
Fe(TCNQ)2 nanorod arrays: an efficient electrocatalyst for electrochemical ammonia synthesis via the nitrate reduction reaction†
Nilmadhab
Mukherjee
a,
Ashadul
Adalder
 a,
Narad
Barman
b,
Ranjit
Thapa
b,
Rajashri
Urkude
c,
Biplab
Ghosh
c and
Uttam Kumar
Ghorai
*a
a,
Narad
Barman
b,
Ranjit
Thapa
b,
Rajashri
Urkude
c,
Biplab
Ghosh
c and
Uttam Kumar
Ghorai
*a
aDepartment of Industrial Chemistry & Applied Chemistry, Swami Vivekananda Research Centre, Ramakrishna Mission Vidyamandira, Belur Math, Howrah - 711202, India. E-mail: uttam.indchem@vidyamandira.ac.in
bDepartment of Physics and Centre for Computational and Integrative Sciences, SRM University – AP, Amaravati 522240, Andhra Pradesh, India
cBeamline Development & Application Section, Bhabha Atomic Research Center, Trombay, Mumbai 400 085, India
First published on 2nd December 2023
Abstract
The electrochemical reduction of nitrate to ammonia (NO3RR) catalyzed by metal organic frameworks (MOFs) is a promising and efficient method for reducing nitrate pollution in water while simultaneously producing a valuable product, ammonia. Herein, we report the 3D nanoarray architecture of the metal organic complex Fe-(tetracyanoquinodimethane)2 Fe(TCNQ)2 as an efficient electrocatalyst that exhibits a high ammonia yield rate of 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 351.6 μg h−1 cm−2 and faradaic efficiency (FE) of 85.2% at −1.1 V vs. RHE and excellent catalytic stability up to 2 days. The excellent catalytic performance is evaluated by ATR-FTIR spectroscopy and a series of control experiments. Density functional-based theoretical calculations are carried out to identify Fe–N4 active sites in metal–organic network structures. This study showcases the advancement of transition metal-based organic frameworks as very effective electrocatalysts for the reduction of nitrate to ammonia (NH3).
351.6 μg h−1 cm−2 and faradaic efficiency (FE) of 85.2% at −1.1 V vs. RHE and excellent catalytic stability up to 2 days. The excellent catalytic performance is evaluated by ATR-FTIR spectroscopy and a series of control experiments. Density functional-based theoretical calculations are carried out to identify Fe–N4 active sites in metal–organic network structures. This study showcases the advancement of transition metal-based organic frameworks as very effective electrocatalysts for the reduction of nitrate to ammonia (NH3).
Introduction
Ammonia (NH3) is a vital chemical that is extensively manufactured on a global scale for its utmost importance in the fertilizer sector as well as a feasible intermediate form of hydrogen energy storage and a carbonless energy vector. Thus, to mitigate the energy crisis NH3 can exclusively serve as a carrier for the conveyance of renewable energy on a worldwide scale.1–3 The industrially exemplary Haber-Bosch process, which is energy demanding and produces an extensive amount of greenhouse gases, is the primary method for the large scale industrial production of NH3.4–6 A sustainable, environment-friendly, and energy efficient strategy for NH3 synthesis is the desperate need of the hour. The production of NH3 from N2via the nitrogen reduction reaction (NRR) has recently attracted an enormous amount of research interest.7–10 The bottlenecks of the NRR persist due to the low N2 solubility implying poor conversion efficiency and slow reaction kinetics because of the high stability of the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond (bond energy 941 kJ mol−1), as well as the competing hydrogen evolution reaction (HER) at the cathode surface. All these factors culminate into extremely low NH3 yield and faradaic efficiency (FE) and prevent the scalability process of the NRR.11–14 On the other end of the spectrum, nitrate (NO3−) ions stand out owing to their high solubility in water and a low dissociation energy (204 kJ mol−1) for the N
N bond (bond energy 941 kJ mol−1), as well as the competing hydrogen evolution reaction (HER) at the cathode surface. All these factors culminate into extremely low NH3 yield and faradaic efficiency (FE) and prevent the scalability process of the NRR.11–14 On the other end of the spectrum, nitrate (NO3−) ions stand out owing to their high solubility in water and a low dissociation energy (204 kJ mol−1) for the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond,2 hence promoting a reduction in the barrier for mass transfer and minimizing energy consumption during electrolysis, therefore making it an energetically more favorable process than the NRR. Nitrate (NO3−) is one of the most important forms of nitrogen in the environment. It is a key water pollutant which poses a serious threat to human health, causing cancer and methemoglobinemia.15–18 The electrochemical conversion of waste NO3− into valuable products in the form of N2/NH3 under mild reaction conditions offers a potential solution to the problem of nitrate and/or nitrite pollution.16,19–24 Additionally, this process presents an environmentally friendly and sustainable alternative to the conventional Haber-Bosch method for ammonia synthesis;25–27 hence, it is a hot topic in the field of electrocatalysis. The eight electron transfer process associated electrochemical NO3− reduction reaction (NO3RR) to NH3 is regarded as a promising means of converting NO3− without causing secondary contamination.15,27–34 Nevertheless, it is possible to mitigate the competitive hydrogen evolution reaction (HER) by designing a suitable electrocatalyst. Transition metal organic complexes, including cobalt phthalocyanine (CoPc),35,36 copper phthalocyanine (CuPc),10,37 and nickel phthalocyanine-based composites,14,38,39 are regarded as promising electrocatalysts where the transition metal center is the active center of the electrochemical reduction reactions and organic ring phthalocyanine inhibits the competing HER. Fe based single atom catalysts were extensively studied by various research groups for the NO3RR to ammonia under ambient conditions.27,28,40 Recently, Fe based conducting metal organic frameworks such as Fe-(tetracyanoquinodimethane)2 (Fe(TCNQ)2) have been studied as oxygen evaluation reaction electrocatalysts.41,42 However, to the best of our knowledge, Fe(TCNQ)2 nanostructures have not yet been studied as an effective electrocatalyst for the electrochemical NO3RR to NH3.
