
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High-pressure electrochemistry: a new frontier in decarbonization†
Nishithan C.
Kani
 a,
Samuel
Olusegun
b,
Rohit
Chauhan
a,
Joseph A.
Gauthier
*b and
Meenesh R.
Singh
*a
a,
Samuel
Olusegun
b,
Rohit
Chauhan
a,
Joseph A.
Gauthier
*b and
Meenesh R.
Singh
*a
aDepartment of Chemical Engineering, University of Illinois Chicago, Chicago, IL 60607, USA
bDepartment of Chemical Engineering, Texas Tech University, Lubbock, TX 79409, USA. E-mail: mrsingh@uic.edu; joe.gauthier@ttu.edu
First published on 28th November 2023
Abstract
The chemical manufacturing of commodity chemicals, responsible for approximately 24% of global carbon emissions, poses a critical environmental challenge. With escalating demand due to economic development, urgent decarbonization strategies are imperative. Traditional electrochemical synthesis encounters hindrances, especially mass transfer limitations at relevant current densities, primarily attributed to gas phase reactants and products. In this Perspective, we explore the viability of high-pressure electrochemistry as a transformative solution. Our analysis reveals that applying pressure can overcome mass transfer limitations, enhance selectivity, and improve overall activity, all while minimizing energy consumption and capital expenditure for distributed production processes. We shed light on the influence of pressure on Pourbaix diagrams, electric double layer, electrolyte activity, conductivity, electrostriction-enhanced selectivities, catalyst activity, and stability. Additionally, insights are provided into the design and operation of existing reactors and tools for high-pressure electrochemistry, along with the imperative for future fundamental studies. In the context of decentralized production, we argue that the marginal differential capital costs associated with high-pressure reactors become inconsequential. Ultimately, our work seeks to pave the way for the decarbonization of the chemical industry by establishing innovative pathways for the electrochemical synthesis of commodity chemicals, presenting high-pressure electrochemical synthesis as a potential paradigm shift in this transformative journey.
Broader contextHigh-pressure electrochemistry is an emerging area with significant promise for commercializing green chemicals. However, little is known about the underlying theories and practices in implementing high-pressure electrochemical reactors. Electrochemical synthesis at high pressures can increase the operating current density and reduce the cell potential, which would decrease the CapEx and OpEx costs, respectively, and thereby reduce barriers to industrial-scale deployment. We review the fundamental theories of high-pressure electrochemistry and discuss how pressure affects the activity, selectivity, stability, and energy efficiency of reactions. We also explore the designs of reactors for synthesis and in situ characterization studies and the economics of high-pressure electrochemical synthesis. |
1. Introduction
The steady increase in anthropogenic greenhouse gas (GHG) emissions has driven decarbonization needs for energy and chemical technologies. To achieve the net-zero carbon emissions target globally by the end of the 21st century,1,2 it is essential to develop and deploy net-zero and net-negative carbon technologies across various sectors. For instance, the decarbonization of power generation necessitates the utilization of solar, wind, and hydropower in conjunction with stable energy storage systems; the decarbonization of transportation can be achieved with electric vehicles concomitant with decarbonization of power generation; and the decarbonization of buildings can be done by implementing heat pumps and carbon-negative concrete. However, much-needed efforts towards decarbonizing chemical manufacturing are still lacking on a large scale. As a result, industrial manufacture of commodity chemicals is one of the most significant contributors to global GHG emissions, and it is predicted to rise from the demand for commodities driven by economic development.3 Combustion of fossil fuels, both for heating needs and for production of feedstocks (e.g., H2 from steam methane reforming and water gas shift reactions), are two significant sources of GHG emissions in producing commodity chemicals.Decarbonization of chemical manufacturing is projected to follow two pathways. First, emissions can be mitigated by capturing emitted CO2 and sequestration or valorization into value-added chemicals (CO, C2H4, CH3OH, etc.). The second approach consists of avoiding the generation of CO2 or other greenhouse gases in the first place. In particular, the second pathway can be achieved in several ways, including electrical heating, using green H2 as a fuel for heating, and electrochemical synthesis of chemicals using renewable energy. A detailed review of the advantages and disadvantages of the technologies mentioned above is given in recent literature.1,3 In brief, most of these technologies rely on photo/electrochemistry to capture and reduce GHG emissions by directly using renewably produced electrons. This work focuses on the electrochemical synthesis of chemicals using renewable energy. Specifically, we investigate the prospect of using high-pressure electrochemical processes for manufacturing commodity chemicals such as NH3, C2H4, and CH3OH while directly utilizing GHGs such as NOx, CO2, and CH4.4
Electrochemical synthesis of the chemicals mentioned above is attractive, as the electric potential drives reactions, without extreme temperatures. In principle, this feature gives a distinct advantage of operating the reactor at ambient conditions using carbon-neutral electricity. Also, electrochemical processes provide opportunities to reduce downstream separation costs as a membrane separates the anode and cathode side products. The reactants used to produce the above-considered chemicals are typically gaseous, for example, N2 from the air, captured CO2, CO electrochemically synthesized from captured CO2, excess CH4 produced in natural gas extraction plants, NOx from exhaust fumes, and H2O.
Some examples of electrochemical conversion of gases include N2 reduction to produce NH3, CO2/CO reduction to C2H4, and partial CH4 oxidation to CH3OH. The studies reported so far on the electrochemical synthesis of NH3 from N2 and H2O under ambient conditions suffer from low yield (<−1 mA cm−2),5–7 though recent efforts in non-aqueous Li-mediated pathways show high selectivity.8–10 As a result, it is difficult to attribute that produced NH3 from the N2 in reactant feed, not from adverse contaminants such as NOx, NO3− and NO2−.11–15 The process is very challenging due to the low solubility of N2 in H2O and the competing hydrogen evolution reaction (HER). In contrast, there has been considerable progress in the synthesis of C2H4 and CH3OH electrochemically by reducing CO2.16–19 The challenges associated with the synthesis of these chemicals from CO2 and CO are (1) tuning the selectivity of the products in a two electrode MEA based setup, as the product selectivity is strongly dependent on the applied potential; (2) low solubility of CO in H2O; (3) competition with the HER. Finally, electrochemical oxidation of CH4 to CH3OH is desired as CH3OH can be easily stored and transported. Several studies20,21 have reported the electrochemical oxidation of CH4 to CH3OH, and this process suffers from low solubility of CH4 in H2O, stability of the catalyst under highly oxidizing conditions, and the competing oxygen evolution reaction (OER). A common aspect of the above-mentioned processes is that they are synthesized from gas-phase reactants and suffer from mass transfer limitations due to the low solubility of the gases in aqueous media. Such challenges can be overcome by operating the reactors at higher pressures. While higher pressure will likely increase the reactor's capital cost, the operating cost to pressurize the reactor is low compared to increasing the reactors temperature.
Fundamental studies involving high-pressure electrochemistry have been reported as far back as the 1930s,22–25 and high pressures are essential in studies involving hydrothermal systems, oceanographic analysis, and marine science.26 High-pressure electrochemistry has been employed to get insights into the thermodynamics and kinetics of redox reactions at elevated pressures. Electrochemical properties such as redox potentials, diffusion, electron transfer rates, ion-solvent interactions, electrical double layer (EDL), etc., depend on pressure.26 Several studies have been reported in the literature that estimate the pressure dependence of these properties.27–31 For example, a theoretical study by Guan et al.32 predicts that metal superhydrides can be synthesized at high pressures simultaneously with an applied bias (known as the pressure-potential approach). Such a method extends the thermodynamic stability regime of super hydrides, which are challenging to form at ambient pressures. Similarly, electrochemical reduction of CO2 at elevated pressures has been previously investigated.33,34 At elevated pressures up to 30 atm, the overpotentials drop with a drastic improvement in the current densities and product selectivity.33 The HER, a parasitic side reaction in electrochemical CO2 reduction, was drastically suppressed around 30 bar, and 100% selectivity towards HCOOH was observed at 60 bar on Pb and In catalysts.34 Ramdin et al.35 used a high-pressure semicontinuous batch electrolyzer with Sn-based electrodes to reduce CO2 to formates with 90% faradaic efficiency. Welch et al.36 performed electrochemical CO2 reduction in ionic liquids at high pressures and established a correlation between the volume expansion of ionic liquid and current densities. Electrochemical oxidation of CH4 to CH3OH at elevated pressures was investigated by Lee et al.37 and Kim et al.,38 who reported improved CH3OH faradaic efficiencies with an increase in pressure. Synthesizing chemicals involving gas phase reactants electrochemically at high pressures would overcome the existing challenges, such as mass transfer limitations at ambient pressures. A brief summary of the literature comparisons is provided in the ESI.† In general, high-pressure electrochemistry and the effects of pressure are not well consolidated in the literature. This review provides a comprehensive and authoritative structure to the subject.
The purpose of high-pressure electrochemistry is (i) to improve the activity and selectivity of the redox reaction when the reactants are in the gas phase. Most reactions of interest, such as N2 reduction, N2 oxidation, CO2 reduction, and CH4 oxidation, involve single gas phase reactants. By pressurizing, we mean pressurizing using the reactant gas; and (ii) to deliver the product gases at high pressure (e.g., high-pressure delivery of H2 from PEM electrolyzers). In this case, the pressure of the anode/cathode chamber is regulated to the desired pressure by either accumulating pure product gases or by charging with a high-pressure sweep gas. The partial pressure will govern the activity of the product gases. Note that the pressure effects discussed in this work are related to the system's overall pressure, which will also be translated to the partial pressure effects. The electrochemical synthesis at high pressures can help increase operating current density and reduce cell potential, which would address a critical weakness inhibiting industrial-scale deployment. PEM electrolyzers are well-established and generally operate at high pressures. A brief description of the advantages of operating PEM electrolyzers at high pressures is provided in the ESI.† As shown below, the energy penalty associated with pressurizing a reactor is significantly lower than the energy required to heat it to extreme temperatures. Simultaneously, the overpotential required to make these chemicals at high pressures would be less than the ones at ambient pressures, thereby improving energy efficiency. The temperature significantly influences the reaction rates and thermodynamics in electrochemistry compared to pressure. Increasing the temperature can accelerate the reaction rates by providing more kinetic energy to the reactant molecules, increasing their collision frequency and enabling more successful collisions. However, in this review/perspective article, we focus only on the effect of pressure on the electrochemical systems, and we do not discuss in detail the role of temperature in improving performance.
This article reviews the fundamental theories of high-pressure electrochemistry and discusses how pressure affects the activity, selectivity, stability, and energy efficiency of reactions (see Fig. 1). We also explore the designs of reactors for synthesis and in situ characterization studies and the economics of high-pressure electrochemical synthesis.
