
An environmentally benign and atom-economical protocol for the regioselective synthesis of isoquinolones from o-alkynylaldehydes†
Muskan
and
Akhilesh K.
Verma
 *
*
Synthetic Organic Chemistry Research Laboratory, Department of Chemistry, University of Delhi, Delhi-110007, India. E-mail: averma@acbr.du.ac.in
First published on 19th December 2023
Abstract
An environmentally benign, transition metal- and base-free, iodine-promoted atom-economical protocol for the synthesis of the privileged isoquinolone scaffold via regioselective intramolecular iodoamidation of alkynes under mild conditions has been developed. The present synthetic approach being metal, additive, and solvent-free adheres to the principles of green chemistry, as it tends to minimize waste production. The synthesized product contains an iodo as well as a free –OH group that is readily accessible for subsequent transformation to afford biologically relevant compounds.
Contemporary synthetic chemistry is witnessing a paradigm shift towards more efficient, sustainable, and environmentally benign protocols for the construction of biologically relevant molecules.1 A sustainable and environmentally accountable approach in the realm of chemistry signifies a commitment to conduct chemical processes and industrial research in a manner that prioritizes the well-being of the planet, human health, and future generations.2 One of the most notable developments in this context is the emergence of metal and solvent-free synthesis as a cornerstone of modern organic chemistry.3 Metal-based catalysts are particularly effective at accelerating chemical reactions, but because of their toxicity and potential for bioaccumulation, they pose significant ecological risks and generate a significant amount of hazardous waste.4 Solvents not only increase the cost of production, but also some solvents especially volatile organic compounds (VOCs) have adverse environmental effects and can contribute to air pollution.5 Metal and solvent-free reactions adhere well with green chemistry principles as they typically generate less waste.6 The reduction in metal and solvent usage directly contributes to a smaller environmental footprint, which is a critical consideration in modern synthetic chemistry.
Isoquinolone, a versatile N-heterocyclic compound, holds a prominent place in the realm of organic chemistry. It serves as a fundamental structural unit in various pharmaceutically relevant as well as naturally occurring compounds, particularly alkaloids (Fig. 1).7 In order to expand the biological spectrum of isoquinolone, great efforts have been made towards the development of new synthetic protocols for the construction of this privileged motif. The metal-catalyzed annulation reaction between 2-halobenzamide derivatives and alkynes is the most prevalent method for the synthesis of isoquinolone derivatives.8 While being efficient and versatile, these transformations are accompanied by certain limitations including pricey and toxic metal-based catalytic systems like palladium,9 rhodium,10 ruthenium,11 nickel,12 copper,13etc. as well as the use of environmentally hazardous organic solvents. Electrophilic annulation of ortho-(1-alkynyl) benzamides is another alternative for the synthesis of isoquinolones.14
 | ||
| Fig. 1 Biologically active isoquinolone cores. | ||
On the other hand, ortho-alkynyl aldehydes as versatile synthons have been well explored for the synthesis of numerous heterocycles and carbocycles (Scheme 1).15 However, the transformation of ortho-alkynyl aldehydes for the construction of isoquinolones is quite rare. Chiba16et al. achieved the synthesis of isoquinolones via cyclocondensation of ortho-alkynyl aldehydes and primary amines in the presence of an excess amount of CuBr·SMe2 and SiO2 along with the benzene–pyridine solvent system (Scheme 1b). The group of Wen17 and co-workers demonstrated the synthesis of isoquinolones through the Cu-salt catalyzed cyclization of 2-(1-alkynyl)benzaldimines under very high-temperature conditions. Hua18 and colleagues reported Zn(OTf)2 catalyzed cyclocondensation of aryl amines with ortho-alkynyl benzaldehydes in the presence of Zn(OTf)2, base (1.2 equiv.) at 120 °C, affording isoquinolones in moderate to good yields (Scheme 1c). Synthetic methodologies available for the synthesis of isoquinolones using ortho-alkynyl benzaldehydes have relied on metal-based catalytic systems, additives, and high temperatures and are limited to aryl amines only. Despite having impressive advances, the developed strategies still require advancement, in terms of environmentally benign conditions (metal-free, solvent-free, atom-economical, wide substrate scope with operational simplicity).