O bond,2 hence promoting a reduction in the barrier for mass transfer and minimizing energy consumption during electrolysis, therefore making it an energetically more favorable process than the NRR. Nitrate (NO3−) is one of the most important forms of nitrogen in the environment. It is a key water pollutant which poses a serious threat to human health, causing cancer and methemoglobinemia.15–18 The electrochemical conversion of waste NO3− into valuable products in the form of N2/NH3 under mild reaction conditions offers a potential solution to the problem of nitrate and/or nitrite pollution.16,19–24 Additionally, this process presents an environmentally friendly and sustainable alternative to the conventional Haber-Bosch method for ammonia synthesis;25–27 hence, it is a hot topic in the field of electrocatalysis. The eight electron transfer process associated electrochemical NO3− reduction reaction (NO3RR) to NH3 is regarded as a promising means of converting NO3− without causing secondary contamination.15,27–34 Nevertheless, it is possible to mitigate the competitive hydrogen evolution reaction (HER) by designing a suitable electrocatalyst. Transition metal organic complexes, including cobalt phthalocyanine (CoPc),35,36 copper phthalocyanine (CuPc),10,37 and nickel phthalocyanine-based composites,14,38,39 are regarded as promising electrocatalysts where the transition metal center is the active center of the electrochemical reduction reactions and organic ring phthalocyanine inhibits the competing HER. Fe based single atom catalysts were extensively studied by various research groups for the NO3RR to ammonia under ambient conditions.27,28,40 Recently, Fe based conducting metal organic frameworks such as Fe-(tetracyanoquinodimethane)2 (Fe(TCNQ)2) have been studied as oxygen evaluation reaction electrocatalysts.41,42 However, to the best of our knowledge, Fe(TCNQ)2 nanostructures have not yet been studied as an effective electrocatalyst for the electrochemical NO3RR to NH3.
In this work, a Fe(TCNQ)2 nanorod array has been fabricated on a copper foam (CF) substrate, which is a binder-free electrocatalyst-electrode assembly for NO3RR studies. FESEM observations confirm the distinct 3-dimensional (3D) nanoarray architecture. Extended X-ray absorption fine structure (EXAFS) spectra identify the distinctive Fe–N4 coordination environment which is the active center for the NO3RR. Hence, the 3D Fe(TCNQ)2/CF system delivered an ammonia yield rate 11351.6 μg h−1 cm−2 and FE 85.2% at −1.1 V vs. RHE along with electrocatalytic stability of more than 2 days. Attenuated total reflection–Fourier transform infrared (ATR-FTIR) spectroscopy confirms the various intermediate species, specifically *NO, *NH, and *NH2 formed during NH3 formation. Furthermore, density functional based theoretical calculations demonstrated the mechanism of the eight electron transfer associated NO3RR. Additionally, several control assays and isotopic labelling experiments confirm the source of nitrogen in the produced NH3, as well.
Results and discussion
The growth mechanism of the catalyst showcases (Fig. 1a) the feasibility of employing cationic exchange under low-temperature conditions to fabricate a nanorod array of Fe(TCNQ)2 on a copper foam (CF) substrate. | ||
| Fig. 1 (a) Growth mechanism of Fe(TCNQ)2/CF, (b, c) FESEM images of CuTCNQ/CF, (d) FESEM image of Fe(TCNQ)2/CF, and (e) TEM image of Fe(TCNQ)2/CF. | ||
Prior to this, a nanorod array of Cu(TCNQ) had been successfully synthesized on a copper foam substrate defined as Cu(TCNQ)/CF. The transformation of Cu(TCNQ)/CF into Fe(TCNQ)2/CF involved a controlled cationic exchange process, where Fe2+ ions effectively exchange Cu+ ions.42 This innovative Fe(TCNQ)2/CF nanorod array configuration served as a conductive electrode in the realm of electrochemical nitrate reduction, yielding notably enhanced performance in the context of the nitrate reduction reaction. The influence of the morphology of the resultant metal-TCNQ composites was systematically examined through scanning electron microscopy (SEM) and transmission electron microscopy (TEM). The FESEM depiction of the Cu(TCNQ) specimen unveils developed growth of Cu(TCNQ) nanorods achieved through the surface coverage of Cu foam, as demonstrated in Fig. 1b and c. Upon investigating the Fe(TCNQ)2 sample subsequent to the cationic exchange process, its distinct nanoarray architecture is observed to persist, as evident in Fig. 1d. And Fig. 1e shows a TEM image of Fe(TCNQ)2 where the nanorods are ∼150–160 nm in width.
To gain insight into the structural properties of the catalyst, we conducted X-ray diffraction (XRD) analysis. Fig. 2a shows the XRD patterns of both Cu(TCNQ)/CF and Fe(TCNQ)2/CF. The distinct peaks in the XRD plot correspond to the X-ray diffraction patterns of Cu(TCNQ) and Fe(TCNQ)2, thereby revealing important information about their crystal structures which is well matched by the previous data.42,43 The X-ray photoelectron spectroscopy (XPS) spectrum of Fe (TCNQ)2/CF is depicted in Fig. S1† and shows that the catalyst is composed of the elements Fe, C, N and Cu. The oxygen (O) element may appear due to material contamination or oxidation on the product surface.42 At about 398.6 eV, an XPS N 1s spectrum peak (Fig. 2b) is observed due to Fe–NC; also a shake-up peak comes at 400.1 eV in the Fe(TCNQ)2/CF system. The peaks with binding energy at 284.58 and 285.94 eV are produced by carbon bonds (C–H) and nitrile carbon bonds (C–N) in the TCNQ system, respectively (as shown in Fig. 