
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsights into the nucleation and growth of BiOCl nanoparticles by in situ X-ray pair distribution function analysis and in situ liquid cell TEM†
Matthew N.
Gordon‡§
 a,
Laura S.
Junkers‡
b,
Jack S.
Googasian
a,
Jette K.
Mathiesen¶
b,
Xun
Zhan||
c,
David Gene
Morgan
c,
Kirsten M. Ø.
Jensen
b and
Sara E.
Skrabalak
*a
a,
Laura S.
Junkers‡
b,
Jack S.
Googasian
a,
Jette K.
Mathiesen¶
b,
Xun
Zhan||
c,
David Gene
Morgan
c,
Kirsten M. Ø.
Jensen
b and
Sara E.
Skrabalak
*a
aDepartment of Chemistry, Indiana University, Bloomington, Indiana 47405, USA. E-mail: sskrabal@indiana.edu
bDepartment of Chemistry and Nanoscience Center, University of Copenhagen, 2100 Copenhagen Ø, Denmark
cElectron Microscopy Center, Indiana University, Bloomington, Indiana 47405, USA
First published on 19th July 2024
Abstract
The synthesis of bismuth oxyhalides as defined nanostructures is hindered by their fast nucleation and growth in aqueous solutions. Using our recently developed single-source precursor, the formation of bismuth oxychloride in such solutions can be slowed significantly. As reported herein, this advance enables BiOCl formation to be investigated by in situ X-ray total scattering and in situ liquid cell transmission electron microscopy. In situ pair distribution function analysis of X-ray total scattering data reveals the local order of atomic structures throughout the synthesis, while in situ liquid cell transmission electron microscopy allows for tracking the growth of individual nanoparticles. Through this work, the precursor complex is shown to give rise to BiOCl upon heating in solution without the observation of structurally distinct intermediates. The emerging nanoparticles have a widened interlayer spacing, which moderately decreases as the particles grow. Mechanistic insights into the formation of bismuth oxyhalide nanoparticles, including the absence of distinct intermediates within the available time resolution, will help facilitate future design of controlled BiOX nanostructures.
Introduction
Solar water splitting is a useful method to store solar energy in chemical bonds.1–10 After a semiconductor absorbs sunlight of energy greater than its bandgap, photogenerated holes and electrons can migrate to the surface of the semiconductor to oxidize and reduce water to O2 and H2, respectively.11–14 The produced hydrogen can then be stored until needed as a fuel.3,5,9,15–20 Tapping into solar energy in such a carbon neutral way makes the use of photocatalytic systems a promising route to replace non-renewable resources like fossil fuels.A photocatalytic material with an appropriate band gap is key for the success of solar water splitting. Heteroanionic materials like oxynitrides, oxyhalides, and oxysulfides commonly have smaller band gaps than their oxide counterparts, as the less electronegative atoms raise the valence band minimum.21–25 This smaller band gap allows for a wider spectrum of wavelengths of light to be used to initiate photocatalysis.
Bismuth oxyhalides of the form BiOX (X = Cl, Br, I) have a band gap that is tunable from only UV absorbing when X = Cl to visible absorbing when X = Br or I.26–29 Their structure furthermore minimizes electron–hole recombination.30–32 BiOX materials are comprised of positively charged [Bi2O2]2+ layers and negatively charged double halide layers, e.g., [Cl]− for BiOCl (Fig. S1†).33 These charged layers give rise to a static internal electric field which, in turn, spatially separates photogenerated holes and electrons to the negatively and positively charged layers, respectively.30–32 This spatial separation minimizes recombination and facilitates an efficient use of charge carriers in photocatalytic redox reactions.
Bismuth oxyhalides can be readily obtained via hydrolysis-based precipitation reactions occurring when bismuth and halide ions are simultaneously present in aqueous solution.27,28,34,35 The general insolubility of Bi3+ compounds in aqueous media (e.g., BiOCl Ksp,298 K = 1.8 × 10−31)36 causes these precipitations to occur so rapidly that there is not enough time for shape-controlling agents to affect the final morphology. Precipitation syntheses therefore often result in large aggregates of polycrystalline particles.30 The resulting speed of BiOCl precipitation furthermore impeded detailed experimental studies on the involved formation mechanism.
Mechanistic understanding can facilitate the design of new syntheses with desired outcomes. Classical nucleation theories, such as the LaMer model, are typical starting points for mechanistic explanations of nanoparticle formation in solution.37,38 The chemistry involved in nucleation and growth processes is, however, vastly underrepresented in these theories.39 One assumption in LaMer's approach is that the resulting nanoparticles are spherical (isotropic) and of constant shape.37 Additionally, classical nucleation theory assumes a single interfacial energy term for the nanoparticle.37
Both assumptions are inconsistent for bismuth oxyhalide systems due to their layered crystal structures. The surface energies of the tetragonal crystal's facets differ vastly: γ{001} ≪ γ{010} < γ{110} (0.026, 0.532, 1.426 J m−2, respectively).40 To minimize the overall surface energy,41 BiOCl particles preferentially grow in-plane, giving rise to a high degree of anisotropy. The resulting nanoplates have highly expressed {001} facets and smaller {010} facets at their sides.
Thus, more complex mechanistic models are needed to account for the intricacies of BiOCl formation. Despite this, the current understanding about the BiOCl formation process is limited. While Bi3+ is known to hydrolyze rapidly in the absence of strongly acidic conditions,42 the structure of the hydrolyzation product involved in BiOCl formation is not agreed upon. One hypothesis assumes an initial hydrolyzation step to yield H+ and covalently bound [Bi2O2]2+.30 By coupling of the [Bi2O2]2+ cations to Cl− anions via electrostatic interactions, individual [Cl–Bi–O–Bi–Cl] nuclei form and subsequently grow in-plane to minimize their overall surface energy. Stacking of these layers in the c direction through van der Waals interactions then gives rise to BiOCl.30,35 This hypothesis is rooted in the layered BiOCl structure and the Bi3+ hydrolysis mechanism proposed by Garnér and Sillén.43 They suggested the formation of covalently bound [BiaOa]a+ layers upon hydrolysis through ‘polymeric’-like growth in 2D. More extensive studies did, however, show that hydrolyzed Bi3+ in solution takes on the form of discrete cluster species, among which the hexamer [Bi6(OH)12]6+ is particularly stable at the high Bi3+ concentrations typically applied in BiOCl syntheses.42,44 Whether this model holds true in the presence of Cl− anions is, however, not clear. Until now, the rapid nature of BiOCl formation prevented direct experimental insight into the influence of Cl− on the hydrolyzation process of Bi3+ and thereby into the mechanism yielding BiOCl. In this work, we take first steps towards such direct evidence aiming to experimentally test the proposed models.
Single-source precursors have been demonstrated to supply the necessary atoms for a chemical process in a single molecule.45,46 Recently, we developed single-source precursors for bismuth oxyhalides which include BiClLac (Fig. 1a), a metal–organic complex composed of a central Bi3+ bound with four bidentate 3-chlorolactate ligands (ClLac).47Via a proposed nucleophilic substitution mechanism, water replaces the chlorine on the ligand.48 Releasing this chloride ion into solution initiates precipitation with Bi3+ as BiOCl. Temperature correlates with the ligand's reactivity and, thereby, provides a way to tune the generation rate of chloride ions in situ, and thus the BiOCl formation rate.
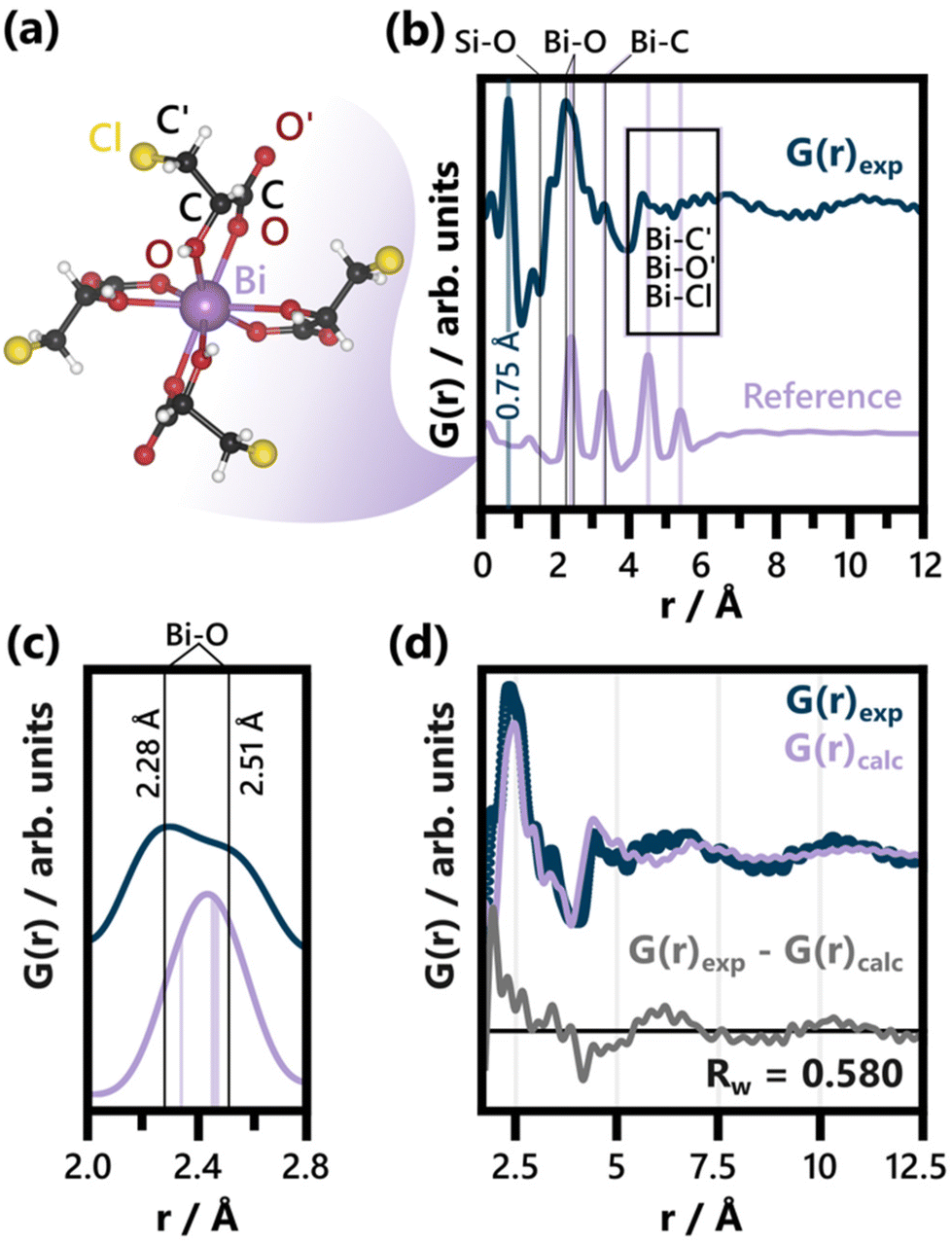 | ||
| Fig. 1 (a) BiClLac complex reference structure as obtained from a cut-out of the full crystal structure (CCDC Deposition 2211246, Refcode AFADEU).47 The respective xyz coordinates are listed in Table S1.† (b) Comparison of PDF of the precursor solution and the reference PDF calculated based on the same parameters. Characteristic peaks are assigned to the respective interatomic distances and the unphysical peak at 0.75 Å is highlighted. A detailed discussion of the background subtraction approach regarding this peak is given in the ESI.† The experimental PDF has an acquisition time of 10 min. (c) Comparison of the experimental PDF and the reference PDF from (b) focusing on the Bi–O peak splitting. The Bi–O distances expected for the BiClLac reference structure (2.34 Å and between 2.45 Å and 2.47 Å)47 are indicated in purple, while estimates for the experimentally observed Bi–O distances in solution are given as black lines. (d) Refinement of the BiClLac complex to the precursor PDF from (b), including a refinement of the chlorine Biso values. A fit range of 1.7 to 20 Å was used. The results of this refinement are listed in Table S2.† | ||
The significantly slowed BiOCl formation provided by these single-source precursors allows the formation and growth processes to be probed. Such studies have not been possible before now. Here, two complementary techniques—Pair Distribution Function (PDF) analysis of in situ X-ray total scattering (TS) data and in situ liquid cell transmission electron microscopy (LCTEM)—are used to provide insights into the early stages of BiOCl formation from the atomic scale to the nanoscale.