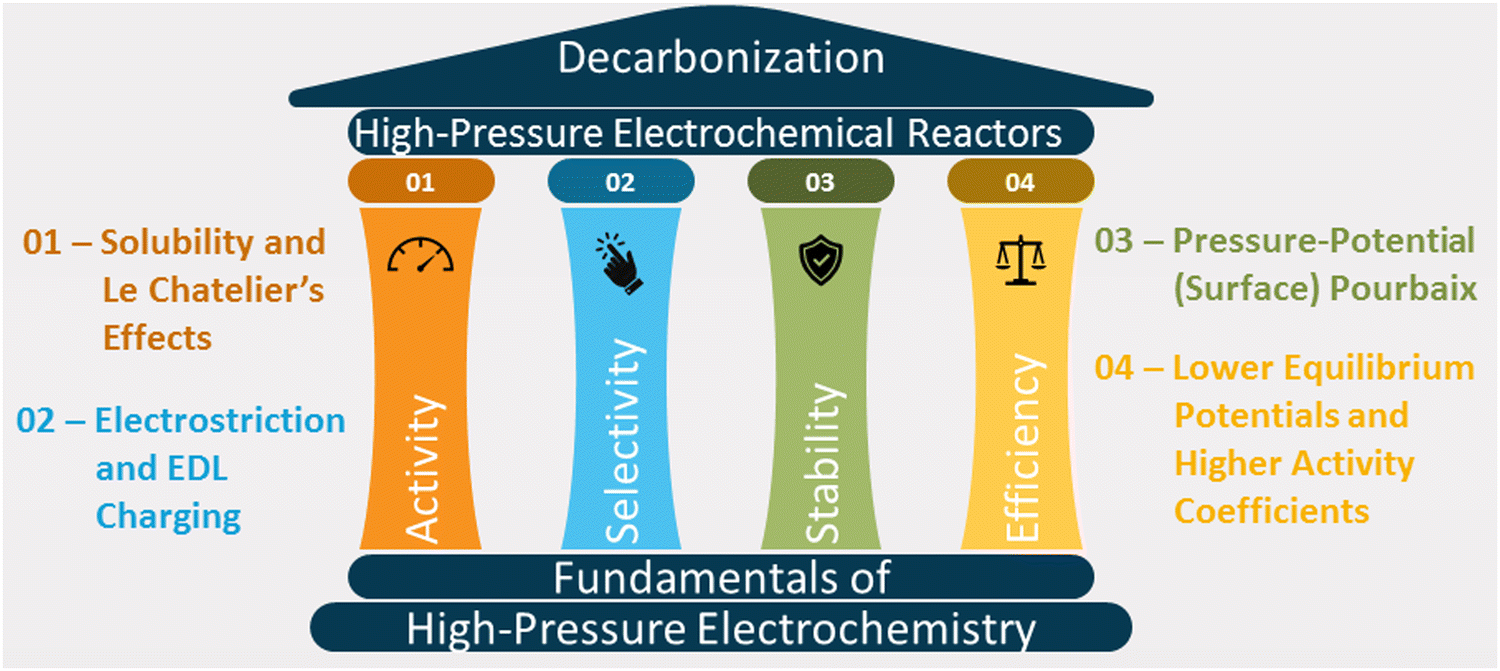 | ||
| Fig. 1 Fundamental factors affecting activity, selectivity, stability, and energy efficiency of high-pressure electrochemical systems. | ||
2. Fundamentals of high-pressure electrochemical systems
Here, we discuss the role played by the pressure of gas phase reactants and products on the activity, selectivity, stability, and efficiency of electrochemical reactors.2.1 Effect of pressure on thermodynamics
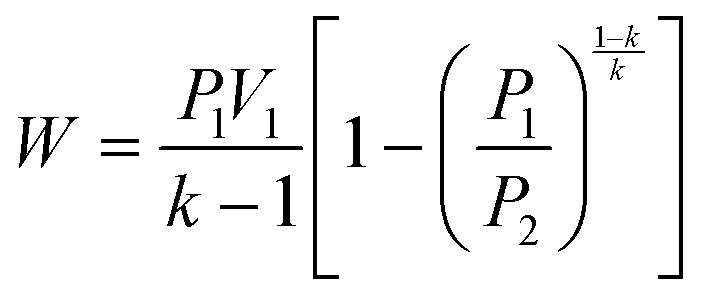 | (1) |
The work done to pressurize an ideal gas can be calculated from the above equation of isentropic expansion, which is normalized with respect to the number of moles of the ideal gas. Here, N2 is taken as the model system. Fig. 2A shows the energy required to pressurize a reactor normalized with respect to the number of moles of gas as a function of the pressure. The energy required to pressurize a reactor to 10 bar is 60% of the energy required to pressurize a 300 bar reactor.
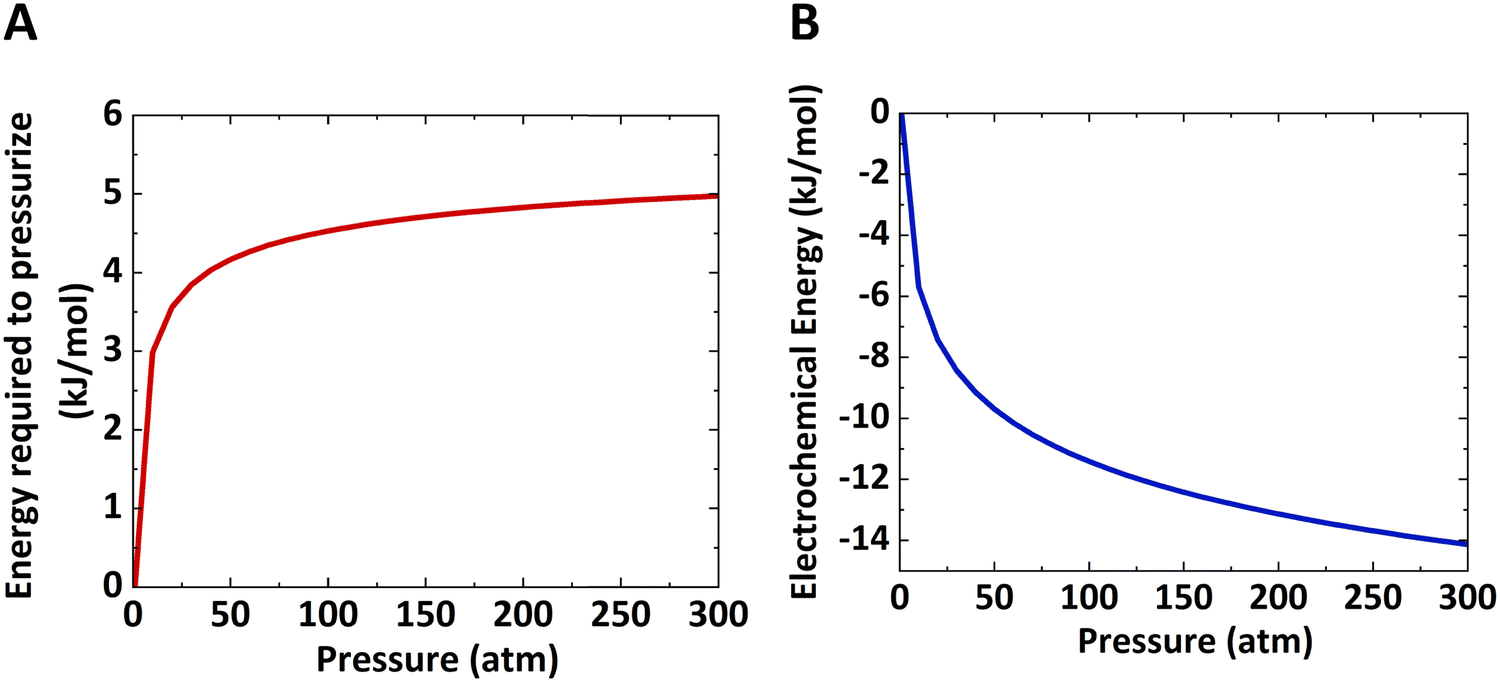 | ||
| Fig. 2 Thermodynamic energy requirements: (A) energy required to pressurize a reactor as a function of pressure assuming isentropic expansion of an ideal gas. (B) Electrochemical energy reduction for any redox reaction as a function of pressure. | ||
| O + ne− ⇌ R | (i) |
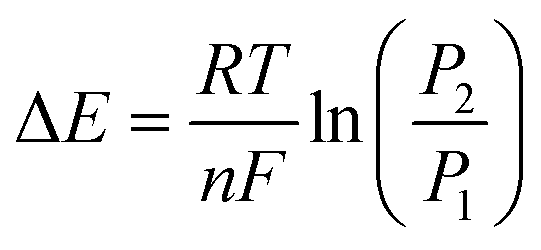 | (2) |
The difference in Gibb's free energy of the systems at pressures P1 and P2 can be obtained from the equilibrium potential difference in eqn (2),
 | (3) |
To summarize, the energy savings from the reduction in electrochemical potential are at least double the energy penalty for pressurizing the reactors. Therefore, the operating costs are lower for pressurized electrochemical reactors. However, the capital cost to build a thick-wall reactor to withstand high pressures will be higher than for a reactor operated at ambient pressure.