2c). In addition, the unsaturated carbon bond produces a satellite at 288.1 eV. The two main binding energies for Fe 2p3/2 and Fe 2p1/2 are shown in Fig. 2d at 712.7 and 724.1 eV, respectively.41,42,44 To elucidate the distinctive Fe–N4 coordination and to unravel the intricate structural attributes of the catalytically active sites within the developed Fe(TCNQ)2/CF catalyst, a comprehensive investigation employing X-ray absorption spectroscopy (XAS), encompassing both X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) spectra, was carried out. Fig. 2e illustrates the XANES absorption edge of Fe in the Fe(TCNQ)2/CF catalyst, positioned intermediate to the reference standards of elemental Fe foil and Fe2O3. This positioning of the absorption edge signifies that the average valence state of Fe within the Fe(TCNQ)2/CF catalyst falls within the range of 0 to +3. The detailed structural insight derived from Fig. 2f showcases the k3-weighted Fourier transform (FT) of the Fe K-edge EXAFS spectra for Fe(TCNQ)2/CF, Fe foil, and Fe2O3. The distinct peak observed near 1.42 Å in the Fe(TCNQ)2/CF sample corresponds to Fe–N coordination, a significant indicator of the active sites for the NO3RR process. Contrastingly, the negligible presence of peaks at 2.19 Å and 2.55 Å, which signify Fe–Fe metal pair interaction, establishes the uniqueness of the Fe(TCNQ)2/CF catalyst.45,46 This analysis collectively points towards a practical coordination number of around 4 for Fe–N4 in the Fe(TCNQ)2/CF catalyst (Table S1†), strongly implicating its role as the dominant active site for the NO3RR.45,46 Additionally, a wavelet transform (WT) analysis was undertaken on the K3-weighted EXAFS signal to offer further clarity on atom backscattering, as depicted in Fig. 2g–i. In this context, the distinctive Fe foil exhibited a WT maximum at 7.3 Å−1, aligned with Fe–Fe interactions. Conversely, the reference Fe2O3 displayed two prominent WT maxima at 7.4 and 3.2 Å−1, corresponding to Fe–Fe and Fe–O interactions, respectively. Also, the Fe(TCNQ)2/CF catalyst showcased a singular WT maximum situated at 2.9 Å−1, unequivocally attributed to Fe–N4 interactions, thereby corroborating its structural significance in the catalytic process.45,46
 | ||
| Fig. 2 (a) XRD patterns of CuTCNQ/CF and Fe(TCNQ)2/CF, (b) high resolution XPS N 1s scan of Fe(TCNQ)2/CF, (c) high resolution XPS C 1s scan of Fe(TCNQ)2/CF, (d) high resolution XPS Fe 2p scan of Fe(TCNQ)2/CF, (e) XANES spectra of Fe(TCNQ)2/CF, Fe foil and Fe2O3, (f) k3-weighted FT-EXAFS spectra (R space fitting curves) corresponding to the Fe K-edge, and (g–i) wavelet transform contour plots for Fe foil, Fe(TCNQ)2/CF and Fe2O3. | ||
Evaluation of the catalytic efficiency of metal-TCNQ/CF electrodes regarding nitrate reduction was performed using a conventional three-electrode system (H-type cell), as detailed in the Experimental methods section. Validation of the electrocatalytic performance for NO3− reduction was established by observing the amplified current density within the linear sweep voltammogram (LSV) profile following the introduction of 0.2 M NaNO3 in 0.1 M Na2SO4 (illustrated in Fig. 3a). Furthermore, electrochemically active surface area (ECSA), obtained via double-layer capacitance (Cdl) assessment (Fig. 3b and S20), revealed significant insights. In particular, the Fe(TCNQ)2/CF configuration exhibited an Cdl and corresponding ECSA of 11.2 mF cm−2 and 252 cm2, which exceeded both CuTCNQ/CF (4.2 mF cm−2 and 94.5 cm2) and CF (0.43 mF cm−2 and 9.7 cm2), thereby confirming its higher internal factoring intrinsic reduction rate. To comprehensively assess the catalyst's catalytic proficiency, electrochemical impedance spectroscopy (EIS) plots were harnessed to elucidate its charge transfer (CT) kinetics. The parameters extracted from these measurements were effectively modeled using a Randles circuit, incorporating solution resistance (Rs) and charge transfer resistance (Rct), yielding an accurate representation of the Nyquist plots' complexities (Fig. 3c). Impressively, Fe(TCNQ)2/CF exhibited notably diminished Rct values in stark comparison to CF and CuTCNQ, indicative of the heightened conductivity and enhanced proton transfer facilitation intrinsic to Fe(TCNQ)2/CF, ultimately underscoring its superior nitrate reduction efficacy. For a comprehensive exploration of the ammonia production efficiency within the catalytic system, chronoamperometry (CA) experiments were performed over 1 h reduction time, as exemplified in Fig. S13.† After 1 h of testing, all electrolyte solutions were used for indophenol blue method for ammonia detection by UV-vis spectroscopy (details in the ESI†). The UV-vis spectra of the electrolyte solutions (after electrolysis) exhibited increasing peak intensities at different applied potentials (Fig. 3d–f). Fe(TCNQ)2/CF manifested an exceptional electrocatalytic prowess in catalyzing the NO3RR, achieving an impressive maximum faradaic efficiency (FE) of 85.2% at −1.1 V vs. RHE (Fig. 3g), accompanied by a peak ammonia yield rate of up to 11351.6 μg h−1 cm−2 at −1.1 V vs. RHE (Fig. 3h). Similarly, both the FE and ammonia yield rate of CuTCNQ/CF (84.8% & 10562 μg h−1 cm−2) and CF (67.3% & 5661 μg h−1 cm−2) outperformed expectations for the NO3RR.
 | ||
