
Acidic CO2 electroreduction for high CO2 utilization: catalysts, electrodes, and electrolyzers
Taemin
Lee†
 ,
Yujin
Lee†
,
Jungsu
Eo
and
Dae-Hyun
Nam
*
,
Yujin
Lee†
,
Jungsu
Eo
and
Dae-Hyun
Nam
*
Department of Energy Science and Engineering, Daegu Gyeongbuk Institute of Science and Technology (DGIST), Daegu 42988, Republic of Korea. E-mail: dhnam@dgist.ac.kr
First published on 19th December 2023
Abstract
The electrochemical carbon dioxide (CO2) reduction reaction (CO2RR) is considered a promising technology for converting atmospheric CO2 into value-added compounds by utilizing renewable energy. The CO2RR has developed in various ways over the past few decades, including product selectivity, current density, and catalytic stability. However, its commercialization is still unsuitable in terms of economic feasibility. One of the major challenges in its commercialization is the low single-pass conversion efficiency (SPCE) of CO2, which is primarily caused by the formation of carbonate (CO32−) in neutral and alkaline electrolytes. Notably, the majority of CO2RRs take place in such media, necessitating significant energy input for CO2 regeneration. Therefore, performing the CO2RR under conditions that minimize CO32− formation to suppress reactant and electrolyte ion loss is regarded an optimal strategy for practical applications. Here, we introduce the recent progress and perspectives in the electrochemical CO2RR in acidic electrolytes, which receives great attention because of the inhibition of CO32− formation. This includes the categories of nanoscale catalytic design, microscale microenvironmental effects, and bulk scale applications in electrolyzers for zero carbon loss reactions. Additionally, we offer insights into the issue of limited catalytic durability, a notable drawback under acidic conditions and propose guidelines for further development of the acidic CO2RR.
 Taemin Lee | Taemin Lee received his B.S. and M.S. degrees from Daegu Gyeongbuk Institute of Science and Technology (DGIST), Republic of Korea. He is currently a Ph.D. candidate in the Department of Energy Science and Engineering, DGIST, under the supervision of Prof. Dae-Hyun Nam. His research interests are focused on electrocatalyst fabrication for carbon upgrading. |
 Yujin Lee | Yujin Lee received her B.S. degree in applied physics from Sookmyung Women's University (2022). Currently, she is an M.S./Ph.D. combined candidate in the Department of Energy Science and Engineering at Daegu Gyeongbuk Institute of Science and Technology (DGIST). Her research focuses on the ionomer effect and controlling the microenvironment of catalyst surfaces for electrochemical CO2 conversion. |
 Jungsu Eo | Jungsu Eo is currently an M.S. candidate in the Department of Energy Science and Engineering, Daegu Gyeongbuk Institute of Science and Technology (DGIST), Republic of Korea. He received his B.S. degree in undergraduate studies from DGIST. His research interests are mainly focused on gas diffusion electrode design and pH effects for the efficient electrochemical CO2 reduction reaction. |
 Dae-Hyun Nam | Dae-Hyun Nam is an assistant professor in the Department of Energy Science and Engineering, Daegu Gyeongbuk Institute of Science and Technology (DGIST), Republic of Korea. He received his B.S. (2011) and Ph.D. degrees (2017) from the Department of Materials Science and Engineering, Seoul National University, Republic of Korea. He worked as a postdoctoral fellow at the Department of Electrical and Computer Engineering, University of Toronto, Canada from 2017 to 2020. His research interests are heterogeneous catalysts for renewable energy conversion including nanomaterials synthesis, electrolysis, and mechanistic studies. |
1. Introduction
The electrochemical carbon dioxide (CO2) reduction reaction (CO2RR) has received great attention in the areas of carbon neutrality and long-term seasonal energy storage. For carbon capture and utilization (CCU), the CO2RR enables the conversion of captured atmospheric CO2 into value-added chemicals such as fuels and feedstocks.1–5 The CO2RR can produce C1 chemicals such as carbon monoxide (CO),6–9 methane (CH4),10–12 and formic acid (HCOOH);13–17 multicarbon (C2+) chemicals such as ethylene (C2H4)18–20 and ethanol (C2H5OH);21,22 and even C3 chemicals by CO2 conversion.23–25However, there are still significant challenges to address in the practical application of the CO2RR, including critical performance metrics often referred to as key figures of merit: product selectivity (Faradaic efficiency, FE), production rate or productivity (partial current density), energy efficiency (overpotential), catalytic stability (long-term operation), and carbon efficiency (CO2 conversion).26 In a favorable light, recent strides in CO2RR studies aim to propel the energy efficiency and selectivity to levels which show the potential to be competitive in commercialization. Such achievements have been attained through the development of heterogeneous catalysts, which enable the steering of the CO2RR pathway by controlling the intermediate binding and careful management of the reaction microenvironment.18,26–29 In addition, the introduction of flow cells comprising gas diffusion electrodes (GDEs) has facilitated the tailored design of electrolyzers to augment reaction rates and scale up the reaction.18,30–33 Moreover, the employment of membrane electrode assemblies (MEAs) has paved the way for enhancements in catalytic stability.20,30,34–36
Even with such technological progress, the cost-effectiveness, below that of product formation through traditional thermochemical techniques, and the single-pass carbon efficiency (SPCE) remain as challenges.26,28,37 Conventionally, CO2RRs are typically designed to occur in neutral or alkaline electrolytes, driven by a dual purpose: to decrease the competitive hydrogen evolution reaction (HER)38,39 and to harness the promoting effect of the hydroxide ion (OH−) on CO2 activation or *CO coupling.40–42 However, CO2RRs under such conditions are perceived to be unfavorable from an economic standpoint due to supplementary external costs. This negative perception primarily stems from product losses attributed to carbonate (CO32−) formation – a consequence of interactions between CO2 and OH− in alkaline or neutral electrolytes – along with the necessity for electrolyte regeneration.28
Recently, operating parameters within acidic electrolytes (pH < 2) have received significant interest, chiefly due to their potential to obviate the energy penalties associated with CO32− formation in the CO2RR.9,42–55 However, this approach is a double-edged sword: on the one hand, it promises enhanced energy efficiency, while on the other hand, it presents challenges such as a lowered HER activation barrier and diminished catalyst stability. To judiciously operate the CO2RR in acidic media, it is imperative to engineer both catalysts and electrodes capable of attenuating proton permeation or sustaining elevated *CO coverage. Additionally, the architecture of the electrolytic cell must be precisely designed to allow operation under regimes that amplify CO2 conversion.
Here, we introduce the progress and perspectives of CO2RRs for high CO2 utilization in acidic electrolytes. Our narrative unfolds through the categories of insights from (1) a nanoscale catalytic perspective, (2) the microenvironment within electrode structures at the microscale, and (3) bulk-scale applications pertaining to electrolyzers (Fig. 1). Furthermore, we endeavor to articulate our insights into the existing hurdles and innovative strides associated with catalytic stability in acidic electrolytes. The acidic CO2RR is still an emerging field of study, and a consolidated overview and fresh perspectives on the relevant strategic concepts are anticipated to provide a broader understanding and open new avenues of research within this domain.
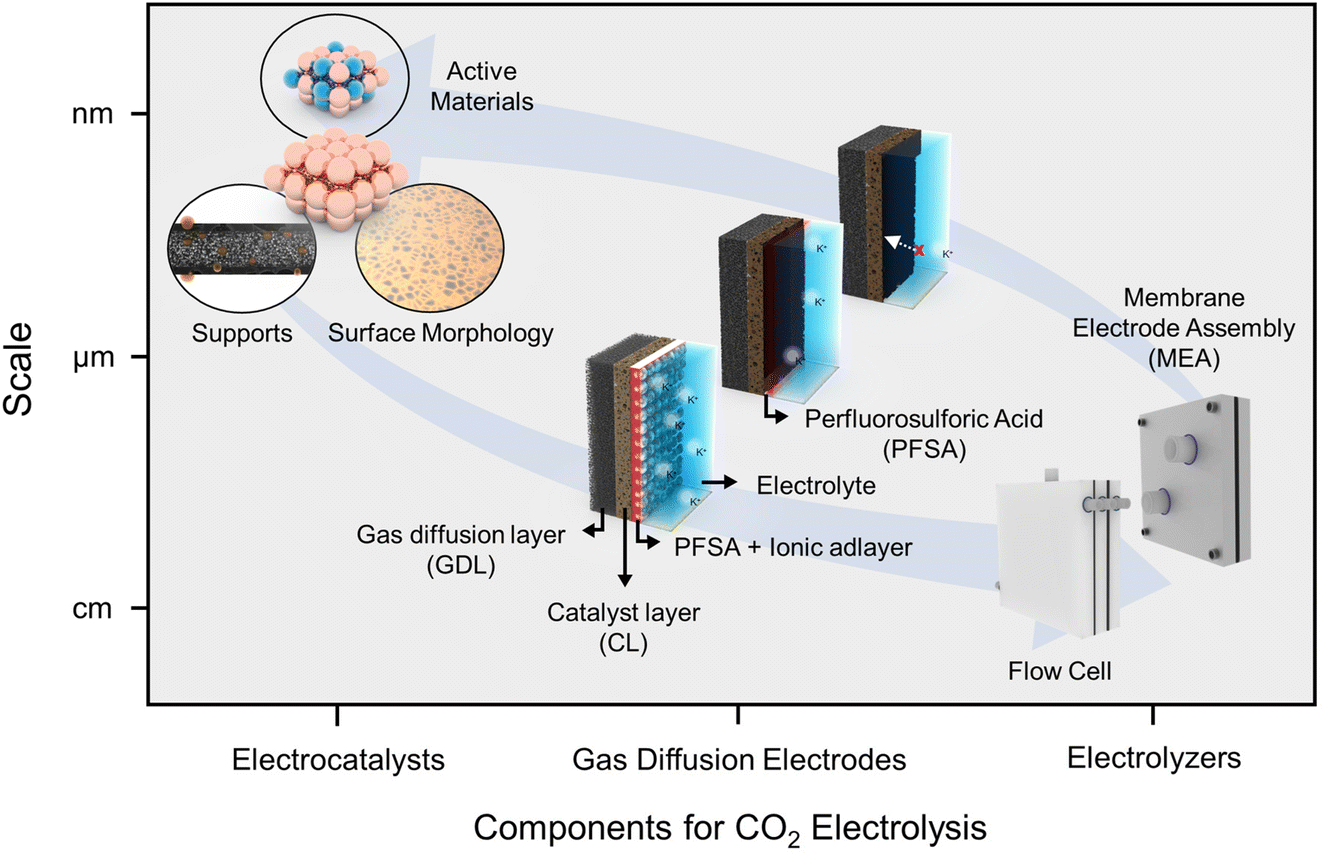 | ||
| Fig. 1 Strategic insights into the acidic CO2RR in terms of scale and components such as electrocatalysts, gas diffusion electrodes, and electrolyzers. | ||
2. Merits of performing the CO2RR in acidic electrolytes
The Bjerrum plot for CO2–bicarbonate (HCO3−)–CO32− systems indicates that the introduction of CO2 into alkaline and neutral electrolytes for the CO2RR can induce severe CO2 consumption through CO32− formation reactions with OH− in aqueous electrolytes (Fig. 2a, eqn (1) and (2)).56| CO2 + OH− → HCO3− | (1) |
| HCO3− + OH− → CO32− + H2O | (2) |
 | ||
| Fig. 2 Comparison of reaction conditions and energy consumption in electrolytes with different pH values. (a) Bjerrum plot for the CO2–HCO3−–CO32− system; carbonate (CO32−) is generated under high pH conditions.56 Reproduced with permission from ref. 56. Copyright 2001, Elsevier. (b) CO32− generation and regeneration in alkaline and acidic conditioned electrolyzer systems.58 Reproduced with permission from ref. 58. Copyright 2023, ACS Publications. (c) Comparison of reaction energy consumption in the CO2RR with acidic, near neutral, and alkaline electrolytes.46 Reproduced with permission from ref. 46. Copyright 2022, Springer Nature. | ||
This has a significant impact on the low SPCE of the CO2RR in alkaline and neutral electrolytes.42,43 Under flow cell operating conditions, theoretical calculations of SPCE for C1 (CO, HCOO−) and C2 (C2H4) chemicals indicate that, in near-neutral electrolytes, the SPCE is 50% for both CO and HCOO− and 25% for C2H4.42,52 In alkaline electrolytes, the SPCE is considerably lower, with theoretical results showing 11.7% for CO and HCOO− and 4.2% for C2H4.46 However, in acidic electrolytes, 100% SPCE can be achieved because of preventing CO32− formation.
