DOI:
10.1039/D4NR02795G
(Review Article)
Nanoscale, 2024,
16, 18274-18294
Unleashing nanotechnology to redefine tumor-associated macrophage dynamics and non-coding RNA crosstalk in breast cancer
Received
6th July 2024
, Accepted 12th September 2024
First published on 18th September 2024
Abstract
Breast cancer is a significant global health issue. Tumor-associated macrophages (TAMs) are crucial in influencing the tumor microenvironment and the progression of the disease. TAMs exhibit remarkable plasticity in adopting distinct phenotypes ranging from pro-inflammatory and anti-tumorigenic (M1-like) to immunosuppressive and tumor-promoting (M2-like). This review elucidates the multifaceted roles of TAMs in driving breast tumor growth, angiogenesis, invasion, and metastatic dissemination. Significantly, it highlights the intricate crosstalk between TAMs and non-coding RNAs (ncRNAs), including microRNAs, long noncoding RNAs, and circular RNAs, as a crucial regulatory mechanism modulating TAM polarization and functional dynamics that present potential therapeutic targets. Nanotechnology-based strategies are explored as a promising approach to reprogramming TAMs toward an anti-tumor phenotype. Various nanoparticle delivery systems have shown potential for modulating TAM polarization and inhibiting tumor-promoting effects. Notably, nanoparticles can deliver ncRNA therapeutics to TAMs, offering unique opportunities to modulate their polarization and activity.
1. Introduction
Cancer has emerged as one of the foremost contributors to mortality rates and a significant obstacle to enhancing longevity across all nations globally. It poses a formidable challenge to public health and ranks among the top causes of death worldwide, irrespective of geographical boundaries or socioeconomic status.1 Breast cancer is one of the most prevalent and life-threatening malignancies worldwide that poses a significant burden on global healthcare systems and affects the well-being of women disproportionately.2 According to recent global statistics (2024), an alarming 2.308 million new cases of breast cancer were diagnosed in the year 2022, and a staggering 665![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 684 deaths were attributed to this disease globally.3 This sobering statistic highlights the severity of the global increase in breast cancer diagnoses among women.3 Based on receptor expression, breast cancer is classified into four molecular subtypes: Luminal A (ER+/PR+, low Ki67, HER2−), Luminal B (ER+/PR+, high Ki67 or HER2+), HER2-positive (HER2+, ER−/PR−), and triple-negative (ER−/PR−/HER2−).4 This subtyping guides treatment selection as the subtypes differ in behaviour, prognosis, and therapy response.5 The therapeutic approach for breast cancer has been heavily influenced by its molecular subtype classification. While substantial advancements have been made in recent years regarding targeted and endocrine therapies for breast cancer, however, the overall prognosis for patients remains suboptimal.6 This unsatisfactory outcome can be attributed to the high rates of cancer recurrence and metastatic spread.7 Emerging research has highlighted the pivotal role played by the tumor microenvironment (TME) in influencing the initiation and growth of breast cancer.8 The TME represents the complex interplay between cancer cells and various immune cells, such as T cells, B cells, natural killer cells, and myeloid-derived suppressor cells within the tumor's immediate surroundings.9 Accumulating evidence suggests that the characteristics and composition of the TME can significantly impact the behaviour and clinical outcomes of breast cancer. A balanced and effective anti-tumor immune response within the TME can potentially suppress tumor growth, invasion, and metastasis.10 On the contrary, an immunosuppressive microenvironment characterized by the presence of myeloid-derived suppressor cells or regulatory T cells can facilitate tumor progression and resistance to therapy.11 The TME's influence on breast cancer is multifaceted and affects various aspects of the disease's development and response to treatment.12 For instance, the presence of high infiltrating activated NK cells, tumor infiltrating lymphocytes, CD4+, CD8+, cytotoxic T cells, B cells, macrophages and dendritic immune cell populations within the TME has been associated with improved prognosis and increased survival rates in HER2+, HER2/neu positive and triple-negative breast cancer subtypes compared to other subtypes. An immunosuppressive microenvironment can contribute to tumor immune evasion and promote cancer cell proliferation, angiogenesis, and metastatic spread.13 An integral part of the TME is macrophages that infiltrate and reside within the tumor niche known as tumor-associated macrophages (TAMs).12 TAMs are often the most abundant immune cell type in breast cancer TME compared to other immune cells. These TAMs exhibit remarkable flexibility to alter their cellular characteristics and functions based on cues received from the surrounding milieu. TAMs can adopt either a phenotype that facilitates the elimination of cancer cells or one that fosters tumor cell proliferation, invasion, and metastatic dissemination depending on the signals in the microenvironment.12 TAMs engage in extensive crosstalk with breast cancer cells and promote tumor growth and invasion more effectively than other immune cells.14 TAMs have also been shown to contribute to developing resistance against various therapeutic approaches, including chemotherapy, endocrine therapy, immunotherapy, and targeted therapy by altering signaling pathways like PI3K/Akt/mTOR, NF-Kb/STAT3/ERK, EGFR/STAT3/SOX2, jagged1-Notch in preclinical breast cancer studies.15 Their presence and functional state can significantly undermine the effectiveness of different treatment modalities. Experimental research utilizing mouse models has revealed that TAMs possess the ability to induce resistance mechanisms, rendering cancer cells less responsive to chemotherapeutic agents, targeted therapies, and immunotherapies.16,17 Non-coding RNAs (ncRNAs) play a crucial role in mediating the crosstalk between cancer cells and TAMs within the TME.18 These ncRNAs, including microRNAs, long ncRNAs, and circular RNAs, regulate macrophage recruitment, polarization, and function, influencing tumor progression.19 Exosomes facilitate the transfer of ncRNAs between cancer cells and macrophages, contributing to bilateral communication.20 The interaction between ncRNAs and macrophages can promote tumor growth, angiogenesis, and metastasis.18 Exosomal ncRNAs in biofluids show promise as non-invasive biomarkers for cancer diagnosis and prognosis.20,21 Understanding the complex relationship between ncRNAs and macrophages in the TME may lead to the development of novel therapeutic strategies targeting these interactions to impede tumor progression.19,21 Nanotechnology involves the design, fabrication, and application of materials at the nanoscale, particularly in delivering therapeutic agents efficiently to specific cells or tissues.22 Nanotechnology-based delivery of ncRNAs represents a significant advancement in the treatment of breast cancer and other diseases through the precision of therapeutic targeting.23 Employment of nanocarriers designed to facilitate the effective distribution of ncRNAs has the potential to influence gene expression and promote desirable cellular responses, especially in modulating macrophage behavior within the TME.24 Integrating nanotechnology with ncRNA therapeutics enables the precise delivery of agents that can modulate macrophage behavior. For instance, nanoparticles can be engineered to encapsulate lncRNA-targeting small molecules or RNA interference (RNAi) which can downregulate detrimental lncRNAs and reprogram macrophages to an M1 phenotype, potentially enhancing immune responses against tumors.25 These nanoparticles can also be crafted to release their payload in response to specific stimuli within the TME, further optimizing treatment efficacy. Delivering ncRNAs using nanotechnology employs several mechanisms to achieve effective targeting and cellular uptake.26 Cationic liposomes, for instance, can encapsulate RNA molecules, protecting them from degradation while facilitating cell membrane fusion through electrostatic.27 Alternatively, RNA nanostructures can be engineered to enhance stability and specificity, ensuring that the therapeutic RNA reaches its intended target within cancer cells or macrophages in the TME.28 This review aims to explore the functional roles of TAMs in driving breast cancer progression and metastasis, with a particular focus on their crosstalk with ncRNAs. We will examine how different ncRNAs, such as microRNAs, lncRNAs, and circular RNAs, modulate TAM phenotypes and pro-tumorigenic functions. Additionally, the review will investigate potential nanotherapeutic strategies targeting TAMs and this ncRNA-mediated crosstalk to reprogram TAMs and inhibit their tumor-promoting effects. By elucidating these mechanisms, this review seeks to provide insights into novel therapeutic approaches that could improve the prognosis and treatment outcomes for breast cancer patients (Fig. 1).
684 deaths were attributed to this disease globally.3 This sobering statistic highlights the severity of the global increase in breast cancer diagnoses among women.3 Based on receptor expression, breast cancer is classified into four molecular subtypes: Luminal A (ER+/PR+, low Ki67, HER2−), Luminal B (ER+/PR+, high Ki67 or HER2+), HER2-positive (HER2+, ER−/PR−), and triple-negative (ER−/PR−/HER2−).4 This subtyping guides treatment selection as the subtypes differ in behaviour, prognosis, and therapy response.5 The therapeutic approach for breast cancer has been heavily influenced by its molecular subtype classification. While substantial advancements have been made in recent years regarding targeted and endocrine therapies for breast cancer, however, the overall prognosis for patients remains suboptimal.6 This unsatisfactory outcome can be attributed to the high rates of cancer recurrence and metastatic spread.7 Emerging research has highlighted the pivotal role played by the tumor microenvironment (TME) in influencing the initiation and growth of breast cancer.8 The TME represents the complex interplay between cancer cells and various immune cells, such as T cells, B cells, natural killer cells, and myeloid-derived suppressor cells within the tumor's immediate surroundings.9 Accumulating evidence suggests that the characteristics and composition of the TME can significantly impact the behaviour and clinical outcomes of breast cancer. A balanced and effective anti-tumor immune response within the TME can potentially suppress tumor growth, invasion, and metastasis.10 On the contrary, an immunosuppressive microenvironment characterized by the presence of myeloid-derived suppressor cells or regulatory T cells can facilitate tumor progression and resistance to therapy.11 The TME's influence on breast cancer is multifaceted and affects various aspects of the disease's development and response to treatment.12 For instance, the presence of high infiltrating activated NK cells, tumor infiltrating lymphocytes, CD4+, CD8+, cytotoxic T cells, B cells, macrophages and dendritic immune cell populations within the TME has been associated with improved prognosis and increased survival rates in HER2+, HER2/neu positive and triple-negative breast cancer subtypes compared to other subtypes. An immunosuppressive microenvironment can contribute to tumor immune evasion and promote cancer cell proliferation, angiogenesis, and metastatic spread.13 An integral part of the TME is macrophages that infiltrate and reside within the tumor niche known as tumor-associated macrophages (TAMs).12 TAMs are often the most abundant immune cell type in breast cancer TME compared to other immune cells. These TAMs exhibit remarkable flexibility to alter their cellular characteristics and functions based on cues received from the surrounding milieu. TAMs can adopt either a phenotype that facilitates the elimination of cancer cells or one that fosters tumor cell proliferation, invasion, and metastatic dissemination depending on the signals in the microenvironment.12 TAMs engage in extensive crosstalk with breast cancer cells and promote tumor growth and invasion more effectively than other immune cells.14 TAMs have also been shown to contribute to developing resistance against various therapeutic approaches, including chemotherapy, endocrine therapy, immunotherapy, and targeted therapy by altering signaling pathways like PI3K/Akt/mTOR, NF-Kb/STAT3/ERK, EGFR/STAT3/SOX2, jagged1-Notch in preclinical breast cancer studies.15 Their presence and functional state can significantly undermine the effectiveness of different treatment modalities. Experimental research utilizing mouse models has revealed that TAMs possess the ability to induce resistance mechanisms, rendering cancer cells less responsive to chemotherapeutic agents, targeted therapies, and immunotherapies.16,17 Non-coding RNAs (ncRNAs) play a crucial role in mediating the crosstalk between cancer cells and TAMs within the TME.18 These ncRNAs, including microRNAs, long ncRNAs, and circular RNAs, regulate macrophage recruitment, polarization, and function, influencing tumor progression.19 Exosomes facilitate the transfer of ncRNAs between cancer cells and macrophages, contributing to bilateral communication.20 The interaction between ncRNAs and macrophages can promote tumor growth, angiogenesis, and metastasis.18 Exosomal ncRNAs in biofluids show promise as non-invasive biomarkers for cancer diagnosis and prognosis.20,21 Understanding the complex relationship between ncRNAs and macrophages in the TME may lead to the development of novel therapeutic strategies targeting these interactions to impede tumor progression.19,21 Nanotechnology involves the design, fabrication, and application of materials at the nanoscale, particularly in delivering therapeutic agents efficiently to specific cells or tissues.22 Nanotechnology-based delivery of ncRNAs represents a significant advancement in the treatment of breast cancer and other diseases through the precision of therapeutic targeting.23 Employment of nanocarriers designed to facilitate the effective distribution of ncRNAs has the potential to influence gene expression and promote desirable cellular responses, especially in modulating macrophage behavior within the TME.24 Integrating nanotechnology with ncRNA therapeutics enables the precise delivery of agents that can modulate macrophage behavior. For instance, nanoparticles can be engineered to encapsulate lncRNA-targeting small molecules or RNA interference (RNAi) which can downregulate detrimental lncRNAs and reprogram macrophages to an M1 phenotype, potentially enhancing immune responses against tumors.25 These nanoparticles can also be crafted to release their payload in response to specific stimuli within the TME, further optimizing treatment efficacy. Delivering ncRNAs using nanotechnology employs several mechanisms to achieve effective targeting and cellular uptake.26 Cationic liposomes, for instance, can encapsulate RNA molecules, protecting them from degradation while facilitating cell membrane fusion through electrostatic.27 Alternatively, RNA nanostructures can be engineered to enhance stability and specificity, ensuring that the therapeutic RNA reaches its intended target within cancer cells or macrophages in the TME.28 This review aims to explore the functional roles of TAMs in driving breast cancer progression and metastasis, with a particular focus on their crosstalk with ncRNAs. We will examine how different ncRNAs, such as microRNAs, lncRNAs, and circular RNAs, modulate TAM phenotypes and pro-tumorigenic functions. Additionally, the review will investigate potential nanotherapeutic strategies targeting TAMs and this ncRNA-mediated crosstalk to reprogram TAMs and inhibit their tumor-promoting effects. By elucidating these mechanisms, this review seeks to provide insights into novel therapeutic approaches that could improve the prognosis and treatment outcomes for breast cancer patients (Fig. 1).
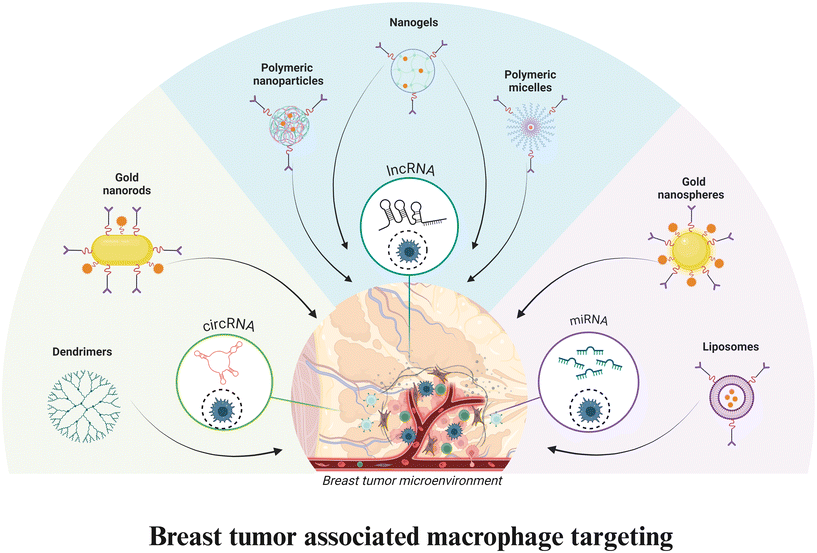 |
| | Fig. 1 Schematic representation for breast tumor associated macrophage targeting scheme. Image created with BioRender.com. | |
2. Macrophages in breast cancer
The mammary gland is a constantly changing tissue that is modified throughout various stages, including embryonic development, puberty, pregnancy, lactation, and involution. Macrophages have been found in the embryonic mammary gland and are located near, though not directly interacting with, the mammary epithelial buds at E14.5.29 Macrophages are located near the terminal end buds during the process of ductal elongation. They play a role in the growth of ducts and in the formation of collagen fibrils around these terminal end buds. Additionally, macrophages may help by engulfing and digesting apoptotic epithelial cells.30 Tissue-resident macrophages are involved not only in ductal elongation but also in various other stages of mammary gland development. They contribute to epithelial remodeling related to the estrous cycle, support changes during pregnancy, and assist in the process of involution.31 Macrophage subsets in the mammary gland play different roles during its development and are found in specific tissue regions. These subsets are crucial for the normal function of the mammary gland. Their presence and positioning in both epithelial and stromal areas suggest that their functions might be utilized or altered during mammary tumor formation.32 Having a brief overview of macrophages in mammary tissues, we must extend our understanding of macrophages in breast tumorigenesis.