| Fig. 3 (a) LSV profile of the catalyst with and without nitrate containing electrolyte, (b) electrochemical double layer capacitance analysis of the electrocatalysts, (c) EIS plot of CF and CuTCNQ/CF, Fe(TCNQ)2/CF, (d–f) UV-vis absorption spectra of ammonia solution at various potentials (after certain dilution) of CF, CuTCNQ/CF and Fe(TCNQ)2/CF respectively, (g) bar chart of the NH3 yield rate at different potentials of the various electrocatalysts, (h) bar chart of FE (%) at various potentials of the various electrocatalysts, and (i) bar chart of TOF at various potentials of Fe(TCNQ)2/CF. | ||
These collective findings serve to underline the remarkable potential of Fe(TCNQ)2/CF as a superior electrocatalytic agent for nitrate reduction, signifying significant strides in the field of electrochemical nitrogen fixation. To quantitatively gauge catalytic activity, the turnover frequency (TOF, s−1) was meticulously calculated based on ammonia production rates at various potentials. The distinctive histograms of TOF plotted against potential in Fig. 3i vividly depict the ammonia generation rates at Fe sites within the Fe (TCNQ)2/CF systems. Specifically, at a potential of −1.1 V, Fe (TCNQ)2/CF exhibited a noteworthy peak TOF of 2.6 s−1. A series of control experiments were systematically conducted to unequivocally confirm the generation of ammonia during the nitrate reduction reaction (NO3RR) (Fig. 4a). Initially, no signal was observed for nitrate free electrolyte solution also and a moderate signal was discerned in a solution composed of 0.2 M NaNO3 and 0.1 M Na2SO4 in the absence of metal-TCNQ catalyst growth copper foam (CF). Subsequently, the CuTCNQ/CF system produced ammonia during the NO3RR. In parallel, the Fe (TCNQ)2/CF system evaluation within a solution composed of 0.1 M Na2SO4 and 0.2 M NaNO3 unambiguously established the occurrence of ammonia production during the NO3RR. To unequivocally establish the provenance of ammonia from NO3− ions, we conducted 1H–nuclear magnetic resonance (NMR) analysis employing a 14N and 15N-labeled NO3− electrolyte solution, showcased in Fig. 4b.16 The resultant 1H-NMR spectra depicted a distinctive doublet peak corresponding to 15NH4+, providing unambiguous confirmation of ammonia's genesis via the electrochemical reduction of nitrate ions (15N-labeled), eliminating any confounding influences of extraneous factors. Equally noteworthy, in experiments using a 14NO3− containing electrolyte, a distinct triplet peak corresponding to 14NH4+ further reaffirmed the specific nitrate-driven ammonia formation. Crucially, in scenarios where nitrate was absent within the electrolyte during the electroreduction process, the absence of detectable ammonia served as a definitive confirmation of the dependency on nitrate for ammonia production. Subsequent to the electrolytic process, we conducted attenuated total reflection-Fourier transform infrared (ATR-FTIR) spectroscopy (Fig. 4c). Evident within the FTIR spectrum was a deformation vibration signal at 1637 cm−1, which is characteristic of H2O. Remarkably, discernible peaks at 1795 cm−1, 1395 cm−1, and 1101 cm−1 were attributed to various intermediate species, specifically *NO, *NH, and *NH2. The primary formation of NH3 was conclusively ascertained through the prominent peak at 1530 cm−1.47–49 Intriguingly, spectral analysis revealed a direct correlation between applied potentials and ammonia intensity, substantiating the influence of potential on product yield. As relevant to catalyst evaluation, stability analysis is paramount. Obviously, a prolonged electrolysis test at −1.1 V was performed to measure the long-term stability of Fe(TCNQ)2/CF (Fig. 4d). Impressively, as depicted in Fig. 4d, a marginal decrease in current density was observed only after 50 h of continuous operation, which underscores the exceptional stability of Fe(TCNQ)2/CF under practical working conditions. Addressing reusability, an important aspect for practical application, the nitrate reduction protocol was subjected to ten consecutive cycles, each lasting one hour (illustrated in Fig. 4e). Also, the catalyst consistently maintained high faradaic efficiency (FE) and yield rates throughout these cycles, firmly establishing its recyclability for the NO3RR. The XRD pattern (Fig. S21†) and FESEM image (Fig. S22†) observed for post-electrolysis Fe(TCNQ)2/CF specimens (as depicted in respective figures) provide further compelling evidence of the extraordinary durability of our catalyst under operational conditions. This comprehensive analysis substantiates the absence of deleterious species and further underscores the catalyst's impeccable stability.
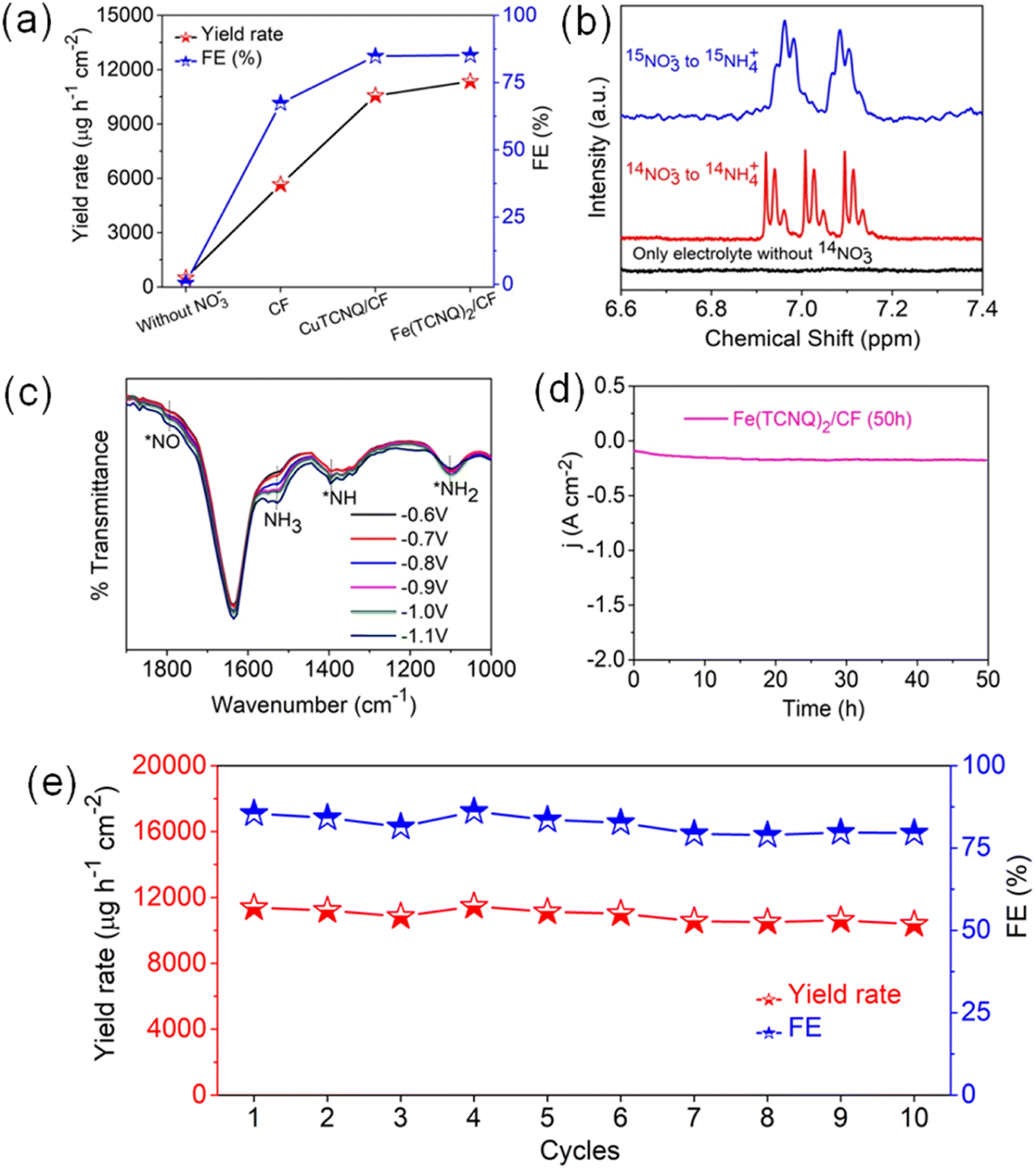 | ||
| Fig. 4 (a) Control experiments without NO3−, CF, CuTCNQ/CF, and Fe(TCNQ)2/CF for the NO3RR, (b) 1H-NMR for nitrate to ammonia production conformation using Fe(TCNQ)2/CF, (c) ATR-FTIR spectra at various potentials of electrolyte solution of Fe(TCNQ)2/CF, (d) long term stability curve of Fe(TCNQ)2/CF, and (e) recyclability test of Fe(TCNQ)2/CF. | ||
Theoretical studies of the NO3RR, on the catalyst Fe-TCNQ, have been carried out by introducing an optimized model structure of Fe-TCNQ (Fig. S23†), where the lattice parameters of the system are 6.88 Å, 11.27 Å and 20 Å along a, b and c axes respectively. Along the c-axis, a length of 20 Å is incorporated to nullify the false interactions due to the virtual image. The theoretical study comprises three sections; the first one is about checking for improved NO3 adsorption on the metal site of the catalyst over hydrogen adsorption and in the next part we have illustrated the reaction mechanism and corresponding free energy diagram with a possible pathway for the NO3RR. In the last section, we have discussed the catalytic activity of the given catalyst.