| Sr. No. | Half cell reaction | Equilibrium potential (V vs. RHE), pH = 0 | |
|---|---|---|---|
| P = 1 bar | P = 200 bar | ||
| 1 | N2 + 6H+ + 6e− → 2NH3 | 0.056 | 0.079 |
| 2 | CH4 + H2O → CH3OH + 2H+ + 2e− | 0.58 | 0.51 |
| 3 | 2CO2 + 12H+ + 12e− → C2H4 + 4H2O | 0.08 | 0.10 |
| 4 | 2CO + 8H+ + 8e− → C2H4 + 2H2O | 0.17 | 0.20 |
| 5 | CO2 + 6H+ + 6e− → CH3OH + H2O | 0.03 | 0.05 |
| 6 | CO + 4H+ + 4e− → CH3OH | 0.08 | 0.11 |
| 7 | 2H2O → O2 + 4H+ + 4e− | 1.23 | 1.23 |
| 8 | 2H+ + 2e− → H2 | 0.00 | 0.00 |
| S. no. | Overall reaction |
|---|---|
| 1 | 2N2 + 6H2O → 4NH3 + 3O2 |
| 2 | CH4 + H2O → CH3OH + H2 |
| 3 | 2CO2 + 2H2O → C2H4 + 3O2 |
| 4 | 2CO + 2H2O → C2H4 + 2O2 |
| 5 | 2CO2 + 4H2O → 2CH3OH + 3O2 |
| 6 | CO + 2H2O → CH3OH + O2 |
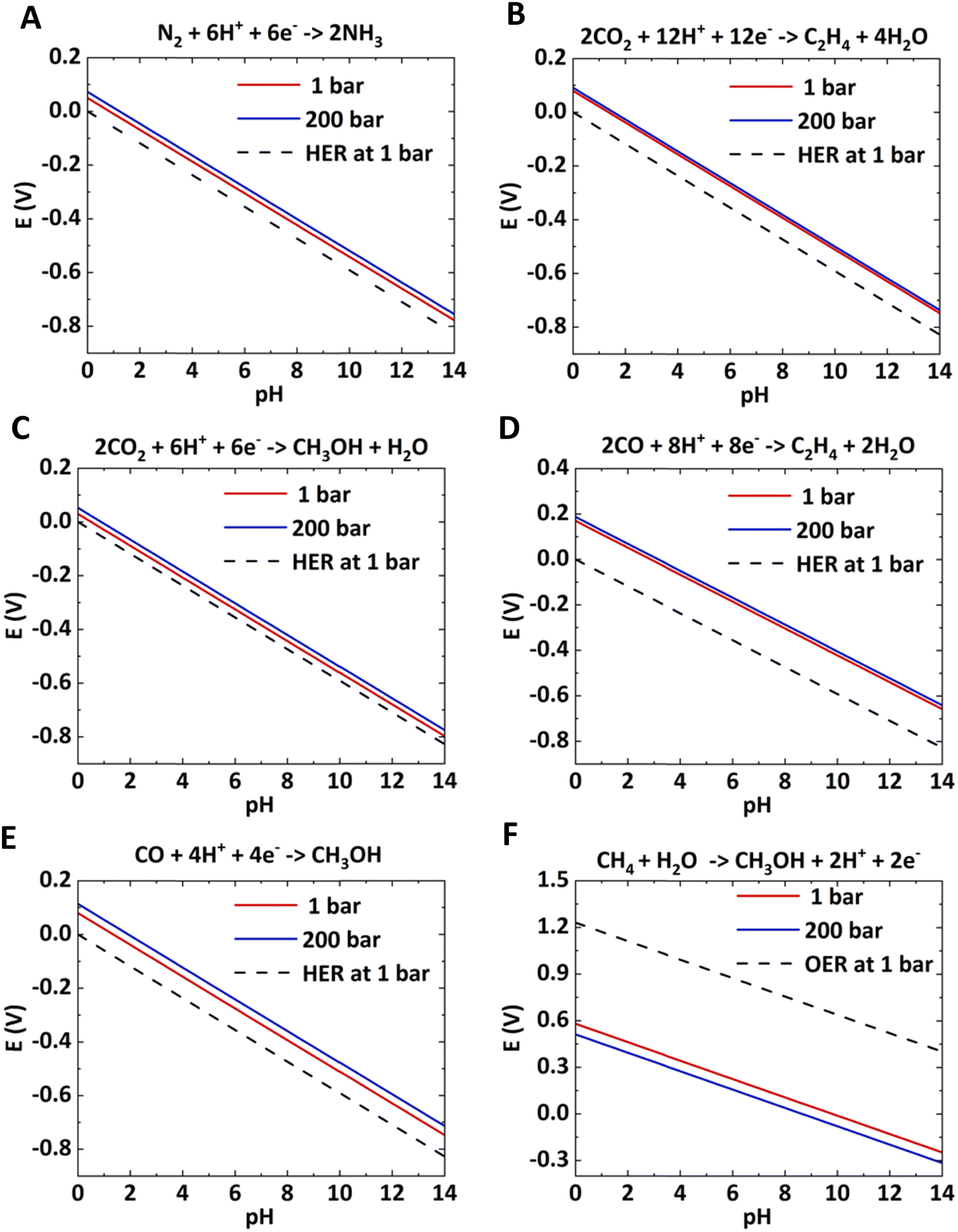 | ||
| Fig. 3 Effect of pressure on the bulk Pourbaix diagram for different half-cell reactions. | ||
With increasing pressure, the Pourbaix lines shift up for reduction reactions and shift down for oxidation reactions. In both half-cell reactions, the overpotential will decrease with increasing pressure. The shift in Pourbaix lines is not significant for reactions with a higher number of electron transfers, such as electrosynthesis of C2H4 from CO2 and CO (< 0.05 V). The electrochemical reactions, such as CH4 to CH3OH, CO2 to CO, CO2 to HCOOH, etc., would be significantly improved with applied pressure.
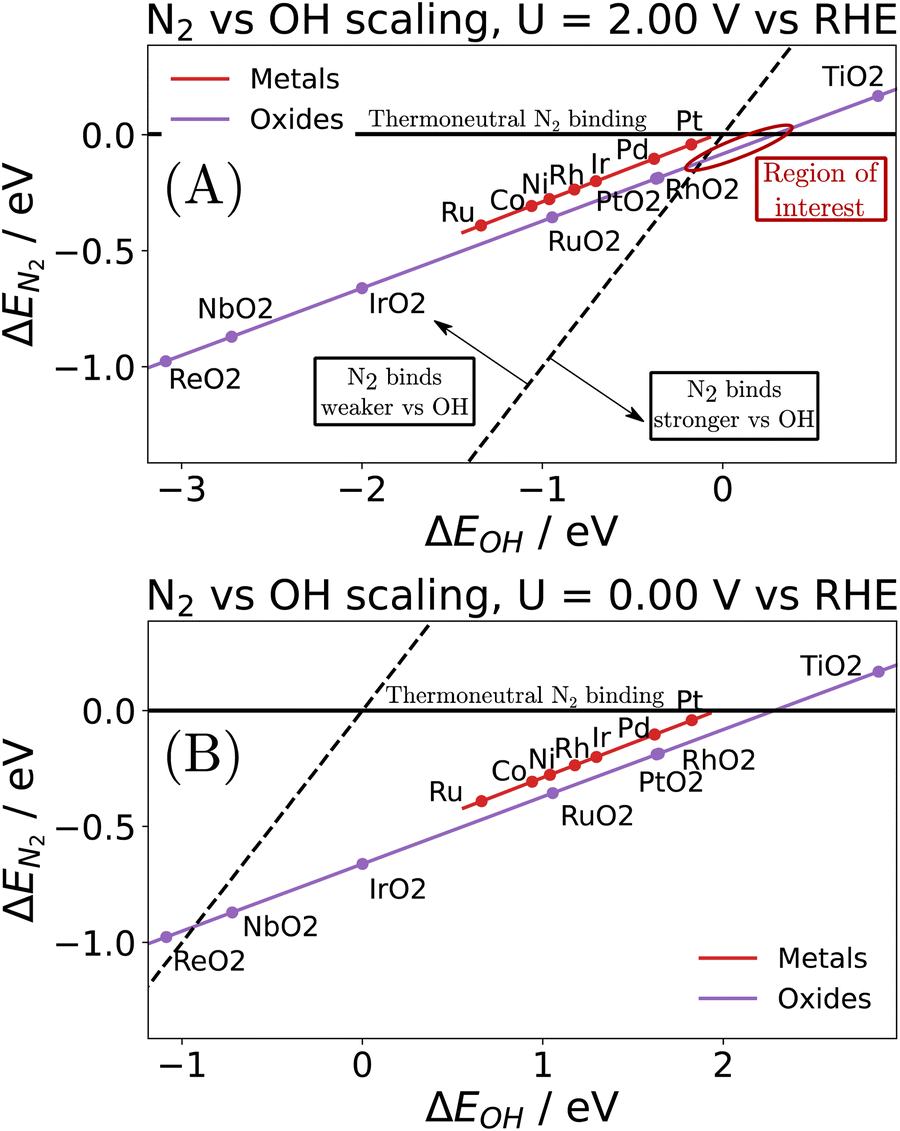 | ||
| Fig. 4 Illustration of the scaling between N2 binding and hydroxide binding on various metals and rutile oxides at an applied bias of (A) U = +2 V vs. RHE, and (B) U = 0.0 V vs. RHE. Note that N2 binding is first order independent of potential, while hydroxide binding depends linearly on potential. | ||
Note that the energetics reported here are (approximately) enthalpies. A standard procedure in estimating entropic effects (where pressure plays the most significant role) for binding (Gibbs free) energy calculations is two-fold: (i) for the adsorbed state, molecules are typically assumed to have only vibrational degrees of freedom, with the overall entropy described by the harmonic oscillator approximation. Note that this approximation has been explored extensively in the literature, with recent findings suggesting that, for small molecules on metal surfaces, the HO approximation provides an excellent approximation for the entropy.39–41 (ii) For the gas-phase state, typically, molecules are approximated using the ideal gas approximation, in which the translational component of the entropy depends on the logarithm of the pressure. Here, the y-axis of Fig. 4 represents the reaction,
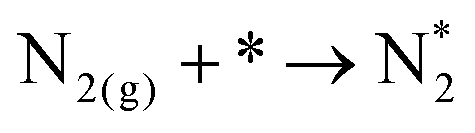 | (ii) |
Although a complete exploration of the energetic landscape of the N2OR has, to our knowledge, not yet been published, in Fig. 5, we illustrate a limiting potential OER volcano and a schematic N2OR limiting potential volcano.
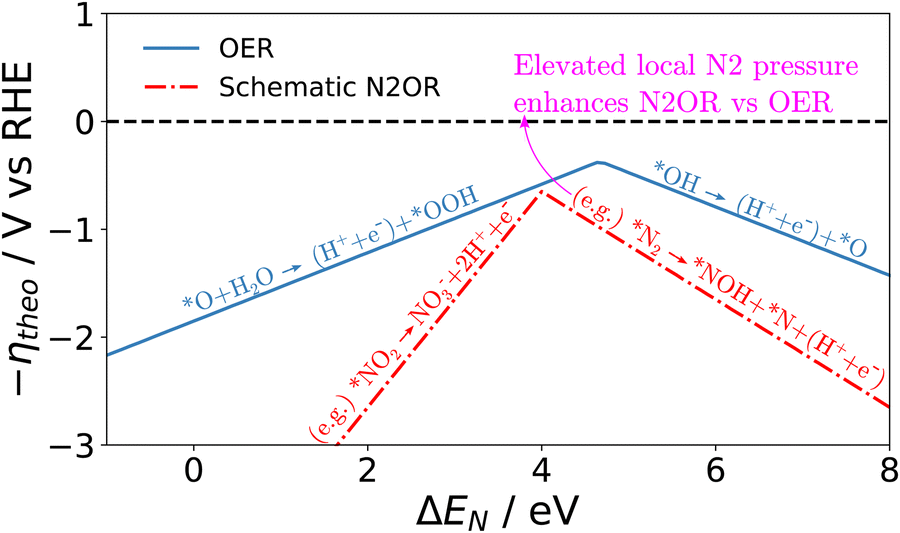 | ||
| Fig. 5 Limiting potential volcanoes for the oxygen evolution reaction (OER) and dinitrogen oxidation reaction (N2OR, schematic shown) with a common descriptor given as the binding energy of atomic nitrogen. | ||
Here, the y-axis represents the negative theoretical limiting potential for the reaction of interest, while the x-axis represents the binding energy of atomic nitrogen, which is linearly correlated with the typical OER descriptor ΔEO − ΔEOH. Here, catalysts closer to the dashed black line are predicted to be more active for the given reaction. The schematic N2OR volcano, based in part on the scaling lines from Fig. 4, shows that no catalyst will be more active towards the N2OR than the competing OER. However, if one of the legs of the N2OR volcano involves N2(g) as a reactant, elevated N2 pressure will enhance the N2OR, while having no effect on the OER. In our schematic N2OR volcano, increasing the N2 pressure effectively raises the right leg, shifting the peak to the right and potentially overcoming the OER-limiting potential volcano.