2.1 Macrophage heterogeneity: origins and functional diversity
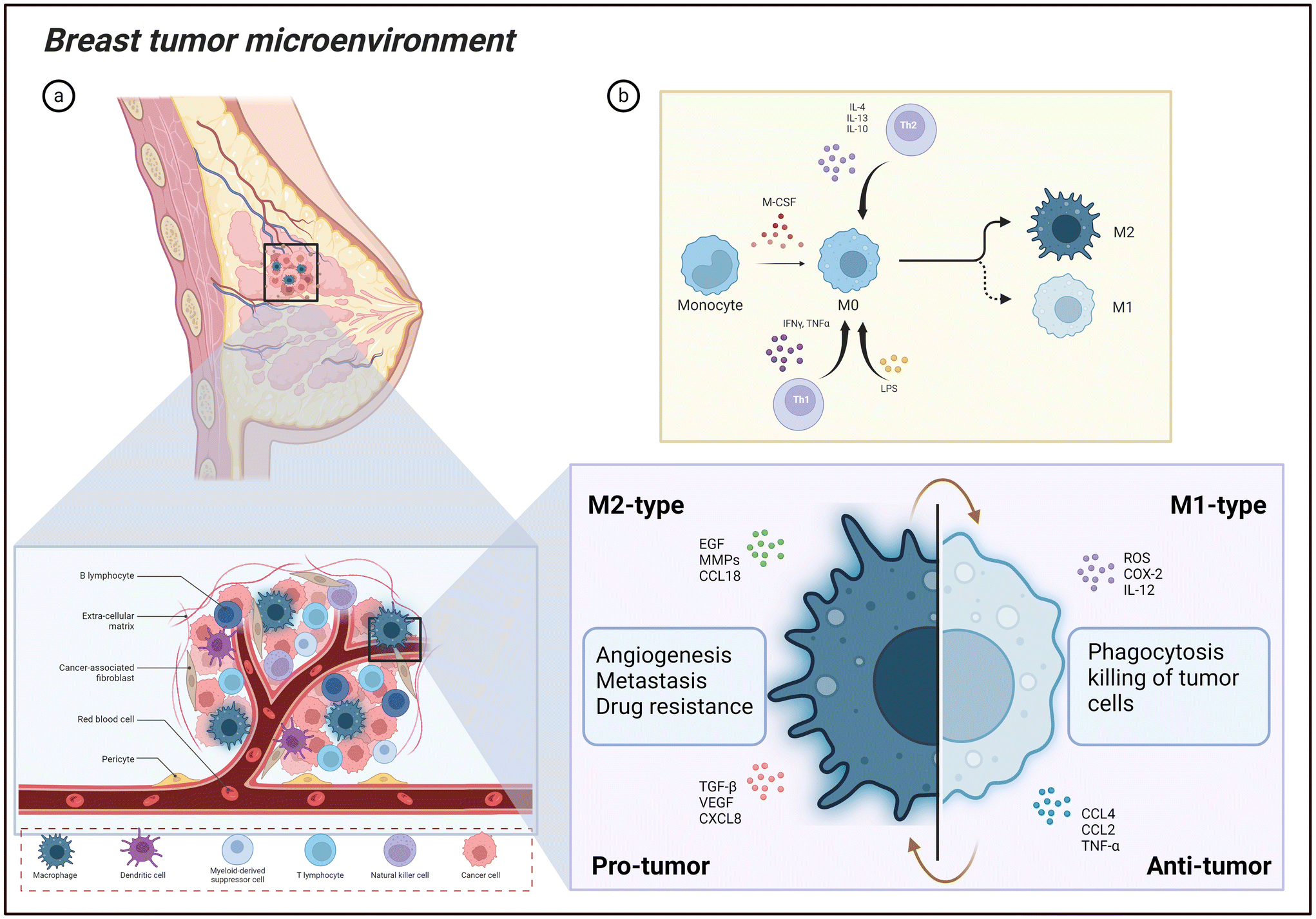
Tumor niches are dynamic and complex ecosystems made up of a variety of molecular and cellular components. Numerous cell types comprise this complex environment, including immune cells, fibroblasts that support the structural framework, cancer stem cells that can self-renew, and endothelial cells that generate blood vessels.33 The complex interactions within this specialized milieu are established by many soluble molecules that include extracellular matrix elements, chemokines, and cytokines present in the TME and the cellular components.34 One common non-cancerous cell type in the TME is macrophages,35 as highlighted in Fig. 2. Their generation begins in the bone marrow, where hematopoietic stem cells (HSCs) differentiate through a series of progenitor stages to form monocytes.36 These monocytes then enter the bloodstream and can further differentiate into macrophages upon migrating into tissues.37 Research suggests that TAMs can come from populations of primitive macrophages generated in the yolk sac during embryonic development or from precursors produced from bone marrow.38 Macrophages can develop into specific subsets in healthy tissues, such as Kupffer cells in the liver, Langerhans cells in the skin, and microglial cells in the brain.39,40 As monocytes enter tissues, they respond to local cues such as cytokines and growth factors, undergoing morphological changes and enhancing their phagocytic and secretory capabilities.41 Mature macrophages perform a wide array of functions critical for maintaining health and combating disease. Their primary roles include phagocytosis of pathogens and cellular debris, antigen presentation to initiate adaptive immune responses, and cytokine production to regulate inflammation.42,43 Beyond immune functions, macrophages contribute to tissue repair, iron homeostasis, metabolic regulation, and even support organ development during embryogenesis.44 TAMs in breast cancer microenvironments are variable, displaying a broad range of polarized behaviors. These polarization states can be broadly divided into two groups: macrophages resembling M1 and M2,45 as shown in Fig. 2. In general, the M1-like subgroup is known as classically activated macrophages. Inflammatory triggers such as lipopolysaccharides (LPS) and cytokines released by type 1 helper T cells (Th1) can activate these cells. Strong cytotoxic and phagocytic properties allow M1-like TAMs to specifically target and destroy tumor cells. Furthermore, they have anti-tumor and pro-inflammatory properties within the TME.46 On the flip hand, the subgroup of M2-like cells is known as alternatively activated macrophages. Type 2 helper T cell (Th2) signals, which include cytokines like interleukin (IL)-4, IL-10, and IL-13, cause them to become polarized (Fig. 2). M2-like TAMs stimulate angiogenesis, tumor growth, and metastatic spread, among other activities that aid in advancing tumors.47 M2-like macrophages usually have high CD163, CD206, CD209, and CCL2 levels, whereas the expression of markers such as HLA-DR and CD197 defines M1-like macrophages.48 Furthermore, M1-like polarization has been linked to transcription factors, including IRF4, while tissue-resident M2-like macrophages have been linked to Trib1.49 These results suggest that macrophage subgroups may reflect a broader and more complex range of phenotypic states beyond the traditional M1/M2 classification.50,51 Notably, the M1-like and M2-like macrophage populations are in a dynamic balance within the TME.52 The equilibrium between these two extreme states has the potential to profoundly affect the course of disease and, eventually, the prognosis of people with breast cancer. It is critical to realize that this M1/M2 classification is an oversimplified model since TAMs display a spectrum of functional states impacted by the intricate interactions between cytokines and growth factors, hypoxic conditions, metabolic factors and extracellular matrix components in the TME.53,54 Macrophages are present in breast cancer samples from different subtypes. Luminal A breast cancer exhibits notably lower levels of CD68+ macrophages compared to luminal B, HER2+, and triple-negative breast cancer (TNBC) subtypes. The latter three subtypes tend to have comparable macrophage counts.30 Dr Jeffrey and team performed pioneering work highlighting the macrophage's specific function for breast tumor progression, demonstrating the importance of macrophages in the metastasis of breast tumors to the lung. This group revealed that the potential of tumor cells to metastasize to the lung is inhibited by macrophage depletion using the mouse mammary tumor virus-polyoma middle T breast tumor model.55 Macrophages are now well-established elements of the TME, and their involvement is understood to extend from the initial stages of cancer to malignant and metastatic states.56
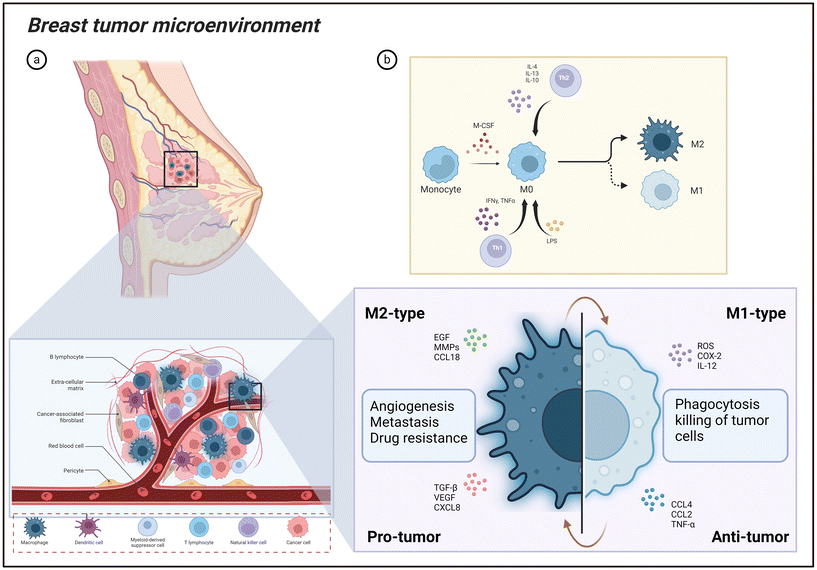 |
| | Fig. 2 Breast tumor microenvironment. (a) Breast tumor microenvironment with highlighted role of tumor associated macrophages and its pro-tumor and anti-tumor functions. (b) The overview of M1 and M2 macrophage differentiation from the monocyte cells. Image created with BioRender.com. | |
2.2 TAMs fuel breast tumor growth
2.2.1 Fostering tumorigenesis: the role of TAMs.
TAMs have a remarkable phenotypic variety, contributing to their diverse involvement in cancer progression and patient outcomes. In vitro and in vivo studies have shown a reciprocal feedback loop near the invasive borders of breast tumors between tumor cells and TAMs, where macrophages supply cytokines and growth factors (TNF-α, IL-6, IL-1β, VEGF), MMPs (MMP-2, MMP-9), ROS, RNS, Chemokines (CCL2, CXCL12) that encourage carcinogenesis.57 The growth of cancer cells is directly promoted by secreting inflammatory mediators and growth factors like TNF-α, IL-6, CCL2, and IL-1α.58 Specifically, the NF-κB pathway in tumor cells is activated by TNF-α produced by TAMs, avoiding cell death and improving invasive capacities.57 Tumor cells in this setting actively seek out and polarize macrophages toward an M2-like phenotype, which triggers the release of M2-related cytokines such as CCL18.59,60 Morphological alterations in breast cancer cells are induced by this cytokine, which suggests an epithelial–mesenchymal transition (EMT) and includes elongation, loss of contact inhibition, and increased expression of the mesenchymal marker vimentin.61 Interestingly, CCL18 and GM-CSF targeting reversed the EMT state of cancer cells in humanized murine models and thereby prevented metastasis.60 These results were further supported by investigations of human breast cancers, which showed a substantial relationship between poor clinical outcomes, metastasis, high GM-CSF expression, and the presence of macrophages that express CCL18 and cancer cells.14
2.2.2 Supporting angiogenesis: the role of TAMs.
Angiogenesis is an important mechanism that promotes tumor growth and progression, the complex process of creating new blood vessels from pre-existing ones. It enables the delivery of essential nutrients and oxygen while facilitating the removal of metabolic waste products.62,63 By secreting different angiogenic factors, macrophages are essential in promoting tumor angiogenesis.60 The process of angiogenesis is a complex and multi-step phenomenon triggered primarily by hypoxia in the TME.64 It begins with the release of pro-angiogenic factors VEGF, bFGF, PDGF.65 These factors initiate a cascade of events starting with the degradation of the basement membrane of existing blood vessels by matrix metalloproteinases (MMPs). This is followed by the activation and migration of endothelial cells towards the angiogenic stimulus to proliferate and form new vessel sprouts.66 M2-like TAMs play a dual role in angiogenesis: they directly produce the crucial cytokine VEGF-A and indirectly activate latent forms of VEGF-A by producing MMP9.67 TAMs also release TGF-α, TGF-β, EGF, and PDGF, among various pro-angiogenic molecules from the family of fibroblast growth factors.14 Additionally, TAMs promote angiogenesis by releasing CCL18, a chemokine that increases angiogenesis in vivo and in vitro. Notably, increased metastasis and tumor invasiveness in breast cancer have also been connected to TAM-secreted CCL18.68 Tie2-expressing monocytes (TEMs), a subpopulation of monocytes that express the angiopoietin receptor Tie2, are essential in inducing tumor angiogenesis in addition to TAMs. These TEMs have been discovered in mouse models as leukocytes displaying specific surface markers within mammary tumors, such as CD11b, Sca-1, and Tie2.69 Human TEMs isolated from breast cancer patients have a distinct surface marker profile that lacks expression of CD62L and CCR2 but expresses CD45, CD11b, CD11c, CD33, CCR5, CD13, and M-CSF-1R.70 Remarkably, TEMs isolated from primary invasive breast carcinomas exhibit antigen presentation-associated markers, including CD80, CD86, HLA-DR, and CD1a along with VEGFR-1, Tie-2, and CD14, indicating their possible tumor-specific immune responses role.70,71
2.2.3 The influence of TAMs on the metastatic potential.
TAMs are essential for the growth and spread of tumors because they produce urokinase (uPA) and matrix metalloproteases (MMPs), two types of matrix-degrading enzymes.72 These enzymatic activities promote the local infiltration of cancer cells and their capacity to dissociate from neighboring epithelial cells and the extracellular matrix (ECM) by disrupting adherens junctions.73 This starts the complex process of invasion-metastasis, which includes cancer cells entering blood and lymphatic vessels, spreading to other tissues, and finally developing metastatic lesions.73 Studies on direct vision with intravital multiphoton imaging have demonstrated the role of perivascular macrophages in promoting cancer cell intravasation in mammary tumors. In a mouse model of breast cancer, genetic ablation of CSF-1, a significant regulator of macrophage development and function, has slowed tumor proliferation and metastasis and decreased macrophage density.74 Through the production of many proteins that facilitate angiogenesis, the epithelial–mesenchymal transition (EMT), and cancer cell migration, TAMs have a role in metastasis. For example, in both mouse and human breast cancer models, CXCL1 production by TAMs has been linked to EMT and breast cancer cell migration.75 The Ets-2 transcription factor in TAMs plays a critical role in regulating angiogenesis and the development of lung metastases as well as primary malignancies.75 It has been demonstrated that clodronate-encapsulated liposomes can deplete macrophages, thereby reducing metastasis in ovarian cancer models and emphasizing their role in the dissemination of metastatic disease. Two transcription factors have been found to be essential for controlling angiogenesis and the development of lung metastases as well as primary malignancies in TAMs.76 TAMs exhibit significant levels of protease activity by cathepsin, which elevates tumor expansion and angiogenesis in the MMTV-PyMT animal model of breast cancer. Notably, IL-4 generated by T cells and tumor cells can stimulate the TAM's cathepsin protease activity, hence facilitating tumor invasion.77 It's crucial to remember that tissue-resident macrophages at metastatic locations could have a variety of phenotypes and roles. For instance, lysyl oxidase (LOX) released from hypoxic primary breast tumor settings can stimulate osteoclastogenesis, which is mediated by the transcription factor NFATc1.78 This procedure promotes bone resorption and allows circulating tumor cells to metastasize. For individuals with high-LOX primary breast cancers, bisphosphonate therapy has been suggested as a possible combination therapy.79 Bisphosphonates decrease osteoclast activity. Targeting TAMs should also take into account a therapeutic agent's capacity to reach metastatic locations.78 Local pulmonary injection of CSF-1R inhibitors dramatically improved the M1/M2 ratio at lower doses than oral administration in a mouse breast cancer model by overcoming physiological barriers and enabling more effective drug delivery to lung-resident macrophages. This emphasizes how crucial it is to investigate site-specific administration pathways in order to successfully target TAMs at metastatic sites.78
3. Macrophage reprogramming
TAMs convert between several polarization phenotypes under the influence of the TME. The promotion of tumor growth and metastasis is aided by TAMs’ M2 polarization.80 The following section discusses the main mechanisms that induce macrophage polarization toward the M2 phenotype in breast cancer tissues and the corresponding modifications to signaling pathways seen in TAMs.