Since the HER is taking part as a major competitor, hampering the reaction phenomena parallelly by blocking active sites, it plays a crucial role while forming NH3 from NO3 reduction. We have found that the adsorption process of NO3, on the Fe-TCNQ catalyst, is more exothermic in nature than the same for hydrogen with free energy changes of −1.04 eV and 0.13 eV respectively as shown in Fig. 5a, indicating that reaction selectivity moves towards the NO3RR.50,51 Now, when NO3 comes and sits on the active metal site, it gets reduced by taking charges from valence orbitals of the metal atom. We have verified this reduction phenomenon quantitatively and qualitatively by using Bader charge analysis and a known parameter, called the d-band centre35 respectively. The charges associated with the metal atom, before and after adsorption of NO3−, are estimated to be 6.82e and 6.686e respectively. The shifting of the d-band centre from 2.73 eV to 2.35 eV, after adsorption of NO3− towards the Fermi level, points out that charge transfer occurred. To visualise the difference in charge density i.e., loss and gain of charges by respective atoms after NO3− adsorption, we have presented Fig. 5b and shifting of the d-band centre is depicted in Fig. 5c and d.
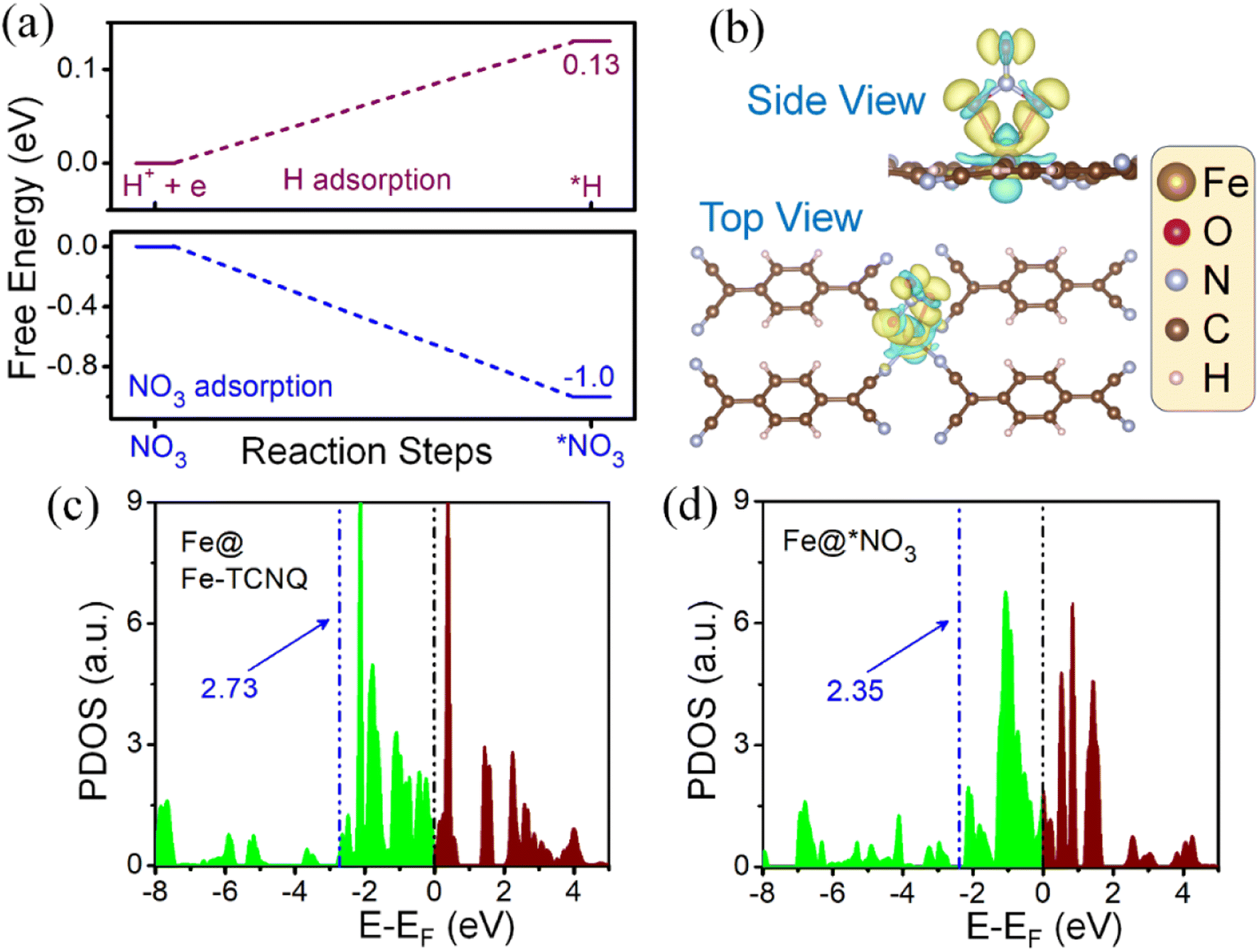 | ||
| Fig. 5 (a) Difference between free energy changes of NO3 and H adsorption respectively, (b) top and side views of the charge density difference with NO3 adsorbed on the metal centre, (c) partial density of states (PDOS) of d-electrons of the Fe atom before NO3 adsorption and (d) PDOS of d-electrons of the Fe atom after NO3 adsorption. *NO3 indicates the adsorbed case of NO3 on the metal centre. | ||
To understand the reaction mechanism of the NO3RR on the given catalyst, we have considered three known possible pathways (Fig. 6a), reported in the literature52 for metal surfaces and single atom catalysts etc., where protonation takes place at the 2nd step of the possible reaction pathway i.e., NO3− → *NO3 → NO3H and all the free energy calculations of the respective intermediates are improved by adding the correction term (EZPE-TS). In Fig. 6b–d, we have demonstrated the reaction mechanism, along the most favourable pathway i.e., Path I (chosen by the lowest ΔG of the potential determining step), and corresponding full free energy profile for the NO3RR respectively. From the free energy diagram, it is observed that NO3− comes and easily gets adsorbed on the active metal site and forms the intermediate *NO, by crossing a series of exothermic steps with a free energy change of −1.04 eV (step 1), −0.26 eV (step 2), −1.62 eV (step 3), −0.35 eV (step 4) and −1.63 eV (step 5) respectively. During this five step process, two H2O molecules are liberated into the electrolyte in the following way: *NO3H + 3H+ + 3e− → *NO + 2H2O. In the next step, protonation takes place in an endothermic way by the (H+ + e−) pair and forms the intermediate *NOH, which is recognized as the potential determining step (PDS),53 with a free energy change of 0.30 eV. After this, the (H+ + e−) pair attacks serially and causes the generation of a series of intermediates that follow Path I (*NOH → *NHOH → *NH → *NH2 → *NH3), where the first step is endothermic and the rest of the intermediates are formed spontaneously with a free energy change of 0.22 eV, −0.63 eV, −1.53 eV, and −0.79 eV respectively. At last, desorption of NH3 also, from the surface of the catalyst, takes place spontaneously. Now, estimation of the highest energy barrier known as the PDS for two pathways, along Path I of *NOH → *NHOH and Path III of *NOH → *N, tells us that Path I is more favourable over Path III.
