
Single and dual-atom catalysts towards electrosynthesis of ammonia and urea: a review
Wenyu
Luo
 a,
Jiawei
Liu
ab,
Yue
Hu
c and
Qingyu
Yan
*ad
a,
Jiawei
Liu
ab,
Yue
Hu
c and
Qingyu
Yan
*ad
aSchool of Materials Science and Engineering, Nanyang Technological University, 639798, Singapore. E-mail: alexyan@ntu.edu.sg
bInstitute of Sustainability for Chemicals, Energy and Environment (ISCE2), Agency for Science, Technology and Research (A*STAR), 1 Pesek Road, Jurong Island, Singapore 627833, Republic of Singapore
cSchool of Mathematics and Physics, University of Science and Technology Beijing, Beijing 100083, China
dInstitute of Materials Research and Engineering (IMRE), Agency for Science, Technology and Research (A*STAR), 2 Fusionopolis Way, Innovis #08-03, Singapore 138634, Republic of Singapore
First published on 22nd October 2024
Abstract
Ammonia and urea represent two important chemicals that have contributed to the rapid development of humanity. However, their industrial production requires harsh conditions, consuming excessive energy and resulting in significant greenhouse gas emission. Therefore, there is growing interest in the electrocatalytic synthesis of ammonia and urea as it can be carried out under ambient conditions. Recently, atomic catalysts (ACs) have gained increased attention for their superior catalytic properties, being able to outperform their micro and nano counterparts. This review examines the advantages and disadvantages of ACs and summarises the advancement of ACs in the electrocatalytic synthesis of ammonia and urea. The focus is on two types of AC – single-atom catalysts (SACs) and diatom catalysts (DACs). SACs offer various advantages, including the 100% atom utilization that allows for low material mass loading, suppression of competitive reactions such as hydrogen evolution reaction (HER), and alternative reaction pathways allowing for efficient synthesis of ammonia and urea. DACs inherit these advantages, possessing further benefits of synergistic effects between the two catalytic centers at close proximity, particularly matching the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond for N2 reduction and boosting C–N coupling for urea synthesis. DACs also possess the ability to break the linear scaling relation of adsorption energy of reactants and intermediates, allowing for tuning of intermediate adsorption energies. Finally, possible future research directions using ACs are proposed.
N bond for N2 reduction and boosting C–N coupling for urea synthesis. DACs also possess the ability to break the linear scaling relation of adsorption energy of reactants and intermediates, allowing for tuning of intermediate adsorption energies. Finally, possible future research directions using ACs are proposed.
1 Introduction
Nitrogen is one of the most abundant elements on Earth, comprising 78% of the Earth's atmosphere. The nitrogen cycle is the conversion of various nitrogen-containing products by nature and is vital to life. With nitrogen being the fourth most common element in the human body, the importance of nitrogen cannot be understated. Two nitrogen-containing products: ammonia (NH3) and urea, are of great importance to humans. The invention of the Haber–Bosch (H–B) process converting atmospheric dinitrogen N2 and hydrogen gas H2 to ammonia facilitated fertiliser production, increasing crop yield rates by almost 2.3 times.1 Ammonia is also a great potential source of carbon-free renewable energy due to its high gravimetric hydrogen content (17.5%),2,3 high energy density (12.92–14.4 MJ L−1) comparable to coal and oil,4,5 cheap and easy liquefaction for transport (−33 °C)6,7 and products of nitrogen and water only from combustion.7,8 In a similar vein, urea is also widely utilised in the agricultural industry as well as having great potential for green energy.9–11The current industrial production of ammonia and urea is by the H–B process and Bazarov process, respectively. In the H–B process, the main source of hydrogen comes from fossil fuel feedstocks, resulting in the H–B process emitting 1.2% of global anthropogenic CO2 emission.12 The energy consumption of H–B is significant as well, a hefty 11% of the total energy consumed by the chemical industry. This massive energy consumption is due to the conditions in which ammonia is synthesized: a temperature of 723–823 K and pressure of 250–350 atm.13 80% of the produced ammonia is then fed to the Bazarov process to synthesize urea,14,15 a process requiring a temperature of 150 °C and 2 MPa to combine CO2 and ammonia supercritical fluid.16,17 Both H–B and Bazarov processes require high temperature and high pressure, consuming more than 2% of the world's energy each.15,18,19 Electrocatalytic synthesis of ammonia and urea is therefore much desired, as it can be done under ambient conditions using energy from renewable sources.20,21 The absence of heat and pressure in the electrocatalytic method results in a significantly lower energy requirement compared with the H–B process or Bazarov process. Much research has been done on the utilisation of catalysts on the microscale and nanoscale involving transition metals such as Zn,21 Co,22,23 Ti,24 Fe,19 Cu,25 In15,26 and Ru.27 Main group metals such as Al28 and even metal-free catalysts were also reported.29,30
Going further down the scale of size, the atomic catalyst (AC) is a relatively new topic that has gained increased attention from the catalytic community.31 Single atomic catalyst (SAC) is the atomic dispersion of a single catalytic material onto a support material such that the active centers are isolated.32,33 One of the most prominent advantages of SACs is the small amount of materials required to achieve the same results as their nano or micro counterparts due to the 100% atom efficiency of AC from the atomically dispersed catalyst, maximising the use of catalytic surface area.34 This atomic dispersion of catalytic material, however, brings in the question of catalyst stability as isolated atoms have high surface energy, resulting in a tendency to aggregate.35,36 There are methods to circumvent this, for example, by tuning the catalyst mass loading37 or by varying the type of support material.38 Further advantages of SACs include the suppression of competitive reactions such as hydrogen evolution reaction (HER) from (1) the ensemble effect, where the lack of adjacent active centers allows for only top site adsorption, while the preferred adsorption of *H is bridge or hollow site which is only possible with more than one active center present in nanomaterials, and (2) the electronic effect, where the metal atom–support interaction induces charge transfer from the metal atom to the support, resulting in the metal active center being positively charged, forming an electrostatic repulsion against H+.39 There is also the possibility of new reaction pathways not possible for nano and micro counterparts, such as the direct oxidation of methane to methanol or ethane when comparing Rh SACs with Rh nanoparticles and a two-electron pathway in the oxygen reduction reaction (ORR) when using Pt SACs compared with the four-electron pathway when using Pt nanoparticles.40
On the other hand, the distance between catalytic centers in SACs is too wide for any cooperative effects. For example, in dinitrogen N2, the NN bond has a bond length of 0.1–0.2 nm.41 The interatomic distance of catalytic centers must be reduced to match that of N2 to have cooperative effects. Diatomic catalysts (DACs) are able to effectively bridge this problem by having two catalyst atoms side by side.42 While inheriting the unique traits of SACs, DACs further exhibit advantages that SACs do not possess, as having two catalytic atoms side by side also results in synergistic effects for the catalytic process. One of the atoms can regulate the spin state and electronic configuration of the other atom, enhancing the catalytic activity.43 Strong interactions between the two atoms can also stabilise each other, preventing agglomerations.44 The Sabatier principle states that an ideal catalyst must have an intermediate binding energy that is neither too strong to the extent that it inhibits desorption of products nor too weak to the extent the reactants cannot be activated.45–47 A linear scaling relation between the adsorption energy of reactants and adsorption energy of intermediate is therefore detrimental to catalytic activities, as while a stronger adsorption energy of reactants will strongly activate the reactants, a corresponding stronger adsorption energy of intermediates will inhibit the desorption of products, potentially resulting in the formation of unwanted side-products. DACs are able to break this scaling relationship, allowing for fine tuning of adsorption energy for intermediates, further improving the catalytic activity.48,49
ACs have been utilised in many various reactions, such as carbon dioxide reduction (CO2RR), the nitrogen reduction reaction (NRR), nitrate reduction reaction (NO3RR) and water splitting reaction. Many reviews were published regarding ACs,50–55 exploring various synthesis methods, introducing the recent advances of SACs in various electrochemical reactions and summarising the characterisation techniques used for ACs. Some reviews were published outlining advancements in using SACs for ammonia synthesis from nitrate56,57 and nitrogen gas.58–60 One review even focused on theoretical DACs.61 These reviews are specific to nitrate reduction or nitrogen reduction to ammonia only. In this review, we emphasize specifically the recent advancements in NH3 and urea electrosynthesis that utilise SACs and DACs (Scheme 1) and compare the various nitrogen sources that are used. Research into the electrochemical synthesis of urea has taken off with atomic catalysts as well. It is about time to step back and look at the various advancements utilizing these catalysts. We will look at the challenges of each reaction and how both SACs and DACs overcome them in their unique ways. We will also explore some theoretical papers that utilise density functional theory (DFT) to screen suitable catalysts for the respective application. Finally, we will give suggestions and outlooks for further research in the area of NH3 and urea synthesis using ACs.
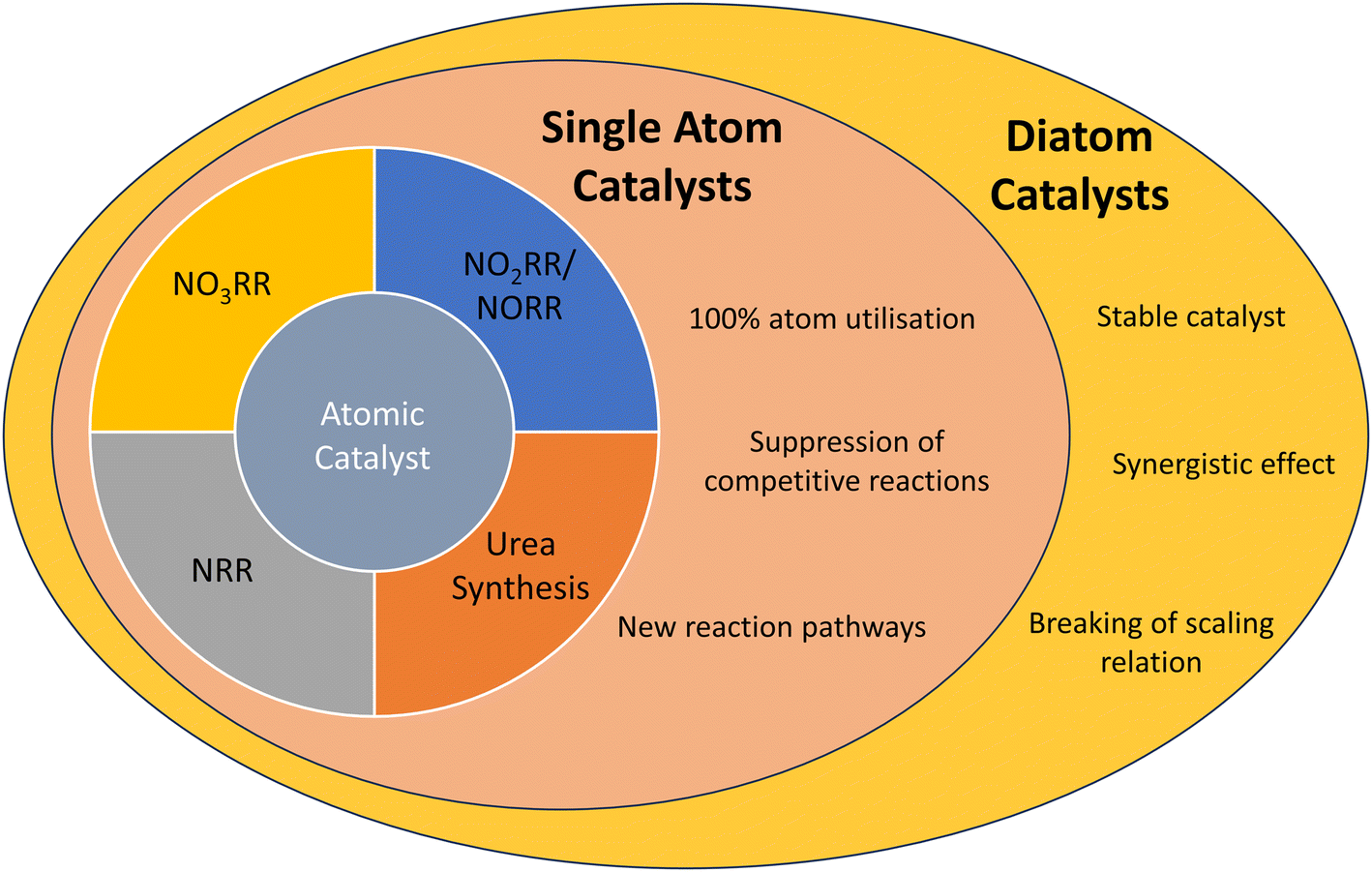 | ||
| Scheme 1 Illustration of atomic catalysts in five different reactions: nitrogen reduction, nitrate reduction, nitrite reduction, nitric oxide reduction and urea synthesis. Advantages of diatom catalysts include those of single-atom catalysts. | ||
2 Ammonia from nitrogen reduction reaction
There are various nitrogen-containing precursors that can be used for the electrochemical synthesis of ammonia. N2 is one such example. As stated in the introduction, N2 is very abundant, making up a major portion of the Earth's atmosphere. However, NRR under ambient conditions is challenging due to a multitude of issues: low solubility in aqueous electrolyte (0.00061 M at 25 °C in water), strong triple bond (941 kJ mol−1), competitive HER affecting the overall efficiency and selectivity of NRR and highly challenging activation of N2 under ambient conditions.62,63 Current catalysts face the issues of large overpotential, low current densities, low efficiency, and yield rate far from what the current industrial H–B process is outputting.64 NRR also has a complex mechanism, with several mechanisms upon which NRR proceeds: associative, dissociative, enzymatic and Mars–van Krevelen (MvK). For associative and dissociative mechanisms, N2 is adsorbed onto the catalyst surface in an end-on position, with only one N atom bonded to the surface. In the associative mechanism, the two N atoms are hydrogenated while they are still bonded. Under the associative mechanism, there are two possible pathways: distal and alternating. In the distal pathway, one N atom is hydrogenated twice successively and the NN bond is cleaved before desorbing as ammonia. The second N atom is then hydrogenated and desorbed as ammonia molecule. For the alternating path, the two N atoms are hydrogenated alternately before NN bond cleavage and desorption as ammonia. In the dissociative mechanism, the NN bond is cleaved in the first step followed by hydrogenation of the N atom.65 For the enzymatic pathway, N2 is adsorbed side-on onto the catalyst surface with both N atoms bonded to the surface. The N atoms are then hydrogenated alternatively to ammonia.66 The MvK mechanism is specially for transition metal nitrides (TMNs), where the N atoms, as part of TMNs, take part in the reaction to form ammonia. This leaves behind a vacancy which N2 will then fill. The dangling N atom is then hydrogenated to ammonia and a singular N atom is left behind, reforming the TMN's structure.64 ACs offer a counter to these issues by being able to suppress competitive HER due to the ensemble effect and are able to achieve higher faradaic efficiencies (FEs) by exposing more active catalyst sites for the same mass of catalyst.
2.1 Single-atom catalysts
Gold SACs on nitrogen-doped porous carbon (AuSAs-NDPCs) were synthesized and found to achieve an NH3 FE of 12.3% and a yield rate of 2.32 μg h−1 cm−2 at −0.2 V vs. reversible hydrogen electrode (RHE) (Fig. 1c and d).67 The catalyst was synthesized via a simple impregnation method, where the as-prepared NDPC was mixed with HAuCl4 under stirring and subsequently annealed under an inert atmosphere. Au mass loading was found to be 0.205 wt%, and the atomically dispersed Au can be seen using high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) in Fig. 1a. The linear sweep voltammetry (LSVs) of the catalyst in electrolyte with and without N2 gas bubbling are compared in Fig. 1b, demonstrating the occurrence of N2 reduction. When mass loading was increased to 0.713 wt%, nanoparticles (NPs) formed. AuSAs-NDPCs exhibited excellent selectivity to ammonia over hydrazine, a common by-product of NRR, with virtually no hydrazine produced at all potentials tested in the work. The excellent NRR performance can be attributed to the synergistic contributions by both Au single sites and NDPCs, where Au single atoms were stabilised by the surrounding N and C species which have high oxidative character and strong polarisation. This resulted in the high selectivity, with N2 being adsorbed onto positively polarized Au and negatively polarized N or C, forming a frustrated Lewis pair-like structure motif. In another work, single Au atoms on carbon nitrite (Au1/C3N4) achieved an NH3 FE of 11.1% and a yield rate of 1305 μg h−1 mgAu−1 at −0.1 V vs. RHE, an even lower potential compared with the previous study.68 An interesting thing to note is that the Au loading mass of this study was only 0.15 wt%, lower than the previous study of 0.205 wt%. These two studies showcased the low catalytic mass loading of SACs. Molybdenum was studied as well due to its presence in biological nitrogen fixation processes in enzymes.69 Single Mo atoms were anchored onto N-doped porous carbon (SA-Mo/NPC), with the most optimal loading mass at 9.54% achieving an NH3 FE of 14.6 ± 1.6% and a yield rate of 34.0 ± 3.6 μg h−1 mgcat−1 at −0.3 V vs. RHE.37 This is one of the highest mass loadings for SACs, achieving close to 10 wt% of catalyst mass loading (Table 1) in contrast with the usual ultra-low loading mass for SACs. The catalyst was synthesized by a facile pyrolysis method, heating to 650 °C for 4 h under Ar atmosphere (Fig. 1e). The NH3 FEs and yield rates of optimised SA-Mo/NPC at various potentials are shown in Fig. 1g. Various Mo loading at different potentials were also compared (Fig. 1h), with mass loading of 9.54 wt% showing the best yield rate. Mo–N active sites can activate N2 molecules, stabilise N2H and destabilise NH2 species. Thus, a higher amount of Mo on N-doped porous carbon will facilitate ammonia synthesis. However, when mass loading was increased further, Mo nanoclusters formed instead of atomically dispersed Mo, thus decreasing the effectiveness of the catalyst. The catalyst was also found to be stable up to 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s (Fig. 1i). In another case, instead of the usual N–C matrix, Yang et al. synthesized Ru SACs on S–C matrix.70 This unique setup resulted in a synergistic “push–push” effect: the adjacent S atom was hydrogenated to form S–H*, pushing electrons to adsorbed N2*; electron donation from H* to S atom enriched electrons on Ru, further pushing Ru electrons to adsorbed N2*, destabilising NN even further. This resulted in an NH3 yield rate of 13.186 μg h−1 mgcat−1, almost twice that of Ru single-atom catalyst on N–C matrix.