2.2 Effect of pressure on the reaction kinetics or current density
Generally, the kinetic rate of redox reactions increases with increasing the pressure of the reactant. This effect is primarily due to the higher solubility of dissolved gases and the lower equilibrium potential of redox reactions at elevated pressures.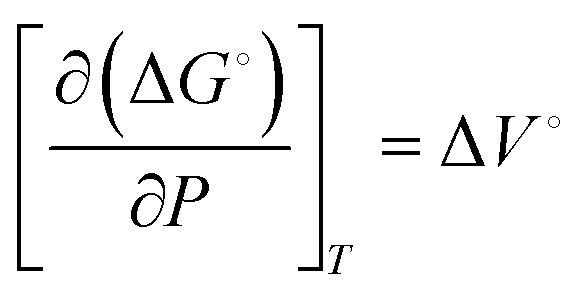 | (4) |
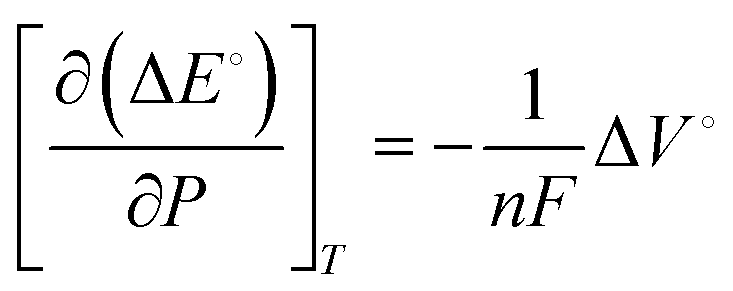 | (5) |
The reaction will be favored by increasing the pressures if the activated complex has a smaller partial volume than the reactants. As per Le-Chatelier's principle, when the pressure of reactant gases such as N2, CO2, CO, and CH4 is increased, they would favor the forward reaction as the reactions proceed by a net decrease in volume. At elevated pressures, the current densities of the products would be drastically higher than at ambient pressure and have higher faradaic efficiencies.
2.3 Effect of pressure on selectivities
A similar electrostriction effect is also applicable to redox reactions. Electrostriction makes the arrangement of solvent molecules more compact around the products than the reactants, which promotes the redox reaction.44 For example, the conversion of N2 (neutral species) to NO3− or NH4+ (charged species) will be promoted due to electrostriction, whereas the competing reactions (OER: OH− to O2, or HER: H+ to H2) involving the formation of neutral (less polar) species will be suppressed due to positive volume of reaction.
The volume change due to electrostriction is driven by higher local electrostatic interaction between the solute and solvent, which is counterbalanced by the internal pressure of the solvated ion at equilibrium. This equilibrium is described by the following model,46
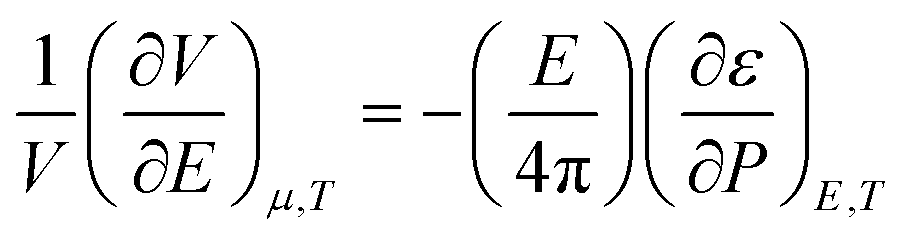 | (6) |
2.4 Effect of pressure on the electrode–electrolyte interface
 | ||
| Fig. 6 Schematic of the electrical double layer (EDL) at extreme pressures induced by either a positive overhead pressure or ultrasound. | ||
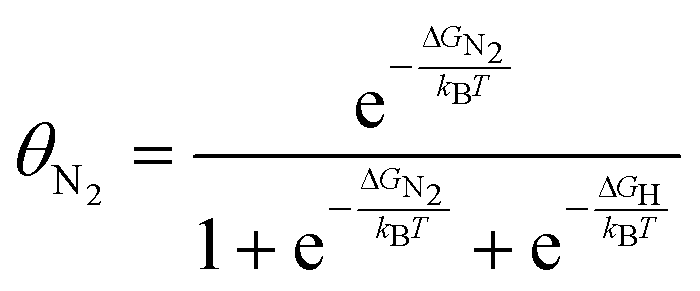 | (7) |
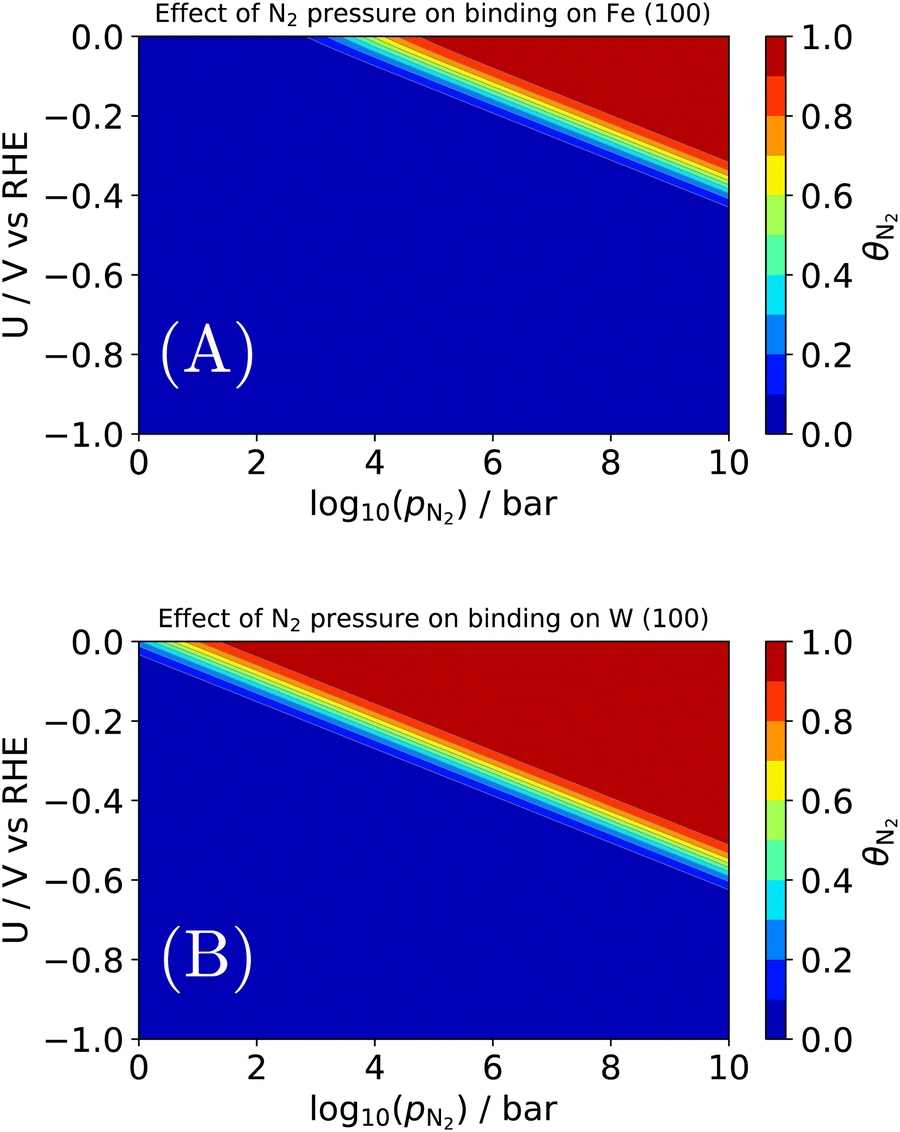 | ||
| Fig. 7 Effect of N2 pressure on the equilibrium coverage of *N2 on two candidates for an electrochemical Haber–Bosch process: (A) the terrace of Fe (100); (B) the terrace of W (100). | ||
As before, binding energies of N2 and H2 were calculated using semi-local DFT. Entropic contributions in the adsorbed state were assumed to be entirely vibrational and approximated as a harmonic oscillator. The gas phase state was approximated as an ideal gas, which may not hold above e.g., 106 bar at room temperature. By construction, the coverages are normalized such that 1 = θN2 + θH + θ*. An interesting observation is seen in Fig. 7A, where we find a pressure of ∼103–4 bar is needed for N2 to appreciably adsorb on Fe (100) at the RHE equilibrium potential. There are several important deviations from our model catalyst system (clean Fe (100)) and e.g., the industrial Haber–Bosch catalyst, which operates at pressures of 100–150 bar. It has long been known that the rate-determining step, i.e., N2 activation, takes place on the steps of catalysts53 (whether Ru or Fe), and in the presence of one of several alkali metal promoters.54 Furthermore, the industrial reaction occurs at high temperature, which would improve the needed pressure for appreciable N2 coverage in Fig. 7A. A more complete picture would be given by a complete N2RR microkinetic model. Such a model would incorporate much stronger binding states, e.g., *N, which may dominate the coverage of an actual Haber–Bosch catalyst.