PDF analysis of in situ TS data monitors the evolution of the local atomic structure throughout the synthesis. This methodology has been widely applied to study the formation of, among others, metal and metal oxide nanoparticles49,50 as well as metal organic frameworks.51 The reduced pair distribution function, G(r), is obtained from TS data via a Fourier transform and both Bragg and diffuse scattering are included in the analysis.52,53In situ studies based on PDF analysis can therefore provide information on the atomic scale structure of species present both prior to and after crystallization. The PDF is, in essence, a histogram of interatomic distances, r. The intensity of a PDF peak is governed by both the atomic number of the involved atoms and the frequency of the respective distance. Thus, PDF makes it possible to extract qualitative information through model free analysis. If more intricate insights are sought, structural models can be refined to the data.52 By applying this technique in situ, precursor structures, amorphous intermediates, and the local structure of emerging particles can be studied for a given synthesis.
Complementarily to in situ PDF, in situ LCTEM provides the opportunity to visually follow the early stages of nanoparticle formation.54–59 LCTEM involves encasing a small volume of solution in a chamber which is then imaged using an electron beam. Here, an aqueous solution of BiClLac was sandwiched between two chips containing silicon nitride membrane windows (Fig. S2†). One of the liquid cell chips contains a resistive coil allowing for in situ heating of the reaction solution. This setup was used to sequentially image the solution with electron microscopy as the solution was heated. Importantly, the electron beam is not an innocuous imaging tool but inherently influences the studied system, so beam effects limit in situ LCTEM experiments. The electron beam has the ability to reduce metal cations, generate radicals from water, and thereby degrade nanostructures.60–65 Such electron beam-induced artifacts are reduced in this study by minimizing the electron dose to replicate ex situ conditions (i.e., no electron beam) as closely as possible.
Previously, in situ TEM experiments have shown the degradation of BiOCl nanocrystals under electron beam irradiation by imaging previously synthesized nanoparticles.66 Those were, however, not liquid cell experiments, nor did they probe the formation process. These differences are important, as particle formation is not simply the reverse of particle degradation. Other TEM experiments have followed the growth of metallic Bi on BiOCl67 and explored metallic Bi growth in solution and ex situ,68,69 but the literature still lacks the foundational understanding and imaging of BiOCl formation and structural evolution.
Experimental
Materials
Bismuth nitrate pentahydrate (98%) and (±)-3-chloropropane-1,2-diol (98%) were purchased from Alfa Aesar. Nitric acid (ACS Plus) and ethyl acetate (ACS) were purchased from Fisher. Chloroform (ACS) was obtained from Macron. NaCl (≥99.0%) was purchased from VWR. MgSO4 (≥98.0%, anhydrous) was obtained from EMD Millipore. All chemicals were used as received. Milli-Q (18.2 MΩ·cm at 25 °C) purified water was used for all experiments.Synthesis of 3-chlorolactic acid
The synthesis of 3-chlorolactic acid has been fully described in our previous publication.70 Briefly, (±)-3-chloropropane-1,2-diol was added to a round bottom flask. Concentrated nitric acid was added with stirring. (CAUTION: handling large amounts of concentrated acids is dangerous, and the reaction produces NOx gases. For these reasons, it is critical to perform these steps in a fume hood.) A condenser was attached, and the flask was heated slowly with stirring to 80 °C until a vigorous reaction started. The reaction was kept at 80 °C for 30 min and then raised to 100 °C for another 30 min. The solution was cooled and then neutralized. The product was extracted using ethyl acetate, washed with brine, dried over MgSO4, and concentrated. Chloroform was added to the crude product and kept at −20 °C overnight to produce 3-chlorolactic acid (ClLac). Fourier-transform infrared spectroscopy (FTIR), 1H nuclear magnetic resonance spectroscopy (NMR), high-resolution mass spectrometry (HRMS), and melting point match previous results.70Synthesis of aqueous BiClLac
The synthesis of BiClLac here is based on our previous publication.47 For in situ LCTEM experiments in TEM mode, 3-chlorolactic acid (0.498 g, 4.00 mmol, 4.00 equiv.) was added to a 10 mL volumetric flask and dissolved by adding a portion of water. Bismuth nitrate pentahydrate (0.485 g, 1.00 mmol, 1.00 equiv.) was added and mixed with vortexing and sonication. The flask was filled to the mark with water and thoroughly mixed for a final concentration of 0.100 M BiClLac. Haziness was observed when the ligand![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :bismuth nitrate pentahydrate ratio was less than 4:1, so any haziness was treated by adding slight excess of 3-chlorolactic acid and mixing. The solution could be further purified by filtering to remove any unreacted bismuth compounds which could cause heterogeneous nucleation. 1H and 13C NMR and FTIR spectra match previous results.47 The pH of this solution was 0.89. For in situ LCTEM experiments in STEM mode, the precursor concentration was decreased to 3.33 mM BiClLac. For in situ PDF experiments, the precursor concentration was 0.2 M BiClLac.
:bismuth nitrate pentahydrate ratio was less than 4:1, so any haziness was treated by adding slight excess of 3-chlorolactic acid and mixing. The solution could be further purified by filtering to remove any unreacted bismuth compounds which could cause heterogeneous nucleation. 1H and 13C NMR and FTIR spectra match previous results.47 The pH of this solution was 0.89. For in situ LCTEM experiments in STEM mode, the precursor concentration was decreased to 3.33 mM BiClLac. For in situ PDF experiments, the precursor concentration was 0.2 M BiClLac.