000 s (Fig. 1i). In another case, instead of the usual N–C matrix, Yang et al. synthesized Ru SACs on S–C matrix.70 This unique setup resulted in a synergistic “push–push” effect: the adjacent S atom was hydrogenated to form S–H*, pushing electrons to adsorbed N2*; electron donation from H* to S atom enriched electrons on Ru, further pushing Ru electrons to adsorbed N2*, destabilising NN even further. This resulted in an NH3 yield rate of 13.186 μg h−1 mgcat−1, almost twice that of Ru single-atom catalyst on N–C matrix.
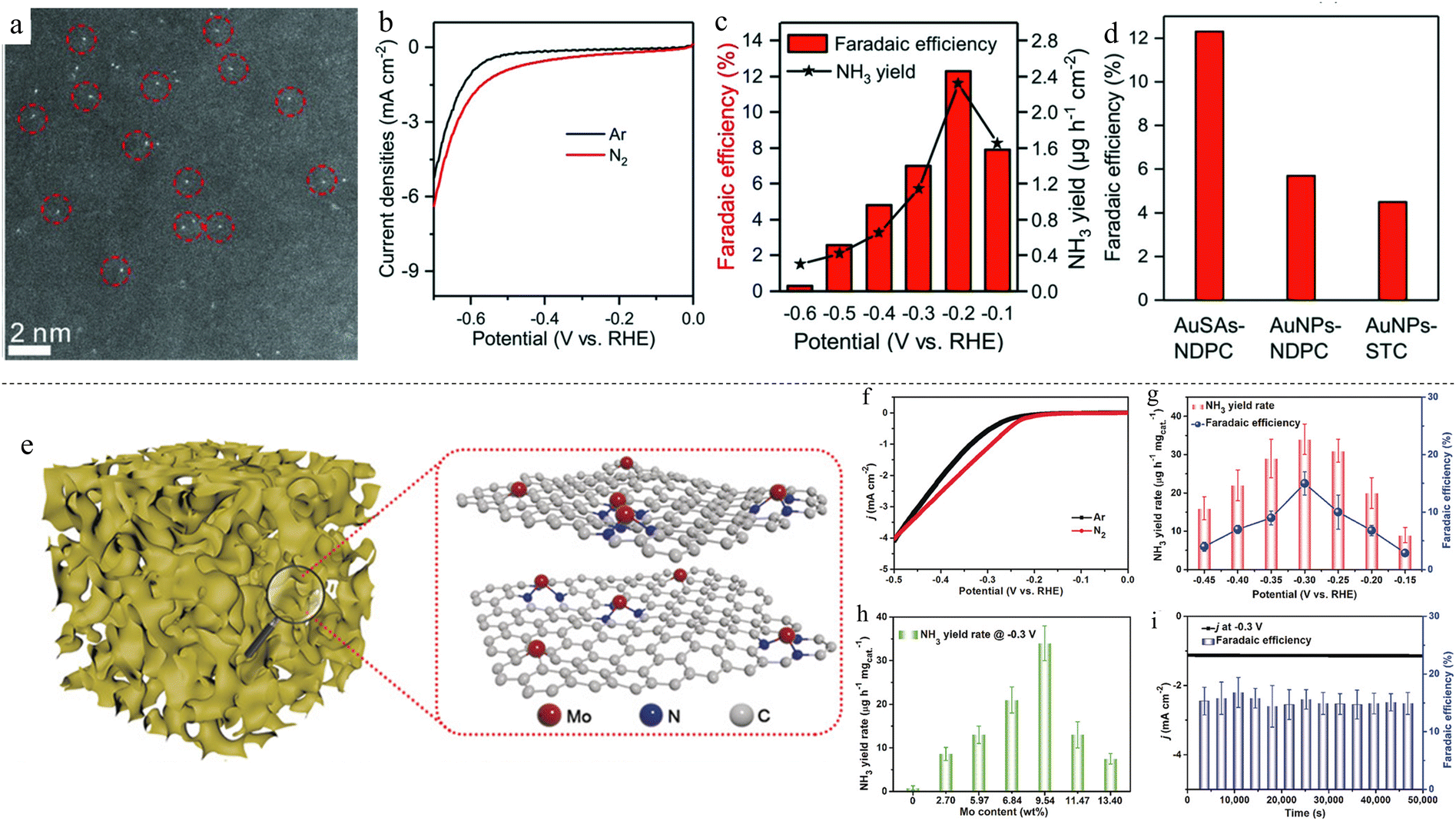 | ||
| Fig. 1 (a) HAADF-STEM image of Au-NDPC. Single Au atoms are circled. (b) LSV curves of Au-NDPC in Ar- and N2-saturated electrolytes. (c) NH3 FEs and yield rates across various potentials. (d) Comparison of FEs for SAC, NP and blank support. Reproduced with permission.67 Copyright 2018, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. (e) Illustration of SA-Mo/NPC. (f) LSV curves of SA-Mo/NPC in Ar- and N2-saturated electrolytes. (g) NH3 FEs and yield rates across different potentials. (h) NH3 FE comparison of various Mo wt% loading. (i) Stability test of SA-Mo/NPC across 50000 s. Reproduced with permission.37 Copyright 2019, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
| Reaction | Classification | Catalysts | Mass loading | Yield rate | FE | Potential vs. RHE | Electrolyte | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Not stated in paper, back calculated. | ||||||||
| NRR | SAC | AuSAs-NDPC | 0.205 wt% | 2.32 μgNH3 h−1 cm−2/1886 μgNH3 h−1 mgAu−1 | 12.30% | −0.2 V | 0.1 M HCl | 67 |
| Au1/C3N4 | 0.15 wt% | 1305 μgNH3 h−1 mgAu−1 | 11.10% | −0.1 V | 5 mM H2SO4 | 68 | ||
| SA-Mo/NPC | 9.54 wt% | 34.0 ± 3.6 μgNH3 h−1 mgcat−1/356 μgNH3 h−1 mgMo−1a |
14.6 ± 1.6% | −0.3 V | 0.1 M KOH | 37 | ||
| Ru–S–C | 0.55 wt%a | 13.17 μgNH3 h−1 mgcat−1/2.4 mgNH3 h−1 mgRu−1 | 6.16% | −0.15 V | 0.1 M H2SO4 | 70 | ||
| BG | 6.2 wt% | 9.8 μgNH3 h−1 cm−2 | 10.80% | −0.5 V | 0.05 M H2SO4 | 71 | ||
| DAC | CNT @C3N4-Fe&Cu | Fe: 7.51 wt%, Cu: 10.77 wt% | 9.86 μgNH3 h−1 mg−1 @ −1.2 V vs. Ag/AgCl | 34% @ −0.8 V vs. Ag/AgCl | — | Not stated | 42 | |
| O–V2-NC | 1.32 wt% | 26 μgNH3 h−1 mg−1/1969 μgNH3 h−1 mgV−1a @ −0.4 V vs. RHE |
77.2% @ 0 V vs. RHE | — | 0.1 M HCl | 73 | ||
| NO3RR | SAC | Fe-Ppy SAC | 2.38 wt% | 30 mmolNH3 h−1 mgFe−1 | 100% | −0.6 V | 0.1 M KOH + 0.1 M KNO3 | 86 |
| Fe1/CN-900 | 0.44 wt% | 1.10 mmolNH3 h−1 mgcat−1/18.8 mgNH3 h−1 mgcat−1/4250 mgNH3 h−1 mgFe−1a at −0.9 V vs. RHE |
86.7% @ −0.7 V vs. RHE | — | 0.1 M K2SO4 + 0.5 M KNO3 | 87 | ||
| Fe SAC | 2.9 wt% | 46 mgNH3 h−1 mgcat−1 @ −0.88 V vs. RHE | 92% @ −0.68 V vs. RHE | — | 0.1 M K2SO4 + 0.5 M KNO3 | 88 | ||
| Cu–N–C | 1.0 wt% | 4.5 mgNH3 h−1 cm−2/212.5 mgNH3 h−1 mgCu−1 | 84.70% | −1 V | 0.1 M KOH + 0.1 M KNO3 | 90 | ||
| ZnSA-MNC | 1.33 wt% | 39 mgNH3 h−1 mgcat−1 @ −1.0 V vs. RHE | 94.8% @ −0.9 V vs. RHE | — | 0.1 M Na2SO4 + 0.5 mM NaNO3 | 91 | ||
| NiO4-CCP | 18 wt% | 1.83 mmolNH3 h−1 mg−1/173 mgNH3 h−1 mgNi−1a |
94.70% | −0.7 V | 1 M Na2SO4 + 0.5 M NO3− | 92 | ||
| VCu-Au1CuSAA | 0.41 wt% | 555 μgNH3 h−1 cm−2/169 mgNH3 h−1 mgAu−1a |
98.70% | −0.2 V | 0.1 M KOH + 7.14 mM KNO3 | 93 | ||
| Ru1–TiOx/Ti | 0.03 wt% | 22.2 mgNH3 h−1 mg−1/74000 mgNH3 h−1 mgRu−1a |
87.60% | −0.3 V | 1 M KOH + 0.5 M NaNO3 | 94 | ||
| DAC | Cu/Ni-NC | Not stated | 5480 μg h−1 mgcat−1 cm−2 | 97% | −0.7 V | 0.5 M Na2SO4 + 100 ppm NaNO3 | 97 | |
| Fe/Cu-HNG | Fe: 3.3 wt%, Cu: 2.8 wt% | 1.08 mmolNH3 h−1 mg−1 @ −0.5 V vs. RHE | 92.51% @ −0.3 V vs. RHE | — | 1 M KOH + 0.1 M KNO3 | 98 | ||
| NO2RR | SAC | Co1Ru | Co: 3.3 wt%a | 476.8 μmol h−1 cm−2 (H-cell)/2379.2 μmol h−1 cm−2 (flow cell) | 94.2%/92% | −0.7 V/−0.8 V | 0.5 M Na2SO4 + 0.1 M NaNO2 | 106 |
| CuRh1 | Rh: 10.4 wt%a | 2191.6 μmol h−1 cm−2 | 98.9% | −0.6 V | 0.5 M Na2SO4 + 0.1 M NaNO2 | 107 | ||
| Rh1/BN | Not stated | 2165.4 μmol h−1 cm−2 | 97.83% | −0.7 V | 0.5 M Na2SO4 + 0.1 M NaNO2 | 108 | ||
| DAC | FeCu DAC | Not stated | 24526 μmol h−1 mgcat−1 |
99.88% | −0.6 V | 0.1 M KOH + 0.1 M NaNO2 | 115 | |
| NORR | SAC | W1/MoO3−x | W: 13.7 wt% | 308.6 μmol h−1 mg−1 @−0.5 V vs. RHE/38.4 mgNH3 h−1 mgW−1 | 91.2% @ −0.4 V vs. RHE | — | 0.5 M Na2SO4 | 109 |
| Cu1/MoS2 | Cu: 1.58 wt% | 337.5 μmol h−1 cm−2 | 90.6% | −0.6 V | 0.5 M Na2SO4 | 111 | ||
| Ir1/a-MoO3 | Ir: 7.6 wt% | 438.8 μmol h−1 cm−2 | 93.2% | −0.47 V | 0.5 M Na2SO4 | 110 | ||
| Urea synthesis | SAC | Cu-GS-800 | 3.9 wt% | 1.8 mgurea h−1 mgcat−1/4.3 nmolurea s−1 cm−2/46.2 mgurea h−1 mgCu−1 | 28% | −0.9 V | 0.1 M KHCO3 + 0.1 M KNO3 + CO2 | 126 |
| L-Cu1–CeO2 | 3.99 wt% | 52.84 mmolurea h−1 gcat−1/79.5 mgurea h−1 mgCu−1 | Not stated | −1.6 V | 0.1 M KHCO3 + 50 mM KNO3 + CO2 | 127 | ||
| DAC | B-FeNi-DASC | Fe: 0.991 wt%, Ni: 0.896 wt% | 20.2 mmolurea h−1 gcat−1 | 17.80% | −1.5 V | 0.1 M KHCO3 + 50 mM KNO3 + CO | 119 | |
| Pd1Cu1–TiO2 | Not stated | 166.67 molurea molPd−1 h−1/10 mmolurea h−1 gcat−1 | 22.54% | −0.5 V | 0.1 M KHCO3 + CO2 + N2 | 129 | ||
Non-metallic catalysts were reported as well. Yu et al. doped 6.2% concentration Boron onto graphene and managed to achieve a high NH3 FE of 10.8%.71 The electronegative difference between Boron (2.04) and Carbon (2.55) results in Boron having a positive charge of +0.59e. This small positive charge allowed Boron to adsorb N2 easily, acting as excellent catalytic centers for NRR. In addition, competitive HER was suppressed due to the unfavourable adsorption of H+ ions, thus further boosting NH3 FE.
Computational studies on SACs have been conducted as well. Various transition metal-porphyrins (TM-PPs) were studied by Xue et al.72 They found that Ti, V, Co, Zr, Ru and Hf should be excluded due to their high first hydrogenation energies. W-PP was found to exhibit the best performance. This could be attributed to the strong interaction between W and N atoms, which was due to the hybridisation of W-5d and Nc-2p orbitals, resulting in W having a charge of +0.51e and a magnetic moment of 2.23μB. This charge and magnetic moment led to good N2 adsorption and HER suppression.
2.2 Diatom catalysts
The application of DACs towards NRR is lacking, with most works being theoretical only. There is still much room for research in DACs for NRR. DAC for NRR has the advantage of the cooperative effect as the catalytic centers are in close proximity to each other, matching the bond length of NN triple bond.
Fe and Cu atoms were synthesized with a ratio of 1:1 on graphitic carbon nitride (CNT@C3N4-Fe&Cu), simultaneously coordinating with each other and surrounding N atoms.42 A two-step synthesis process was developed: the precursor solution was first lyophilised followed by annealing under an inert atmosphere (Fig. 2a). These confined atoms formed a “sub-nano” reactor with three atoms (Fig. 2b–d), achieving a high NH3 yield rate of 9.86 μg h−1 mg−1 at −1.2 V vs. Ag/AgCl. A significantly higher FE of 34.0% vs. −0.8 V vs. Ag/AgCl was obtained as compared with single Fe catalyst (15.3%) and single Cu catalyst (22.0%). This excellent performance can be attributed to the triple metal atoms consisting of both Fe and Cu. An adsorbed N2 molecule is stabilised by all three atoms at once, resulting in a weaker N–metal bond and NH3 formed can be desorbed with lower energy. The sub-nano reactor consisting of both Fe and Cu atoms catalyst also changed the potential-determining step (PDS) to the first hydrogenation step with a low energy barrier of 0.58 eV (Fig. 2e), compared with Fe-only catalyst (0.71 eV) and Cu-only catalyst (0.86 eV). The metal–metal coordination between Fe and Cu facilitated electron transfer better compared with metal–N coordination in conventional SAC. In another paper by Wang et al., a DAC consisting of dual vanadium atoms with oxygen bridge on N-doped carbon (O–V2-NC) was synthesized by a template-assisted pyrolysis method.73 The catalyst exhibited an extremely high NH3 FE of 77.2% at 0 V vs. RHE and a yield rate of 26 μg h−1 mg−1 at −0.4 V vs. RHE. Extended X-ray absorption fine structure (EXAFS) showed the presence of V–V bond with an unusually long bond length of 2.70 Å. The authors attributed this anomaly to the O atom bridging the two V atoms. This was further proved by treatment of the catalyst in 5% H2 at 250 °C for 2 hours, where the V–O bond disappeared. The catalyst also exhibited good stability after 10 consecutive cycling tests, showing V leaching of only 5 parts per billion (ppb) after the cycling tests. Post-test characterisations also showed no changes to the catalyst structure. On the other hand, DFT study found that during the protonation process, V–O–V broke down to V–O and isolated V due to hindrance of *NNH, leaving vanadium connected to *NNH alone instead of V–O–V*NNH. This transformed structure remained until nitrogen species were released from vanadium and it was regenerated back to V–O–V.