In general, competition between N2 and H adsorption can be understood by the scaling shown in Fig. 8. Here, we see a poor scaling between these two states, though nearly all of the tested transition metals favor H adsorption over dinitrogen. An ideal catalyst candidate for the N2RR process will bind N2 more strongly than H, but without binding ammonia so strongly that the catalyst becomes poisoned by products. If the slope of the (black dashed) scaling line holds, then perhaps weak binding catalysts (i.e., with high d band centers55) will favor N2 binding over H at lower pressures. Naturally, weak binding catalysts like Cu will struggle to dissociate N2 at room temperature, but if a strong reducing bias can be applied without compromising N2 coverage, such a reaction may not be necessary like in the thermochemical process. Instead, dissociation of *N2H or *N2H2 may be more facile. Alternatively, weak binding catalysts doped with single atoms of a strong binding catalyst may be a promising system for future investigation.
 | ||
| Fig. 8 Scaling of dinitrogen binding with hydrogen binding energetics. | ||
2.5 Effect of pressure on the electrolyte conductivity
ηP = η0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(αP) exp(αP) | (8) |
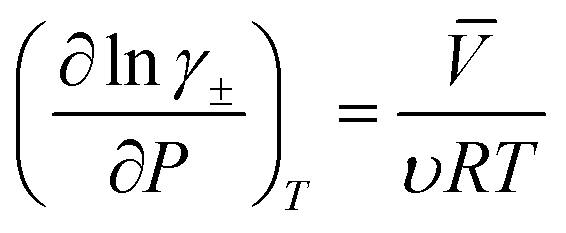 | (9) |
![[V with combining macron]](https://www.rsc.org/images/entities/i_char_0056_0304.gif) denotes the partial molar volume and υ denotes the no. of moles of ions into which a mole of electrolyte dissociates. For 1–1 electrolyte (e.g., HCl and HNO3) of molar volume 30 cm3 mol−1 at 25 °C, the change in activity coefficient with pressure is ∼6 × 10−4 bar−1. This suggests that the activity coefficient increases by ∼6% with an increase in pressure to 100 bar. The increase in activity coefficient will increase the conductivity of the electrolyte by increasing the concentration of free ions in the solution.
denotes the partial molar volume and υ denotes the no. of moles of ions into which a mole of electrolyte dissociates. For 1–1 electrolyte (e.g., HCl and HNO3) of molar volume 30 cm3 mol−1 at 25 °C, the change in activity coefficient with pressure is ∼6 × 10−4 bar−1. This suggests that the activity coefficient increases by ∼6% with an increase in pressure to 100 bar. The increase in activity coefficient will increase the conductivity of the electrolyte by increasing the concentration of free ions in the solution.
3. Experimental tools for high-pressure electrochemistry
In contrast to ambient-pressure electrochemical studies, high-pressure electrochemical studies require a new frontier in instrumentation defined by innovative tools that allow observation, understanding, and control of complex, dynamic, and hierarchical catalytic processes. Performing electrochemical experiments at elevated pressures (>200 bar) is challenging, and reactors/instruments must be designed for safe operation. Realizing extreme conditions in the confines of a laboratory setting is a formidable challenge, especially characteristics that include extreme pressures coupled with corrosive or contaminating fluids and gases, high electric fields, and highly heterogeneous sample environments. The development of capabilities to evaluate processes under extreme conditions is needed across the disciplines of materials and chemistry. To study high-pressure electrochemistry, progress is required to develop novel electrochemical reactors operating at extreme pressures. For example, we do not have (i) high-pressure electrochemical reactors and their stacks that can withstand >200 bar, (ii) high-throughput screening tools for electrochemical synthesis at high pressures, and (iii) high-pressure electrochemical cells for operando FTIR, Raman, NMR, XAS, ambient XPS, and neutron scattering experiments.A range of high-pressure electrochemical reactors with different capacities and capabilities are available (see Fig. 9). These are – (i) high-pressure (<0.04 GPa/400 bar) batch and continuous reactors of 10 to 1000 mL volumes with better gas management, (ii) very high pressure (1–10 GPa) Paris–Edinburgh reactors of 0.1–1 mL volume, and (iii) ultrahigh pressure (10–100 GPa) Diamond Anvil Cell (DAC) reactors with < 0.1 nL volumes.
 | ||
| Fig. 9 Range of high-pressure electrochemical reactors with varying volume and pressure levels. | ||
As shown in Fig. 9, various apparatuses are available to push the capabilities of electrochemistry to very high pressure, above 1 GPa, such as the cubic anvil press,60 Paris–Edinburgh (PE) cell,61 and the piston-cylinder.62–64 Such apparatus is used to study the electrochemical properties at elevated pressures for systems involving extreme pressures, such as oceanographic, geothermal, and exoplanet explorative studies. Systematic adaptation of these apparatuses to an electrochemical cell stack and simultaneous characterization probes that can be used to conduct the electrochemical synthesis of chemicals at elevated pressures is required. The high-pressure electrochemical cells must be configured to place electrodes connected to a potentiostat to control potentials.
Fig. 10A shows a detailed schematic of the piston-based high-pressure cell, and Fig. 10B shows a schematic of the modified autoclave to perform high-pressure electrochemical synthesis. The schematic of these designs is taken from Cruanes et al.,44 and Hara et al.33 Autoclaves usually are designed to carry out reactions at high temperatures and pressures. The existing autoclaves must be modified to insert three electrodes to carry out the electrochemical reactions. Autoclaves are generally made of stainless steel; hence, they cannot be used directly to perform electrochemical reactions. Either the walls of the autoclave have to be coated with an insulating material like Teflon, or a separate electrochemical cell made of glass or polypropylene has to be placed inside the autoclave. The electrochemical cell has to be designed based on the autoclave dimensions. The other approach to pressurizing is by using a piston, which is commonly employed to pressurize liquid-based systems.
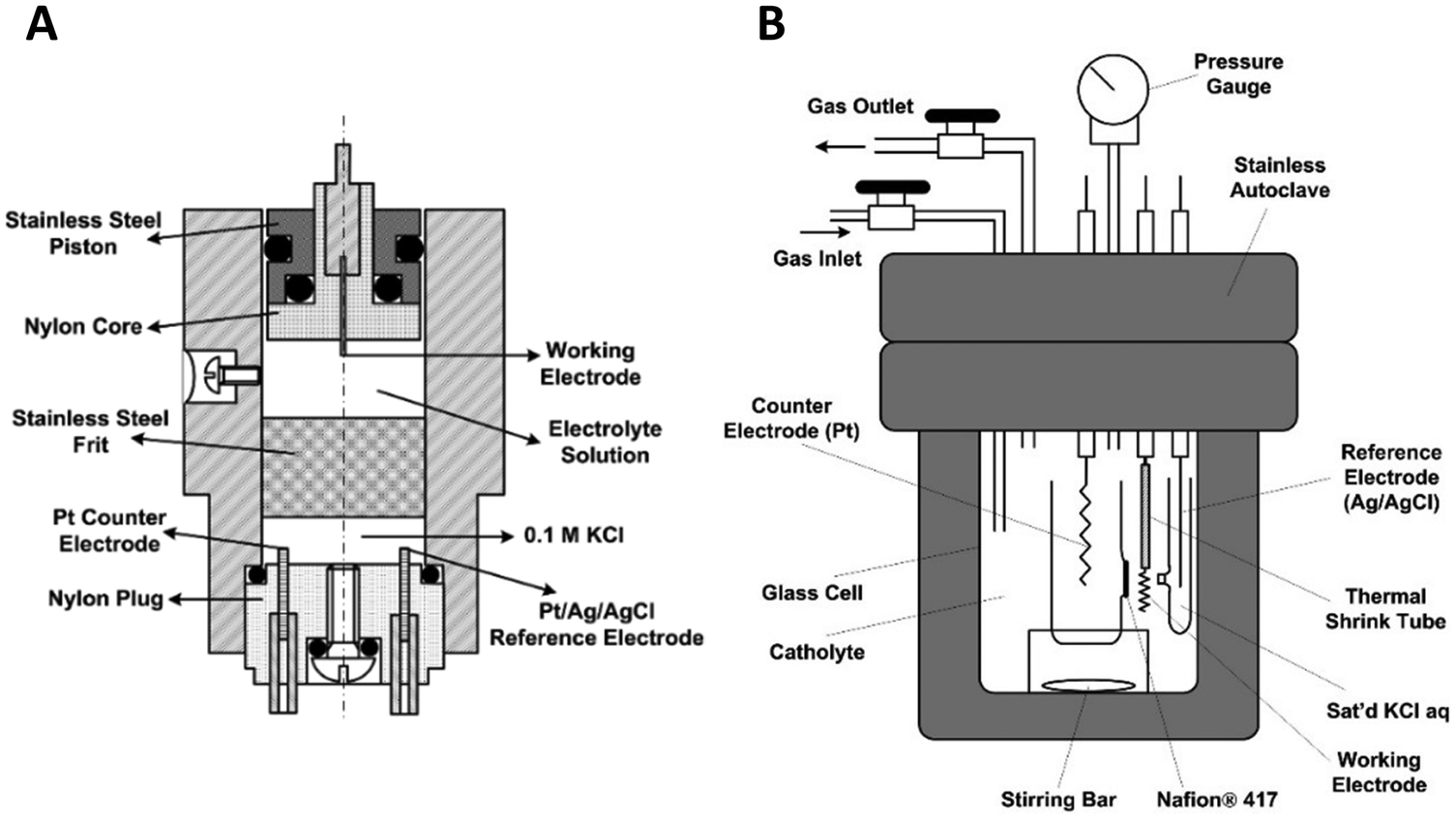 | ||
| Fig. 10 Schematic of a modified autoclave to place the electrodes for high-pressure electrochemical synthesis (adapted from (Cruanes et al.,44); and (Hara et al.,33 1995)). Copyright 1992 American Chemical Society, and Copyright 1995 Elsevier B.V. The electrochemical cell is placed inside the autoclave and the gas is pressurized using a compressor. | ||
Increased pressure in situ/operando electrochemical arrangements for Fourier transform IR (FTIR) spectroscopy, X-ray absorption spectroscopy (XAS), ambient pressure X-ray photoelectron spectroscopy (AP-XPS), and nuclear magnetic resonance (NMR) spectroscopy are not currently available. AZO materials sell a high-pressure, high-temperature FTIR cell to analyze solid samples (denoted by Fig. S3A, ESI†), which can be modified to perform operando electrochemical reactions at high pressures. A setup used in (a) FTIR and (b) UV-Vis determinations showing the high-pressure view cell coupled to the respective spectrophotometers is shown in Fig. 11a and b, respectively. A schematic of the custom-built operando Raman system with the high-pressure H-cell is shown in Fig. 11c. A flat and beveled diamond anvil for generating high pressures is shown in Fig. 11d. In addition, a range of diamond anvil cells (DACs) are sold by Almax easyLab Inc. (Fig. S3B, ESI†) which can be used for testing the properties of materials using electrochemical impedance spectroscopy as described by Han et al.65 The method can be modified and extended for other fundamental electrochemical studies. Fig. 11e represents an assembled rotational tomographic Paris Edinburgh cell device, which is a portable press for micro-tomographic 4-D imaging at extreme pressure conditions and Paris-Edinburgh cells developed at Oakridge National Laboratory (Fig. S3C, ESI†) are used for neutron diffraction studies, and such cells can be modified for in situ XAS and operando ambient pressure XPS studies at high pressures. Parr Instrument Company sells several modified autoclave designs for high-pressure electrochemical synthesis. Fig. S4 (ESI†) show a few of the commercially available high-pressure reactors, including a high-pressure batch electrochemical cell, a high-pressure rotating disc electrode (RDE) setup, and a high-pressure continuous electrochemical cell. Commercial autoclave setups available for high-pressure electrochemical studies like electrochemical reduction of CO2 to formic acid/formate and electrochemical synthesis of ammonia are shown in Fig. 12.