Recently, using an acidic electrolyte in the CO2RR has emerged as a potential avenue for minimizing CO2 losses (Fig. 2b). The intrinsic low pH of acidic electrolytes appears to decelerate CO32− formation rates.57 Intriguingly, even in instances where CO32− forms due to a slightly elevated pH near the electrode interface, this CO32− is prone to reconversion to CO2 upon encountering protons in the bulk solution. This mechanism offers a groundbreaking strategy in the CO2RR for high CO2 utilization that is theoretically capable of achieving nearly 100% SPCE.
Beyond the issue of CO2 loss, the acidic CO2RR is perceived as a more economical reaction due to its higher energy efficiency compared to alkaline and neutral CO2RRs.46 This distinction is attributed to the differences in the reaction environments between these CO2RR conditions (Fig. 2b).58 In alkaline electrolyte environments, the regeneration of CO32− in the catholyte back to CO2 represents a dominant fraction of the overall energy expenditure (Fig. 2c). The excessive energy demand can be determined from the forward reaction and reverse reaction energy costs for CO32− formation.28,42,59 While the interaction of CO2 with OH− consumes −56 kJ mol−1 to produce CO32−, regenerating CO2 and OH− from CO32− requires an energy expenditure of 230 kJ mol−1, over four times the absolute value of the energy.59 Additional systematic penalties in alkaline or neutral electrolytes are induced by anodic separation of the oxygen evolution reaction (OER). This is because of CO2 gas generation due to the crossover of CO32− through an anion-exchange membrane (AEM)42,43,49,53 and the formation of metal–CO32− species as a consequence of side reactions between CO32− and metal cations that accumulate on the electrode surface, compromising catalytic efficiency.32 The cost for electrolyte regeneration due to CO32− formation is critical in the CO2RR with alkaline electrolytes. In contrast, in an acidic electrolyte environment, there is no economic loss attributed to catholyte regeneration or anodic separation. Additionally, there is no decrease in catalytic efficiency due to the formation of metal–CO32− complexes, making acidic conditions a more commercially viable option (Fig. 2b and c).
3. Strategies for the acidic CO2RR: electrocatalysts, electrodes, and electrolyzers
The economic feasibility of the acidic CO2RR is highly regarded, but to date, early research outcomes in this field have been relatively limited in comparison with research conducted in alkaline or neutral electrolyte systems. Enhancing the catalyst performance under proton-dominant and harsh acidic conditions remains a challenge in the acidic CO2RR. In acidic electrolytes, achieving a fine balance between selectivity and catalytic durability is a pressing challenge. However, the dominance of the HER under acidic conditions often overshadows CO2 electrolysis. This phenomenon becomes clearer when inspecting the E-pH (or Pourbaix) diagram of water: heightened acidity lowers the voltage window where the HER occurs. Consequently, this constricted voltage window implies that the HER is kinetically more favored than the CO2RR in acidic electrolytes.42Another CO2RR issue in harsh acid environments is metal corrosion. Numerous investigations have been undertaken to address these challenges in water electrolysis employing acidic electrolytes.60–62 Previous studies employed noble metals adept at resisting corrosion under acidic conditions, but these metals unfortunately have significantly lower CO2RR activity than alternative metals. Consequently, when transition metal-based efficient CO2RR catalysts in alkaline or neutral electrolytes are employed in the acidic CO2RR, their stability and CO2RR activity can degrade. Given the aforementioned challenges, this section will present the latest trends in acidic CO2RR research focusing on ways to overcome the limitations of CO2 electrolysis in acidic electrolytes. We introduce the challenges and progress of the acidic CO2RR in terms of electrocatalysts, electrodes, and electrolyzers (Fig. 1).
3. 1. Electrocatalysts for the acidic CO2RR
In the CO2RR, the selectivity of C1 and C2+ chemicals can be modulated by the design of heterogeneous catalysts that control the adsorption energy of intermediates on the surface of metal active sites.63–66 Supports as well as active materials can help in designing the active sites to control the binding of intermediates over the scaling relationship during CO2 electrolysis.67–72 In the acidic CO2RR, although the partial current density for C1 chemicals is generally lower than that in alkaline electrolytes, the product selectivity of CO has increased to near 100% FE with the aid of electrocatalyst development (Fig. 3a). In the acidic CO2RR, various carbon supports have been investigated to enhance the selectivity and reaction rate of C1 chemicals by hybridization with active materials (Fig. 3b).51–54 Manipulating the morphology of carbon supports to hinder proton penetration is a notable methodology in acidic electrolytes. This approach might promote the binding of CO2RR intermediates instead of protons. | ||
| Fig. 3 Catalyst strategies for a successful acidic CO2RR toward C1 and C2+ chemicals. (a) CO selectivity and the corresponding total current density in acidic electrolytes.9,45,46,109–117 (b) Carbon support-based structural variation in catalysts in acidic media for selective C1 chemicals. (c) C2+ chemical selectivity and the corresponding total current density in acidic electrolytes.29,41–44,112,118–124 (d) Composition and morphology variations in Cu-based catalysts for C2 chemical production in the acidic CO2RR. | ||
Carbon nanotubes (CNTs) have a structural advantage due to their large surface area, which provides a high number of CO2 adsorption sites and promotes the dispersion of active materials.45,54 The CO2RR performances of highly dispersed Ni3N nanoparticles/multiwalled carbon nanotubes (MCNTs) and aggregated Ni(OH)2 nanosheets/MCNTs were compared to identify the structural effect of the catalysts. Under neutral electrolyte conditions, Ni3N/MCNTs exhibited an FE of 89% for CO and 10% for hydrogen (H2), whereas Ni(OH)2/MCNTs exhibited a CO FE of 10% in 0.5 M NaHCO3 electrolyte with an applied potential of −0.73 V (vs. RHE), corresponding to an approximately 8-fold increase in CO partial current density from 0.2 to 1.6 mA cm−2. To exploit their structural advantages, Ni3N/MCNTs were used in the CO2RR under acidic conditions by adding HCl and NaHCO3 for pH adjustment to NaCl solution with an applied potential of −0.9 to −1.0 (vs. RHE); the system exhibited a CO FE of 85.7% at pH 3.7 and 50.1% at pH 2.5 with an applied total current density of 20 mA cm−2. This demonstrates the acid pH tolerance and HER competing ability of Ni3N/MCNTs under acidic conditions. Additionally, CNTs have been used as supports for highly dispersed and finely controlled metal single atom catalysts (SACs).45 In the acidic CO2RR, a β-tetra methoxy-substituted nickel phthalocyanine molecularly dispersed electrocatalyst (NiPc-OMe MDE) exhibited a 98% CO FE at a 400 mA cm−2 total current density at pH 1 in 0.05 M H2SO4 + K2SO4 electrolyte. This superior performance is attributed to the large surface area of CNTs and the sophisticated structure of SACs with molecular supports.
Furthermore, a Ni in N-doped carbon nanocage (Ni5@NCN) material was used in the acidic CO2RR to hinder the HER.51 To verify the effect of the carbon support, the CO2RR activities of Ni5@NCN and Ni nanoparticles supported on N-doped carbon spheres without cavities (Ni5/NCS) were compared. In the CO2RR with 0.25 mM H2SO4 + 0.25 M Na2SO4 electrolyte (pH 1), Ni5@NCN showed a CO FE of 60%, while Ni5/NCS produced only H2 during electrolysis. The suppression of the HER by Ni5@NCN was also observed in water electrolysis conducted in an Ar-saturated electrolyte, which exclusively permits the HER. In contrast to the gradual increase in the current density of Ni5/NCS with increasing potential, water electrolysis in Ni5@NCN induced current density saturation.
The effect of carbon supports in the acidic CO2RR is not limited to transition metal-based active materials. It was reported that Ag nanoparticles confined in hollow carbon nanospheres (Ag/h-CNS) lead to effective CO production.52 Ag/h-CNS exhibited a CO FE of 95% and a 46.2% SPCE in 0.05 M H2SO4 + 0.5 M K2SO4 acidic electrolyte (pH 1.1). They found that h-CNS can finely regulate ion diffusion, particularly by maintaining local alkaline conditions through the control of H+ and OH− diffusivity. A mass transport simulation of Ag/h-CNS confirmed that a substantial amount of OH− can penetrate the core of h-CNS, leading to the establishment of a highly localized alkaline environment. Additionally, the nanocage structure was found to promote a high local concentration of cations through continuous proton consumption via buffer effects and proton coupled electro transfer (PCET). Overall, it can be deduced that the structural attributes of the carbon support exert a pivotal influence on the production of C1 chemicals with high selectivity. Utilizing a variety of structured carbon matrices plays a pivotal role in regulating ions and confining active materials. Additionally, fine-tuning the attributes of active catalyst materials, ranging from their composition to intricate structural details, can significantly improve *CO coverage or amplify cation concentrations. This, in turn, facilitates the CO2RR while mitigating the HER.
Converting CO2 into C2+ chemicals with higher energy density is important for the acidic CO2RR.18–20,22,24–26,28–31,36,42 Cu, which enables *CO dimerization for C–C coupling, has been widely used as a CO2RR active material for C2+ chemical production.73–75 While numerous studies have made substantial advances in the acidic CO2RR with Cu-based catalysts, the FEs of C2 chemicals remain notably lower than those achieved in alkaline electrolytes (Fig. 3c). This discrepancy can be attributed to the influx of protons from the surrounding environment, despite the occurrence of localized proton consumption in high current ranges.