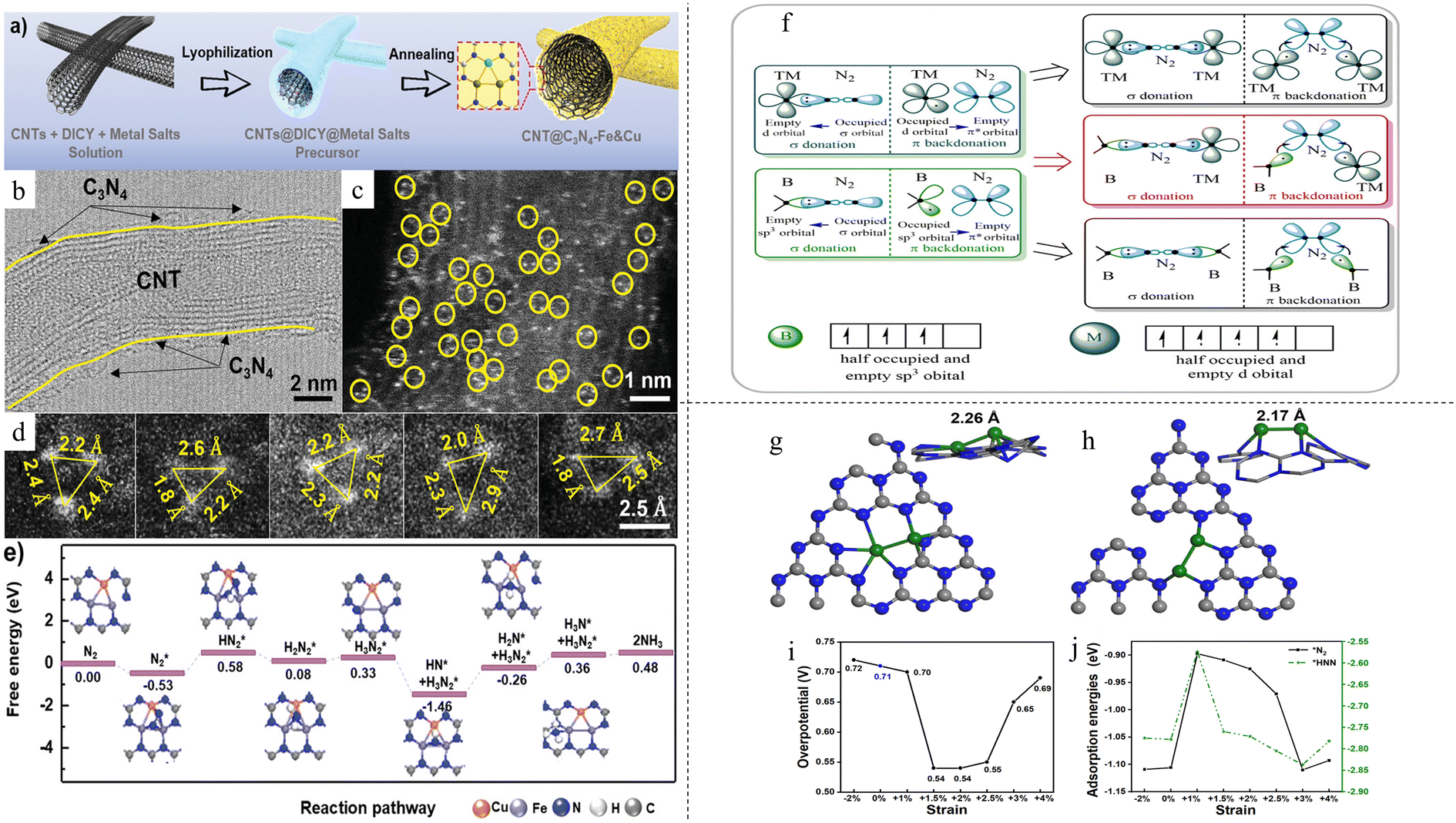 | ||
| Fig. 2 (a) Schematic diagram for synthesis process and morphology of CNT@C3N4-Fe&Cu (b) TEM image of CNT@C3N4-Fe&Cu, (c and d) STEM image of CNT@C3N4-Fe&Cu. Reproduced with permission.42 Copyright 2020, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. (f) Illustration of catalytic mechanism of SAC and DAC. Reproduced with permission.74 Copyright 2021 American Chemical Society. (g) Fe2 in-plane of g-C3N4 (h) Fe2 on the side of the g-C3N4 (i) and (j) effect of strain on overpotential and adsorption energy, respectively. Reproduced with permission.75 Copyright 2021 American Chemical Society. | ||
The local environment of the active centers for DACs can be easily tuned via various strategies, such as changing the substrate that the DACs are anchored on, utilisation of doping and even changing the DACs themselves. A theoretical study was done to test the feasibility of using boron and transition metals (TMs) (B-TM@g-CN) as DACs.74 As illustrated in Fig. 2f, both B and TMs can act as the catalytic active centers for NRR because d orbitals of TMs and sp3 hybrid orbitals of B are able to accept the lone-pair electrons of N2. The difference in charge transfer ability of the two centers can enhance the polarisation of adsorbed N2 and reduce the N–N bond strength. From this, a hypothesis was put forward: “The greater the difference in valence electron distribution between the two atoms in the DAC, the more favourable it is to achieve low potential nitrogen fixation”. It was found that electrons were more likely to enter the antibonding orbitals on B-TM@g-CN and the difference in electron supply caused N2 to polarise greatly, easily pulling N2 apart. 29 TMs were tested, and it was found that Ti and Ta showed the best results with low limiting potentials of 0.13 V and 0.11 V, respectively, for NRR and high potentials of 0.54 V and 0.82 V, respectively, for competitive HER. In another paper, investigation was done on iron dimer Fe2 on g-C3N4 monolayer.75 Two most stable geometries were found, with both geometries comprising the two iron atoms containing positive charges and located side by side – together in the same plane as g-C3N4 or on one side of the g-C3N4 matrix, as shown in Fig. 2g and h, respectively. It was postulated that during the electrocatalytic process, the two geometries may interchange. Further study on the strain effect was done, as it is known that strain can tune the catalytic properties of transition metals. When a tensile strain of 2% was applied, the rate-determining step changed with a decrease in overpotential (Fig. 2i). This was attributed to the change in geometry due to strain. One Fe–N bond was shortened due to the strain, weakening the binding between the two Fe atoms. This resulted in a much stronger interaction between the other Fe atom and adsorbates. Most importantly, the scaling relation was shown to be broken. Adsorption energies of *N2 and *HNN were expected to be monotonic and smooth with the magnitude of applied stress. However, for iron dimer DAC, the adsorption energy of *N2 exhibited a hysteresis instead (Fig. 2j). The effect of coordination on DAC was studied by Wu et al.76 They found that when B or O was doped, the adsorption free energy of N2 increased, enhancing the activation of reactants. Linear scaling of adsorption energy between intermediates was also found to be poor in both cases. In particular, Mn2ON5/G was found to be the most active DAC, with a limiting potential of −0.27 V, much lower than that of MnN6/G with a limiting potential of −0.48 V. Mn2ON5/G was found to adopt the enzymatic pathway instead of the distal pathway that Mn2N6/G adopted. Alkaline earth (AE) metals are typically not used as catalysts as they lack partially filled d-orbitals for the acceptance-donation reaction with reactants. However, a study by Wu et al. showed that AE metals can form partially filled d orbitals by the diffusion of electrons from the ns orbital to low-lying (n − 1)d orbitals.77 This was illustrated in Ca2 dimer, where the d-orbital gained 1.08 electrons when forming Ca2@WS2 DAC, allowing for adsorption and activation of reactants. AE metals were thought to have difficulty catalysing NRR as they lack partially occupied d-orbitals for the acceptance-donation reaction with N2. It was also found that the adsorption strengths of the intermediates have poor scaling relation, with R2 values of 0.80 for adsorption energies between *N2 and *NH2 and 0.79 for adsorption energies between *NH2 and *NH3, further improving NRR activity. Further testing found that CaBa@MoSe2 DAC has the lowest limiting potential of −0.6 V owing to the synergistic effect between Ca, which has a stronger adsorption for NxHy, and Ba which has a weaker adsorption for NxHy, facilitating N2 adsorption and activation while at the same time reducing the binding strength of *NH2, reducing the amount of energy required to convert it to *NH3.
3 Ammonia from nitrate reduction reaction
Compared with NRR, NO3RR can achieve a much higher NH3 FE and yield rate owing to the high solubility of nitrate in water and the lower amount of energy required for bond dissociation (N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond requires 204 kJ mol−1)78 compared with dinitrogen (NN bond requires 941 kJ mol−1)79 However, the mechanism of nitrate reduction is much more complex, requiring a total of eight electrons and nine protons. This brings about the issue of selectivity as nitrate (with an N valence state of +5) can be reduced to other N species with lower valence state of N instead of fully reduced to ammonia. There are two different classifications of reaction mechanism for NO3RR: direct association vs. dissociation–association and electron transfer reduction vs. atomic hydrogen reduction. Nitrate can be adsorbed mainly via two configurations: one oxygen atom bonded to the catalytic surface or two oxygen atoms bonded to the catalytic surface. In the direct association pathway, NO3− is adsorbed and hydrogenated to *NO3H intermediate, which is then sequentially hydrogenated to ammonia, losing oxygen atoms first before gaining hydrogen atoms. In the dissociation–association mechanism, after NO3− is adsorbed, it is dissociated into *O and *NO2 and adsorbed on two different active sites. The two adsorbed moieties are then hydrogenated sequentially. In addition, while going past the step of *NO intermediate, there are four further possible pathways: O-end, O-side, N-end and N-side.80 In the electron transfer reduction mechanism, the adsorbed nitrate is first reduced to nitrite, which is the rate-determining step. Nitrite, which is highly reactive, will be quickly reduced to *NO, which is the determining point of what product is produced. While *NO can be further reduced to ammonia, it can also desorb and ultimately form N2. In the atomic hydrogen reduction mechanism, atomic hydrogen, being a strong reducing agent, can reduce the reactant NO3− and other intermediate species including NO2− and NO. Formation of the N–H bond is more kinetically favourable as compared with the N–N bond when mediated by atomic hydrogen, therefore the dominant product of this mechanism is ammonia.81 On the other hand, since nitrate, as a common pollutant of wastewater, poses a toxic threat to the ecosystem and humans, there are a multitude of studies that focus on removing nitrate from water as nitrogen gas.82–84 Some other issues with current NO3RR to ammonia include difficulty with achieving industrial current densities at low overpotentials, difficulty achieving high ammonia yield rate and FE across a wide range of pH of electrolyte and nitrate concentrations, and finally considering whether, if the nitrate source used is from the wastewater industry, there exist other components which may poison the catalyst.85 SACs can increase the selectivity of ammonia over nitrogen due to the atomic catalysts being isolated with an absence of adjacent active sites. This lack of adjacent sites inhibits N–N coupling, thus reducing the formation of N2 during nitrate reduction.
O bond requires 204 kJ mol−1)78 compared with dinitrogen (NN bond requires 941 kJ mol−1)79 However, the mechanism of nitrate reduction is much more complex, requiring a total of eight electrons and nine protons. This brings about the issue of selectivity as nitrate (with an N valence state of +5) can be reduced to other N species with lower valence state of N instead of fully reduced to ammonia. There are two different classifications of reaction mechanism for NO3RR: direct association vs. dissociation–association and electron transfer reduction vs. atomic hydrogen reduction. Nitrate can be adsorbed mainly via two configurations: one oxygen atom bonded to the catalytic surface or two oxygen atoms bonded to the catalytic surface. In the direct association pathway, NO3− is adsorbed and hydrogenated to *NO3H intermediate, which is then sequentially hydrogenated to ammonia, losing oxygen atoms first before gaining hydrogen atoms. In the dissociation–association mechanism, after NO3− is adsorbed, it is dissociated into *O and *NO2 and adsorbed on two different active sites. The two adsorbed moieties are then hydrogenated sequentially. In addition, while going past the step of *NO intermediate, there are four further possible pathways: O-end, O-side, N-end and N-side.80 In the electron transfer reduction mechanism, the adsorbed nitrate is first reduced to nitrite, which is the rate-determining step. Nitrite, which is highly reactive, will be quickly reduced to *NO, which is the determining point of what product is produced. While *NO can be further reduced to ammonia, it can also desorb and ultimately form N2. In the atomic hydrogen reduction mechanism, atomic hydrogen, being a strong reducing agent, can reduce the reactant NO3− and other intermediate species including NO2− and NO. Formation of the N–H bond is more kinetically favourable as compared with the N–N bond when mediated by atomic hydrogen, therefore the dominant product of this mechanism is ammonia.81 On the other hand, since nitrate, as a common pollutant of wastewater, poses a toxic threat to the ecosystem and humans, there are a multitude of studies that focus on removing nitrate from water as nitrogen gas.82–84 Some other issues with current NO3RR to ammonia include difficulty with achieving industrial current densities at low overpotentials, difficulty achieving high ammonia yield rate and FE across a wide range of pH of electrolyte and nitrate concentrations, and finally considering whether, if the nitrate source used is from the wastewater industry, there exist other components which may poison the catalyst.85 SACs can increase the selectivity of ammonia over nitrogen due to the atomic catalysts being isolated with an absence of adjacent active sites. This lack of adjacent sites inhibits N–N coupling, thus reducing the formation of N2 during nitrate reduction.
3.1 Single-atom catalysts
Several types of metallic SACs using different metal elements have been reported for nitrate reduction. Fe is a commonly used TM for its abundance and low cost. Li et al. synthesized Fe SACs (Fe-PPy SAC) using a polymer-hydrogel strategy, achieving an NH3 FE of almost 100% and a yield rate of 30 molNH3 h−1 gFe−1 at −0.6 V vs. RHE.86 Fe content was found to be 2.38 wt%. The formation of isolated Fe atoms resulted in d band energy splitting and a decrease in electron density near the Fermi level. NO3− adsorbed on Fe-PPy SAC was more active compared with Fe NPs due to the pronounced hybridization between oxygen d band and Fe band as well as the overlapping electrons at the antibonding orbitals.63 Liu et al. found that Fe on N-doped carbon, synthesized under a pyrolysis temperature of 900 °C, yielded the best NO3RR-to-NH3 performance due to the dominant active sites of Fe–N3.87 Pyrolysis at 800 °C and 1000 °C was found to yield dominant active sites of Fe–N4–OH and Fe–N4, respectively, which produced poorer results. NH3 FE of 86.7% at −0.7 V vs. RHE and NH3 yield rate of 1.10 mmolNH3 h−1 mgcat−1 at −0.9 V vs. RHE was achieved. Zhang et al. synthesized Fe SACs with O and N coordination (FeN2O2) and a metal loading of 2.0 wt%.88 The catalyst was prepared by pyrolyzing the pre-synthesized metal–organic framework (MOF) precursor (Fig. 3a). The catalyst showed an NH3 FE of 92% at −0.68 V vs. RHE and a yield rate of 46 mg h−1 mgcat−1 at −0.88 V vs. RHE. LSV curves of nanoparticles, Fe SAC with a lower loading of 1.6 wt% and blank support showed the synthesized Fe SACs exhibited the highest increase in NH3 partial current density. The catalyst exhibited good stability over 15 cycles of testing as well, showing stable FE and yield rate (Fig. 3e). DFT study showed that nitrate was more easily adsorbed on FeN2O2 and formation of nitrite had a higher energy barrier when compared with FeN4 (Fig. 3f). | ||
| Fig. 3 (a) Schematic for the synthesis of Fe SAC from MOF precursor. (b) LSV curves of Fe SAC in electrolytes with and without KNO3. (c) NH3 FEs of Fe SAC across different potentials. (d) LSV curves of Fe SAC (2.0 wt%), Fe SAC (1.6 wt%), Fe nanoparticles and blank support. (e) Stability test of Fe SAC over 15 cycles. (f) Gibbs free energy diagram of NO3RR on FeN2O2 and FeN4. Reproduced with permission from ref. 88, copyright 2022 Elsevier B.V. (g) Operando first-order derivatives of XANES at various potentials. (h) LCF results of Cu K-edge XANES spectra at various potentials. (i) Mechanism of Cu SAC aggregation and dispersion. Reprinted with permission from ref. 90. Copyright 2022 American Chemical Society. | ||
While copper is cheap and abundant, there are various issues when using Cu in electrocatalysis: deactivation of catalyst after long operation due to passivation, leaching and corrosion, and accumulation of toxic secondary product of nitrite.89 Cu SACs can circumvent these issues. Zhu et al. synthesized Cu atoms supported on nitrogenated carbon nanosheets, demonstrating good activity and stability towards NO3RR.89 The catalyst synthesized at 800 °C showed the best (minimum) selectivity towards nitrite of only 5.0%, a stark contrast to Cu plate showing 60.6% selectivity. Furthermore, stability testing after 20 cycles showed an electrocatalytic activity drop of only 7.2%. The drop was due to the formation of Cu nanoparticles, but this aggregation was not significant based on the absence of additional peaks in the X-ray diffraction (XRD) pattern. Further DFT studies showed the established rate-determining step of adsorption was much lower for that of Cu SAC: 6.13 eV for Cu(111) in bulk copper vs. −4.06 eV for Cu–N2 and −2.05 eV for Cu–N4 in Cu SAC. In another instance, Cu SAC on N-doped carbon was synthesized and phase transformation was observed as the potential was changed from 0 to −1 V vs. RHE as evident in the operando X-ray absorption spectroscopy (XAS) measurements.90 The first-order derivatives X-ray absorption near edge structure (XANES) spectra and linear combination fitting (LCF) results of Cu K-edge XANES spectra in Fig. 3g and h revealed that the Cu–N4 structure transformed from Cu–N3 to near free Cu0 single atoms to eventually aggregated Cu0 nanoparticles. The NH3 FE and yield rate were enhanced throughout the transformation, reaching a maximum FE of 84.7% and yield rate of 0.264 mmolNH3 h−1 mgcat−1 at −1 V vs. RHE, indicating that these nanoparticles are active sites that contribute greatly to the NO3RR process. It was also found that the nanoparticles can be restored back to the SAC state of Cu–N4. However, the restoration is not as straightforward as the forward process by simply changing the potential from −1 V back to 0. It must be done so in an oxidative environment, via the formation of [Cu(OH)4]2− intermediate which is captured by the pyridinic nitrogen on the carbon support (Fig. 3i).