 | ||
| Fig. 6 (a) Three possible pathways of forming NH3 from the NO3 reduction reaction on the Fe-TCNQ catalyst, (b) optimized structure of the reporting catalyst (Fe-TCNQ), (c) reaction mechanism of the NO3RR through Path I with optimized structures of intermediates and (d) the full free energy profile for the NO3RR to form NH3 by catalysing Fe-TCNQ along with three possible pathways. | ||
On the other side, the thermodynamic step *NHOH → *NH is more spontaneous than the step *NHOH → *NH2OH; hence there would be very little chance for Path II to occur. Therefore, based on the above discussion we can conclude that reduction of NO3− to NH3 on the Fe-TCNQ catalyst takes place smoothly through Path I over Path II and Path III. To find the catalytic activity of the proposed catalyst, there are so many factors to be investigated, e.g., limiting potential or the PDS, HER selectivity, change in free energy during desorption of NH3 from the catalyst surface etc. In this DFT study, we have obtained a lower value of limiting potential 0.30 V (Fig. 6d) and the given catalyst is observed to suppress the side reaction, HER phenomena that we already mentioned in the above section, resulting in higher selectivity towards the NO3RR. Besides, the change in free energy in the case of the desorption process of NH3 from the surface of the host catalyst is negative, −0.09 eV, which signifies that Fe-TCNQ is an efficient catalyst making the reduction process more feasible.
Conclusion
In summary, a 3D nanorod array of Fe(TCNQ)2 has been fabricated on a copper foam (CF) substrate using a controlled cationic exchange procedure for the electrochemical NO3RR. The phase purity, chemical constituents, and morphology of the nanostructures were confirmed by XRD, XPS, and FESEM studies, respectively. The local Fe–N4 coordination environment of the Fe(TCNQ)2 electrocatalyst was studied by EXAFS analysis. The chemically synthesized 3D Fe(TCNQ)2/CF system showed electrocatalytic stability for 2 days, with a maximum ammonia yield rate of 11351.6 μg h−1 cm−2 and FE of 85.2% at −1.1 V vs. RHE. Different intermediate species, including *NO, *NH, and *NH2, were confirmed by attenuated total reflection Fourier transform infrared (ATR-FTIR) spectroscopy. Density-functional-based theoretical computations confirmed active sites responsible for the NO3RR and possible reaction mechanisms too. Multiple tests confirming the source of nitrogen in the produced NH3 include control experiments and isotopic labeling experiments. This finding opens up new avenues to explore other transition metal-TCNQ-based electrocatalysts for the electrochemical NO3RR and NRR to NH3 synthesis.
Experimental section
Materials
7,7,8,8-Tetracyanoquinodimethane (TCNQ) (Merck), copper foam (Merck), FeSO4 (Merck), acetonitrile (Merck), methanol (Merck), and ethanol (Merck), HCl (Merck) were used. A Millipore system was utilised to purify the water used in each experiment.Preparation of CuTCNQ/CF
A meticulously pre-treated piece of copper foam that had its surface oxide and impurities removed using 1N HCl, ethanol, and deionized water multiple times was wetted in 6 mL of acetonitrile containing 8.58 mg of TCNQ for 2.5 hours at 60 °C. The colour of the solution changed to green, signifying the production of CuTCNQ. Before using, the sample was dried and washed multiple times with deionized water.Preparation of Fe(TCNQ)2/CF
By means of cationic exchange, Fe(TCNQ)2 nanorods on copper foam were synthesised. For four hours at 60 °C, the precursors were submerged in 20 mL of methanol that contained 20 mg of FeSO4. Deionized water was used to rinse the sample multiple times before it was dried.Materials characterization and electrochemical set-up
A Bruker D8 Advance Eco X-ray powder diffractometer apparatus with Cu-Kα radiation was used to perform the X-ray diffraction (XRD) investigation. A Shimadzu UV-3600 plus UV-vis spectrophotometer was used to acquire absorbance data. Shimadzu's IRAffinity-1S was used to get FTIR data. The surface morphology of the synthesized electrocatalysts was monitored using an FESEM (Zeiss-Germany) and transmission electron microscope (JEOL-JEM 2100F). All measurements involving the electrocatalytic reduction of nitrate to synthesise ammonia were performed using a CHI 760E electrochemical instrument, which featured a three-electrode system with Fe(TCNQ)2/CF acting as the working electrode, platinum (Pt) foil acting as the counter electrode, and Ag/AgCl (in 3.5 M KCl) acting as the reference electrode.Author contributions
UKG conceived the idea and designed the experiments. NM synthesized the catalyst and performed XRD, FTIR, and UV-vis. NM and AA carried out all the electrochemical measurements and analyzed the results. NB and RT performed the theoretical (DFT) calculations. RT, NB, UKG and NM analyzed the DFT results. NM and AA analyzed the NMR result. NM, RU, BG and AA analyzed the XANES and EXAFS. AA and NM analyzed the XPS results. NM and UKG wrote the original manuscript. All authors contributed to writing the final version of the manuscript. UKG supervised the project.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
NM acknowledges the DST INSPIRE fellowship for JRF (IVR Number: 202300000148 [IF220079]). AA acknowledges the SERG-CRG for JRF. NB thanks the University Grants Commission (UGC) for supporting the Fellowship under NFSC (No. F. 82-44/2020 (SA-III)). This work was financially supported by SERB (CRG/2022/009427) and SERB (CRG/2022/005423) research grants. The authors thank the High-Performance Computing Center, SRM Institute of Science and Technology, for providing computational facilities.References
- S. Giddey, S. P. S. Badwal, C. Munnings and M. Dolan, ACS Sustain. Chem. Eng., 2017, 5, 10231–10239 CrossRef CAS.