3.1 Influence of TME on macrophage polarization
Within breast cancer tumors, TAMs are subjected to a complicated microenvironment. TAM heterogeneity and function are modulated by cooperative signals from multiple sources, including tumor cells, lymphocytes, extracellular matrix components, and cancer-associated fibroblasts (CAFs).81 The impact of various cellular and matrix elements on macrophage polarization inside the TME will be discussed in this section. There is growing evidence that tumor cells can secrete particular substances, like lactic acid and cytokines, that can induce macrophages to become activated in an M2-like manner.82 M2 TAM polarization enhances the proliferation, invasiveness, and migration potential of tumor cells. Furthermore, it has been shown that a few cytokines associated with macrophage recruitment—CCL2, CCL5, CSF1, and CXCL12 assist in TAM M2 polarization.83 Moreover, tumor-derived TGF-β promotes M2-type polarization by inhibiting macrophages’ activation and production of the transcription factor EB (TFEB).84 It has been discovered that macrophages polarized towards the M2 phenotype due to TGF-β influence aid in the development of tumor growth. Reprogramming energy metabolism is one of the hallmarks of breast cancer.85 Even in the existence of normal oxygen levels, breast cancer cells preferentially rely on glycolysis over oxidative phosphorylation, eventually increasing the formation of lactic acid.85 According to preclinical research, lactic acid produced by tumor cells in the TME can cause macrophage M2 polarization by activating signaling pathways in breast cancer, including ERK/STAT3, PKA/CREB, and HIF-1α/STAT3.86 It has been demonstrated that lactic acid-induced M2 polarization of macrophages promotes the migration, angiogenesis, and proliferation of breast cancer cells as well as their resistance to the treatment drug tamoxifen.87 The lactic acid generated by the tumor cells’ preferential glycolytic metabolism enriches the surrounding tissue in the tumor, providing an environment that is favorable for macrophage polarization towards an M2 phenotype. This, in turn, supports multiple aspects of tumor development and resistance to treatment.88 Differential cytokine profiles secreted by T helper 1 (Th1) and T helper 2 (Th2) cells affect macrophage polarization in different ways. Th2 cytokines have been linked to the promotion of TAMs M2 polarization and breast cancer metastasis. These cytokines include interleukin-4 (IL-4), IL-6, and IL-13. In particular, in inflammatory breast cancer models, IL-13 or IL-4 can cause STAT6 phosphorylation (Tyr641), which causes macrophages to become M2 polarized.89 It has been demonstrated that blocking IL-13 and IL-4 lowers the quantity of M2-like macrophages and possibly lessens radioresistance in inflammatory breast cancer. Moreover, it has been shown that IL-6 mediates M2 polarization via the mTORC2-Akt1 signaling axis, ultimately leading to a rise in distant metastasis.89 It has been demonstrated that Th1 cytokines polarize macrophages toward the M1 phenotype, in contrast to T helper 2 (Th2) cytokines. In this phase, one important Th1 cytokine is interferon-gamma (IFN-γ). According to research by Sun et al.,90 TAMs displaying the M2 marker CD206 may be reprogrammed to take on an M1-like phenotype, which is indicated by the production of inducible nitric oxide synthase (iNOS) by treating them with monophosphoryl lipid A and IFN-γ together. The cytokines interleukin-12 (IL-12) and tumor necrosis factor-alpha (TNF-α) released by these reprogrammed macrophages stimulated cytotoxic T cells. This depicted a system-wide immune response against tumors that led to decreased tumor development and metastasis in the PyMT and 4T1 mouse models.90 The interaction between CAFs and TAMs is essential to mediate macrophage polarization and tumor expansion. Research clarified this complex relationship; in patients with triple-negative breast cancer (TNBC), CAFs were linked to a higher infiltration of CD163+ macrophages, a hallmark of M2-like TAMs.91 According to a study by Yavuz et al., CAFs can actively attract monocytes and use the release of stromal cell-derived factor-1 (SDF-1) and CCL2 to polarize them towards an M2 phenotype. These M2-polarized TAMs inhibit the immune system, possibly through the PD-1 axis.91 CAFs can alter the cytokine environment, which in turn can indirectly aid in the polarization of macrophages. Chi3L1 derived from CAF upregulates the expression of factors linked with T helper 2 (Th2) cells, such as IL-10, IL-4, IL-13, and Gata3. The M2 polarization of TAMs is indirectly encouraged by this Th2-skewed cytokine milieu, which creates an immunosuppressive TME in breast cancer.92
3.2 Conventional breast cancer treatments and macrophage reprogramming
Breast cancer treatment has mainly targeted tumor cells directly using chemotherapy, radiotherapy, and hormone therapy. However, the TME, especially the immune landscape shaped by TAMs, is now recognized as an important factor affecting treatment outcomes.93 While these treatments can modulate TAM polarization, they often do so in ways that inadvertently promote tumor survival and progression.94 Chemotherapy is a key treatment for breast cancer, but its effect on TAM polarization can create challenges that reduce its effectiveness.95 The interaction between conventional cancer treatments, like chemotherapy and radiotherapy, and TAM polarization presents major challenges. Although TAMs play a crucial role in immune responses and tumor progression, traditional therapies often fail to influence macrophage polarization due to their limitations effectively.96 Conventional chemotherapy may inadvertently promote the polarization of TAMs toward the M2 phenotype, which is associated with immunosuppression and tumor progression. This phenomenon complicates therapeutic outcomes, as M2 macrophages can enhance tissue repair processes that support cancer survival and metastasis. Studies have shown that the increased presence of M2 TAMs correlates with poorer patient prognosis following chemotherapy.97 Therefore, while the immediate effects of chemotherapy may reduce tumor cell populations, the long-term repercussions may augment tumor aggressiveness by facilitating a supportive TME via M2 polarization.98 While effective inducing tumor cell death, certain chemotherapeutic agents can also inadvertently promote M2 polarization of TAMs. This pro-tumorigenic shift is particularly evident with agents such as doxorubicin and cyclophosphamide.99 Doxorubicin induces a hypoxic environment within tumors, leading to the upregulation of hypoxia-inducible factor 1-alpha (HIF-1α). HIF-1α, in turn, promotes the expression of VEGF and TGF-β, key drivers of M2 polarization.100 These M2-polarized TAMs contribute to an immunosuppressive TME, supporting tumor growth, angiogenesis, and metastasis, thereby reducing the overall effectiveness of chemotherapy. While using paclitaxel, it has been observed that initial M1 polarization does not prevent the eventual emergence of M2-polarized TAMs that contribute to chemoresistance.101 The ability of TAMs to switch phenotypes in response to treatment stresses the need for more targeted approaches that can lock TAMs into an M1 state or prevent the M2 transition. Radiotherapy-induced hypoxia is another critical factor influencing TAM polarization. High doses of radiation can exacerbate hypoxic conditions within the tumor, which are well-known drivers of M2 polarization. Hypoxia leads to the stabilization of HIF-1α, which promotes the expression of factors that drive M2 polarization.102 Radiotherapy-induced hypoxia significantly increases the recruitment and polarization of M2 TAMs, creating a feedback loop that enhances the tumor's resistance to further radiation. This hypoxia-induced shift towards M2 polarization presents a significant limitation, as it undermines the intended therapeutic effects of radiotherapy by promoting an environment conducive to tumor survival and proliferation.103 Tamoxifen and other therapies can influence TAM behavior, but they do not fully shift the TAM population towards an M1 phenotype. While novel strategies aimed at reprogramming TAMs have emerged, conventional treatments often lack specificity in modulating macrophage polarization. NcRNAs, including miRNAs, lncRNAs, and circRNAs, play crucial roles in the regulation of gene expression and cellular processes, including immune cell polarization. These ncRNAs can either promote or inhibit TAM polarization by targeting specific signaling pathways and transcription factors involved in macrophage activation.104 Utilizing ncRNA regulation strategies to modulate TAM polarization offers a novel avenue for overcoming the limitations of conventional breast cancer treatments.
4. Decoding the enigmatic influence of non-coding RNAs on TAM dynamics in breast cancer
NcRNAs, such as miRNAs, lncRNAs, and circRNAs do not code for proteins but play crucial roles in gene regulation and cellular processes. They are involved in post-transcriptional regulation, chromatin remodeling, and transcriptional control, making them key players in the TME.105 Cancer cell-derived ncRNAs can modulate the recruitment of monocytes to the TME and influence their differentiation into either pro-inflammatory M1 or anti-inflammatory M2 macrophages. These ncRNAs exert their effects by regulating the expression of genes involved in inflammatory responses within macrophages.18
4.1 miRNAs regulating TAM functions in breast TME
4.1.1 Recruitment and polarization of TAMs: the miRNA Nexus.
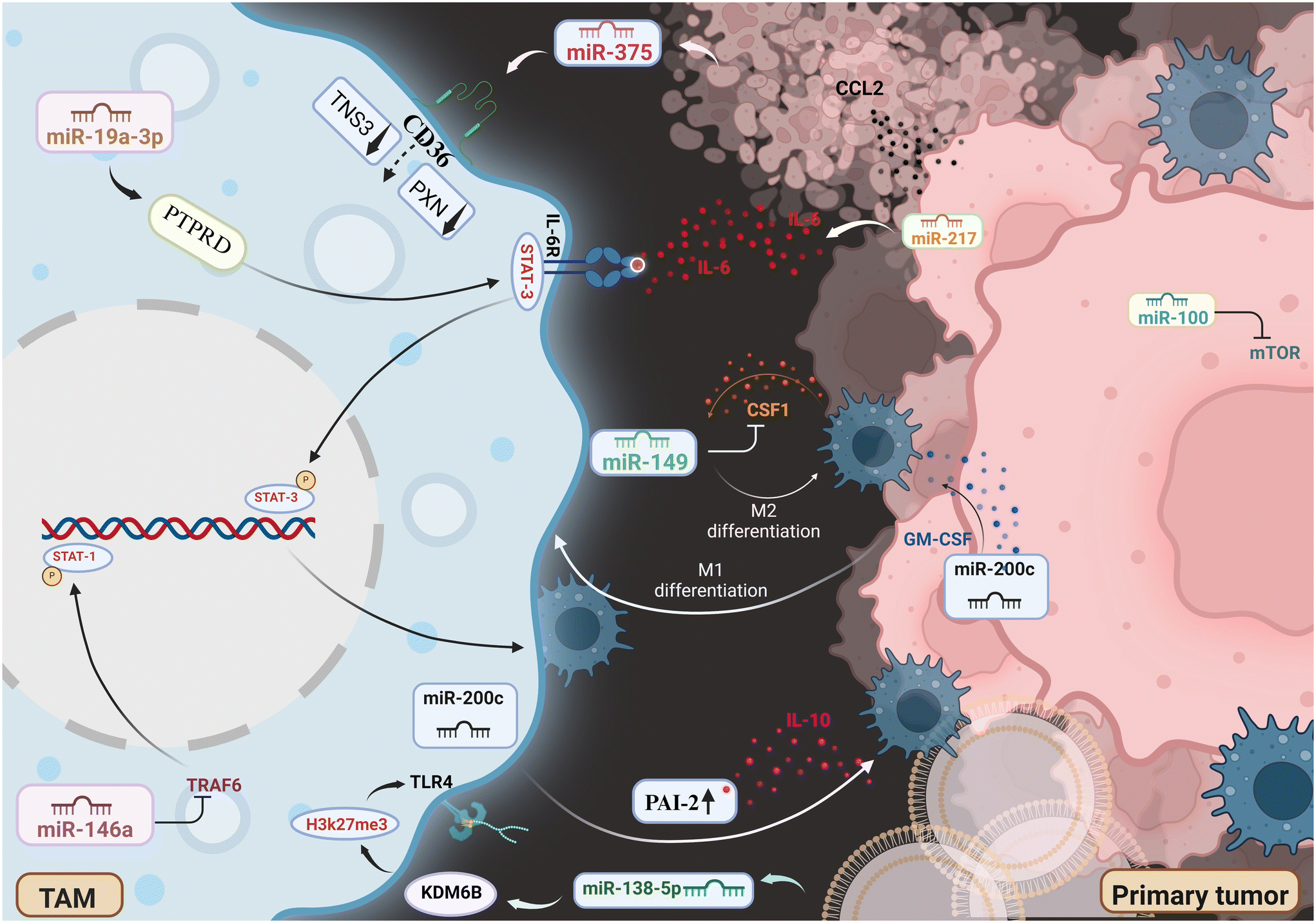
miRNAs are small RNA molecules that do not code for proteins but regulate gene expression after transcription. They are widely studied for their roles in cancer and immune regulation, including their influence on TAM polarization.106 Tumor progression and metastasis dissemination are impacted by the recruitment, polarization, and functional dynamics of TAMs within the breast cancer microenvironment, all of which are primarily controlled by miRNAs.18 Chemotactic cues from cancer cells direct the recruitment of monocytes, the progenitors of TAMs, and CCL2, which is one of the most influential factors that is expressed at an elevated level in a variety of cancers.107 Notably, it has been demonstrated that miR-375, a miRNA released by breast cancer cells that have undergone apoptosis, causes the production of CCL2 in additional cancer cells via as-yet-unidentified pathways. Moreover, miR-375 has the ability to bind to macrophages’ CD36 receptor, which promotes its uptake into TAMs and suppresses the production of TNS3 and PXN.107 These two proteins are known to impede cell motility. This miRNA-mediated regulation eventually encourages the growth of cancer by increasing macrophages’ capacity to infiltrate the tumor bulk.107 It's interesting to note that CCL2 also facilitates macrophage migration to metastatic locations.108 The cytokine colony-stimulating factor-1 (CSF1) is essential for promoting macrophage recruitment and survival, and higher production of this cytokine has been linked to higher mortality rates in breast cancer patients.109,110 The production of CSF1 mRNA in breast tumors is directly targeted by miRNA miR-149, which inhibits it. This limits macrophage appointment to the main tumor location. Notably, in a mouse model of breast cancer, when MDA-MB-231 cells were injected orthotopically, this lower TAM infiltration has been linked to a decreased risk of lung metastasis development.111 Furthermore, two crucial indicators linked to the M2 macrophage phenotype-arginase-1 (ARG1) and mannose receptor C-type 1 (MRC1) are also downregulated by miR-149. The underlying mechanism of miR-149's action is yet unknown. However, it is thought to function in a paracrine fashion by controlling the amount of soluble substances produced by breast cancer cells, which in turn affects macrophage polarization.111 Numerous different miRNAs can either promote or repress the pro-tumoral M2 phenotype by influencing the polarization of TAMs (Fig. 3). The miR-200 family, which is well-known for its tumor-suppressive properties and capacity to adversely influence the epithelial–mesenchymal transition (EMT) process, can modify macrophage polarization through different pathways.112 The elevation of the Nos2 (M1 marker) and the downregulation of the Arg1 (M2 marker), leading to the upregulation of miR-200c in breast cancer cells, triggers the production of GM-CSF, which is a potent inducer of M1-macrophage polarization.112 However, other studies showed that miR-200c can also upregulate the functions of PAI-2 in breast tumor cells, directly inducing M2 polarization by promoting the release of the anti-inflammatory cytokine IL-10.113
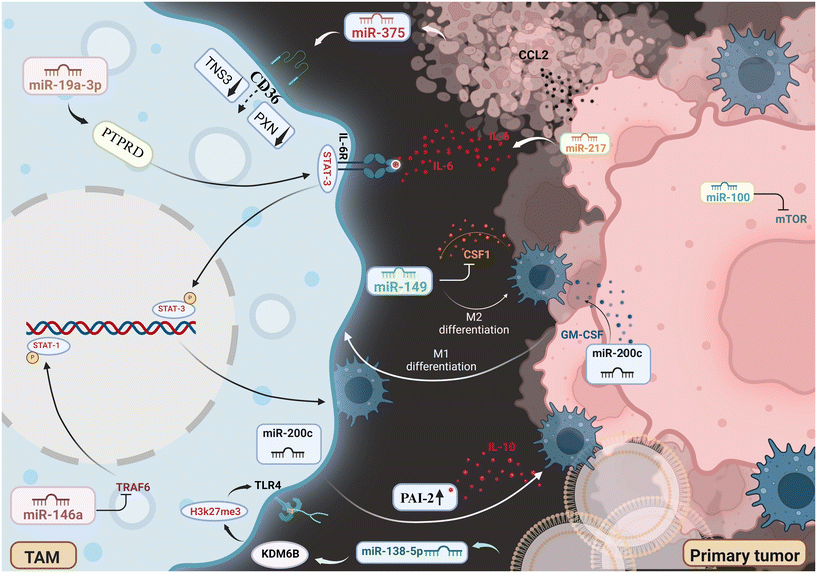 |
| | Fig. 3 Schematic representation of the miRNAs interplay in breast tumor microenvironment, tumor associated macrophages and breast cancer cells. Image created with BioRender.com. | |
4.1.2 Orchestrating the TME: miRNAs at the Helm.
Macrophage polarization is partly shaped by the TME, and miRNAs are important mediators in this complex process. The cytokine IL-6, which is generated by many cell types in the TME, has been linked to lower survival rates in individuals with breast cancer. Remarkably, by suppressing the expression of miR-19a-3p in TAMs, IL-6 released by breast tumors can cause macrophages to polarize towards the M2 phenotype. This, in turn, activates the STAT3 pathway, which is a significant driver of the M2 polarization program.114 Furthermore, TAMs isolated from breast tumors and the neighboring tumor tissue show high miR-100 expression levels. The M2 polarization phenotype is supported by the increased expression of miR-100 in TAMs since it inhibits the mTOR pathway and upregulates the expression of CD206, a recognized M2 marker.115 Macrophage polarization has been found to be regulated by exosomes produced by cancer cells. By reducing the KDM6B expression in macrophages, miR-138-5p, an exosomal overexpressing miRNA derived from breast cancer cell lines like MDA-MB-231 and T47D, suppresses M1 polarization and promotes the M2 phenotype.116 Interestingly, exosomal miR-138-5p-treated macrophages have been demonstrated to enhance lung metastasis in breast cancer models. Modulating macrophage polarization is also possible using miR-222, another exosomal miRNA produced from adriamycin-resistant MCF-7 cells.116 It has been shown that several miRNAs control important mechanisms linked to the spread of metastatic breast cancer, including angiogenesis, invasion, and metastatic seeding. For example, it has been demonstrated that the transcription factor ID4, which is produced at elevated levels in TNBC, can rewire TAMs by upregulating the functions of angiogenesis-related genes and downregulating the anti-angiogenic miRNA miR-15b/107.117 On the other hand, miR-497 has been shown to block the angiogenesis of breast cancer by focusing on VEGFR2.118 Notably, miR-375 is a miRNA that is generated by breast cancer cells that have undergone apoptosis. TAMs subsequently bind to this miRNA via the CD36 receptor, facilitating macrophage migration and infiltration into the tumor mass, ultimately creating an environment supporting tumor growth.107 Furthermore, it has been observed that breast cancer tumors isolated TAMs exhibit considerable downregulation of both miR-222 and miR-146a. The suppression of miR-146a is associated with a decrease in M2 macrophage markers and a corresponding attenuation of tumor progression. On the contrary, CXCL12 and CXCR4 are the targets of miR-222 overexpression in TAMs, which inhibits tumor growth and macrophage migration.119
4.1.3 miRNA-mediated regulation of TAM function and BC progression.