SACs composed of other cheap and abundant transition metals such as Ni and Zn have also been used for NO3RR to NH3. Zhao et al. synthesized Zn SACs with 1.33 wt% on microporous N-doped carbon (ZnSA-MNC).91 They managed to achieve an NH3 yield rate of 39000 μg h−1 mgcat−1 at −1.0 V vs. RHE, 4 times that of the corresponding nanoparticles. A high FE of 94.8% at −0.9 V vs. RHE was achieved as well. The selectivity of ammonia was also very high at 97.2% due to the lack of adjacent sites in the isolated active metal centers preventing N–N coupling and reducing N2 selectivity. Further DFT study showed Zn single atoms possessed a positive charge of +1.58e which was conducive for the adsorption of negatively charged NO3−. Zhang et al. synthesised Ni SAC with either NiO4 or NiN4 coordination on conjugated coordination polymers (CCP) using a one-step solvothermal method.92 CCP was selected due to the enhancement of electrical conductivity via interactions between the π system of aromatic domains in CCP and the d orbitals of transition metals. Both configurations exhibited a high metal loading of around 18 wt%. In contrast to NiN4-CCP, which exhibited an NH3 FE of 61.8% and a yield rate of 0.86 mmol h−1 mg−1, NiO4-CCP was able to deliver a superior NH3 FE of 94.7% and a yield rate of 1.83 mmol h−1 mg−1 at −0.7 V vs. RHE. Additionally, the catalyst demonstrated good stability over the course of three days of continuous chronoamperometry testing, exhibiting no structural alterations and no obvious decline in both current density and NH3 FE.
Precious metals have been used for their generally excellent catalytic activity. Au single atoms were anchored on two-dimensional (2D) Cu nanosheets with (111) single crystal surface and Cu vacancies (VCu-Au1Cu SAA) via a three-step process (Fig. 4a).93 Single-crystal Cu nanosheets were first prepared by chemical reduction, then single Au atoms were deposited via galvanic replacement. Finally, the resulting product was dispersed in acetic acid for etching to generate vacancies in the copper nanosheet. High-resolution TEM (HRTEM) and energy dispersive X-ray spectroscopy (EDS) (Fig. 4c and d) showed the structure and distribution of Au on Cu nanosheet with Au loading of 0.41 wt%. HAADF-STEM (Fig. 4b) revealed the presence of both Au single atoms and Cu vacancies. NH3 FE of 98.7% and yield rate of 555 μg h−1 cm−2 were achieved at −0.2 V vs. RHE, superior to those of pristine copper nanosheet, copper nanosheet with vacancies and Au NPs (Fig. 4e–g). Mechanism study showed that Au atoms and Cu vacancies can prevent HER by inhibiting the Heyrovsky step: *H + H2O + e− → H2 + HO−. Single Ru atoms anchored on an amorphous layer of monolithic Ti support was synthesized,94 exhibiting an NH3 FE of 87.6% and a yield rate of 22.2 mol g−1 h−1. The authors tested the scaling capability of their synthesized catalyst and found that Ru atoms were evenly dispersed with no aggregation even when the titanium-based monolithic single-atom electrode was increased from 2 × 2 cm to 25 × 15 cm. The synthesis method was found to be applicable to other single-atom electrodes, such as Sn, Bi, Pd, and Pt, demonstrating the versatility of the synthesis method.
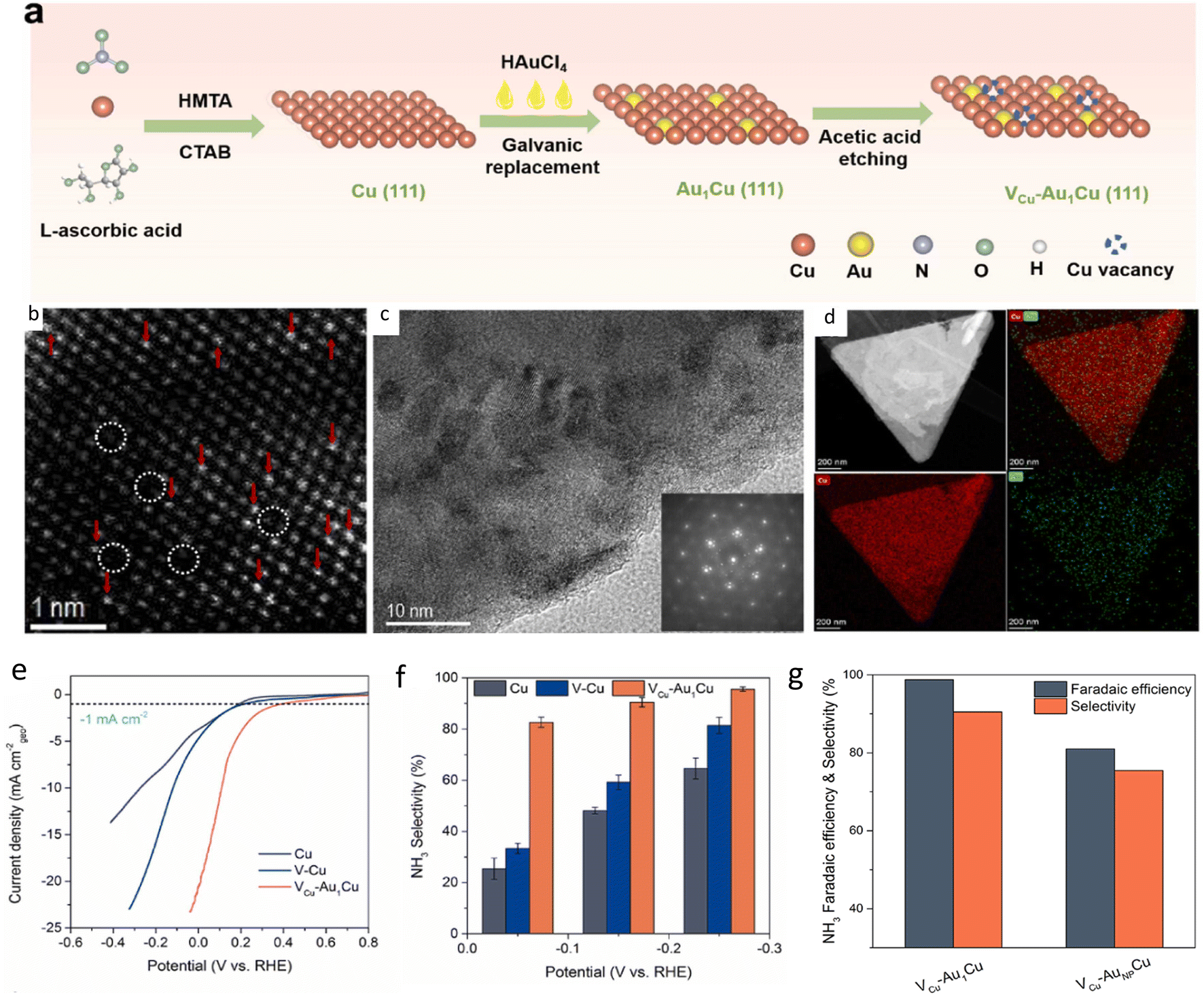 | ||
| Fig. 4 (a) Scheme illustrating synthesis of VCu-Au1Cu SAA. (b) HAADF-STEM image. Single Au atoms indicated by red arrows while Cu vacancies are circled in white. (c) HRTEM image and fast Fourier transformation (FFT) pattern of VCu-Au1Cu SAA. (d) EDS mapping showing distribution of Cu and Au elements. (e) LSV curves comparison of various synthesized catalysts. (f) Selectivity of various samples. (g) NH3 FE and selectivity of SAC compared with NP. Reproduced with permission from ref. 93. Copyright 2022 Elsevier B.V. | ||
In a theoretical work by Wu et al., AE metals were screened for their feasibility to be used as SACs for NO3RR.95 Similar to NRR, it had been thought that the lack of partially filled d-orbitals renders AE metals catalytically inactive. However, it was proved experimentally that AE metals have catalytic properties, being able to catalyse ORR. Nitrate can adsorb onto AE metals with two O atoms binding to the AE active site, with an adsorption energy similar to that of transition metals. The change in Gibbs free energy from binding of nitrate was also found to be more negative than that of hydrogen, indicating a more favourable NO3RR instead of competitive HER. The limiting potential of SrN3C and BaN3C was found to be −0.05 V, even lower than those of TM SACs. Interestingly, AE metal was stabilised by ionic bond interactions with the N-doped support. Similarly, NO3− was mainly adsorbed via ionic bond interaction. The N-doped graphene support acted as an electron reservoir while the AE metal acted as a transmitter, allowing for the mimicking of TM's donation–back donation mechanism of activating nitrate.
3.2 Diatom catalysts
Similar to DACs for NRR, the application of DACs for NO3RR is lacking as well. As stated in the introduction, SACs have the issue of agglomeration, resulting in nanoparticles that may reduce the product FE and selectivity, which is especially significant for NO3RR. DACs have a different coordination that can change the electronic environment to alter the production selectivity and reaction activity.96 Therefore, there is much potential to be seen from the advancement of DACs for NO3RR.In what might be the first synthesis of DACs for NO3RR to NH3, Wang and colleagues synthesized Cu–Ni DAC anchored in N-doped carbon (Cu/Ni-NC) by pyrolyzing ZIF-8 MOF precursor97 (Fig. 5a). The synthesized materials consisted of both Ni–Cu dual atoms as well as Cu and Ni single atoms, as evident in the atomic resolution HAADF-STEM image (Fig. 5b). They were able to achieve a stable NH3 FE of ∼97% and yield rate of 5480 μg h−1 mgcat−1 cm−2 at −0.70 V vs. RHE (Fig. 5d). LSV comparison with Cu SAC and Ni SAC in Fig. 5c showcased the superiority of the synthesized Cu/Ni-NC as it had the greatest increase in current density. DFT study showed that the optimal adsorption site of nitrate was N-coordinated Cu–Ni dual–single-atom rather than single Cu or Ni atom due to the much lower adsorption energy of 0.5 eV. The rate-determining step of the deoxidation process of HNO* to N* on Cu–Ni (−0.37 eV) was also much lower than those on Cu (1.03 eV) and Ni (1.30 eV) (Fig. 5e). Stronger hybridisation was also found between Cu–Ni and O of adsorbed nitrate, effectively forming chemical bonds of Cu–O–NO3−*–O–Ni and promoting electron transfer from the DAC to NO3−*. Zhang et al. synthesized Fe/Cu DACs anchored on holey edge sites of nitrogen-doped graphene (HNG).98 Holes were made in graphene to create a large number of edge sites which were then nitrified to bind Fe/Cu atoms, forming a “Y-type” ML3 structure (Fe/Cu-HNG). Compared with homogeneous diatomic catalysts (Fe/Fe-HNG and Cu/Cu-HNG) and SACs (Fe-HNG and Cu-HNG), Fe/Cu-HNG exhibited the best performance with an NH3 FE of 92.51% at −0.3 V vs. RHE and a yield rate of 1.08 mmol h−1 mg−1 at −0.5 V vs. RHE. DFT study showed that two oxygen atoms of NO3− were adsorbed to Fe/Cu dual sites, with a higher binding energy (−1.19 eV) compared with Fe SACs (−0.89 eV) and Cu SACs (−0.56 eV). This high binding energy led to a lower energy barrier of the first discharge step: * + NO3− → NO3* + e−. Subsequent adsorption energies of intermediate products were also shown to have medium binding energies compared with Fe and Cu SAC, with all steps showing a downhill trend (Fig. 2d).
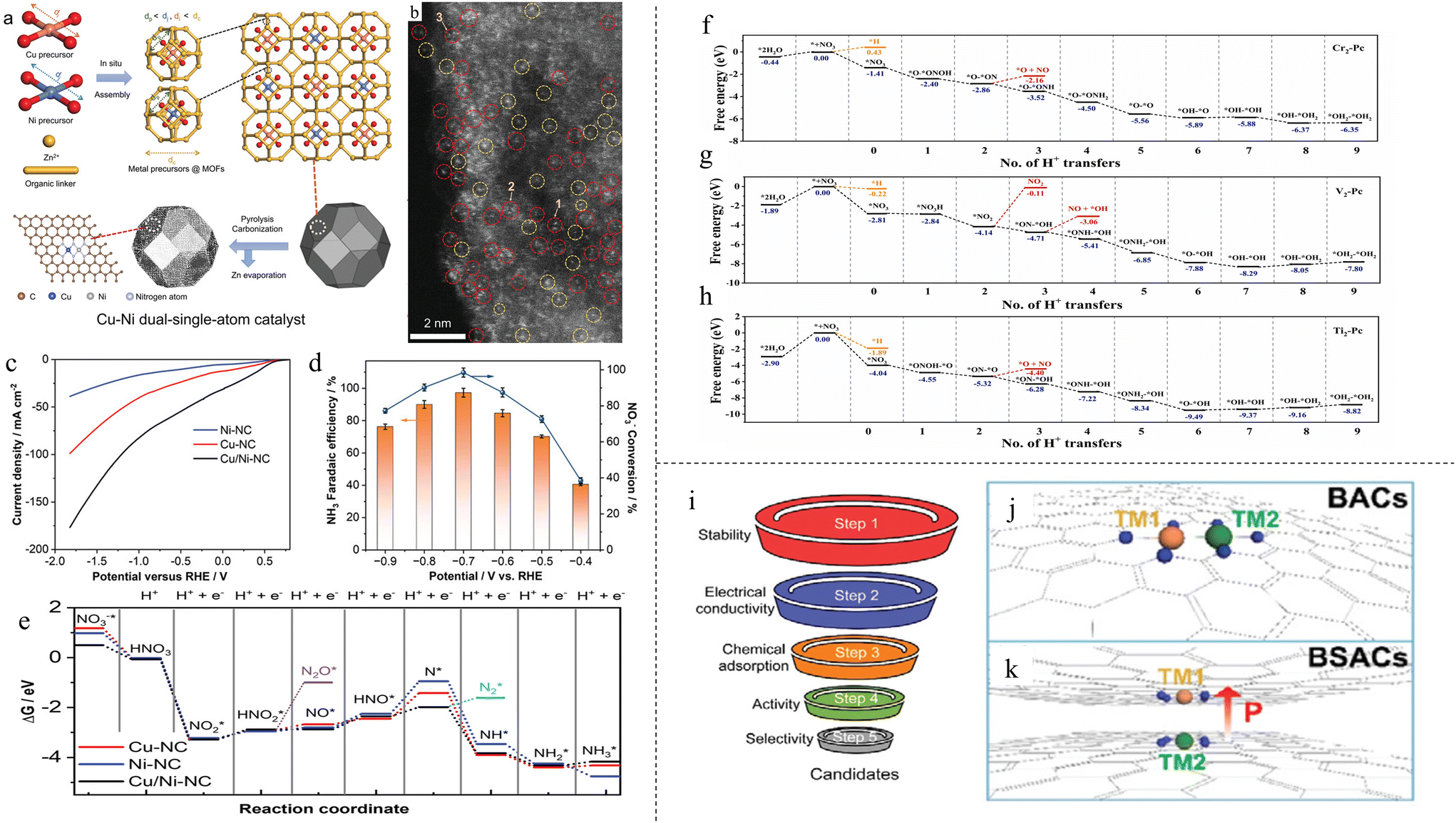 | ||
| Fig. 5 (a) Schematic of the synthesis of Cu/Ni-NC. (b) Atomic resolution HAADF-STEM image of Cu/Ni-NC. Cu/Ni DACs are circled in red; Cu or Ni SACs are circled in yellow. (c) LSV curves of Ni SAC, Cu SAC and Cu/Ni DAC. (d) NH3 FEs and yield rates of Cu/Ni-NC at various potentials. (e) Free energy diagram of NO3RR. Reproduced with permission.97 Copyright 2023, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. (f–h) Free energy diagrams for Cr2, V2 and Ti2, respectively. Reproduced with permission.99 Copyright 2022, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. (i) Screening methods for filtering elements suitable for BSAC. (j and k) Illustrations of conventional BACs and proposed BSACs, respectively. Reproduced with permission.101 Copyright 2022, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
The effect of varying substrate on DACs was introduced in the following theoretical studies, followed by a novel DACs structure with an entirely different mechanism. Rehman et al. conducted a theoretical study on DACs for NO3RR to NH3 using metal catalysts in 2D expanded phthalocyanine (Pc) substrate.99 Pc was used as it has a strong ability to bind metal atoms, being able to accommodate both TMs and main group metals. It was found that Cr2-Pc, V2-Pc, Ti2-Pc and Mn2-Pc DAC showed the best potential for NO3RR, with incredibly complex mechanisms. As shown in the free energy diagrams (Fig. 5f–h), the limiting potentials were found to be −0.02, −0.25, −0.34 and −0.41 V, respectively, significantly lower than other metals. Nitrate ion binds strongly to the dual-atom site compared with proton, indicating better selectivity to NO3RR over competitive HER. In another paper, catalysts on N-doped graphene were theoretically studied.100 N-doped graphene (N6-G) was chosen due to its higher electrical conductivity compared with graphene and higher tunability to improve electrochemical activity in a coordinated environment. The paper found that Cr2/N6-G, Mn2/N6-G and Cu2/N6-G had the best potential due to their high stability, selectivity (FEs of 100, 61.26 and 99.99%, respectively) and relatively low limiting potentials of −0.46, −0.45 and −0.36 V, respectively. Cr, Mn and Cu were able to effectively adsorb NO3− ions and activate them by the ease of charge transfer from d-orbitals of TMs to the empty p* orbital of the *NO3 molecule. Free energy calculations also showed that hydrogen poisoning can be avoided. In a novel approach to the topic of DAC, Lv et al. proposed an entirely different DAC structure. Instead of the usual focus on “in-plane” DACs where the two catalytic atoms are located in the same plane and dispersed on one support material (Fig. 5j), they proposed a bilayer SAC (BSAC), shown in Fig. 5k.101 This structure has many advantages: prevention of metal aggregation, lack of neighbouring sites in BSAC reducing undesirable N–N coupling as it reduces NH3 selectivity, and preservation of the ability to tune reactivity. This tunability comes from BSAC composed of different catalytic atoms, or heterogeneous BSAC. Heterogeneous BSAC can induce polarisation perpendicular to the surface, resulting in an electric field that can accumulate a higher concentration of NOx, and adjust the binding strength between reactants and catalysts. With this, DFT calculations for homogeneous and heterogeneous BSAC were carried out on a support of N-doped graphene for its structural flexibility and thermal stability. After several rounds of screening and filtering (Fig. 5i), only V2-N4-GN was found to be a promising homogeneous BSAC. It was discovered that there are two ways polarisation can affect the mechanism of NO3RR: (1) Polarisation does not change the mechanism of NO3RR, instead altering the free energy change of each step. This can be seen in the case of NbV-N4-GN. (2) Polarisation completely changes the NO adsorption pattern and reaction pathway, which can be seen in the example of Hf and Zr. TiV-N4, NbV-N4 and GaV-N4-GN were found to be the best three systems to suppress HER, with a limiting potential of −0.32 V, −0.20 V and −0.25 V, respectively.