- S. Paul, A. Adalder and U. K. Ghorai, Mater. Chem. Front., 2023, 7, 3820–3854 RSC.
- S. L. Foster, S. I. P. Bakovic, R. D. Duda, S. Maheshwari, R. D. Milton, S. D. Minteer, M. J. Janik, J. N. Renner and L. F. Greenlee, Nat. Catal., 2018, 1, 490–500 CrossRef.
- S. Paul, S. Sarkar, D. Dolui, D. Sarkar, M. Robert and U. K. Ghorai, Dalton Trans., 2023, 52, 15360–15364 RSC.
- Nat. Synth. 2023, 2, 581–582, DOI:10.1038/s44160-023-00362-y.
- A. Adalder, S. Paul, B. Ghorai, S. Kapse, R. Thapa, A. Nagendra and U. K. Ghorai, ACS Appl. Mater. Interfaces, 2023, 15, 34642–34650 CrossRef CAS PubMed.
- Y. Fang, Y. Xue, L. Hui, X. Chen and Y. Li, J. Mater. Chem. A, 2022, 10, 6073–6077 RSC.
- A. Adalder, S. R. Waghela, S. A. Shelukar, N. Mukherjee, S. Das and U. K. Ghorai, Eng. Reports, 2023, e12705 Search PubMed.
- A. Biswas, S. Nandi, N. Kamboj, J. Pan, A. Bhowmik and R. S. Dey, ACS Nano, 2021, 15, 20364–20376 CrossRef CAS PubMed.
- J. Mukherjee, A. Adalder, N. Mukherjee and U. K. Ghorai, Catal. Today, 2023, 423, 113905 CrossRef CAS.
- B. H. R. Suryanto, H. L. Du, D. Wang, J. Chen, A. N. Simonov and D. R. MacFarlane, Nat. Catal., 2019, 24(2), 290–296 CrossRef.
- A. Adalder, S. Paul and U. K. Ghorai, J. Mater. Chem. A, 2023, 11, 10125–10148 RSC.
- C. Lee and Q. Yan, Curr. Opin. Electrochem., 2021, 29, 100808 CrossRef CAS.
- S. Murmu, S. Paul, S. Kapse, R. Thapa, S. Chattopadhyay, A. Nagendra, S. N. Jha, D. Bhattacharyya and U. K. Ghorai, J. Mater. Chem. A, 2021, 9, 14477–14484 RSC.
- G. F. Chen, Y. Yuan, H. Jiang, S. Y. Ren, L. X. Ding, L. Ma, T. Wu, J. Lu and H. Wang, Nat. Energy, 2020, 58(5), 605–613 CrossRef.
- S. Paul, S. Sarkar, A. Adalder, S. Kapse, R. Thapa and U. K. Ghorai, ACS Sustain. Chem. Eng., 2023, 11, 6191–6200 CrossRef CAS.
- W. J. Rogan, M. T. Brady, H. J. Binns, J. A. Forman, C. J. Karr, K. Osterhoudt, J. A. Paulson, J. R. Roberts, M. T. Sandel, J. M. Seltzer, R. O. Wright, E. Blackburn, M. Anderson, S. Savage, N. B. Ragan, P. Spire, J. A. Bocchini, H. H. Bernstein, J. S. Bradley, C. L. Byington, P. H. Dennehy, M. C. Fisher, R. W. Frenck, M. P. Glode, H. L. Keyserling, D. W. Kimberlin, W. A. Orenstein, L. G. Rubin, R. S. Baltimore, J. A. McMillan, B. Bell, R. Bortolussi, R. D. Clover, M. A. Fischer, R. L. Gorman, R. D. Pratt, J. S. Read, B. Gellin, J. R. Starke, J. Swanson, C. J. Baker, S. S. Long, L. K. Pickering, E. O. Ledbetter, H. C. Meissner and J. Frantz, Pediatrics, 2009, 123, e1123–e1137 CrossRef PubMed.
- M. Xie, S. Tang, Z. Li, M. Wang, Z. Jin, P. Li, X. Zhan, H. Zhou and G. Yu, J. Am. Chem. Soc., 2023, 145, 13957–13967 CrossRef CAS PubMed.
- S. Zhao, Z. Zheng, L. Qi, Y. Xue and Y. Li, Small, 2022, 18, 2107136 CrossRef CAS PubMed.
- Y. Li, J. Ma, Z. Wu and Z. Wang, Environ. Sci. Technol., 2022, 56, 8673–8681 CrossRef CAS PubMed.
- Y. Li, J. Ma, T. D. Waite, M. R. Hoffmann and Z. Wang, Environ. Sci. Technol., 2021, 55, 10695–10703 CrossRef CAS PubMed.
- J. Liang, Z. Li, L. Zhang, X. He, Y. Luo, D. Zheng, Y. Wang, T. Li, H. Yan, B. Ying, S. Sun, Q. Liu, M. S. Hamdy, B. Tang and X. Sun, Chem, 2023, 9, 1768–1827 CAS.
- Y. Li, X. He, J. Chen, X. Fan, Y. Yao, L. Ouyang, Y. Luo, Q. Liu, S. Sun, Z. Cai, S. Alfaifi, J. Du, B. Zheng and X. Sun, Chem. Commun., 2023, 59, 10805–10808 RSC.
- A. Adalder, S. Paul, N. Barman, A. Bera, S. Sarkar, N. Mukherjee, R. Thapa and U. K. Ghorai, ACS Catal., 2023, 13516–13527 CrossRef CAS.
- K. Fan, W. Xie, J. Li, Y. Sun, P. Xu, Y. Tang, Z. Li and M. Shao, Nat. Commun., 2022, 131(13), 1–13 Search PubMed.