Consequently, it is valuable to manipulate the composition or morphology of Cu electrodes to give preference to *CO over *H in competitive processes. This leads to substantial *CO coverage, which facilitates C–C coupling kinetics and constrains proton binding sites, ultimately impeding the HER. Recent research into the generation of C2 chemicals via the modification of Cu catalysts under acidic conditions has involved the control of intermediate binding kinetics through alloying with heterometals to facilitate C–C coupling and inhibit the HER; alternatively, the catalyst's porosity has been fine-tuned to regulate cation concentrations through a confining effect (Fig. 3d). The stabilization of *CO is a crucial factor in C2+ chemical formation because the C–C coupling of *CO intermediates on the catalyst is fundamental for initiating the conversion of CO2 into C2+ chemicals. Consequently, there is a significant focus on improving *CO formation by controlling the adsorption energy to facilitate the formation of C2+ chemicals.
A Pd–Cu alloy catalyst was synthesized to increase the coverage of both *CO and *CO2 on the surface of the catalyst.43 Density functional theory (DFT) calculations confirmed that the Pd–Cu catalyst can decrease the adsorption energy barrier of CO2 and CO, resulting in an increased *CO affinity over *H and subsequently diminishing the availability of active sites for *H adsorption due to its higher *CO coverage. The Gibbs free energy for ΔGOCCOH* indicates a tendency to form C2+ chemicals, and ΔGOCCOH* − ΔGCHO* provides insight into the reason for CO2RR selectivity for C2+ over C1 chemicals, showing that the Cu–Pd alloy has good potential for *CO dimerization. Therefore, the Cu–Pd alloy suppresses the HER and improves C–C coupling, which increases the C2+ chemical FE to approximately 89% at 500 mA cm−2.
Manipulation of the catalyst morphology can adjust the local cation concentration, which is the main factor stabilizing intermediates and influencing the selectivity toward C2+ chemicals. The local cation concentration can be modulated by adjusting the porosity of the catalyst.44 An electrochemically reduced Cu porous nanosheet (ER-CuNS) demonstrated a cation-confining effect, as confirmed by inductively coupled plasma (ICP) analysis of the electrolyte-loaded catalyst, compared with a flat CuO-NS. ER-CuNS exhibited a 4.5 times higher potassium cation (K+) concentration than pristine CuO-NS. The electrochemical CO2RR in acidic electrolytes showed a morphologically confining effect; ER-CuNS showed 84% FE and a partial current density of 560 mA cm−2 for C2+ chemicals, whereas CuO-NS resulted in HER dominance.
3. 2. Microenvironment engineering in a gas diffusion electrode
The utilization of cations in acidic electrolytes can affect the microenvironment of electrocatalysts.76–78 In electrochemical reduction processes, cationic migration is driven from the bulk electrolyte toward the cathode surface by coulombic forces, culminating in the formation of the outer Helmholtz plane (OHP). This, in turn, gives rise to an electric double layer (EDL) field, which plays a critical role in modulating the adsorption energetics of key reaction intermediates. Modifications to the EDL field augment the efficiency of the CO2RR, with pronounced selectivity for multicarbon products and mitigation of HER activity.In the CO2RR process, the presence of an EDL field can boost the reaction activity by enhancing the CO2 absorption rate and stabilizing *CO, which serves as the primary intermediate for the formation of valuable C2 chemicals. Additionally, it reduces the energy barrier associated with C–C coupling, thereby facilitating the synthesis of C2 chemicals. Interestingly, an electric field exerts minimal effects on the adsorption of *H, resulting in selectivity favoring the CO2RR over the HER. Given these observations, the introduction of cationic effects within acidic media is paramount for the activation of CO2RR intermediates and the inhibition of the HER (Fig. 4a).46,79
 | ||
| Fig. 4 Enhancing the CO2RR performance in acidic electrolytes through microenvironment control. (a) Schematics for controlling the catalyst microenvironment by applying the cation effect and adlayers. (b) Effect of radius and concentration control of hydrated cations on the CO2RR performance.55 Reproduced with permission from ref. 55. Copyright 2022, ACS Publications. (c) Enhanced local pH in adlayer-augmented GDEs by limiting proton mass transport.53 Reproduced with permission from ref. 53. Copyright 2023, Springer Nature. | ||
The cation effect on the acidic CO2RR has been studied with K+.42 Upon introducing K+ into 1 M H3PO4 electrolyte at concentrations ranging from 0 to 3 M, the H2 FE declined from 100% to 50%. Moreover, the maximum FE for CH4 reached 28% at 1 M K+, and the maximum FE for C2H4 was 9.3% at 3 M K+. Furthermore, to optimize the influence of K+, the authors introduced a perfluorosulfonic acid (PFSA) ionomer onto the catalyst surface. This ionomer layer serves a dual purpose: it hinders the diffusion of K+ away from the active sites of the catalyst and increases the local pH by obstructing the diffusion of OH− generated during the reaction from the catalyst surface to the bulk electrolyte. These results indicate the significance of controlling the proximity of cations to the electrode surface and applying an ionomer layer for cation adhesion in acidic environments to enhance the efficiency of the CO2RR.
In terms of cation interactions within the EDL, applying cation species with different radii and adjusting their concentrations enable regulation of the cation effect in acidic media (Fig. 4b).9,50,55,80 On a Ag/PTFE electrode, the application of 10 mM LiClO4 in HClO4 electrolyte led to a notable enhancement in the CO FE of 70%, whereas the CO FE remained at 10% when no Li+ was present in the HClO4 electrolyte. This improvement can be attributed to the role played by Li+ in augmenting the EDL field on the catalyst side.53 Since the cation effect is intricately linked to the formation of the OHP on the catalyst side, the size of the hydrated cation crucially influences this effect. When the size of hydrated ions decreases, a greater number of ions aggregate within the OHP, consequently strengthening the EDL field.
The influence of hydrated ion dimensions becomes evident upon the introduction of K+, which has a greater hydrated ion radius than Li+. The introduction of 10 mM KClO4 into the HClO4 electrolyte resulted in a marked increase in the CO FE, reaching almost 100%. This underscores the pivotal influence that differences in hydrated cationic radii have on CO2RR performance. Moreover, a decrease in the K+ concentration directly correlates with a decline in the CO FE. Notably, a peak CO FE of 40% was observed with 1 mM KClO4, in stark contrast to the value of 100% achieved with 10 mM KClO4. To gain a more precise understanding of the cation effect, a generalized modified Poisson–Nernst–Planck (GMPNP) simulation was conducted. The simulation results indicated that the generation of the EDL is closely associated with the radius and concentration of the hydrated cation, whereas the inhibition of proton migration and alterations in diffusion layer thickness are primarily influenced by the ion concentration.
Another approach involves the application of an adlayer on the catalyst surface to elevate the local pH, effectively suppressing the HER through the modulation of proton mass transport and optimization of the EDL. Thus, the adlayer acts as a protective layer for the catalyst under rigorous conditions, suggesting an efficient strategy for the acidic CO2RR.17,48,49,53,81,82 Adlayers commonly used for the acidic CO2RR are ionomers that consist of a hydrophobic backbone, side chains and hydrophilic ionic groups. Hydrated cations migrate through the hydrophilic channel established by the SO3− ionic groups of the ionomer, which exhibit a greater affinity for K+ than for protons. Moreover, the hydrophobic segments of the ionomer create a gas diffusion pathway. This implies that the ionomer can enhance the efficiency of the CO2RR while impeding the HER by facilitating the mass transport of K+ and CO2 gas. Nonetheless, a limitation exists in the uneven distribution of the ionomer on the catalyst surface.
To address this concern, a PFSA ionomer was utilized in conjunction with covalent organic frameworks (COFs) (Fig. 4c).53 The presence of imine groups within COFs contributes to the uniform distribution of the ionomer, thus enhancing its effectiveness and remarkably constraining proton permeability. To demonstrate proton inhibition by COFs, the pH value was measured by monitoring the permeates traveling from the bulk catholyte to the backside of an electrode consisting of PFSA/COFs/Cu/hydrophilic PTFE. When the ionomer was applied in conjunction with COFs, the pH of the permeate increased significantly to pH 12 at a current density of 250 mA cm−2, whereas in the absence of COFs, it increased to only pH 7. Furthermore, the FE for C2+ chemicals reached approximately 75% at a current density of 200 mA cm−2, and a stability of 30 hours was observed. Improving the distribution of ionomers by applying COFs efficiently hinders proton mass transport and induces a high local pH near the electrode surface, which increases C2+ chemical selectivity.
In summary, the mass transport of protons from the bulk electrolyte to the catalyst, the diffusion of OH− from the catalyst surface to the bulk electrolyte, and the local concentration of cations can all be manipulated by controlling the microenvironment of the catalyst. Consequently, managing the local microenvironment by adjusting hydrated ion radii or ion concentrations to optimize the cation effect and by applying an adlayer on the electrode surface can be a highly impactful strategy for enhancing the efficiency of the acidic CO2RR.