4 Ammonia from nitric and nitric oxide reduction reaction
Other N-containing compounds such as nitrite (NO2−) and nitric oxide (NO) were also studied as the nitrogen source in synthesizing ammonia via NO2−RR and NRR, respectively. Both compounds are key intermediates present during the NO3RR process. As stated, reduction of nitrate to nitrite is commonly known to be the rate-determining step of NO3RR. Using nitrite as the starting reactant will skip this energy barrier, potentially resulting in better catalytic activity and higher yield rate of ammonia. On the other hand, NO is the intermediate at which the final product is determined. Having an in-depth study of NORR will allow an insight into the selectivity mechanism and result in a potential improvement of ammonia selectivity. The reaction mechanism of NO2− follows the same path as NO3−.102 NORR, on the other hand, has a more complex mechanism. NO can adsorb onto the catalyst via O-end, N-end and side-on. Afterwards, the reaction takes place via the associative or dissociative pathway, similar to that of NO3RR. The hydrogenation can follow a Tafel-type path in which protons are adsorbed onto the catalyst surface before hydrogenation takes place, or a Heyrovsky-type path in which *NO and intermediates are protonated directly. The associative pathway is further split into distal and alternating pathways. In the distal pathway, the N or O atom is fully hydrogenated before the other atom is hydrogenated, whereas in the alternating pathway the N and O atoms are hydrogenated alternately.103 In addition, NO is hazardous to both health and the environment: it can cause photochemical smog, oxidise in atmosphere to form acid rain resulting in ecosystem acidification and eutrophication, and deplete the Earth's ozone layer.104,105 Therefore, the electrocatalytic NORR also represents a sustainable route for upgrading NO wastes into valuable NH3.4.1 Single-atom catalysts
Starting with NO2RR, the SACs exhibited excellent stability; Wang et al. anchored single Co atoms on Ru (Co1Ru) and achieved an ammonia FE of 92% and a yield rate of 2379.2 μmol h−1 cm−2 in a flow cell.106 The catalyst displayed good stability over 10 cycles and 30 h of chronoamperometry in an H-cell while maintaining a current density of 410 mA cm−2 over 100 h in a flow cell. The rate-determining step of *NH2O → *NH2OH on Co1Ru was found to have a lower energy barrier compared with Ru (0.5 eV vs. 0.77 eV). HER was also found to be suppressed as the binding free energy of NO2 was −1.55 eV, more negative than that of *H of −0.71 eV. In another paper, Rh atoms anchored on Cu (CuRh1) was synthesized and an ammonia FE of 98.9% and yield rate of 2191.6 μmol h−1 cm−2 at −0.6 V vs. RHE was achieved.107 Stability test in a flow cell also showed good stability over 15 cycles and 100 h of chronoamperometry testing. DFT calculations showed that the addition of Rh1 into Cu improved electron transfer to *NO2−, improving catalyst/reactant interactions. CuRh1 was also found to facilitate H2O dissociation for proton generation, improving hydrogenation of *NO2. From these improvements, a tandem pathway for NO2RR was proposed: Rh1 activated the initial adsorption and hydrogenation of NO2− to *NO. Afterwards, *NO was transferred to the Cu substrate, where the rest of the hydrogenation steps occurred. In another work utilising Rh, Rh single atoms were anchored on defective BN nanosheets via a simple two-step procedure: firstly, defective BN nanosheets were obtained by liquid phase exfoliation. Then, isolated Rh atoms were anchored by the impregnation method (Fig. 6a).108 An impressive ammonia FE of 97.83% and a yield rate of 2165.4 μmol h−1 cm−2 at −0.7 V vs. RHE were achieved using a flow cell. The current density increase using flow cell showed a drastic improvement over H cell (Fig. 6b). DFT studies showed that Rh1 displayed a significant electron-deficient state, showing a strong stabilisation on BN substrate. The stability of the catalyst was also experimentally tested through a continuous chronoamperometry test of 200 h in a flow cell and a current density of 355.7 mA cm−2 was maintained throughout (Fig. 6c). Competitive HER was also shown to be highly suppressed, with *H having a much higher binding free energy of 0.62 eV compared with −0.15 eV for NO2−. The radical distribution function (RDF) curve also showed catalyst/*H interaction to be much weaker than catalyst/NO2− interaction.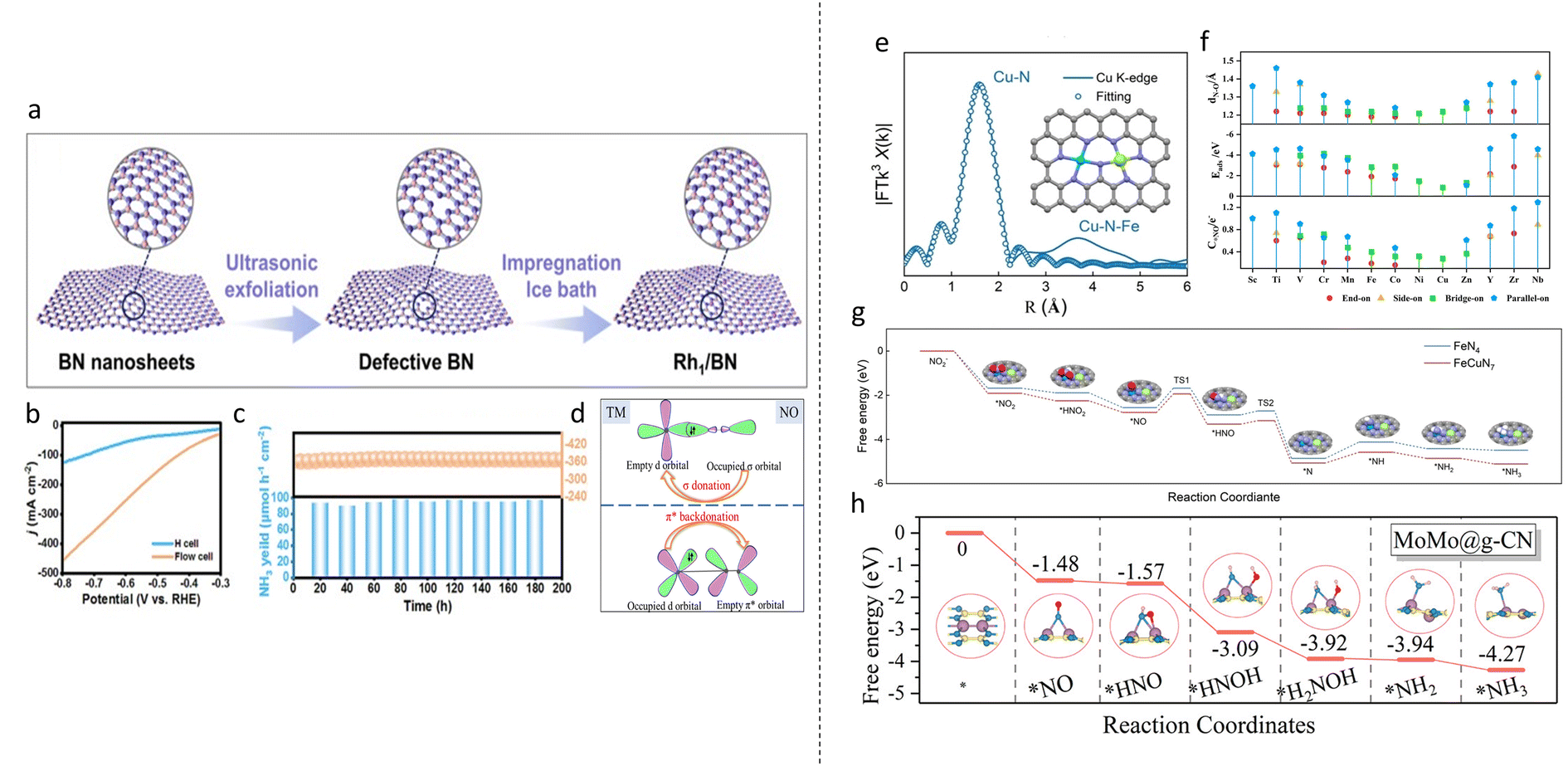 | ||
| Fig. 6 (a) Synthesis of single Rh atoms on defective BN nanosheet. (b) LSV curves of Rh1/BN in H cell and flow cell. (c) Stability test of Rh1/BN over 200 h. Reproduced with permission.108 Copyright 2024, Elsevier Inc. (d) The donation–backdonation mechanic of NORR. Reproduced with permission.113 Copyright 2023 American Chemical Society. (e) R-space curve fitting of FeCu DAC with the proposed structure of FeCuN7. Reproduced with permission.115 Copyright 2023 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. (f) Bader charge number obtained by NO in various configurations. (g) Reaction pathway for FeCuN7. Reproduced with permission.116 Copyright 2024 Elsevier Ltd. (h) Gibbs free energy diagram for NORR on MoMo@g-CN. Reproduced with permission.117 Copyright 2023 American Chemical Society. | ||
Moving on to NORR, the following papers studied the usage of different elements and their different substrates. Chen et al. confined W single atoms in amorphous MoO3−x nanosheet (W1/MoO3−x) with W1–O5 motifs for NORR.109 They managed to achieve a NH3 FE of 91.2% at −0.4 V vs. RHE and yield rate of 308.6 μmol h−1 cm−2 at −0.5 V vs. RHE. Compared with pristine MoO3 on which NO adsorbed weakly, OV-induced coordinatively unsaturated Mo1–O5 and W1–O5 could effectively adsorb and dissociate the NO bond, with W1–O5 further promoting H2O dissociation and retarding *H dimerization to supply adequate protons. W1–O5 motifs also exhibited a dual function forming a self-tandem NORR mechanism, where some W1–O5 activated NO to form intermediates while others dissociated H2O and supplied *H for intermediate protonation. In two other studies done by the same group, Ir SACs110 and Cu SACs111 were synthesized for NORR. In both studies, NH3 FE exceeded 90% and showcased high NH3 yield rates of 337.5 μmol h−1 cm−2 for Cu SACs and 438.8 μmol h−1 cm−2 for Ir SACs. Similar to Rh SACs, the respective SACs showcased unique moieties (Cu1–S3 and Ir1–O5) which boosted the selectivity of NH3 and suppressed HER. In another study by Zhao et al., they demonstrated that the active metal center is pivotal in product selectivity.112 When Ni was anchored on a metalloporphyrin framework epitaxially grown on graphene, NH3 FE reached 81.2% and a yield rate of 1.6 mmol mg−1 h−1 was achieved at −0.51 V vs. RHE, whereas if Fe was used instead, NH2OH was selectively produced with a FE of 83.5% and a yield rate of 3.1 mmol mg−1 h−1 at −0.14 V vs. RHE.
In a theoretical paper published by Hu et al., TMs on 1,3,5-triethynnylbenzene (TM–TEB) were screened as candidates for NORR.113 The TM–TEB system was an assembly of TM atoms and TEB molecules through twofold TM–alkynyl coordination bonds (–CC–TM–CC–), combining the superior properties of SACs and graphynes, such as unsaturated metal coordination, electrochemical stability, high electrical conductivity, easy synthesis and separation, fast mass transfer and effective charge transport. After extensive screening and selection, Cr, Pd and Pt were identified as potential candidates. Cr-TEB was found to be the best candidate, having high NH3 selectivity and low limiting potential. Cr-TEB interacted with *NO by a “donation–backdonation” mechanism (Fig. 6d). The empty d orbitals of TM accepted the electrons of NO-σ orbitals, forming the bonding states to strengthen the adsorption of NO molecules. The occupied d orbitals of TM then back-donated electrons to the empty π* orbitals of NO, leading to the activation of *NO. Protonation of *N2O2 was the potential-determining step with an energy barrier of 0.867 eV for NORR to N2. With such a high energy barrier, N2 selectivity was thus low, improving ammonia selectivity. In another DFT study, Li et al. looked at graphene-based Fe SACs for NORR. 26 systems were investigated, including PD-FeN4 (coordinated N atom replacing one C atom of the six-membered ring), PR-FeN4 (coordinated N atom replacing one C atom of the five-membered ring) and the C, N and O, N coordinated cases.114 Similar to the previous paper, Fe SACs could adsorb and activate NO via the same electron donation–backdonation mechanism. PD-FeN3O and PR-FeN4 were found to have excellent NORR properties with low limiting potentials of −0.33 and −0.26 V, respectively. They also exhibited good selectivity towards NH3, not only suppressing HER but also prohibiting the formation of N2O and N2. Finally, Qian et al. looked at the effect of pH and potential on the product selectivity of NORR. It was found that before −1.60 V, *NO adsorption dominated, whereas any potential more negative than −1.60 V became *H dominated. The branching reaction of *NO was deterministic of what N-containing products would form in the end: if *HNO intermediate was formed, single-N products tended to form (e.g. NH3 or NH2OH). If *ONNO intermediate was formed, N–N coupled products tended to form instead (e.g. N2O or N2). In a highly acidic solution, a lower NO concentration and a more negative applied potential selectively promoted the formation of *HNO, while in a highly alkaline solution, increased NO concentration and a moderately negative applied potential favourably benefited the formation of *ONNO.