In situ and ex situ X-ray total scattering
The in situ X-ray total scattering (TS) experiments were conducted at beamline P21.1, PETRAIII, Deutsches Electronen-Synchrotron (DESY) in Germany, in a custom-made setup for in situ studies similar to one previously used by Mathiesen et al.49 The setup is composed of an aluminum block with cut out slots for NMR tubes with the precursor solutions. The precursor solution was injected into a thin NMR tube (Wilmad) with a 3 mm diameter and a wall thickness of 0.29 mm, ensuring a high transmission of X-rays. The NMR tubes were positioned vertically in the in situ setup and fixed by springs. When the experiments were initiated, the entire aluminum block was heated to 90 °C at a heating rate of 3 °C min−1. The temperature was monitored using a thermocouple, which was placed in an adjacent NMR tube filled with sand. TS data were collected in transmission geometry using a PerkinElmer XRD1621 area detector with a pixel size of 200 × 200 μm in the RA-PDF setup mode.71 A wavelength of λ = 0.122 Å and a sample-to-detector distance of ca. 400 mm were used. The latter was calibrated using a LaB6 standard and Fit2D.72–74 The collected 2D data had a time resolution of 10 s per frame and were integrated using Dioptas.75X-ray powder diffraction
Ex situ X-ray powder diffraction (PXRD) data of the reaction product was collected at room temperature right after an in situ TS experiment conducted by first heating to 70 °C and subsequently heated up to 90 °C. The sample-to-detector distance was changed to ca. 1400 mm and calibrated with Fit2D72–74 using a LaB6 standard. Apart from that, setup and conditions remained the same.PDF analysis
The pair distribution functions (PDFs) were obtained with the ad hoc approach of PDFgetX3,76 using the following parameters: Qmax,inst = 23.7 Å−1, Qmin = 1.5 Å−1, Qmax = 18 Å−1, rpoly = 0.9 Å. In line with the stoichiometry of the precursor complex single crystal, Bi(C3H4O3Cl)3(C3H5O3Cl), a composition estimate of BiC12H17O12Cl4 was used when calculating the PDFs.47 Reference TS data acquired from a 0.8 M aqueous 3-chlorolactic acid solution in the in situ setup was subtracted as background: for the in situ data, TS data measured during the heating of this 3-chlorolactic acid solution from RT to 90 °C functioned as the background. For ex situ data, TS data of the empty NMR tube was additionally used for a separate subtraction of the glass and the RT solution background. Qdamp and Qbroad were determined based on the PDF of a LaB6 standard. The PDFs of crystalline BiOCl particles were analyzed using real space Rietveld refinements in PDFgui.77 The precursor structure did not exhibit long-range order and was therefore modeled using the DebyePDFCalculator in DiffPy-CMI.78 The sequential refinement of the in situ PDF data was conducted backwards, i.e., starting from the last frame (76.66 min of heating) of the data set. Nyquist sampling was applied for all the sequentially refined data, except the frame used for the initial refinement (76.66 min of heating). The individual modeling parameters and parameter restrictions of each refinement are listed in the ESI.†PXRD data analysis
The PXRD data was fitted using Topas Academic79 and the Rietveld method.80 The instrumental resolution function was determined based on a LaB6 standard. Apart from scaling factors and lattice parameters, the peak shape parameters of the BiOCl phase were refined using the Thompson–Cox–Hastings Pseudo Voigt function (U, V, W, X, Y, and Z refined). The preferred orientation of the BiOCl particles was accounted for using the March–Dollase approach.81 Peak asymmetry was refined with the Simple Axial Model for both phases. Background scattering caused by solvent and glass contributions was fitted with a polynomial function. VESTA82 was used to visualize the structural models used for both reciprocal and real space refinements.In situ liquid cell transmission electron microscopy
LCTEM studies were performed using a Protochips Poseidon Select liquid cell holder with a PJAS tip. The liquid cell was assembled by stacking a perfluoroelastomer gasket, small e-chip (part EPB-52DNF, 550 μm × 20 μm × 50 nm silicon nitride window with built-in 150 nm spacer between windows), sample solution droplet (<1 μL), liquid heating large e-chip (part EHT-45ZZ, 550 μm × 50 μm × 50 nm silicon nitride window), and lid onto the holder tip and sealing with screws (Fig. S2†). The small and large e-chip windows were arranged perpendicular to each other (crossed) for an overall viewable volume of 50 μm × 20 μm × 150 nm (ca. 0.15 pL). Prior to assembly, the liquid cell e-chips were cleaned for 2 min in acetone followed by 2 min in methanol to remove the photoresist layer, followed by plasma cleaning for 2 min on high (30 W; Harrick Plasma PDC-001) to increase the hydrophilicity of the chips. A leak check was then performed using a Gatan Model 655 Dry Pumping Station. The solution was heated using a resistive coil in the large e-chip and a Keithley 2450 power supply attached to the holder which was controlled by the Poseidon Select software (V10.1.0.0). The chip/solution was heated from room temperature at 5 K min−1 in Movie 1† (TEM) and at 3 K min−1 in Movie 2, S1, and S2† (STEM). The heating program and TEM recording were started simultaneously so time and temperature could be correlated with individual frames.Transmission electron microscopy
Sample analysis by TEM and selected area electron diffraction (SAED) were collected using a JEOL JEM 1400Plus, equipped with a LaB6 source and Gatan OneView CMOS camera, operating at 120 kV. Frames were acquired using the In Situ Acquisition feature in DigitalMicrograph. All alignments, settings, and calibrations were conducted prior to the liquid cell experiments using standard gold nanoparticles or a diffraction grating waffle TEM sample. TEM electron dose rate was estimated by first comparing the total intensities of two sets of images under identical conditions (similar to the experimental conditions), one with a liquid cell with sample solution, and another with nothing in the beam path. This gave an attenuation factor due to the presence of the liquid sample and holder. Total intensities were measured from experimental frames before any nucleation occurred. Using the attenuation factor, the incident dose of the beam was roughly estimated.Scanning transmission electron microscopy
Sample analysis by scanning transmission electron microscopy (STEM) and energy-dispersive X-ray spectroscopy (EDS) were conducted on a JEOL JEM 3200FS (S)TEM, equipped with a thermal field emission gun source and Gatan 4k × 4k UltraScan 4000 CCD camera, operating at 300 kV interfaced with an Oxford Instruments X-maxN 100 TLE silicon drift detector. STEM frames were acquired using an in-house SerialEM83 script that acquired frames either continuously or with a set delay time between frames. STEM probe current was calculated by measuring the total intensity of the STEM probe at the appropriate spot size over an exposure time of 0.05 s (Fig. S3†). The total counts were converted to electrons using a conversion factor of 6.74 counts per electron for the microscope and camera determined using a Faraday cup and then finally converted from electrons per s to nA. Pixel dwell times were between 3.8 and 8 μs. The instrument interfaces with an Oxford INCA detector for EDS mapping.Image processing
Automated particle size measurements were achieved through step-by-step processing of the in situ STEM movies in ImageJ84 and Fiji.85 First, particles/clusters suitable for tracking (isolated, in frame throughout) were identified using TrackMate.86 The movies were stabilized87 and then cropped to a representative area containing the particle(s) of interest. Next, the boundary between particle and background was defined using an appropriate thresholding algorithm. The TryAll function at various timepoints, (beginning, middle, and end), allowed for direct comparisons between the thresholding algorithms over the whole timescale. Based on visual comparison of the thresholds with the microscopy movie, optimal thresholding was achieved using the Triangle88 or Yen89 algorithms. The processed binary video was inverted (to be black particles on white background) for further analysis. Finally, the particle size was automatically measured using the Analyze Particles function of Fiji with no circularity limits.85 A size limitation was selected to capture relevant particle information while removing background noise. The movie may be further cropped to isolate the measurements of single particle if there are other particles nearby. Sample temperatures from Poseidon Select were correlated to image frames and then the labeled90 particle video was then exported. Particle sizes were plotted as 2D size vs. time. Videos of analysis steps were assembled for export using Blender.Results and discussion
In situ pair distribution function
The formation of BiOCl from BiClLac was probed by in situ PDF. The precursor structure in solution was first studied prior to heating and compared to a single Bi(C3H4O3Cl)3(C3H5O3Cl) complex taken from the BiClLac crystal structure (CCDC Deposition 2211246, Refcode AFADEU, cutout Fig. 1a) reported previously.47 Due to the nature of the setup and the aqueous solution surrounding the precursor species, a significant amount of background scattering is observed. To extract meaningful structural insight from the acquired TS data we subtracted TS data of an NMR tube filled with 0.8 M ClLac solution acquired under identical conditions as the actual experiments as the background. This approach assumes that the local solvent structure is not affected by the presence of BiClLac. Possible solvent restructuring, as it has been observed in close proximity of nanoparticle surfaces,91 is thereby neglected.Comparing the precursor solution PDF in Fig. 1b to the calculated PDF of a single Bi(C3H4O3Cl)3(C3H5O3Cl) complex (Fig. 1a) shows that the experimentally observed structure has a similar correlation length as the structural model. Both species are smaller than 10 Å. The experimental PDF exhibits an intense peak at 0.75 Å (highlighted in Fig. 1b). Since this distance is too short for it to belong to an actual interatomic distance, it is likely an artefact of the Fourier transform53 or the background subtraction. The background subtraction strategy is therefore further discussed in the ESI.† Apart from that, the key distances of the complex align well with the experimental PDF. Significantly, no Bi–Bi distance is evident in the experimental BiClLac PDF, which would appear as intense peaks between 3.6 Å and 3.8 Å for bismuth oxo species.92–94 Thus, the formation of multinuclear Bi-based cluster structures in solution can be excluded. The main peak of the PDF at ca. 2.45 Å can be assigned to Bi–O bonds. Peak splitting points towards more varied Bi–O distances between ca. 2.3 and 2.5 Å (Fig. 1c) that are not expected for the reference cluster obtained from the solid state BiClLac. Although this deviation from the reference initially seems to indicate significant structural differences, it might be caused by inherent differences between complexes in solution and solid state instead. Most prominently, the degree of protonation in solution can differ from the one in solid state. Unlike the BiClLac complexes in solid state (CCDC Deposition 2211246, Refcode AFADEU), BiClLac in solution does not need to be partially deprotonated to satisfy charge balance. A 0.1 M aqueous solution of BiOCl exhibits a pH of 0.89, indicating that a large share of the ligand's carboxy groups are protonated. This in turn affects the ability of these groups to function as electron donors to the Bi3+ and thereby impacts the Bi–O bond distance. Moreover, the observed Bi–O peak splits into two peaks of around equal intensity (Fig. 1b). This ratio matches well with the bidentate nature of the ClLac ligands and indicates that our general hypothesis regarding the precursor structure holds despite the Bi–O peak splitting. Nevertheless, a partial replacement of the ClLac ligands with aqua or hydroxy ligands cannot be ruled out based on our PDF data. A large degree of ligand exchange with water can, however, be excluded, as this would cause the precipitation of bismuth, as seen for aqueous Bi(NO3)3 solutions without ClLac ligands.
Apart from the Bi–O peak, the Bi–C distances in the structural model match well with an observed peak at ca. 3.35 Å. Ligand atoms located spatially further away from Bi (notated as O′, C′ and Cl in Fig. 1a), on the other hand, experience more freedom of movement relative to the Bi atom. Therefore, the broad experimental features observed between 4.0 Å and 6.5 Å can be assigned to the distances of O′, C′ and Cl to the central Bi atom. The Cl atoms, especially, can move significantly with respect to the central Bi as the terminal –C′H2Cl group can rotate around the C–C′ single bond.
Fig. 1d shows a fit of the BiClLac complex structure to the experimental PDF. The fit makes clear that the key features of the experimental PDF can be described by the model both qualitatively regarding the peak positions and quantitatively regarding the peak intensity. This is, however, only achievable by allowing for very high atomic displacement parameters (ADP) of the Cl atoms, i.e., 9.27 Å2. Fig. S5† shows a systematic comparison of refinements conducted with different treatments of the Cl position and ADPs. The observation that the fit improves substantially when the Biso(Cl) values are refined (as in Fig. 1d) is in line with the previous argument regarding the rotational degree of freedom of the terminal –C′H2Cl groups. Freeing the Cl positions as well (Fig. S5c†), results in an improved fit but unphysical C–Cl bond distances (up to 3.48 Å). Since these shortcomings of the structural model are, however, limited to the Cl atom positions, they do support the overall model choice.