Through modifying transcription factor expression and binding proteins involved in macrophage development and functional dynamics, miRNAs have been linked to the regulation of macrophage polarization. As an illustration, it has been demonstrated that the transcription factor PU.1 targets miR-146a, which is essential for hematopoietic stem cell differentiation into peritoneal macrophages in mice models.120 Additionally, by targeting Kruppel-like factor 6 (KLF6) and CCAAT/enhancer binding protein-α (C/EBPα),121 a downregulating transcription factor in primary breast cancers and associated with the dysregulation of various genes implicated in tumor progression.121 Higher expression levels of miRNA-181a in M2 macrophages inhibit the M1 phenotype and induce the M2 polarization program. Annexin A1 (ANXA1), an immunomodulatory protein, has also been linked to controlling the polarization of macrophages in breast cancer. Lower production levels of ANXA1 in ER + MCF-7 cells are negatively related to greater production levels of the oncogenic miR-196a and suppression of NFκβ and c-myc.122 However, the lack of ANXA1 inhibits the growth and spread of tumors in vivo by enhancing the polarization of M1 macrophages through the modulation of the FPR2 (formyl peptide receptor2)-ERK-CCL5 signaling pathway.123 Numerous investigations have documented the practical application of miRNA-based strategies aimed at reprogramming TAM activity and polarization of macrophages. MiR-223,124 miR-155,125 and miR-511-3p,126 for example, have been demonstrated to rewire macrophage activation and reestablish the equilibrium between pro- and anti-tumoral functions. Furthermore, miRNAs have been linked to the regulation of tumor drug resistance, a significant obstacle in the treatment of cancer. Notably, it has been observed that suppressing miR-21 enhances chemosensitivity by upregulating the expression of the tumor suppressor PTEN, whilst miR-21 itself has been shown to decrease the sensitivity of cancer cells to chemotherapeutic agents.127
4.2 circRNAs regulating TAM functions in breast TME
4.2.1 Circular RNAs: the hidden conductors of macrophage fate.
Single-stranded RNA molecules with a distinct closed circular structure are known as circRNAs or circular RNAs. These RNA molecules have expression patterns specific to tissues and developmental stages that are controlled by a variety of trans- and cis-acting factors.128–130 Studies have shown that there is a connection between macrophages and circRNAs. TAMs can be influenced by circ-ASAP1 by acting as a competitive endogenous RNA (ceRNA) for miR-326 and miR-532-5p. It has been demonstrated that circ-ASAP1 specifically mediates TAM invasion by controlling the miR-326/miR-532-5PCSF-1 pathway.131 Furthermore, circRNA-CDR1as has been connected to processes of cell penetration and tumor tissue immunology, including the infiltration of M2 macrophages, activated natural killer (NK) cells, and CD8+ T cells.132 The process of macrophage polarization is dependent on a number of components, including signal transducers and activators of transcription (STATs) and interferon regulatory factors (IRFs).133,134 M1 macrophage differentiation is induced by lipopolysaccharide (LPS) stimulation through the activation of STAT1-α and STAT1-β, which interact with the TLR-4 receptor.135 Through the circPPM1F–HUr–PPM1F–NF-kB axis, circular RNA molecules such as circPPM1F can actively contribute to the LPS-induced M1 macrophages’ activation.136 In a similar vein, circCdy can stimulate M1 polarization by preventing IRF4 from entering the nucleus.137 However, TGF-β, CSF-1, IL-13, IL-10, and IL-4 all promote the polarization of the M2 macrophage subpopulation.43,138 Certain circular RNAs, such as has-circ-0005567, can be overexpressed to promote M2 macrophage differentiation and prevent M1 macrophage polarization.139 It's interesting to note that M1 macrophages express circRNAs like circRNA-010231, circRNA-003780, and circRNA-010056 at higher levels than M2 macrophages, whereas M1 macrophages express circRNAs like circRNA-018127, circRNA-001489, circRNA-013630 and circRNA-003424 at lower levels.140 Exosomal circRHCG (hsa_circ_ 0104852) promotes M2 polarization and accelerates TNBC progression by BTRC-mediated ubiquitination and degradation of TFEB.141 Tumor-derived exosomal cSERPINE2 increases MALT1 levels and IL-6 release in TAMs.142
4.3 LncRNAs regulating TAM functions in breast TME
LncRNAs are longer transcripts that do not code for proteins but can regulate gene expression at multiple levels, including chromatin remodeling, transcription, and post-transcriptional processing.143 Xing et al.144 found that increased expression of LINC00337 improved the viability and proliferated BC cells along with drug resistance to paclitaxel (PAX). Overexpression of LINC00337 decreased the level of GM-CSF and increased levels of M2 TAM markers and M-CSF.BC cell viability was markedly decreased by PAX, which also down-regulated LINC00337. Moreover, LINC00337 strengthened the impact of M2-type macrophages and effectively generated M2-type macrophages to support BC cell activity, migration, and EMT protein expression. Silencing the LINC00337 had the opposite effect on BC cells.144 LINC00657 is distinctly upregulated in breast cancer exosomes, which is associated with increased m6A methylation. LINC00657 depletion diminishes BC cell proliferation, migration, and invasion and accelerates apoptosis (Table 1). LINC00657 activates TGF-β signaling in macrophages by sequestering miR-92b-3p and contributing to a tumor-promoting microenvironment and M2 macrophage polarization.145
Table 1 Roles of non-coding RNAs in tumor associated macrophage regulation
| ncRNA |
Targeting pathway/signaling |
Functional effect |
TAM polarization state |
Ref. |
|
miRNA
|
|
| miR-149 |
CSF-1 mRNA inhibition/ARG1 & MRC1 downregulation |
Limits TAM recruitment to BC cells |
Promotes M1-polarization |
111
|
| miR-375 |
Binds to CD36 on macrophage/decreased production of TNS3 & PXN/CCL2 production |
Promotes TAM recruitment to BC cells |
Promotes M2-polarization |
107
|
| miR-19a-3p |
Downregulated expression of FRA1 & STAT-3 |
Suppressed expression in TAMs |
Facilitates M1-polarization |
114
|
| miR-200c |
Increased Nos expression/ARG1 downregulated |
Expressed in BC cells |
Drives M1-polarization |
112
|
|
|
Upregulated PAI-2 in BC cells |
Drives M2-polarization |
113
|
| miR-138-5p |
Restricts KDM6B activity |
Expression in TAMs |
Promotes M2-polarization |
116
|
| MiR-222 |
Promotes CXCL12 & CXCR4 downregulation of PTEN |
Promotes M2-polarization |
119 and 146 |
| miR-100 |
Inhibits mTOR pathway upregulates expression of CD206 |
Induces M2-polarization |
115
|
| miR-183-5p |
Inhibits PPP2CA inducing NF κB activation |
Promotes an anti-tumorigenic TME |
147
|
| miR-15b |
Highly expressed ID4 |
Anti-angiogenic TAM functions |
142
|
| miR-497 |
Downregulation of VEGFR |
118
|
|
lncRNA
|
|
| Linc00337 |
Decreased level of GM-CSF, increased chemoresistance |
|
Increased M2 TAM polarization |
144
|
| LINC00657 |
Increased m6A methylation, activates TGF-β signaling |
Expressed by BC exosomes |
Increased M2 TAM polarization |
145
|
| LncRNA_109 |
Blocked the ubiquitin-mediated degradation of FUBP1, activates c-Myc |
|
Promotes M2-polarization |
148
|
| LncRNA_GNAS-AS1 |
Suppressed the expression of GATA3, act as a miR-433-3p sponge |
|
Stimulates M2 repolarization |
149
|
| Linc00514 |
Boosted the expression of Jagged1 |
|
Promotes M2-polarization |
150
|
A study conducted by Zhang et al. revealed that lncRNA_109, through blocking the ubiquitin-mediated degradation of FUBP1 and activating c-Myc, induced the M2-like polarization of macrophages. Moreover, lncRNA_109 is upregulated by c-Myc. This lncRNA_109/FUBP1/c-Myc loop is a positive feedback system that forms a tumor-promoting, M2-like TAM microenvironment. These discoveries reveal the new pathways by lncRNA_109 controlling TAM M2 polarization.148 It was discovered that ER+ breast cancer and M2 macrophages both have elevated levels of the lncRNA GNAS-AS1. Importantly, GNAS-AS1 stimulated M2 macrophage polarization by controlling the miR-433-3p/GATA3 axis, which in turn enhanced the growth, migration, and invasion of ER+ breast cancer cells. The lncRNA suppressed the expression of GATA3 by acting as a miR-433-3p sponge (Fig. 4).149 Linc00514 is one such lncRNA that has a crucial role in M2 activation. Overexpression of linc00514 in BC tissues and cell lines revealed a high proportion of the M2 markers CD206 and CD163. This overexpression was facilitated by increased phosphorylation of the STAT3 transcription factor, which boosted the transcriptional expression of Jagged1.150 The Notch signaling pathway regulated by Jagged1 then stimulated the release of IL-4 and IL-6 from breast cancer cells, which in turn caused the macrophages to become M2 polarized.150
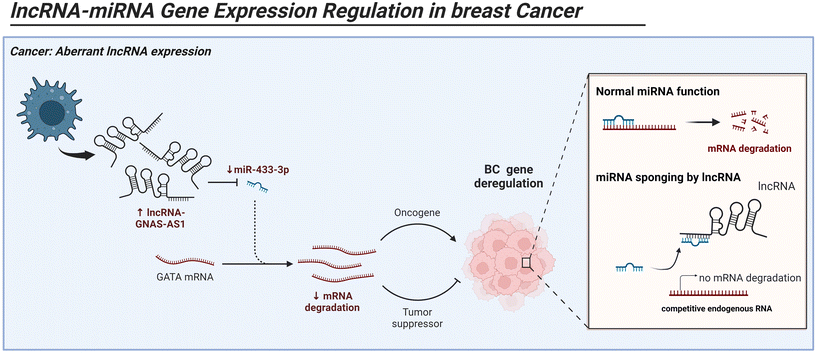 |
| | Fig. 4 Mechanism of action and role of lncRNA in regulation of tumor associated macrophage and breast cancer by acting as miRNA sponge. Image created with BioRender.com. | |
4.4 Delivery challenges of ncRNA therapeutics
The development of ncRNA-based therapies holds great promise for treating breast cancer, offering innovative approaches to modulate tumor progression and immunity. However, effective delivery of these molecules presents significant challenges that must be addressed to realize their therapeutic potential.151 The delivery of ncRNAs into target cells is a critical step for their therapeutic application. Traditional methods, such as lipid-based transfection or electroporation, can be inefficient or damaging, especially in the clinical setting.152 The inherent instability of ncRNAs poses a significant challenge for their therapeutic use. In biological fluids, ncRNAs are susceptible to degradation by nucleases, which can limit their bioavailability and efficacy.153 Strategies such as chemical modifications like 2′-O-methyl modifications and nanoparticle encapsulation can improve ncRNAs’ stability.154 Yet optimizing these approaches remains an area of active research. Off-target interactions are another concern when employing ncRNA-based therapies. While the specificity of ncRNA action is advantageous, unintended effects on other genes can result in deleterious outcomes. Careful design and screening of ncRNA sequences and delivery systems that enhance targeting to specific tissues or cells are essential to minimize this risk. The efficiency of ncRNA delivery remains a hurdle in achieving desired therapeutic outcomes. Factors such as cellular uptake, endosomal escape, and intracellular trafficking significantly impact the effectiveness of delivery systems. Current strategies often require optimizing these processes, particularly in heterogeneous tumor environments where access to all cancer cells may be limited. Continued innovation in nanoparticle technology is essential to improve the efficiency and specificity of ncRNA delivery.151 Therefore, developing efficient strategies for delivering ncRNA-based therapeutics to their target sites is crucial. This can be achieved by leveraging advanced nanocarrier systems, which offer promising avenues for the precise and effective transport of ncRNAs to specific cells or tissues, for instance, non-coding RNAs, such as miRNAs and their mimics, have been successfully conjugated to the surfaces of metal nanoparticles, including gold nanoparticles.155 The following section explores various nanocarrier-based approaches and their potential applications in enhancing the delivery and therapeutic efficacy of ncRNA-based treatments.
5. Nanotechnology-based TAM targeting strategies for breast cancer
Nanoparticle-based targeting strategies have emerged as a promising approach for modulating TAM polarization, a crucial aspect of cancer immunotherapy.156 Nanoparticle-based targeted therapy offers a promising approach for macrophage reprogramming, significantly impacting antitumor and inflammatory diseases. Macrophages are also being targeted for various chronic inflammatory diseases for advanced novel treatments for its transition from M1 to M2 state for its effective anti-inflammatory role.157 One of the studies explores a combinational approach for targeting RAW264.7 macrophage reprogramming via delivery of the methotrexate and RELA siRNA using the folate receptor targeting liposomes via NF-κB pathway inhibition.158 These nanoscale delivery systems offer unique opportunities to overcome the limitations of conventional therapies and selectively reprogram TAMs towards an anti-tumor phenotype.