4.2 Diatom catalysts
There are several theoretical studies on the topic of DACs for NO2RR and NORR, but experimental studies are severely lacking, with only one paper DACs for NO2RR found. In the experimental paper for NO2RR, an impressive FE and yield rate were obtained, surpassing that of SACs.Cu was introduced into Fe–N–C (iron–metal–carbon) to form FeCu DAC with a configuration of FeCuN7 (Fig. 6e) for NO2RR.115 An impressive NH3 FE of 99.88% and a yield rate of 24526 μg h−1 mgcat−1 at −0.6 V vs. RHE were achieved. DFT study showed that NO2 adsorption was enhanced by the introduction of Cu atom as the d-band center shifted upwards from −1.55 eV (FeN4) to −1.48 eV (FeCuN7). The energy barrier of PDS (*N → *NH) also decreased from 0.74 eV to 0.49 eV (Fig. 6g). The electronic property of the Fe atom was altered upon incorporation of the Cu atom, with a better electron redistribution promoting catalytic conversion of the intermediates, reducing the thermodynamic energy barriers of key steps for NO2RR.
While the experimental works regarding DACs for NORR are limited, several theoretical investigations have been conducted. The effects of substrate on DACs were investigated. Wu et al. published a study on SACs and DACs for NORR, looking at TMs doped on graphdiyne (GDY).118 SACs were found to be thermodynamic process controlled, i.e., NH3 and H2O molecule desorption, while DACs were found to be electrochemical process controlled i.e., hydrogenation steps. For SACs, Cu@GDY was found to have the lowest limiting potential of −0.1 V with a NH3 desorption energy of 2.81 eV. Cu2@GDY, on the other hand, was found to have a limiting potential of −1.04 V. However, Cu2@GDY showed a better selectivity towards ammonia compared with H2. In another paper, TMs on nitrogen-doped graphite was screened and Ti2–N6 was found to exhibit the best catalytic activity and selectivity towards NH3 for NORR, with a low limiting potential of −0.24 V and strongly repulsive against the HER process.116 This addresses the issue that while SACs exhibited good catalytic ability in catalysing NORR, they are not selective and are inclined to generate H2. The origin of the enhanced NH3 selectivity and activity comes from the introduction of adjacent metal atoms in active sites leading to enhanced charge communication. This allows for unique parallel-on structure adsorption of NO on DACs, which was found to be the most favourable and activated configuration of NO adsorption. Bader charge analysis also showed an impressive 0.9e charge donation from substrate to adsorbate, higher than all other configurations (Fig. 6f). Sun et al. conducted a similar study to screen candidates for heterogeneous DACs on graphitic carbon nitride (TM1TM2@g-CN) for NORR.117 TiCr, TiMo and MnMo@g-CN with limiting potentials of −0.37, −0.37 and −0.43 V, respectively, were screened as potential candidates. In particular, MoMo@g-CN was found to be able to reduce NO to NH3 spontaneously. Mo SAC has a 3d orbital that is localised and closer to Fermi level. A second Mo atom is able to modulate the orbital distribution and local electronic structure, making the d-band center in MoMo@g-CN more negative. This resulted in antibonding states between MoMo and NO having an electron occupancy greater than Mo and NO, weakening NO adsorption on MoMo@g-CN. This allowed for the thermodynamically spontaneous reduction of NO to NH3 as the PDS of Mo@g-CN stemmed from hydrogenation of *NO to *NOH (Fig. 6h).
5 Urea synthesis
Electrochemical synthesis of urea is realised with nitrogen-containing compounds (dinitrogen, nitrate, ammonia, etc.) and carbon-containing compounds (usually CO2). This synthesis route is extremely challenging as it requires the simultaneous activation and reduction of nitrogen-containing precursor (NO3RR and NRR etc.) and carbon-containing precursor (usually CO2RR) while needing to deal with competitive reactions such as HER all at the same time. There also lies the problem of side-products such as ammonia and other carbon-containing products being generated from the two reactants. Additionally, the electrochemical synthesis of urea is not purely a electrochemical reaction, with the possible involvement of chemical steps as well.119 These result in a wide range of side products that are C-based, N-based and C–N-based.120 Therefore, with all these challenges, current electrochemical synthesis of urea faces the issues of poor selectivity, poor FE, with most works <50%, and poor yield rate.120 If the N-precursor selected is N2, the reaction mechanism is as follows: CO2 and N2 are adsorbed onto the surface of the catalyst and activated. The adsorbed CO2 is reduced to *CO, which will then migrate to the adsorbed *N2 to form a tower-like *NCON precursor. This precursor is then hydrogenated by either the distal or alternate pathway to urea.14,121 If nitrate/nitrite is chosen as the N-precursor, there are multiple possible mechanisms. In one mechanism, *CO is generated by reduction of CO2 while *NH2 is produced by reduction of NOx.*NH2 then binds to *CO in a tandem reaction, forming urea.122 In another mechanism, *CO is still produced from reducing CO2. Multiple N intermediates are suggested to couple with *CO, such as *NO226,123 and *NH.124 In both cases, the general steps follow that the N-containing moiety is reduced to *NH2 before another N-precursor is coupled with the carbon atom and reduced to the second *NH2, forming urea. If NH3 is selected as the N-precursor, *CO2− intermediate is formed from CO2via an electron transfer process. C–N coupling then occurs as NH3 acts as a nucleophilic reagent and attacks CO2−, forming the *COOHNH2 intermediate. Finally, NH3 and *COOHNH2 undergo dehydration-condensation to form urea.125 Therefore, ACs exhibiting high selectivity and activity will be a good choice in this reaction.5.1 Single-atom catalysts
Very few papers were found pertaining to the use of SACs towards urea electrosynthesis. Leverette et al. did pioneering work to study the application of SACs for urea electrosynthesis from CO2 and NO3, investigating the local environment of the SAC.126 Cu single atoms confined in graphene sheets (Cu-GS) were prepared (Fig. 7a–e). Briefly, lyophilisation with NaCl template was done, followed by pyrolysis, acid washing to remove NaCl and a second pyrolysis under the same condition as the first pyrolysis. Three different pyrolysis temperatures were used, and the synthesized catalysts were denoted as Cu-GS-800, Cu-GS-900 and Cu-GS-1000, respectively. It was found that by varying the pyrolysis temperature, the local environment of the Cu SAC can be changed (Fig. 7h). As the temperature increased, Cu–N4 sites were converted to Cu–N4−x–Cx sites. Individual testing with CO2RR and NO3RR found that FE for CO was able to reach 59% for Cu-GS-800, and FE for NH4+ for Cu-GS-1000 was able to reach 98% at −0.8 V vs. RHE. The ability to carry out both reactions implies that Cu-SAC can synthesize urea as it can couple *CO2 and *NO3. Further experiments confirmed this, as FE for CO reduced to almost 0 as it was consumed to produce urea (Fig. 7f). A urea yield rate of 1.8 mg h−1 mgcat−1 and an FE of 28% were achieved at −0.9 V vs. RHE. The free energy diagrams of urea synthesis on the various Cu–N4−x–Cx are shown in Fig. 7(i). In another instance, Cu was also used as the catalyst metal, except it was decorated on CeO2 support (Cu1–CeO2) (Fig. 7j–o).127 The catalyst showed a urea yield rate of 52.84 mmol h−1 gcat−1 at −1.6 V vs. RHE. It was also found with operando XAS experiments that the single Cu atoms (Cu1) reconstituted to clusters (Cu4) during electrolysis, similar to the Cu SAC for NO3RR discussed previously (Fig. 7r–t). Stability testing in Fig. 7q comparing between Cu1–CeO2 and CuO–CeO2 showed the rapid degradation of catalytic activity of CuO–CeO2 as compared with Cu1–CeO2. DFT calculations showed that the adsorption and activation of NO3− on Cu1–CeO2 took place spontaneously without any external energy, with N–O bonds elongating to 1.502 Å. Furthermore, CO2 adsorption energy was low, with the CO2 electron cloud destabilised as evident by the bond angle changing from 180° to 124.4°. The replacement of high-coordinated Ce atoms in CeO2 with low-coordinated Cu also resulted in frustrated Lewis Pairs, being able to activate CO2 more efficiently.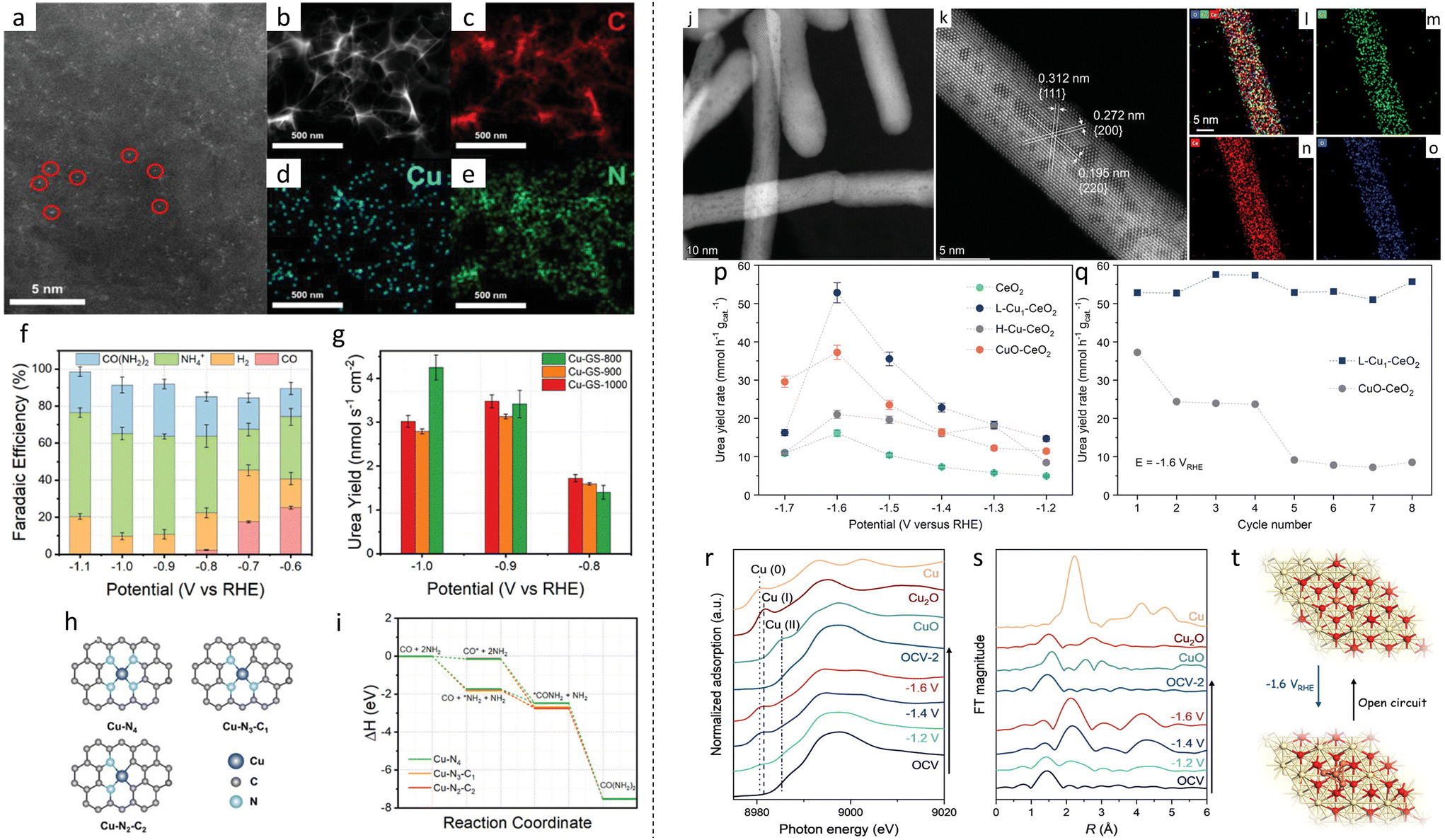 | ||
| Fig. 7 (a) HAADF-STEM image of Cu SACs. Isolated Cu atoms are marked with red circles. (b–e) EDS mapping showing distribution of C, Cu and N elements. (f) FEs of various products across different potentials. (g) Yield rates of urea across potentials comparing the 3 different Cu SACs synthesized. (h) Illustration of various Cu SAC local environments. (i) Free energy diagrams of urea synthesis on the various Cu SACs. Reproduced with permission.126 Copyright 2022. Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. (j) Aberration-corrected TEM (AC-TEM) image. (k) AC-HAADF-STEM of Cu SAC on CeO2. (l–o) EDS mapping showing distribution of Cu, Ce and O, respectively. (p) Urea yield rates of various catalysts across a range of potentials. (q) Stability testing over 8 cycles comparing between Cu SAC and CuO on CeO2. (r) Cu K-edge XANES spectra and (s) Cu R space EXAFS of Cu SACs on CeO2 at various potentials. (t) Illustration of the reversible transformation between Cu SACs and Cu clusters. Reproduced with permission.127 Copyright 2023. Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Boron has an empty sp2 orbital and an occupied p-orbital, allowing the acceptance of lone-pair electrons from N2 and back-donation to the antibonding orbital of N2. Therefore, Kong et al. theoretically studied single atoms anchored on porous boron nitride (p-BN) for urea synthesis.128 After a four-step strategy to screen and filter elements, Fe and Co were found to satisfy all four conditions. In both cases when N2 was adsorbed, the NN bond length increased from 1.12 Å to 1.35 Å, indicating the activation of N2. Both catalysts were also shown to have higher selectivity over HER. The PDS for both Fe and Cu was shown to be the hydrogenation step of NHCONH2* to NH2CONH2*, with limiting potentials of −0.63 V and −0.66 V, respectively.
5.2 Diatom catalysts
There are a multitude of theoretical studies on the topic of DACs for electrochemical synthesis of urea. Experimental studies, on the other hand, are lacking. In the few experimental papers, it had been shown that DAC for urea synthesis exhibited superior activity and FE compared with SAC, further proving the benefits of DAC.Using a host–guest strategy, Zhang et al. synthesized Fe and Ni DACs (Fig. 8a–c).119 Experiments with individual Fe and Ni SACs found that NO3RR and CO2RR dominated, respectively, leading to low yield rates and FEs for urea. Isolated diatomic Fe–Ni pair (I-FeNi-DASC) showed improved results, showing much higher FEs of 82.5% for CO2RR and 7.9% for NO3RR, resulting in a yield rate of 10.7 mmol h−1 g−1 and FE of 3.8% for urea. On the other hand, bonded Fe–Ni (B-FiNi-DASC) showed far superior results, delivering a urea FE of 17.8% and a yield rate of 20.2 mmol h−1 g−1 at −1.5 V vs. RHE. DFT calculations were then done to identify the origin of activity. Electrons were mainly concentrated over Ni sites and electron-deficient Fe atoms served as the Lewis acidic sites to enhance the adsorption and activation of *NO, resulting in high urea FE and yield rate. Pan et al. compared single Pd1 and dual Pd1Cu1 atoms on TiO2 nanosheet for urea synthesis (Fig. 8f).129 As shown in Fig. 8g, DACs were shown to far outperform SACs, having a urea yield rate of 166.67 molurea molpd−1 h−1 with a FE of 22.54% at −0.5 V vs. RHE compared with that of SAC (urea yield rate of 29.36 molurea molpd−1 h−1, FE 3.79%). DFT study elucidated a synergistic effect of DAC on TiO2. Pd1Cu1 was electron-depleted, matching the electron-rich N atom in N2, while oxygen vacancies in TiO2 were electronegative, attracting CO2. The potential barrier to form *NCON was also much lower for DACs (−0.265 eV) compared with SACs (1.360 eV). Competitive NRR was also suppressed due to the barrier of *NCON formation being lower than that of *NNH (Fig. 8h).
 | ||
| Fig. 8 (a) TEM image of FeNi DAC. (b) HAADF-STEM image with the FeNi DACs circled in red. (c) HAADF-STEM image profile intensity with atomic resolution EELS mapping of FeNi DAC. (d) FE comparisons of various catalysts in different reactions. (e) Free energy diagram of urea synthesis for FeNi DAC. Reproduced with permission.119 Copyright 2022, Springer Nature. (f) HAADF-STEM images of single Pd and dual PdCu catalysts on TiO2 with insets of their respective atomic structure. (g) Urea yield rate comparison of PdCu DAC and Pd SAC. (h) Free energy diagrams of NO3RR on Pd SAC and PdCu DAC. Reproduced with permission129 copyright 2023, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Various starting precursor reactants were tested in theoretical studies and the best candidates were screened for each reaction. Zhang and Guo did a DFT study on the feasibility of using DACs to synthesize urea from CO2 and N2.130 Corrole was chosen as the base for its ability to modify catalytic activity by introducing different substituents at the median and β positions. The structure of V2N6C was found to have the best projected density of state with a d-band value of −0.40 eV, which showed the closest value to the Fermi level compared with other structures such as V2N4C. This improved interactions between catalyst and adsorbed molecules, enhancing catalyst activity. Competitive HER was also found to be inhibited, further increasing urea selectivity. In a continuation of this study, the authors published another paper studying heterogeneous DACs, with vanadium as one of the components (MVN6C).131 Nb was found to have the lowest d-band center closest to the Fermi level, making it the most catalytically active metal with a small limiting voltage of −0.37 V. Competitive HER on MVN6C catalyst was also found to have no effect on urea synthesis because the adsorption energy of hydrogen is weaker than that of CO2 and N2. Ren et al. explored the feasibility of urea formation from N2O and CO, both of which are toxic to human health and potent greenhouse gases.132 An entirely new mechanism for the reaction was proposed, where CO is directly inserted into *NN formed from the reduction of *N2O to form the key intermediate *NCON. TMs anchored on porous g-CN (TM2/g-CN) as DACs were screened. Cr2/g-CN, Fe2/g-CN and Co2/g-CN were identified to be the best TMs with high stability, activity and selectivity. Cr and Co presented low limiting potentials (−0.19 V and −0.15 V, respectively), while Fe presented the most efficient catalytic activity with no thermodynamic energy barriers. Competitive NRR and HER were found to be inhibited as well.