The fit in Fig. 1d is based on a PDF from TS data acquired for 10 minutes. Applying the BiClLac model to a PDF with the acquisition time used for the in situ study (10 s, see Fig. S6†) yielded similar results, although with an even higher result for Biso(Cl) (10 Å2), reaching the upper limit of the applied Biso(Cl) parameter range. Collectively, the analysis of the precursor solution PDF hints towards a precursor structure in water closely related to that of the mononuclear BiClLac complex structure deduced via single-crystal XRD. The PDFs clearly show that prior to heating, no BiOCl is present.
To study BiOCl formation, the temperature of the precursor solution was increased from 30 °C in a rate of 3 °C min−1 until reaching 90 °C. The temperature was then held over a total heating time of 75 minutes (Fig. 2d), while collecting TS data at a rate of 10 s per frame (460 frames in total). Fig. 2 and Fig. S8† give an overview of the data gathered throughout the BiOCl synthesis. Both the reciprocal space (F(Q), in Fig. 2a and c) and the real space representation (G(r), in Fig. 2b and e) of the TS data are shown. This data makes it possible to follow the evolution in both the long range and local structure. In the first 30 minutes, no significant changes are observed, neither in the reciprocal nor the real space data. Afterwards, Bragg peaks appear (Fig. 2a and c) and grow gradually in intensity as time progresses. A comparison of these Bragg peaks to the calculated F(Q) of the reference structures can be found in Fig. S7.† A closer inspection of Fig. 2a reveals that prior to the emergence of the BiOCl reflection at 2.31 Å−1,33 a small Bragg peak appears at the slightly lower Q of 2.19 Å−1. This peak is emphasized in more detail in Fig. S9,† where a higher time resolution of 3 min is employed in the Q range of interest. The peak position matches with the (040) and (131) Bragg peaks of Bi(C2O4)OH,95 which is visualized in Fig. S10.† To identify this side product, more reliable PXRD data was acquired. The respective 2D detector images (Fig. S11†) show spotty Debye Scherrer rings, indicating large Bi(C2O4)OH crystals. A two-phase Rietveld refinement of this data using Bi(C2O4)OH and BiOCl is given (Fig. S11†) and further discussed in the ESI.† The impact of the spotty Debye Scherrer rings on the resulting PDF quality is discussed there as well.
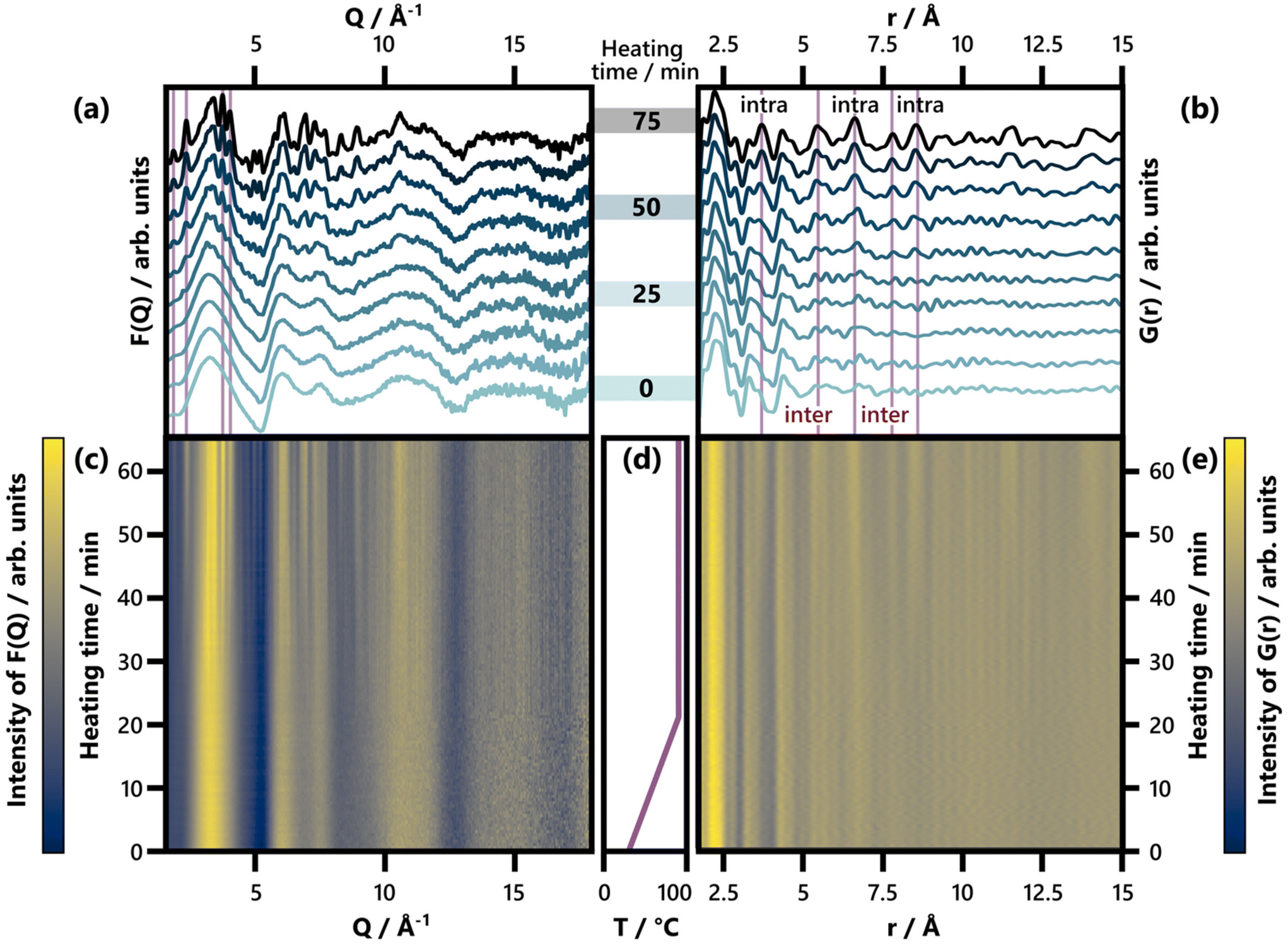 | ||
| Fig. 2 (a) Selected F(Q) patterns obtained between 0 and 75 min of heating, shown in increments of 8.3 min. Characteristic Bragg peaks of BiOCl (ICSD code 74502)33 are highlighted. For a comparison of the last F(Q) frame with calculated reference patterns, see Fig. S7.† (b) Selected G(r) patterns obtained between 0 and 75 min of heating, shown in increments of 8.3 min. Characteristic interatomic distances (3.73 Å, 5.50 Å, 6.64 Å, 7.81 Å, and 8.62 Å) of BiOCl (ICSD code 74502)33 are highlighted and indicated as belonging to intra- or interlayer Bi–Bi distances. For an analogous plot covering an r range up to 30 Å, see Fig. S8.† (c) and (e) show the full set of time-resolved F(Q) and G(r) patterns respectively with a color scale where yellow and blue represent high and low intensity, respectively. (d) Temperature with respect to heating time. | ||
The Bi(C2O4)OH side product was initially surprising as it had not been observed in the PXRD data obtained from products of comparable syntheses conducted outside of the synchrotron beam.47 It is therefore most likely the effects of the synchrotron irradiation that initiate the side phase formation.96 Oxalate anions can be produced through the oxidation of glyceric acid, which forms from the ClLac ligand upon halide release. The oxidation of glyceric acid to oxalate requires strong oxidizing agents such as sodium periodate or nitric acid when performed on the bench.97,98 In the case of our in situ study, however, the oxidizing agent is likely hydroxyl radicals produced from beam-induced water radiolysis.99–102 Such radiolysis can occur at synchrotron sources because of their high flux. Since hydroxyl radicals are among the strongest known oxidizing agents, they are capable of oxidizing the ligand to yield oxalate anions.96 Furthermore, lactate, a compound structurally similar to the ClLac ligand has been shown to be an effective free radical scavenger of hydroxyl radicals.103 The observed Bi(C2O4)OH formation is, therefore, most likely a result of the measurement technique.
Returning to the studied BiOCl formation pathway, the PDFs obtained from the total scattering data are shown in Fig. 2b and e. Fig. 2e shows how the characteristic interatomic distances of BiOCl begin to emerge clearly in the PDFs after ca. 50 min and grow gradually in intensity as time progresses. Meanwhile, the precursor complex remains the dominating structure in the G(r) throughout the reaction. These observations are consistent with the BiClLac precursor being designed to facilitate reaction control by intentionally slowing down the BiOCl formation.