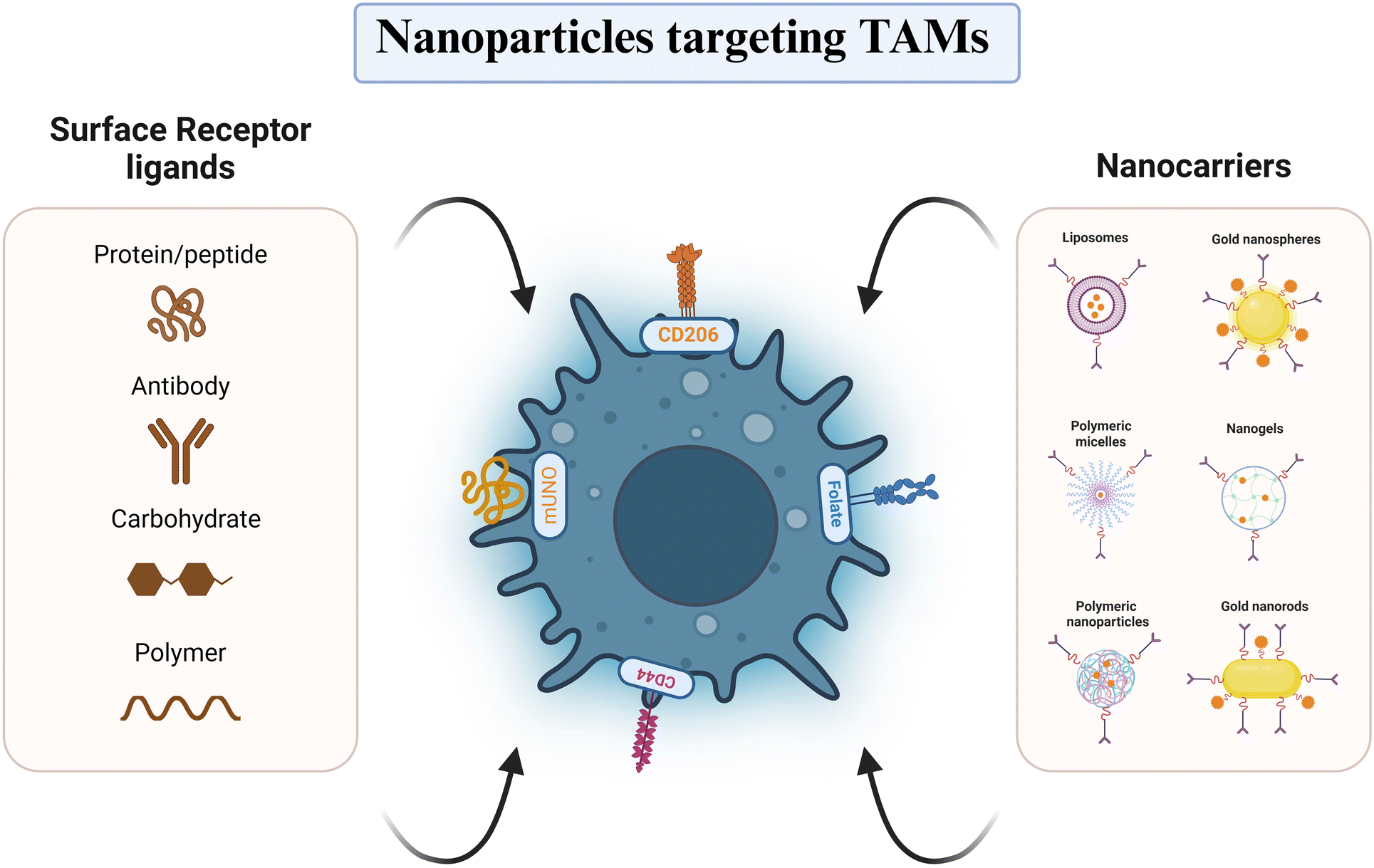
5.1 Active targeting based on specific receptors
Active-targeting nanocarrier-based delivery systems (NCDSs) exploit the overexpression of specific receptors on the TAM membrane, facilitating the selective delivery of drugs by the ligand's interaction on the nanoparticle surface with these receptors (Fig. 5). Nanoparticles can be designed to target specific receptors overexpressed on TAMs, allowing for selective delivery of immunomodulatory agents. One widely explored target is the mannose receptor (CD206), which is highly expressed on M2-like TAMs. Kumari and her team159 prepared self-assembled amphiphilic PEGylated galactomannan (GM) nanoparticles containing hydrazinocurcumin (HC), targeting the CD206 receptor on M2-like macrophage cells. Treatment with these PSGM-HCNPs led to increased reactive oxygen species (ROS) levels, decreased expressions of Arg-1 and CD206, and increased secretion of pro-inflammatory cytokines, suggesting successful repolarization of TAMs from pro-tumorigenic to anti-tumorigenic characteristic.159 Li and his team created porous hollow iron oxide nanoparticles (PHNPs) attached with mannose to facilitate the delivery of the PI3Kγ small molecule inhibitor 3-methyladenine (3-MA) to TAMs.160 These PHNPs depicted enhanced cellular uptake in RAW 264.7 macrophages compared to mannose-unlinked nanoparticles, likely due to their ability to target the overexpressing mannose receptors on M2-like TAM. Importantly, PHNPs outperformed their non-targeted counterparts in the repolarization of M2-like TAM and tumor progression inhibition, highlighting their potential as nanocarriers for cancer therapy targeting TAMs.160 Similarly, Figueiredo et al. employed the specific mannose receptor binding “mUNO” peptide to target and reprogram M2-like macrophages. By conjugating mUNO to lignin nanoparticles loaded with the immunomodulatory agent resiquimod, they observed enhanced cellular uptake in M2-like TAMs and significant tumor proliferation suppression, offering a promising approach for breast cancer treatment.161 In addition to mannose receptors, other membrane overexpressing proteins on TAMs, like scavenger receptors, CD44, and folate receptors (FRs), have been exploited to improve the target capabilities of NCDSs.162 CD44, a receptor for hyaluronic acid (HA) and chondroitin sulfate, is highly expressed on both TAMs and BC cells, making it an attractive target for NCDSs aiming to simultaneously target both cell types.163,164 Gong et al. developed HA-modified iron oxide nanoparticles (Fe3O4-DOX-HA) to deliver doxorubicin (DOX) to CD44-positive TAMs and 4T1 breast tumor cells, demonstrating enhanced tumor accumulation and antitumor efficacy compared to non-targeted nanoparticles.163 Folate receptors (FRs), with folic acid (FA) as the ligand, are overexpressed on tumor cells and macrophages, making them another potential target for NCDSs.162 Li et al. ideated FA-modified lipid nanoemulsions (PTX/DHA-FA-LNs) to co-deliver docosahexaenoic acid (DHA) and paclitaxel (PTX). These FA-targeted nanoparticles displayed increased accumulation in M2-like macrophages and MCF-7 BC cells with enhanced MCF-7 cell cytotoxicity and the capability to modulate TAM programming (Table 2).165 Dual-targeting strategies, where distinct ligands are conjugated to NCDSs to target varied receptors on TAMs and breast tumor cells, respectively, have also been explored. A terpolymer-lipid hybrid nanoparticle (TPLN) system co-loaded with mitomycin C (MMC) and DOX developed by Zhang et al.166 was conjugated with the cyclic internalizing peptide iRGD, which targets integrins abundantly expressed on BC cell membranes, while also exhibiting the ability to target TAMs through low-density lipoprotein receptor (LDLR)-mediated endocytosis and apolipoprotein E (ApoE) attraction. This dual-targeting approach enhanced cellular uptake in both BC cells and TAMs, demonstrating its potential for TAM-targeted therapy.166 It is important to note that active-targeting NCDSs may also bind to and be phagocytosed by macrophages present in other tissues, potentially reducing their accumulation in tumor tissues. To address this limitation, researchers have explored strategies to initially target NCDSs to tumor tissues before their subsequent uptake by TAMs. Peng and colleagues167 developed a docetaxel (DTX) loaded dual-targeting nanoparticle system (ATpep-NPs) to treat BC. ATpep, comprising of the phagocytosis-triggering tuftsin peptide (Tpep) and the legumain-activated substrate peptide alanine-alanine-asparagine (AAN) in the TME, facilitates endocytosis by tumor cells and TAMs via Fc receptors or neuropilin-1. This TME-responsive nanoplatform successfully prevents non-specific absorption of actively-targeting NCDSs during blood circulation.167
Table 2 Various nanoparticles based delivery systems for tumor associated macrophage targeting and regulation
| NCDS |
Active therapeutic agent |
Target |
Targeting ligand/gene |
Functional effect |
BC tumor model |
Ref. |
| PEG-galactomannan NPs (PSGM-HCNP) |
Hydrazinocurcumin |
CD206 |
Mannose |
Increased ROS & decreased Arg-1, CD206 expression |
4T1 |
159
|
| Mannose-linked porous hollow iron oxide nanoparticles (PHNPs@DPA-S-S-BSA-MA@3-MA) |
PI3Kγ small molecule inhibitor 3-methyladenine (3-MA) |
Activation of macrophage inflammatory factor NF-κB p65, inhibited PI3Kγ |
MDA-MB-231 |
160
|
| Lignin nanoparticles (LNPs) |
Resiquimod |
CD206 |
mUNO peptide |
Enhanced M2 uptake/increased M1 type polarization |
4T1 |
161
|
| HA-modified iron oxide nanoparticles (Fe3O4-DOX-HA) |
Doxorubicin |
CD44 |
HA |
Enhanced tumor efficacy |
4T1 |
163
|
| FA-modified lipid nanoemulsions (PTX/DHA-FA-LNs) |
PTX + DHA |
Folate receptor |
Folic acid |
Facilitated M2 reprogramming |
MCF-7 |
165
|
| Terpolymer-lipid hybrid nanoparticle (TPLN) |
MMC + DOX |
LDLR |
ApoE |
Enhanced uptake in BC cells and TAM depletion |
MDA-MB-231 |
166
|
| ATpep-NPs |
Docetaxel |
FC + neuropilin-1 |
ATpep |
Facilitating endocytosis by dual targeting |
4T1 |
167
|
| NP-180 |
|
siCCL-18 |
CCL-18 |
Enhanced cellular uptake and efficient silencing of CCL-18 inhibiting EMT and metastasis |
MDA-MB-231 |
169
|
| PEG-PLA NPs |
|
siCCR-2 |
CCR-2 |
Macrophage recruitment inhibition |
4T1 |
170
|
| BG34-10-Re-I |
|
siMIF |
MIF |
Reduction of factors for M2 polarization |
MDA-MB-231 |
171
|
| PEG = MT/PC NPs |
|
siPIGF + siVEGF |
VEGF + PIGF |
Reduced expression of M2 surface marker, increase in TNF-α & IL-2 |
MDA-MB-231/4T1 |
172
|
| Polydopamine-coupled magnetite nanoparticles (PDA-MNPs) |
|
siPERK |
PERK |
Downregulated PERK pathway and M2 markers |
|
175
|
| Dextran-g-poly(histidine) copolymer (DH@ECm) |
BLZ-945 |
CD206 |
Dextran |
Hybrid membrane system enhanced tumor specificity |
4T1 |
177
|
| Dendrigraft poly-L-lysines-zoledronic acid NPs (DGL-ZA)n |
Zolendronic acid (ZA) |
|
|
Depleted and repolarized TAMs and increased M1/M2 ratio |
|
178
|
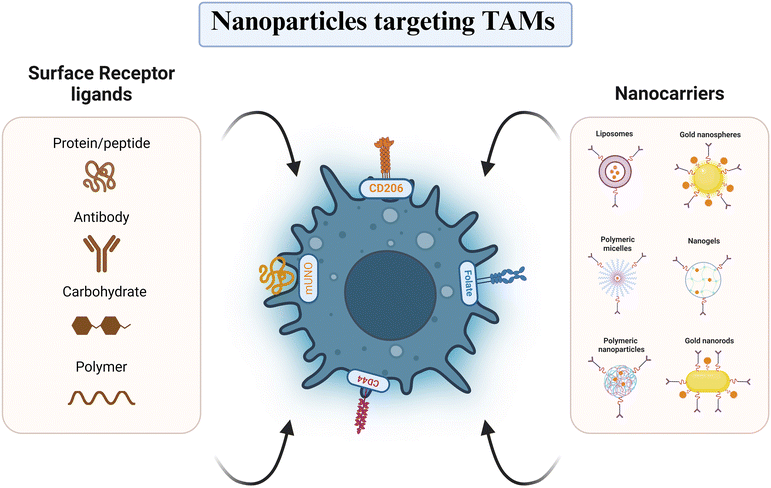 |
| | Fig. 5 Schematic representation of nanocarrier-based delivery system targeting strategy for tumor associated macrophage modulation in breast cancer. Image created with BioRender.com. | |
5.2 Nanoparticle-based delivery systems for ncRNA delivery
Nanoparticle-based delivery systems have emerged as a promising strategy for delivering nucleic acid therapeutics, such as siRNA, shRNA, and miRNA, to TAMs to modulate their polarization and activity.168 These nucleic acid therapies offer a unique approach to control macrophage function by silencing specific genes or regulating the expression of key miRNAs involved in TAM polarization and tumor progression. One of the widely explored approaches involves the delivery of small siRNA to TAMs using nanoparticle carriers. Liang et al.169 developed 180 nm nanoparticles composed of biodegradable poly (ethylene glycol)-b-poly (ε-caprolactone) (PEG-b-PCL), poly (ε-caprolactone)-b-poly (2-aminoethyl ethylene phosphate) (PCL-b-PPEEA), and PCL homopolymer. These nanoparticles can load and transport siRNA targeting CCL-18, a chemokine secreted by TAMs that promotes breast cancer metastasis through electrostatic interactions. The results demonstrated effective cellular uptake of the siCCL-18 encapsulated nanoparticles compared to free siCCL-18, resulting in restraining BC cell metastasis.169 PEG-PLA nanoparticles developed by Rafael et al. have a positive charge to deliver siRNA for CCR2 to inflammatory macrophages. CCR2 is a key transmembrane protein involved in the recruitment of macrophages to the tumor site. The delivery of CCR2 siRNA facilitated the inhibition of macrophage recruitment, transforming the immunosuppressive TME into an immunostimulatory one.170 Co-administration of multiple siRNAs to TAMs has also been explored to attain synergic anti-cancer effects. Zhang et al. utilized the glucan-based siRNA carrier technology BG34-10-Re-I to co-deliver siRNA targeting migration inhibitory factor (MIF) to both tumor cells and TAMs. While MIF silencing in tumor cells led to increased cell death and reduced proliferation, MIF silencing in TAMs significantly reduced factors associated with M2 polarization.171 Song et al. employed polyethylene glycol (PEG) and mannose-modified trimethyl chitosan (PEG = MT), as well as poly(allylamine hydrochloride) (PC) grafted with citraconic anhydride-based nanoparticles (PEG = MT/PC NPs) with dual pH-responsiveness. These were used to co-deliver VEGF siRNA (siVEGF) and PIGF siRNA (siPIGF), which are upregulated in both tumor cells and TAMs. The co-delivery of siVEGF and siPIGF using these nanoparticles demonstrated superior suppression of tumor progression and lung metastasis in contrast to administering each siRNA separately.172 In addition to siRNA delivery, nanoparticle systems have been explored for the delivery of CRISPR-based gene editing tools to TAMs. Leonard and colleagues173 utilized CRISPR-RICTOR-Liposomes to disable RICTOR, an adaptor protein in the mTORC2 complex, thereby preventing M2 polarization.95 The use of CRISPR-RICTOR-Liposomes in breast cancer models decreased the percentage of M2-like TAMs and enhanced the effectiveness of paclitaxel treatment.173 As discussed in the previous sections, miRNAs have also been investigated as potential targets for modulating TAM polarization and tumor progression. Hu et al.174 developed a concurrent and cascade control framework for the efficient transfection of miR-125b, which regulates TAM polarization and enhances T-cell activation. This system combined anionic magnetic nanoparticles (MNPs) and cationic lipid peptides with an arginine-rich boundary (RLS). The high transfection efficiency of MNPs aided in polarizing breast cancer cells into M1-like macrophages, thereby impeding tumor growth and metastasis.174 D'Urso et al.175 used Fe3O4 magnetic nanoparticles coated in polydopamine, which can bind siRNAs, to deliver siRNA that targets protein kinase R-like ER kinase (PERK), playing a major role in unfolded protein response in the ER of TAM, in primary mouse macrophages. Significant downstream effectors of the PERK pathway that aided in UPR (CHOP and ATF4) were also downregulated when PERK was inhibited. Additionally, M2 markers—particularly CD206—were downregulated, whereas M1 markers such as CD86 and inflammatory cytokines—were upregulated.175 The authors of the work176 coupled tetrahedral framework nucleic acid with an agonist of TLR9 cytosine-guanosine oligodeoxynucleotide (CpG ODN) and anti-PI3Kγ (phosphatidylinositol triphosphate kinase) siRNA. Acquired NPs facilitated TLR9 activation in TAMs, that leads to the initiation of the NF-κB signaling cascade. This cascade is associated with the activation of transcription of inflammatory genes (M1 phenotype). Concurrent downregulation of PI3Kγ amplified this outcome.176
5.3 Other strategic nanoparticles
5.3.1 Polymer-based nanoparticles.
Polymer-based nanoparticles have gained significant attention due to their versatility, biocompatibility, and ability to encapsulate a wide range of therapeutic agents. One such example is the dextran-g-poly(histidine) copolymer (DH@ECm) developed by Wang et al.177 This nanocarrier exploits cancer cell hybrid membranes to disguise and deliver the CSF1R inhibitor BLZ-945 specifically to M2-type macrophages. The dextran moiety facilitates targeting to CD206, a receptor overexpressed on TAMs, while the hybrid membrane camouflage enhances tumor specificity. Notably, DH@ECm exhibited superior anti-tumor activity, achieving a 64.5% inhibition rate, three times higher than the free drug.177 Dendrigraft poly-L-lysines (DGLs) have also been employed to encapsulate zoledronic acid (ZA), a nitrogen-containing bisphosphonate known for depleting and repolarizing TAMs. Guo et al. developed (DGL-ZA)n nanoparticles to overcome the limited tumor accumulation of ZA due to its high affinity for bone. Treatment with these nanoparticles significantly increased the proportion of M1 (CD16/32+) to M2 (CD206+) TAMs, demonstrating their potent ability to reprogram M2-type macrophages.178 Ann-Kathrin et al. discovered that while carboxyl-modified polystyrene nanoparticles and amino-modified ones did not alter the M1 markers, they were successful in preventing macrophages from polite programming to M2 by downregulating the CD200R, CD163, and IL-10 expression.179 Combined with anti-PD-1 therapy, R848 encapsulated β-cyclodextrin nanoparticles were successful in controlling TAMs and enhancing the efficiency of growth inhibition of cancer cells in a variety of scenarios.180 A lignin nanoparticle with particular peptides altered on the surface was also a potential carrier of R848 and targeted M2 that expressed CD206.161In vitro, the nanomedicine effectively decreased the tumor burden in mice nearly twenty times more than the control when it came to the M1 marker TNF-α.161 To distribute R837, other group produced acetylated chondroitin sulfate protoporphyrin polymer.181 It inhibited the growth of 4T1 in mice in conjunction with different Dox-loaded polymeric micelle.181