- L. Xie, L. Hu, Q. Liu, S. Sun, L. Zhang, D. Zhao, Q. Liu, J. Chen, J. Li, L. Ouyang, A. A. Alshehri, Q. Kong and X. Sun, Inorg. Chem. Front., 2022, 9, 3392–3397 RSC.
- Z. Y. Wu, M. Karamad, X. Yong, Q. Huang, D. A. Cullen, P. Zhu, C. Xia, Q. Xiao, M. Shakouri, F. Y. Chen, J. Y. (Timothy) Kim, Y. Xia, K. Heck, Y. Hu, M. S. Wong, Q. Li, I. Gates, S. Siahrostami and H. Wang, Nat. Commun., 2021, 12, 1–10 CrossRef PubMed.
- P. Li, Z. Jin, Z. Fang and G. Yu, Energy Environ. Sci., 2021, 14, 3522–3531 RSC.
- X. Luan, L. Qi, Z. Zheng, S. Zhao, Y. Gao, Y. Xue and Y. Li, Chem. Commun., 2023, 59, 7611–7614 RSC.
- J. Xia, H. Du, S. Dong, Y. Luo, Q. Liu, J. S. Chen, H. Guo and T. Li, Chem. Commun., 2022, 58, 13811–13814 RSC.
- Y. Gao, H. Liu, Z. Zheng, X. Luan, Y. Xue and Y. Li, NPG Asia Mater., 2023, 151(15), 1–10 Search PubMed.
- L. Zhong, Q. Chen, H. Yin, J. Song Chen, K. Dong, S. Sun, J. Liu, H. Xian and T. Li, Chem. Commun., 2023, 59, 8973–8976 RSC.
- X. Fan, C. Liu, Z. Li, Z. Cai, L. Ouyang, Z. Li, X. He, Y. Luo, D. Zheng, S. Sun, Y. Wang, B. Ying, Q. Liu, A. Farouk, M. S. Hamdy, F. Gong, X. Sun and Y. Zheng, Small, 2023, 2303424 CrossRef CAS PubMed.
- S. Dong, A. Niu, K. Wang, P. Hu, H. Guo, S. Sun, Y. Luo, Q. Liu, X. Sun and T. Li, Appl. Catal., B, 2023, 333, 122772 CrossRef CAS.
- U. K. Ghorai, S. Paul, B. Ghorai, A. Adalder, S. Kapse, R. Thapa, A. Nagendra and A. Gain, ACS Nano, 2021, 15, 5230–5239 CrossRef CAS PubMed.
- S. Paul, S. Sarkar, A. Adalder, A. Banerjee and U. Kumar Ghorai, J. Mater. Chem. A, 2023, 11, 13249–13254 RSC.
- J. Mukherjee, S. Paul, A. Adalder, S. Kapse, R. Thapa, S. Mandal, B. Ghorai, S. Sarkar and U. K. Ghorai, Adv. Funct. Mater., 2022, 32, 2200882 CrossRef CAS.
- Y. Li, L. Ren, Z. Li, T. Wang, Z. Wu and Z. Wang, ACS Appl. Mater. Interfaces, 2022, 14, 53884–53892 CrossRef CAS PubMed.
- S. Murmu, S. Paul, A. Santra, M. Robert and U. K. Ghorai, Catal. Today, 2023, 423, 113938 CrossRef CAS.
- J. Xu, S. Zhang, H. Liu, S. Liu, Y. Yuan, Y. Meng, M. Wang, C. Shen, Q. Peng, J. Chen, X. Wang, L. Song, K. Li and W. Chen, Angew. Chem., Int. Ed., 2023, 62, e202308044 CrossRef CAS PubMed.
- M. Xie, X. Xiong, L. Yang, X. Shi, A. M. Asiri and X. Sun, Chem. Commun., 2018, 54, 2300–2303 RSC.
- X. Guo, S. Zhu, R. M. Kong, X. Zhang and F. Qu, ACS Sustain. Chem. Eng., 2018, 6, 1545–1549 CrossRef CAS.
- A. Pearson, R. Ramanathan, A. P. O’mullane, V. Bansal, A. Pearson, R. Ramanathan, V. Bansal and A. P. O’mullane, Adv. Funct. Mater., 2014, 24, 7570–7579 CrossRef CAS.
- P. Li, Y. Xuan, B. Jiang, S. Zhang and C. Xia, Electrochem. Commun., 2022, 134, 107188 CrossRef CAS.
- Z. Miao, X. Wang, M.-C. Tsai, Q. Jin, J. Liang, F. Ma, T. Wang, S. Zheng, B.-J. Hwang, Y. Huang, S. Guo, Q. Li, Z. Miao, J. Liang, F. Ma, T. Wang, Y. Huang, Q. Li, X. Wang, M. Tsai, B. Hwang, Q. Jin, S. Zheng and S. Guo, Adv. Energy Mater., 2018, 8, 1801226 CrossRef.
- Z. Liang, D. Yang, P. Tang, C. Zhang, J. Jacas Biendicho, Y. Zhang, J. Llorca, X. Wang, J. Li, M. Heggen, J. David, R. E. Dunin-Borkowski, Y. Zhou, J. R. Morante, A. Cabot and J. Arbiol, Adv. Energy Mater., 2021, 11, 2003507 CrossRef CAS.
- D. Pavia, G. Lampman, G. Kriz and J. Vyvyan, Introduction to spectroscopy Cengage Learning, 2008 Search PubMed.
- K. Chen, Z. Ma, X. Li, J. Kang, D. Ma and K. Chu, Adv. Funct. Mater., 2023, 33, 2209890 CrossRef CAS.
- Y. Yao, S. Zhu, H. Wang, H. Li, M. Shao, Y. Yao, S. Zhu, M. Shao, H. Li and H. Wang, Angew. Chem., Int. Ed., 2020, 59, 10479–10483 CrossRef CAS PubMed.
- A. Biswas, S. Kapse, B. Ghosh, R. Thapa and R. S. Dey, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2204638119 CrossRef CAS PubMed.
- S. Kapse, S. Narasimhan and R. Thapa, Chem. Sci., 2022, 13, 10003–10010 RSC.
- Z. Shu, H. Chen, X. Liu, H. Jia, H. Yan and Y. Cai, Adv. Funct. Mater., 2023, 33, 2301493 CrossRef CAS.
- S. Sinthika and R. Thapa, RSC Adv., 2015, 5, 93215–93225 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta05300h |
| This journal is © The Royal Society of Chemistry 2024 |