No structurally distinct intermediates are observed in either of the two contour plots (Fig. 2c and e). This is noteworthy, as possible intermediates like Bi2O3, Bi(OH)3, BiOOH, BiONO3, (BiO)2CO3, and other BiaObClc stoichiometries might influence the emerging BiOCl. Since [BiaOa]a+ layers are structurally closely related to BiOCl, the assessment of whether single [Cl–Bi–O–Bi–Cl] or [Bi2O2]2+ layers are observed requires further analysis of the product structure. This was facilitated by refining BiOCl (ICSD code 74502)33 to one of the final frames (75 min heating) of the in situ PDF data set shown in Fig. 2e. The resulting fit is given in Fig. 3a and the refinement details in Table S5.† Due to the low relative intensity of the BiOCl peaks in the final G(r), we limited the refinement to the lattice parameters, the scale factor, and a parameter fitting the particle size to the PDF dampening by assuming a spherical morphology. This size parameter was only included to enable damping of the refined G(r)calc. As the layered structure of BiOCl favors anisotropic morphologies like platelets, the approximation of BiOCl nanoparticles as a sphere is inadequate. Accordingly, the obtained ‘size’ should not be trusted. The relatively simple model describes the data well with regard to the inter- and intralayer Bi–Bi peaks (highlighted in Fig. 3a), that give rise to the most intense PDF features of BiOCl.
 | ||
| Fig. 3 Refinement of BiOCl (ICSD code 74502)33 to the PDF obtained after 75 min of heating. The lattice parameters, scale factor and spherical particle size were refined in an r range from (a) 2 to 40 Å (shown from 2 to 20 Å) and (b) from 6 to 40 Å. The results of both refinements are listed in Table S5.† Intra- and interlayer Bi–Bi distance peaks are differentiated in (a). For comparison with the difference curve resulting from the full range fit (G(r)exp, 75 min − G(r)calc), the PDFs of the heated precursor solution (25 min heating) and the unheated one (0 min heating) are given. | ||
Disagreements between the experimental and calculated PDF in Fig. 3 occur below 6 Å. A closer inspection of the resulting difference curve (G(r)exp, 75 min − G(r)calc) reveals strong similarities to the initial precursor complex structure. The PDFs from both the heated and unheated precursor solution are given for reference. The proximity of a Bi–Cl distance (3.47 Å) in BiOCl and the Bi–C distance (ca. 3.4 Å) in BiClLac as well as slight imperfections in refinement regarding the fitting of the first intralayer Bi–Bi peak (3.8 Å) cause minor deviations between the difference curve and these references. The overall similarity is nevertheless apparent. It indicates a significant amount of precursor complex remaining in the reaction mixture even after 75 min of heating. For the sake of completeness, the difference curve from Fig. 3a (fit range 2 to 40 Å) was subsequently refined with the BiClLac precursor model (Fig. S12†). Despite the high level of noise and the mentioned mismatch between model and data (between 3.4 and 3.8 Å), the qualitative similarity can be confirmed through this refinement.
By excluding the r range below 6 Å from the refinement of BiOCl to the 75 min frame, a significantly better fit is achieved (Fig. 3b), and more quantitative information is extracted from the structural refinement. The refined lattice parameter of a = 3.8829 Å only differs from the reference value of a = 3.8855 Å by −0.07%.33 The c parameter, on the other hand, is significantly enlarged, c = 7.4899 Å, compared to the value expected for BiOCl, c = 7.3489 Å (1.92% larger).33 Accordingly, the interlayer spacing of BiOCl after 75 min of heating in solution is wider than reported for the bulk BiOCl structure.
To follow the temporal development of the lattice parameters, we sequentially refined the BiOCl model to our in situ PDF data. Fig. 4 shows the results of these refinements plotted against heating time. Frames acquired before 55 min of heating could not be modelled reliably and were therefore excluded from Fig. 4. The choice of this cut-off time is discussed further in the ESI,† where Fig. S13† shows individual fits conducted for every 5 min step between 40 min and 75 min. The complete set of sequential refinement results is furthermore visualized in Fig. S14.†
 | ||
| Fig. 4 Results of the sequential refinement of BiOCl (ICSD code 74502)33 to the in situ PDF data, with a fit range from 6 Å to 40 Å, plotted against heating time. (a) The normalized scaling factor, the lattice parameters (b) a and (c) c, as well as (d) the fit agreement parameter Rw are shown. The spherical particle size was refined as well but was omitted here as it does not carry physical meaning for anisotropic BiOCl particles. Yellow reference lines and their position are included in (b) and (c) to highlight the development of the lattice parameters. Frames below 55 min of heating are excluded here, as BiOCl could not be reliable refined to them. This is discussed further in the ESI,† where individual refinements for every 5 min step between 40 min and 75 min of heating is shown in Fig. S13.† The results of the whole sequential refinement are shown in Fig. S14.† The results for the initial frame of the sequential refinement are listed in Table S9.† | ||
Fig. 4b shows that the a parameter remains constant throughout the 20 min period probed in the shown data. This robustness is in line with the covalent in-layer bonds of BiOCl. The c parameter, on the other hand, relies on weaker van der Waals forces between the [Cl–Bi–O–Bi–Cl] layers. These forces increase with the layer size, as larger layers allow for stronger induced dipoles. Accordingly, Fig. 4c shows a decrease in c from ca. 7.515 Å to 7.489 Å with prolonged heating. Up until 75 min of heating, the c parameter does not stabilize and might still decrease further upon further particle growth. Based on our results, it remains uncertain if other factors such as potential ion or water intercalation impact this process.
The normalized scale factor results plotted in Fig. 4a make clear that this structural change of the product phase coincides with ongoing particle growth. Since spherical particle size approximations fall short for BiOCl particles, the increasing normalized scaling factor throughout the sequential refinement is our best measure for this process. Qualitatively, the growth of Bragg peaks belonging to BiOCl can be seen in Fig. 2c and Fig. S9.†
The presence of strong BiClLac scattering contributions throughout the experiment make it challenging to determine whether single [Cl–Bi–O–Bi–Cl] or [Bi2O2]2+ layers are present as intermediates. The available data quality makes it impossible to distinguish between [Cl–Bi–O–Bi–Cl] and [Bi2O2]2+ using PDF. Due to the similarity between [Cl–Bi–O–Bi–Cl] single layers and BiOCl, these two species can, furthermore, only be differentiated through the presence and position of interlayer Bi–Bi peaks. Especially the first, most intense interlayer Bi–Bi peak is, however, not clearly defined up until the end of our experiment. In the final PDF of the reaction the intralayer peak (highlighted in Fig. 3a) still overlaps with the broad features caused by the Bi-Cl distances of remaining BiClLac complex. Finally, single layers of [Cl–Bi–O–Bi–Cl] are expected to be present in significantly lower concentrations than BiOCl, which further impedes the identification of single [Cl–Bi–O–Bi–Cl] layers in the data.
All in all, the in situ PDF study shows that the BiClLac complex structure in aqueous media is closely related to that observed in the solid state.47 Upon heating, the precursor gives rise to gradually increasing concentration of BiOCl with a widened interlayer spacing. Throughout the synthesis, no intermediates in the form of distinct complexes or crystal structures have been observed. Instead, the product phase undergoes a gradual structural change in its interlayer spacing, which is in line with increasing van der Waals forces upon in-layer growth. Furthermore, Bi(C2O4)OH formed as a side product, likely induced by the synchrotron beam. The absence of distinct intermediates in our observations is a significant finding, which restrains the hypotheses on BiOCl formation proposed before. We can, for instance, be certain that structurally distinct Bi clusters such as the hexamer are not involved in the formation of BiOCl from BiClLac. As single [Cl–Bi–O–Bi–Cl] or [Bi2O2]2+ layers are more challenging to differentiate from BiOCl nanoparticles, their presence cannot be ruled out based on our data. If present, though, they have a collective lifetime below the data's acquisition time of 10 s or appear in low concentrations relative to the formed BiOCl. This knowledge may help to guide future synthesis strategies for well-defined nanostructures of BiOX.
In situ liquid cell transmission electron microscopy
LCTEM experiments were performed to directly visualize formation of BiOCl as it occurs in situ. An aqueous solution of BiClLac was encased between two silicon nitride membrane windows for imaging using TEM. The solution was heated from room temperature at a rate of 5 K min−1 in Movie 1† (TEM) and 3 K min−1 in Movies 2, S1, and S2† (STEM). We note that the reaction times for the LCTEM experiments are shorter than the in situ PDF analysis as TEM is best for observing individual nuclei and small particles in their initial seconds to minutes; PDF, on the other hand, probes an ensemble of both precursor complexes and particles and requires more particles to form before the PDF features of the product phase can be identified.Movie 1† and Fig. 5a show an in situ experiment using the BiClLac precursor in TEM mode. The field of view is initially blank as the sample is heated until particles appear at 53.4 °C/6:14.8. Fig. 5a highlights individual frames of Movie 1† showing the particles have a plate-like morphology and further develop with continued heating and time. The particles initially do not have much difference in contrast from the background, but with increasing time, the particles grow laterally in the ab plane resulting in a larger particle area and the contrast increases indicating a thickening of the nanoplate (stacking in the c-direction). Additionally, further heating seems to make the {010} edge faceting more defined as square/rectangular plate morphology develops and persists.