5.3.2 Inorganic nanoparticles.
Inorganic nanoparticles, such as iron oxide nanoparticles, have demonstrated remarkable potential in modulating TAM polarization.182 The FDA-approved ferumoxytol nanoparticle can polarize M2-like TAMs toward an anti-tumor M1 phenotype, initiating an anti-cancer response.183 However, its limited efficacy prompted the development of mannose-bound porous hollow iron nanoparticles (PHNPs) by Li et al. These nanoparticles were loaded with 3-methyladenine (3-MA), a PI3Kγ small molecule inhibitor, and displayed enhanced therapeutic efficacy and TAM repolarization. Higher therapeutic effectiveness and TAM repolarization were attained with PHNPs@DPA-S-S-BSA-MA@3-MA therapy. The hollow nanomedicine prevented the growth of tumors in the MDA-MB-231 mouse model of human breast cancer.160 Another innovative approach involves the use of metal–organic framework nanoparticles (MOFs). Gu et al. developed an iron-based MOF (MIL88B) capable of loading RSL3, a potent inhibitor of mitochondrial activity. By forcing macrophages to undergo glycolytic metabolism, the RSL3-loaded MIL88B nanoparticles effectively inhibited tumor growth and metastasis.184 It has been demonstrated that coating iron oxide coating with a SIRPα expressing biological membrane from genetically altered cells functions efficiently. After the “do not eat me” signal was blocked, magnetic orientation caused the nanoparticles containing iron to assemble in the TME. This caused TAMs to repolarize towards M1, evoking strong immune reactions and inhibiting the proliferation of both 4T1 cancer cells and B16F10 with T lymphocytes.185 The NF-κB p65 expression was effectively increased, and TAMs were reprogrammed to M1 by hollow iron oxide treated with mannose and carrying a PI3Kγ inhibitor payload.160 Mucin-1 peptide-bound gold nanoparticles were successful in inducing polarization to the M1 state in response to rising cytokines levels, including TNF-α, IL-6, IL-10, and IL-12.186 When albumin and paclitaxel were wrapped around gold nanorods, Li et al. were able to modify the TME and decrease M2 polarization in animals having tumors.187 The application of manganese dioxide NPs includes delivering repolarization factors. Chih-Chuh Chang's research team developed a nanomedicine with a core–shell structure containing manganese dioxide in the core and PLGA and lipid in the shell to target the TME.188 The successful repolarization of bone marrow-derived macrophages was demonstrated by the downregulation of M2 markers and the increased expression of M1 markers.188
5.3.3 DNA-based nanocarriers.
DNA scaffolds have emerged as unique nano-drug carriers capable of delivering small molecules to specific cellular compartments. Cui et al.189 employed a DNA duplex of a 38-base pair to deliver the cysteine protease inhibitor E64 to the TAM lysosomes via scavenger receptor-mediated uptake. By inhibiting lysosomal cysteine protease activity, this approach facilitated antigen cross-presentation and activation of CD8+ T lymphocytes, leading to effective tumor burden management without altering the overall TAM phenotype.189
5.3.4 Multi-drug co-delivery systems.
NCDSs offer the unique advantage of co-delivering multiple therapeutic agents, potentially leading to synergistic effects in TAM repolarization. Yang et al.190 prepared a poly(lactic-co-glycolic acid) (PLGA) nanocarrier loaded with the anti-malarial drug chloroquine (CQ) and a polysaccharide, both known to re-educate TAMs through distinct signaling pathways.191 In a 4T1-M2 co-culture model, the MP-ss-PLGA@CQ nanoparticles were preferentially taken up by M2-type macrophages, resulting in increased M1 macrophage polarization and enhanced inhibition of both primary and metastatic tumors.190 Similarly, in order to administer a MAPK regulator and a CSF1R inhibitor simultaneously, Ramesh et al. developed supramolecular nanoparticles (DSNs) loaded with dual kinase inhibitors. Co-delivery of these inhibitors in an intense 4T1 cancer model led to superior tumor suppression and repolarization of M2-type macrophages to a tumor-fighting M1 type compared to single-agent delivery.192
5.3.5 Immunomodulatory nanoparticles.
Nanoparticles can also be engineered to modulate the “eat me” and “don't eat me” signals, thereby enhancing TAM phagocytosis of tumor cells. Zhang et al.193 developed azide-modified silica nanoparticles (SNPAs) covalently conjugated with calreticulin (CALR) and an anti-CD47 antibody (aCD47). By simultaneously presenting the CALR “eat me” signal and blocking the CD47 “don't eat me” signal, the SNPA CALR&aCD47 nanoparticles significantly enhanced the proportion of tumor cells ingested by macrophages and exhibited potent anti-tumor activity in 4T1 cancer model.193
6. Conclusion and perspectives
Breast cancer is a highly heterogeneous disease with TME playing a crucial role in its progression and metastasis. TAMs are a significant component of the TME which regulate the expression of ncRNAs in breast tumors and vice versa, which in turn promote TAM recruitment and polarization to the tumor niche. This ncRNA crosstalk between TAMs and the TME represents a promising therapeutic target for breast cancer treatment. Various studies have demonstrated the potential of targeting TAMs for breast cancer therapy. However, the clinical translation of nanoparticle-based approaches to target TAMs and modulate the TME is hindered by the complexity and heterogeneity of cells in the TME. One of the major challenges is the potential off-target effects of TAM-targeting nanoparticles which needs to be carefully addressed. The efficacy of nanoparticle-based therapies depends significantly on their ability to selectively target macrophages within the body. Specific targeting can be achieved by functionalizing nanoparticles with ligands that bind to receptors overexpressed on macrophages. Receptor-mediated endocytosis is a critical mechanism through which macrophages internalize nanoparticles. Functionalized nanoparticles that engage with specific macrophage receptors promote efficient cellular uptake, which is essential for intracellular therapeutic payloads. For example, coating nanoparticles with galactose or mannose moieties can significantly enhance their recognition and uptake by macrophages, enabling more effective drug delivery. Particle size is also a crucial factor influencing nanoparticle biodistribution and cellular uptake. Nanoparticles in the range of 100 to 200 nm are optimal for macrophage targeting, as this size range can facilitate efficient endocytosis through phagocytic pathways. Larger particles may be recognized and eliminated by the mononuclear phagocyte system, while smaller particles may fail to engage adequately with macrophages. It is crucial to consider the molecular subtype of breast cancer, the diagnostic technologies available, and the TME's response to therapeutic interventions to improve the ability to accurately target TAMs and modulate the TME. A deeper understanding of the intricate interplay between TAMs, ncRNAs, and the TME is essential for developing effective and personalized treatment strategies. Future perspectives in nanoparticle-based targeting of TAMs and modulating ncRNA crosstalk should prioritize several key aspects. Developing more specific and targeted nanocarrier systems is crucial to minimize off-target effects and enhance the selective delivery of therapeutic agents to TAMs within the TME. This could involve exploring novel targeting ligands, stimuli-responsive nanocarriers, or multi-stage delivery systems. Combination therapies that simultaneously target TAMs and other key components of the TME, such as cancer stem cells or immune cells, should be explored to achieve synergistic anti-tumor effects. The complex interplay between TAMs, ncRNAs, and other cellular players within the TME necessitates a multi-pronged approach for effective therapeutic intervention. The development of more efficient and stable delivery systems is crucial in the field of ncRNA-based interventions to overcome the challenges associated with nucleic acid degradation and off-target effects. The incorporation of chemical modifications, the exploration of novel ncRNA targets, and the development of multiplexed ncRNA delivery systems could offer a more comprehensive approach to modulating TAM dynamics and their crosstalk with other cellular components within the TME. Furthermore, integrating advanced diagnostic techniques like liquid biopsies or single-cell genomics could aid in better characterizing the molecular heterogeneity of breast tumors. This improved understanding could facilitate the tailoring of TAM-targeting strategies to specific tumor subtypes, which enhance their efficacy and personalization. Identifying potential targets and mechanisms involved in this crosstalk could pave the way for overcoming therapy resistance, which is a significant challenge in breast cancer management. Ultimately, a multifaceted approach combining targeted nanocarrier development, combination therapies, advanced diagnostics, clinical evaluation, and elucidation of resistance mechanisms holds promise in unlocking the full potential of TAM-targeting strategies and improving patient outcomes. Conducting comprehensive clinical trials to evaluate the safety and efficacy of these TAM-targeting nanoparticle therapies across different breast cancer subtypes and stages is also imperative for translating these approaches into clinical practice. It is crucial to investigate the role of ncRNA-mediated crosstalk between TAMs and the TME in regulating treatment resistance. By addressing these challenges and pursuing a multi-disciplinary approach, researchers and clinicians can unlock the full potential of TAM-targeting therapies and ncRNA-based strategies for improving the outcomes of breast cancer patients.
Author contributions
Hardik Patni: Writing and image curation, Ramesh Chaudhary: writing and editing, Ashutosh Kumar: conceptualization, supervision, editing and funding acquisition.
Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Indian Council of Medical Research (ICMR), Government of India [grant number: – EM/Dev/SG/217/1078/2023].
References
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, CA-Cancer J. Clin., 2021, 71, 209–249 CrossRef.
- G. Collaborators, K. B. Tran, J. J. Lang, M. Compton, R. Xu, A. R. Acheson, H. J. Henrikson, J. M. Kocarnik, L. Penberthy and A. Aali, The Lancet, 2022, 400, 563–591 CrossRef.
- F. Bray, M. Laversanne, H. Sung, J. Ferlay, R. L. Siegel, I. Soerjomataram and A. Jemal, CA-Cancer J. Clin., 2024, 74, 229–263 CrossRef.
- J. Y. Tsang and M. T. Gary, Adv. Anat. Pathol., 2020, 27, 27–35 CrossRef CAS.
- S. Loibl, P. Poortmans, M. Morrow, C. Denkert and G. Curigliano, Lancet, 2021, 397, 1750–1769 CrossRef CAS.
- C. W. Tong, M. Wu, W. C. Cho and K. K. To, Front. Oncol., 2018, 8, 227 CrossRef.
- J. K. Jallah, T. J. Dweh, A. Anjankar and O. Palma, Cureus, 2023, 15, 10 Search PubMed.
- T. Akinsipe, R. Mohamedelhassan, A. Akinpelu, S. R. Pondugula, P. Mistriotis, L. A. Avila and A. Suryawanshi, Front. Immunol., 2024, 15, 1302587 CrossRef CAS.
- K. G. K. Deepak, R. Vempati, G. P. Nagaraju, V. R. Dasari, S. Nagini, D. N. Rao and R. R. Malla, Pharmacol. Res., 2020, 153, 104683 CrossRef CAS.
- Q. Wang, X. Shao, Y. Zhang, M. Zhu, F. X. Wang, J. Mu, J. Li, H. Yao and K. Chen, Cancer Med., 2023, 12, 11149–11165 CrossRef.
- K. Li, H. Shi, B. Zhang, X. Ou, Q. Ma, Y. Chen, P. Shu, D. Li and Y. Wang, Signal Transduction Targeted Ther., 2021, 6, 362 CrossRef.
- R. Noy and J. W. Pollard, Immunity, 2014, 41, 49–61 CrossRef CAS.
- A. Shafqat, M. H. Omer, E. N. Ahmed, A. Mushtaq, E. Ijaz, Z. Ahmed, K. Alkattan and A. Yaqinuddin, Front. Immunol., 2023, 14, 1252998 CrossRef CAS.
- S. Su, Q. Liu, J. Chen, J. Chen, F. Chen, C. He, D. Huang, W. Wu, L. Lin, W. Huang, J. Zhang, X. Cui, F. Zheng, H. Li, H. Yao, F. Su and E. Song, Cancer Cell, 2014, 25, 605–620 CrossRef CAS.
- M. Xiao, J. He, L. Yin, X. Chen, X. Zu and Y. Shen, Front. Immunol., 2021, 12, 799428 CrossRef CAS.
- D. G. DeNardo, D. J. Brennan, E. Rexhepaj, B. Ruffell, S. L. Shiao, S. F. Madden, W. M. Gallagher, N. Wadhwani, S. D. Keil, S. A. Junaid, H. S. Rugo, E. S. Hwang, K. Jirström, B. L. West and L. M. Coussens, Cancer Discovery, 2011, 1, 54–67 CrossRef CAS.
- S. L. Shiao, B. Ruffell, D. G. DeNardo, B. A. Faddegon, C. C. Park and L. M. Coussens, Cancer Immunol. Res., 2015, 3, 518–525 CrossRef CAS.
- A. Benedetti, C. Turco, G. Fontemaggi and F. Fazi, Non-Coding RNA, 2022, 8, 16 CrossRef CAS.
- Z. Zhou, Z. Wang, J. Gao, Z. Lin, Y. Wang, P. Shan, M. Li, T. Zhou and P. Li, Mol. Ther. – Oncolytics, 2022, 25, 98–120 CrossRef CAS.
- W. Li, X. Wang, C. Li, T. Chen and Q. Yang, Mol. Ther., 2021, 22, 36 Search PubMed.
- M. Entezari, M. Sadrkhanloo, M. Rashidi, S. E. Asnaf, A. Taheriazam, M. Hashemi, M. Ashrafizadeh, A. Zarrabi, N. Rabiee, K. Hushmandi, S. Mirzaei and G. Sethi, Crit. Rev. Oncol. Hematol., 2022, 103680 CrossRef.
- S. Sim and N. K. Wong, Biomed. Rep., 2021, 14, 42 CrossRef CAS.
- S. Toden, T. J. Zumwalt and A. Goel, Biochim. Biophys. Acta, Rev. Cancer, 2021, 1875, 188491 CrossRef CAS.
- D. Parashar, A. Singh, S. Gupta, A. Sharma, M. K. Sharma, K. K. Roy, S. C. Chauhan and V. K. Kashyap, Genes, 2022, 13, 1254 CrossRef CAS.
- X. D. Mao, X. Wei, T. Xu, T. P. Li and K. S. Liu, Am. J. Cancer Res., 2022, 12, 3208–3222 CAS.
- P. Sharma and M. Otto, Bioact. Mater., 2024, 31, 440–462 CAS.
- M. T. Vitor, P. C. Bergami-Santos, J. A. Barbuto and L. G. de la Torre, Recent Pat. Drug Delivery Formulation, 2013, 7, 99–110 CrossRef CAS PubMed.
- L. Teodori, M. Omer and J. Kjems, RNA Biol., 2024, 21, 1–19 CrossRef CAS.
- S. K. Biswas, S. Banerjee, G. W. Baker, C. Y. Kuo and I. Chowdhury, Int. J. Mol. Sci., 2022, 23, 3883 CrossRef CAS PubMed.
- A. K. Elfstrum, A. S. Bapat and K. L. Schwertfeger, Cancer Med., 2024, 13, e7053 CrossRef CAS PubMed.
- M. D. Rojo, I. Bandyopadhyay, C. M. Burke, A. D. Sturtz, E. S. Phillips, M. G. Matherne, S. J. Embrey, R. LaRue, Y. Qiu, K. L. Schwertfeger and H. L. Machado, Life Sci. Alliance, 2024, 7, e202302516 CrossRef PubMed.
- Y. Yang, J. Hou, J. Liu, S. Bhushan and G. Wu, Int. Immunopharmacol., 2022, 110, 109047 CrossRef CAS.
- C. E. Weber and P. C. Kuo, Surg. Oncol., 2012, 21, 172–177 CrossRef.
- S. Mittal, N. J. Brown and I. Holen, Expert Rev. Mol. Diagn., 2018, 18, 227–243 CrossRef CAS PubMed.
- S. Vinogradov, G. Warren and X. Wei, Nanomedicine, 2014, 9, 695–707 CrossRef CAS PubMed.
- F. Geissmann, M. G. Manz, S. Jung, M. H. Sieweke, M. Merad and K. Ley, Science, 2010, 327, 656–661 CrossRef CAS.
- F. Ginhoux and M. Guilliams, Immunity, 2016, 44, 439–449 CrossRef CAS.
- M. T. Munir, M. K. Kay, M. H. Kang, M. M. Rahman, A. Al-Harrasi, M. Choudhury, N. Moustaid-Moussa, F. Hussain and S. M. Rahman, Int. J. Mol. Sci., 2021, 22, 6526 CrossRef CAS.
- N. J. Horwood, Clin. Rev. Allergy Immunol., 2016, 51, 79–86 CrossRef CAS.
- E. Gomez Perdiguero, K. Klapproth, C. Schulz, K. Busch, E. Azzoni, L. Crozet, H. Garner, C. Trouillet, M. F. de Bruijn, F. Geissmann and H.-R. Rodewald, Nature, 2015, 518, 547–551 CrossRef.
- S. Gordon and A. Plüddemann, BMC Biol., 2017, 15, 53 CrossRef.
- T. A. Wynn, A. Chawla and J. W. Pollard, Nature, 2013, 496, 445–455 CrossRef CAS.