3. 3. Theoretical understanding of the electrolyte pH effect on the CO2RR
Most DFT computations in the acidic CO2RR have focused on the effect of catalysts or microenvironments on the *CO2 adsorption or *COOH stabilization.43,47 However, the effect of electrolyte pH on tuning the CO2RR pathways has not been studied frequently. Under acidic conditions with pH 1, it was reported that the formation of *COH and *CHOH intermediates hinders the kinetic pathway for C2 chemical formation (Fig. 5a).83 This facilitates the C1 chemical formation pathway. Under neutral conditions with pH 7, they suggest that *COH and *CO–COH exhibit nearly equivalent energy preferences, implying a conducive environment for the formation of both C1 and C2 chemicals (Fig. 5b). Conversely, under the alkaline conditions with pH 12, the CO2RR pathway involving *CO–CO is preferred and this enables an increase in selectivity towards C2 chemical formation (Fig. 5c).83 In all modeled systems, the C1 pathway was calculated at the low *CO coverage (θCO = 1/9 or 1/16), resulting in preference for the HER.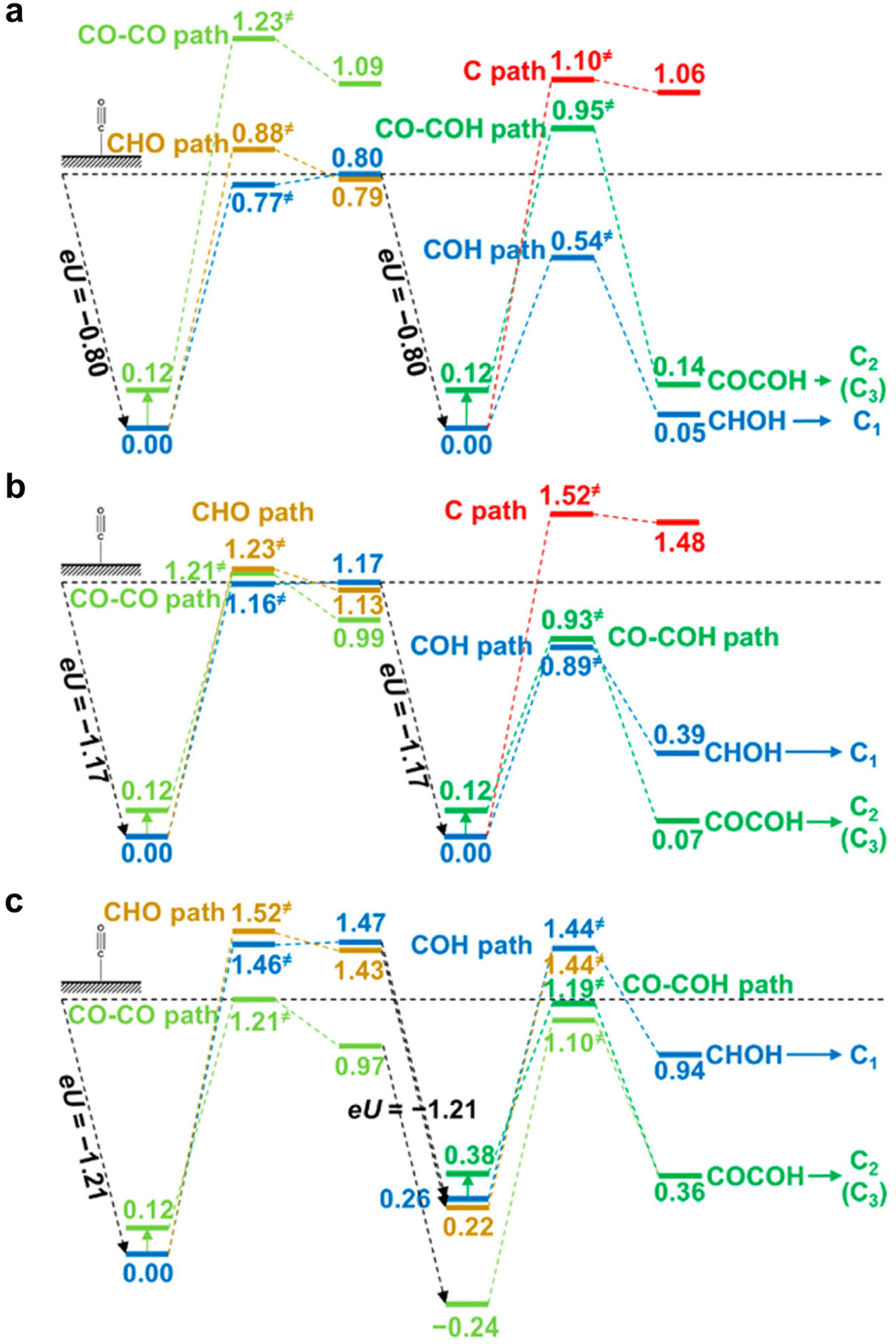 | ||
| Fig. 5 DFT-based theoretical free energy diagrams of CO2RR intermediates according to the electrolyte pH.83 Free energy diagrams under (a) acidic conditions with pH 1, (b) neutral conditions with pH 7, and (c) alkaline conditions with pH 12. Reproduced with permission from ref. 83. Copyright 2016, ACS Publications. | ||
Theoretical CO2RR pathways according to the electrolyte pH reveals that H2 can be the major product by the HER in the acidic CO2RR. Therefore, comprehensive strategies from nanoscale-to-bulk scale are required to address the challenges of the acidic CO2RR. They include strategies such as catalyst development to enhance *CO binding and microenvironment engineering to control the metal cations and local OH− concentration to sustain a high local pH near the catalysts.
3. 4. Catalyst degradation and stability in the acidic CO2RR
The harsh environment of acidic electrolytes promotes the dissolution of metal ions in heterogeneous catalysts, causing corrosion or reconstruction with dissolution and electrodeposition.48 Indeed, the rate of metal extraction in the reaction environment, as influenced by the electrolyte pH, plays a pivotal role in determining the catalyst's lifespan. Catalyst stabilities of over 150 hours in alkaline or neutral electrolyte environments were reported previously, whereas significantly shorter stability has been observed in acidic media (Fig. 6a, Table 1). Catalyst stability in an acidic environment has already been studied with respect to the HER or OER via Pt- or Ir-based catalysts.60–62 Unfortunately, these noble metals are not active in the CO2RR.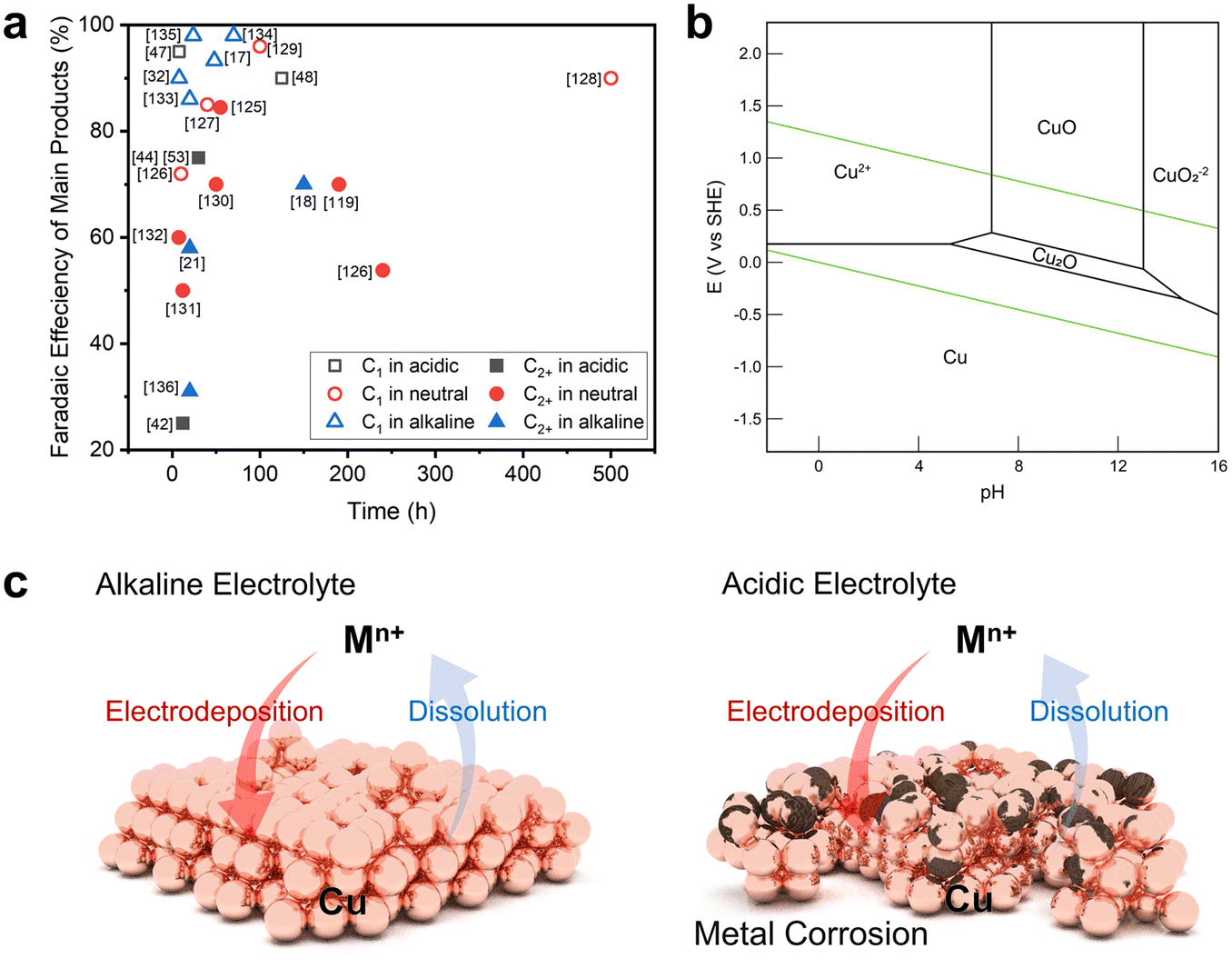 | ||
| Fig. 6 Catalytic stability issues in the acidic CO2RR. (a) Selectivity versus stability of CO2RR catalysts in acidic, neutral, or alkaline electrolytes.17,18,21,32,42,44,47,48,53,119,125–136 (b) Pourbaix diagram of Cu.84 Reproduced with permission from ref. 84. Copyright 2021, Elsevier. (c) Schematics comparing Cu reconstruction behavior during the CO2RR in alkaline and acidic electrolytes. | ||
| Electrolyte | Catalyst | Electrolyzer | Total J (mA cm−2) | Potential | FE (%) | Stability (h) | Ref. | |
|---|---|---|---|---|---|---|---|---|
| Acidic | 0.05 M H2SO4 + KCl | ER-CuNS (reduced Cu porous nanosheet) | Flow cell | 700 | −1.48 VRHE | 75 (C2+) | 30 | 44 |
| 1 M H3PO4 + 3 M KCl | COF: PFSA-modified PTFE–Cu | Flow cell | 200 | −2.4 VAg/AgCl | 75 (C2+) | 30 | 53 | |
| 1 M H3PO4 + 3 M KCl | Cation-augmenting layer-modified Cu nanoparticles | Flow cell | 1200 | 4.2 V (full cell) | 25 (C2H4) | 12 | 42 | |
| 0.05 M H2SO4 + 3 M KCl | SiC-Nafion™/SnBi/PTFE | Flow cell | 100 | −1.5 VRHE | 90 (HCOOH) | 125 | 48 | |
| 0.5 M K2SO4 + H2SO4 | Ni–N–C | MEA | 500 | 3.7 V (full cell) | 95 (CO) | 8 | 47 | |
| Neutral | 0.5 M KCl | Cu nanostructure | Flow cell | 150 | 4.5 V (full cell) | 84.5 (C2H4) | 55 | 125 |
| 0.1 M KHCO3 | Ligand-doped ZIF-8 | H-cell | 3 | −1 VRHE | 72 (CO) | 10 | 126 | |
| 0.1 M KHCO3 | Co3O4 atomic layer with oxygen vacancies | H-cell | 2.7 | −0.87 VSCE | 85 (HCOOH) | 40 | 127 | |
| 0.1 M KHCO3 | CuOHFCl | H-cell | 15 | −1.00 VRHE | 53.8 (C2+) | 240 | 126 | |
| 0.5 M KHCO3 | Bismuthene (Bi-ene-NW) | H-cell | 100 | −1.17 VRHE | 90 (HCOOH) | 500 | 128 | |
| 0.5 M KHCO3 | CoPc@MWCNTs | H-cell | 1 | −0.7 VRHE | 96 (CO) | 100 | 129 | |
| 1 M KHCO3 | OD-Cu-III | Flow cell | 300 | −1 VRHE | 70 (C2+) | 50 | 130 | |
| 0.1 M KHCO3 | CuAg | Custom cell | 45 | −1.05 VRHE | 50 (C2H4) | 12 | 131 | |
| 0.1 M KHCO3 | Cu and iridium oxide supported on a titanium mesh | Flow cell | 600 | ∼ 3.3 V (full cell) | 70 (C2H4) | 190 | 119 | |
| 1 M KHCO3 | DMAN-modified CuNPs | Flow cell | 100 | −1 VRHE | 60 (C2+) | 7.5 | 132 | |
| Alkaline | 7 M KOH | Graphite/carbon NPs/Cu/PTFE | Flow cell | 80 | −0.55 VRHE | 70 (C2H4) | 150 | 18 |
| 1 M KOH | Bi rhombic dodecahedra | Flow cell | 200 | −0.68 VRHE | 86 (HCOOH) | 20 | 133 | |
| 1 M KOH | Sn | MEA | 40 | 2.2 V (full cell) | 93.3 (HCOOH) | 48 | 17 | |
| 1 M KOH | N-coordinated Ni on CNTs | MEA | 100 | 2.1 V (full cell) | 98 (CO) | 70 | 134 | |
| 1 M KOH | CNT-sulfamethoxazole(SMX) | Flow cell | 100 | −0.7 VRHE | 98 (CO) | 24 | 135 | |
| 1 M KOH | BaO/Cu | Flow cell | 400 | −0.75 VRHE | 58 (alcohol) | 20 | 21 | |
| 2 M KOH | MWNTs/PyPBI/Au | Flow cell | 100 | −0.44 VRHE | 90 (CO) | 8 | 32 | |
| 1 M KOH | Cu–Cl–Cs | Flow cell | 1000 | −0.75 VRHE | 31 (C2H5OH) | 20 | 136 | |
Metal catalysts mainly used in the CO2RR, especially Cu, exist in the form of oxides or ions in acidic environments that cause corrosion or detachment from the electrode surface due to their ionization kinetics (Fig. 6b).84 The corrosion phenomenon observed in these non-noble metal catalysts not only prevents achieving the desired catalytic performance because of catalytic reconstruction, but also poses a substantial hindrance to commercialization due to their limited lifetime.85–89 Hence, we propose a catalytic approach to sustain catalytic stability in acidic electrolytes; this is based on emphasizing factors that are expected to exert a substantial influence on catalyst durability instead of employing an indirect approach to elevate the local pH.
