The kinetic isotope effect (KIE) experiments were conducted using CD4 to investigate a rate-determining step in the CPOX reaction over LSO. The KIE values are estimated to be 2.00 and 1.47 at 600 °C and 700 °C, respectively, from the ratio between the CH4 and CD4 oxidation rates. The KIE values larger than unity indicate that C–H bond breaking is the rate-determining step during the CPOX reaction over the LSO catalyst.36 This finding is consistent with past research on CPOX catalysts.37–39 Based on this finding, improving methane activation, for example, by adding transition metals, would further enhance the catalytic activity of LSO. Transition metals, such as nickel, can effectively facilitate the C–H bond breaking, thereby lowering the activation energy and increasing the reaction rate.40
Conclusion
To explore potential candidate materials for catalytic partial oxidation of methane (CPOX), the present work investigated the catalytic activity of La9.33Si6O26 (LSO) with an apatite-type structure. The LSO catalyst demonstrated relatively high CPOX activity at temperatures from 500 to 700 °C, with CH4 conversion reaching 22.1% at 700 °C and CO and H2 selectivities of 34.0% and 19.2%, respectively. Notably, the CPOX activity of LSO is higher than that of La2SiO5 with a non-apatite-type structure despite their similar specific surface areas, highlighting the structural superiority of LSO containing mobile oxide ions in its crystal structure. The reaction kinetic study reveals that lattice oxygens in LSO contribute to the catalytic reaction. Furthermore, the kinetic isotope effect (KIE) study suggests that C–H bond breaking is the rate-determining step of CPOX over LSO.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This study was partly supported by JSPS KAKENHI Grant Number JP22H05143 (Transformative Research Area (A) “Supra-ceramics”).
References
- R. Detz, M. Beerse, N. Meulendijks, P. Buskens and B. van der Zwaan, ChemSusChem, 2024, e202400059 CrossRef CAS PubMed.
- L. Sun, Y. Wang, N. Guan and L. Li, Energy Technol., 2020, 8, 1900826 CrossRef CAS.
- M. E. Turano, E. A. Jamka, M. Z. Gillum, K. D. Gibson, R. G. Farber, W. Walkosz, S. J. Sibener, R. A. Rosenberg and D. R. Killelea, J. Phys. Chem. Lett., 2021, 12, 5844–5849 CrossRef CAS PubMed.
- N. Nunotani, J. Ceram. Soc. Jpn., 2022, 130, 825–831 CrossRef CAS.
- J. Zhu, J. G. van Ommen, H. J. M. Bouwmeester and L. Lefferts, J. Catal., 2005, 233, 434–441 CrossRef CAS.
- J. C. Jung, H. Kim, A. S. Choi, Y. M. Chung, T. J. Kim, S. J. Lee, S. H. Oh and I. K. Song, Catal. Commun., 2007, 8, 625–628 CrossRef CAS.
- B. Yan, L. Wang, B. Wang, F. Alam, Z. Xiao, J. Li and T. Jiang, Appl. Catal., A, 2019, 572, 71–79 CrossRef CAS.
- H. Borchert and M. Baerns, J. Catal., 1997, 168, 315–320 CrossRef CAS.
- M. Salazar, D. A. Berry, T. H. Gardner, D. Shekhawat and D. Floyd, Appl. Catal., A, 2006, 310, 54–60 CrossRef CAS.
- Y. Ogura, T. Yokoi, K. Fujii, M. Yashima and K. Matsunaga, Solid State Ion., 2021, 373, 115793 CrossRef CAS.
- C. A. Fuller, M. J. Gutmann, C. D. Ling, C. H. Wang, W. Zhang, P. S. Halasyamani, I. R. Evans and J. S. O. Evans, J. Mater. Chem. A, 2022, 10, 14576–14584 RSC.
- A. Mineshige, A. Saito, M. Kobayashi, H. Hayakawa, M. Momai, T. Yazawa, H. Yoshioka, M. Sakao, R. Mori, Y. Takayama, Y. Kagoshima and J. Matsui, J. Power Sources, 2020, 475, 228543 CrossRef CAS.
- S. Okada, F. Maekawa, R. Tago, Y. Kobayashi, I. Terao, A. Mineshige, D. Urushihara, T. Asaka and K. Fukuda, Solid State Ion., 2023, 399, 116309 CrossRef CAS.
- K. Kobayashi, K. Hirai, T. Uchikoshi, Y. Sakka, T. Akashi and T. S. Suzuki, Open Ceram., 2021, 6, 100100 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- H. Okudera, Y. Masubuchi, S. Kikkawa and A. Yoshiasa, Solid State Ion., 2005, 176, 1473–1478 CrossRef CAS.