In a step away from the conventional use of transition metals as catalysts for urea synthesis, Roy et al. examined the usage of dual-Si-doped graphitic carbon nitride (g-C6N6) in urea synthesis from N2 and CO2.133 g-C6N6 is known to activate N2 easily. N2 is adsorbed onto dual Si via side-on configuration and then becomes activated. Subsequently CO2 is adsorbed onto the activated N2 and converted to CO via proton-coupled electron transfer. CO is then directly inserted into the activated N–N bond, essentially cleaving the N–N bond as charge is transferred from the nonbonding orbital of CO to antibonding N–N π* orbital. Competitive HER and NRR were found to be inhibited, as onset potentials for both reactions were higher than that of urea synthesis (−1.31 V, −0.92 V and −0.79 V, respectively).
In summary, SACs and DACs are able to improve the yield rate and FE of NRR, NO3RR, NO2RR, NORR and urea synthesis. For SAC:
(i) Synergistic effects between atomically dispersed catalyst and support lead to better activation of reactants – examples include Ru–S bond leading to a “push–push” effect where N2 is destabilised by two contributions and has better electrical conductivity.70
(ii) Easily tuneable local environment of atomic sites, leading to better optimisation of reaction. This can be observed in Fe SACs for NO3RR, where changing the temperature of pyrolysis results in different moieties – FeN3, Fe–N4–OH and FeN4, giving rise to different electrosynthesis performance.87
(iii) Better electronic properties that are more conducive for the various reactions. This is evident in the Fe-PPy SAC,86 where it has a more pronounced hybridisation between oxygen and Fe with overlapping electrons at antibonding orbitals.
(iv) Good stability over micro/nano materials, as seen in the case of Cu SAC on CeO2, where urea yield rate of CuO–CeO2 was found to rapidly decrease over a few cycles.127
(v) Lack of adjacent sites resulting in difficulty of N–N coupling, preventing N2 from forming and improving selectivity towards ammonia, as evident when Zn SAC was used in NO3RR91 and the novel BSAC proposed by Lv et al.101
For DAC:
(i) Reactants are better stabilised by multiple catalytic centers, resulting in better activation of reactants and lowering the desorption energy of products, which can be seen in the case of FeCu DACs for NRR42 and NO3RR.98
(ii) Synergistic effects between the two catalytic centers for urea synthesis: one site allows for efficient NO3RR while the other allows for CO2RR, enabling both reactions to occur simultaneously. Zhang et al. showcased this in their work with Fe–Ni DAC, where Fe dominated in NO3RR while Ni dominated in CO2RR and a bonded Fe–Ni DAC showed the most superior C–N coupling activity.119
(iii) The proximity at which the reactions take place makes it conducive for the intermediates to combine to form urea. This is apparent in the theoretical work done by Roy et al. in which *CO is inserted directly into the activated N2 to form urea.133
The FEs and yield rates of NH3 and urea synthesized via NRR, NO3RR, NO2RR, NORR and N–C coupling over various SACs and DACs are summarised in Table 1. The catalysts showed excellent FEs and high yield rates especially when normalised to the loading mass of catalytically active metals.
6 Conclusion
In conclusion, AC is an up and rising topic that has proved its excellent performance in various reactions, far outperforming its nano/micro counterparts in terms of activity, selectivity and stability. This is due to the 100% atom utilisation, the ensemble effect, new synthesis routes, synergistic effects and breaking of the linear scaling relation. This is a big step towards green and sustainable ammonia and urea synthesis. There are still many ways that AC can be further improved upon.The topic of DAC for nitrate and nitrogen reduction can be further explored as it was already proved to be superior to SAC and nanoparticles in some cases due to its synergistic effects of having two atoms. For urea synthesis, theoretical works on it are lacking, let alone experimental. For theoretical works, more focus can be placed on exploring how the two atoms work synergistically to catalyse the reduction reaction more effectively. Investigation of the effects of homogeneous and heterogeneous DACs is another great direction to work on.
Catalyst mass loading can be further increased as well. Most works on SACs and DACs have a mass loading of <1%, with some as low as 0.15%.68 Only a few works have focused on increasing the mass loading to beyond 10%. A higher catalyst mass loading indicates a higher number of active sites, increasing product yield rate for the same amount of catalyst added.
The effects of various scaffolds for SACs and DACs upon which they are deposited on can be further investigated as well. Most SACs and DACs utilise N-doped carbon as scaffolding due to its great electrical conductivity and tunability. Other scaffolding such as chalcogenides, boron nitride, MXenes, MOFs and many others can be explored. Taking the example of chalcogenides, Zheng et al. showed that single transition metal atoms can achieve a high loading, with Fe reaching up to 10 wt% on transition metal chalcogenides (TMCs).134 Other substrates on which catalytic atoms are deposited may have other synergistic effects worth investigating as well. Li et al. conducted a study of Pt single atom on various supports, including Co3O4, CeO2, ZrO2 and graphene, and found that Pt on Co3O4 (Pt1/Co3O4) showed the best activity and stability in the dehydrogenation of ammonia borane.135
There could be attempts to increase the size of the atomic catalyst by synthesizing clusters of 3 or more atoms. When increasing the number of atoms, it also has the effect of increasing mass loading. A DFT study by Chen et al. postulated a possible 35.8 wt% of catalytic Fe3 on a stacked heterostructure of graphdiyne and graphene.136 In addition, considering the electrosynthesis of urea from CO2 and nitrogen oxides involves the coupling of two nitrogen and one carbon atoms, a triple-atom catalyst (TAC) may prove to have better adsorption than DACs.137 A TAC has been synthesized before for other functions: Ru3 synthesized for oxidation of alcohol, exhibiting 100% conversion and 100% efficiency.138 This proves the feasibility of the idea and the potential merit of synthesizing triple or larger atomic cluster catalysts.
Finally, there is a need for advancements on in operando/in situ techniques to observe the catalysts as the reaction occurs. It has already been proved that Cu SACs underwent structural changes from SAC to NP at a negative applied potential in NO3RR90 and urea synthesis.127 The real catalytic sites were therefore the clusters that formed under negative potentials, rather than the single-atom sites. Apart from operando XAS, other techniques such as operando TEM had been utilised to observe the structural changes of catalysts under reaction conditions.139
Data availability
No new data were generated or analysed in support of this research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Yan, Q. acknowledges Singapore MOE AcRF Tier 1 project under Grant No. RT6/22, the SCARCE project supported by the National Research Foundation, Singapore, and National Environment Agency, Singapore under its Closing the Waste Loop Funding Initiative (Award No. USS-IF-2018-4) and Singapore RIE2025 USS LCER PHASE 2 PROGRAMME 2305D4001.References
- J. W. Erisman, M. A. Sutton, J. Galloway, Z. Klimont and W. Winiwarter, Nat. Geosci., 2008, 1, 636–639 CrossRef CAS
.
- R. Lan and S. Tao, Front. Energy Res., 2014, 2, 35 CrossRef
- A. F. Dalebrook, W. Gan, M. Grasemann, S. Moret and G. Laurenczy, Chem. Commun., 2013, 49, 8735–8751 RSC
- R. Lan, J. T. S. Irvine and S. Tao, Int. J. Hydrogen Energy, 2012, 37, 1482–1494 CrossRef CAS
- S. Chatterjee, R. K. Parsapur and K.-W. Huang, ACS Energy Lett., 2021, 6, 4390–4394 CrossRef CAS
- S. Wang, F. Ichihara, H. Pang, H. Chen and J. Ye, Adv. Funct. Mater., 2018, 28, 1803309 CrossRef
- C. Zamfirescu and I. Dincer, J. Power Sources, 2008, 185, 459–465 CrossRef CAS
- M. Iwamoto, M. Akiyama, K. Aihara and T. Deguchi, ACS Catal., 2017, 7, 6924–6929 CrossRef CAS
- A. Rollinson, J. M. Jones, V. Dupont and M. Twigg, Energy Environ. Sci., 2011, 4, 1216–1224 RSC
- R. Lan and S. Tao, J. Power Sources, 2011, 196, 5021–5026 CrossRef CAS
- R. Lan, S. Tao and J. T. S. Irvine, Energy Environ. Sci., 2010, 3, 438–441 RSC
- C. Smith, A. K. Hill and L. Torrente-Murciano, Energy Environ. Sci., 2020, 13, 331–344 RSC
- J. Lim, C. A. Fernández, S. W. Lee and M. C. Hatzell, ACS Energy Lett., 2021, 6, 3676–3685 CrossRef CAS
- C. Chen, X. Zhu, X. Wen, Y. Zhou, L. Zhou, H. Li, L. Tao, Q. Li, S. Du, T. Liu, D. Yan, C. Xie, Y. Zou, Y. Wang, R. Chen, J. Huo, Y. Li, J. Cheng, H. Su, X. Zhao, W. Cheng, Q. Liu, H. Lin, J. Luo, J. Chen, M. Dong, K. Cheng, C. Li and S. Wang, Nat. Chem., 2020, 12, 717–724 CrossRef CAS PubMed
- C. Lv, L. Zhong, H. Liu, Z. Fang, C. Yan, M. Chen, Y. Kong, C. Lee, D. Liu, S. Li, J. Liu, L. Song, G. Chen, Q. Yan and G. Yu, Nat. Sustainable, 2021, 4, 868–876 CrossRef
- Y. Manaka, Y. Nagatsuka and K. Motokura, Sci. Rep., 2020, 10, 2834 CrossRef CAS PubMed
- C. A. Tsipis and P. A. Karipidis, J. Phys. Chem. A, 2005, 109, 8560–8567 CrossRef CAS PubMed
- N. Meng, Y. Huang, Y. Liu, Y. Yu and B. Zhang, Cell Rep., 2021, 2, 100697 Search PubMed
- J. Kong, A. Lim, C. Yoon, J. H. Jang, H. C. Ham, J. Han, S. Nam, D. Kim, Y.-E. Sung, J. Choi and H. S. Park, ACS Sustainable Chem. Eng., 2017, 5, 10986–10995 CrossRef CAS
- R. Zhao, H. Xie, L. Chang, X. Zhang, X. Zhu, X. Tong, T. Wang, Y. Luo, P. Wei, Z. Wang and X. Sun, J. Energy Chem., 2019, 1, 100011 Search PubMed
- P. Huang, T. Fan, X. Ma, J. Zhang, Y. Zhang, Z. Chen and X. Yi, ChemSusChem, 2022, 15, e202102049 CrossRef CAS PubMed
- L. Mi, Q. Huo, J. Cao, X. Chen, H. Yang, Q. Hu and C. He, Adv. Energy Mater., 2022, 12, 2202247 CrossRef CAS
- J. Wang, C. Cai, Y. Wang, X. Yang, D. Wu, Y. Zhu, M. Li, M. Gu and M. Shao, ACS Catal., 2021, 11, 15135–15140 CrossRef CAS
- R. Jia, Y. Wang, C. Wang, Y. Ling, Y. Yu and B. Zhang, ACS Catal., 2020, 10, 3533–3540 CrossRef CAS
- Y. Hu, J. Liu, C. Lee, W. Luo, J. Dong, Z. Liang, M. Chen, E. Hu, M. Zhang, X. Y. D. Soo, Q. Zhu, F. Li, R. S. Rawat, M.-F. Ng, L. Zhong, B. Han, D. Geng and Q. Yan, ACS Nano, 2023, 17, 23637–23648 CrossRef CAS PubMed
- C. Lv, C. Lee, L. Zhong, H. Liu, J. Liu, L. Yang, C. Yan, W. Yu, H. H. Hng, Z. Qi, L. Song, S. Li, K. P. Loh, Q. Yan and G. Yu, ACS Nano, 2022, 16, 8213–8222 CrossRef CAS PubMed
- J. Li, G. Zhan, J. Yang, F. Quan, C. Mao, Y. Liu, B. Wang, F. Lei, L. Li, A. W. M. Chan, L. Xu, Y. Shi, Y. Du, W. Hao, P. K. Wong, J. Wang, S.-X. Dou, L. Zhang and J. C. Yu, J. Am. Chem. Soc., 2020, 142, 7036–7046 CrossRef CAS PubMed
- B. Han, L. Zhong, C. Chen, J. Ding, C. Lee, J. Liu, M. Chen, S. Tso, Y. Hu, C. Lv, Y. Han, B. Liu and Q. Yan, Small, 2024, 20, 2307506 CrossRef CAS PubMed
- C. Lv, N. Jia, Y. Qian, S. Wang, X. Wang, W. Yu, C. Liu, H. Pan, Q. Zhu, J. Xu, X. Tao, K. P. Loh, C. Xue and Q. Yan, Small, 2023, 19, 2205959 CrossRef CAS PubMed
- J. Kundu, T. Kwon, K. Lee and S.-I. Choi, Exploration, 2024, 4, 20220174 CrossRef CAS PubMed
- H. Niu, Z. Zhang, X. Wang, X. Wan, C. Shao and Y. Guo, Adv. Funct. Mater., 2021, 31, 2008533 CrossRef CAS
- L. Zhang, Y. Ren, W. Liu, A. Wang and T. Zhang, Natl. Sci., 2018, 5, 653–672 CAS
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed
- S. Ghoshal, A. Ghosh, P. Roy, B. Ball, A. Pramanik and P. Sarkar, ACS Catal., 2022, 12, 15541–15575 CrossRef CAS
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed
- Y. Xue, B. Huang, Y. Yi, Y. Guo, Z. Zuo, Y. Li, Z. Jia, H. Liu and Y. Li, Nat. Commun., 2018, 9, 1460 CrossRef PubMed
- L. Han, X. Liu, J. Chen, R. Lin, H. Liu, F. Lü, S. Bak, Z. Liang, S. Zhao, E. Stavitski, J. Luo, R. R. Adzic and H. L. Xin, Angew. Chem., Int. Ed., 2019, 58, 2321–2325 CrossRef CAS PubMed