To discuss catalyst stability, it is necessary to understand the catalyst reconstruction behavior during the CO2RR (Fig. 6c). Catalyst reconstruction takes place when metal ions are released into the electrolytes during the reaction and are subsequently redeposited onto the electrode surface due to the reduction potential, resulting in alterations in the morphology based on the rate of metal ionization.85,89 Catalyst reconstruction will occur more actively in an acidic environment with a higher ionization rate than in a neutral or alkaline electrolyte with a lower metal ionization rate.90,91 Understanding this behavior is advantageous for increasing catalyst stability because the reconstructed catalyst structure will actively participate in the reaction.
Nonetheless, there is less understanding about the reconstruction of CO2RR catalysts in an acidic electrolyte, and predicting alterations in the catalyst morphology during the catalytic reaction remains challenging. Accordingly, proposing a method to mitigate the extraction of active metals could represent a significant innovation. For this kind of catalyst design strategy, sacrificial metals can be utilized. When two metals with different ionization tendencies come into contact within a bulk electrolyte, the metal with the greater ionization tendency will undergo oxidation in place of the other metal. In other words, by locating the sacrificial metal around the active metal in acidic media, it is possible to inhibit the dissolution of the active metal during electrochemical operation. Consequently, employing metals with higher oxidation tendencies, such as Fe, Mg, and Al, alongside Cu as the active site for CO2 conversion into C2+ chemicals, could enhance the stability of Cu active materials due to the dopant materials acting as sacrificial agents. However, it is crucial to optimize the dopant species and its concentration, as these materials may also function as active sites for the HER.
3. 5. CO2RR electrolyzer strategy for high CO2 utilization
GDE-based electrolyzers have been developed for the CO2RR to overcome low CO2 solubility and mass transport. The flow cell, where gaseous CO2 is directly supplied to catalyst layer, enables to overcome mass transport limitations in an H-type cell and is the most actively used electrolytic cell (Fig. 7a).92 However, due to the low catalytic stability and iR loss induced by liquid catholytes, this is inadequate in terms of long-term operation and energy efficiency. | ||
| Fig. 7 Application of the acidic CO2RR in a tandem electrolyzer system for achieving high SPCE. Schematics of the (a) flow cell and (b) MEA. (c) Zero carbon loss tandem MEA system for MEA to MEA, CO2 to CO, and CO to C2H4.47 Reproduced with permission from ref. 47. Copyright 2023, The Royal Chemical Society. | ||
To address the challenges of energy loss and low stability, a zero gap MEA has been used for the CO2RR. The MEA consists of a two-electrode system capable of extracting protons and products from humidified CO2, thereby mitigating ohmic losses, and is well suited for long-term operation (Fig. 7b).92,93 Nonetheless, within the MEA setup, CO32− that permeate through the AEM can result in the loss of reactants and increased costs associated with anodic separation. This energy loss can be mitigated by utilizing an acid electrolyte in both electrolyzer systems, thereby enabling the development of a zero carbon loss electrolysis system.
To develop a high carbon efficiency electrochemical reaction system, a flow cell to flow-cell electrolysis system was constructed; this system achieved a high efficiency CO2RR by creating a significantly elevated local pH using a SiC-Nafion™ additive layer on a Sn–Bi alloyed metal, even in a harsh electrolyte environment (pH = 1).48 The HCOOH FE of SiC-Nafion™/SnBi/PTFE was over 92% in all current density ranges (100–800 mA cm−2). The cathodic energy efficiency (CEE) corresponding to the CO2RR performance was calculated, revealing a notably high CEE of 50% at a current density of 100 mA cm−2. Furthermore, for an economic evaluation, the SPCE was calculated in an acidic environment at a CO2 flow rate of 3 sccm and an efficiency of 65% was achieved that was unattainable with an alkaline electrolyte. To enhance the SPCE, two flow cells based on acidic electrolytes were connected, enabling the conversion of unreacted CO2 from the first electrolyzer into a product. As a result, a 76% SPCE was achieved through the reaction in the second electrolytic cell. During this process, the proportion of H2 produced in the first electrolytic cell remained below 10% and had a negligible impact on the CO2RR in the second electrolyzer.
The adoption of acidic anolytes in the MEA can dramatically diminish carbon loss by suppressing the penetration of CO32− generated in the cathodes through a cation-exchange membrane (CEM). Furthermore, the proton crossover through the CEM enables the regeneration of CO32−, potentially decreasing the CO2 loss and enhancing the SPCE.47,94 Similar to the neutral MEA, there is an increase of local pH in the cathode of the acidic MEA due to the continuous consumption of protons and the generation of OH− due to PCET. However, the local pH in the acidic MEA is around pH 7, significantly lower compared to over pH 12 in the neutral anolyte MEA.94 This is due to the large amount of proton crossover from the anode to cathode side in the acidic MEA. Lower pH in the acidic MEA can suppress the formation of CO32− more effectively than the neutral MEA. Additionally, the penetration of metal cations along with the protons through the CEM modulates the EDL, stabilizes the intermediates and enhances the CO2RR performance.47
DFT calculations suggest that the concurrent penetration of protons and cations through the CEM stabilizes CO2RR intermediates.46,47 This is attributed to a decrease in OH−, which otherwise interferes with the interactions between intermediates and cations, an effect amplified by coexisting protons. Taking advantage of this point, the MEA system was developed into a zero carbon loss reaction system via cascade electrolysis (Fig. 7c). A CO2RR–CORR tandem electrolysis system was designed with a CO2-to-CO conversion in the acidic MEA and a CO-to-C2H4 conversion in the alkaline MEA in sequence, which promoted C2+ chemical formation.47 A Ni–N–C single-atom catalyst was utilized to facilitate CO formation in an acidic MEA with a substantial reaction area of 25 cm2. The residual CO2 was captured by a high-concentration alkaline solution trap, and C2+ chemicals were produced at a 4 cm2 MEA in alkaline electrolytes as a second electrolyzer by converting CO from the first electrolyzer. The formation rate of C2 chemicals was 0.35 mmol min−1 without any carbon loss since CO does not form CO32− even under alkaline conditions. Therefore, designing an electrolyzer that leverages the advantages of the acidic CO2RR allows for significantly enhanced carbon efficiency and price competitiveness that aligns closely with commercialization goals.
In terms of full cell operation in the acidic MEA, addressing the issues for overpotential, catalyst stability, and leaching in the anode is important for ensuring economic feasibility. These challenges have been extensively studied in acidic water electrolysis.95–101 Despite the advantages of acidic water electrolysis, such as low ohmic loss, high energy efficiency, and high hydrogen purity, the harsh acidic conditions require strategies to solve low stability issues for both cathode and anode designs. Such strategies include (1) metal alloy active sites and (2) carbon supports in electrocatalysts for the acidic OER.
In acidic water electrolysis, core/shell structured catalysts including Ru-based catalysts (core) for high OER activity with Ir oxide (shell) for high-stability exhibited superior catalytic performances.97 At a current density of 10 mA cm−2, they required only 282 mV overpotential and showed high OER stability for 24 hours at 1.55 V (vs. RHE). Furthermore, the use of low-cost, highly conductive, structurally modifiable, and acid corrosion-resistant carbon supports can provide an avenue toward the stable OER. For example, Zn-doped RuO2 embedded in carbon cloth catalysts, resulted in 179 mV overpotential at a current density of 10 mA cm−2 and sustained long-term operation for 100 hours.100 There have been various efforts to achieve high activity and long-term stability in OERs within acidic water electrolysis. CO2RR studies have mainly focused on half-cell cathodic reactions, with less attention being given to the anode side. Therefore, we envision that applying the rational design principles of OER electrocatalysts in water electrolysis to the CO2RR might be helpful in achieving highly efficient and stable anodes for the acidic MEA.
4. Requirement of nanoscale catalysts for the acidic CO2RR
Acidic electrolytes for the CO2RR induce the HER with abundant protons and decay the catalyst due to the harsh environment by the low pH. To overcome these shortcomings of the acidic CO2RR, it is necessary to interrupt the proton transport toward the catalyst surface and block the absorption of protons on the active site for the suppression of the HER. In addition, the cation effect, which lowers the reaction energy barrier and increases the selectivity of the CO2RR, can be maximized by confining the ions near the surface of catalysts. This confinement increases the local ion concentration with the structural factor of the electrode.102 Thus, the electrochemical CO2RR performance can be affected by the behaviors of ions and intermediates.103,104Structural control of the nanoscale catalysts allows to modulate microenvironments while maintaining the large active surface area.105–107 For instance, nanostructured Cu catalysts can maximize the cation effect by regulating the electric field distribution near the electrode surface. According the multiphysics simulations (COMSOL), the high curvature of Cu nanoneedles (CuNNs) increased the local electric field to 1.19 × 105 kV m−1 near the tip, while a small electric field intensity of about 0.6 × 104 kV m−1 was induced on a Cu plate.107 This increased the tip-adsorbed K+ concentration to 4.22 M, which exceeds the solubility limit of 3 M KCl solution (3.5 M). The promoted K+ solubility by CuNNs was also identified by ion chromatography. CuNNs showed over 4 times higher K+ concentration than that of the Cu film.
It is also important to apply nanotechnology for the design of supports because they enable to confine ions near the surface of the catalyst.41,108 The carbon nanoparticle layer on the Cu-based catalyst increased the local OH− concentration in both neutral and acidic electrolytes.41 OH− ions generated during the CO2RR were confined near the catalyst surface by the carbon layer and the increased local OH− concentration improved the C2H4 FE to 64.5% at a current density of 300 mA cm−2 in 0.5 M K2SO4 solution with H2SO4 (pH = 2.0). Nanoscale catalysts are crucial for the CO2RR under acidic conditions because they enhance catalytic performances by optimizing the structures of active sites/supports and induce the cation effect which controls the local pH and suppresses the HER.