- P. J. Murray, J. E. Allen, S. K. Biswas, E. A. Fisher, D. W. Gilroy, S. Goerdt, S. Gordon, J. A. Hamilton, L. B. Ivashkiv, T. Lawrence, M. Locati, A. Mantovani, F. O. Martinez, J.-L. Mege, D. M. Mosser, G. Natoli, J. P. Saeij, J. L. Schultze, K. A. Shirey, A. Sica, J. Suttles, I. Udalova, J. A. van Ginderachter, S. N. Vogel and T. A. Wynn, Immunity, 2014, 41, 14–20 CrossRef CAS.
- D. M. Mosser and J. P. Edwards, Nat. Rev. Immunol., 2008, 8, 958–969 CrossRef CAS.
- Y. Fan and S. He, Cancer Manage. Res., 2022, 14, 1–17 CrossRef CAS.
- C. D. Mills, Crit. Rev. Immunol., 2012, 32, 463–488 CrossRef CAS.
- D. Laoui, K. Movahedi, E. Van Overmeire, J. Van den Bossche, E. Schouppe, C. Mommer, A. Nikolaou, Y. Morias, P. De Baetselier and J. A. Van Ginderachter, Int. J. Dev. Biol., 2011, 55, 861–867 CrossRef.
- A. K. Mishra, S. Banday, R. Bharadwaj, A. Ali, R. Rashid, A. Kulshreshtha and S. K. Malonia, Vaccines, 2022, 11, 55 CrossRef.
- T. Satoh, H. Kidoya, H. Naito, M. Yamamoto, N. Takemura, K. Nakagawa, Y. Yoshioka, E. Morii, N. Takakura and O. Takeuchi, Nature, 2013, 495, 524–528 CrossRef CAS.
- L. Gao, F.-Q. Wang, H.-M. Li, J.-G. Yang, J.-G. Ren, K.-F. He, B. Liu, W. Zhang and Y.-F. Zhao, Oncotarget, 2016, 7, 87037–87051 CrossRef.
- C. Feriotti, F. V. Loures, E. Frank de Araújo, T. A. da Costa and V. L. G. Calich, PLoS One, 2013, 8, e54845 CrossRef CAS.
- R. Huang, T. Kang and S. Chen, J. Cancer Res. Clin. Oncol., 2024, 150, 238 CrossRef CAS.
- C. Wang, M. Cao, X. Jiang, Y. Yao, Z. Liu and D. Luo, Int. Immunopharmacol., 2021, 97, 107682 CrossRef CAS.
- J. Steenbrugge, K. Breyne, K. Demeyere, O. De Wever, N. N. Sanders, W. Van Den Broeck, C. Colpaert, P. Vermeulen, S. Van Laere and E. Meyer, J. Exp. Clin. Cancer Res., 2018, 37, 191 CrossRef.
- E. Y. Lin, A. V. Nguyen, R. G. Russell and J. W. Pollard, J. Exp. Med., 2001, 193, 727–740 CrossRef CAS.
- K. E. de Visser and J. A. Joyce, Cancer Cell, 2023, 41, 374–403 CrossRef CAS.
- C.-H. Lee, C.-L. Wu and A.-L. Shiau, J. Immunother., 2010, 33, 73–82 CrossRef CAS.
- L. Yang and Y. Zhang, J. Hematol. Oncol., 2017, 10, 58 CrossRef.
- L. She, Y. Qin, J. Wang, C. Liu, G. Zhu, G. Li, M. Wei, C. Chen, G. Liu, D. Zhang, X. Chen, Y. Wang, Y. Qiu, Y. Tian, X. Zhang, Y. Liu and D. Huang, Cancer Cell Int., 2018, 18, 120 CrossRef.
- R. D. Leek and A. L. Harris, J. Mammary Gland Biol. Neoplasia, 2002, 7, 177–189 CrossRef.
- T. N. Augustine, R. Duarte and G. P. Candy, Anticancer Res., 2020, 40, 6179–6193 CrossRef CAS.
- N. Nishida, H. Yano, T. Nishida, T. Kamura and M. Kojiro, Vasc. Health Risk Manage., 2006, 2, 213–219 CrossRef CAS.
- P. Carmeliet and R. K. Jain, Nature, 2000, 407, 249–257 CrossRef CAS.
- Y. Huang, S. Goel, D. G. Duda, D. Fukumura and R. K. Jain, Cancer Res., 2013, 73, 2943–2948 CrossRef CAS.
- M. Potente, H. Gerhardt and P. Carmeliet, Cell, 2011, 146, 873–887 CrossRef CAS.
- P. Carmeliet and R. K. Jain, Nature, 2011, 473, 298–307 CrossRef CAS.
- S. Quintero-Fabián, R. Arreola, E. Becerril-Villanueva, J. C. Torres-Romero, V. Arana-Argáez, J. Lara-Riegos, M. A. Ramírez-Camacho and M. E. Alvarez-Sánchez, Front. Oncol., 2019, 9, 1370 CrossRef.
- L. Lin, Y.-S. Chen, Y.-D. Yao, J.-Q. Chen, J.-N. Chen, S.-Y. Huang, Y.-J. Zeng, H.-R. Yao, S.-H. Zeng and Y.-S. Fu, Oncotarget, 2015, 6, 34758 CrossRef.
- C. E. Lewis, M. De Palma and L. Naldini, Cancer Res., 2007, 67, 8429–8432 CrossRef CAS.
- M. Ibberson, S. Bron, N. Guex, E. Faes-van't Hull, A. Ifticene-Treboux, L. Henry, H.-A. Lehr, J.-F. Delaloye, G. Coukos, I. Xenarios and M.-A. Doucey, Clin. Cancer Res., 2013, 19, 3439–3449 CrossRef CAS PubMed.
- F. Pucci, M. A. Venneri, D. Biziato, A. Nonis, D. Moi, A. Sica, C. Di Serio, L. Naldini and M. De Palma, Blood, 2009, 114, 901–914 CrossRef CAS.
- J. E. Talmadge and I. J. Fidler, Cancer Res., 2010, 70, 5649–5669 CrossRef CAS.
- S. D. Mason and J. A. Joyce, Trends Cell Biol., 2011, 21, 228–237 CrossRef CAS.
- E. Y. Lin, V. Gouon-Evans, A. V. Nguyen and J. W. Pollard, J. Mammary Gland Biol. Neoplasia, 2002, 7, 147–162 CrossRef PubMed.
- N. Wang, W. Liu, Y. Zheng, S. Wang, B. Yang, M. Li, J. Song, F. Zhang, X. Zhang, Q. Wang and Z. Wang, Cell Death Dis., 2018, 9, 1–18 CrossRef CAS.
- T. M. Robinson-Smith, I. Isaacsohn, C. A. Mercer, M. Zhou, N. Van Rooijen, N. Husseinzadeh, M. M. McFarland-Mancini and A. F. Drew, Cancer Res., 2007, 67, 5708–5716 CrossRef CAS PubMed.
- T. Zabuawala, D. A. Taffany, S. M. Sharma, A. Merchant, B. Adair, R. Srinivasan, T. J. Rosol, S. Fernandez, K. Huang, G. Leone and M. C. Ostrowski, Cancer Res., 2010, 70, 1323–1333 CrossRef CAS.
- S. S. Alhudaithi, R. M. Almuqbil, H. Zhang, E. R. Bielski, W. Du, F. S. Sunbul, P. D. Bos and S. R. P. da Rocha, Mol. Pharmaceutics, 2020, 17, 4691–4703 CrossRef CAS.
- X. Wang, X. Zhong, Z. Liu and L. Cheng, Nano Today, 2020, 35, 100946 CrossRef CAS.
- C. M. Neophytou, M. Panagi, T. Stylianopoulos and P. Papageorgis, Cancers, 2021, 13, 2053 CrossRef CAS.
- Y. Yang, J. Qin, L. Lan, N. Li, C. Wang, P. He, F. Liu, H. Ni and Y. Wang, Cancer Biol. Ther., 2014, 15, 99–107 CrossRef CAS.
- S. Babazadeh, S. M. Nassiri, V. Siavashi, M. Sahlabadi, M. Hajinasrollah and M. Zamani-Ahmadmahmudi, Cell. Mol. Biol. Lett., 2021, 26, 30 CrossRef CAS.
- Y.-F. Wang, L. Yu, Z.-L. Hu, Y.-F. Fang, Y.-Y. Shen, M.-F. Song and Y. Chen, Cell Death Dis., 2022, 13, 748 CrossRef CAS.
- L. Fang, J. Hodge, F. Saaoud, J. Wang, S. Iwanowycz, Y. Wang, Y. Hui, T. D. Evans, B. Razani and D. Fan, OncoImmunology, 2017, 6, e1312042 CrossRef.
- D. Hanahan and R. A. Weinberg, Cell, 2011, 144, 646–674 CrossRef CAS.
- X. Mu, W. Shi, Y. Xu, C. Xu, T. Zhao, B. Geng, J. Yang, J. Pan, S. Hu, C. Zhang, J. Zhang, C. Wang, J. Shen, Y. Che, Z. Liu, Y. Lv, H. Wen and Q. You, Cell Cycle, 2018, 17, 428–438 CrossRef CAS.
- X. Niu, J. Ma, J. Li, Y. Gu, L. Yin, Y. Wang, X. Zhou, J. Wang, H. Ji and Q. Zhang, Cell Death Dis., 2021, 12, 509 CrossRef CAS.
- H. Jiang, H. Wei, H. Wang, Z. Wang, J. Li, Y. Ou, X. Xiao, W. Wang, A. Chang, W. Sun, L. Zhao and S. Yang, Cell Death Dis., 2022, 13, 206 CrossRef CAS PubMed.
- R. Shrivastava, M. Asif, V. Singh, P. Dubey, S. A. Malik, M.-U. D. Lone, B. N. Tewari, K. S. Baghel, S. Pal, G. K. Nagar, N. Chattopadhyay and S. Bhadauria, Cytokine, 2019, 118, 130–143 CrossRef CAS.
- L. Sun, T. Kees, A. S. Almeida, B. Liu, X.-Y. He, D. Ng, X. Han, D. L. Spector, I. A. McNeish, P. Gimotty, S. Adams and M. Egeblad, Cancer Cell, 2021, 39, 1361–1374 CrossRef CAS PubMed.
- B. Gok Yavuz, G. Gunaydin, M. E. Gedik, K. Kosemehmetoglu, D. Karakoc, F. Ozgur and D. Guc, Sci. Rep., 2019, 9, 3172 CrossRef PubMed.
- N. Cohen, O. Shani, Y. Raz, Y. Sharon, D. Hoffman, L. Abramovitz and N. Erez, Oncogene, 2017, 36, 4457–4468 CrossRef CAS PubMed.
- A. Kotsifaki, N. Alevizopoulos, V. Dimopoulou and A. Armakolas, Int. J. Mol. Sci., 2023, 24, 15332 CrossRef CAS.
- S. Shao, H. Miao and W. Ma, Front. Immunol., 2023, 14, 1295684 CrossRef CAS.
- C. Zhan, Y. Jin, X. Xu, J. Shao and C. Jin, Cancer Med., 2023, 12, 11049–11072 CrossRef CAS.
- C. Belgiovine, E. Digifico, C. Anfray, A. Ummarino and F. Torres Andón, J. Clin. Med., 2020, 9, 3226 CrossRef CAS.
- P. Moeini and P. Niedźwiedzka-Rystwej, Int. J. Mol. Sci., 2021, 22, 7239 CrossRef CAS.
- E. Cendrowicz, Z. Sas, E. Bremer and T. P. Rygiel, Cancers, 2021, 13, 1946 CrossRef CAS.
- I. Larionova, N. Cherdyntseva, T. Liu, M. Patysheva, M. Rakina and J. Kzhyshkowska, OncoImmunology, 2019, 8, 1596004 CrossRef.
- B. P. Bui, P. L. Nguyen, K. Lee and J. Cho, Cancers, 2022, 14, 6054 CrossRef CAS.
- C. W. Wanderley, D. F. Colon, J. P. M. Luiz, F. F. Oliveira, P. R. Viacava, C. A. Leite, J. A. Pereira, C. M. Silva, C. R. Silva and R. L. Silva, Cancer Res., 2018, 78, 5891–5900 CrossRef CAS.
- R. Bai, Y. Li, L. Jian, Y. Yang, L. Zhao and M. Wei, Mol. Cancer, 2022, 21, 177 CrossRef CAS.
- C. Becherini, A. Lancia, B. Detti, S. Lucidi, D. Scartoni, G. Ingrosso, M. G. Carnevale, M. Roghi, N. Bertini, C. Orsatti, M. Mangoni, G. Francolini, S. Marani, I. Giacomelli, M. Loi, S. Pergolizzi, E. Bonzano, C. Aristei and L. Livi, Strahlenther. Onkol., 2023, 199, 1173–1190 CrossRef.
- X. Zhang, X. Xu, J. Song, Y. Xu, H. Qian, J. Jin and Z. f. Liang, Biomed. Pharmacother., 2023, 167, 115527 CrossRef CAS.
- K. R. Uppaluri, H. J. Challa, A. Gaur, R. Jain, K. Krishna Vardhani, A. Geddam, K. Natya, K. Aswini, K. Palasamudram and K. Sri Manjari, Transl. Oncol., 2023, 35, 101730 CrossRef CAS.
- L. A. Macfarlane and P. R. Murphy, Curr. Genomics, 2010, 11, 537–561 CrossRef CAS.
- A.-C. Frank, S. Ebersberger, A. F. Fink, S. Lampe, A. Weigert, T. Schmid, I. Ebersberger, S. N. Syed and B. Brüne, Nat. Commun., 2019, 10, 1135 CrossRef.
- S. Wang, K. Liang, Q. Hu, P. Li, J. Song, Y. Yang, J. Yao, L. S. Mangala, C. Li, W. Yang, P. K. Park, D. H. Hawke, J. Zhou, Y. Zhou, W. Xia, M.-C. Hung, J. R. Marks, G. E. Gallick, G. Lopez-Berestein, E. R. Flores, A. K. Sood, S. Huang, D. Yu, L. Yang and C. Lin, J. Clin. Invest., 2017, 127, 4498–4515 CrossRef.
- M. A. Cannarile, M. Weisser, W. Jacob, A.-M. Jegg, C. H. Ries and D. Rüttinger, J. Immunother. Cancer, 2017, 5, 53 CrossRef.
- E. Richardsen, R. D. Uglehus, S. H. Johnsen and L.-T. Busund, Anticancer Res., 2015, 35, 865–874 Search PubMed.
- I. Sánchez-González, A. Bobien, C. Molnar, S. Schmid, M. Strotbek, M. Boerries, H. Busch and M. A. Olayioye, Cancer Res., 2020, 80, 1330–1341 CrossRef.
- M. M. Williams, J. L. Christenson, K. I. O'Neill, S. A. Hafeez, C. L. Ihle, N. S. Spoelstra, J. E. Slansky and J. K. Richer, npj Breast Cancer, 2021, 7, 64 CrossRef CAS.
- Z. Meng, R. Zhang, Y. Wang, G. Zhu, T. Jin, C. Li and S. Zhang, Int. Immunopharmacol., 2020, 81, 106028 CrossRef CAS.
- J. Yang, Z. Zhang, C. Chen, Y. Liu, Q. Si, T. H. Chuang, N. Li, A. Gomez-Cabrero, R. A. Reisfeld, R. Xiang and Y. Luo, Oncogene, 2014, 33, 3014–3023 CrossRef CAS.
- W. Wang, Y. Liu, J. Guo, H. He, X. Mi, C. Chen, J. Xie, S. Wang, P. Wu, F. Cao, L. Bai, Q. Si, R. Xiang and Y. Luo, Oncogenesis, 2018, 7, 1–17 CrossRef.
- J. Xun, L. Du, R. Gao, L. Shen, D. Wang, L. Kang, C. a. Chen, Z. Zhang, Y. Zhang, S. Yue, S. Feng, R. Xiang, X. Mi and X. Tan, Theranostics, 2021, 11, 6847–6859 CrossRef CAS.
- S. Donzelli, E. Milano, M. Pruszko, A. Sacconi, S. Masciarelli, I. Iosue, E. Melucci, E. Gallo, I. Terrenato, M. Mottolese, M. Zylicz, A. Zylicz, F. Fazi, G. Blandino and G. Fontemaggi, Breast Cancer Res., 2018, 20, 59 CrossRef.
- Y. Tu, L. Liu, D. Zhao, Y. Liu, X. Ma, Y. Fan, L. Wan, T. Huang, Z. Cheng and B. Shen, Sci. Rep., 2015, 5, 13827 CrossRef.
- Y. Li, L. Zhao, B. Shi, S. Ma, Z. Xu, Y. Ge, Y. Liu, D. Zheng and J. Shi, Sci. Rep., 2015, 5, 18648 CrossRef CAS.