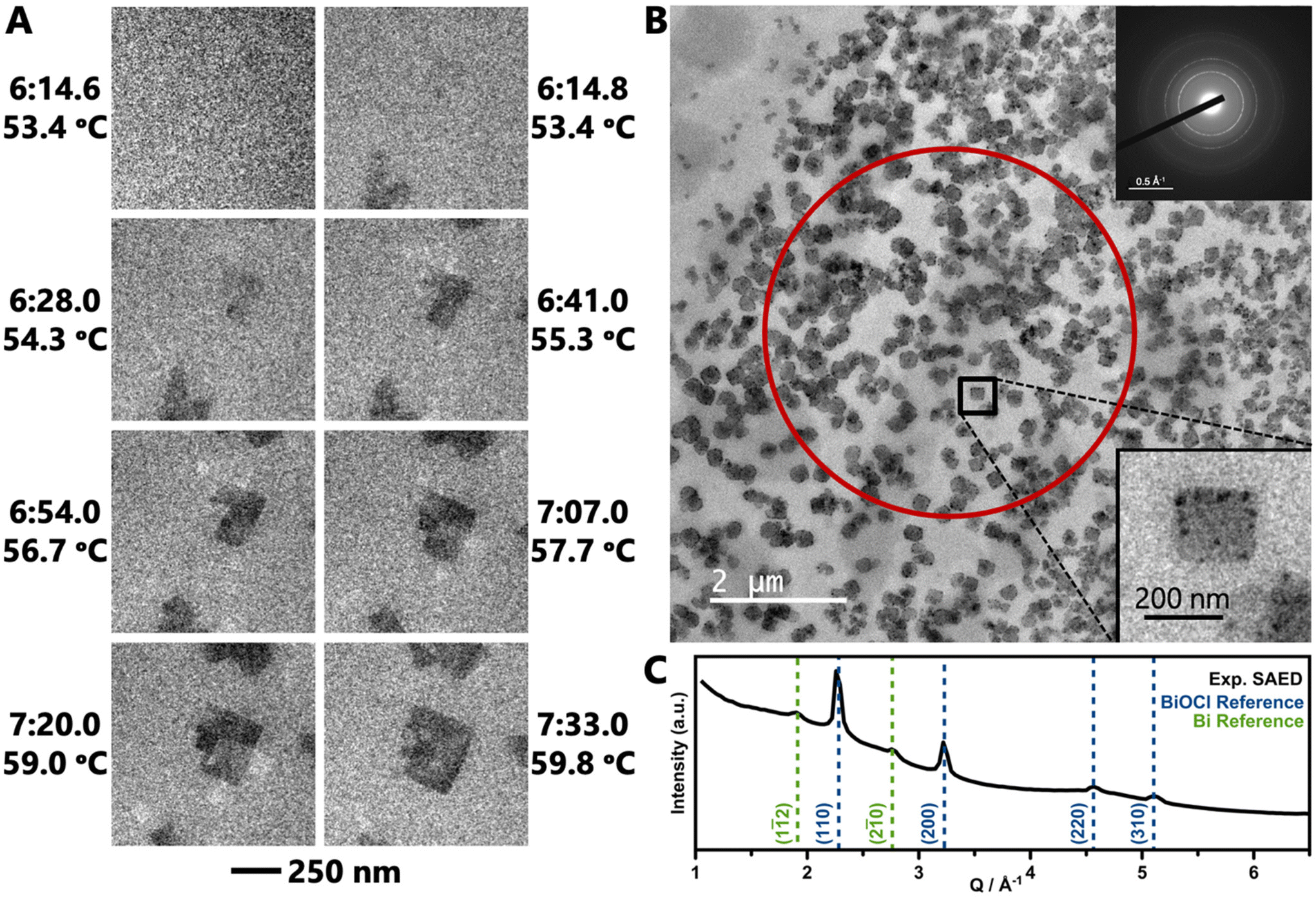 | ||
| Fig. 5 (a) Selected TEM images from Movie 1 showing BiOCl formation and growth from BiClLac. Images are spaced ca. 13 s apart, except for first two images which are 0.2 s apart, with times and temperatures indicated. The scale bar at bottom applies to all eight images. (b) TEM image of BiOCl nanoplates after a heating experiment. The lower-right inset shows a higher magnification of the indicated particle. The area within the red circle was used to collect the SAED pattern in the upper-right inset. Radial integration of this pattern gives the 1D diffraction pattern in (c). BiOCl and Bi reference patterns are JCPDS 85-0861 and 85-1329, respectively. | ||
After a similar heating experiment, a SAED pattern was collected from the nanoplates (Fig. 5b inset). Radial integration to a 1D diffraction pattern shows strong peaks characteristic of BiOCl and weak, broad peaks characteristic to Bi metal (Fig. 5c). The Bi metal is likely the dark black spots on the nanoplates. The reduction of Bi is an artifact of the extended imaging, where the electron beam itself acts as a reducing agent. Such metal deposition does not occur in benchtop experiments (without the electron beam).
Post-heating STEM-EDS elemental mapping of a particle after a TEM experiment shows an appropriate homogenous distribution of Bi, O, and Cl across the particle (Fig. 6a), consistent with the formation of BiOCl nanoparticles. The oxygen signal is high outside of the particle due to the oxygen in water. The SAED pattern of the particle shows that it is single-crystalline with slight mosaicity (Fig. 6b).
 | ||
| Fig. 6 (a) STEM image and EDS mapping of a synthesized BiOCl particle, with bismuth in green, oxygen in blue, and chlorine in red. The oxygen signal is high outside of the particle due the oxygen in water. Scale bar applies for all four images. STEM image field of view is 289 nm × 289 nm (802 × 802 pixels) with an EDS signal binning factor of 2. (b) SAED pattern of particle in (a) with reflections indexed. | ||
Returning to Movie 1† and Fig. 5a, this TEM movie shows no particles in one frame (6:14.6) and then formed particles in the next frame (0.2 s later, 6:14.8). Thus, the exact nucleation process remains unclear despite the formation of well-defined, isolated nanoplates. Therefore, higher magnification STEM images were collected to record the earliest moments of particle formation at a slightly slower heating rate to further slow the formation process. Compared to TEM, STEM imaging offers a higher signal-to-noise ratio for a high-Z sample (BiOCl) in a thick, low-Z medium (H2O), as image contrast is proportional to Z2.104–107
Movie 2† and Fig. 7 show a set of particle clusters imaged in STEM mode at a higher magnification. Image processing measured the 2D area of particle clusters in individual frames (full image processing details are described in the experimental section). Note that these measurements are 2D measurements of a 3D particle cluster, so exact values should be used cautiously. Automation of this process minimizes the potential for bias to be introduced. Particle clusters are tracked until the thresholding algorithm is no longer able to distinguish the particle cluster signal from background noise.
 | ||
| Fig. 7 Selected STEM images from Movie 2† showing BiOCl formation and growth from BiClLac. Images are spaced ca. 11 s apart with times and temperatures indicated. The black area in the last frame is where the particle clusters drifted near the edge of the field of view and was applied during drift correction. Scale bar applies to all images. Below the images is a plot of the clusters’ 2D area over time. Image processing steps for Movie 2 are shown in Movie S1.† | ||
The STEM video shows some variability in particle cluster development with time and temperature. The three adjacent particle clusters in Movie 2† and Fig. 7 each have unique growth trajectories, despite being spatially close and having identical imaging conditions. While the three particle clusters begin with similar morphology and size at the 8:09 time point, they soon follow different growth trajectories. The left particle cluster begins its growth first and enlarges in size steadily. The right particle cluster growth begins second at 8:53 and grows more quickly, surpassing the size of the left particle cluster by 8:59. The middle particle cluster begins its growth last but becomes the largest of the three soon after. Interestingly, the size of the middle cluster appears to decrease from 9:21 to 9:27. This decrease is believed to be an artifact of the measurement as the area represents the 2D projection of a 3D object. As the cluster moves and rotates within the liquid cell a smaller profile view may be observed despite the overall particle cluster continuing to grow.
While the nucleation time/temperature differs slightly compared to the TEM video, the results demonstrate that the application heat was still required to induce nucleation, despite the presence of the beam. A control experiment of imaging the precursor solution in STEM mode without the application of heat for 45 min showed no formation of BiOCl (Movie S2†), indicating that heating is necessary and suggesting that the electron beam alone is unable to overcome the nucleation barrier. After beginning heating at 45 min, particle formation was observed at 54 min (9 min later) at 50 °C. Because the measurements are only 2D projections, extracting a true particle growth rate (volume/time or mass/time) is not possible. The consistent particle growth under the applied heating program suggests that further control of BiOX growth can be achieved by modulating temperature and ramp rate. By controlling these variables, the substitution reaction rate could be tuned and thus the particle growth rate could be controlled directly using temperature as a synthetic lever.
The combined results of the in situ PDF and in situ TEM experiments reveal initial insights into the formation and growth of BiOCl, which can hopefully function as a stepping stone for future investigations. Meanwhile, this study highlights the limitations faced when studying BiOCl formation as well as the inherent challenges faced when combining in situ PDF and in situ TEM in general. Both the synchrotron and electron beam did, for instance, influence the studied reaction, by causing the formation of the side products Bi(C2O4)OH and Bi metal, respectively. Furthermore, a highly concentrated BiClLac solution is needed to obtain high quality in situ PDF data, while in situ TEM requires a high degree of dilution. Considering the lack of experimental insight into BiOCl formation up until now, the combination of the two techniques did nevertheless prove to be useful. Furthering the mechanistic understanding of BiOCl formation relies on our ability to combine advanced analytical techniques and knowledge about the instrumental challenges faced in this study may prove helpful for the future design of mechanistic studies.
Conclusions
Using in situ PDF and in situ LCTEM evidence, the genesis of BiOCl nanoparticles was followed for the first time. The in situ PDF results characterized the BiClLac structure in aqueous media at room temperature and upon heating. Notably, no structurally distinct intermediates were observed during its decomposition to produce BiOCl during heating. The absence of known Bi cluster species such as [Bi6(OH)12]6+ is a first indication that the Cl− counter ion significantly influences the outcome of Bi3+ hydrolysis itself. Our results furthermore indicate that single [Cl–Bi–O–Bi–Cl] or [Bi2O2]2+ layers, if they occur, are present in low concentrations relative to the BiOCl product or they exhibit a lifetime below the applied acquisition time prior to stacking along the c direction of the crystal structure. Moreover, we found that the interlayer spacing of the emerging BiOCl particles gradually decreases with prolonged heating. While a widened interlayer spacing persists until the end of our TS experiment, this development highlights the link between in-plane particle size and the resulting van der Waals forces. In situ LCTEM movies directly show the formation and rapid growth of BiOCl nanomaterials and further illustrate the role of heat in the formation pathway starting from a single-source precursor. Prior to this work, the formation and growth sequence of BiOCl was only hypothesized based on little experimental evidence as the rapid formation kinetics of precipitation reactions impeded further insights. Such insights are now enabled using a single-source precursor with slow release of halide ions. Our study serves as a first step towards a detailed mechanistic understanding of the BiOX formation and the future development of controlled BiOX nanostructures. It furthermore highlights the intrinsic challenges faced when moving towards this goal. Profound understanding of the BiOX nucleation and growth processes will be key as photocatalytic performance is significantly influenced by particle faceting, crystal defects, and morphology.Author contributions
M. N. G. and L. S. J. contributed equally with M. N. G. conceptualizing the project and leading the LCTEM analysis and L. S. J. conducting the PDF analysis. J. S. G. performed LCTEM image processing and supported LCTEM analysis. J. K. M. performed the total scattering experiments at the beamline and supported the PDF analysis. X. Z. and D. G. M. collected LCTEM data. K. M. Ø. J. and S. E. S. provided guidance and support for the project. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Data availability
Data for this article, including all electron microscopy and total scattering data are available at The Materials Data Facility108,109 at https://doi.org/10.18126/vzbt-a750.110Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. N. G. thanks Yanyao Liu and Dr Kevin Brown for ligand synthesis and characterization. We acknowledge DESY (Hamburg, Germany), a member of the Helmholtz Association HGF, for the provision of experimental facilities. Parts of this research were carried out at PETRAIII, and we would like to thank Dr Ann-Christin Dippel and Dr Soham Banerjee for assistance in using the P21.1 beamline as part of Dr Soham Banerjee's in-house beamtime. Dr Olivia Aalling-Frederiksen, Dr Baiyu Wang, Dr Andrea Kirsch, and Dr Emil Thyge Skaaning Kjær are thanked for conducting this in situ X-ray total scattering beamtime together with J. K. M. L. S. J. thanks Dr Nicolas P. L. Magnard and Dr Andrea Kirsch for assistance with the Rietveld refinement in TOPAS. M. N. G. thanks Dr Madeline Dukes for in situ LCTEM advice and Dr Kaustav Chatterjee, Dr Trevor Douglas, Dr Jeremy Smith, and Dr Romualdo deSouza for fruitful conversations. The authors thank the IU – Bloomington Electron Microscopy Center, for access to instrumentation and training.This work was supported by Indiana University and US NSF DMR-2113536. J. K. M. is grateful for funding from the VILLUM FONDEN through a Villum International Postdoc Grant (VIL41388). The Danish Research Council is acknowledged for covering travel expenses in relation to the synchrotron experiment (DanScatt).