- B. Qiao, J.-X. Liang, A. Wang, C.-Q. Xu, J. Li, T. Zhang and J. J. Liu, Nano Res., 2015, 8, 2913–2924 CrossRef CAS
- C. Choi, S. Back, N.-Y. Kim, J. Lim, Y.-H. Kim and Y. Jung, ACS Catal., 2018, 8, 7517–7525 CrossRef CAS
- H. Jeong, S. Shin and H. Lee, ACS Nano, 2020, 14, 14355–14374 CrossRef CAS PubMed
- C. Lv, Y. Qian, C. Yan, Y. Ding, Y. Liu, G. Chen and G. Yu, Angew. Chem., Int. Ed., 2018, 57, 10246–10250 CrossRef CAS PubMed
- X. Wang, S. Qiu, J. Feng, Y. Tong, F. Zhou, Q. Li, L. Song, S. Chen, K.-H. Wu, P. Su, S. Ye, F. Hou, S. X. Dou, H. K. Liu, G. Q. Lu, C. Sun, J. Liu and J. Liang, Adv. Mater., 2020, 32, 2004382 CrossRef CAS PubMed
- Z. Chen, X. Liao, C. Sun, K. Zhao, D. Ye, J. Li, G. Wu, J. Fang, H. Zhao and J. Zhang, Appl. Catal., B, 2021, 288, 120021 CrossRef CAS
- C. Brea and G. Hu, ACS Catal., 2023, 13, 4992–4999 CrossRef CAS
- A. J. Medford, A. Vojvodic, J. S. Hummelshøj, J. Voss, F. Abild-Pedersen, F. Studt, T. Bligaard, A. Nilsson and J. K. Nørskov, J. Catal., 2015, 328, 36–42 CrossRef CAS
- H. Ooka, J. Huang and K. S. Exner, Front. Energy Res., 2021, 9, 654460 CrossRef
- Z. W. Chen, J. Li, P. Ou, J. E. Huang, Z. Wen, L. Chen, X. Yao, G. Cai, C. C. Yang, C. V. Singh and Q. Jiang, Nat. Commun., 2024, 15, 359 CrossRef CAS PubMed
- L. Li, K. Yuan and Y. Chen, Acc. Mater. Res., 2022, 3, 584–596 CrossRef CAS
- Y. Ouyang, L. Shi, X. Bai, Q. Li and J. Wang, Chem. Sci., 2020, 11, 1807–1813 RSC
- J. Li, Z. Yang, Y. Li and G. Zhang, J. Hazard. Mater., 2022, 429, 128285 CrossRef CAS PubMed
- J. Kim, H.-E. Kim and H. Lee, ChemSusChem, 2018, 11, 104–113 CrossRef CAS PubMed
- M. Flytzani-Stephanopoulos, Chin. J. Catal., 2017, 38, 1432–1442 CrossRef CAS
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS
- M. Kottwitz, Y. Li, H. Wang, A. I. Frenkel and R. G. Nuzzo, Chem.: Methods, 2021, 1, 278–294 CAS
- T. Tang, Z. Wang and J. Guan, Exploration, 2023, 3, 20230011 CrossRef CAS PubMed
- L. Liu and S. Zheng, ChemCatChem, 2024, 16, e202301641 CrossRef CAS
- Z. Mo, J. Mu and B. Liu, J. Electroanal. Chem., 2024, 969, 118533 CrossRef CAS
- X. Peng, M. Zhang, T. Zhang, Y. Zhou, J. Ni, X. Wang and L. Jiang, Chem. Sci., 2024, 15, 5897–5915 RSC
- J. Wan, J. Zheng, H. Zhang, A. Wu and X. Li, Catal. Sci. Technol., 2022, 12, 38–56 RSC
- X. Long, F. Huang, Z. Yao, P. Li, T. Zhong, H. Zhao, S. Tian, D. Shu and C. He, Small, 2024, 20, 2400551 CrossRef CAS PubMed
- D. Wu, B. He, Y. Wang, P. Lv, D. Ma and Y. Jia, J. Phys. D: Appl. Phys., 2022, 55, 203001 CrossRef
- M. I. Ahmed, D. B. Hibbert and C. Zhao, Green Energy Environ., 2023, 8, 1567–1595 CrossRef CAS
- H. Shen, C. Choi, J. Masa, X. Li, J. Qiu, Y. Jung and Z. Sun, Chem, 2021, 7, 1708–1754 CAS
- M. Ismael and M. Wark, Appl. Mater. Today, 2024, 39, 102253 CrossRef
- J. Hou, M. Yang and J. Zhang, Nanoscale, 2020, 12, 6900–6920 RSC
- A. Jain, M. Bar-Sadan and A. Ramasubramaniam, J. Phys. Chem. C, 2021, 125, 19980–19990 CrossRef CAS
- Q. Qin, T. Heil, M. Antonietti and M. Oschatz, Small Methods, 2018, 2, 1800202 CrossRef
- X. Wang, W. Wang, M. Qiao, G. Wu, W. Chen, T. Yuan, Q. Xu, M. Chen, Y. Zhang, X. Wang, J. Wang, J. Ge, X. Hong, Y. Li, Y. Wu and Y. Li, Sci. Bull., 2018, 63, 1246–1253 CrossRef CAS PubMed
- L. C. Seefeldt, B. M. Hoffman and D. R. Dean, Annu. Rev. Biochem., 2009, 78, 701–722 CrossRef CAS PubMed
- L. Yang, C. Cheng, X. Zhang, C. Tang, K. Du, Y. Yang, S.-C. Shen, S.-L. Xu, P.-F. Yin, H.-W. Liang and T. Ling, Chin. J. Catal., 2022, 43, 3177–3186 CrossRef CAS
- X. Yu, P. Han, Z. Wei, L. Huang, Z. Gu, S. Peng, J. Ma and G. Zheng, Joule, 2018, 2, 1610–1622 CrossRef CAS
- X. Yao, Z.-W. Chen, Y.-R. Wang, X.-Y. Lang, Y.-F. Zhu, W. Gao and Q. Jiang, Appl. Surf. Sci., 2020, 529, 147183 CrossRef CAS
- L. Wang, Y. Liu, H. Wang, T. Yang, Y. Luo, S. Lee, M. G. Kim, T. T. T. Nga, C.-L. Dong and H. Lee, ACS Nano, 2023, 17, 7406–7416 CrossRef CAS PubMed
- Y. Wu, C. He and W. Zhang, ACS Appl. Mater. Interfaces, 2021, 13, 47520–47529 CrossRef CAS PubMed
- B. Wang, S. Huang, L. Yang, Q. Fu and Y. Bu, J. Phys. Chem. C, 2021, 125, 14253–14262 CrossRef CAS
- J. Wu, D. Wu, H. Li, Y. Song, W. Lv, X. Yu and D. Ma, Nanoscale, 2023, 15, 16056–16067 RSC
- D. Wu, J. Wu, H. Li, W. Lv, Y. Song, D. Ma and Y. Jia, J. Mater. Chem. A, 2024, 12, 4278–4289 RSC
- K. Fan, W. Xie, J. Li, Y. Sun, P. Xu, Y. Tang, Z. Li and M. Shao, Nat. Commun., 2022, 13, 7958 CrossRef CAS PubMed
- H. Xu, Y. Ma, J. Chen, W.-X. Zhang and J. Yang, Chem. Soc. Rev., 2022, 51, 2710–2758 RSC
- X. Zheng, Y. Yan, X. Li, Y. Liu and Y. Yao, J. Hazard. Mater., 2023, 446, 130679 CrossRef CAS PubMed
- X. Zhang, Y. Wang, C. Liu, Y. Yu, S. Lu and B. Zhang, J. Chem. Eng., 2021, 403, 126269 CrossRef CAS
- J. Ingle and U. D. Patel, J Water Process Eng, 2022, 49, 103082 CrossRef
- H. Lan, X. Liu, H. Liu, R. Liu, C. Hu and J. Qu, Catal. Lett., 2016, 146, 91–99 CrossRef CAS
- Z. Zhang, Y. Xu, W. Shi, W. Wang, R. Zhang, X. Bao, B. Zhang, L. Li and F. Cui, J. Chem. Eng., 2016, 290, 201–208 CrossRef CAS
- X. Fu, Chin. J. Catal., 2023, 53, 8–12 CrossRef CAS
- P. Li, Z. Jin, Z. Fang and G. Yu, Energy Environ. Sci., 2021, 14, 3522–3531 RSC
- L. Liu, T. Xiao, H. Fu, Z. Chen, X. Qu and S. Zheng, Appl. Catal., B, 2023, 323, 122181 CrossRef CAS
- W.-D. Zhang, H. Dong, L. Zhou, H. Xu, H.-R. Wang, X. Yan, Y. Jiang, J. Zhang and Z.-G. Gu, Appl. Catal., B, 2022, 317, 121750 CrossRef CAS
- T. Zhu, Q. Chen, P. Liao, W. Duan, S. Liang, Z. Yan and C. Feng, Small, 2020, 16, 2004526 CrossRef CAS PubMed
- J. Yang, H. Qi, A. Li, X. Liu, X. Yang, S. Zhang, Q. Zhao, Q. Jiang, Y. Su, L. Zhang, J.-F. Li, Z.-Q. Tian, W. Liu, A. Wang and T. Zhang, J. Am. Chem. Soc., 2022, 144, 12062–12071 CrossRef CAS PubMed
- J. Zhao, X. Ren, X. Liu, X. Kuang, H. Wang, C. Zhang, Q. Wei and D. Wu, J. Chem. Eng., 2023, 452, 139533 CrossRef CAS
- Y. Zhang, H. Zheng, K. Zhou, J. Ye, K. Chu, Z. Zhou, L. Zhang and T. Liu, Adv. Mater., 2023, 35, 2209855 CrossRef CAS PubMed
- Y. Zhang, X. Chen, W. Wang, L. Yin and J. C. Crittenden, Appl. Catal., B, 2022, 310, 121346 CrossRef CAS
- Y. Yao, L. Zhao, J. Dai, J. Wang, C. Fang, G. Zhan, Q. Zheng, W. Hou and L. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202208215 CrossRef CAS PubMed
- D. Wu, P. Lv, J. Wu, B. He, X. Li, K. Chu, Y. Jia and D. Ma, J. Mater. Chem. A, 2023, 11, 1817–1828 RSC
- X. Zheng, J. Yang, Z. Xu, Q. Wang, J. Wu, E. Zhang, S. Dou, W. Sun, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2022, 61, e202205946 CrossRef CAS PubMed
- Y. Wang, H. Yin, F. Dong, X. Zhao, Y. Qu, L. Wang, Y. Peng, D. Wang, W. Fang and J. Li, Small, 2023, 19, 2207695 CrossRef CAS PubMed
- S. Zhang, J. Wu, M. Zheng, X. Jin, Z. Shen, Z. Li, Y. Wang, Q. Wang, X. Wang, H. Wei, J. Zhang, P. Wang, S. Zhang, L. Yu, L. Dong, Q. Zhu, H. Zhang and J. Lu, Nat. Commun., 2023, 14, 3634 CrossRef CAS PubMed
- F. Rehman, S. Kwon, C. B. Musgrave, M. Tamtaji, W. A. Goddard and Z. Luo, Nano Energy, 2022, 103, 107866 CrossRef CAS
- N. Sathishkumar and H.-T. Chen, J. Phys. Chem. C, 2023, 127, 994–1005 CrossRef CAS
- X. Lv, T. Mou, J. Li, L. Kou and T. Frauenheim, Adv. Funct. Mater., 2022, 32, 2201262 CrossRef CAS
- L. Bai, F. Franco, J. Timoshenko, C. Rettenmaier, F. Scholten, H. S. Jeon, A. Yoon, M. Rüscher, A. Herzog, F. T. Haase, S. Kühl, S. W. Chee, A. Bergmann and R. C. Beatriz, J. Am. Chem. Soc., 2024, 146, 9665–9678 CrossRef CAS PubMed
- J. Long, S. Chen, Y. Zhang, C. Guo, X. Fu, D. Deng and J. Xiao, Angew. Chem., Int. Ed., 2020, 59, 9711–9718 CrossRef CAS PubMed
- V. I. Pârvulescu, P. Grange and B. Delmon, Catal. Today, 1998, 46, 233–316 CrossRef
- N. Gruber and J. N. Galloway, Nature, 2008, 451, 293–296 CrossRef CAS PubMed
- F. Wang, J. Xiang, G. Zhang, K. Chen and K. Chu, Nano Res., 2024, 17, 3660–3666 CrossRef CAS
- J. Xiang, C. Qiang, S. Shang, K. Chen, C. Kang and K. Chu, Adv. Funct. Mater., 2024, 34, 2401941 CrossRef CAS
- J. Xiang, H. Zhao, K. Chen, X. Yang and K. Chu, J. Colloid Interface Sci., 2024, 659, 432–438 CrossRef CAS PubMed
- K. Chen, J. Wang, H. Zhang, D. Ma and K. Chu, Nano Lett., 2023, 23, 1735–1742 CrossRef CAS PubMed
- K. Chen, G. Wang, Y. Guo, D. Ma and K. Chu, Nano Res., 2023, 16, 8737–8742 CrossRef CAS
- K. Chen, G. Zhang, X. Li, X. Zhao and K. Chu, Nano Res., 2023, 16, 5857–5863 CrossRef CAS
- S. Zhao, M. Chang, J. Liu, G. Shi, Y. Yang, H. Gu, J. Zhang, C. Yang, H. Tong, C. Zhu, K. Cao, S. Li and L. Zhang, Chem Catal., 2023, 3, 100598 CrossRef CAS
- X. Hu, N. Q. Su and W.-H. Fang, J. Phys. Chem. C, 2023, 127, 11026–11039 CrossRef CAS
- H. Li, D. Wu, J. Wu, W. Lv, Z. Duan and D. Ma, Nanoscale, 2024, 16, 7058–7067 RSC
- Z. Bi, J. Hu, M. Xu, H. Zhang, Y. Zhou and G. Hu, Angew. Chem., Int. Ed., 2024, 63, e202313434 CrossRef CAS PubMed
- X. Zhang, L. Xia, Y. Li, H. Feng, X. Wang and J. Yu, Fuel, 2024, 366, 131432 CrossRef CAS
- X. Sun, Y. Dai, B. Huang and W. Wei, J. Phys. Chem. Lett., 2023, 14, 11684–11690 CrossRef CAS PubMed
- Y. Wu, J. Lv, F. Xie, R. An, J. Zhang, H. Huang, Z. Shen, L. Jiang, M. Xu, Q. Yao and Y. Cao, J. Colloid Interface Sci., 2024, 656, 155–167 CrossRef CAS PubMed
- X. Zhang, X. Zhu, S. Bo, C. Chen, M. Qiu, X. Wei, N. He, C. Xie, W. Chen, J. Zheng, P. Chen, S. P. Jiang, Y. Li, Q. Liu and S. Wang, Nat. Commun., 2022, 13, 5337 CrossRef CAS PubMed
- Y. Kohlhaas, Y. S. Tschauder, W. Plischka, U. Simon, R.-A. Eichel, M. Wessling and R. Keller, Joule, 2024, 8, 1579–1600 CrossRef CAS
- X. Zhu, X. Zhou, Y. Jing and Y. Li, Nat. Commun., 2021, 12, 4080 CrossRef CAS PubMed
- N. Cao, Y. Quan, A. Guan, C. Yang, Y. Ji, L. Zhang and G. Zheng, J. Colloid Interface Sci., 2020, 577, 109–114 CrossRef CAS PubMed
- Y. Zhao, Y. Ding, W. Li, C. Liu, Y. Li, Z. Zhao, Y. Shan, F. Li, L. Sun and F. Li, Nat. Commun., 2023, 14, 4491 CrossRef CAS PubMed
- X. Liu, Y. Jiao, Y. Zheng, M. Jaroniec and S.-Z. Qiao, Nat. Commun., 2022, 13, 5471 CrossRef CAS PubMed
- P. Li, Z. Zhang, X. Yang, Y. Zhu, Z. Zhou, X. Jiang, Q. Wang, X. Gao, X. Yang, Y. Shen and M. Wang, ChemCatChem, 2024, 16, e202301302 CrossRef CAS
- J. Leverett, T. Tran-Phu, J. A. Yuwono, P. Kumar, C. Kim, Q. Zhai, C. Han, J. Qu, J. Cairney, A. N. Simonov, R. K. Hocking, L. Dai, R. Daiyan and R. Amal, Adv. Energy Mater., 2022, 12, 2201500 CrossRef CAS
- X. Wei, Y. Liu, X. Zhu, S. Bo, L. Xiao, C. Chen, T. T. T. Nga, Y. He, M. Qiu, C. Xie, D. Wang, Q. Liu, F. Dong, C.-L. Dong, X.-Z. Fu and S. Wang, Adv. Mater., 2023, 35, 2300020 CrossRef CAS PubMed
- L. Kong, D. Jiao, Z. Wang, Y. Liu, Y. Shang, L. Yin, Q. Cai and J. Zhao, J. Chem. Eng., 2023, 451, 138885 CrossRef CAS
- L. Pan, J. Wang, F. Lu, Q. Liu, Y. Gao, Y. Wang, J. Jiang, C. Sun, J. Wang and X. Wang, Angew. Chem., Int. Ed., 2023, 62, e202216835 CrossRef CAS PubMed
- Z. Zhang and L. Guo, Dalton Trans., 2021, 50, 11158–11166 RSC
- Z. Zhang, L. Guo, J. Du and Y. Hou, New J. Chem., 2022, 46, 5278–5287 RSC
- Z. Ren, X. Wang, S. Wang, H. Zhang, B. Huang, Y. Dai and W. Wei, J. Mater. Chem. A, 2023, 11, 11507–11516 RSC
- P. Roy, A. Pramanik and P. Sarkar, J. Phys. Chem. Lett., 2021, 12, 10837–10844 CrossRef CAS PubMed
- J. Zheng, K. Lebedev, S. Wu, C. Huang, T. Ayvalı, T.-S. Wu, Y. Li, P.-L. Ho, Y.-L. Soo, A. Kirkland and S. C. E. Tsang, J. Am. Chem. Soc., 2021, 143, 7979–7990 CrossRef CAS PubMed
- J. Li, Q. Guan, H. Wu, W. Liu, Y. Lin, Z. Sun, X. Ye, X. Zheng, H. Pan, J. Zhu, S. Chen, W. Zhang, S. Wei and J. Lu, J. Am. Chem. Soc., 2019, 141, 14515–14519 CrossRef CAS PubMed
- Z. W. Chen, L. X. Chen, M. Jiang, D. Chen, Z. L. Wang, X. Yao, C. V. Singh and Q. Jiang, J.
Mater. Chem. A, 2020, 8, 15086–15093 RSC
- L. Chen, C. Tang, Y. Zheng, K. Davey and Y. Jiao, Sci. China Mater., 2023, 66, 2346–2353 CrossRef CAS
- S. Ji, Y. Chen, Q. Fu, Y. Chen, J. Dong, W. Chen, Z. Li, Y. Wang, L. Gu, W. He, C. Chen, Q. Peng, Y. Huang, X. Duan, D. Wang, C. Draxl and Y. Li, J. Am. Chem. Soc., 2017, 139, 9795–9798 CrossRef CAS PubMed
- P. Grosse, A. Yoon, C. Rettenmaier, A. Herzog, S. W. Chee and B. R. Cuenya, Nat. Commun., 2021, 12, 6736 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2024 |