- S. Ghani, P. Riemke, J. Schönheit, D. Lenze, J. Stumm, M. Hoogenkamp, A. Lagendijk, S. Heinz, C. Bonifer, J. Bakkers, S. Abdelilah-Seyfried, M. Hummel and F. Rosenbauer, Blood, 2011, 118, 2275–2284 CrossRef CAS.
- S. Gery, S. Tanosaki, S. Bose, N. Bose, J. Vadgama and H. P. Koeffler, Clin. Cancer Res., 2005, 11, 3184–3190 CrossRef CAS.
- Y. Yuan, D. Anbalagan, L. H. Lee, R. P. Samy, M. K. Shanmugam, A. P. Kumar, G. Sethi, P. E. Lobie and L. H. K. Lim, Oncotarget, 2016, 7, 27007–27020 CrossRef.
- L. A. Moraes, S. Kar, S. L. Foo, T. Gu, Y. Q. Toh, P. B. Ampomah, K. Sachaphibulkij, G. Yap, O. Zharkova, H. M. Lukman, A.-M. Fairhurst, A. P. Kumar and L. H. K. Lim, Sci. Rep., 2017, 7, 17925 CrossRef.
- M. Yang, J. Chen, F. Su, B. Yu, F. Su, L. Lin, Y. Liu, J.-D. Huang and E. Song, Mol. Cancer, 2011, 10, 117 CrossRef CAS.
- X. Cai, Y. Yin, N. Li, D. Zhu, J. Zhang, C.-Y. Zhang and K. Zen, J. Mol. Cell Biol., 2012, 4, 341–343 CrossRef CAS.
- M. L. Squadrito, F. Pucci, L. Magri, D. Moi, G. D. Gilfillan, A. Ranghetti, A. Casazza, M. Mazzone, R. Lyle, L. Naldini and M. De Palma, Cell Rep., 2012, 1, 141–154 CrossRef CAS.
- M. Mei, Y. Ren, X. Zhou, X.-b. Yuan, L. Han, G.-x. Wang, Z. Jia, P.-y. Pu, C.-s. Kang and Z. Yao, Technol. Cancer Res. Treat., 2010, 9, 77–86 CrossRef CAS.
- S. Memczak, M. Jens, A. Elefsinioti, F. Torti, J. Krueger, A. Rybak, L. Maier, S. D. Mackowiak, L. H. Gregersen, M. Munschauer, A. Loewer, U. Ziebold, M. Landthaler, C. Kocks, F. le Noble and N. Rajewsky, Nature, 2013, 495, 333–338 CrossRef CAS.
- J. Greene, A.-M. Baird, L. Brady, M. Lim, S. G. Gray, R. McDermott and S. P. Finn, Front. Mol. Biosci., 2017, 4, 38 CrossRef.
- J. Salzman, C. Gawad, P. L. Wang, N. Lacayo and P. O. Brown, PLoS One, 2012, 7, e30733 CrossRef CAS.
- Z.-Q. Hu, S.-L. Zhou, J. Li, Z.-J. Zhou, P.-C. Wang, H.-Y. Xin, L. Mao, C.-B. Luo, S.-Y. Yu, X.-W. Huang, Y. Cao, J. Fan and J. Zhou, Hepatology, 2020, 72, 906 CrossRef CAS.
- X. Li, W. Yao, Y. Yuan, P. Chen, B. Li, J. Li, R. Chu, H. Song, D. Xie, X. Jiang and H. Wang, Gut, 2017, 66, 157–167 CrossRef CAS PubMed.
- Z. Zhong, Z. Wen and J. E. Darnell, Science, 1994, 264, 95–98 CrossRef CAS PubMed.
- K. Shuai, G. R. Stark, l. M. Kerr and J. E. Darnell, Science, 1993, 261, 1744–1746 CrossRef CAS.
- V. Toshchakov, B. W. Jones, P.-Y. Perera, K. Thomas, M. J. Cody, S. Zhang, B. R. G. Williams, J. Major, T. A. Hamilton, M. J. Fenton and S. N. Vogel, Nat. Immunol., 2002, 3, 392–398 CrossRef CAS PubMed.
- C. Zhang, X. Han, L. Yang, J. Fu, C. Sun, S. Huang, W. Xiao, Y. Gao, Q. Liang, X. Wang, F. Luo, W. Lu and Y. Zhou, Theranostics, 2020, 10, 10908–10924 CrossRef CAS PubMed.
- H. Song, Y. Yang, Y. Sun, G. Wei, H. Zheng, Y. Chen, D. Cai, C. Li, Y. Ma, Z. Lin, X. Shi, W. Liao, Y. Liao, L. Zhong and J. Bin, Mol. Ther., 2022, 30, 915–931 CrossRef CAS PubMed.
- S. J. Jenkins, D. Ruckerl, G. D. Thomas, J. P. Hewitson, S. Duncan, F. Brombacher, R. M. Maizels, D. A. Hume and J. E. Allen, J. Exp. Med., 2013, 210, 2477–2491 CrossRef CAS.
- J. Zhang, F. Cheng, G. Rong, Z. Tang and B. Gui, Bioengineered, 2021, 12, 8920–8930 CrossRef CAS.
- Y. Zhang, Y. Zhang, X. Li, M. Zhang and K. Lv, Int. J. Mol. Med., 2017, 39, 373–379 CrossRef CAS.
- H.-y. Shen, J.-l. Xu, W. Zhang, Q.-n. Chen, Z. Zhu and Y. Mao, npj Precis. Oncol., 2024, 8, 1–16 CrossRef.
- B. Zhou, Z. Mo, G. Lai, X. Chen, R. Li, R. Wu, J. Zhu and F. Zheng, J. Exp. Clin. Cancer Res., 2023, 42, 48 CrossRef CAS.
- J. S. Mattick, P. P. Amaral, P. Carninci, S. Carpenter, H. Y. Chang, L.-L. Chen, R. Chen, C. Dean, M. E. Dinger, K. A. Fitzgerald, T. R. Gingeras, M. Guttman, T. Hirose, M. Huarte, R. Johnson, C. Kanduri, P. Kapranov, J. B. Lawrence, J. T. Lee, J. T. Mendell, T. R. Mercer, K. J. Moore, S. Nakagawa, J. L. Rinn, D. L. Spector, I. Ulitsky, Y. Wan, J. E. Wilusz and M. Wu, Nat. Rev. Mol. Cell Biol., 2023, 24, 430–447 CrossRef CAS.
- Z. Xing, M. Zhang, J. Liu, G. Liu, K. Feng and X. Wang, Mol. Immunol., 2021, 138, 1–9 CrossRef CAS.
- J. Chen, Y. Zhou, M. Wu, Y. Yuan and W. Wu, Clin. Breast Cancer, 2023, 23, 546–560 CrossRef CAS.
- W.-X. Chen, D.-D. Wang, B. Zhu, Y.-Z. Zhu, L. Zheng, Z.-Q. Feng and X.-H. Qin, Aging, 2021, 13, 10415 CrossRef CAS.
- J. Guo, Z. Duan, C. Zhang, W. Wang, H. He, Y. Liu, P. Wu, S. Wang, M. Song, H. Chen, C. Chen, Q. Si, R. Xiang and Y. Luo, J. Immunol., 2020, 205, 2916–2925 CrossRef CAS.
- C. Zhang, S. Wei, S. Dai, X. Li, H. Wang, H. Zhang, G. Sun, B. Shan and L. Zhao, J. Immunother. Cancer, 2023, 11, e006230 CrossRef.
- S.-Q. Liu, Z.-Y. Zhou, X. Dong, L. Guo and K.-J. Zhang, Biosci. Rep., 2020, 40, BSR20200626 CrossRef CAS.
- S. Tao, Q. Chen, C. Lin and H. Dong, J. Exp. Clin. Cancer Res., 2020, 39, 1–17 CrossRef.
- R. Chaudhari, V. Patel and A. Kumar, Nanoscale Adv., 2024, 6, 2270–2286 RSC.
- Z. X. Chong, S. K. Yeap and W. Y. Ho, PeerJ, 2021, 9, e11165 CrossRef.
- J. Wang, T. Tian, X. Li and Y. Zhang, Molecules, 2022, 27, 6717 CrossRef CAS.
- S. Anwar, F. Mir and T. Yokota, Pharmaceutics, 2023, 15, 1130 CrossRef CAS.
- R. Chaudhari, S. Nasra, N. Meghani and A. Kumar, Sci. Rep., 2022, 12, 4713 CrossRef CAS.
-
S. Nasra, R. Chaudhari and A. Kumar, in Nanomedicine for Cancer Diagnosis and Therapy, ed. A. Malik, S. Afaq and M. Tarique, Springer Singapore, Singapore, 2021, pp. 115–132, DOI:10.1007/978-981-15-7564-8_6.
- S. Nasra, T. Shah, M. Bhatt, R. Chaudhari, D. Bhatia and A. Kumar, ACS Appl. Bio Mater., 2023, 6, 2886–2897 CrossRef CAS.
- S. Nasra, D. Bhatia and A. Kumar, Adv. Healthc. Mater., 2024, 13, 2400679 CrossRef CAS.
- M. Kumari, M. P. Purohit, R. Pahuja, S. Patnaik, Y. Shukla, P. Kumar and K. C. Gupta, Drug Delivery Transl. Res., 2019, 9, 1159–1188 CrossRef CAS.
- K. Li, L. Lu, C. Xue, J. Liu, Y. He, J. Zhou, Z. Xia, L. Dai, Z. Luo, Y. Mao and K. Cai, Nanoscale, 2020, 12, 130–144 RSC.
- P. Figueiredo, A. Lepland, P. Scodeller, F. Fontana, G. Torrieri, M. Tiboni, M.-A. Shahbazi, L. Casettari, M. A. Kostiainen, J. Hirvonen, T. Teesalu and H. A. Santos, Acta Biomater., 2021, 133, 231–243 CrossRef CAS.
- C. Chen, J. Ke, X. E. Zhou, W. Yi, J. S. Brunzelle, J. Li, E.-L. Yong, H. E. Xu and K. Melcher, Nature, 2013, 500, 486–489 CrossRef CAS.
- T. Gong, Z. Dong, Y. Fu, T. Gong, L. Deng and Z. Zhang, J. Mater. Chem. B, 2019, 7, 5861–5872 RSC.
- X. Li, J. Pan, Y. Li, F. Xu, J. Hou, G. Yang and S. Zhou, ACS Nano, 2022, 16, 5778–5794 CrossRef CAS.
- B. Li, T. Tan, W. Chu, Y. Zhang, Y. Ye, S. Wang, Y. Qin, J. Tang and X. Cao, Drug Delivery, 2022, 29, 75–88 CrossRef CAS.
- T. Zhang, H. Lip, C. He, P. Cai, Z. Wang, J. T. Henderson, A. M. Rauth and X. Y. Wu, Adv. Healthc. Mater., 2019, 8, e1900543 CrossRef.
- H. Peng, J.-H. Wang, F. Guo, F.-F. Zhu, Z.-J. Wen, H.-J. Zhong and D.-S. Liang, Colloids Surf., B, 2021, 197, 111442 CrossRef CAS.
- R. Kanasty, J. R. Dorkin, A. Vegas and D. Anderson, Nat. Mater., 2013, 12, 967–977 CrossRef CAS.
- S. Liang, J. Zheng, W. Wu, Q. Li, P. E. Saw, J. Chen, X. Xu, H. Yao and Y. Yao, Front. Pharmacol., 2018, 9, 1465 CrossRef CAS.
- S. Shen, Y. Zhang, K.-G. Chen, Y.-L. Luo and J. Wang, Mol. Pharmaceutics, 2018, 15, 3642–3653 CrossRef CAS.
- M. Zhang, L. Yan and J. A. Kim, Cancer Gene Ther., 2015, 22, 463–474 CrossRef CAS.
- Y. Song, C. Tang and C. Yin, Biomaterials, 2018, 185, 117–132 CrossRef CAS.
- F. Leonard, L. T. Curtis, A. R. Hamed, C. Zhang, E. Chau, D. Sieving, B. Godin and H. B. Frieboes, Cancer Immunol. Immunother., 2020, 69, 731–744 CrossRef CAS.
- A. Hu, X. Chen, Q. Bi, Y. Xiang, R. Jin, H. Ai and Y. Nie, Nanoscale, 2020, 12, 22615–22627 RSC.
- A. D'Urso, F. Oltolina, C. Borsotti, M. Prat, D. Colangelo and A. Follenzi, Pharmaceutics, 2023, 15, 1711 CrossRef PubMed.
- H. Qian, T. Zhou, Y. Fu, M. Guo, W. Yang, D. Zhang, W. Fang, M. Yao, H. Shi, C. Chai, W. Cheng, S. Ding and T. Chen, Mol. Ther. – Nucleic Acids, 2022, 27, 763–773 CrossRef CAS.
- Y. Wang, Z. Luan, C. Zhao, C. Bai and K. Yang, Eur. J. Pharm. Sci., 2020, 142, 105136 CrossRef CAS PubMed.
- Q. Guo, X. He, C. Li, Y. He, Y. Peng, Y. Zhang, Y. Lu, X. Chen, Y. Zhang, Q. Chen, T. Sun and C. Jiang, Adv. Sci., 2019, 6, 1901430 CrossRef CAS PubMed.
- A.-K. Fuchs, T. Syrovets, K. A. Haas, C. Loos, A. Musyanovych, V. Mailänder, K. Landfester and T. Simmet, Biomaterials, 2016, 85, 78–87 CrossRef CAS PubMed.
- C. B. Rodell, S. P. Arlauckas, M. F. Cuccarese, C. S. Garris, R. Li, M. S. Ahmed, R. H. Kohler, M. J. Pittet and R. Weissleder, Nat. Biomed. Eng., 2018, 2, 578–588 CrossRef CAS.
- X. Wei, L. Liu, X. Li, Y. Wang, X. Guo, J. Zhao and S. Zhou, J. Controlled Release, 2019, 313, 42–53 CrossRef CAS.
- X. Wang, X. Zhong, J. Li, Z. Liu and L. Cheng, Chem. Soc. Rev., 2021, 50, 8669–8742 RSC.
- S. Zanganeh, G. Hutter, R. Spitler, O. Lenkov, M. Mahmoudi, A. Shaw, J. S. Pajarinen, H. Nejadnik, S. Goodman, M. Moseley, L. M. Coussens and H. E. Daldrup-Link, Nat. Nanotechnol., 2016, 11, 986–994 CrossRef CAS PubMed.
- Z. Gu, T. Liu, C. Liu, Y. Yang, J. Tang, H. Song, Y. Wang, Y. Yang and C. Yu, Nano Lett., 2021, 21, 6471–6479 CrossRef CAS PubMed.
- L. Rao, S.-K. Zhao, C. Wen, R. Tian, L. Lin, B. Cai, Y. Sun, F. Kang, Z. Yang, L. He, J. Mu, Q.-F. Meng, G. Yao, N. Xie and X. Chen, Adv. Mater., 2020, 32, e2004853 CrossRef.
- T. Mocan, C. Matea, F. Tabaran, C. Iancu, R. Orasan and L. Mocan, J. Cancer, 2015, 6, 583–592 CrossRef.
- D. Li, M. Zhang, F. Xu, Y. Chen, B. Chen, Y. Chang, H. Zhong, H. Jin and Y. Huang, Acta Pharm. Sin. B, 2018, 8, 74–84 CrossRef.
- C.-C. Chang, T. K. Dinh, Y.-A. Lee, F.-N. Wang, Y.-C. Sung, P.-L. Yu, S.-C. Chiu, Y.-C. Shih, C.-Y. Wu, Y.-D. Huang, J. Wang, T.-T. Lu, D. Wan and Y. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 44407–44419 CrossRef CAS.
- C. Cui, K. Chakraborty, X. A. Tang, K. Q. Schoenfelt, A. Hoffman, A. Blank, B. McBeth, N. Pulliam, C. A. Reardon, S. A. Kulkarni, T. Vaisar, A. Ballabio, Y. Krishnan and L. Becker, Nat. Nanotechnol., 2021, 16, 1394–1402 CrossRef CAS.
- Y. Yang, T. Guo, J. Xu, Y. Xiong, X. Cui, Y. Ke and C. Wang, Int. J. Biol. Macromol., 2021, 189, 577–589 CrossRef CAS.
- D. Chen, J. Xie, R. Fiskesund, W. Dong, X. Liang, J. Lv, X. Jin, J. Liu, S. Mo, T. Zhang, F. Cheng, Y. Zhou, H. Zhang, K. Tang, J. Ma, Y. Liu and B. Huang, Nat. Commun., 2018, 9, 873 CrossRef.
- A. Ramesh, S. Kumar, D. Nandi and A. Kulkarni, Adv. Mater., 2019, 31, e1904364 CrossRef.
- Y.-R. Zhang, J.-Q. Luo, J.-Y. Zhang, W.-M. Miao, J.-S. Wu, H. Huang, Q.-S. Tong, S. Shen, K. W. Leong, J.-Z. Du and J. Wang, Small, 2020, 16, e2004240 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ,
Ramesh
Chaudhary
,
Ramesh
Chaudhary