- S. Yan, D. Yang, S. Chen, J. Wen, W. He, S. Ji, Y. Xia, Y. Wang, L. Zhou and Y. Li, Dalton Trans., 2020, 49, 2578–2588 RSC.
- K. Fujii, M. Yashima, K. Hibino, M. Shiraiwa, K. Fukuda, S. Nakayama, N. Ishizawa, T. Hanashima and T. Ohhara, J. Mater. Chem. A, 2018, 6, 10835–10846 RSC.
- N. Nunotani, N. Moriyama, K. Matsuo and N. Imanaka, J. Ceram. Soc. Jpn., 2017, 125, 773–775 CrossRef CAS.
- K. Matsuo, N. Nunotani and N. Imanaka, J. Asian Ceram. Soc., 2021, 9, 1466–1472 CrossRef.
- P. K. Pandis, D. E. Perros and V. N. Stathopoulos, Catal. Commun., 2018, 114, 98–103 CrossRef CAS.
- K. Fukuda, T. Iwata and E. Champion, Powder Diffr., 2006, 21, 300–303 CrossRef CAS.
- J. E. H. Sansom, E. Kendrick, J. R. Tolchard, M. S. Islam and P. R. Slater, J. Solid State Electrochem., 2006, 10, 562–568 CrossRef CAS.
- H. Li, T. Baikie, S. S. Pramana, J. F. Shin, P. J. Keenan, P. R. Slater, F. Brink, J. Hester, T. An and T. J. White, Inorg. Chem., 2014, 53, 4803–4812 CrossRef CAS PubMed.
- S. Rehman, H. Kim, M. F. Khan, J. H. Hur, A. D. Lee and D. K. Kim, Sci. Rep., 2019, 9, 19387 CrossRef CAS PubMed.
- K. T. de C. Roseno, M. Schmal, R. Brackmann, R. M. B. Alves and R. Giudici, Int. J. Hydrogen Energy, 2019, 44, 8166–8177 CrossRef.
- J. C. Jung, H. Lee, H. Kim, Y. M. Chung, T. J. Kim, S. J. Lee, S. H. Oh, Y. S. Kim and I. K. Song, Catal. Commun., 2008, 9, 943–949 CrossRef CAS.
- J. C. Jung, H. Lee, H. Kim, Y. M. Chung, T. J. Kim, S. J. Lee, S. H. Oh, Y. S. Kim and I. K. Song, Catal. Lett., 2008, 124, 262–267 CrossRef CAS.
- A. Matsuda, H. Tateno, K. Kamata and M. Hara, Catal. Sci. Technol., 2021, 11, 6987–6998 RSC.
- A. Matsuda, K. Obara, A. Ishikawa, M. H. Tsai, C. H. Wang, Y. C. Lin, M. Hara and K. Kamata, Catal. Sci. Technol., 2023, 13, 5180–5189 RSC.
- J. Tian, J. Tan, Z. Zhang, P. Han, M. Yin, S. Wan, J. Lin, S. Wang and Y. Wang, Nat. Commun., 2020, 11, 5693 CrossRef CAS PubMed.
- P. Han, R. Yan, Y. Wei, L. Li, J. Luo, Y. Pan, B. Wang, J. Lin, S. Wan, H. Xiong, Y. Wang and S. Wang, J. Am. Chem. Soc., 2023, 145, 10564–10575 CrossRef CAS PubMed.
- K. Murata, K. Arai, N. Kondo, R. Manabe, T. Yumura and S. Hosokawa, Catal. Sci. Technol., 2024, 14, 3253–3264 RSC.
- M. C. J. Bradford and M. A. Vannice, Appl. Catal., A, 1996, 142, 73–96 CrossRef CAS.
- A. Oda, R. Ichihashi, Y. Yamamoto, K. Sawabe and A. Satsuma, J. Mater. Chem. A, 2023, 11, 23854–23866 RSC.
- K. C. Westaway, J. Labelled Compd. Radiopharm., 2007, 50, 989–1005 CrossRef CAS.
- C. Elmasides, T. Ioannides and X. E. Verykios, AIChE J., 2000, 46, 1260–1270 CrossRef CAS.
- A. G. Steghuis, J. G. Van Ommen and J. A. Lercher, Catal. Today, 1998, 46, 91–97 CrossRef CAS.
- J. Zhu, J. G. Van Ommen and L. Lefferts, J. Catal., 2004, 225, 388–397 CrossRef CAS.
- Z. Ding, S. Chen, T. Yang, Z. Sheng, X. Zhang, C. Pei, D. Fu, Z. J. Zhao and J. Gong, Nat. Commun., 2024, 15, 4636 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Yuta
Goto
a,
Kazumasa
Murata
a,
Yuta
Goto
a,
Kazumasa
Murata