References
- IEA ( 2019), The Future of Hydrogen, IEA, https://www.iea.org/reports/the-future-of-hydrogen Search PubMed.
- D. G. Nocera, Acc. Chem. Res., 2017, 50, 616–619 CrossRef CAS.
- N. P. Brandon and Z. Kurban, Philos. Trans. R. Soc., A, 2017, 375, 20160400 CrossRef PubMed.
- F. Eljack and M.-K. Kazi, Front. Sustainable, 2021, 1, 612762 CrossRef.
- K. W. A. Guy, Trans. Inst. Chem. Eng., 2000, 78, 324–327 CAS.
- J. Rifkin, The Hydrogen Economy: The Creation of the Worldwide Energy Web and the Redistribution of Power on Earth, J. P. Tarcher/Putnam, New York, 2002 Search PubMed.
- M. A. Rosen and S. Koohi-Fayegh, Energy Ecol. Environ., 2016, 1, 10–29 CrossRef.
- I. Staffell, D. Scamman, A. Velazquez Abad, P. Balcombe, P. E. Dodds, P. Ekins, N. Shah and K. R. Ward, Energy Environ. Sci., 2019, 12, 463–491 RSC.
- S. van Renssen, Nat. Clim. Change, 2020, 10, 799–801 CrossRef.
- Q. Wang and K. Domen, Chem. Rev., 2020, 120, 919–985 CrossRef CAS.
- K. Maeda, ACS Catal., 2013, 3, 1486–1503 CrossRef CAS.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- F. E. Osterloh, Top. Curr. Chem., 2016, 371, 105–142 CrossRef CAS.
- H. Kisch, Semiconductor Photocatalysis: Principles and Applications, Wiley-VCH, Weinheim, Germany, 2015 Search PubMed.
- The Fuel Cell and Hydrogen Energy Association (2020), Road Map to a US Hydrogen Economy: Reducing emmission and driving growth across the nation, https://www.fchea.org/us-hydrogen-study.
- A. I. Osman, N. Mehta, A. M. Elgarahy, M. Hefny, A. Al-Hinai, A. A. H. Al-Muhtaseb and D. W. Rooney, Environ. Chem. Lett., 2021, 20, 153–188 CrossRef.
- U. Bossel, B. Eliasson and G. Taylor, Cogener. Distrib. Gener. J., 2003, 18, 29–70 CrossRef.
- M. M. Ayyub, R. Singh and C. N. R. Rao, Sol. RRL, 2020, 4, 2000050 CrossRef CAS.
- H. Nishiyama, T. Yamada, M. Nakabayashi, Y. Maehara, M. Yamaguchi, Y. Kuromiya, Y. Nagatsuma, H. Tokudome, S. Akiyama, T. Watanabe, R. Narushima, S. Okunaka, N. Shibata, T. Takata, T. Hisatomi and K. Domen, Nature, 2021, 598, 304–307 CrossRef CAS PubMed.
- P. Ganguly, M. Harb, Z. Cao, L. Cavallo, A. Breen, S. Dervin, D. D. Dionysiou and S. C. Pillai, ACS Energy Lett., 2019, 4, 1687–1709 CrossRef CAS.
- Y. Subramanian, A. Dhanasekaran, L. A. Omeiza, M. R. Somalu and A. K. Azad, Catalysts, 2023, 13, 173 CrossRef CAS.
- K. Chatterjee and S. E. Skrabalak, ACS Appl. Mater. Interfaces, 2021, 13, 36670–36678 CrossRef CAS.
- B. Ohtani, J. Photochem. Photobiol., C, 2010, 11, 157–178 CrossRef CAS.
- Q. Wang, M. Nakabayashi, T. Hisatomi, S. Sun, S. Akiyama, Z. Wang, Z. Pan, X. Xiao, T. Watanabe, T. Yamada, N. Shibata, T. Takata and K. Domen, Nat. Mater., 2019, 18, 827–832 CrossRef CAS PubMed.
- T. Takata, C. Pan and K. Domen, Sci. Technol. Adv. Mater., 2015, 16, 033506 Search PubMed.
- L. Ye. BiOX (X = Cl, Br, and I) Photocatalysts, in Semicond. Photocatal.: Mater., Mech. Appl., 2016, ch. 10, pp. 273–299 Search PubMed.
- L. Ye, Y. Su, X. Jin, H. Xie and C. Zhang, Environ. Sci.: Nano, 2014, 1, 90–112 RSC.
- X. Lv, F. L. Y. Lam and X. Hu, Front. Catal., 2022, 2, 839072 Search PubMed.
- S. Garg, M. Yadav, A. Chandra and K. Hernadi, J. Nanosci. Nanotechnol., 2019, 19, 280–294 CrossRef CAS PubMed.
- J. Li, Y. Yu and L. Zhang, Nanoscale, 2014, 6, 8473–8488 RSC.
- J. Li, H. Li, G. Zhan and L. Zhang, Acc. Chem. Res., 2017, 50, 112–121 CrossRef CAS.
- K. Zhang, C. Liu, F. Huang, C. Zheng and W. Wang, Appl. Catal., B, 2006, 68, 125–129 CrossRef CAS.
- K. G. Keramidas, G. P. Voutsas and P. I. Rentzeperis, Z. Kristallogr., 1993, 205, 35–40 CAS.
- M. N. Gordon, K. Chatterjee, N. Christudas Beena and S. E. Skrabalak, ACS Sustainable Chem. Eng., 2022, 10, 15622–15641 CrossRef CAS.
- W.-W. Liu and R.-F. Peng, J. Electron. Sci. Technol., 2020, 18, 100020 CrossRef.
- J. Geng, W.-H. Hou, Y.-N. Lv, J.-J. Zhu and H.-Y. Chen, Inorg. Chem., 2005, 44, 8503–8509 CrossRef CAS PubMed.
- V. K. LaMer and R. H. Dinegar, J. Am. Chem. Soc., 1950, 72, 4847–4854 CrossRef CAS.
- V. K. La Mer and G. M. Pound, J. Am. Chem. Soc., 1952, 74, 2323–2332 CrossRef.
- E. D. Bøjesen and B. B. Iversen, CrystEngComm, 2016, 18, 8332–8353 RSC.
- H. Zhang, L. Liu and Z. Zhou, RSC Adv., 2012, 2, 9224–9229 RSC.
- G. Wulff, Z. Kristallogr. – Cryst. Mater., 1901, 34, 449–530 CrossRef CAS.
- P. L. Brown and C. Ekberg, Bismuth and Polonium, in Hydrolysis of Metal Ions, Wiley-VCH, Weinheim, Germany, 2016, vol. 1, ch. 15, pp. 873–891 Search PubMed.
- F. Granér and L. G. Sillén, Acta Chem. Scand., 1947, 1, 631–655 CrossRef.
- Å. Olin, Acta Chem. Scand., 1957, 11, 1445–1456 CrossRef.
- W. J. Hunks and G. A. Ozin, Adv. Funct. Mater., 2005, 15, 259–266 CrossRef CAS.
- B. D. Dhanapala, H. N. Munasinghe, L. Suescun and F. A. Rabuffetti, Inorg. Chem., 2017, 56, 13311–13320 CrossRef CAS.
- M. N. Gordon, Y. Liu, M. K. Brown and S. E. Skrabalak, Inorg. Chem., 2023, 62, 9640–9648 CrossRef CAS PubMed.
- M. N. Gordon, K. Chatterjee, A. L. Lambright, S. L. A. Bueno and S. E. Skrabalak, Inorg. Chem., 2020, 60, 4218–4225 CrossRef PubMed.
- J. K. Mathiesen, J. Quinson, S. Blaseio, E. T. S. Kjær, A. Dworzak, S. R. Cooper, J. K. Pedersen, B. Wang, F. Bizzotto, J. Schröder, T. L. Kinnibrugh, S. B. Simonsen, L. T. Kuhn, J. J. K. Kirkensgaard, J. Rossmeisl, M. Oezaslan, M. Arenz and K. M. Ø. Jensen, J. Am. Chem. Soc., 2023, 145, 1769–1782 CrossRef CAS PubMed.
- M. Juelsholt, T. L. Christiansen and K. M. Ø. Jensen, J. Phys. Chem. C, 2019, 123, 5110–5119 CAS.
- H. Xu, S. Sommer, N. L. N. Broge, J. Gao and B. B. Iversen, Chem. – Eur. J., 2019, 25, 2051–2058 CrossRef CAS.
- T. L. Christiansen, S. R. Cooper and K. M. Ø. Jensen, Nanoscale Adv., 2020, 2, 2234–2254 RSC.
- T. Egami and S. J. L. Billinge, Underneath the Bragg Peaks: Structural Analysis of Complex Materials, Pergamon, Oxford/Amsterdam/San Diego, 2nd edn, 2012 Search PubMed.
- J. E. Evans, K. L. Jungjohann, N. D. Browning and I. Arslan, Nano Lett., 2011, 11, 2809–2813 CrossRef CAS PubMed.
- F. M. Alcorn, P. K. Jain and R. M. van der Veen, Nat. Rev. Chem., 2023, 7, 256–272 CrossRef CAS PubMed.