5. Conclusion and outlook
We have highlighted the potential of the acidic CO2RR in contrast to the use of alkaline and neutral electrolytes. The reaction and energy losses induced by CO32− formed during the CO2RR in alkaline or neutral electrolytes can be suppressed in acidic electrolytes. Despite these advantages, the acidic CO2RR faces challenges such as a high HER preference and low catalyst stability, for which we have outlined strategies across reaction scales from the nanoscale to the bulk scale. At the nanoscale, refined catalyst design enables the enhancement of CO2RR selectivity in acidic electrolytes. The morphology of the carbon support and the porosity of the active material can be manipulated to enhance CO2RR activity via ion diffusion control and an expanded reaction area. Additionally, alloying of the active metal facilitates an increase in *CO binding energy, enabling the removal of *H binding sites. At the microscale, the proximity of cations to the electrode, coupled with a protective adlayer, affects an EDL, serving to stabilize the CO2RR intermediate while also impeding proton mass transport. At the bulk scale, designing electrolyzers suitable for acidic electrolytes or constructing a cascade reaction system can remarkably increase the SPCE. Similarly, the CO2RR in acidic media not only retains the advantage of zero carbon loss but also offers economic benefits via minimized energy loss, thereby showcasing potential for the development of the acidic CO2RR. However, the stability of catalyst materials remains limited in acidic electrolytes. We anticipate that future work aimed at the stabilization of catalysts under harsh acidic conditions could markedly elevate the competitiveness of the acidic CO2RR in terms of both cost and energy efficiency and make an impact in the field of carbon neutrality technologies.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (NRF-2021R1C1C1013784 and NRF-2021M3D1A2047041).References
- D. R. Kauffman, J. Thakkar, R. Siva, C. Matranga, P. R. Ohodnicki, C. Zeng and R. Jin, ACS Appl. Mater. Interfaces, 2015, 7, 15626–15632 CrossRef CAS PubMed
.
- M. Li, H. Wang, W. Luo, P. C. Sherrell, J. Chen and J. Yang, Adv. Mater., 2020, 32, e2001848 CrossRef PubMed
- Y. Pei, H. Zhong and F. Jin, Energy Sci. Eng., 2021, 9, 1012–1032 CrossRef CAS
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC
- D. T. Whipple and P. J. A. Kenis, J. Phys. Chem. Lett., 2010, 1, 3451–3458 CrossRef CAS
- M. Liu, Y. Pang, B. Zhang, P. De Luna, O. Voznyy, J. Xu, X. Zheng, C. T. Dinh, F. Fan, C. Cao, F. P. de Arquer, T. S. Safaei, A. Mepham, A. Klinkova, E. Kumacheva, T. Filleter, D. Sinton, S. O. Kelley and E. H. Sargent, Nature, 2016, 537, 382–386 CrossRef CAS PubMed
- X. Li, W. Bi, M. Chen, Y. Sun, H. Ju, W. Yan, J. Zhu, X. Wu, W. Chu, C. Wu and Y. Xie, J. Am. Chem. Soc., 2017, 139, 14889–14892 CrossRef CAS PubMed
- C. Hiragond, H. Kim, J. Lee, S. Sorcar, C. Erkey and S.-I. In, Catalysts, 2020, 10, 98 CrossRef CAS
- M. C. O. Monteiro, M. F. Philips, K. J. P. Schouten and M. T. M. Koper, Nat. Commun., 2021, 12, 4943 CrossRef CAS PubMed
- J. Cai, Q. Zhao, W. Y. Hsu, C. Choi, Y. Liu, J. M. P. Martirez, C. Chen, J. Huang, E. A. Carter and Y. Huang, J. Am. Chem. Soc., 2023, 145, 9136–9143 CrossRef CAS PubMed
- X. Wang, A. Xu, F. Li, S. F. Hung, D. H. Nam, C. M. Gabardo, Z. Wang, Y. Xu, A. Ozden, A. S. Rasouli, A. H. Ip, D. Sinton and E. H. Sargent, J. Am. Chem. Soc., 2020, 142, 3525–3531 CrossRef CAS PubMed
- L. Xiong, X. Zhang, L. Chen, Z. Deng, S. Han, Y. Chen, J. Zhong, H. Sun, Y. Lian, B. Yang, X. Yuan, H. Yu, Y. Liu, X. Yang, J. Guo, M. H. Rummeli, Y. Jiao and Y. Peng, Adv. Mater., 2021, 33, e2101741 CrossRef PubMed
- N. Han, Y. Wang, H. Yang, J. Deng, J. Wu, Y. Li and Y. Li, Nat. Commun., 2018, 9, 1320 CrossRef PubMed
- S. Gao, Y. Lin, X. Jiao, Y. Sun, Q. Luo, W. Zhang, D. Li, J. Yang and Y. Xie, Nature, 2016, 529, 68–71 CrossRef CAS PubMed
- D. Ewis, M. Arsalan, M. Khaled, D. Pant, M. M. Ba-Abbad, A. Amhamed and M. H. El-Naas, Sep. Purif. Technol., 2023, 316, 123811 CrossRef CAS
- C. Oloman and H. Li, ChemSusChem, 2008, 1, 385–391 CrossRef CAS PubMed
- W. Lee, Y. E. Kim, M. H. Youn, S. K. Jeong and K. T. Park, Angew. Chem., Int. Ed., 2018, 57, 6883–6887 CrossRef CAS PubMed
- C.-T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. García de Arquer, A. Kiani, J. P. Edwards, P. De Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783–787 CrossRef CAS PubMed
- D. H. Nam, O. Shekhah, A. Ozden, C. McCallum, F. Li, X. Wang, Y. Lum, T. Lee, J. Li, J. Wicks, A. Johnston, D. Sinton, M. Eddaoudi and E. H. Sargent, Adv. Mater., 2022, 34, e2207088 CrossRef PubMed
- J. Y. Kim, D. Hong, J. C. Lee, H. G. Kim, S. Lee, S. Shin, B. Kim, H. Lee, M. Kim, J. Oh, G. D. Lee, D. H. Nam and Y. C. Joo, Nat. Commun., 2021, 12, 3765 CrossRef CAS PubMed
- A. Xu, S.-F. Hung, A. Cao, Z. Wang, N. Karmodak, J. E. Huang, Y. Yan, A. Sedighian Rasouli, A. Ozden, F.-Y. Wu, Z.-Y. Lin, H.-J. Tsai, T.-J. Lee, F. Li, M. Luo, Y. Wang, X. Wang, J. Abed, Z. Wang, D.-H. Nam, Y. C. Li, A. H. Ip, D. Sinton, C. Dong and E. H. Sargent, Nat. Catal., 2022, 5, 1081–1088 CrossRef CAS
- T. Jaster, A. Gawel, D. Siegmund, J. Holzmann, H. Lohmann, E. Klemm and U. P. Apfel, iScience, 2022, 25, 104010 CrossRef CAS PubMed
- M. Esmaeilirad, Z. Jiang, A. M. Harzandi, A. Kondori, M. Tamadoni Saray, C. U. Segre, R. Shahbazian-Yassar, A. M. Rappe and M. Asadi, Nat. Energy, 2023, 8, 891–900 CrossRef CAS
- Z. Gu, H. Shen, Z. Chen, Y. Yang, C. Yang, Y. Ji, Y. Wang, C. Zhu, J. Liu, J. Li, T.-K. Sham, X. Xu and G. Zheng, Joule, 2021, 5, 429–440 CrossRef CAS
- S. A. Francis, J. M. Velazquez, I. M. Ferrer, D. A. Torelli, D. Guevarra, M. T. McDowell, K. Sun, X. Zhou, F. H. Saadi, J. John, M. H. Richter, F. P. Hyler, K. M. Papadantonakis, B. S. Brunschwig and N. S. Lewis, Chem. Mater., 2018, 30, 4902–4908 CrossRef CAS
- A. Somoza-Tornos, O. J. Guerra, A. M. Crow, W. A. Smith and B. M. Hodge, iScience, 2021, 24, 102813 CrossRef CAS PubMed
- M. Jouny, W. Luc and F. Jiao, Ind. Eng. Chem. Res., 2018, 57, 2165–2177 CrossRef CAS
- J. Sisler, S. Khan, A. H. Ip, M. W. Schreiber, S. A. Jaffer, E. R. Bobicki, C.-T. Dinh and E. H. Sargent, ACS Energy Lett., 2021, 6, 997–1002 CrossRef CAS
- F. P. García de Arquer, C.-T. Dinh, A. Ozden, J. Wicks, C. McCallum, A. R. Kirmani, D.-H. Nam, C. Gabardo, A. Seifitokaldani, X. Wang, Y. C. Li, F. Li, J. Edwards, L. J. Richter, S. J. Thorpe, D. Sinton and E. H. Sargent, Science, 2020, 367, 661–666 CrossRef PubMed
- T.-T. Zhuang, Z.-Q. Liang, A. Seifitokaldani, Y. Li, P. De Luna, T. Burdyny, F. Che, F. Meng, Y. Min, R. Quintero-Bermudez, C. T. Dinh, Y. Pang, M. Zhong, B. Zhang, J. Li, P.-N. Chen, X.-L. Zheng, H. Liang, W.-N. Ge, B.-J. Ye, D. Sinton, S.-H. Yu and E. H. Sargent, Nat. Catal., 2018, 1, 421–428 CrossRef CAS
- T. T. H. Hoang, S. Verma, S. Ma, T. T. Fister, J. Timoshenko, A. I. Frenkel, P. J. A. Kenis and A. A. Gewirth, J. Am. Chem. Soc., 2018, 140, 5791–5797 CrossRef CAS PubMed
- S. Verma, Y. Hamasaki, C. Kim, W. Huang, S. Lu, H.-R. M. Jhong, A. A. Gewirth, T. Fujigaya, N. Nakashima and P. J. A. Kenis, ACS Energy Lett., 2017, 3, 193–198 CrossRef
- S. Ma, R. Luo, J. I. Gold, A. Z. Yu, B. Kim and P. J. A. Kenis, J. Mater. Chem. A, 2016, 4, 8573–8578 RSC