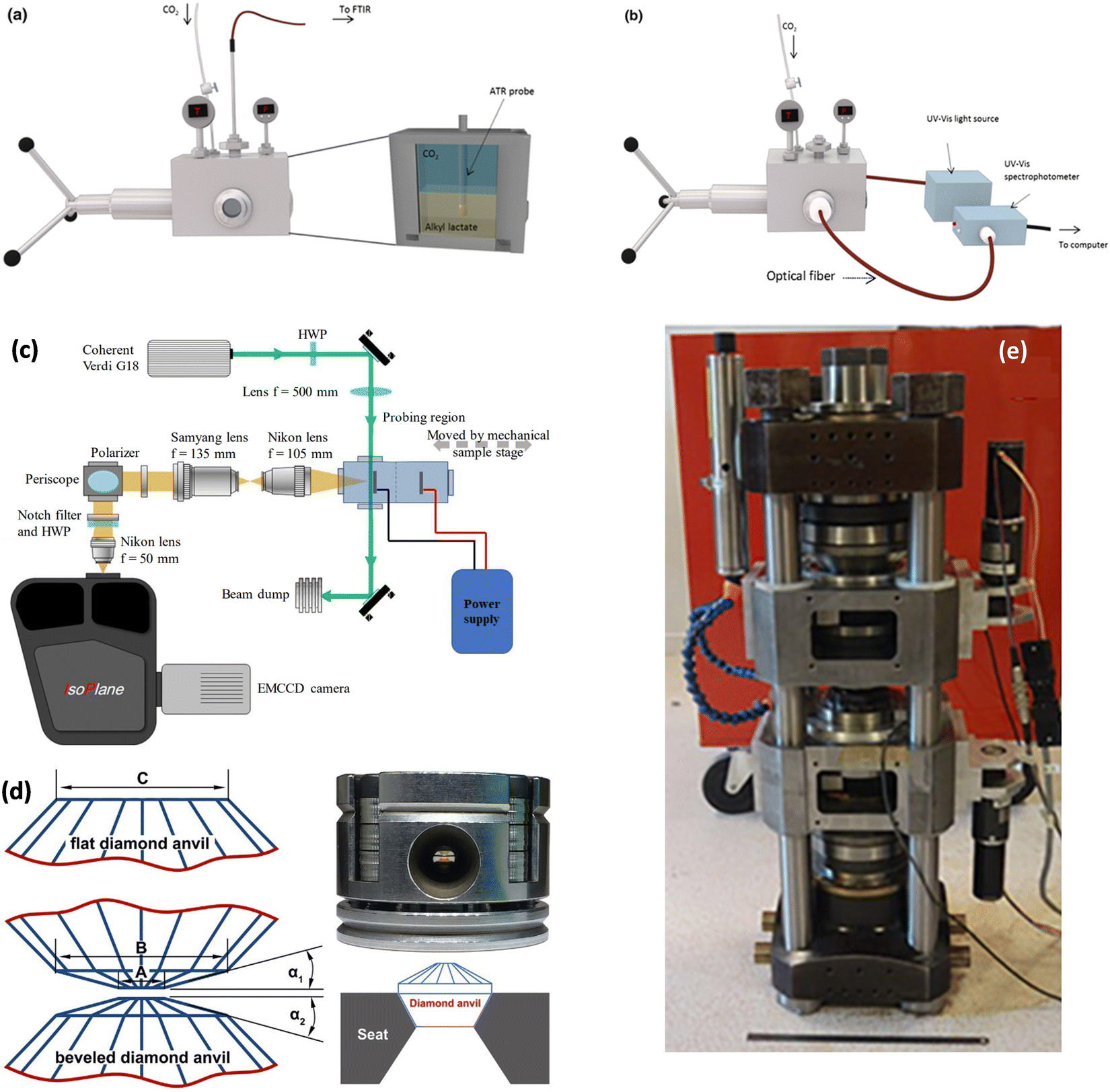 | ||
| Fig. 11 Instrumentation for fundamental electrochemical studies at higher pressures: setup used in (a) FTIR and (b) UV-Vis determinations showing the high-pressure view cell coupled to the respective spectrophotometers, Adapted from Medina-Gonzalez et al.66 Copyright 2017 Springer Science Business Media New York. (c) Schematic of the custom-built operando Raman system with the high-pressure H-cell, Adapted from Ramdin et al.35 Copyright 2019 American Chemical Society. (d) Flat and beveled diamond anvils for generating high pressures, Adapted from Li et al.67 Copyright 2018 Published under the PNAS license. (e) Assembled rotational tomographic Paris Edinburgh cell device which is a portable press for micro-tomographic 4-D imaging at extreme pressure conditions, Adapted from Philippe et al.68 Copyright 2016 The Author(s). Published by Informa UK Limited, trading as Taylor & Francis Group. | ||
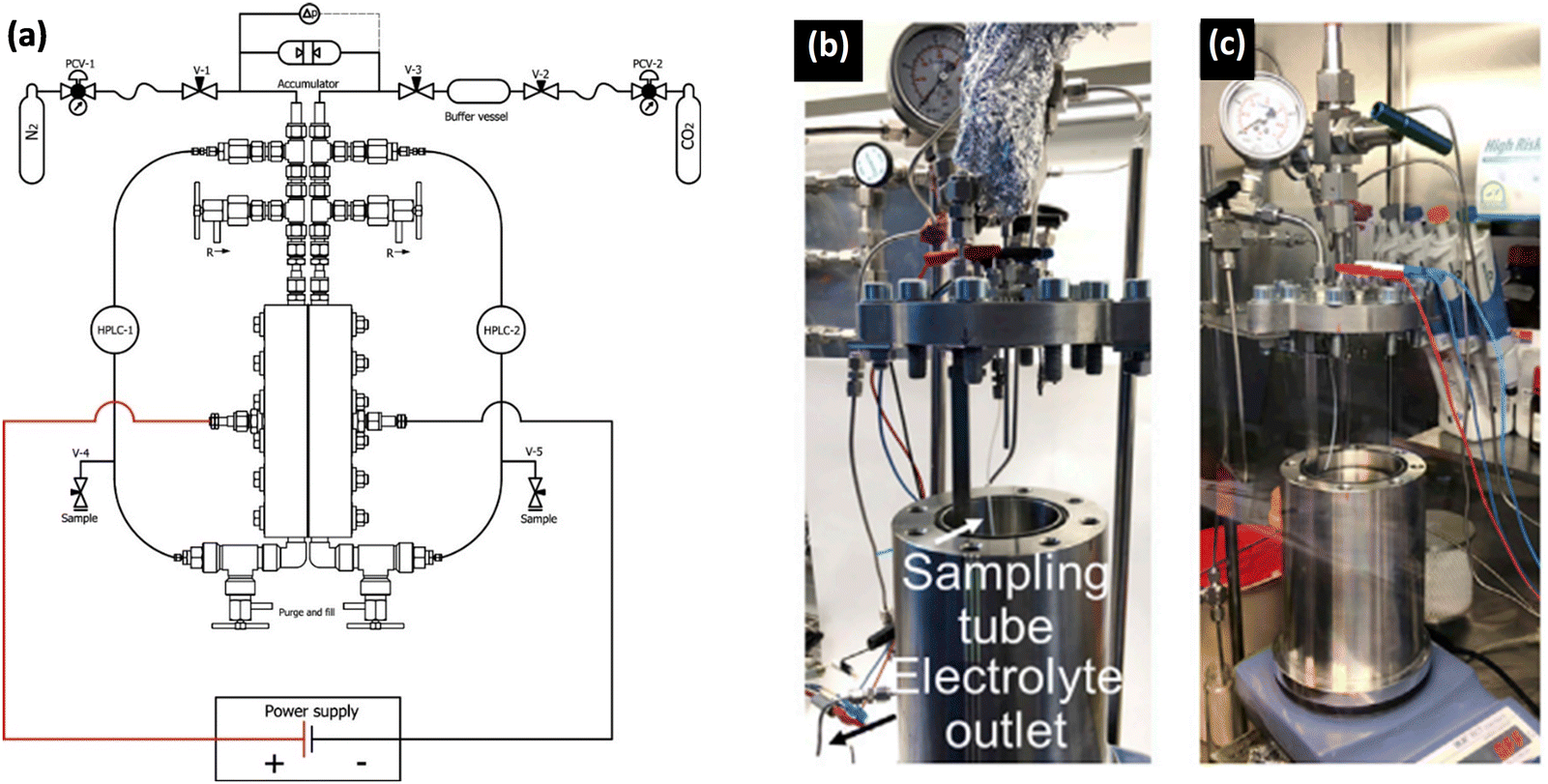 | ||
| Fig. 12 Examples of autoclave setups developed for high-pressure electrochemical studies. (a) A schematic of a continuous-flow high-pressure setup for electrochemical reduction reaction,1 Adapted from Ramdin et al.35 Copyright 2019 American Chemical Society. Photos of batch autoclave reactors placed in (b) the fume hood and (c) the Ar glovebox for electrochemical synthesis of ammonia. Adapted from Li et al.69 Copyright 2022 The Author(s). Published by Elsevier Inc. | ||
4. Operation of high-pressure electrochemical reactors
Differential pressure operation
Differential pressure operation involves maintaining different pressures in the anode and the cathode chambers of the electrolyzers. Differential pressure operation has numerous advantages as well as disadvantages. One of the significant advantages of differential pressure operation is the reduction in product crossover, especially at low current densities. By maintaining a higher pressure at the anode than the cathode, the migration of products from the anode to the cathode will be mitigated, resulting in improved selectivity and purity of the product gases. Differential pressure operation can help retain the electrolyte within the cell by reducing the risk of electrolyte loss due to evaporation or other factors. This helps improve the selectivity of the gas evolution reactions, leading to improved control over the desired product generation. One of the disadvantages of differential pressure operation is the complicated system design and the requirement of additional pressure control mechanisms and safety measures needed to maintain pressure differentials. Differential pressure operation has a risk of H2 crossover, where H2 can permeate through the membrane and impact the cell efficiency and product purity. Differential pressure operation could lead to variation in the electrolyte concentration between the compartments, affecting the reaction rates and the overall system performance. Pressurization of both compartments ensures uniform conditions and simplifies the system design. However, differential pressure operation has unique advantages, as mentioned above.Dynamic operation
Dynamic operation of an electrochemical system refers to dynamically changing the operating conditions based on the price and availability of renewable energy sources. Pressurization affects the dynamic operation of electrochemical processes such as PEM electrolyzers. Pressurization affects the response time and efficiency during dynamic operation. The system requires more controls for rapidly shifting between high and low operating current densities. Pressurization can improve electrode stability by minimizing the concentration polarization and electrode kinetics and could maintain consistent H2 production rates during fluctuating energy inputs. The relationship between pressure, current densities, and the cost of renewable electricity must be carefully analyzed to get a pressure range that offers the best efficiency during the dynamic operation. It can impact heat generation and dissipation within the electrochemical system, and efficient heat management systems are important for stable operation and preventing overheating during dynamic load changes. Pressurization has several advantages for operating the systems dynamically but requires better system control and heat management techniques.Safety precautions for operating high-pressure electrochemical systems
Safety requirements are essential when dealing with high-pressure electrochemical systems. A comprehensive discussion of the safety measures is provided in this section to ensure that early-stage researchers who might be less experienced understand the potential hazards and carry out the experiments safely and responsibly.Material compatibility and efficient design: It needs to be ensured that the reactors are built using materials that can withstand operating conditions and resist corrosion from the reactants and products. All reactor vessels, fittings, sealants, and other components need to be designed and constructed to withstand beyond the operating pressure to ensure safe operation. Refer to ASTM standards while designing reactors and other components like sealants and fittings to withstand high pressures. And always operate a high-pressure reactor in a blast-proof chamber.