 | ||
| Scheme 1 Previous and present work. | ||
The synthesis of isoquinolones from ortho-alkynylbenzaldehydes using aliphatic amino alcohols has remained elusive. Considering the notable lack of procedures and in continuation of our ongoing work19 using ortho-alkynyl benzaldehydes, herein we report an environmentally benign, transition metal- and base-free, atom-economical protocol for the synthesis of the privileged isoquinolone scaffold via regioselective intramolecular iodoamidation of alkynes under mild conditions. The present methodology is advantageous over the hitherto known strategies as, here, the free-OH group remains intact and is readily accessible for subsequent transformation.
We started our investigation using o-alkynyl aldehyde 1a, 4-amino-1-butanol 2a, and iodine as our model substrates (Table 1). A reaction of substrate 1a with amino alcohol 2a (3.0 equiv.) using 1.0 equiv. of I2 and 2.0 equiv. of K2CO3 in DCM for 2 h at 25 °C gave the product 3a in 40% yield (entry 1). The use of 2.0 equiv. of iodine provided product 3a in 60% yield (entry 2); however a further increase of iodine and the time of the reaction did not improve the yield of product 3a (entries 3 and 4). When the reaction was conducted at an elevated temperature, a sluggish reaction developed, and only 35% yield of product 3a was formed (entry 5). The use of Cs2CO3, KHCO3, and NaHCO3 was found to be inferior for the reaction (entries 6–8). A reaction without using a base provided the desired product 3a in 70% yield (entry 9). Subsequently, we conducted the reaction without any base, and surprisingly, the yield improved. Following this, in our quest to identify the most suitable solvent, we performed a series of reactions under different solvent conditions. Regrettably, no positive impact on the yield of the desired product was observed (entries 10–14). Consequently, we attempted the reaction under neat conditions and observed the product 3a in 82% yield (entry 15). The use of ICl resulted in a lower yield of the cyclized product (entry 16).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Reagent (equiv.) | Solvent | Base | Temp./time (°C)/(h) | Yieldb (%) 3a |
| a Reaction conditions: reactions were carried out using o-alkynyl benzaldehyde 1a (0.25 mmol), 4-amino-1-butanol 2a (0.75 mmol), and I2 in an appropriate solvent (2 mL). b Isolated yield. | |||||
| 1 | I2 (1.0) | DCM | K2CO3 | 25/2 | 40 |
| 2 | I2 (2.0) | DCM | K2CO3 | 25/2 | 60 |
| 3 | I2 (3.0) | DCM | K2CO3 | 25/2 | 60 |
| 4 | I2 (2.0) | DCM | K2CO3 | 25/4 | 58 |
| 5 | I2 (2.0) | DCM | K2CO3 | 50/2 | 35 |
| 6 | I2 (2.0) | DCM | Cs2CO3 | 25/2 | 52 |
| 7 | I2 (2.0) | DCM | KHCO3 | 25/2 | 35 |
| 8 | I2 (2.0) | DCM | NaHCO3 | 25/2 | 30 |
| 9 | I2 (2.0) | DCM | — | 25/2 | 70 |
| 10 | I2 (2.0) | THF | — | 25/2 | Trace |
| 11 | I2 (2.0) | CH3CN | — | 25/2 | 55 |
| 12 | I2 (2.0) | Toluene | — | 25/6 | Trace |
| 13 | I2 (2.0) | DCE | — | 25/6 | 60 |
| 14 | I2 (2.0) | Dioxane | — | 25/6 | Trace |
| 15 | I 2 (2.0) | — | — | 25/2 | 82 |
| 16 | ICl (2.0) | — | — | 25/2 | 40 |

Having established the optimal conditions, we then probed the scope of substrates with respect to readily available ortho-alkynyl aldehydes 1 using 4-amino-1-butanol 2 for the synthesis of the corresponding quinolones 3a–k (Scheme 2). The reaction does not show any profound electronic effect, and both electron-rich 1b–d, 5a, 5g (4-Me, 4-OMe, 4-Et, 3-Me, 4-nBu) and electron-deficient 1f, 1h (4-F, 4-NO2) substituents on the phenyl/aryl ring attached to the alkyne were found to be suitable to afford the desired products in good to excellent yields (78%–90%). Next, a diverse array of starting substrates possessing the sterically hindered alkynyl group 1i–k were examined. To our delight, steric effects did not appear to impact the iodo-cyclization, as we obtained the respective products 3i–k in appreciable yields. The X-ray crystal structure20 of 3e confirmed that the product formed is exclusively a 6-endo-dig cyclized product. Incredibly, a thienyl heterocyclic motif was also tolerated to afford 3g in 75% yield.