- R. Sinclair, MRS Bull., 2013, 38, 1065–1071 CrossRef CAS.
- J. Sung, Y. Bae, H. Park, S. Kang, B. K. Choi, J. Kim and J. Park, Annu. Rev. Chem. Biomol. Eng., 2022, 13, 167–191 CrossRef CAS PubMed.
- H. G. Liao and H. Zheng, Annu. Rev. Phys. Chem., 2016, 67, 719–747 CrossRef CAS.
- S. Pu, C. Gong and A. W. Robertson, R. Soc. Open Sci., 2020, 7, 191204 CrossRef CAS.
- D. A. Welch, R. Faller, J. E. Evans and N. D. Browning, Ultramicroscopy, 2013, 135, 36–42 CrossRef CAS.
- T. J. Woehl, K. L. Jungjohann, J. E. Evans, I. Arslan, W. D. Ristenpart and N. D. Browning, Ultramicroscopy, 2013, 127, 53–63 CrossRef CAS PubMed.
- N. M. Schneider, M. M. Norton, B. J. Mendel, J. M. Grogan, F. M. Ross and H. H. Bau, J. Phys. Chem. C, 2014, 118, 22373–22382 CrossRef CAS.
- J. M. Grogan, N. M. Schneider, F. M. Ross and H. H. Bau, Nano Lett., 2014, 14, 359–364 CrossRef CAS.
- T. J. Woehl and P. Abellan, J. Microsc., 2017, 265, 135–147 CrossRef CAS PubMed.
- T. H. Moser, T. Shokuhfar and J. E. Evans, Micron, 2019, 117, 8–15 CrossRef CAS.
- S. Wu, J. Sun, S. Z. Yang, Q. He, L. Zhang and L. Sun, Inorg. Chem., 2018, 57, 8988–8993 CrossRef CAS PubMed.
- X. Chang, S. Wang, Q. Qi, M. A. Gondal, S. G. Rashid, S. Gao, D. Yang, K. Shen, Q. Xu and P. Wang, Dalton Trans., 2015, 44, 15888–15896 RSC.
- L. Liu, H. Wang, Z. Yi, Q. Deng, Z. Lin and X. Zhang, Micron, 2018, 105, 30–34 CrossRef CAS PubMed.
- H. L. Xin and H. Zheng, Nano Lett., 2012, 12, 1470–1474 CrossRef CAS PubMed.
- M. N. Gordon, Y. Liu, I. H. Shafei, M. K. Brown and S. E. Skrabalak, Acta Crystallogr., Sect. C: Struct. Chem., 2022, 78, 257–264 CrossRef CAS.
- P. J. Chupas, X. Qiu, J. C. Hanson, P. L. Lee, C. P. Grey and S. J. L. Billinge, J. Appl. Crystallogr., 2003, 36, 1342–1347 CrossRef CAS.
- A. Hammersley, J. Appl. Crystallogr., 2016, 49, 646–652 CrossRef CAS.
- A. P. Hammersley, S. O. Svensson, A. Thompson, H. Graafsma, Å. Kvick and J. P. Moy, Rev. Sci. Instrum., 1995, 66, 2729–2733 CrossRef CAS.
- A. P. Hammersley, S. O. Svensson and A. Thompson, Nucl. Instrum. Methods Phys. Res., Sect. A, 1994, 346, 312–321 CrossRef CAS.
- C. Prescher and V. B. Prakapenka, High Pressure Res., 2015, 35, 223–230 CrossRef CAS.
- P. Juhás, T. Davis, C. L. Farrow and S. J. L. Billinge, J. Appl. Crystallogr., 2013, 46, 560–566 CrossRef.
- C. L. Farrow, P. Juhás, J. W. Liu, D. Bryndin, E. S. Božin, J. Bloch, T. Proffen and S. J. L. Billinge, J. Phys.: Condens. Matter, 2007, 19, 335219 CrossRef CAS PubMed.
- P. Juhás, C. L. Farrow, X. Yang, K. R. Knox and S. J. Billinge, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 562–568 CrossRef.
- A. Coelho, J. Appl. Crystallogr., 2018, 51, 210–218 CrossRef CAS.
- H. Rietveld, J. Appl. Crystallogr., 1969, 2, 65–71 CrossRef CAS.
- E. Zolotoyabko, J. Appl. Crystallogr., 2009, 42, 513–518 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2008, 41, 653–658 CrossRef CAS.
- D. N. Mastronarde, Microsc. Microanal., 2003, 9, 1182–1183 CrossRef.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J. Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
- J. Y. Tinevez, N. Perry, J. Schindelin, G. M. Hoopes, G. D. Reynolds, E. Laplantine, S. Y. Bednarek, S. L. Shorte and K. W. Eliceiri, Methods, 2017, 115, 80–90 CrossRef CAS.
- N. M. Schneider, NMS_fixTranslation_ver1.ijm, 2014, https://github.com/NMSchneider/fixTranslation-Macro-for-ImageJ/blob/master/NMS_fixTranslation_ver1.ijm (accessed 2023-05-31).
- G. W. Zack, W. E. Rogers and S. A. Latt, J. Histochem. Cytochem., 1977, 25, 741–753 CrossRef CAS PubMed.
- J.-C. Yen, F.-J. Chang and S. Chang, IEEE Trans. Image Process., 1995, 4, 370–378 CrossRef CAS PubMed.
- V. P. Sharma, LabelStackFromATextFile.ijm, 2021, https://gist.github.com/ved-sharma/8b6ff203af59827086f1cc1e0775f6d7/revisions (accessed 2023-05-31).
- M. Zobel, R. B. Neder and S. A. J. Kimber, Science, 2015, 347, 292–294 CrossRef CAS PubMed.
- J. Brugger, B. Tooth, B. Etschmann, W. Liu, D. Testemale, J.-L. Hazemann and P. V. Grundler, J. Solution Chem., 2014, 43, 314–325 CrossRef CAS.
- L. Miersch, T. Rüffer, H. Lang, S. Schulze, M. Hietschold, D. Zahn and M. Mehring, Eur. J. Inorg. Chem., 2010, 2010, 4763–4769 CrossRef.
- A. S. Anker, T. L. Christiansen, M. Weber, M. Schmiele, E. Brok, E. T. S. Kjær, P. Juhás, R. Thomas, M. Mehring and K. M. Ø. Jensen, Angew. Chem., Int. Ed., 2021, 60, 20407–20416 CrossRef CAS PubMed.
- M. Rivenet, P. Roussel and F. Abraham, J. Solid State Chem., 2008, 181, 2586–2590 CrossRef CAS.
- R. A. Mayanovic, A. J. Anderson, H. A. Dharmagunawardhane, S. Pascarelli and G. Aquilanti, J. Synchrotron Radiat., 2012, 19, 797–805 CrossRef CAS PubMed.
- A. S. Perlin, Adv. Carbohydr. Chem. Biochem., 2006, 60, 183–250 CrossRef CAS PubMed.
- H. Kiliani, Ber. Dtsch. Chem. Ges., 1921, 54, 456–472 CrossRef.
- J. Lee, D. Nicholls, N. D. Browning and B. L. Mehdi, Phys. Chem. Chem. Phys., 2021, 23, 17766–17773 RSC.
- I. G. Draganić and Z. D. Draganić, Primary Products of Water Radiolysis: Oxidizing Species—The Hydroxyl Radical and Hydrogen Peroxide, in The Radiation Chemistry of Water, ed. E. M. Loebl, Academic Press, New York and London, 1971, ch. 4, pp. 91–121 Search PubMed.
- N. K. V. Leitner and M. Dore, J. Photochem. Photobiol., A, 1996, 99, 137–143 CrossRef.
- L. M. Dorfman and G. E. Adams, Reactivity of the Hydroxyl Radical in Aqueous Solutions, NSRDS-NBS 46, National Standard Reference Data System - National Bureau of Standards, U.S. Department of Commerce, Washington, DC, 1973.
- C. Groussard, I. Morel, M. Chevanne, M. Monnier, J. Cillard and A. Delamarche, J. Appl. Physiol., 2000, 89, 169–175 CrossRef CAS.
- N. de Jonge, N. Poirier-Demers, H. Demers, D. B. Peckys and D. Drouin, Ultramicroscopy, 2010, 110, 1114–1119 CrossRef CAS.
- N. de Jonge, D. B. Peckys, G. J. Kremers and D. W. Piston, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 2159–2164 CrossRef CAS.
- N. de Jonge, Ultramicroscopy, 2018, 187, 113–125 CrossRef CAS.
- N. de Jonge and F. M. Ross, Nat. Nanotechnol., 2011, 6, 695–704 CrossRef CAS.
- B. Blaiszik, K. Chard, J. Pruyne, R. Ananthakrishnan, S. Tuecke and I. Foster, JOM, 2016, 68, 2045–2052 CrossRef.
- B. Blaiszik, L. Ward, M. Schwarting, J. Gaff, R. Chard, D. Pike, K. Chard and I. Foster, MRS Commun., 2019, 9, 1125–1133 CrossRef CAS.
- M. N. Gordon, L. S. Junkers, J. S. Googasian, J. K. Mathiesen, X. Zhan, D. G. Morgan, K. M. Ø. Jensen and S. E. Skrabalak, The Materials Data Facility, 2024, DOI:10.18126/vzbt-a750.
Footnotes |
| † Electronic supplementary information (ESI) available: Four in situ LCTEM movies (.mov), supplemental discussion of background subtraction strategy, supplemental discussion of side product Bi(C2O4)OH, movie captions, supplemental figures, and supplemental tables (.pdf). See DOI: https://doi.org/10.1039/d4nr01749h |
| ‡ Equal authorship. |
| § Present address: Savannah River National Laboratory, Aiken, SC 29808, USA. |
| ¶ Present address: Surface Physics & Catalysis (SURFCAT), Department of Physics, Technical University of Denmark, 2800 Kgs Lyngby, Denmark. |
| || Present address: Texas Materials Institute, University of Texas at Austin, Austin, Texas 78712, USA. |
| This journal is © The Royal Society of Chemistry 2024 |