Pressure relief devices: Fail-safe mechanisms such as rupture discs and pressure relief valves must be installed to prevent over-pressurization. Over-pressure protection systems need to be set to release at a pressure slightly above the operating pressure of the reactor. The pressure relief systems must be routinely tested and maintained to ensure proper functioning. Refer to ASTM standards for adequate choice for fail-safe mechanisms.
Documentations: A detailed standard operating procedure (SOP) must be developed to operate the high-pressure electrochemical reactors. A detailed record of all the experiments performed, safety checks, maintenance checks, operating conditions, and any deviations from the SOP must be maintained.
Operator training: Comprehensive training must be provided to all personnel operating the high-pressure electrochemical reactor to follow the SOP. It needs to ensure that the operators understand the principles of electrochemistry, pressurization, and the potential hazards associated with the processes.
Emergency response plan: A detailed emergency response plan should be in place that outlines the steps to be followed in case of leaks, fires, and explosions. Regular drills and training exercises must be conducted to ensure all personnel are familiar with the emergency response procedures.
Regular maintenance and inspection: A regular maintenance schedule for high-pressure electrochemical reactors and associated instruments must be conducted. Also, regular inspections, calibration of the pressure sensors, and replacement of damaged or worn-out components need to be performed.
It is essential to involve safety engineers, design experts, and other authorities to ensure that the design, operation, and maintenance of high-pressure electrochemical reactors are performed at the highest level of safety for the operating personnel and the environment.
5. Economic considerations
The primary motivation for high-pressure electrochemistry is to realize higher current density and reduce the energy consumption (or cell voltage) of the process. Fig. 2 shows that there could be a net reduction in electrical energy requirement to operate an electrochemical reactor at high pressure compared to ambient pressures. However, the high-pressure operation requires thick-walled reactors, which are more expensive than ambient-pressure reactors.The higher operating current density at high pressure also helps in reducing the number of reactors and, thereby, capital cost. For example, consider an electrochemical reaction operating at 1 mA cm−2 at 1 atm that is scaled up using 100 electrochemical cells of 10 cm2 area each to attain a total operating current of 1 A for 100 cells. Since the current density varies linearly with the pressure of reactants, increasing the pressure to 100 atm is expected to increase the current density to 100 mA cm−2. Now, scaling up the 100 atm reactors to attain an overall current of 1 A will require 1 electrochemical cell of 10 cm2 area. This linear dependence of current density on the pressure results in two capital (or reactor) choices – the first choice is 100 cells operating at 1 atm and the second choice is 1 cell operating at 100 atm. Choosing a high-pressure reactor can result in a significant reduction in footprint and some savings in capital investment.
Fig. 13 shows the estimated cost of mild steel as a function of the size of the reactor that can withstand up to 100 bar (adapted from the book on Chemical Engineering Economics by Garrett).70 It can be observed that the cost of the material and the reactor increases exponentially as the size of the reactor increases. For decentralized production, the volume of the reactor will be less than 100 gallon and the estimated cost of the material is much less. We note that the original estimate made in the book is based on the 1989 data and inflation has to be taken into account, but the order of reduction in cost would be the same as the ones reported.
 | ||
| Fig. 13 Cost of mild steel as a function of the size of the reactor and operation pressures. Figure adapted from Chemical Engineering Economics by Garrett.70 | ||
6. Challenges of high-pressure electrochemistry
High-pressure electrochemistry requires specialized and often complex equipment, including pressure vessels, seals, and safety measures. These components can be costly to acquire, maintain, and operate. Additionally, the complexity of the system may introduce additional sources of experimental variability and potential points of failure. High-pressure experiments entail inherent safety risks. The presence of pressurized gases or liquids increases the potential for leaks, ruptures, or explosions, posing risks to researchers and the laboratory environment. Proper safety protocols, training, and infrastructure are crucial to mitigate these hazards effectively. A detailed safety precautions procedure is provided in Section 4. Not all electrochemical cells, electrodes, or catalyst materials may be compatible with high-pressure conditions. The corrosive nature of certain electrolytes or the instability of electrode materials at high pressures can limit the range of materials that can be used in high-pressure electrochemistry experiments. Elevated pressures can significantly affect the transport of reactants and products within the electrochemical cell. Increased viscosity and reduced diffusion coefficients can impede mass transport, leading to slower reaction rates and limiting the overall performance of the system. High-pressure electrochemical reactions may involve intricate kinetic pathways and complex reaction mechanisms. Understanding and modeling these reactions can be challenging, requiring advanced theoretical approaches and computational resources. Conducting experiments at high pressures introduces technical challenges. Ensuring uniform pressure distribution, preventing leaks, and accurately measuring and controlling reaction parameters are complex tasks that require specialized expertise. These challenges can impact data quality and reliability. Scaling up high-pressure electrochemical processes for practical applications can be challenging. Designing and constructing large-scale high-pressure systems, along with ensuring safety and reliability, pose significant engineering and operational hurdles that need to be addressed. Gas crossover is one of the key challenges associated with pressurization. Gas crossover refers to the unintended transfer of gases across the membranes in electrochemical systems. This could be predominant if one of the cathode/anode sides is pressurized. This would lead to reduced efficiency (due to undesired side reactions) and higher energy consumption, and affect the purity of the product gases. Potential safety concerns can arise if the gas crossing could lead to mixing reactive gases on the opposite sides of the cell. The gas crossover can be prevented by choosing an appropriate membrane material with low gas permeability or via further processing, such as annealing.717. Conclusions and outlook
High-pressure electrochemical synthesis of chemicals is in the nascent stages of development. The prospect of using elevated pressures is to overcome the existing challenges in the electrochemical synthesis of chemicals that involve gas phase reactants, which tend to have lower activities and selectivities. High pressures require lesser energy compared to high-temperature operation; hence, high-pressure electrochemical synthesis of chemicals can be scaled up for the decentralized production of chemicals, and the capital cost will be considerably less for the decentralized production as the volume of the reactor is an exponential function of the cost. The theory behind the improvement in the electrochemical reaction performance, including activity, selectivity, stability, and energy efficiency at elevated pressures, is discussed. For high-pressure electrochemistry – (i) the energy requirement is lower due to the Nernst effect and selective absorption of gases, (ii) the current densities are higher due to higher solubility, lower overpotential, and high exchange current density, (iii) the selectivities can be better due to electrostriction, a higher separation between reduction lines or oxidation lines in the Pourbaix diagram, higher stabilization of intermediates, and higher surface coverages of reactants, and (iv) improved stability due to the increase in the oxidation potential of the catalyst. The electrical conductivity does decrease with increasing pressure, which could result in higher ohmic losses.We emphasize the need for novel high-pressure reactors for the synthesis of chemicals and operando tools for understanding the reaction mechanisms. High-pressure electrochemical synthesis could be a paradigm shift in the decarbonization of chemical manufacturing, and the justifications provided in this work would motivate researchers to explore the field.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based on the work performed in the Materials and Systems Engineering Laboratory at the University of Illinois Chicago in collaboration with Dr. Gauthier's research group. This research used resources of the National Energy Research Scientific Computing Center (NERSC), a U.S. Department of Energy Office of Science User Facility located at Lawrence Berkeley National Laboratory, operated under Contract No. DE-AC02-05CH11231 using NERSC award BES-ERCAP0022848.References
- R. Sharifian, R. Wagterveld, I. Digdaya, C. Xiang and D. Vermaas, Energy Environ. Sci., 2021, 14, 781–814 RSC.
- M. Fajardy, A. Köberle and N. Macdowell, BECCS deployment: a reality check. Report for the Grantham Institute, Imperial College London, Report, 2018.
- D. S. Mallapragada, Y. Dvorkin, M. A. Modestino, D. V. Esposito, W. A. Smith, B.-M. Hodge, M. P. Harold, V. M. Donnelly, A. Nuz and C. Bloomquist, Joule, 2023, 7, 23–41 CrossRef CAS.
- Z. J. Schiffer and K. Manthiram, Joule, 2017, 1, 10–14 CrossRef.
- N. C. Kani, A. Prajapati, B. A. Collins, J. D. Goodpaster and M. R. Singh, ACS Catal., 2020, 10, 14592–14603 CrossRef CAS.
- A. R. Singh, B. A. Rohr, J. A. Schwalbe, M. Cargnello, K. Chan, T. F. Jaramillo, I. Chorkendorff and J. K. Nørskov, ACS Catal., 2017 DOI:10.1021/acscatal.6b03035.
- M. A. Shipman and M. D. Symes, Catal. Today, 2017, 286, 57–68 CrossRef CAS.
- N. Lazouski, M. Chung, K. Williams, M. L. Gala and K. Manthiram, Nat. Catal., 2020, 3, 463–469 CrossRef CAS.
- K. Li, S. Z. Andersen, M. J. Statt, M. Saccoccio, V. J. Bukas, K. Krempl, R. Sažinas, J. B. Pedersen, V. Shadravan and Y. Zhou, Science, 2021, 374, 1593–1597 CrossRef CAS PubMed.
- H.-L. Du, M. Chatti, R. Y. Hodgetts, P. V. Cherepanov, C. K. Nguyen, K. Matuszek, D. R. MacFarlane and A. N. Simonov, Nature, 2022, 609, 722–727 CrossRef CAS PubMed.
- J. Choi, H.-L. Du, C. K. Nguyen, B. H. Suryanto, A. N. Simonov and D. R. MacFarlane, ACS Energy Lett., 2020, 5, 2095–2097 CrossRef CAS.
- S. Z. Andersen, V. Čolić, S. Yang, J. A. Schwalbe, A. C. Nielander, J. M. McEnaney, K. Enemark-Rasmussen, J. G. Baker, A. R. Singh and B. A. Rohr, Nature, 2019, 570, 504–508 CrossRef CAS PubMed.
- N. C. Kani, A. Prajapati and M. R. Singh, ACS ES&T Engg., 2022, 2(6) DOI:10.1021/acsestengg.1c00484.
- N. C. Kani, J. A. Gauthier, A. Prajapati, J. Edgington, I. Bordawekar, W. Shields, M. Shields, L. C. Seitz, A. R. Singh and M. R. Singh, Energy Environ. Sci., 2021, 14(12) 10.1039/D1EE01879E.
- N. C. Kani, N. H. L. Nguyen, K. Markel, R. R. Bhawnani, B. Shindel, K. Sharma, S. Kim, V. P. Dravid, V. Berry, J. A. Gauthier and M. R. Singh, Adv. Energy Mater., 2023, 13(17) DOI:10.1002/aenm.202204236.
- W. Liu, P. Zhai, A. Li, B. Wei, K. Si, Y. Wei, X. Wang, G. Zhu, Q. Chen and X. Gu, Nat. Commun., 2022, 13, 1877 CrossRef CAS PubMed.
- H. S. Jeon, S. Kunze, F. Scholten and B. Roldan Cuenya, ACS Catal., 2018, 8, 531–535 CrossRef CAS.
- A. Prajapati, N. C. Kani, J. A. Gauthier, R. Sartape, J. Xie, I. Bessa, M. T. Galante, S. L. Leung, M. H. Andrade and R. T. Somich, Cell Rep. Phys. Sci., 2022, 3, 101053 CrossRef CAS.
- D. Yang, Q. Zhu, C. Chen, H. Liu, Z. Liu, Z. Zhao, X. Zhang, S. Liu and B. Han, Nat. Commun., 2019, 10, 677 CrossRef CAS PubMed.
- A. H. B. Mostaghimi, T. A. Al-Attas, M. G. Kibria and S. Siahrostami, J. Mater. Chem. A, 2020, 8, 15575–15590 RSC.
- A. Prajapati, R. Sartape, N. C. Kani, J. A. Gauthier and M. R. Singh, ACS Catal., 2022, 12, 14321–14329 CrossRef CAS.
- L. Adams, J. Am. Chem. Soc., 1931, 53, 3769–3813 CrossRef CAS.
- L. Adams and R. Hall, J. Washington Acad. Sci., 1931, 21, 183–194 CAS.
- L. Adams and R. Hall, J. Phys. Chem., 2002, 35, 2145–2163 CrossRef.
- E. Cohen, ZS. Physik. Chem. (A), 1934, 167, 365 Search PubMed.
- D. Giovanelli, N. S. Lawrence and R. G. Compton, Electroanalysis, 2004, 16, 789–810 CrossRef CAS.
- G. Schneider, Berichte der Bunsengesellschaft für physikalische Chemie, 1972, 76, 325–331 CrossRef CAS.
- G. Hills and D. Kinnibrugh, J. Electrochem. Soc., 1966, 113, 1111 CrossRef CAS.
- A. Gancy and S. Brummer, J. Electrochem. Soc., 1968, 115, 804 CrossRef CAS.
- A. Ewald and S. Lim, J. Phys. Chem., 1957, 61, 1443–1445 CrossRef CAS.
- M. B. Towns, R. S. Greeley and M. Lietzke, J. Phys. Chem., 1960, 64, 1861–1863 CrossRef CAS.
- P.-W. Guan, R. J. Hemley and V. Viswanathan, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2110470118 CrossRef CAS PubMed.
- K. Hara, A. Kudo and T. Sakata, J. Electroanal. Chem., 1995, 391, 141–147 CrossRef.
- M. Todoroki, K. Hara, A. Kudo and T. Sakata, J. Electroanal. Chem., 1995, 394, 199–203 CrossRef.
- M. Ramdin, A. R. Morrison, M. De Groen, R. Van Haperen, R. De Kler, L. J. Van Den Broeke, J. M. Trusler, W. De Jong and T. J. Vlugt, Ind. Eng. Chem. Res., 2019, 58, 1834–1847 CrossRef CAS PubMed.
- L. M. Welch, M. Vijayaraghavan, F. Greenwell, J. Satherley and A. J. Cowan, Faraday Discuss., 2021, 230, 331–343 RSC.
- J. Lee, J. Yang and J. H. Moon, ACS Energy Lett., 2021, 6, 893–899 CrossRef CAS.
- R. S. Kim and Y. Surendranath, ACS Cent. Sci., 2019, 5, 1179–1186 CrossRef CAS PubMed.
- E. M. Lee, T. Ludwig, B. Yu, A. R. Singh, F. Gygi, J. K. Nørskov and J. J. de Pablo, J. Phys. Chem. Lett., 2021, 12, 2954–2962 CrossRef CAS PubMed.
- M. Jørgensen and H. Gronbeck, J. Phys. Chem. C, 2017, 121, 7199–7207 CrossRef.
- A. Bajpai, P. Mehta, K. Frey, A. M. Lehmer and W. F. Schneider, ACS Catal., 2018, 8, 1945–1954 CrossRef CAS.
- P. K. Frolich, E. Tauch, J. Hogan and A. Peer, Ind. Eng. Chem., 1931, 23, 548–550 CrossRef CAS.
- R. Nakhaei-Kohani, E. Taslimi-Renani, F. Hadavimoghaddam, M.-R. Mohammadi and A. Hemmati-Sarapardeh, Sci. Rep., 2022, 12, 3625 CrossRef CAS PubMed.
- M. T. Cruanes, H. G. Drickamer and L. R. Faulkner, J. Phys. Chem., 1992, 96, 9888–9892 CrossRef CAS.
- T. Han, H. Liu, J. Wang, C. Gao and Y. Han, J. Phys. Chem. C, 2021, 125, 8788–8793 CrossRef CAS.
- J. Desnoyers, R. Verrall and B. Conway, J. Chem. Phys., 1965, 43, 243–250 CrossRef CAS.
- M. T. Cruanes, H. G. Drickamer and L. R. Faulkner, Langmuir, 1995, 11, 4089–4097 CrossRef CAS.
- Z. Yang, B. Kan, J. Li, Y. Su and L. Qiao, Int. J. Hydrogen Energy, 2017, 42, 27446–27457 CrossRef CAS.
- F. Sun, S. Ren, Z. Li, Z. Liu, X. Li and C. Du, Mater. Sci. Eng., A, 2017, 685, 145–153 CrossRef CAS.
- D. D. Macdonald, J. Supercritical Fluids, 2004, 30, 375–382 CrossRef CAS.
- J. Newman and K. E. Thomas-Alyea, Electrochemical systems, John Wiley & Sons, 2012 Search PubMed.
- D. Brazier and G. Freeman, Can. J. Chem., 1969, 47, 893–899 CrossRef CAS.
- S. Dahl, A. Logadottir, R. Egeberg, J. Larsen, I. Chorkendorff, E. Törnqvist and J. K. Nørskov, Phys. Rev. Lett., 1999, 83, 1814 CrossRef.
- A. Cao, V. J. Bukas, V. Shadravan, Z. Wang, H. Li, J. Kibsgaard, I. Chorkendorff and J. K. Nørskov, Nat. Commun., 2022, 13, 2382 CrossRef CAS PubMed.
- B. Hammer and J. K. Norskov, Nature, 1995, 376, 238–240 CrossRef CAS.
- R. Horne and D. Johnson, J. Phys. Chem., 1967, 71, 1147–1149 CrossRef.
- S. Sawamura, Y. Yoshimura, K. Kitamura and Y. Taniguchi, J. Phys. Chem., 1992, 96, 5526–5529 CrossRef CAS.
- L. B. Sargent Jr, ASLE Trans., 1983, 26, 1–10 CrossRef.
- J. Newman and N. P. Balsara, Electrochemical systems, John Wiley & Sons, 2021 Search PubMed.
- H. Saitoh, A. Machida and K. Aoki, Chin. Sci. Bull., 2014, 59, 5290–5301 CrossRef CAS.
- J. Valenta, T. Naka, M. Diviš, M. Vališka, P. Proschek, K. Vlášková, M. Klicpera, J. Prokleška, J. Custers and J. Prchal, J. Phys.: Condens. Matter, 2020, 32, 425601 CrossRef CAS PubMed.
- S. Gabáni, M. Orendáč, J. Kušnír, E. Gažo, G. Pristáš, T. Mori and K. Flachbart, J. Low Temp. Phys., 2017, 187, 559–564 CrossRef.
- Y. Lu, H. Kono, T. Larkin, A. Rost, T. Takayama, A. Boris, B. Keimer and H. Takagi, Nat. Commun., 2017, 8, 1–7 CrossRef PubMed.
- T. Suski, J. Zulawnik and M. Malinowski, J. Phys. E: Sci. Instrum., 1982, 15, 1016 CrossRef CAS.
- Y. Han, S. Axnanda, E. J. Crumlin, R. Chang, B. Mao, Z. Hussain, P. N. Ross, Y. Li and Z. Liu, J. Phys. Chem. B, 2018, 122, 666–671 CrossRef CAS PubMed.
- Y. Medina-Gonzalez, A. Jarray, S. Camy, J.-S. Condoret and V. Gerbaud, J. Solution Chem., 2017, 46, 259–280 CrossRef CAS.
- B. Li, C. Ji, W. Yang, J. Wang, K. Yang, R. Xu, W. Liu, Z. Cai, J. Chen and H.-K. Mao, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 1713–1717 CrossRef CAS PubMed.
- J. Philippe, Y. Le Godec, M. Mezouar, M. Berg, G. Bromiley, F. Bergame, J. Perrillat, M. Alvarez-Murga, M. Morand and R. Atwood, High Press. Res., 2016, 36, 512–532 CrossRef CAS.
- S. Li, Y. Zhou, K. Li, M. Saccoccio, R. Sažinas, S. Z. Andersen, J. B. Pedersen, X. Fu, V. Shadravan and D. Chakraborty, Joule, 2022, 6, 2083–2101 CrossRef CAS PubMed.
- D. E. Garrett, Chemical engineering economics, Springer Science & Business Media, 2012 Search PubMed.
- C. M. Evans, M. R. Singh, N. A. Lynd and R. A. Segalman, Macromolecules, 2015, 48, 3303–3309 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ey00284e |
| This journal is © The Royal Society of Chemistry 2024 |