 | ||
| Scheme 2 Scope of ortho-alkynylaldehydes. Reactions were performed using 1a (0.25 mmol), 2a (0.75 mmol), and I2 (0.50 mmol) at 25 °C for 2 h. Isolated yield. | ||
Next, we investigated how substituents on the alkyne-tethered phenyl ring influenced the reaction (Scheme 3). Substrates bearing 5-Cl, 5-Br, and 5-CF3, 5-NO2 groups on the phenyl were all compatible with this conversion, giving the corresponding products 5a–k in 87%–95% yields. It is noteworthy that substrates with –NO2 and –CF3 at the C-5 position, with respect to the phenyl ring, delivered products with slightly high yields. Furthermore, cycloalkyl substituted o-alkynyl aldehyde 4b–c also worked well and afforded the desired product 5b and 5c in 80% and 75% yields, respectively. The substrate-bearing methoxy at the 4- and 5-positions of the ring afforded the desired product 5i in 70% yield. Even more challenging functional handles such as 4j and 4k were tolerated and provided the desired products albeit in lower yields.
 | ||
Scheme 3 Scope of arylsubstituted ortho-alkynylaldehydes. Reactions were performed using 4a (0.25 mmol), 2a (0.75 mmol), and I2 (0.50 mmol) at 25 °C for 2 h. Isolated yield. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) For 4 h. For 4 h. | ||
Upon completion of the study of the o-alkynyl aldehyde scope, we then looked to examine the tolerance of the system to an array of amino alcohols for the synthesis of diverse products 6a–e (Scheme 4). It was observed that all amino alcohols (2b–d) provided the required products in yields ranging from 65–82%. It is worth mentioning here that long-chain nucleophiles were also amenable to the reaction conditions which provided a diminished reaction yield.
 | ||
| Scheme 4 Scope of amino alcohols. Reactions were performed using 1a (0.25 mmol), 2a (0.75 mmol), and I2 (0.50 mmol) at 25 °C for 2 h. Isolated yield. | ||
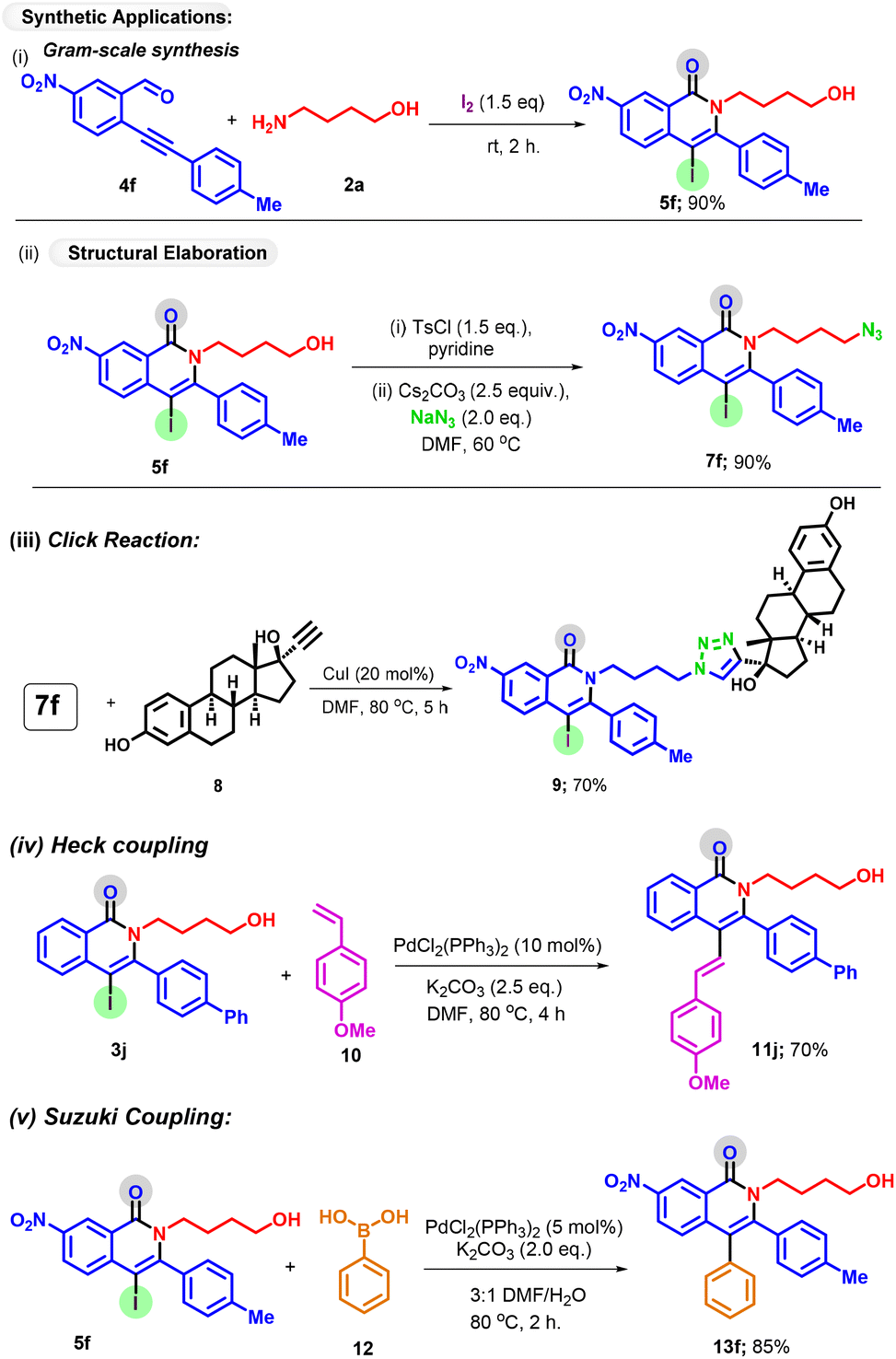
Furthermore, to illustrate the practical utility of the reaction, we performed a gram-scale reaction, and to our delight, the desired isoquinolone was obtained in 76% yield (Scheme 5i). The robustness of the protocol was further reflected by synthetic elaboration and late-stage modification of drugs. Product 5f bearing a free OH group was easily transformed into azide 7f in 90% yield (Scheme 5ii). A further reaction of azide substrate 7f with alkyne 8 afforded the triazole hybrid 9 in 70% yield by a click reaction. Considering the synthetic versatility of alkene and aryl groups, the synthetic utility of the iodo handle was further explored by conducting the Heck (Scheme 5iv) and Suzuki coupling (Scheme 5v) reactions, resulting in the formation of coupling products 11j and 13f in good yields.
 | ||
| Scheme 5 Synthetic utility of products. | ||
To validate the mechanism of the regio- and chemoselective iodine-mediated 6-endo-dig ring closure, several control experiments were performed. In our first control experiment, we carried out the reaction of ortho-alkynyl aldehyde 1a with amino alcohol 2a in the absence of iodine at room temperature for 2 h and 24 h. The desired iodocyclized product 3a was not observed, instead the formation of an imine 14 was observed only in 26% (after 2 h) and 30% (after 24 h) yields (Scheme 6a(i)), as verified by crude NMR (see the ESI† for details). This result suggests that the formation of the product proceeds via aminol formation. Next, to confirm the use of 2.0 equiv. of iodine, we performed two sets of reactions using 1.0 and 2.0 equiv. of iodine under standard reaction conditions (Scheme 6a(ii)). On using 1.0 equiv. of iodine, we observed the formation of product 3a in 55% yield; however, on using 2.0 equiv. of iodine, product 3a was observed in 82% yield. This control experiment suggests the use of the second equiv. of iodine for the autooxidation of intermediate B into product 3a.
 | ||
| Scheme 6 Control experiments and a plausible reaction pathway. | ||
On the basis of control experiments and literature studies, we presented a plausible mechanistic pathway for the regio- and chemoselective formation of product 3a along with the free OH group in Scheme 6b. The reaction was initiated by the nucleophilic attack of the amine 2a to the CHO group of 1a to give the unstable species (hemiaminal) A. Subsequently, hemiaminal A undergoes a regioselective intramolecular 6-endo-dig iodoamidation with an electrophilically activated alkyne, leading to the generation of iodo species B (Scheme 6b). The presumed intermediate B, upon subsequent oxidation, yielded the desired product 3a. I2 serves dual purposes in this context, acting as the catalyst for N–C bond-forming cyclization as well as the oxidant (species B to product 3a) under the current reaction conditions.
Conclusions
In conclusion, we have unveiled a straightforward transition-metal, additive, and solvent-free strategy for the synthesis of the pharmaceutically important isoquinolone motif via regioselective intramolecular iodoamidation of alkynes. Synthetically, the strategy appears to hold much promise as this protocol provides an environmentally benign and atom-economical route for the facile construction of functionalized isoquinolones that have not been easily accessible. Notably, it is the first report that involves the use of amino alcohols as the coupling partner with ortho-alkynyl aldehydes to synthesize diverse isoquinolones. The incorporation of the free-OH group opens the gateway for a more sustainable and efficient synthesis of diverse isoquinolone derivatives, fostering the development of novel drug candidates and expanding the scope of chemical synthesis in the pharmaceutical industry.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the IoE-FRP (2023) for financial support and USIC, the University of Delhi for providing instrumentation facilities. M. is thankful to CSIR for a fellowship.References
- (a) M. O. Simon and C. J. Li, Chem. Soc. Rev., 2012, 41, 1415–1427 RSC; (b) E. A. A. Hafez, S. M. Al-Mousawi, M. S. Moustafa, K. U. Sadek and M. H. Elnagdi, Green Chem. Lett. Rev., 2013, 6, 189–210 CrossRef.
- (a) J. H. Clark, Green Chem., 1999, 1, 1–8 RSC; (b) T. Deligeorgiev, N. Gadjev, A. Vasilev, S. Kaloyanova, J. J. Vaquero and J. Alvarez-Builla, Mini-Rev. Org. Chem., 2010, 7, 44–53 Search PubMed.
- (a) S. Zangade and P. Patil, Curr. Org. Chem., 2019, 23, 2295–2318 CrossRef; (b) K. Tanaka and F. J. C. R. Toda, Chem. Rev., 2000, 100, 1025–1074 CrossRef PubMed; (c) C. I. Herrerías, T. Y. Zhang and C. J. Li, Tetrahedron Lett., 2006, 47, 13–17 CrossRef.
- (a) T. M. Benn and P. Westeroff, Environ. Sci. Technol., 2008, 42, 4133–4139 CrossRef CAS; (b) F. Gottschalk and B. Nowack, J. Environ. Monit., 2011, 13, 1145–1155 RSC.
- F. I. Khan and A. K. Ghoshal, J. Loss Prev. Process Ind., 2000, 13, 527–545 CrossRef.
- M. B. Gawande, V. D. Bonifacio, R. Luque, P. S. Branco and R. S. Varma, ChemSusChem, 2014, 7, 24–44 CrossRef CAS PubMed.
- (a) H. Heaney and K. F. Shuhaibar, Synlett, 1995, 47–48 CrossRef CAS; (b) M. Berger, H. Rehwinkel, N. Schmees, H. Schacke, K. Edman, L. Wissler, A. Reichel and S. Jaroch, Bioorg. Med. Chem. Lett., 2017, 27, 437–442 CrossRef CAS PubMed; (c) T. Bosanac, E. R. Hickey, J. Ginn, M. Kashem, S. Kerr, S. Kugler, X. Li, A. Olague, S. Schlyer and E. R. R. Young, Bioorg. Med. Chem. Lett., 2010, 20, 3746–3749 CrossRef CAS.
- (a) X. Yu, K. Chen, S. Guo, P. Shi, C. Song and J. Zhu, Org. Lett., 2017, 19, 5348–5351 CrossRef CAS PubMed; (b) S. Lee, S. Mah and S. Hong, Org. Lett., 2015, 17, 3864–3867 CrossRef CAS PubMed.
- (a) G.-D. Xu and Z.-Z. Huang, Org. Lett., 2017, 19, 6265–6267 CrossRef CAS; (b) S. Lee, S. Mah and S. Hong, Org. Lett., 2015, 17, 3864–3867 CrossRef CAS.
- (a) N. J. Webb, S. P. Marsden and S. A. Raw, Org. Lett., 2014, 16, 4716–4721 CrossRef PubMed; (b) Y. Wu, P. Sun, K. Zhang, T. Yang, H. Yao and A. Lin, J. Org. Chem., 2016, 81, 2166–2173 CrossRef CAS.
- (a) H. Huang, S. Nakanowatari and L. Ackermann, Org. Lett., 2017, 19, 4620–4623 CrossRef CAS; (b) S. Allu and K. K. Swamy, J. Org. Chem., 2014, 79, 3963–3972 CrossRef CAS PubMed.
- C. C. Liu, K. Parthasarathy and C. H. Cheng, Org. Lett., 2010, 12, 3518–3521 CrossRef CAS.
- M. S. Mayo, X. Yu, X. Feng, Y. Yamamoto and M. Bao, J. Org. Chem., 2015, 80, 3998–4002 CrossRef CAS.
- T. Yao and R. C. Larock, J. Org. Chem., 2005, 70, 1432–1437 CrossRef CAS.
- (a) A. K. Verma, D. Choudhary, R. K. Saunthwal, V. Rustagi, M. Patel and R. K. Tiwari, J. Org. Chem., 2013, 78, 6657–6669 CrossRef PubMed; (b) S. Kumar, R. K. Saunthwal, K. M. Saini and A. K. Verma, Chem. Commun., 2019, 55, 10721–10724 RSC; (c) P. K. Mishra and A. K. Verma, Green Chem., 2016, 18, 6367–6372 RSC; (d) A. K. Verma, T. Aggarwal, V. Rustagi and R. C. Larock, Chem. Commun., 2010, 46, 4064–4066 RSC; (e) V. Rustagi, T. Aggarwal and A. K. Verma, Green Chem., 2011, 13, 1640–1643 RSC; (f) H. Wang, Y. Kuang and J. Wu, Asian J. Org. Chem., 2012, 1, 302–312 CrossRef.
- P. C. Too and S. Chiba, Chem. Commun., 2012, 48, 7634–7636 RSC.
- M. Zhang, H. J. Zhang, W. Ruan and T. B. Wen, Eur. J. Org. Chem., 2015, 5914–5918 CrossRef CAS.
- D. M. Khan and R. Hua, Catalysts, 2020, 10, 683 CrossRef CAS.
- (a) A. K. Verma, S. K. R. Kotla, D. Choudhary, M. Patel and R. K. Tiwari, J. Org. Chem., 2013, 78, 4386–4401 CrossRef CAS; (b) S. Kumar, C. Cruz-Hernández, S. Pal, R. K. Saunthwal, M. Patel, R. K. Tiwari, E. Juaristi and A. K. Verma, J. Org. Chem., 2015, 80, 10548–10560 CrossRef CAS PubMed; (c) P. Kumar, T. Aggarwal and A. K. Verma, J. Org. Chem., 2017, 82, 6388–6397 CrossRef CAS; (d) S. Verma, M. Kumar and A. K. Verma, Org. Lett., 2019, 21, 5059–5063 CrossRef CAS PubMed; (e) D. Thakur, T. Aggarwal, Muskan, Sushmita and A. K. Verma, J. Org. Chem., 2023, 88, 2474–2486 CrossRef CAS PubMed.
- Crystallographic data for compound 3e has been deposited with the Cambridge Crystallographic Data Centre. CCDC deposit number for compound 3e is 2301423.†.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2301423. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3gc04096h |
| This journal is © The Royal Society of Chemistry 2024 |