- Z. Liu, H. Yang, R. Kutz and R. I. Masel, J. Electrochem. Soc., 2018, 165, J3371–J3377 CrossRef CAS
- I. A. Digdaya, I. Sullivan, M. Lin, L. Han, W. H. Cheng, H. A. Atwater and C. Xiang, Nat. Commun., 2020, 11, 4412 CrossRef CAS PubMed
- N. R. de Tacconi, W. Chanmanee, B. H. Dennis and K. Rajeshwar, J. Mater. Res., 2017, 32, 1727–1734 CrossRef CAS
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Norskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed
- P. An, L. Wei, H. Li, B. Yang, K. Liu, J. Fu, H. Li, H. Liu, J. Hu, Y.-R. Lu, H. Pan, T.-S. Chan, N. Zhang and M. Liu, J. Mater. Chem. A, 2020, 8, 15936–15941 RSC
- X.-Z. Wang, S. Liu, Q. Liu and J.-L. Luo, Electrochem. Commun., 2019, 107, 106531 CrossRef CAS
- Y. Jiang, X. Wang, D. Duan, C. He, J. Ma, W. Zhang, H. Liu, R. Long, Z. Li, T. Kong, X. J. Loh, L. Song, E. Ye and Y. Xiong, Adv. Sci., 2022, 9, e2105292 CrossRef PubMed
- Z. Wang, Y. Li, X. Zhao, S. Chen, Q. Nian, X. Luo, J. Fan, D. Ruan, B. Q. Xiong and X. Ren, J. Am. Chem. Soc., 2023, 145, 6339–6348 CrossRef CAS PubMed
- J. E. Huang, F. Li, A. Ozden, A. Sedighian Rasouli, F. P. García de Arquer, S. Liu, S. Zhang, M. Luo, X. Wang, Y. Lum, Y. Xu, K. Bertens, R. K. Miao, C.-T. Dinh, D. Sinton and E. H. Sargent, Science, 2021, 372, 1074–1078 CrossRef CAS PubMed
- Y. Xie, P. Ou, X. Wang, Z. Xu, Y. C. Li, Z. Wang, J. E. Huang, J. Wicks, C. McCallum, N. Wang, Y. Wang, T. Chen, B. T. W. Lo, D. Sinton, J. C. Yu, Y. Wang and E. H. Sargent, Nat. Catal., 2022, 5, 564–570 CrossRef CAS
- Z. Ma, Z. Yang, W. Lai, Q. Wang, Y. Qiao, H. Tao, C. Lian, M. Liu, C. Ma, A. Pan and H. Huang, Nat. Commun., 2022, 13, 7596 CrossRef CAS PubMed
- Z. Jiang, Z. Zhang, H. Li, Y. Tang, Y. Yuan, J. Zao, H. Zheng and Y. Liang, Adv. Energy Mater., 2023, 13, 2203603 CrossRef CAS
- J. Gu, S. Liu, W. Ni, W. Ren, S. Haussener and X. Hu, Nat. Catal., 2022, 5, 268–276 CrossRef CAS
- H. Li, H. Li, P. Wei, Y. Wang, Y. Zang, D. Gao, G. Wang and X. Bao, Energy Environ. Sci., 2023, 16, 1502–1510 RSC
- L. Li, Z. Liu, X. Yu and M. Zhong, Angew. Chem., Int. Ed., 2023, 62, e202300226 CrossRef CAS PubMed
- M. Fan, J. E. Huang, R. K. Miao, Y. Mao, P. Ou, F. Li, X.-Y. Li, Y. Cao, Z. Zhang, J. Zhang, Y. Yan, A. Ozden, W. Ni, Y. Wang, Y. Zhao, Z. Chen, B. Khatir, C. P. O’Brien, Y. Xu, Y. C. Xiao, G. I. N. Waterhouse, K. Golovin, Z. Wang, E. H. Sargent and D. Sinton, Nat. Catal., 2023, 6, 763–772 CrossRef CAS
- M. C. O. Monteiro, F. Dattila, N. Lopez and M. T. M. Koper, J. Am. Chem. Soc., 2022, 144, 1589–1602 CrossRef CAS PubMed
- Z. Liu, T. Yan, H. Shi, H. Pan, Y. Cheng and P. Kang, ACS Appl. Mater. Interfaces, 2022, 14, 7900–7908 CrossRef CAS PubMed
- X. Li, P. Zhang, L. Zhang, G. Zhang, H. Gao, Z. Pang, J. Yu, C. Pei, T. Wang and J. Gong, Chem. Sci., 2023, 14, 5602–5607 RSC
- Y. Zhao, L. Hao, A. Ozden, S. Liu, R. K. Miao, P. Ou, T. Alkayyali, S. Zhang, J. Ning, Y. Liang, Y. Xu, M. Fan, Y. Chen, J. E. Huang, K. Xie, J. Zhang, C. P. O'Brien, F. Li, E. H. Sargent and D. Sinton, Nat. Synth., 2023, 2, 403–412 CrossRef
- Z. Wang, P. Hou, Y. Wang, X. Xiang and P. Kang, ACS Sustainable Chem. Eng., 2019, 7, 6106–6112 CrossRef CAS
- H.-G. Qin, F.-Z. Li, Y.-F. Du, L.-F. Yang, H. Wang, Y.-Y. Bai, M. Lin and J. Gu, ACS Catal., 2022, 13, 916–926 CrossRef
- N. Pismenskaya, E. Laktionov, V. Nikonenko, A. El Attar, B. Auclair and G. Pourcelly, J. Membr. Sci., 2001, 181, 185–197 CrossRef CAS
- Y. T. Yuen, P. N. Sharratt and B. Jie, Environ. Sci. Pollut. Res. Int., 2016, 23, 22309–22330 CrossRef CAS PubMed
- B. C. Kash, R. J. Gomes and C. V. Amanchukwu, J. Phys. Chem. Lett., 2023, 14, 920–926 CrossRef CAS PubMed
- J. A. Rabinowitz and M. W. Kanan, Nat. Commun., 2020, 11, 5231 CrossRef CAS PubMed
- Q. Dang, H. Lin, Z. Fan, L. Ma, Q. Shao, Y. Ji, F. Zheng, S. Geng, S. Z. Yang, N. Kong, W. Zhu, Y. Li, F. Liao, X. Huang and M. Shao, Nat. Commun., 2021, 12, 6007 CrossRef CAS PubMed
- Q. Li, J. Li, J. Xu, N. Zhang, Y. Li, L. Liu, D. Pan, Z. Wang and F. L. Deepak, ACS Appl. Energy Mater., 2020, 3, 3736–3744 CrossRef CAS
- J. Kim, Y. Hong, K. Lee and J. Y. Kim, Adv. Energy Mater., 2020, 10, 2002049 CrossRef CAS
- S. Kattel, B. Yan, Y. Yang, J. G. Chen and P. Liu, J. Am. Chem. Soc., 2016, 138, 12440–12450 CrossRef CAS PubMed
- Z. Pan, K. Wang, K. Ye, Y. Wang, H.-Y. Su, B. Hu, J. Xiao, T. Yu, Y. Wang and S. Song, ACS Catal., 2020, 10, 3871–3880 CrossRef CAS
- Y. Wang, P. Han, X. Lv, L. Zhang and G. Zheng, Joule, 2018, 2, 2551–2582 CrossRef CAS
- K. Larmier, W. C. Liao, S. Tada, E. Lam, R. Verel, A. Bansode, A. Urakawa, A. Comas-Vives and C. Coperet, Angew. Chem., Int. Ed., 2017, 56, 2318–2323 CrossRef CAS PubMed
- R. M. Arán-Ais, F. Scholten, S. Kunze, R. Rizo and B. Roldan Cuenya, Nat. Energy, 2020, 5, 317–325 CrossRef
- Y. Chen, J. A. Wrubel, A. E. Vise, F. Intia, S. Harshberger, E. Klein, W. A. Smith, Z. Ma, T. G. Deutsch and K. C. Neyerlin, Chem. Catal., 2022, 2, 400–421 CrossRef CAS
- D. Hursan, A. A. Samu, L. Janovak, K. Artyushkova, T. Asset, P. Atanassov and C. Janaky, Joule, 2019, 3, 1719–1733 CrossRef CAS PubMed
- X. Zhang, R. Sa, F. Zhou, Y. Rui, R. Liu, Z. Wen and R. Wang, CCS Chem., 2021, 3, 199–207 CrossRef CAS
- Z. Yan and T. Wu, Int. J. Mol. Sci., 2022, 23, 14381 CrossRef CAS PubMed
- H. Shang, D. Kim, S. K. Wallentine, M. Kim, D. M. Hofmann, R. Dasgupta, C. J. Murphy, A. Asthagiri and L. R. Baker, Chem. Sci., 2021, 12, 9146–9152 RSC
- S. Y. Lee, S. Y. Chae, H. Jung, C. W. Lee, D. L. T. Nguyen, H.-S. Oh, B. K. Min and Y. J. Hwang, J. Mater. Chem. A, 2020, 8, 6210–6218 RSC
- Y. Zhou, F. Che, M. Liu, C. Zou, Z. Liang, P. De Luna, H. Yuan, J. Li, Z. Wang, H. Xie, H. Li, P. Chen, E. Bladt, R. Quintero-Bermudez, T. K. Sham, S. Bals, J. Hofkens, D. Sinton, G. Chen and E. H. Sargent, Nat. Chem., 2018, 10, 974–980 CrossRef CAS PubMed
- X. Wang, Z. Wang, F. P. García de Arquer, C.-T. Dinh, A. Ozden, Y. C. Li, D.-H. Nam, J. Li, Y.-S. Liu, J. Wicks, Z. Chen, M. Chi, B. Chen, Y. Wang, J. Tam, J. Y. Howe, A. Proppe, P. Todorović, F. Li, T.-T. Zhuang, C. M. Gabardo, A. R. Kirmani, C. McCallum, S.-F. Hung, Y. Lum, M. Luo, Y. Min, A. Xu, C. P. O'Brien, B. Stephen, B. Sun, A. H. Ip, L. J. Richter, S. O. Kelley, D. Sinton and E. H. Sargent, Nat. Energy, 2020, 5, 478–486 CrossRef CAS
- M. Akira and H. Yoshio, Bull. Chem. Soc. Jpn., 1991, 64, 123–127 CrossRef
- J. Resasco, L. D. Chen, E. Clark, C. Tsai, C. Hahn, T. F. Jaramillo, K. Chan and A. T. Bell, J. Am. Chem. Soc., 2017, 139, 11277–11287 CrossRef CAS PubMed
- S. Ringe, E. L. Clark, J. Resasco, A. Walton, B. Seger, A. T. Bell and K. Chan, Energy Environ. Sci., 2019, 12, 3001–3014 RSC
- Z. Xu, M. Sun, Z. Zhang, Y. Xie, H. Hou, X. Ji, T. Liu, B. Huang and Y. Wang, ChemCatChem, 2022, 14, e202200052 CrossRef CAS
- M. C. O. Monteiro, F. Dattila, B. Hagedoorn, R. García-Muelas, N. López and M. T. M. Koper, Nat. Catal., 2021, 4, 654–662 CrossRef CAS
- M. Zhao, H. Tang, Q. Yang, Y. Gu, H. Zhu, S. Yan and Z. Zou, ACS Appl. Mater. Interfaces, 2020, 12, 4565–4571 CrossRef CAS PubMed
- C. Lu, Y. Su, J. Zhu, J. Sun and X. Zhuang, Chem. Commun., 2023, 59, 6827–6836 RSC
- H. Xiao, T. Cheng, W. A. Goddard 3rd and R. Sundararaman, J. Am. Chem. Soc., 2016, 138, 483–486 CrossRef CAS PubMed
- I. Hamidah, A. Solehudin, A. Hamdani, L. Hasanah, K. Khairurrijal, T. Kurniawan, R. Mamat, R. Maryanti, A. B. D. Nandiyanto and B. Hammouti, Alexandria Eng. J., 2021, 60, 2235–2243 CrossRef
- J. Vavra, T. H. Shen, D. Stoian, V. Tileli and R. Buonsanti, Angew. Chem., Int. Ed., 2021, 60, 1347–1354 CrossRef CAS PubMed
- G. Jiang, D. Han, Z. Han, J. Gao, X. Wang, Z. Weng and Q.-H. Yang, Trans. Tianjin Univ., 2022, 28, 265–291 CrossRef CAS
- Y. Hori, H. Konishi, T. Futamura, A. Murata, O. Koga, H. Sakurai and K. Oguma, Electrochim. Acta, 2005, 50, 5354–5369 CrossRef CAS
- D. Kim, C. S. Kley, Y. Li and P. Yang, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 10560–10565 CrossRef CAS PubMed
- N. Hodnik, G. Dehm and K. J. Mayrhofer, Acc. Chem. Res., 2016, 49, 2015–2022 CrossRef CAS PubMed
- S. Seethammaraju and M. Rangarajan, IOP Conf. Ser.: Mater. Sci. Eng., 2019, 577, 012188 CAS
- I. V. Makarova, D. S. Kharitonov, I. B. Dobryden’ and A. A. Chernik, Russ. J. Appl. Chem., 2018, 91, 1441–1450 CrossRef CAS
- D. M. Weekes, D. A. Salvatore, A. Reyes, A. Huang and C. P. Berlinguette, Acc. Chem. Res., 2018, 51, 910–918 CrossRef CAS PubMed
- D. Higgins, C. Hahn, C. Xiang, T. F. Jaramillo and A. Z. Weber, ACS Energy Lett., 2018, 4, 317–324 CrossRef
- B. Pan, J. Fan, J. Zhang, Y. Luo, C. Shen, C. Wang, Y. Wang and Y. Li, ACS Energy Lett., 2022, 7, 4224–4231 CrossRef CAS
- M. Carmo, D. L. Fritz, J. Mergel and D. Stolten, Int. J. Hydrogen Energy, 2013, 38, 4901–4934 CrossRef CAS
- S. Shiva Kumar and V. Himabindu, Mater. Sci. Energy Technol., 2019, 2, 442–454 Search PubMed
- J. Shan, C. Guo, Y. Zhu, S. Chen, L. Song, M. Jaroniec, Y. Zheng and S.-Z. Qiao, Chem, 2019, 5, 445–459 CAS
- L. Li, P. Wang, Q. Shao and X. Huang, Adv. Mater., 2021, 33, e2004243 CrossRef PubMed
- Y. Lin, Y. Dong, X. Wang and L. Chen, Adv. Mater., 2023, 35, e2210565 CrossRef PubMed
- R. Ge, L. Li, J. Su, Y. Lin, Z. Tian and L. Chen, Adv. Energy Mater., 2019, 9, 1901313 CrossRef
- J. Gao, C. Q. Xu, S. F. Hung, W. Liu, W. Cai, Z. Zeng, C. Jia, H. M. Chen, H. Xiao, J. Li, Y. Huang and B. Liu, J. Am. Chem. Soc., 2019, 141, 3014–3023 CrossRef CAS PubMed
- J. Yu, M. Sun, J. Wang, Y. Wang, Y. Li, P. Lu, Y. Ma, J. Zhou, W. Chen, X. Zhou, C.-S. Lee, B. Huang and Z. Fan, Cell Rep. Phys. Sci., 2023, 4, 101366 CrossRef CAS
- X. Deng, D. Alfonso, T.-D. Nguyen-Phan and D. R. Kauffman, ACS Catal., 2022, 12, 5921–5929 CrossRef CAS
- S. Khan, J. Hwang, Y.-S. Horn and K. K. Varanasi, Cell Rep. Phys. Sci., 2021, 2, 100318 CrossRef CAS
- J. J. Lv, M. Jouny, W. Luc, W. Zhu, J. J. Zhu and F. Jiao, Adv. Mater., 2018, 30, e1803111 CrossRef PubMed
- S. Yu, S. Louisia and P. Yang, JACS Au, 2022, 2, 562–572 CrossRef CAS PubMed
- X. Zi, Y. Zhou, L. Zhu, Q. Chen, Y. Tan, X. Wang, M. Sayed, E. Pensa, R. A. Geioushy, K. Liu, J. Fu, E. Cortes and M. Liu, Angew. Chem., Int. Ed., 2023, 62, e202309351 CrossRef CAS PubMed
- W. Zhang, S. Yang, M. Jiang, Y. Hu, C. Hu, X. Zhang and Z. Jin, Nano Lett., 2021, 21, 2650–2657 CrossRef CAS PubMed
- H. Seong, M. Choi, S. Park, H.-w. Kim, J. Kim, W. Kim, J. S. Yoo and D. Lee, ACS Energy Lett., 2022, 7, 4177–4184 CrossRef CAS
- H. Zhang, C. Xu, X. Zhan, Y. Yu, K. Zhang, Q. Luo, S. Gao, J. Yang and Y. Xie, Nat. Commun., 2022, 13, 6029 CrossRef CAS PubMed
- Z. Yan, J. L. Hitt, Z. Zeng, M. A. Hickner and T. E. Mallouk, Nat. Chem., 2021, 13, 33–40 CrossRef CAS PubMed
- C.-T. Dinh, F. P. García de Arquer, D. Sinton and E. H. Sargent, ACS Energy Lett., 2018, 3, 2835–2840 CrossRef CAS
- B. Endrődi, E. Kecsenovity, A. Samu, T. Halmágyi, S. Rojas-Carbonell, L. Wang, Y. Yan and C. Janáky, Energy Environ. Sci., 2020, 13, 4098–4105 RSC
- C. Cai, B. Liu, K. Liu, P. Li, J. Fu, Y. Wang, W. Li, C. Tian, Y. Kang, A. Stefancu, H. Li, C. W. Kao, T. S. Chan, Z. Lin, L. Chai, E. Cortes and M. Liu, Angew. Chem., Int. Ed., 2022, 61, e202212640 CrossRef CAS PubMed
- J. Lee, J. Lim, C.-W. Roh, H. S. Whang and H. Lee, J. CO2 Util., 2019, 31, 244–250 CrossRef CAS
- J. P. Edwards, Y. Xu, C. M. Gabardo, C.-T. Dinh, J. Li, Z. Qi, A. Ozden, E. H. Sargent and D. Sinton, Appl. Energy, 2020, 261, 114305 CrossRef CAS
- Q. Wang, K. Liu, J. Fu, C. Cai, H. Li, Y. Long, S. Chen, B. Liu, H. Li, W. Li, X. Qiu, N. Zhang, J. Hu, H. Pan and M. Liu, Angew. Chem., Int. Ed., 2021, 60, 25241–25245 CrossRef CAS PubMed
- X. Zhang, J. Li, Y. Y. Li, Y. Jung, Y. Kuang, G. Zhu, Y. Liang and H. Dai, J. Am. Chem. Soc., 2021, 143, 3245–3255 CrossRef CAS PubMed
- F. Li, A. Thevenon, A. Rosas-Hernandez, Z. Wang, Y. Li, C. M. Gabardo, A. Ozden, C. T. Dinh, J. Li, Y. Wang, J. P. Edwards, Y. Xu, C. McCallum, L. Tao, Z. Q. Liang, M. Luo, X. Wang, H. Li, C. P. O'Brien, C. S. Tan, D. H. Nam, R. Quintero-Bermudez, T. T. Zhuang, Y. C. Li, Z. Han, R. D. Britt, D. Sinton, T. Agapie, J. C. Peters and E. H. Sargent, Nature, 2020, 577, 509–513 CrossRef CAS PubMed
- H. Zhang, Y. Qiao, Y. Wang, Y. Zheng and H. Huang, Sustainable Energy Fuels, 2022, 6, 4860–4865 RSC
- H. Li, T. Liu, P. Wei, L. Lin, D. Gao, G. Wang and X. Bao, Angew. Chem., Int. Ed., 2021, 60, 14329–14333 CrossRef CAS PubMed
- W. Ma, S. Xie, T. Liu, Q. Fan, J. Ye, F. Sun, Z. Jiang, Q. Zhang, J. Cheng and Y. Wang, Nat. Catal., 2020, 3, 478–487 CrossRef CAS
- X. Yan, C. Chen, Y. Wu, S. Liu, Y. Chen, R. Feng, J. Zhang and B. Han, Chem. Sci., 2021, 12, 6638–6645 RSC
- C. Reller, R. Krause, E. Volkova, B. Schmid, S. Neubauer, A. Rucki, M. Schuster and G. Schmid, Adv. Energy Mater., 2017, 7 CAS
- W. Liu, P. Zhai, A. Li, B. Wei, K. Si, Y. Wei, X. Wang, G. Zhu, Q. Chen, X. Gu, R. Zhang, W. Zhou and Y. Gong, Nat. Commun., 2022, 13, 1877 CrossRef CAS PubMed
- M. Li, Y. Ma, J. Chen, R. Lawrence, W. Luo, M. Sacchi, W. Jiang and J. Yang, Angew. Chem., Int. Ed., 2021, 60, 11487–11493 CrossRef CAS PubMed
- S. Gao, Z. Sun, W. Liu, X. Jiao, X. Zu, Q. Hu, Y. Sun, T. Yao, W. Zhang, S. Wei and Y. Xie, Nat. Commun., 2017, 8, 14503 CrossRef CAS PubMed
- M. Zhang, W. Wei, S. Zhou, D.-D. Ma, A. Cao, X.-T. Wu and Q.-L. Zhu, Energy Environ. Sci., 2021, 14, 4998–5008 RSC
- C. Sun, Y. Hou, N. Lüdi, H. Hu, M. de Jesús Gálvez-Vázquez, M. Liechti, Y. Kong, M. Liu, R. Erni, A. V. Rudnev and P. Broekmann, J. Catal., 2022, 407, 198–205 CrossRef CAS
- Z. Z. Wu, X. L. Zhang, Z. Z. Niu, F. Y. Gao, P. P. Yang, L. P. Chi, L. Shi, W. S. Wei, R. Liu, Z. Chen, S. Hu, X. Zheng and M. R. Gao, J. Am. Chem. Soc., 2022, 144, 259–269 CrossRef CAS PubMed
- J. Gao, H. Zhang, X. Guo, J. Luo, S. M. Zakeeruddin, D. Ren and M. Gratzel, J. Am. Chem. Soc., 2019, 141, 18704–18714 CrossRef CAS PubMed
- L. Fan, C.-Y. Liu, P. Zhu, C. Xia, X. Zhang, Z.-Y. Wu, Y. Lu, T. P. Senftle and H. Wang, Joule, 2022, 6, 205–220 CrossRef CAS
- H. Xie, T. Zhang, R. Xie, Z. Hou, X. Ji, Y. Pang, S. Chen, M. M. Titirici, H. Weng and G. Chai, Adv. Mater., 2021, 33, e2008373 CrossRef PubMed
- Y. E. Kim, Y. N. Ko, B.-S. An, J. Hong, Y. E. Jeon, H. J. Kim, S. Lee, J. Lee and W. Lee, ACS Energy Lett., 2023, 8, 3288–3296 CrossRef CAS
- Y. Gang, B. Li, S. Fang, J. Pellessier, L. Fang, F. Pan, Z. Du, Y. Hang Hu, T. Li, G. Wang and Y. Li, Chem. Eng. J., 2023, 453, 139712 CrossRef CAS
- C. Peng, S. Yang, G. Luo, S. Yan, M. Shakouri, J. Zhang, Y. Chen, Z. Wang, W. Wei, T. K. Sham and G. Zheng, Small, 2023, 19, e2207374 CrossRef PubMed
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |