
N–N atropisomer synthesis via electrolyte- and base-free electrochemical cobalt-catalysed C–H annulation†
Jiating
Cai‡
,
Linzai
Li‡
,
Chuitian
Wang
,
Shi
Qin
,
Yuanyuan
Li
,
Si-Yan
Liao
,
Shengdong
Wang
 ,
Hui
Gao
,
Zhi
Zhou
,
Yugang
Huang
*,
Wei
Yi
* and
Zhongyi
Zeng
*
,
Hui
Gao
,
Zhi
Zhou
,
Yugang
Huang
*,
Wei
Yi
* and
Zhongyi
Zeng
*
Guangzhou Municipal and Guangdong Provincial Key Laboratory of Molecular Target & Clinical Pharmacology, the NMPA and State Key Laboratory of Respiratory Disease, School of Pharmaceutical Sciences and the Fifth Affiliated Hospital, Guangzhou Medical University, Guangzhou, Guangdong 511436, China. E-mail: hyug@gzhmu.edu.cn; yiwei@gzhmu.edu.cn; zzeng@gzhmu.edu.cn
First published on 30th October 2024
Abstract
Merging electrochemistry with asymmetric C–H activation has proven to be an advantageous alternative to build valuable enantiopure molecules. However, established methods require a stoichiometric use of supporting electrolytes to promote the electron transfer in solution and often additionally serve as a base to assist C–H bond cleavage, which are hazardous and would produce additional waste. Herein, we described an exogenous electrolyte- and base-free electrocatalytic atroposelective C–H annulation, providing facile and sustainable access to N–N axially chiral isoquinolinones in excellent enantioselectivities and good yields. This protocol is enabled by a combination of simple Co(OAc)2·4H2O and readily available chiral salicyloxazoline (Salox), which proceeds well with 13 classes of alkynes, including highly challenging polarized either internal or terminal alkynes, and tolerates a wealth of functional groups for streamlined transformations.
In recent years, organic electrosynthesis has witnessed a remarkable renaissance as an efficient and powerful method to construct valuable molecules.1–6 Electrochemistry is often considered as an environmentally benign technique for organic synthesis as it uses electric current as an inexpensive, waste-free, and prospectively renewable redox reagent. Therefore, diverse electrochemical reactions have been established, e.g., alkene/alkyne functionalization,7 cross-couplings,8,9 radical cyclization,10 and C–H functionalization.11–15 Despite impressive progress, it continues to be a challenge to realize precise selectivity control in terms of asymmetric electrochemical synthesis. Toward this goal, routine enantiocontrol approaches including metal catalysis, organocatalysis, and enzymatic catalysis have been gradually merged with electrochemistry to boost the development of this burgeoning field.16–18 While some asymmetric electrocatalytic systems have been reported,19–35 their synthetic utilizations to construct axially chiral skeletons,36–48 especially with stereogenic N–N linkages,49 lag far behind (Fig. 1A). This could be ascribed to several major issues concerning atroposelective manifolds: (a) shorter N–N bond distance posing steric hindrance during new bond formation around this axis; (b) relatively poor configurational stability of N–N atropisomers; (c) electrochemical degradation of chiral ligands/catalysts or even related N-heterocyclic substrates/products; (d) unfavourable interactions of the supporting electrolyte within the enantio-determining transition state. Given the ubiquity of N–N atropisomers in natural bioactive molecules,50,51 chiral ligands52 as well as functional materials (Fig. 1C),53 it is highly desirable to explore an efficient catalytic system and novel asymmetric transformation to construct N–N axially chiral architectures.
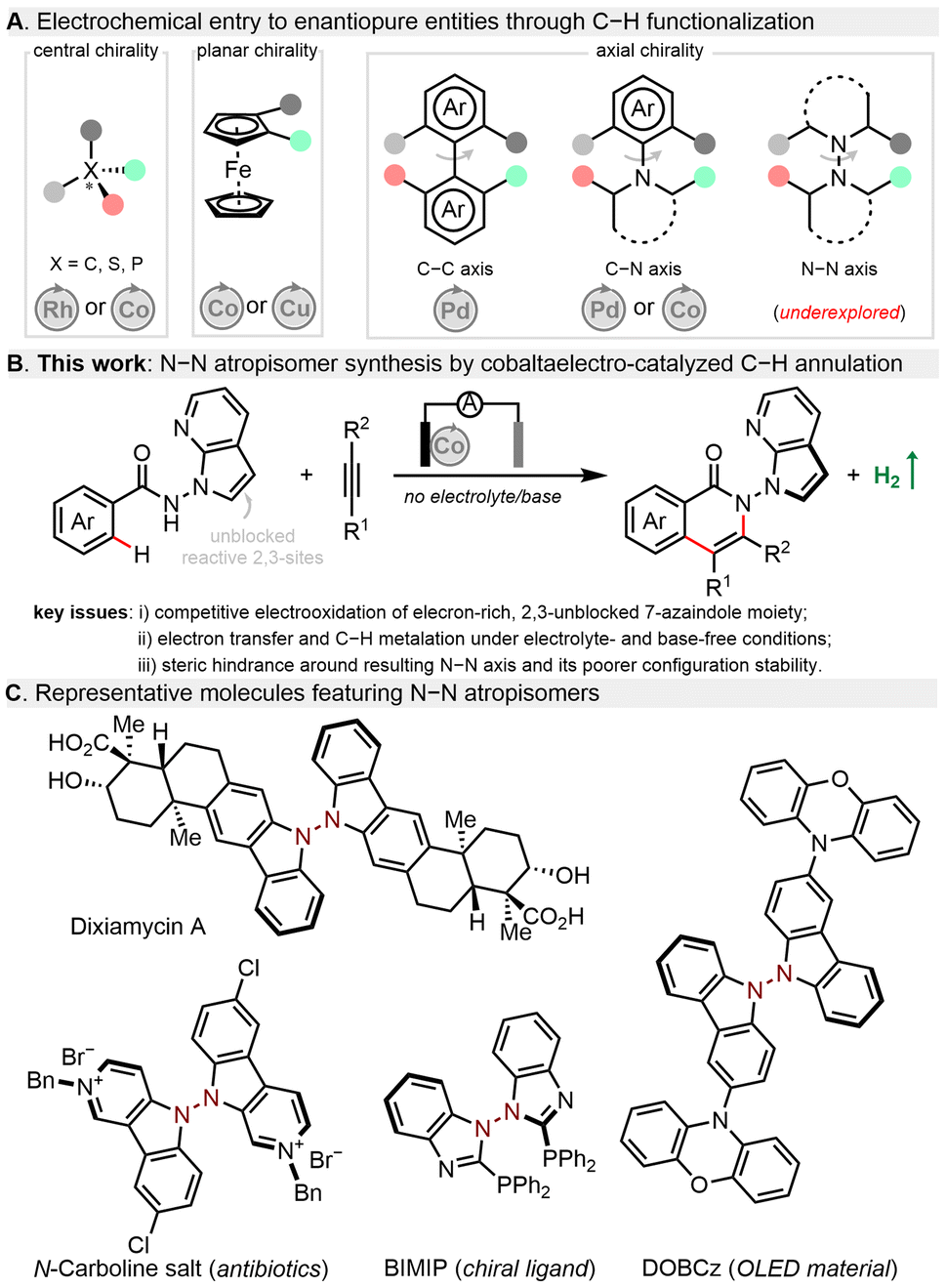 | ||
| Fig. 1 Background and design blueprint. | ||
In fact, enantioselective transition-metal-catalysed C–H activation provides facile and step-economical access to axially chiral motifs.54–64 Since the pioneering work by Ackermann,36 asymmetric electrocatalytic C–H activation has proved to be an environmentally benign and sustainable alternative to prepare C–C39,40 and C–N41–48 axially chiral compounds via dihydrogen evolution. By contrast, electrochemical manifolds toward stereogenic N–N counterparts remain elusive. Notable yet often overlooked is that all reported enantioselective electrocatalytic C–H activations require stoichiometric amounts of a hazardous supporting electrolyte.
Herein, we present an unprecedented electrolyte- and base-free electrocatalytic atroposelective C–H annulation, providing facile and sustainable access to N–N axially chiral isoquinolinones in excellent enantioselectivities (mostly >90% ee) and good yields (Fig. 1B).49 Salient features of this protocol comprise (a) electrocatalytic entry to N–N atropisomers enabled by simple and cost-effective Co(II)/Salox catalytic system; (b) no need of any exogenous supporting electrolyte and base to effectively reduce chemical waste production; (c) a broad range of benzamides and 13 classes of alkynes bearing diverse functionalities for streamlined diversifications.
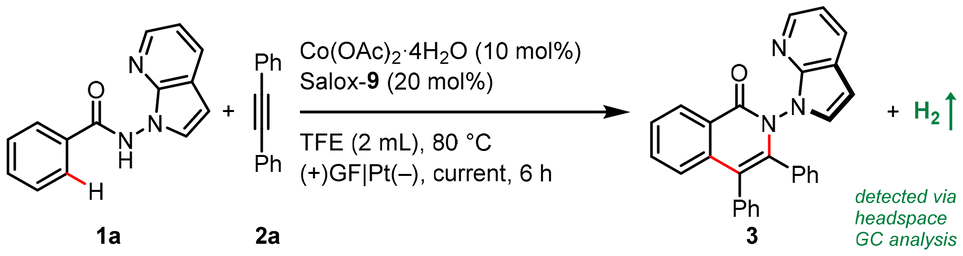
Our proof-of-concept work was commenced with enantioselective electrocatalytic C–H/N–H annulation of N-(7-azaindole)benzamide 1a with diphenylacetylene (2a) in an undivided cell equipped with a graphite felt (GF) anode and a platinum cathode (Table 1). Notably, benzamide 1a bearing a reactive and versatile 2,3-unsubstituted azaindolyl handle is a demanding reactant for electrochemical transformations in that it may be prone to oxidation. Pleasingly, the atroposelective N–N isoquinolinone 3 was obtained in 40% yield and 96% enantioselective excess (ee) when Co(OAc)2·4H2O and Salox-9 were employed as the catalytic combination with NaOPiv·H2O as both an electrolyte and base in heated 2,2,2-trifluoroethanol (TFE) under a 2.0 mA galvanostatic electrolysis (entry 1). Catalytic performance was enhanced at a lower current density and further improved with one equivalent of NaOPiv·H2O (entries 2 and 3). Running the reaction under dioxygen atmosphere led to an increased yield of 71% (entries 4 and 5). Control experiments confirmed the indispensability of the cobalt salt and electricity to spur this net-oxidation reaction (entries 11 and 12). Surprisingly, the addition of NaOPiv·H2O was discovered to be unnecessary (entries 5 and 6). This is remarkable, to our knowledge, as there has been no precedent on electrocatalytic asymmetric C–H functionalization under electrolyte-free conditions.17
|
|
|||
|---|---|---|---|
| Entry | Reaction condition | 3 | |
| Yield, % | ee, % | ||
| a Conditions: 1a (0.08 mmol), 2a (0.16 mmol), Co(OAc)2·4H2O (10 mol%), Salox-9 (20 mol%), TFE (2 mL), undivided cell with graphite felt (GF) anode (10 mm × 20 mm × 2 mm) and platinum plate (Pt) cathode (10 mm × 20 mm × 0.3 mm), constant current, 80 °C, 6 h; isolated yields and enantioselective excess (ee) determined by HPLC with a chiral stationary phase. b NaOPiv·H2O (1 equiv.). | |||
| 1 | NaOPiv·H2O (2 equiv.), 2.0 mA, air | 40 | 96 |
| 2 | NaOPiv·H2O (2 equiv.), 1.0 mA, air | 49 | 96 |
| 3 | NaOPiv·H2O (1 equiv.), 1.0 mA, air | 63 | 96 |
| 4 | NaOPiv·H2O (1 equiv.), 1.0 mA, N2 | 65 | 95 |
| 5 | NaOPiv·H2O (1 equiv.), 1.0 mA, O2 | 71 | 96 |
| 6 | 1.0 mA, O2 | 71 | 96 |
| Further variation from optimal condition (entry 6) | |||
| 7 | C as anode | 8 | 96 |
| 8 | GF as cathode | 25 | 96 |
| 9 | Running at 60 °C | 26 | 96 |
| 10 | Salox-1–10 | As below | |
| 11 | 15 mol% Salox-9 | 25 | 96 |
| 12 | 25 mol% Salox-9 | 70 | 96 |
| 13 | MeOH, HFIP, MeCN, DMSO | <1 | — |
| 14 | No Co(OAc)2·4H2O | <1 | — |
| 15 | No electricity | <1 | — |
|
|||
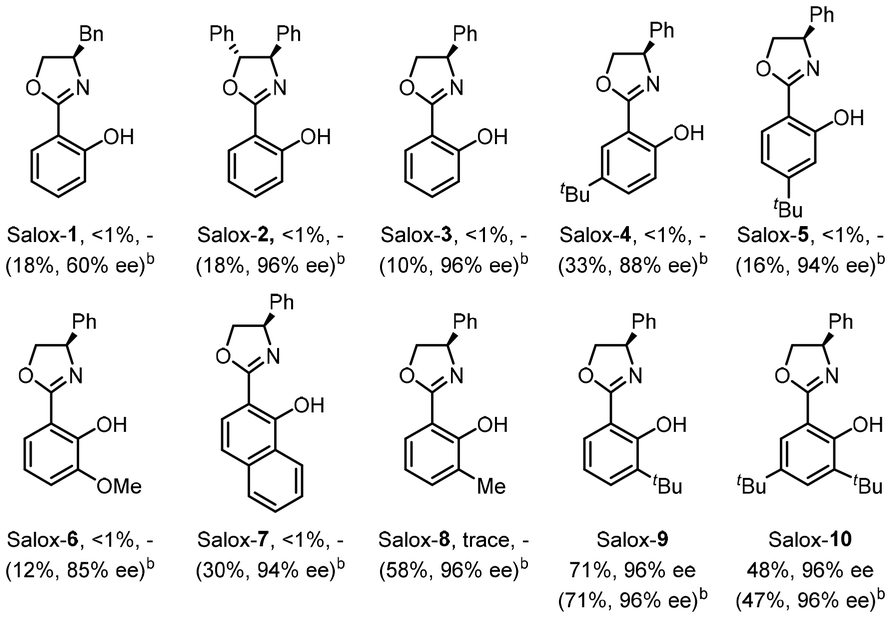
Switching the electrode to other materials gave unsatisfactory results (entries 7 and 8). It might be rationalized by (1) the large surface area of the GF anode for cobalt contact, and (2) favourable dihydrogen evolution on a Pt surface, which was detected via headspace gas chromatography (GC) analysis. Running the reaction at 60 °C led to much lower efficiency (entry 9). Lower efficiency was observed by decreasing the amount of Salox-9 ligand while its increase led to no further improvement (entries 11 and 12). Nevertheless, excellent enantioselectivity was constantly observed during reaction optimization, suggesting the robust enantiocontrol of the Co(OAc)2·4H2O/Salox-9 catalytic system. Fine-tuning of the ligand design revealed an extremely high sensitivity of the catalyst performance toward the substituents on both the phenol and oxazoline moieties, with only Salox-9 and Salox-10 uniformly bearing a bulky tert-butyl group at the ortho-position of the phenol ring being catalytically active (entry 10).65 Cyclic voltammograms of three representative Salox ligands (including Salox-3, Salox-8, Salox-9) and their combination with Co(OAc)2·4H2O indicates that compared to Salox-3 and Salox-8, Salox-9 is less likely to decompose under electrolyte-free electrolysis and facilitates the oxidation of CoII to CoIII, speculatively owing to such steric congestion (Fig. S11 and S12 in ESI†). To further validate this hypothesis, we conducted the ligand screenings again yet in the presence of a supporting electrolyte, NaOPiv·H2O. This way, all tested Salox ligands could leverage the reaction, albeit with varied yields and enantioselectivities. TFE seems to be the sole medium for this conversion (entry 13), speculatively owing to (1) improved solubility of Co(OAc)2·4H2O; (2) function as a proton shuttle; (3) the resulting ROH/RO− buffer system behaving like a supporting electrolyte to mediate electron transfer as well as a base to facilitate N–H and C–H bond cleavage. The electrooxidative process has a good current efficiency of 51%, underlining the sustainability of the overall process.
With optimized experimental protocols in hand, we sought to evaluate the generality of our electrocatalytic atroposelective C–H annulation (Scheme 1). First, scope with respect to alkyne partners was investigated by coupling with benzamide 2a (Scheme 1a/b). The electrochemical protocol was found to be robust and general: enantioselective C–H annulation could be achieved with 13 classes of alkynes. A broad spectrum of unactivated and specially activated alkynes was engaged in the reaction. The newly established method is applicable to symmetrical diarylacetylenes bearing electron-donating and electron-withdrawing groups as well as dialkylacetylenes (11, 12). A wealth of common functional groups, including methoxy (4), fluoro (5), bromo (6), ester (8), trifluoromethyl (9) as well as even oxidation-sensitive formyl (7) and thienyl (10), are tolerated. In addition, nonsymmetric aryl–alkyl alkynes (13), terminal alkynes (14) as well as 1,3-diynes (15) were coupled in a highly regioselective and enantioselective manner. Impressively, this electrochemical reaction also works well with diversly functionalized π-systems, ranging from electron-neutral propargyl alcohols (16) and alkynyl silanes (17), electron-rich alkynyl sulfides (19, 19′), to electron-deficient chloroalkynes (18), alkynyl nitriles (20), alkynyl esters (21, 21′), alkynyl phosphine oxides (22), and α,α-difluoromethylene alkynes (23). The presence of these functionalities can not only guide the reaction regioselectivity, but also offer opportunities for follow-up modifications.
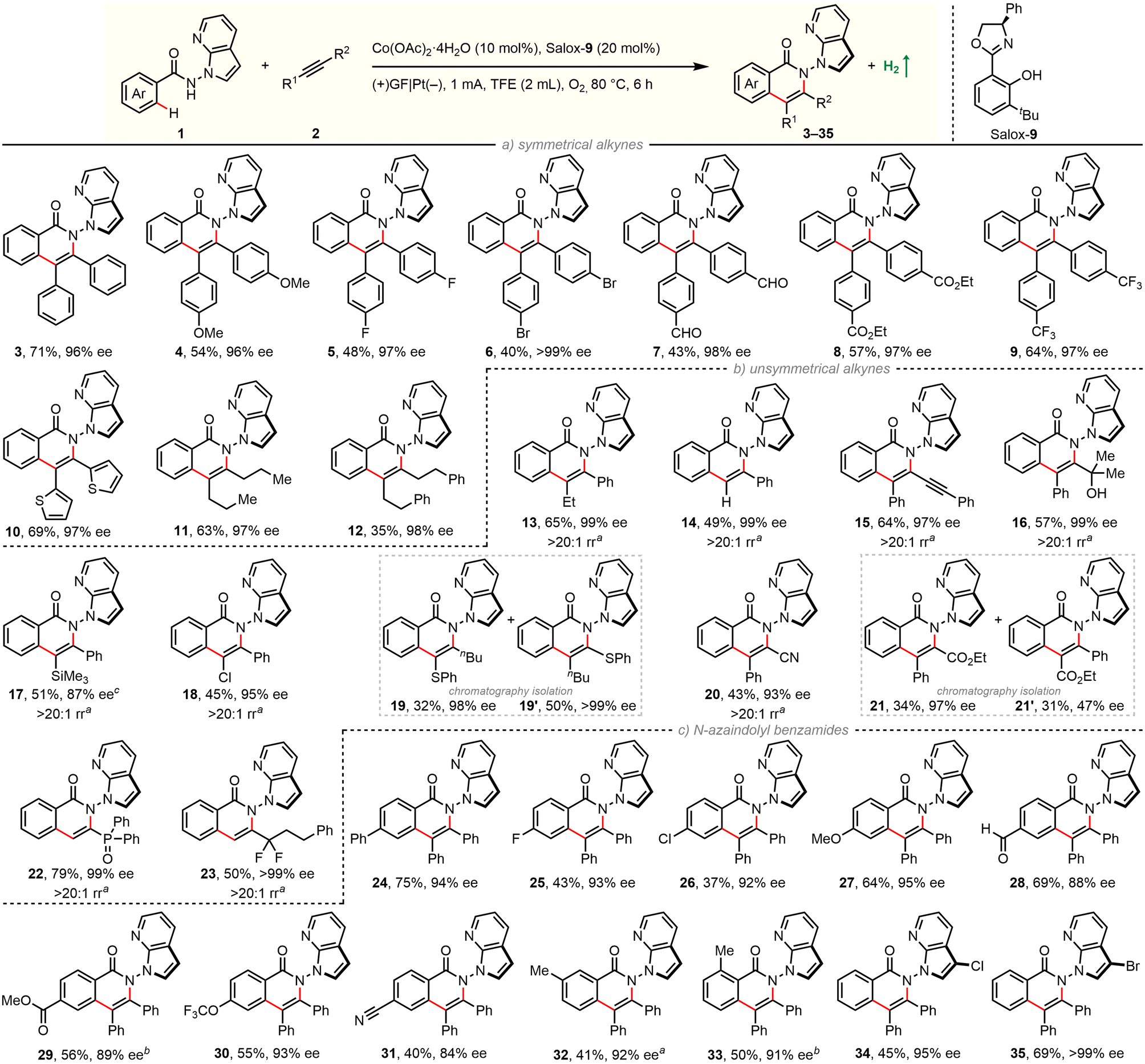 | ||
Scheme 1 Scope of electrocatalytic atroposelective C–H annulation. Conditions: 1 (0.08 mmol), 2 (0.16 mmol), Co(OAc)2·4H2O (10 mol%), Salox-9 (20 mol%), TFE (2 mL), undivided cell with GF (10 mm × 20 mm × 2 mm) and Pt cathode (10 mm × 20 mm × 0.3 mm), constant current of 1 mA (ca. 2.8 F mol−1), 80 °C, 6 h under O2 (balloon); isolated yields given, and enantioselective excess (ee) determined by HPLC with a chiral stationary phase. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Regioselective ratio (rr) determined by 1H NMR. 15 h. Regioselective ratio (rr) determined by 1H NMR. 15 h. | ||
Next, substrate scope of benzamides was examined with diphenylacetylene (Scheme 1c). A set of electronically and sterically varied benzamides proceeds C–H/N–H annulation with good efficiency and excellent enantiocontrol. Again, it tolerates diverse functional groups including –F (25), –Cl (26, 34), –Br (35), –OMe (27), –CHO (28), –CO2Me (29), –OCF3 (30), and –CN (31). It is worth noting that 3-substituted benzamide undergoes selective coupling in the less-hindered position (33). Benzamides bearing a C3-functionalized azaindole moiety were also applicable in our electrochemical protocol (34, 35). It is worth mentioning that a significant amount of unconsumed benzamides remained for reactions with low yields. The product/conversion selectivities were rather high since almost no other new spots were observed on TLC traces. The yields might be improved when running the reaction longer (17, 29, 33) (Scheme 2).
 | ||
| Scheme 2 Synthetic utilizations. | ||
Further derivatization experiments were performed to showcase the synthetic utility. Indeed, the unsubstituted azaindolyl unit allows for consecutive C–H bond functionalization. For instance, such handle in enantiopure product 3 could be selectively chlorinated (34), brominated (35), and iodinated (36) by choosing suitable electrophilic halogenating sources. These introduced halogen atoms would enable numerous subsequent bond-forming reactions, e.g., via classical cross-couplings. Besides, the carbonyl group of skeletal isoquinolinone was facilely translated into a thiocarbonyl group in the presence of Lawesson's reagent (37). Due to the harsh reaction conditions, decayed enantiopurity was observed. This is consistent with the Li's result that racemization of such N–N axial chirality would occur at 120 °C.66
Mechanistic studies were performed to shed light on this electrochemical process. H/D scrambling was not found in the presence of D2O, which points toward an irreversible C–H metalation step (Scheme 3A). In cyclic voltammetry (CV, Scheme 3B), benzamide 1a showed an oxidation peak at +1.14 V (vs. Ag/AgCl, curve b), being supportive of its tendency to anodic oxidation. Although Salox-9 exhibited two pairs of reversible redox peaks and Co(OAc)2·4H2O seemed redox inert (curve c/d), their combination led to a shift forward of the oxidation wave with a potential of 0.60 V (vs. Ag/AgCl, curve e). This implies that the in situ coordination of CoII salt with Salox-9 could not only prevent the ligand decomposition, but also facilitate the oxidation of CoII to CoIII. Such facilitation effect can be further observed in the presence of 1a (curve f). Based on our results and literature precedents,28,47 a plausible catalytic cycle was proposed for this electrochemical enantioselective process (Scheme 3C). Initially, single electron oxidation of the cobalt(II) salt by the anode or dioxygen in the presence of Salox-9 delivers a chiral octahedral Co(III) species A. Subsequent ligand exchange with benzamide 1a followed by a base-assisted C–H activation leads to a five-membered cabaltacycle C. Coordination and migratory insertion with the alkyne gives rise to cabaltacycle D, which reductively eliminate to furnish the final N–N axially chiral isoquinolinone 3. The simultaneously resulting Co(I) species E undergoes an anodic oxidation for the regeneration of the active Co(III) catalyst. At the cathode, the dihydrogen byproduct is released.
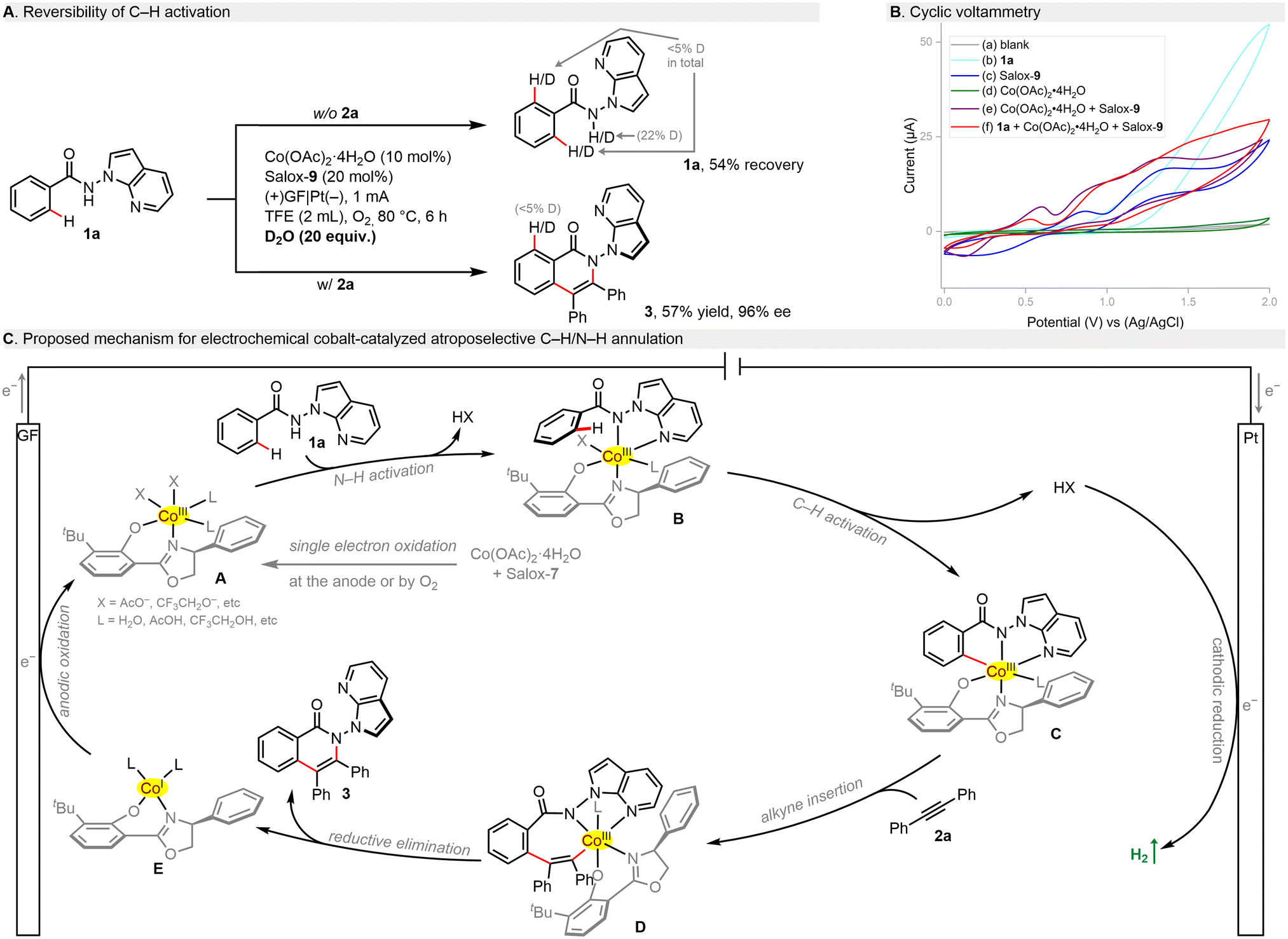 | ||
| Scheme 3 Mechanistic insights and plausible pathway. | ||
Conclusions
In conclusion, we disclosed an exogenous electrolyte- and base-free electrochemical cobalt-catalysed atroposelective C–H annulation to construct N–N axially chiral isoquinolinones with excellent enantiocontrol. This transformation is amenable to a wide set of structurally diverse non-polar and polarized, internal and terminal alkynes with remarkable functional group compatibility. We believe that this work can inspire further development of electrolyte-free electrocatalytic asymmetric reactions and find their robust applications in synthetic and medicinal chemistry.Author contributions
Z. Zeng and W. Y. conceived the project and designed the experiments. J. C. and L. L. performed the experiments and interpreted the data with equal contributions with the assistance of C. W., S. Q., and Y. L. The manuscript was written by Z. Zeng, W. Y., and Y. H. with insightful discussion and proofreading of S. W., H. G., Z. Zhou. All authors have given approval to the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from National Natural Science Foundation of China (no. 82273795, 22201051), Guangdong Basic and Applied Basic Research Foundation (no. 2024A1515030179, 2024A1515010260), Science and Technology Program of Guangzhou (no. 2023A04J0074), and Plan on Enhancing Scientific Research at GMU.References
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed
.
- X. Cheng, A. Lei, T.-S. Mei, H.-C. Xu, K. Xu and C. Zeng, CCS Chem., 2022, 4, 1120–1152 CrossRef CAS
- S. B. Beil, D. Pollok and S. R. Waldvogel, Angew. Chem., Int. Ed., 2021, 60, 14750–14759 CrossRef CAS PubMed
- Y. Guo, W. An, X. Tian, L. Xie and Y.-L. Ren, Green Chem., 2022, 24, 9211–9219 RSC
- X. Tian, Y. Guo, W. An, Y.-L. Ren, Y. Qin, C. Niu and X. Zheng, Nat. Commun., 2022, 13, 6186 CrossRef CAS PubMed
- W.-K. An, X. Xu, S.-J. Zheng, Y.-N. Du, J. Ouyang, L.-X. Xie, Y.-L. Ren, M. He, C.-L. Fan, Z. Pan and Y.-H. Li, ACS Catal., 2023, 13, 9845–9856 CrossRef CAS
- J. C. Siu, Acc. Chem. Res., 2020, 53, 547–560 CrossRef CAS PubMed
- H. Wang, X. Gao, Z. Lv, T. Abdelilah and A. Lei, Chem. Rev, 2019, 119, 6769–6787 CrossRef CAS PubMed
- C. A. Malapit, M. B. Prater, J. R. Cabrera-Pardo, M. Li, T. D. Pham, T. P. McFadden, S. Blank and S. D. Minteer, Chem. Rev., 2022, 122, 3180–3218 CrossRef CAS PubMed
- P. Xiong and H.-C. Xu, Acc. Chem. Res., 2019, 53, 3339–3350 CrossRef PubMed
- K.-J. Jiao, Acc. Chem. Res., 2020, 53, 300–310 CrossRef CAS PubMed
- Z. Zeng, J. F. Goebel, X. Liu and L. J. Gooßen, ACS Catal., 2021, 11, 6626–6632 CrossRef CAS
- J. F. Goebel, Z. Zeng and L. J. Gooßen, Synthesis, 2022, 565–569 CAS
- Y. Wang, S. Dana, H. Long, Y. Xu, Y. Li, N. Kaplaneris and L. Ackermann, Chem. Rev., 2023, 123, 11269–11335 CrossRef CAS PubMed
- S. Qin, M. Yang, M. Xu, Z.-H. Peng, J. Cai, S. Wang, H. Gao, Z. Zhou, A. S. K. Hashmi, W. Yi and Z. Zeng, Nat. Commun., 2024, 15, 7428 CrossRef CAS PubMed
- X. Chang, Q. Zhang and C. Guo, Angew. Chem., Int. Ed., 2020, 59, 2–13 CrossRef
- K.-J. Jiao, Chem. Catal., 2022, 2, 3019–3047 CrossRef CAS
- J. Rein, Chem. Soc. Rev., 2023, 52, 8106–8125 RSC
- X. Huang, Q. Zhang, J. Lin, K. Harms and E. Meggers, Nat. Catal., 2018, 2, 34–40 CrossRef
- Q. Zhang, X. Chang, L. Peng and C. Guo, Angew. Chem., Int. Ed., 2019, 58, 6999–7003 CrossRef CAS PubMed
- P.-S. Gao, X.-J. Weng, Z.-H. Wang, C. Zheng, B. Sun, Z.-H. Chen, S.-L. You and T.-S. Mei, Angew. Chem., Int. Ed., 2020, 59, 15254–15259 CrossRef CAS PubMed
- Y.-Q. Huang, Z.-J. Wu, L. Zhu, Q. Gu, X. Lu, S.-L. You and T.-S. Mei, CCS Chem., 2021, 3, 3501–3509 Search PubMed
- Z.-H. Wang, P.-S. Gao, X. Wang, J.-Q. Gao, X.-T. Xu, Z. He, C. Ma and T.-S. Mei, J. Am. Chem. Soc., 2021, 143, 15599–15605 CrossRef CAS PubMed
- W. Wei, Chem. Sci., 2022, 13, 2783–2788 RSC
- Q. Zhang, K. Liang and C. Guo, Angew. Chem., Int. Ed., 2022, 61, e202210632 CrossRef CAS PubMed
- K. Liang, Q. Zhang and C. Guo, Nat. Synth., 2023, 2, 1184–1193 CrossRef
- T. Liu, W. Zhang, C. Xu, Z. Xu, D. Song, W. Qian, G. Lu, J. Zhang, W. Zhong and F. Ling, Green Chem., 2023, 25, 3606–3614 RSC
- Q.-J. Yao, F.-R. Huang, J.-H. Chen, M.-Y. Zhong and B. Shi, Angew. Chem., Int. Ed., 2023, 62, e202218533 CrossRef CAS PubMed
- G. Zhou, J.-H. Chen, Q.-J. Yao, F.-R. Huang, Z.-K. Wang and B.-F. Shi, Angew. Chem., Int. Ed., 2023, 62, e202302964 CrossRef CAS PubMed
- P.-Y. Chen, C. Huang, L.-H. Jie, B. Guo, S. Zhu and H.-C. Xu, J. Am. Chem. Soc., 2024, 146, 7178–7184 CrossRef CAS PubMed
- Y. Li, J. Xu, J. C. A. Oliveira, A. Scheremetjew and L. Ackermann, ACS Catal., 2024, 14, 8160–8167 CrossRef CAS PubMed
- X. Xia, C. Zheng, Y. Hang, J. Guo, T. Liu, D. Song, Z. Chen, W. Zhong and F. Ling, Green Chem., 2024, 26, 8323–8329 RSC
- G. Zhou, T. Zhou, A.-L. Jiang, P.-F. Qian, J.-Y. Li, B.-Y. Jiang, Z.-J. Chen and B.-F. Shi, Angew. Chem., Int. Ed., 2024, 63, e202319871 CrossRef CAS PubMed
- F.-R. Huang, P. Zhang, Q.-J. Yao and B.-F. Shi, CCS Chem., 2024, 1–11 Search PubMed
- Z.-Z. Zhang, G. Zhou, Q. Yue, Q.-J. Yao and B.-F. Shi, ACS Catal., 2024, 14, 4030–4039 CrossRef CAS
- U. Dhawa, C. Tian, T. Wdowik, J. C. A. Oliveira, J. Hao and L. Ackermann, Angew. Chem., Int. Ed., 2020, 59, 13451–13457 CrossRef CAS PubMed
- H. Qiu, B. Shuai, Y.-Z. Wang, D. Liu, Y.-G. Chen, P.-S. Gao, H.-X. Ma, S. Chen and T.-S. Mei, J. Am. Chem. Soc., 2020, 142, 9872–9878 CrossRef CAS PubMed
- L.-C. Xu, J. Frey, X. Hou, S.-Q. Zhang, Y.-Y. Li, J. C. A. Oliveira, S.-W. Li, L. Ackermann and X. Hong, Nat. Synth., 2023, 2, 321–330 CrossRef
- X. Hou, S. Li, J. Frey, X. Hong and L. Ackermann, Chem, 2024, 10, 2283–2294 Search PubMed
- S.-S. Xu, H. Qiu, P.-P. Xie, Z.-H. Wang, X. Wang, C. Zheng, S.-L. You and T.-S. Mei, CCS Chem., 2024 DOI:10.31635/ccschem.024.202403939
- U. Dhawa, T. Wdowik, X. Hou, B. Yuan, J. C. A. Oliveira and L. Ackermann, Chem. Sci., 2021, 12, 14182–14188 RSC
- J. Frey, X. Hou and L. Ackermann, Chem. Sci., 2022, 13, 2729–2734 RSC
- Y. Lin, T. Von Münchow and L. Ackermann, ACS Catal., 2023, 13, 9713–9723 CrossRef CAS PubMed
- T. Von Münchow, S. Dana, Y. Xu, B. Yuan and L. Ackermann, Science, 2023, 379, 1036–1042 CrossRef PubMed
- X. Wang, X.-J. Si, Y. Sun, Z. Wei, M. Xu, D. Yang, L. Shi, M.-P. Song and J.-L. Niu, Org. Lett., 2023, 25, 6240–6245 CrossRef CAS PubMed
- L. Zhang, D. Liang, Y. Wang, D. Li, J. Zhang, L. Wu, M. Feng, F. Yi, L. Xu, L. Lei, Q. Du and X. Tang, Chem. Sci., 2023, 9, 44–51 RSC
- T. Von Münchow, Y. Liu, R. Parmar, S. E. Peters, S. Trienes and L. Ackermann, Angew. Chem., Int. Ed., 2024, 63, e202405423 CrossRef PubMed
- Y. Zhang, S.-L. Liu, T. Li, M. Xu, Q. Wang, D. Yang, M.-P. Song and J.-L. Niu, ACS Catal., 2024, 14, 1–9 CrossRef CAS
- Y. Sun, T. Yang, Q. Wang, L. Shi, M.-P. Song and J.-L. Niu, Org. Lett., 2024, 26, 5063–5068 CrossRef CAS PubMed
- Z. Xu, M. Baunach, L. Ding and C. Hertweck, Angew. Chem., Int. Ed., 2012, 51, 10293–10297 CrossRef CAS PubMed
- K. Suzuki, I. Nomura, M. Ninomiya, K. Tanaka and M. Koketsu, Bioorg. Med. Chem. Lett., 2018, 28, 2976–2978 CrossRef CAS PubMed
- T. Benincori, E. Brenna, F. Sannicolò, L. Trimarco, P. Antognazza, E. Cesarotti, F. Demartin, T. Pilati and G. Zotti, J. Organomet. Chem., 1997, 529, 445–453 CrossRef CAS
- X. Liu, Y. Zhang, X. Fei, L. Liao and J. Fan, Chem. – Eur. J., 2019, 25, 4501–4508 CrossRef CAS PubMed
- C.-X. Liu, W.-W. Zhang, S.-Y. Yin, Q. Gu and S.-L. You, J. Am. Chem. Soc., 2021, 143, 14025–14040 CrossRef CAS PubMed
- Z. Zeng, H. Gao, Z. Zhou and W. Yi, ACS Catal., 2022, 12, 14754–14772 CrossRef CAS
- J. Feng and R. Liu, Chem. – Eur. J., 2024, 30, e202303165 CrossRef CAS PubMed
- Z. Zeng, H. Xu, H. Gao, Z. Zhou and W. Yi, Coord. Chem. Rev., 2025, 522, 216244 CrossRef CAS
- X.-M. Wang, P. Zhang, Q. Xu, C.-Q. Guo, D.-B. Zhang, C.-J. Lu and R.-R. Liu, J. Am. Chem. Soc., 2021, 143, 15005–15010 CrossRef CAS PubMed
- Q. Xu, H. Zhang, F.-B. Ge, X.-M. Wang, P. Zhang, C.-J. Lu and R.-R. Liu, Org. Lett., 2022, 24, 3138–3143 CrossRef CAS PubMed
- X. Zhu, H. Wu, Y. Wang, G. Huang, F. Wang and X. Li, Chem. Sci., 2023, 14, 8564–8569 RSC
- W. Yao, C. Lu, L. Zhan, Y. Wu, J. Feng and R. Liu, Angew. Chem., Int. Ed., 2023, 62, e202218871 CrossRef CAS PubMed
- Y. Wang, X. Zhu, D. Pan, J. Jing, F. Wang, R. Mi, G. Huang and X. Li, Nat. Commun., 2023, 14, 4661 CrossRef CAS PubMed
- T. Li, L. Shi, X. Wang, C. Yang, D. Yang, M.-P. Song and J.-L. Niu, Nat. Commun., 2023, 14, 5271 CrossRef CAS PubMed
- S. Yin, Q. Zhou, C. Liu, Q. Gu and S. You, Angew. Chem., Int. Ed., 2023, 62, e202305067 CrossRef CAS PubMed
- W. Chen, H. Xu, F. Liu, K. Chen, Z. Zhou and W. Yi, Angew. Chem., Int. Ed., 2024, 63, e202401498 CrossRef CAS PubMed
- X. Zhu, H. Wu, Y. Wang, G. Huang, F. Wang and X. Li, Chem. Sci., 2023, 14, 8564–8569 RSC
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, compound characterization, and copies of NMR spectra and HPLC traces. See DOI: https://doi.org/10.1039/d4gc04390a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |